Theranostics
13.3
Impact Factor
 Global reach, higher impact
Global reach, higher impact
Theranostics 2011; 1:263-273. doi:10.7150/thno/v01p0263 This volume Cite
Research Paper
Investigation of In Vivo Targeting Kinetics of αvβ3-Specific Superparamagnetic Nanoprobes by Time-Resolved MRI
Chase W. Kessinger1, Osamu Togao2, Chalermchai Khemtong1, Gang Huang1, Masaya Takahashi2, Jinming Gao1, ![]()
1. Department of Pharmacology, Harold C. Simmons Comprehensive Cancer Center;
2. Advanced Imaging Research Center, University of Texas Southwestern Medical Center, 5323 Harry Hines Boulevard, Dallas, Texas 75390, USA
Received 2011-2-9; Accepted 2011-3-7; Published 2011-4-24
Citation:
Kessinger CW, Togao O, Khemtong C, Huang G, Takahashi M, Gao J. Investigation of In Vivo Targeting Kinetics of αvβ3-Specific Superparamagnetic Nanoprobes by Time-Resolved MRI. Theranostics 2011; 1:263-273. doi:10.7150/thno/v01p0263. https://www.thno.org/v01p0263.htm
Other stylesAbstract
Nanoparticulate imaging probes have become an increasingly important arsenal in the visualization of molecular markers for early diagnosis and post-therapy assessment of diseases. Surface functionalization of these nanoparticles has led to the development of a variety of targeted nanoprobes for various imaging modalities (e.g. PET, MRI, optical). Despite these advances, detailed understanding of the nanoparticle targeting kinetics, particularly at the early time points immediately after injection, is still lacking. In this study, we report the combination of a T2*-weighted time-resolved-MRI (TR-MRI) method with ultra-sensitive superparamagnetic polymeric micelle (SPPM) nanoprobes to quantify the targeting kinetics of cyclic (RGDfK) (cRGD)-encoded SPPM to angiogenic endothelium in subcutaneous human tumor xenograft models in mice. TR-MRI analyses of the αvβ3-targeted and non-targeted SPPMs allowed for the subtraction of blood volume and extravascular signal components from the cRGD-SPPM data, resulting in a specific measurement of the accumulation kinetics of nanoprobes in lung, breast and brain cancer preclinical models. In all three models, αvβ3-specific accumulation of SPPM nanoprobes was observed in the first 5 mins after intravenous injection (first order rate constants were in the range of 0.22-0.24 min-1). Similar αvβ3-targeting kinetics was observed for cRGD-SPPM nanoprobes in different tumor xenograft models, consistent with the targeting of mouse angiogenic endothelium despite tumor inoculation from different human cancer cell lines. Results from this study offer new opportunities in the quantitative characterization of the targeting kinetics of cancer-specific nanoparticles to their intended biological targets in an intact animal, which provides fundamental insights on molecular recognition processes in vivo for further development of these nanoprobes.
Keywords: Cancer molecular imaging, αvβ3, time-resolved magnetic resonance imaging, nanoparticle targeting kinetics
INTRODUCTION
Nanosized imaging probes (e.g. quantum dots, iron oxide nanoparticles, radiolabeled dendrimers) have greatly impacted the field of molecular and cellular imaging for the early diagnosis and assessment of therapeutic efficacy in a variety of diseases [1-4]. Among many clinical imaging modalities (e.g. PET, SPECT, ultrasound), recent advances of ultra-sensitive nanoprobes have greatly broadened the capability of magnetic resonance imaging (MRI) in cancer molecular imaging applications. Compared to low-molecular weight T1 contrast agents, superparamagnetic iron oxide nanoparticles (i.e. Fe3O4, MnFe2O4, FeCo) have substantially larger molar relaxivities with detection limits in the nanomolar to picomolar concentration range [5-10]. Imaging of various cancer-specific biomarkers, such as αvβ3 and αvβ5 integrins, Her2-neu, transferrin and folate receptors have been reported [9, 11-17]. In most of these applications, pre-contrast images are first obtained, which is followed by injection of targeted nanoprobes and after certain time (typically several hours), post-contrast images are acquired by conventional T2/T2*-weighted methods to evaluate the accumulation of nanoprobes in the targeted tissues. Despite many successful reports on such contrast changes between pre- and post-injection images, very few studies have examined the fundamental kinetic processes of molecular targeting of cancer biomarkers in vivo. Characterization of the early binding events and uptake of the nanoprobes in targeted cancer cells will provide important information on the critical factors that affect nanoparticle accumulation and tumor/normal tissue contrast in vivo. This knowledge will be particularly important for the development of vascular-targeted contrast agents, where nanoprobes can directly bind to the cell surface receptors of the tumor endothelium.
Dynamic contrast enhanced (DCE) MRI has been clinically used in cancer detection, diagnosis, stratification and assessment of anti-angiogenic therapy [18]. By acquiring T1-weighted images at a temporal resolution of seconds during the administration of non-specific, low-molecular weight T1 contrast agents (e.g. Gd-DTPA), DCE-MRI allows for measurements of pharmacokinetic parameters which represent the volume transfer constant (Ktrans, a combination of vascular flow, vessel surface area and permeability), the fractional volume of the extravascular extracellular space (ve) and other transport parameters in benign and malignant tumor tissues [19, 20]. Although Gd-based small molecular agents work well in tumor perfusion studies, these agents are not very sensitive, requiring millimolar (10-3 M) concentrations for detection, and therefore are ineffective for the specific visualization of protein biomarkers for molecular imaging in vivo [21].
In this study, we describe the use of T2*-weighted time-resolved (TR)-MRI method with 1.3 s temporal resolution over 30 mins to investigate the early phase of the targeting kinetics of an αvβ3-specific superparamagnetic nanoprobes in different tumor xenografts in athymic nude mice. The nanoprobe design is based on the superparamagnetic polymeric micelles (SPPM) that have been previously established in our lab as αvβ3 integrin-specific molecular imaging probes [9, 22]. Micelle nanoparticles have several unique advantages in their application as targeted MRI nanoprobes. Their core-shell architecture allows for the loading of a cluster of superparamagnetic iron oxide (SPIO) nanoparticles that were shown to increase the transverse relaxivity of the nanoprobe to over 400 Fe mM-1 s-1 at 7T [22-24].This increase in T2 relaxivity is 4 fold higher than that of Feridex®, a dextran-coated SPIO formulation currently used in the clinics [25]. Integrin αvβ3 is an established biomarker of angiogenesis, which is highly expressed on active endothelial cells during tumor angiogenesis, and has low to no expression in resting endothelium. Using nanoprobes conjugated with a cyclic RGDfK (cRGD) peptide, an αvβ3-specific ligand, or a non-targeting cyclic RADfK (cRAD) control, we evaluated the change of MR signal intensity (SI) over time at different angiogenic “hot spots” in subcutaneous tumor xenografts of human lung, breast and glioblastoma cancers. Comparison of the targeted and non-targeted SPPM data allows for the subtraction of signal intensity contributions from blood concentration, clearance and enhanced permeability and retention (EPR) effect. The net TR-MRI temporal profiles permit the assessment of the specific targeting kinetics of cRGD-encoded SPPM to αvβ3-expressing tumor endothelial cells in vivo.
MATERIALS AND METHODS
Production of cRGD- and cRAD-SPPM. Methoxy- and maleimide-terminated poly(ethylene glycol)-b-poly(D,L-lactide) (MeO-PEG-PLA and Mal-PEG-PLA), tetramethylrhodamine (TMR)-conjugated MeO-PEG-PLA-TMR copolymers and SPIO nanoparticles were synthesized following previously published procedures [9, 14, 26, 27]. All copolymers had a molecular weight of 10 kD with 5 kD PEG and PLA blocks. The diameter of SPIO nanoparticles was 9.0 ± 0.9 nm by transmission electron microscopy (TEM, JEOL 1200 EX at 120 KeV). SPPMs were prepared by a solvent evaporation method [14]. Briefly, a mixture of MeO-PEG-PLA, MeO-PEG-PLA-TMR, Mal-PEG-PLA and SPIO in THF was added dropwise to 0.05 M HEPES buffer containing 0.01 M EDTA (pH 7.4) under sonication. The micelle suspension was then equally divided into two parts, and then shaken on an orbital shaker for 4 hrs to allow THF to evaporate. A solution of either thiol-terminated cRGD or cRAD was added to the micelle solution. The solutions were then shaken overnight for conjugation of peptides to maleimide-terminated micelles. Thiol-terminated cRGD and cRAD were obtained by deprotections of thioacetate-protected cRGDfK and cRADfK (Peptides International, Louisville, KY), followed by preparative HPLC purifications. All SPPM formulations were filtered through 1.0 µm nylon syringe filters and concentrated using centrifugal filters (100 kDa cutoff, Millipore, Billerica, MA).
Animal tumor models. Animal studies were approved by the Institutional Animal Care and Use Committee at UT Southwestern Medical Center and carried out according to its guidelines. Female athymic nude mice (27-30g) were used. The subcutaneous tumor xenografts were formed from non-small cell A549 lung cancer, MDA-MB-231 breast cancer, or U87 glioblastoma cell lines. For each tumor model, cells were injected subcutaneously into the dorsal flank of the mouse. Five million A549, five million matrigel-supplemented (20%) MDA-MB-231, and one million U87 cells were injected in 100 µL of phosphate buffered saline (PBS) to start tumor inoculation in their respective groups. Tumors were allowed to grow to 300-500 mm3 at which time they were randomized into cRGD- and cRAD-SPPM treatment groups (n = 3 each) for MR imaging. Statistical analysis was calculated using the Student's two-tailed t test with P values ≤ 0.05 considered statistically significant.
MR imaging. MRI studies were conducted in a 7T horizontal bore small animal MRI scanner (Varian Inc., Palo Alto, CA). All mice were anesthetized with 1-2% isoflurane mixed with pure oxygen via a nose cone and were placed in a stretched supine position with a respiratory sensor. SPPM was administered via a tail vein catheter at a dose of 10 mg Fe/kg or 0.18 mmol Fe/kg. The catheter was made in-house with a 27G 0.5 inch butterfly needle with PE10 tubing (50 cm in length) to allow for injection during the TR-MR imaging acquisition. The average dead volume of the catheter was 100-125 µL. The total injected volume for these experiments ranged from 400-500 µL.
Axial and coronal two-dimensional (2D) fast spin-echo sequence images were first acquired as scout imaging to ensure the imaging position of the implanted tumor. The imaging parameters were: TR/TE =1000/5.3 ms, bandwidth = 100 kHz, FOV = 35 x 35 mm, matrix size = 128 x 128, slice thickness = 1 mm, 17 slices with no gap, and NEX = 1. The image slice with the maximal tumor cross-section was chosen for subsequent TR-MRI studies.
TR-MRI images were acquired at a single slice position using the T2*-weighted method with a temporal resolution of 1.3 s over 30 mins. The other imaging parameters were: TR/TE = 10/3 ms, flip angle = 45°, FOV = 35 x 35 mm, matrix size = 128 x 128, slice thickness = 1 mm, and NEX = 1. Thirty seconds after the initial start of the TR-MRI sequence, a SPPM bolus was administered manually over 30-45 s, after which the line was flushed with saline (total injection time about 75 s). The serial images, In, were normalized by dividing signal intensity (SI) of each image by an averaged value from 5 consecutive pre-injection images collected within the first 15 s of the TR-MRI sequence (Figure 1). Hot spot regions-of-interest (ROIs) were selected in tumor sections where SI changed greater than 20% over the 30 min scan time compared to pre-injection SI values. For targeting kinetic analysis, pre-injection images within the first 15 s were averaged to calculate the initial SI(0). Post-injection images yielded SI(t) at any given time. The nanoprobe concentrations (CSPPM) were estimated using the following equation: CSPPM ∝ ΔR2* = -ln[SI(t)/SI(0)]/TE [28]. Three ROIs from each tumor xenograft were analyzed and averaged. Three animals were used in each imaging subgroup. Images were analyzed using ImageJ software (National Institutes of Health, NIH).
In vivo pharmacokinetic and TEM studies of SPPM. Experiments involving radioactive materials were approved by the Radiation Safety Committee at UT Southwestern Medical Center. Female athymic nude mice with MDA-MB-231 tumors (same as those above) were used for all radioactive pharmacokinetic studies. 3H (or T)-labeled cRGD- and cRAD-encoded SPPMs were prepared from 75% MeO-PEG-PLA-C(O)CT3 and 25% MAL-PEG-PLA. For the longer term (i.e. 24 hrs) pharmacokinetic studies, 3H-labeled SPPM solutions were injected via the tail vein (n=3 for each SPPM group). Blood was collected via ocular vein at 1 min, 1, 2 , 4 , 8 , 12, and 24 hrs after the injection. Plasma was isolated from whole blood by centrifugation at 1000 rpm for 10 mins. The plasma was subsequently mixed with a tissue solubilizer solution (1 mL, BTS-450, Beckman, CA) at room temperature for 5 hrs followed by an addition of a liquid scintillation cocktail (10 mL, Ready Organic™, Beckman, CA) for 12 hrs. Amount of radioactive isotope was measured by a liquid scintillation counter (Beckman LS 6000 IC).
For the short term pharmacokinetic studies, 20 µL plasma samples from the mouse were obtained at 1, 10, 20, 40 and 60 mins after injection of cRGD-SPPM (10 mg Fe/kg) (n=4). Samples were digested in concentrated HCl overnight and analyzed for Fe content using atomic absorption spectroscopy (Varian SpectrAA 50, Varian) using Fe standards as a calibration curve. Plasma samples for TEM analyses were collected 40 mins post-injection and placed on the carbon grid, blotted and imaged at 120 keV. The negatively stained TEM samples were prepared with 2% phosphotungstic acid (PTA) prior to analysis.
Histological analysis. After MR imaging, mice were injected with Hoechst 33342 (10 mg/kg) via the tail vein. The dye was allowed to circulate for 1 minute. The tumor tissue was resected and embedded in optimal cutting temperature medium and flash frozen. Tissue sections were collected at 8 µm thickness on a Leica cryostat (model 3050S) and then fixed with -20 °C acetone, mounted and coverslipped. Fluorescence micrographs were taken on an upright Leica microscope (model 5500DM) with proper excitation and emission filters for tetramethylrhodamine dye (λex = 515-560 nm, λem = 580-610 nm) and Hoechst (λex = 340-380 nm, λem = 450-490 nm).
RESULTS
SPPM characterization. Spherical SPPM nanoprobes encoded with cRGD or cRAD peptides were produced and characterized according to published procedures (Fig. 1A; ref. [9]). Nanoprobes had a size distribution of 57±12 and 53±10 nm for cRGD- and cRAD-SPPM, respectively by dynamic light scattering analysis (data not shown). All cRGD- and cRAD-SPPM formulations had a mean transverse relaxivity of 406.6 ± 9.1 Fe mM-1s-1 at 7 T and less than 10% difference in fluorescence emission properties.
Representative in vivo TR-MRI data. Figure 1B illustrates representative dataset in mice bearing subcutaneous A549 non-small cell lung tumor xenografts. The images are marked with color-coded regions-of-interest (ROIs) that correspond to the like-colored signal intensity curves with the tumor margins outlined in yellow (Fig. 1B, lower panel). Baseline images showed little to no SI difference in all tissues before injection. After injection (Fig. 1B, lower panel, dashed line), major blood vessels (systemic circulation) showed a maximum SI loss for both cRGD- (Fig. 1B, green line) and cRAD-SPPM (Fig. 1B, purple line) treated mice. These vascular ROIs continued to show similar SI curves for the entire study, returning to ~ 40% normalized SI at 30 mins (Fig. 1B, lower panel right graph). An initial decrease of SI in tumor ROIs was observed in both cRGD- (Fig. 1B, red line) and cRAD-SPPM (Fig. 1B, blue line) treated animals. cRGD-SPPM tumor ROI showed decrease to 40% SI and a sustained negative slope over the 30 min time span indicating more accumulation of SPPM in that ROI, i.e. tumor angiogenic endothelium, whereas the cRAD-SPPM tumor ROI showed an initial decrease to 70% SI and then a positive slope towards returning to baseline. This could be due to the clearance of the cRAD-SPPM particle from the blood and the tumor vasculature. Muscle tissues, as a reference, in cRGD- and cRAD-SPPM treated animals showed smaller initial deceases in SI and then a return to baseline over the 30 min imaging period (Fig. 1B, lower panel, brown and orange lines, respectively).
Blood clearance half-lives of SPPM by 3H polymer and Fe analyses. We modified the hydroxyl (-OH) terminal group of MeO-PEG-PLA copolymer with a 3H radioactive moiety (-C(O)CT3) for the quantitative measurement of SPPM pharmacokinetics in vivo. Two groups of animals were injected with cRGD- and cRAD-encoded SPPM. Plasma clearance studies showed a two-phase behavior over 24 hrs (Fig. 2A). The plasma half-lives (t1/2,α) for the α-phase were 0.41±0.11 and 0.32±0.08 hrs for the cRGD- and cRAD-SPPM (n=3 for each SPPM formulation, mean ± s.d.), respectively. The t1/2,β values for the β-phase were 2.26±0.32 and 3.85±0.50 hrs for the cRGD- and cRAD-SPPM (n=3), respectively (Fig. 2A). Pharmacokinetic analysis of the Fe content in the plasma, using atomic absorption spectroscopy, also showed the clearance of the SPPM from blood (Fig. 2B, black open circles). Data points at 1 and 60 mins from the 3H polymer pharmacokinetic study (Fig.2B, red open squares) overlap with the Fe data indicating that SPPM nanoparticles were intact after dilution into the blood. TEM analysis of plasma samples showed spherical clusters of SPIO (Fig. 2C). Moreover, the core-shell architecture was maintained and elucidated in the 2% PTA negatively stained TEM sample showing SPIO encapsulation in the white hydrophobic core of the SPPM (Fig. 2C, right panel). Polymer and Fe pharmacokinetic blood clearance data and TEM micrographs corroborate the SI data from the TR-MRI studies, and indicate that the SPPM nanoprobe was still intact within 60 mins post-injection. At early time points, differences in cRGD-SPPM and cRAD-SPPM blood vessel SI curves from TR-MRI (Fig. 1B, lower panel, green and purple lines) are not statistically significant (e.g. P = 0.36 at 30 mins), which is consistent with the 3H polymer pharmacokinetic data (Fig. 2A, green and purple lines) at 60 mins (P = 0.15).
Targeting kinetics of αvβ3-specific SPPM in lung, breast and brain tumor xenografts. Further TR-MRI studies of SPPM in multiple subcutaneous tumor xenografts models were examined and quantified. SI values were converted to ΔR2* values to reflect the non-linear relationship of MRI SI to SPPM concentration. Tumor ROIs from human A549 lung cancer, MDA-MB-231 breast cancer and U87 glioblastoma cancer showed similar ΔR2* profiles with cRGD-SPPM (black lines in Fig. 3, each symbol type, i.e. circle, square, triangle, represents the mean of 3 tumor ROIs from one animal) having a greater decrease in ΔR2* compared to cRAD-SPPM (Fig. 3, gray lines). In different animal tumor models, cRGD-SPPM revealed a similar trend with a continuing decreasing ΔR2* over the 30 min imaging time. cRAD-SPPM treated animals showed a returning trend to baseline or a sustained ΔR2* (U87 model) over the 30 min imaging acquisition. To assess the targeting kinetics of the cRGD-SPPM to αvβ3-expressing tumor endothelium, a compartmental model was proposed to measure their accumulation kinetics (ka) (Fig. 4A).
Figure 1
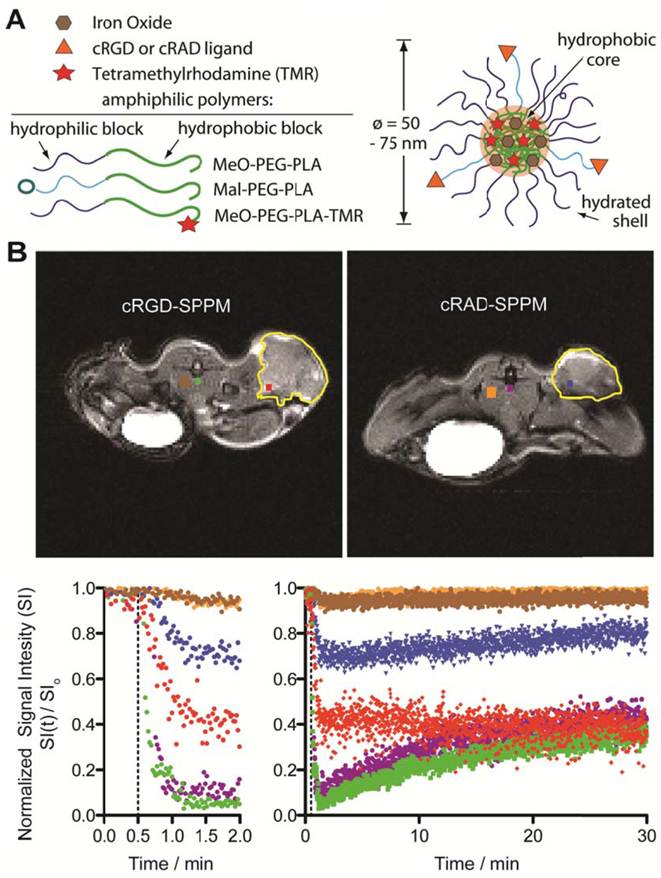
Schematic of superparamagnetic polymeric micelles (SPPM, A) and representative T2*-weighted images of tumor-bearing mice (transverse image) after cRGD- or cRAD-SPPM injection (B, upper panel). ROIs are color coded with the temporal SI curves. Major blood vessels are green or purple, tumor ROIs are red or blue, with muscle brown or orange, for cRGD- and cRAD-SPPM treated animals, respectively. Tumor margins are outlined yellow. The corresponding signal intensity curves show the varied temporal responses recorded by each ROI over a period of 30 mins (B, lower panel). The left graph showing the first 2 min of the study and the right graph showing 0-30 min of the same study. Vertical dashed lines indicate the start of the SPPM bolus (10 mg Fe/kg).
Figure 2
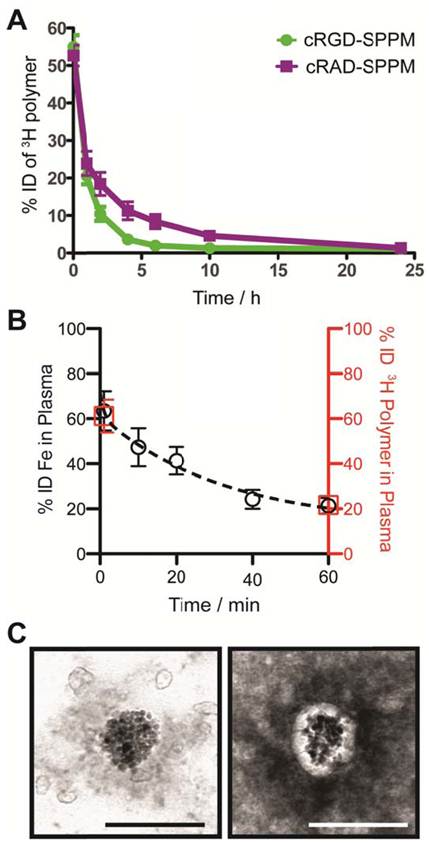
Measurement of the pharmacokinetics and structural integrity of SPPM nanoprobes. (A) Plasma concentration-time relationships (n=3 for each group) for cRGD-SPPM (green line) and cRAD-SPPM (purple line) using 3H-labeled polymers over 24 hrs. (B) Short-term plasma concentration-time relationships measured by Fe content (black open circles) after a bolus injection of cRGD-SPPM (10 mg Fe/kg). Data from radiolabeled polymers (1 min and 60 mins) are shown as red open squares. (C) TEM micrographs of plasma samples that were collected 40 mins after bolus injection of cRGD-SPPM. The sample was negatively stained with 2% PTA solution (right micrograph). Scale bars = 100 nm.
Figure 3
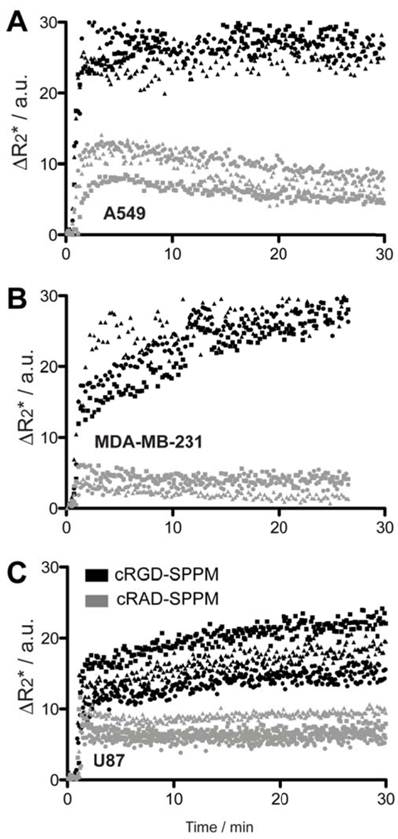
Mean ΔR2* curves of tumor ROI from cRGD- (black symbols) and cRAD-SPPM (gray symbols) in mice bearing A549 (A), MDA-MB-231 (B) or U87 (C) subcutaneous xenografts (n=3 for each xenograft group). Squares, circles and triangles indicate the mean of three measured ROIs from one animal in each group. cRGD-SPPM tumor hot-spot ROIs (black symbols) show a continuous increase in the ΔR2* over 30 mins, indicating SPPM accumulation in tumor tissues. In comparison, the ΔR2* values from cRAD-SPPM treated animals (gray symbols) show a maintained or trend toward baseline indicating clearance from the tumor tissue.
Figure 4
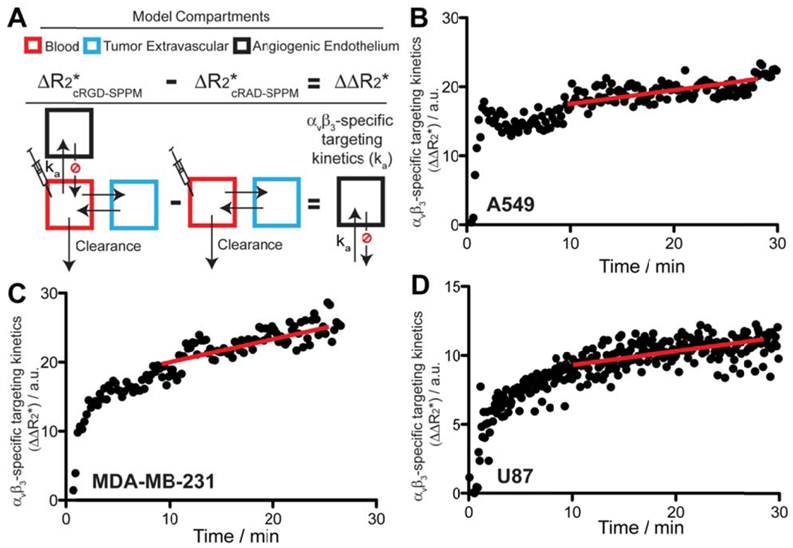
Scheme of the pharmacokinetic model (A) and experimental data (B-D) that describe the αvβ3-targeting kinetics of cRGD-SPPM in tumor-bearing mice. (A) Angiogenic endothelium compartment (black box) is proposed to model the accumulation rate (ka) of cRGD-SPPM. The tumor extravascular compartment (blue box) and blood compartment (red box) can be subtracted from cRAD-SPPM control, yielding only the angiogenic endothelium compartment for analysis of cRGD-SPPM nanoprobes. (B-D) cRGD-SPPM targeting kinetic curves in A549, MDA-MB-231 and U87 tumors, respectively. When fitted to a one-compartment pharmacokinetic model, cRGD-SPPM showed accumulation rates (ka) of 0.24 (R2=0.51), 0.22 (R2=0.87) and 0.24 (R2=0.83) min-1 in A549, MDA-MB-231 and U87 tumors, respectively. The slopes of the linear fit (red lines) from data points after 10 mins also indicate a continuous but slower accumulation of the cRGD-SPPM, with slopes of 0.19 (R2=0.88), 0.18 (R2=0.57) and 0.20 (R2=0.90) min-1 for A549, MDA-MB-231 and U87 tumors, respectively.
Specifically, the targeting kinetics of the cRGD-SPPM (Fig. 4A) was calculated by subtracting the mean cRGD-SPPM tumor ROI ΔR2* curve (Fig. 3, black lines) from its corresponding cRAD-SPPM control (Fig. 3, gray lines). When fitted to a one compartment model of distribution, cRGD-SPPM showed accumulation rates of 0.24 (R2=0.51), 0.22 (R2=0.87) and 0.24 (R2=0.83) min-1 in A549, MDA-MB-231 and U87 tumors, respectively (Fig. 4B-D). The data points after 10 mins also indicated a continuous accumulation of the cRGD-SPPM in tumors, with slopes (linear regression) of 0.19 (R2=0.88), 0.18 (R2=0.57) and 0.20 (R2=0.90) min-1 for A549, MDA-MB-231 and U87 models, respectively (Fig. 4B-D, red lines).
To histologically verify that the ΔR2* in the TR-MRI datasets were due to SPPM accumulation in the tumor tissue, fluorescence micrographs of the tumor ROIs were evaluated. By secondarily injecting Hoechst 33342 intravenously, nuclei of well-perfused vessels were identified (blue nuclei by white arrows, Fig. 5). Red fluorescence was observed in close proximity to these vessels in cRGD-SPPM treated mice (Fig. 5A), whereas a lesser and more diffusive punctate pattern was found in the cRAD-SPPM treated tumors (Fig. 5B). These data support previously published fluorescence and Prussian Blue histological data that show cRGD-SPPM nanoprobes specifically targeting αvβ3 on angiogenic vascular endothelium [9, 22].
Figure 5
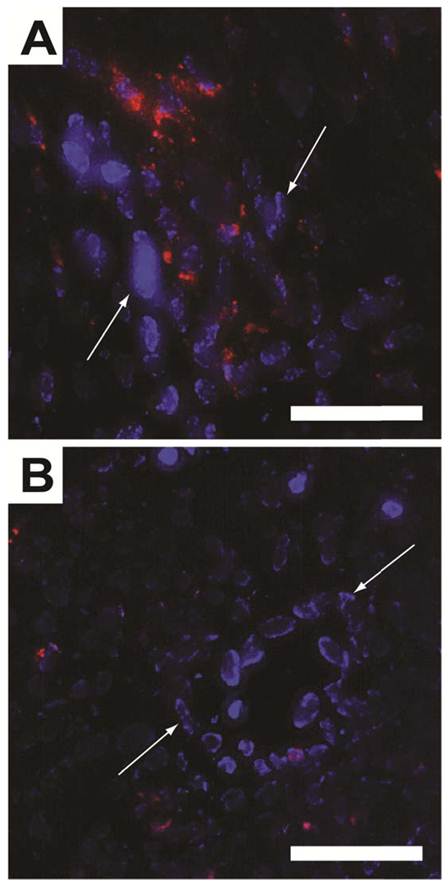
Fluorescence micrographs of cryo-sectioned A549 tumor xenografts from cRGD- (A) and cRAD-SPPM (B) treated animals. Endothelial cells from highly perfused vessels (white arrows) are stained with Hoechst (blue). This staining is caused by the limited circulation time (1 min) of the Hoechst dye. cRGD-SPPMs are shown tightly associated with these vessels (A, red fluorescence), whereas cRAD-SPPMs are dispersed in a lesser content throughout the tumor parenchyma (B, red fluorescence). Scale bars = 50 µm.
DISCUSSION
In molecular imaging studies, the surface of nanoprobes is frequently functionalized with target-specific ligands (e.g. peptides, mAbs, etc), and contrast specificity primarily arises from the difference in payload accumulation of targeted nanoparticles in the corresponding cells or tissues over the non-targeted control. This strategy has resulted in the successful imaging of multiple cancer-specific biomarkers, such as αvβ3 and αvβ5 integrins, Her2-neu, transferrin and folate receptors [9, 11-17]. However, several recent studies have raised serious questions on the efficacy of targeting ligands on the nanoparticle accumulation in tumor tissues. Multiple reports have shown that targeted nanoparticles did not lead to increased tumor accumulation over the non-targeted controls, although increased cellular uptake was observed. For example, Park and coworkers reported that liposomes functionalized with anti-Her2 antibody did not result in significant difference in nanoparticle accumulation in subcutaneous BT474 breast xenografts over the stealth pegylated control over 170 hrs [29]. Similar observations were also reported in transferin- and folate receptor-targeted nanoparticles in different animal tumor models [16, 29]. More recently, Nie and coworkers have shown that gold nanorods that were functionalized with cRGD, EGFR- and uPAR-ligands did not lead to significantly different uptake in A549 lung tumor xenografts over the non-targeted PEG-control 24 hrs after injection [30]. In contrary, for cRGD-encoded nanoparticles, tumor accumulation was found to decrease with the increase of cRGD density on the nanoparticle surface. Most of these studies attributed “passive” targeting from the EPR effect as the predominant mechanism of nanoparticle accumulation in tumors. Although this mechanism may aid in the overall tumor imaging effect, it is detrimental in obtaining targeted, biomarker-specific contrast in cancer molecular imaging applications. This is particularly important if the imaging goal is to monitor the dynamic expression of a disease biomarker (e.g. EGFR, αvβ3) after chemotherapy (e.g. Erbitux, Cilengitide) to assess the therapeutic efficacy.
Due to the conflicting reports on the ligand effect on nanoparticle targeting specificity, more mechanistic studies on the active targeting mechanism are necessary. Most current imaging studies are carried out by comparing the pre- and post-contrast images of targeted nanoparticles over non-targeted control. These data provide only the “snap shot” information of targeting events at later time points. For many molecular imaging processes, particularly for vascular-targeted nanoparticles, the initial receptor binding and nanoparticle internalization steps may occur at much earlier times. Acquiring the early phase information on nanoparticle targeting to their intended biomarkers is important to understand the in vivo molecular recognition process for the rational design and development of effective molecular imaging probes and their clinically translatable applications.
This study proposes a time-resolved MRI method for the non-invasive characterization of targeted nanoprobes to their intended biological targets in vivo. Compared to nuclear imaging techniques (e.g. PET, SPECT), MRI has higher spatial resolution (<mm pixel size), which allows for the use of vascular “hot spots” to analyze the specific SPPM targeting kinetics. In a series of experiments, we measured the targeting kinetics of the cRGD-SPPM nanoprobes to αvβ3 integrins in three tumor xenograft models in vivo using single slice T2*-weighted TR-MRI. The high temporal resolution of this study (1.3s) has not been attempted before with targeted SPIO-based nanoprobes. Similar accumulation rates (ka) were found in the initial phase (<5 mins) across all three subcutaneous tumor types, and furthermore, a sustained accumulation of cRGD-SPPM over 30 mins was also observed.
SPPM nanoprobes used in this study are 50-75 nm in diameter with estimated molar mass around 107-108 Da. Compared to small molecular agents (<1 kD), nanoparticulates such as SPPM are limited in the diffusion, extravasation and penetration from blood vessels into tumor tissues, and are therefore, well suited to target angiogenic endothelium compartments. Although the current TR-MRI pharmacokinetic model was not as elaborate as conventional T1 DCE-MRI models, the dynamic contrast profiles allow for direct comparisons of targeted versus non-targeted T2 contrast agents.
Results from this study showed that αvβ3 targeting kinetics by cRGD-SPPM had a significantly different profile from the non-targeted cRAD-SPPM control in all three different tumor models. A continuous accumulation of the cRGD-SPPM (i.e. SI decrease) was observed over the 30 min time span (Fig. 1B). The cRAD-SPPM profiles, however, showed an initial decrease in SI and then clearance from the tumor ROI that matched the slope of the systemic blood vessel ROI (Fig. 1B, lower panel, purple and blue), indicating the wash-out of the non-targeted SPPM from the tumor tissue. Control ROIs from muscle tissues showed a slight decrease in SI that returned to baseline after 7-9 mins, which indicates that the perfusion of normal tissue does not cause similar kinetic profiles as observed in the cRGD- and cRAD-SPPM treated tumor tissue.
To quantify the targeting kinetics of cRGD-SPPM to αvβ3 integrins in vivo, we subtracted the ΔR2* curve of the non-targeted cRAD-SPPM control from that of cRGD-SPPM. By subtracting the non-targeted ΔR2* data, we hypothesized that blood clearance, extravascular-extracellular components and other passive accumulation mechanisms (e.g. EPR effect) are excluded (Fig. 4A). The use of a single compartment pharmacokinetic model revealed similar accumulation kinetics between the tumor models with first-order rate constants of 0.24, 0.22 and 0.24 min-1 in A549, MDA-MB-231 and U87 tumors, respectively. Although U87 cells are also known to express αvβ3 integrins [31, 32], the similar kinetic data suggest that only vascular targeting events were observed in U87 tumors. This is likely due to the short time frame (<30 mins) of TR-MRI studies, where limited nanoprobe extravasation from tumor vasculature is expected. It is further supported by the fact that the angiogenic endothelial cells in tumor xenografts are mouse endothelial cells in origin (i.e. independent of the injected human cancer cells), which may share similar expression levels of αvβ3 integrins. In addition to single compartment analysis, we also performed linear regression analysis at later times between 10 and 30 mins. Data show an increasing trend in the cRGD-SPPM targeting kinetic profile (Fig. 4B-D, red lines), indicating a continuous, albeit slower, cRGD-SPPM uptake in tumor endothelial cells. This two-phase behavior may explain the fast binding and uptake of the cRGD-SPPM during the initial contact with angiogenic endothelium, followed by the slower steady-state kinetics as controlled by αvβ3 recycling and subsequent αvβ3-mediated endocytosis.
These data clearly demonstrate that increased tumor accumulation of cRGD-encoded SPPM was observed at the vascular “hot spots” in tumors in the first 30 mins after SPPM injection. This is further corroborated by 3H-labeled SPPM studies, where a two-fold increase of nanoparticle accumulation was observed for cRGD-encoded SPPM over the non-targeted control 60 mins post-injection [9]. Although these data may appear to contradict with those reported by Nie and coworkers [30], it should be noted that the two studies were carried out at completely different time scales. In Nie's study, the nanoparticle accumulation was examined 24 hrs after nanoparticle injection. It is entirely feasible that longer interval times (i.e. 24 hrs) will favor non-targeted PEG-particles (with longer blood circulation times) to “passively” accumulate in the tumor parenchyma, despite clear advantages of targeted nanoparticles at the earliest stage of tissue targeting in vivo. For molecular imaging applications of vascular biomarkers, it will be advantageous to image as early as possible to obtain target-specific information. However, this will be limited by the prolonged circulation times of nanoparticulates in blood, which will present significant background signals at early times. Therefore, one future consideration for the target-specific nanoprobes should include an activatable design, where the imaging signals should be suppressed in blood but can be activated upon recognizing a specific biomarker of interest. This is currently under investigation in our lab.
Several considerations are warranted from the current study. First, a single slice MR acquisition method was employed to ensure fast temporal resolution (1.3 s). Multi-slice images using the same MRI parameters are possible to provide a more global understanding of SPPM targeting kinetics, tumor distribution and describing the heterogeneity of angiogenic tumor endothelium[22]. For this purpose, however, a compromise in temporal resolution is anticipated. It should also be noted that the current kinetic constants were obtained without the use of an arterial input function or blood pool data in our compartmental analysis. In conventional DCE-MRI, using the T1 agents, the blood pool data is critical in estimating blood dynamic parameters such as Ktrans and others. However, in our study the drastic loss of SI in the blood ROI kinetic curves exceeded the sensitivity limit of the T2* acquisition, which prevents us from obtaining accurate estimation of the SPPM concentrations in the first minutes of analysis. Finally, a partial volume effect exists where the measured pixel (hotspot ROI) may contain a combined contribution from both tumor parenchyma and vascular compartments. In this study, we used the same ROI pixel size (4 x 4, or 1.1x1.1 mm) for the identified hot spot ROIs for cRGD-SPPM and cRAD-SPPM groups. By keeping the same pixel size, we consider that the partial volume effect will have similar contributions to the SIs of vascular hot spots in both groups, and therefore, won't affect the relative kinetic analysis of the binding rates of cRGD-SPPM to αvβ3-expressing tumor vasculatures. Despite these complexities and limitations that may affect the accuracy of the measurement, the proposed study nevertheless provides a useful first attempt to quantitatively investigate the early phase of nanoparticle targeting to its intended molecular targets (i.e. αvβ3) in the tumor vasculature in vivo.
In summary, this study demonstrates the feasibility to quantitatively measure the targeting kinetics of cancer-specific superparamagnetic nanoprobes to their biological targets in vivo. In particular, cRGD-SPPM targeting kinetics to αvβ3 integrins on the tumor endothelium was evaluated in tumor-bearing mice using the T2*-weighted TR-MRI sequence. αvβ3-specific accumulation of cRGD-SPPM nanoprobes in tumor vasculature was observed at early times with similar kinetic constants in three distinctive tumor xenograft models (A549, MDA-MB-231, and U87). These data indicate that broadened tumor specificity can be achieved in targeting angiogenic vasculature of tumors where vascular-targeted nanoprobes are less dependent on the specific tumor types. Furthermore, the data demonstrate for the first time that specific vascular targeting can be observed in as early as the first 10 mins post-injection of nanoprobes (although not surprisingly due to the direct blood-endothelium contact). These results provide useful mechanistic insights for the development of future vascular-targeted nanoprobes to capture the early events of receptor targeting kinetics (e.g. activatable nanoprobes).
Acknowledgements
This work is supported by NIH Grants EB005394, CA129011 (J. Gao) and P41 RR002584 (M. Takahashi), DOD BCRP Multidisciplinary Postdoctoral Award W81XWH-06-1-0751 (C. Khemtong), and Susan G. Komen foundation postdoctoral fellowship PDF0707216 (G. Huang). The authors would also like to acknowledge the Southwestern Small Animal Imaging Resource (NCI U24 CA126608) and the Simmons Cancer Center Support Grant (P30CA142543) for imaging infrastructure support.
CONFLICT OF INTEREST
The authors have declared that no conflict of interest exists.
References
1. Cai WB, Chen XY. Multimodality molecular imaging of tumor angiogenesis. Journal of Nuclear Medicine. 2008;49:113S-28S
2. Cheon J, Lee JH. Synergistically Integrated Nanoparticles as Multimodal Probes for Nanobiotechnology. Accounts of Chemical Research. 2008;41:1630-40
3. Corot C, Robert P, Idee JM, Port M. Recent advances in iron oxide nanocrystal technology for medical imaging. Advanced Drug Delivery Reviews. 2006;58:1471-504
4. Nie SM, Xing Y, Kim GJ, Simons JW. Nanotechnology applications in cancer. Annual Review of Biomedical Engineering. 2007;9:257-88
5. Bulte JWM, Kraitchman DL. Iron oxide MR contrast agents for molecular and cellular imaging. Nmr Biomed. 2004;17:484-99
6. Thorek DLJ, Chen A, Czupryna J, Tsourkas A. Superparamagnetic iron oxide nanoparticle probes for molecular imaging. Ann Biomed Eng. 2006;34:23-38
7. Lee JH, Huh YM, Jun Y, Seo J, Jang J, Song HT. et al. Artificially engineered magnetic nanoparticles for ultra-sensitive molecular imaging. Nat Med. 2007;13:95-9
8. Seo WS, Lee JH, Sun X, Suzuki Y, Mann D, Liu Z. et al. FeCo/graphitic-shell nanocrystals as advanced magnetic-resonance-imaging and near-infrared agents. Nat Mater. 2006;5:971-6
9. Khemtong C, Kessinger CW, Gao J. Polymeric nanomedicine for cancer MR imaging and drug delivery. Chem Commun (Camb). 2009:3497-510
10. Mills PH, Ahrens ET. Theoretical MRI contrast model for exogenous T2 agents. Magn Reson Med. 2007;57:442-7
11. Artemov D, Mori N, Okollie B, Bhujwalla ZM. MR molecular imaging of the Her-2/neu receptor in breast cancer cells using targeted iron oxide nanoparticles. Magn Reson Med. 2003;49:403-8
12. Hogemann-Savellano D, Bos E, Blondet C, Sato F, Abe T, Josephson L. et al. The transferrin receptor: a potential molecular imaging marker for human cancer. Neoplasia. 2003;5:495-506
13. Mulder WJ, Strijkers GJ, Habets JW, Bleeker EJ, van der Schaft DW, Storm G. et al. MR molecular imaging and fluorescence microscopy for identification of activated tumor endothelium using a bimodal lipidic nanoparticle. FASEB J. 2005;19:2008-10
14. Nasongkla N, Bey E, Ren J, Ai H, Khemtong C, Guthi JS. et al. Multifunctional polymeric micelles as cancer-targeted, MRI-ultrasensitive drug delivery systems. Nano Lett. 2006;6:2427-30
15. Schmieder AH, Caruthers SD, Zhang H, Williams TA, Robertson JD, Wickline SA. et al. Three-dimensional MR mapping of angiogenesis with alpha5beta1(alpha nu beta3)-targeted theranostic nanoparticles in the MDA-MB-435 xenograft mouse model. FASEB J. 2008;22:4179-89
16. Sonvico F, Mornet S, Vasseur S, Dubernet C, Jaillard D, Degrouard J. et al. Folate-conjugated iron oxide nanoparticles for solid tumor targeting as potential specific magnetic hyperthermia mediators: synthesis, physicochemical characterization, and in vitro experiments. Bioconjug Chem. 2005;16:1181-8
17. Zhang C, Jugold M, Woenne EC, Lammers T, Morgenstern B, Mueller MM. et al. Specific targeting of tumor angiogenesis by RGD-conjugated ultrasmall superparamagnetic iron oxide particles using a clinical 1.5-T magnetic resonance scanner. Cancer Res. 2007;67:1555-62
18. O'Connor JP, Jackson A, Parker GJ, Jayson GC. DCE-MRI biomarkers in the clinical evaluation of antiangiogenic and vascular disrupting agents. Br J Cancer. 2007;96:189-95
19. Tofts PS, Brix G, Buckley DL, Evelhoch JL, Henderson E, Knopp MV. et al. Estimating kinetic parameters from dynamic contrast-enhanced T(1)-weighted MRI of a diffusable tracer: standardized quantities and symbols. J Magn Reson Imaging. 1999;10:223-32
20. Su MY, Wang Z, Carpenter PM, Lao X, Muhler A, Nalcioglu O. Characterization of N-ethyl-N-nitrosourea-induced malignant and benign breast tumors in rats by using three MR contrast agents. J Magn Reson Imaging. 1999;9:177-86
21. Ahrens ET, Rothbacher U, Jacobs RE, Fraser SE. A model for MRI contrast enhancement using T1 agents. Proc Natl Acad Sci U S A. 1998;95:8443-8
22. Kessinger CW, Khemtong C, Togao O, Takahashi M, Sumer BD, Gao J. In vivo angiogenesis imaging of solid tumors by alpha(v)beta(3)-targeted, dual-modality micellar nanoprobes. Exp Biol Med (Maywood). 2010;235:957-65
23. Ai H, Flask C, Weinberg B, Shuai X, Pagel MD, Farrell D. et al. Magnetite-loaded polymeric micelles as ultrasensitive magnetic resonance probes. Adv Mater. 2005;17:1949-52
24. Khemtong C, Kessinger CW, Ren J, Bey EA, Yang SG, Guthi JS. et al. In vivo off-resonance saturation magnetic resonance imaging of alphavbeta3-targeted superparamagnetic nanoparticles. Cancer Res. 2009;69:1651-8
25. Barcena C, Sra AK, Chaubey GS, Khemtong C, Liu JP, Gao J. Zinc ferrite nanoparticles as MRI contrast agents. Chem Commun (Camb). 2008:2224-6
26. Luo L, Tam J, Maysinger D, Eisenberg A. Cellular internalization of poly(ethylene oxide)-b-poly(epsilon-caprolactone) diblock copolymer micelles. Bioconjug Chem. 2002;13:1259-65
27. Sun S, Zeng H. Size-controlled synthesis of magnetite nanoparticles. J Am Chem Soc. 2002;124:8204-5
28. Rosen BR, Belliveau JW, Vevea JM, Brady TJ. Perfusion imaging with NMR contrast agents. Magn Reson Med. 1990;14:249-65
29. Kirpotin DB, Drummond DC, Shao Y, Shalaby MR, Hong K, Nielsen UB. et al. Antibody targeting of long-circulating lipidic nanoparticles does not increase tumor localization but does increase internalization in animal models. Cancer Res. 2006;66:6732-40
30. Huang X, Peng X, Wang Y, Shin DM, El-Sayed MA, Nie S. A reexamination of active and passive tumor targeting by using rod-shaped gold nanocrystals and covalently conjugated peptide ligands. ACS Nano. 2010;4:5887-96
31. Monferran S, Skuli N, Delmas C, Favre G, Bonnet J, Cohen-Jonathan-Moyal E. et al. Alphavbeta3 and alphavbeta5 integrins control glioma cell response to ionising radiation through ILK and RhoB. Int J Cancer. 2008;123:357-64
32. Benedetto S, Pulito R, Crich SG, Tarone G, Aime S, Silengo L. et al. Quantification of the expression level of integrin receptor alpha(v)beta3 in cell lines and MR imaging with antibody-coated iron oxide particles. Magn Reson Med. 2006;56:711-6
Author contact
![]() Corresponding author: Prof. Jinming Gao, Simmons Comprehensive Cancer Center, University of Texas Southwestern Medical Center, 5323 Harry Hines Blvd., Dallas, TX 75390. Phone: 214-645-6370; Fax: 214-645-6347; E-mail: jinming.gaoedu.
Corresponding author: Prof. Jinming Gao, Simmons Comprehensive Cancer Center, University of Texas Southwestern Medical Center, 5323 Harry Hines Blvd., Dallas, TX 75390. Phone: 214-645-6370; Fax: 214-645-6347; E-mail: jinming.gaoedu.
Citation styles
APA
Kessinger, C.W., Togao, O., Khemtong, C., Huang, G., Takahashi, M., Gao, J. (2011). Investigation of In Vivo Targeting Kinetics of αvβ3-Specific Superparamagnetic Nanoprobes by Time-Resolved MRI. Theranostics, 1, 263-273. https://doi.org/10.7150/thno/v01p0263.
ACS
Kessinger, C.W.; Togao, O.; Khemtong, C.; Huang, G.; Takahashi, M.; Gao, J. Investigation of In Vivo Targeting Kinetics of αvβ3-Specific Superparamagnetic Nanoprobes by Time-Resolved MRI. Theranostics 2011, 1, 263-273. DOI: 10.7150/thno/v01p0263.
NLM
Kessinger CW, Togao O, Khemtong C, Huang G, Takahashi M, Gao J. Investigation of In Vivo Targeting Kinetics of αvβ3-Specific Superparamagnetic Nanoprobes by Time-Resolved MRI. Theranostics 2011; 1:263-273. doi:10.7150/thno/v01p0263. https://www.thno.org/v01p0263.htm
CSE
Kessinger CW, Togao O, Khemtong C, Huang G, Takahashi M, Gao J. 2011. Investigation of In Vivo Targeting Kinetics of αvβ3-Specific Superparamagnetic Nanoprobes by Time-Resolved MRI. Theranostics. 1:263-273.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) License. See http://ivyspring.com/terms for full terms and conditions.