Impact Factor
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
INTRODUCTION
MATERIALS AND METHODS
RESULTS
DISCUSSION
CONCLUSIONS
SUPPLEMENTARY MATERIAL
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2011; 1:220-229. doi:10.7150/thno/v01p0220 This volume Cite
Research Paper
18F-Labeled GRPR Agonists and Antagonists: A Comparative Study in Prostate Cancer Imaging
Min Yang1,2*, Haokao Gao2, 3*, Yaru Zhou4, Ying Ma2, Qimeng Quan2, Lixin Lang2, Kai Chen2, Gang Niu2, Yongjun Yan2 ![]() , Xiaoyuan Chen2
, Xiaoyuan Chen2 ![]()
1. Key Laboratory of Nuclear Medicine, Ministry of Health, Jiangsu Key Laboratory of Molecular Nuclear Medicine, Jiangsu Institute of Nuclear Medicine, Wuxi, Jiangsu, 214063, China
2. Laboratory of Molecular Imaging and Nanomedicine (LOMIN), National Institute of Biomedical Imaging and Bioengineering (NIBIB), National Institutes of Health (NIH), Bethesda, MD 20892, USA
3. Department of Cardiology, Xijing Hospital, the Fourth Military Medical University, Xi'an, 710032, China
4. Department of Endocrinology, The Third Affiliated Hospital of Hebei Medical University, Shijiazhuang, 050051, China
* These authors contributed equally in this work.
Received 2011-1-17; Accepted 2011-3-6; Published 2011-3-7
Abstract
Radiolabeled bombesin analogs are promising probes for cancer imaging of gastrin-releasing peptide receptor (GRPR). In this study, we developed 18F-labeled GRPR agonists and antagonists for positron emission tomography (PET) imaging of prostate cancer. GRPR antagonists ATBBN (D-Phe-Gln-Trp-Ala-Val-Gly-His-Leu-NHCH2CH3) and MATBBN (Gly-Gly-Gly-Arg-Asp-Asn-D-Phe-Gln-Trp-Ala-Val-Gly-His-Leu-NHCH2CH3), and agonists AGBBN (Gln-Trp-Ala-Val-Gly-His-Leu-MetNH2) and MAGBBN (Gly-Gly-Gly-Arg-Asp-Asn-Gln-Trp-Ala-Val-Gly-His-Leu-MetNH2) were radiolabeled with 18F via 4-nitrophenyl 2-18F-fluoropropionate. The in vitro receptor binding, cell uptake, and efflux properties of the radiotracers were studied on PC-3 cells. An in vivo PET study was performed on mice bearing PC-3 tumors. Direct 18F-labeling of known GRPR antagonist ATBBN and agonist AGBBN did not result in good tumor targeting or appropriate pharmacokinetics. Modification was made by introducing a highly hydrophilic linker Gly-Gly-Gly-Arg-Asp-Asn. Higher receptor binding affinity, much higher cell uptake and slower washout were observed for the agonist 18F-FP-MAGBBN over the antagonist 18F-FP-MATBBN. Both tracers showed good tumor/background contrast, with the agonist 18F-FP-MAGBBN having significantly higher tumor uptake than the antagonist 18F-FP-MATBBN (P < 0.01). In conclusion, Gly-Gly-Gly-Arg-Asp-Asn linker significantly improved the pharmacokinetics of the otherwise hydrophobic BBN radiotracers. 18F-labeled BBN peptide agonists may be the probes of choice for prostate cancer imaging due to their relatively high tumor uptake and retention as compared with the antagonist counterparts.
Keywords: gastrin-releasing peptide receptor (GRPR), bombesin (BBN), agonist, antagonist, positron emission tomography (PET), 18F
INTRODUCTION
Peptide-based molecular imaging probes that target receptors over-expressed on cancer cells have attracted intensive research attention due to their fast clearance, excellent tissue penetration and low immunogenicity [1-3]. Among various tumor targeting peptides, bombesin (BBN) analogs are of particular interest because BBN targets gastrin-releasing peptide receptor (GRPR), which is overexpressed in a wide spectrum of human tumors, including prostate cancer, breast cancer, small cell lung cancer, ovarian cancers, endometrial cancers, and gastrointestinal stromal tumors [4-8]. In particular, radiolabeled BBN analogs [9-11] have been developed for imaging of prostate cancer, which has long been the leading cause of cancer related death in men in the US and Europe.
Positron emission tomography (PET) using radiolabeled peptides is of great interest in diagnostic nuclear medicine [12-13]. Compared to single-photon emission computed tomography (SPECT), PET is more quantitative and provides better spatial and temporal resolution. 18F with almost 100% positron efficiency, low β+ energy (0.64 MeV) and relatively short physical half-life (t1/2 = 109.7 min) is ideally suited for peptide labeling and PET imaging. Despite many advantages of PET and 18F-labeled molecular imaging probes, most BBN radiotracers reported to date are labeled with radiometals, such as 99mTc or 64Cu, for either SPECT or PET imaging [9-10]. We and others developed a number of 18F-labeled BBN radiotracers [14-17]. Unfortunately, the potential for clinical translation of these probes is limited due to their poor tumor/background contrast and unfavorable renal and hepatobiliary clearance.
Recent studies concluded that radiometal-labeled GRPR antagonists are remarkably better than the agonists. For example, the tumor uptake in PC-3 tumor-bearing mice is 4-fold, and the tumor/kidney ratio is 7.5-fold, for the 99mTc-labeled BBN receptor antagonist at 4 h post injection as compared with the corresponding 99mTc-labeled BBN receptor agonist [18]. A few other studies also focused on radiometal labeled GRPR antagonists [19-22]. Because the physical properties and labeling strategies of non-metallic radioisotopes, such as 18F, are quite different from those of radiometals, the generality of the conclusion that antagonists are better than agonists needs to be verified for other non-metal radioisotope-labeled BBN analogs. For this purpose, we developed both 18F-labeled BBN antagonists and agonists for side-by-side comparative studies. In contrast to the reported radiometal-labeled BBN derivatives, our 18F-labeled receptor agonist had much higher tumor uptake than the receptor antagonist. Herein, we would like to report the development of these radiotracers (Fig. 1) including comparative in vitro receptor-binding, cell uptake, and efflux studies on PC-3 cells and in vivo small PET imaging study of PC-3 tumor mice.
Structures of 18F-radiolabeled bombesin (BBN) receptor antagonist 18F-FP-MATBBN and agonist 18F-FP-MAGBBN.
MATERIALS AND METHODS
All chemicals were purchased, of analytical grade, and used as received without further purification, unless otherwise stated. BBN receptor antagonists, ATBBN (D-Phe-Gln-Trp-Ala-Val-Gly-His-Leu-NHCH2CH3) and MATBBN (Gly-Gly-Gly-Arg-Asp-Asn-D-Phe-Gln-Trp-Ala-Val-Gly-His-Leu-NHCH2CH3), and agonists AGBBN (Aca-Gln-Trp-Ala-Val-Gly-His-Leu-MetNH2) and MAGBBN (Gly-Gly-Gly-Arg-Asp-Asn-Gln-Trp-Ala-Val-Gly-His-Leu-MetNH2) were prepared via standard solid-phase Fmoc chemistry with an automatic peptide synthesizer (CS Bio). No-carrier-added 18F-F- was produced from an in-house GE Healthcare PETtrace cyclotron. C18 Sep-Pak cartridges (Waters) were pretreated with ethanol (5 mL) and water (10 mL) before use. The syringe filter and polyethersulfone membranes (pore size, 0.22 mm; diameter, 13 mm) were purchased from Nalge Nunc International. 125I-[Tyr4]BBN (74 TBq/mmol (2,000 Ci/mmol)) was purchased from Perkin-Elmer. Analytical and semi-preparative RP-HPLC (reversed-phase high-performance liquid chromatography) were performed on a Waters 600 chromatography system with a Waters 996 photodiode array detector and Beckman170 radioisotope detector. A C18 Vydac protein and peptide column (218TP510; 5 μm, 250 × 10 mm) was used for peptide purification. The flow was set at 5 mL/min using a gradient system starting from 95% solvent A (0.1% TFA [trifluoroacetic acid] in water) and 5% solvent B (0.1% TFA in ACN [acetonitrile]) (0-2 min) and ramped to 35% solvent A and 65% solvent B at 32 min. The flow rate of the analytical HPLC was set at 1 mL/min using the same gradient system, but with a C18 Vydac column (218TP54, 5 μm, 250 × 4.6 mm). The ultraviolet (UV) absorbance was monitored at 218 nm and the identification of the peptides was confirmed based on the UV spectrum acquired using a photodiode array detector and mass spectrometry.
Synthesis of FP-BBN Peptide Conjugates
Representative procedure: O-(N-succinimidyl)-1,1,3,3-tetramethyluronium tetrafluoroborate (TSTU, 13.7 μmol) and DIPEA (20 μL) were added to a solution of 2-fluoropropionic acid (FPA, 13.7 μmol) in DMF (200 μL). The mixture was heated at 60ºC for 30 min and the BBN peptide (13.7 μmol) was added. After 30 min, the reaction mixture was quenched with 2 mL of 5% HOAc. The crude FP-BBN peptide conjugate was purified by semi-preparative HPLC. The desired fractions were collected and lyophilized to give a white powder in >85% yield. The product identity and purity were confirmed by mass spectrometry (ESI-MS) and analytical HPLC: (FP-ATBBN, m/z 1058.58 for [MH]+ (C52H73FN13O10, calculated molecular weight [MW] 1058.21, HPLC retention time 23.4 min); FP-MATBBN, m/z 1614.91 for [MH]+ (C72H105FN23O19, calculated molecular weight [MW] 1614.79, HPLC retention time 19.4 min); FP-MAGBBN, m/z 1571.14 for [MH]+ (C66H101FN23O19S, calculated molecular weight [MW] 1570.71), HPLC retention time 17.2 min).
Radiochemistry
Representative procedure: 4-nitrophenyl 2-18F-fluoropropionate (18F-NFP) was prepared according to a previously reported procedure [23-24]. BBN peptide (500 μg) solution in DMSO (200 μL) was added to dried 18F-NFP in a 5 mL reaction vial, followed by the addition of DIPEA (20 μL). The reaction mixture was heated for 15 min at 40 ºC and was then quenched with 800 μL of 5% HOAc. The crude 18F-labeled peptide was purified by semi-preparative HPLC. The desired fractions were collected and diluted with 15 mL of water. The dilution was passed through a C18 cartridge, and the cartridge was washed with 5 mL of water. The purified 18F-labeled peptide was eluted with 1 mL of ethanol. The elution was blown dry with a stream of nitrogen at 40ºC. The dried product was formulated in normal saline and passed through a 0.22-μm Millipore filter into a sterile multidose vial for in vitro and in vivo experiments. The radiochemical yields from18F-NFP were 36.1 ± 2.8% (n = 3), 90.4 ± 4.6% (n = 3) and 78.0 ± 8.0% (n = 3) for 18F-FP-ATBBN, 18F-FP-MATBBN and 18F-FP-MAGBBN, respectively.
Cell Lines and Animal Models
A PC-3 human prostate carcinoma cell line purchased from American Type Culture Collection (ATCC) was used for in vitro study and preparation of animal models. PC-3 cells were grown in F-12K nutrient mixture (Kaighn's modification) (Invitrogen) which was supplemented with 10% (v/v) fetal bovine serum (Invitrogen) at 37ºC with 5% CO2. Animal procedures were performed according to a protocol approved by the Institutional Animal Care and Use Committee of the Clinical Center, National Institutes of Health. Subcutaneous injection of 5 × 106 tumor cells into the front flank of male athymic nude mice (Harlan) generated the PC-3 tumor model. When the tumor volume reached 100-300 mm3 (3-4 weeks after inoculation), the mice underwent small animal PET studies.
In Vitro Cell Binding Assay
The in vitro GRPR binding affinity of FPA-BBN peptide conjugates was measured via displacement cell-binding assays using 125I-[Tyr4]BBN as the radioligand. Experiments were performed on GRPR-expressing PC-3 human prostate carcinoma cells following a previously described method [25]. IC50 (the best-fit 50% inhibitory concentration) values were determined using GraphPad Prism 4 (GraphPad Software, Inc.) by fitting the data with nonlinear regression. Experiments were performed with triplicate samples.
Intracellular Calcium Mobilization
To ensure that the BBN analogs maintained their original agonist or antagonist characteristics after modification and 18F-labeling, an intracellular calcium mobilization study was conducted. Intracellular calcium mobilization was measured in PC-3 cells using Fluorometric Imaging Plate Reader (FLIPR) Calcium Assay Kits (Molecular Devices) according to the manufacturer's protocol. In brief, PC-3 cells (30,000 cells per well) were seeded in 96-well Costar black clear bottom plates (Corning) and cultured for 1 d at 37°C in a humidified atmosphere containing 5% CO2. After removal of the growth medium, cells were loaded with 30 μL per well FLIPRTETRA® dye containing 2.5 mM probenecid for 60 min at 37°C and 5% CO2. Changes in intracellular Ca2+ were monitored using FLIPR after different concentrations of BBN derivatives were added. The IC50 values were determined using GraphPad Prism 4 (GraphPad Software, Inc.) by fitting the data with nonlinear regression. Experiments were performed with triplicate samples.
Cell Uptake and Washout Studies
Cell uptake and washout studies of 18F-labeled BBN antagonists and agonists were performed with PC-3 cells. In the cell uptake experiment, PC-3 cells were seeded into 24-well plates at a density of 1 × 105 cells per well for overnight incubation. After incubation, PC-3 cells were washed 3 times with PBS (phosphate buffered saline) and then 18F-labeled BBN peptide was added in triplicate (~0.5 μCi/well). Re-incubation was followed at 37 °C. At 15, 30, 60, and 120 min time points, the PC-3 cells were rinsed 3 times with PBS and lysed with 0.1 M NaOH. The cell lysate was collected and the remaining radioactivity was measured in a γ counter (Packard, Meriden, CT). In the washout experiment, PC-3 cells were seeded into 24-well plates at a density of 1 × 105 cells per well before overnight incubation. After incubation, the PC-3 cells were rinsed 3 times with PBS and then 18F-labeled BBN peptide was added in triplicate (~0.5 μCi/well). After 2 h incubation at 37 ºC, the cells were washed with PBS, and then re-incubated in serum-free medium. At 0, 15, 30, 60 and 120 min, the cells were washed with PBS and lysed with 0.1 M NaOH. The cell lysate was collected and the remaining radioactivity was measured with a γ counter (Packard, Meriden, CT). The cell uptake and washout values were normalized in terms of added radioactivity and expressed as the percent added radioactivity. All experiments were performed with triplicate wells.
Small Animal PET Studies
Small animal PET scans and image data analysis were performed using an Inveon microPET (Siemens Medical Solutions). Mice bearing PC-3 tumors were injected under isoflurane anesthesia with ~3.7 MBq (100 μCi) of 18F-labeled BBN receptor antagonist or agonist via tail vein. Five-minute static PET images were acquired at 30 min, 1 h, and 2 h postinjection. The image reconstruction was done by the 2-dimensional ordered subsets expectation maximum (OSEM) algorithm without attenuation or scatter correction. For GRPR receptor-blocking experiment, [Lys3]BBN (15 mg/kg) was co-injected with 3.7 MBq of 18F-labeled BBN peptide into PC-3 tumor mice. The 5-min static microPET scans were acquired at 1 h post injection. Regions of interest (ROIs) over the tumor, normal tissue, and major organs were drawn on decay-corrected whole-body coronal images using vendor software (ASI Pro 5.2.4.0) for each microPET scan. The radioactivity concentration (accumulation) within a tumor or an organ was obtained from mean pixel values within the multiple ROI volume, which had been converted to MBq/mL/min by using a conversion factor. The conversion to MBq/g/ min assumed a tissue density of 1 g/mL. Imaging ROI-derived % ID/g was calculated by dividing the ROIs by the administered activity.
Statistical Analysis
Quantitative data are expressed as mean ± SD. Means were compared using 1-way ANOVA and the Student's t test. P values of < 0.05 were considered statistically significant.
RESULTS
Chemistry and Radiochemistry
Solid-phase peptide syntheses of BBN receptor antagonists ATBBN (D-Phe-Gln-Trp-Ala-Val-Gly-His-Leu-NHCH2CH3) and MATBBN (Gly-Gly-Gly-Arg-Asp-Asn-D-Phe-Gln-Trp-Ala-Val-Gly-His-Leu-NHCH2CH3), and agonists AGBBN (Aca-Gln-Trp-Ala-Val-Gly-His-Leu-MetNH2) and MAGBBN (Gly-Gly-Gly-Arg-Asp-Asn-Gln-Trp-Ala-Val-Gly-His-Leu-MetNH2) were performed using standard Fmoc chemistry. The FPA-peptide conjugates were synthesized by reacting TSTU activated 2-fluoropropionic acid with corresponding peptides in 70-95% yields. The resulting FPA-antagonist or FPA-agonist conjugates were purified by RP-HPLC. The purity and identity of products were confirmed by HPLC and ESI mass spectrometry. The 18F radiolabeling was conducted using previously described methods [26]. The radiolabeling yield was around 30% from 18F-NPF for unmodified ATBBN and 90% for MATBBN. The main reason that 18F-labeling of Gly-Gly-Gly-Arg-Asp-Asn linker-modified peptides gave higher radiolabeling yields than the unmodified peptides was that acylation of 18F-NPF with the amino group of phenylalanine in ATBBN encountered high steric hindrance, while the N-terminal glycine amine group in MATBBN was very reactive towards acylation. High chemical and radiochemical purity (>95%) were observed for all radiotracers. The specific activities at the end of synthesis were determined to be 40.5 ± 3.9 TBq/mmol, 58.2 ± 7.3 TBq/mmol and 48.3 ± 4.0 TBq/mmol for 18F-FP-ATBBN, 18F-FP-MATBBN and 18F-FP-MAGBBN, respectively.
Cell Binding Assay
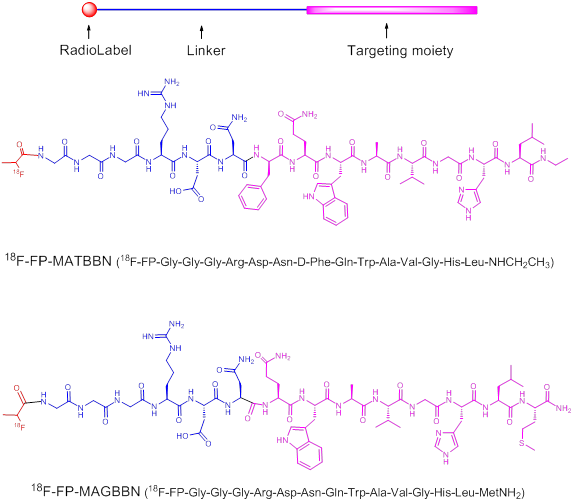
The binding affinities of BBN derivatives to GRPR were evaluated using 125I-[Tyr4]BBN as the radioligand in GRPR-positive PC-3 cells. Results of the cell-binding assay were plotted as sigmoid curves showing the displacement of 125I-[Tyr4]BBN from PC-3 cells as a function of increasing concentration of the BBN analogs. The IC50 values were determined to be 16.4, 65.3, 55.7, 94.3, 46.7 and 55.9 nM on PC-3 cells for ATBBN, MATBBN, FP-ATBBN, FP-MATBBN, MAGBBN and FP-MAGBBN, respectively (Fig. 2). Modification of the original peptide sequences with Gly-Gly-Gly-Arg-Asp-Asn linker and conjugation with FPA somewhat decreased the binding affinity of peptide analogs to GRPR.
Cell binding assay of BBN receptor antagonists and agonists (n = 3). Inhibition of 125I-[Tyr4]BBN (GRPR-specific) binding to GRPR on PC-3 cells by BBN receptor antagonists ATBBN, FP-ATBBN, MATBBN, FP-MATBBN (A) and agonists MAGBN and FP-MAGBBN (B).
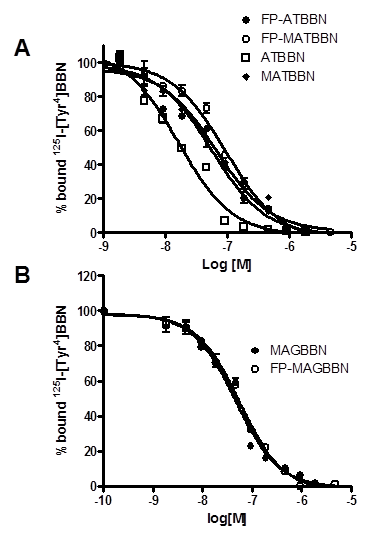
Intracellular Calcium Mobilization
The compounds were evaluated for their effect on signaling by using a calcium mobilization assay in PC-3 cells. As seen in Figure 3, MAGBBN and FP-MAGBBN stimulated calcium mobilization and behaved as receptor agonists with IC50 values of 0.18 and 0.17 µM, respectively. ATBBN, MATBBN and FP-MATBBN behaved as receptor antagonists without stimulating calcium mobilization. These results indicated that modification and 18F-labeling did not change the agonistic or antagonistic characteristics of the BBN analogs.
Intracellular calcium mobilization of ATBBN, MATBBN, FP-MATBBN, MAGBBN and FP-MAGBBN (n = 3).
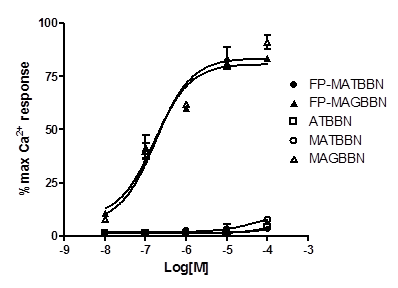
Cell Uptake Studies
The cell uptake of 18F-FP-MATBBN and 18FP-MAGBBN was evaluated in GRPR-expressing PC-3 tumor cells. Figure 4A shows the cell uptake results at 4 °C and 37 °C. All tracers had rapid binding to the cells. The uptake reached maximum at around 15 min and plateaued after a decrease. For BBN receptor agonist 18F-FP-MAGBBN, the cell uptake value at 37 °C was 2-fold higher than that at 4 °C (P < 0.01), suggesting that receptor-mediated internalization occurred at higher temperature. For BBN receptor antagonist 18F-FP-MATBBN, the cell uptake at different temperatures (4 °C and 37 °C) showed no significant difference (P > 0.05), which was consistent with previous reports that receptor antagonists do not internalize into cells. The PC-3 cell uptake peak values (at 37 °C, 15 min post-injection) of 18F-FP-MAGBBN agonist and 18F-FP-MATBBN antagonist were 1.2 ± 0.07% and 0.3 ± 0.07% (n = 3, mean ± SD), respectively (P < 0.01). The cell uptake values of BBN receptor agonist were significantly higher than those of antagonist, likely due to the receptor-mediated internalization of the agonists into PC-3 cells.
Cell uptake and washout assays of BBN receptor antagonists and agonists. (A) PC-3 cell uptake 18F-FP-MATBBN and 18F-FP-MAGBBN on PC-3 cells at 4°C and 37°C. (B) Loss of 18F-FP-MATBBN and 18F-FP-MAGBBN from PC-3 cells at 37°C. (n = 3, mean ± SD).
Much faster washout rate was observed for the BBN receptor antagonist 18F-FP-MATBBN compared to that of the agonist 18F-FP-MAGBBN when the labeled cells were incubated in serum-free medium devoid of radioactivity (Fig. 4B). The slower washout of the receptor agonist 18F-FP-MAGBBN was also likely due to the presence of internalized activity that did not readily efflux.
MicroPET Imaging
Around 3.7 MBq (100 μCi) of either 18F-FP-ATBBN, 18F-FP-MATBBN or 18F-FP-MAGBBN was injected intravenously into PC-3 tumor-bearing mice (n = 3/group for each tracer). The lipophilic 18F-FP-ATBBN antagonist showed almost no tumor uptake, but rather rapid and prominent activity accumulation in the liver and intestines (Fig. 5).
Decay-corrected whole-body coronal PET images of athymic male nude mice bearing PC-3 tumors at 1 h after injection of 3.7 MBq (100 μCi) of 18F-FP-ATBBN (A), 18F-FP-MATBBN (B), 18F-FP-AGBBN (C), and 18F-FP-MAGBBN (D). Arrows indicate PC-3 tumors.
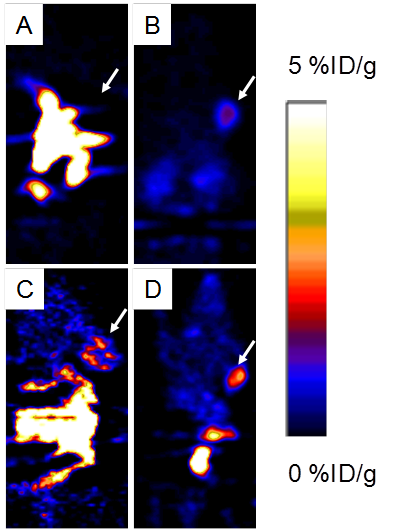
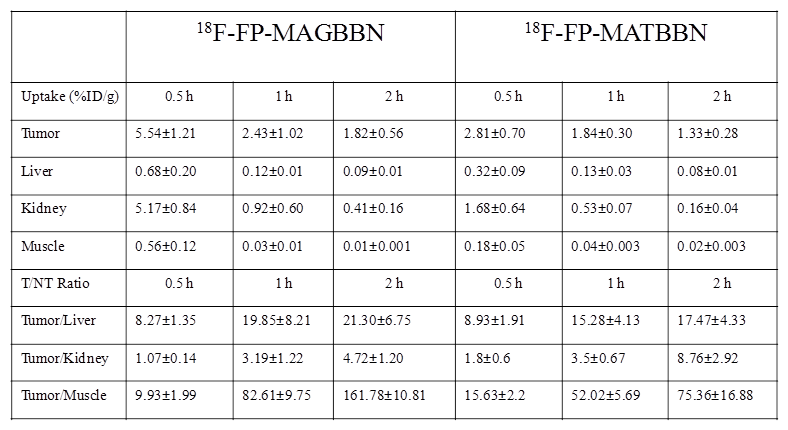
18F-FP-AGBBN agonist had some tumor accumulation, but also very high liver, bile and intestinal uptake. After the insertion of Gly-Gly-Gly-Arg-Asp-Asn linker, both 18F-FP-MATBBN (antagonist) and 18F-FP-MAGBBN (agonist) showed excellent images with high tumor background contrast. The distinct difference between linker-modified tracer and unmodified tracers clearly demonstrated that the Gly-Gly-Gly-Arg-Asp-Asn linker significantly improved the pharmacokinetics of the tracer. The ROI analysis results are summarized in Table 1. Both 18F-FP-MAGBBN and 18F-FP-MATBBN cleared rapidly from normal nontargeted organs with comparably high tumor/liver, tumor/kidney and tumor/muscle ratios. However, 18F-FP-MAGBBN agonist showed much higher tumor uptake than 18F-FP-MATBBN antagonist, especially at 30 min p.i. (P < 0.01).
Comparison of radioactivity accumulation (data obtained from microPET) in selected organs of PC-3 tumor-bearing nude mice after injection of 3.7 MBq (100 μCi) 18F-FP-MATBBN or 18F-FP-MAGBBN at different time points (n = 3/group, mean ± SD).
An in vivo blocking study also demonstrated that 18F-FP-MATBBN and 18F-FP-MAG BBN specifically target GRPR. Tumor uptake of 18F-FP-MAGBBN and 18F-FP-MATBBN in the presence of 15 mg/kg [Lys3]BBN decreased to 0.18 ± 0.01 and 0.08 ± 0.02% ID/g at 1 h time point, respectively.
DISCUSSION
Androgen-independent prostate cancer expresses GRPR. Molecular imaging probes based on GRPR targeting peptides, such as BBN derivatives, have thus attracted intensive research attention in the past decade. Although numerous radiolabeled BBN tracers have been well documented for prostate cancer imaging, very few 18F labeled BBN tracers have been reported, and none have translated successfully into the clinic. Previously, we developed an 18F-labeled BBN agonist tracer for prostate cancer imaging. However, its image quality was not very good in terms of tumor/background contrast [16]. We recently developed several hybrid peptide tracers based on dual GRPR and integrin receptor targeting peptide heterodimers [15, 17, 23, 27]. These heterodimers generally showed improved imaging qualities, but their applications were also limited, due to nonspecific targeting and tedious peptide synthesis.
The natural BBN peptide is a GRPR agonist, originally isolated from frog skin. Logically, most of the BBN-peptide-derived molecular probes were developed as receptor agonists. Recently a number of comparative studies of radiometal labeled BBN receptor antagonists and agonists have been reported [18, 20-21, 28-31], all of which suggest that the in vivo behavior of receptor antagonist is superior to receptor agonist, despite the fact that the antagonist has a lower receptor-mediated cell uptake and internalization in vitro. Similar behavior was also observed in other G-protein coupled receptor targeting systems such as somatostatin analogs [32]. We initially chose to label a known BBN receptor antagonist ATBBN (D-Phe-Gln-Trp-Ala-Val-Gly-His-Leu-NH2CH2CH3) [18, 20, 29-31, 33] with 18F-NFP. Unexpectedly, 18F-FP-ATBBN had virtually no PC-3 tumor uptake, even though this tumor is known to express high levels of GRPR. The agonist 18F-FP-AGBBN had some tumor uptake, but with much higher accumulation in the gallbladder and intestines, similar to what was reported for the same peptide labeled with an 18F-4-fluorobenzoyl (18F-FB) prosthetic group [16]. The poor in vivo fate of the 18F-labeled bombesin agonists and antagonists called for modification of the peptides to improve their pharmacokinetics and tumor targeting efficacy. Introduction of PEG, sugar moiety and oligo-glycine linkers has been applied to improve the in vivo kinetics of peptides [2-3]. Representative examples include Galacto-RGD [34] and Fluciclatide [35], which are currently being tested clinically. Previously, we have also successfully used PEG and oligo-Glycine linkers to improve the pharmacokinetics of RGD peptides [36-40]. We applied the similar strategy in this study and synthesized a number of linker-modified antagonists and agonists. Some of the synthesized compounds showed poor imaging quality and thus were discarded after the pilot experiments (see supporting information for the microPET imaging results of BBN tracers screened). A new peptide linker, Gly-Gly-Gly-Arg-Asp-Asn, with 6 hydrophilic amino acids and no net charge provided best results. This linker has 1) oligo-Glycine to reduce steric hindrance encountered during labeling; 2) oppositely charged Arg-Asp pair to increase hydrophilicity; and 3) Asn to act as a hydrophilic spacer. The attachment of this linker onto the known BBN derivatives and further radiolabeling led to somewhat less receptor binding and cell uptake in vitro. To our delight, this linker also led to much increased tumor uptake with relatively low background, as clearly delineated in the PET images.
Our findings from this study based on 18F-labeled BBN derivatives are different from the literature reports on 99mTc and 111In-labeled analogs. Radiometal (99mTc or 111In) labeled BBN receptor antagonists appear to have higher in vivo tumor uptake than their agonist analogs. In the case of 18F-labeled BBN agonists and antagonists, we found that the agonists showed greater receptor mediated internalization, significantly higher tumor uptake, and prolonged retention than did the antagonists. It is also of note that 18F-FP-MAGBBN and 18F-FP-MATBBN had similar tumor-to-background contrast, but 18F-FP-MAGBBN had beneficial, higher tumor uptake than 18F-FP-MATBBN, especially at early time points. With the same tumor/background contrast, higher tumor uptake means that less radioactivity is needed and a patient will be subjected to less radiation exposure in a clinical setting.
CONCLUSIONS
Unlike the case of radiometal-labeled chelator-conjugated bombesin derivatives, where antagonists outperform agonists, 18F-labeled agonists presented in this work are better than antagonists in terms of receptor-mediated accumulation in tumor areas. By using the new hydrophilic Gly-Gly-Gly-Arg-Asp-Asn linker, we are able to improve the PET image quality of both 18F-labeled agonists and antagonists. The same linker strategy may be applied to improve the pharmacokinetics of other hydrophobic peptide tracers.
SUPPLEMENTARY MATERIAL
Supplementary InformationAcknowledgements
This project was supported in part by the Intramural Research Program (IRP) of the National Institute of Biomedical Imaging and Bioengineering (NIBIB), National Institutes of Health (NIH), the International Cooperative Program of the National Science Foundation of China (NSFC) (81028009), and the Outstanding Professionals Foundation of the Jiangsu Health Bureau (Grant No. RC 2007096). We acknowledge the NIH/CC cyclotron facility for isotope production and Dr. Henry S. Eden for proof-reading this manuscript.
CONFLICT OF INTEREST
The authors have declared that no conflict of interest exists.
References
1. Benedetti E, Morelli G, Accardo A, Mansi R, Tesauro D, Aloj L. Criteria for the design and biological characterization of radiolabeled peptide-based pharmaceuticals. BioDrugs. 2004;18:279-95
2. Lee S, Xie J, Chen X. Peptide-based probes for targeted molecular imaging. Biochemistry. 2010;49:1364-76
3. Lee S, Xie J, Chen X. Peptides and peptide hormones for molecular imaging and disease diagnosis. Chem Rev. 2010;110:3087-111
4. Markwalder R, Reubi JC. Gastrin-releasing peptide receptors in the human prostate: relation to neoplastic transformation. Cancer Res. 1999;59:1152-9
5. Reubi JC, Korner M, Waser B, Mazzucchelli L, Guillou L. High expression of peptide receptors as a novel target in gastrointestinal stromal tumours. Eur J Nucl Med Mol Imaging. 2004;31:803-10
6. Fleischmann A, Waser B, Gebbers JO, Reubi JC. Gastrin-releasing peptide receptors in normal and neoplastic human uterus: involvement of multiple tissue compartments. J Clin Endocrinol Metab. 2005;90:4722-9
7. Sun B, Schally AV, Halmos G. The presence of receptors for bombesin/GRP and mRNA for three receptor subtypes in human ovarian epithelial cancers. Regul Pept. 2000;90:77-84
8. Fleischmann A, Waser B, Reubi JC. Overexpression of gastrin-releasing peptide receptors in tumor-associated blood vessels of human ovarian neoplasms. Cell Oncol. 2007;29:421-33
9. Ananias HJ, de Jong IJ, Dierckx RA, van de Wiele C, Helfrich W, Elsinga PH. Nuclear imaging of prostate cancer with gastrin-releasing-peptide-receptor targeted radiopharmaceuticals. Curr Pharm Des. 2008;14:3033-47
10. Schroeder RP, van Weerden WM, Bangma C, Krenning EP, de Jong M. Peptide receptor imaging of prostate cancer with radiolabelled bombesin analogues. Methods. 2009;48:200-4
11. Smith CJ, Volkert WA, Hoffman TJ. Radiolabeled peptide conjugates for targeting of the bombesin receptor superfamily subtypes. Nucl Med Biol. 2005;32:733-40
12. Reubi JC, Maecke HR. Peptide-based probes for cancer imaging. J Nucl Med. 2008;49:1735-8
13. Riccabona G, Decristoforo C. Peptide targeted imaging of cancer. Cancer Biother Radiopharm. 2003;18:675-87
14. Mu L, Honer M, Becaud J, Martic M, Schubiger PA, Ametamey SM. et al. In vitro and in vivo characterization of novel 18F-labeled bombesin analogues for targeting GRPR-positive tumors. Bioconjug Chem. 2010;21:1864-71
15. Liu Z, Yan Y, Chin FT, Wang F, Chen X. Dual integrin and gastrin-releasing peptide receptor targeted tumor imaging using 18F-labeled PEGylated RGD-bombesin heterodimer 18F-FB-PEG3-Glu-RGD-BBN. J Med Chem. 2009;52:425-32
16. Zhang X, Cai W, Cao F, Schreibmann E, Wu Y, Wu JC. et al. 18F-labeled bombesin analogs for targeting GRP receptor-expressing prostate cancer. J Nucl Med. 2006;47:492-501
17. Yan Y, Chen K, Yang M, Sun X, Liu S, Chen X. A new 18F-labeled BBN-RGD peptide heterodimer with a symmetric linker for prostate cancer imaging. Amino Acids. 2010 [Epub ahead of print]
18. Cescato R, Maina T, Nock B, Nikolopoulou A, Charalambidis D, Piccand V. et al. Bombesin receptor antagonists may be preferable to agonists for tumor targeting. J Nucl Med. 2008;49:318-26
19. Abiraj K, Mansi R, Tamma ML, Forrer F, Cescato R, Reubi JC. et al. Tetraamine-derived bifunctional chelators for technetium-99m labelling: synthesis, bioconjugation and evaluation as targeted SPECT imaging probes for GRP-receptor-positive tumours. Chemistry. 2010;16:2115-24
20. Abd-Elgaliel WR, Gallazzi F, Garrison JC, Rold TL, Sieckman GL, Figueroa SD. et al. Design, synthesis, and biological evaluation of an antagonist-bombesin analogue as targeting vector. Bioconjug Chem. 2008;19:2040-8
21. Mansi R, Wang X, Forrer F, Kneifel S, Tamma ML, Waser B. et al. Evaluation of a 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid-conjugated bombesin-based radioantagonist for the labeling with single-photon emission computed tomography, positron emission tomography, and therapeutic radionuclides. Clin Cancer Res. 2009;15:5240-9
22. Schroeder RP, Muller C, Reneman S, Melis ML, Breeman WA, de Blois E. et al. A standardised study to compare prostate cancer targeting efficacy of five radiolabelled bombesin analogues. Eur J Nucl Med Mol Imaging. 2010;37:1386-96
23. Liu S, Liu Z, Chen K, Yan Y, Watzlowik P, Wester HJ. et al. 18F-labeled galacto and PEGylated RGD dimers for PET imaging of αvβ3 integrin expression. Mol Imaging Biol. 2010;12:530-8
24. Sun X, Yan Y, Liu S, Cao Q, Yang M, Neamati N. et al. 18F-FPPRGD2 and 18F-FDG PET of response to Abraxane therapy. J Nucl Med. 2011;52:140-6
25. Chen X, Park R, Hou Y, Tohme M, Shahinian AH, Bading JR. et al. microPET and autoradiographic imaging of GRP receptor expression with 64Cu-DOTA-[Lys3]bombesin in human prostate adenocarcinoma xenografts. J Nucl Med. 2004;45:1390-7
26. Liu S, Liu Z, Chen K, Yan Y, Watzlowik P, Wester HJ. et al. 18F-labeled galacto and PEGylated RGD dimers for PET imaging of αvβ3 integrin expression. Mol Imaging Biol. 2010;12:530-8
27. Liu Z, Yan Y, Liu S, Wang F, Chen X. 18F, 64Cu, and 68Ga labeled RGD-bombesin heterodimeric peptides for PET imaging of breast cancer. Bioconjug Chem. 2009;20:1016-25
28. Abiraj K, Mansi R, Tamma ML, Forrer F, Cescato R, Reubi JC. et al. Tetraamine-derived bifunctional chelators for technetium-99m labelling: synthesis, bioconjugation and evaluation as targeted SPECT imaging probes for GRP-receptor-positive tumours. Chemistry. 2010;16:2115-24
29. Nock B, Nikolopoulou A, Chiotellis E, Loudos G, Maintas D, Reubi JC. et al. [99mTc]Demobesin 1, a novel potent bombesin analogue for GRP receptor-targeted tumour imaging. Eur J Nucl Med Mol Imaging. 2003;30:247-58
30. Schroeder RP, Muller C, Reneman S, Melis ML, Breeman WA, de Blois E. et al. A standardised study to compare prostate cancer targeting efficacy of five radiolabelled bombesin analogues. Eur J Nucl Med Mol Imaging. 2010;37:1386-96
31. Nock BA, Nikolopoulou A, Galanis A, Cordopatis P, Waser B, Reubi JC. et al. Potent bombesin-like peptides for GRP-receptor targeting of tumors with 99mTc: a preclinical study. J Med Chem. 2005;48:100-10
32. Ginj M, Zhang H, Waser B, Cescato R, Wild D, Wang X. et al. Radiolabeled somatostatin receptor antagonists are preferable to agonists for in vivo peptide receptor targeting of tumors. Proc Natl Acad Sci U S A. 2006;103:16436-41
33. Maina T, Nock BA, Zhang H, Nikolopoulou A, Waser B, Reubi JC. et al. Species differences of bombesin analog interactions with GRP-R define the choice of animal models in the development of GRP-R-targeting drugs. J Nucl Med. 2005;46:823-30
34. Haubner R, Kuhnast B, Mang C, Weber WA, Kessler H, Wester HJ. et al. [18F]Galacto-RGD: synthesis, radiolabeling, metabolic stability, and radiation dose estimates. Bioconjug Chem. 2004;15:61-9
35. Kenny LM, Coombes RC, Oulie I, Contractor KB, Miller M, Spinks TJ. et al. Phase I trial of the positron-emitting Arg-Gly-Asp (RGD) peptide radioligand 18F-AH111585 in breast cancer patients. J Nucl Med. 2008;49:879-86
36. Wu Z, Li ZB, Cai W, He L, Chin FT, Li F. et al. 18F-labeled mini-PEG spacered RGD dimer (18F-FPRGD2): synthesis and microPET imaging of αvβ3 integrin expression. Eur J Nucl Med Mol Imaging. 2007;34:1823-31
37. Chen X, Park R, Hou Y, Khankaldyyan V, Gonzales-Gomez I, Tohme M. et al. MicroPET imaging of brain tumor angiogenesis with 18F-labeled PEGylated RGD peptide. Eur J Nucl Med Mol Imaging. 2004;31:1081-9
38. Chen X, Hou Y, Tohme M, Park R, Khankaldyyan V, Gonzales-Gomez I. et al. Pegylated Arg-Gly-Asp peptide: 64Cu labeling and PET imaging of brain tumor αvβ3-integrin expression. J Nucl Med. 2004;45:1776-83
39. Shi J, Kim YS, Zhai S, Liu Z, Chen X, Liu S. Improving tumor uptake and pharmacokinetics of 64Cu-labeled cyclic RGD peptide dimers with Gly3 and PEG4 linkers. Bioconjug Chem. 2009;20:750-9
40. Liu Z, Niu G, Shi J, Liu S, Wang F, Chen X. 68Ga-labeled cyclic RGD dimers with Gly3 and PEG4 linkers: promising agents for tumor integrin αvβ3 PET imaging. Eur J Nucl Med Mol Imaging. 2009;36:947-57
Author contact
![]() Corresponding author: Email: shawn.chengov (X.C.); yyan26edu (Y. Y)
Corresponding author: Email: shawn.chengov (X.C.); yyan26edu (Y. Y)