Theranostics
13.3
Impact Factor
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Special Issues
Top
1. Introduction
2. PGC: synthesis and structure
3. Physico-chemical and...
4. Applications in imaging
5. Applications in drug delivery
6. Conclusions and perspectives
Acknowledgements
References
1. Introduction
2. PGC: synthesis and structure
3. Physico-chemical and...
4. Applications in imaging
5. Applications in drug delivery
6. Conclusions and perspectives
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2012; 2(6):553-576. doi:10.7150/thno.4070 This issue Cite
Review
Protected Graft Copolymer (PGC) in Imaging and Therapy: A Platform for the Delivery of Covalently and Non-Covalently Bound Drugs
Alexei A. Bogdanov Jr1 ![]() , Mary Mazzanti1, Gerardo Castillo2, Elijah Bolotin2
, Mary Mazzanti1, Gerardo Castillo2, Elijah Bolotin2
1. University of Massahcusetts Medical School, Worcester MA;
2. PharmaIn Corp, Seattle WA, Massachusetts General Hospital and Harvard Medical School.
Received 2012-1-10; Accepted 2012-2-17; Published 2012-6-4
Citation:
Bogdanov Jr AA, Mazzanti M, Castillo G, Bolotin E. Protected Graft Copolymer (PGC) in Imaging and Therapy: A Platform for the Delivery of Covalently and Non-Covalently Bound Drugs. Theranostics 2012; 2(6):553-576. doi:10.7150/thno.4070. https://www.thno.org/v02p0553.htm
Other stylesAbstract
Initially developed in 1992 as an MR imaging agent, the family of protected graft copolymers (PGC) is based on a conjugate of polylysine backbone to which methoxypoly(ethylene glycol) (MPEG) chains are covalently linked in a random fasion via N-ε-amino groups. While PGC is relatively simple in terms of its chemcial composition and structure, it has proved to be a versatile platform for in vivo drug delivery. The advantages of poly amino acid backbone grafting include multiple available linking sites for drug and adaptor molecules. The grafting of PEG chains to PGC does not compromise biodegradability and does not result in measurable toxicity or immunogenicity. In fact, the biocompatablility of PGC has resulted in its being one of the few 100% synthetic non-proteinaceous macromolecules that has suceeded in passing the initial safety phase of clinical trials. PGC is capable of long circulation times after injection into the blood stream and as such found use early on as a carrier system for delivery of paramagnetic imaging compounds for angiography. Other PGC types were later developed for use in nuclear medicine and optical imaging applications in vivo. Recent developments in PGC-based drug carrier formulations include the use of zinc as a bridge between the PGC carrier and zinc-binding proteins and re-engineering of the PGC carrier as a covalent amphiphile that is capabe of binding to hydrophobic residues of small proteins and peptides. At present, PGC-based formulations have been developed and tested in various disease models for: 1) MR imaging local blood circulation in stroke, cancer and diabetes; 2) MR and nuclear imaging of blood volume and vascular permeability in inflammation; 3) optical imaging of proteolytic activity in cancer and inflammation; 4) delivery of platinum(II) compounds for treating cancer; 5) delivery of small proteins and peptides for treating diabetes, obesity and myocardial infarction. This review summarizes the experience accumulated by various research groups that chose to use PGC as a drug delivery platform.
Keywords: contrast agent, gaft-copolymer, poly(ethylene glycol), chelate, paramagnetic, gadolinium.
1. Introduction
PGCs are relatively simple covalent conjugates composed of a poly-l-lysine (PLL) backbone which is covalently grafted with a plurality of carboxylated methoxypoly(ethylene glycol) (MPEG) side chains via the N-ε-amino groups of PLL (Fig. 1). The structure of graft copolymers that will be reviewed below is diferent from di- and multi- head-to-tail block copolymers that were previously used for drug delivery applications [1-4]. While the latter usually form micelles as a result of interaction between the hydrophobic blocks, PGC was specifically designed as a non-aggregating covalent macromolecular garft-copolymer conjugate. As a non-modified copolymer, PGC (also known as PEG-gPLL or MPEGS-PL) carries multiple positive charges and can potentially form polyionic complexes with polyanions, e.g. plasmid DNA. A host of other copolymers, structurally and chemcially similar to PGC, were under consideration as potential non-viral gene delivery vehicles in the late 1990s [1-5]. However, despite the efficient formation of homogenous populations of small (<100 nm) complexes with plasmid DNA, the in vitro and in vivo efficacy of intracellular delivery was insufficiently high to grant further development of protected graft polycations for gene delivery [4].
Figure 1
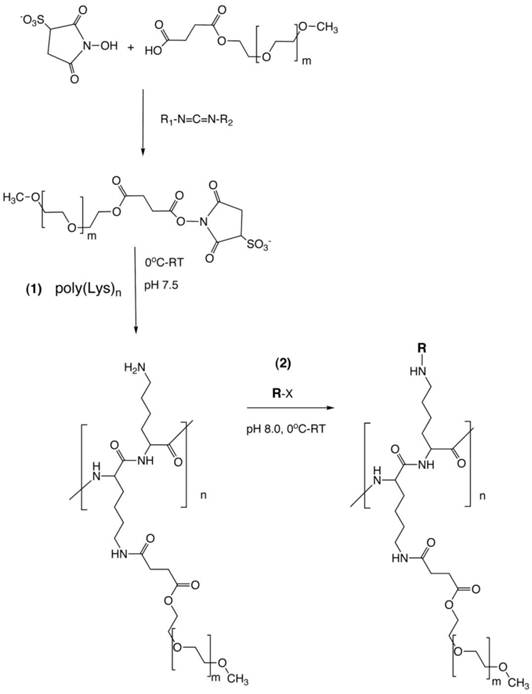
PGC synthesis. Synthesis of a graft copolymer of polylysine and methoxypolyethylene glycol succinate (MPEG)S with subsequent modification of free amino groups with diagnostic labels [18] or adaptor molecules for loading of therapeutics (R). The synthesis consits of a synthesis of MPEG sulfosuccinimide ester in the presence of water-soluble carbodiimide, acylation of 20-30% of total available amin groups of poly-lysine followed by the acylation of amino groups with an activated analog of R (R-x), usually N-hydroxysuccinimide ester.
The adaptability of PGCs is in large due to the existence of multiple sites on the molecule that can be readily modified to carry theraputic or diagnostic agents (Fig 1, step 2). A second important characteristic of PGCs is that they escape the uptake during the multiple passes through the reticuloendothethial system (RES) i.e., they have long circulation times and thus can be described as 'long circulating agents'. This characteristic lead to their initial use as a carrier system for delivery of paramagnetic imaging compounds for magentic resonance imaging (MRI) [6]. As such, the use of PGCs for in vivo imaging of the blood pool enabled visualization of localized morphological and funtional abnormalities in vascular permeability that allowed detection of local inflammation and blood supply of tumors. The initial success of PGC as an MR vascular imaging agent lead to its further developemnt of the PGC as a drug delivery platform as will be summarized in part two of this review.
2. PGC: synthesis and structure
In general, the synthesis of PGC involves covalent grafting of a polyamino acid with multiple MPEG ester chains in weakly alkaline buffered aqeous solutions. There are several potential synthetic pathways that lead to the desired PGC product, i.e. a copolymer of a polyamino acid and covalently grafted MPEG chains. They are defined by the reactive side chain groups of the polyamino acid and specific reactivity of functionalized MPEG (Fig. 1). For example, several classes of biocompatible polycarboxylic acids can be used as the initial backbones for further conjugation with terminated MPEG chains. Some of these polycarboxylic backbones are biocompatible and biodegradable, such as poly-L-glutamic [7] or poly-(α,β)-DL-aspartic acids derivatives [8]. Others, such as uncharged N-(2-hydroxypropyl) methacrylamide copolymers, which are frequently used as backbone polymers, are non-biodegradable but are biocompatible and non-immunogenic [9].
Polylysine can be synthesized and purified to yield homegenous preparations with various polydispersity indeces (PDI) and narrow mass distribution (i.e. low PD=Mw/Mn). While poly-L-lysine (PLL) is readily degradable into smaller oligomers by many serine endopeptidases, its poly D-enantiomer is not, and thus can be used for synthesizing PGCs with limited biodegradability. Currently, PLLs with a polydispersity factor of 1.1 are commercially available. The synthesis of PLL has been perfected through many years of extensive research. The synthesis includes a catalytically initiated ionic polymerization process of ring opening in N-carboxy-(N-ε-benzyloxycarbonyl)-L-lysine anhydride (CBZ-L-lysine NCA) [10]. Most commonly, the synthesis is carried out in polar organic solvents in the presence of diethylamine as an initiator. Therefore, the synthetic product commonly incorporates diethylamide bridges and the resultant PLL polymers (especially those with low degree of polymerization) are composed of two different monomers. Poly(N-ε-benzyloxycarbonyl) lysines can then be reacted with other polymers to yield the formation of copolymers due to the availability of free reactive N-α-amino groups. One of the popular blocks is activated PEG diester [11]. A copolymerization of poly CBZ-L-lysine results in linear block copolymers that are structurally distinct from PGC. These copolymers can then be deprotected and the liberated amino groups used for covalent modification, if needed. Multiple examples of the use of di- tri- and poly- block PLL-PEG copolymers for drug delivery systems exist in the literature [12-14].
Carboxybenzyl protective groups on poly CBZ-L-lysine can be efficiently removed using HBr and the resultant purified hydrobromides can be reacted with MPEG derivatives in water since PLL has a high water solubility at pH<11. At pH 8.5 PLL, exists in extended coil conformation with fewer than 10% deprotonated N-ε-amino groups [15]. Extended coil conformation of PLL enables efficient access to PLL side chain amino groups by hydroxysuccinimide esters of MPEG-succinate. To prevent extensive hydrolysis of activated esters of MPEG succinate which occur at pH 8.5, we used N-hydroxysulfosuccinimide (NHSS) esters of MPEG-succinate which were obtained in water solution after modifying MPEG-succinate (MPEGS) with water soluble carbodiimide [16]. The obtained esters are more stable than N-hydroxusuccinimide esters at pH>7.5. Our experience shows that a 25% modification of PLL amino groups with MPEGS is sufficient for obtaining highly water soluble graft copolymers that can be isolated from the reaction mixture by using ultrafiltration [17]. The covalent linking of multiple MPEGS chains to PLL results in a large increase of the hydrodynamic diameter. This enables purification based on ultrafiltration through hollow fibers with a 100-300 kD cut-off, depending on the sizes of the initial PLL and MPEGS. Such purification results in a complete removal of non-reacted hydrolyzed MPEG esters (i.e. MPEG succinate and NHSS). However, the purification of the obtained graft copolymer (MPEGS-PL) can be delayed until all modifications to the amino groups of PLL are completed. This is especially convenient if the second (or later) component that needs to be covalently linked to MPEGS-PL is soluble in water or water-miscible solvent. For example, an excess cyclic anhydride of diethylenetriaminepentaacetic acid (DTPA) dissolved in DMSO was used in our original protocol for obtaining an MPEG-PL-DTPA conjugate that can be used for further modification of the remaining amino groups of PL as a means of obtaining a long-circulating chelating polymer [18]. Notably, on the second stage of the synthesis, undesirable cross linking of PL with bifunctional DTPA derivatives may be greatly reduced or avoided due to the presence of grafted MPEGS chains.
The second and other reactants that are to be attached to the backbone do not have to be soluble in water for successful linking to MPEGS-PL. The above conjugate forms stable suspensions in chloroform due to MPEG solubility in some non-polar solvents, and the amino groups remaining on PL after the modification with MPEGS esters can be further covalently modified in the presence of an organic base (e.g. triethylamine). The modified product as a rule is more soluble in chloroform. For example, for synthesis of PGC carrying hydrophobic fatty acid chains or cholesteryl hemisuccinate residues, MPEGS-PL can be treated with chloroform-soluble activated esters of fatty acids [19]. Insolubility of MPEGS in diethyl ether and ethyl acetate can be further used for precipitating the obtained PGC conjugates from choroform which greatly simplifies the purification from the excess of hydrophobic modifier.
3. Physico-chemical and biological properties of MPEGS-PL-DTPA
3.1. Molecular mass and size (hydrodynamic radius)
Highly-soluble polycationic graft copolymer MPEGS-PL was initially used by our group to obtain MPEGS-PL-GdDTPA (from here on - abbreviated as PGC-DTPAGd) designed as a biocomaptible non-toxic and long-circulating contrast agent (CA) for MRI [6]. The molecule consited of MPEG5 succinate (5 kD) linked to poly-lysine with a degree of polymerization of 250, which was then conjugated to DTPA and labeled with Gd3+ using transchelation. Protective MPEG chains constitute the bulk of PGC-DTPAGd molecular weight (up to 80% by weight in the case of PL with 40% amino groups modified with 5kD MPEG) [18]. MPEG protective chains in diluted water solutions were not expected to form coordination bonds with free paramagnetic ions. Therefore, the percentage of Gd by weight in PGC-DTPAGd is always lower than in PLL-GdDTPA prepared from PLL with the same degree of polymerization and ranges between 5-6% of dry copolymer weight. The calculated longitudinal molar proton relaxivity of Gd was similar in the presence or in the absence of MPEG chains (i.e. the efficiency of Gd to increase longitudinal (spin-lattice) relaxation rate of water protons, r1= R1/[Gd]= 12-14 [mmol.s.l]-1 for poly(l-lysine)-GdDTPA and PGC-DTPAGd at 20 MHz, 37○C) [18].
The estimation of mass and effective hydrodynamic radius of the obtained graft copolymer was necessary for understanding the in vivo behavior. Niether linear PEG standards nor protein standards were suitable for estimating the mass of PGC-DTPAGd. For example, the mass determined from elemental analysis is approximately 2-times less than the mass determined by size-exclusion chromatography (SEC) [20]. The difference between the calculated mass of PGC from protein standard calibrated-SEC and true mass of PGC is due to the significant effect of highly hydrated large MPEGS side chains around PL on hydrodynamic diameter of PGC (see Fig. 2A).
3.2. Immunogenicity and toxicity
Safety is a major concern in the case of all polymers potentially useful for medical applications, and in the case of long-circulating diagnostic agents in particular [21]. A number of large macromolecules including proteins (serum albumin), synthetic polypeptides, polyamidoamines (poly-l-lysine), polyimines and polysaccharides (dextran) can carry diagnostic labels and exhibit slow renal filtration (which is dependent on the size of the final molecule). Serum albumin (e.g. BSA) is a 66 kD protein and has 30-35 N--lysine amino groups that can be potentially chemically modified, whereas poly-l-lysine of the same mass has more than 500 -lysine residues, with the majority of them accessible for chemical modification. And although a variety of macromolecules can be modified to carry diagnostic labels, many of them will have undesirable imaging characteristics because of rapid opsonization (i.e. binding of complement proteins or immunocomplexes), removal by phagocytic cells (e.g. monocytes or Kupfer cell of the liver with the aid of scavenger or complement receptors), and breakdown and early excretion.
Poly(ethylene glycol) is not toxic for animals even at high doses after systemic administration [22]. Remarkably, poly(ethylene glycol) and its derivatives substantially decrease immunogenicity when chemically attached to proteins [23]. It has been hypothesised that the modification of surface -aminogroups by highly hydrated polymer leads to the masking of potential antigenic determinants of proteins (reviewed in [24, 25]. We observed a similar effect with polymers containing poly(ethylene glycol) grafted to synthetic backbones. Presumably, grafted polymeric chains can potentially provide a "hydrated shell" for the central polymeric carrier. While protein-linked DTPAGd is immunogenic [18, 21], we did not observe any significant immune response against DTPAGd at either 8 or 15 days after PGC-DTPAGd intravenous (I.V.) injection in mice. To test the immune response to PGC-DTPAGd components, MPEG and DTPAGd were conjugated to ovalbumin, which was used for A low-affinity response detected on day 8 had diminished by day 15 (Fig. 2B). Whereas BSA-DTPAGd, used as a positive control and injected at a dose similar to Gd, elicited a substantial response to DTPAGd.
Gadolinium-labeled PGC-DTPA did not show any detectable toxic effects in two rodent species under study. We detected no weight loss or renal toxicity in mice injected at 30 times the projected MR imaging dose (0.6 mmol Gd/kg, a total of 50 mg PGC-DTPAGd/animal, one month observation) or in rats injected with a total dose of 0.2 mmol/kg (a total of 125 mg of PGC-DTPAGd/animal, 5-6 consecutive injections for 2 weeks) [18, 26].
3.3. Biokinetics and biodistribution of PGC labeled with hydrophilic groups
The grafting of long-chain MPEGs results in a significant increase in the blood half-life of a backbone polyamino acid (e.g. PLL) when compared to non-modified PL (Table 1). However, the presence of high amounts of MPEG in the aqueus solution of PGCs may increase viscosity and hamper i.v. administration of concentrated formulations. In an attempt to decrease the size of protective chains while preserving longievity in blood, we prepared PGC with MPEG ranging in mass from 0.75 to 5 kD, and tested these compounds in vivo. Our results indicate that 2-5 kD MPEGs definitely prolong the circulation in vivo while 0.75 kD MPEG does not [20]. A notable decrease in renal uptake was observed with the increase of MPEG chain length. A biphasic pattern of elimination was typical for 2-5 kD MPEG, however, 0.75kD MPEG-modified PGC exhibited a very rapid blood clearance similar to that of PL-DTPAGd with a major short phase (blood half-life t1/2= 0.6 h). A monoexponential blood clearance was only seen for MPEG5-containing PGC. The use of MPEG succinyl succinate for grafting allows the introduction of additional biodegradable ester bonds into the protective chains which tend to break down after prolonged incubation with plasma, while metal cation chelation to DTPA showed excellent stability even in the presence of an excess of apo-transferrin [17] (Fig. 3A,B). A typical time course of PGC-DTPAGd biodistribution in rats harboring ectopic mammary R3230 adenocracinomas is shown in Fig. 3C. The biodistribution confirms: 1) long circulation time in blood; 2) some residual uptake of PGC in the kidneys; 3) the uptake of PGC in tumors due to the enhanced permeability and retention (EPR) effect.
Table 1
Properties of PGC-DTPA(Gd) synthesized using poly-l-lysine hydrobromide with a degree of polymerization of 252 and a MW(LALLS) of 41.8 kD.
| MPEGS mass | Number MPEGS residues/ PGC molecule | Calculated average mass of PGC-DTPAGd | Apparent mass of PGC-DTPAGd, by SECa | Hydrodynamic diameter, mean±SD, nmb | Gd, % by weight | Blood half-life, hc | |
|---|---|---|---|---|---|---|---|
| Calculated | Elemental analysis | ||||||
| 0 | 0 | 136,000 | 315,000 | NM | 24.7 | 16.6±0.5 | 0.3d |
| 750 | 90 | 180,000 | 400,000 | NM | ND | 0.6 15e | |
| 2000 | 96 | 300,000 | 602,000 | 4.0±2.0 | 12.0 | 13.4±0.3 | 1.1 8.2e |
| 5000 | 92 | 560,000 | 1160,000 | 10.3±2.0 | 6.0 | 7.0±0.2 | 14.1d |
a- determined using protein mass standards for SEC;
b- determined using photon correlative spectroscopy
c- determined in rats (n=3/ time point), mean values are presented;
d- monoexponential fitting;
e- biexponential fitting of experimental data; first value - short component, second value - long
component
Figure 2
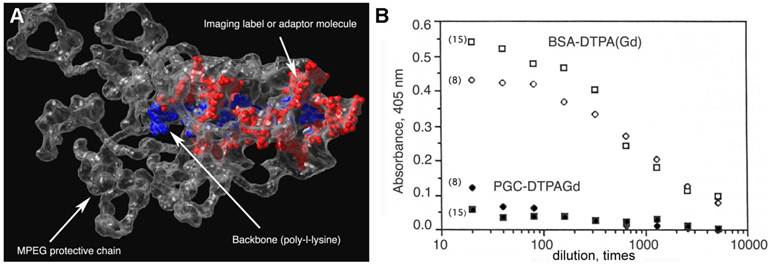
Protective elements in PGC and low immunogenicity. A - an approximate 3D-model of a PGC fragment showing the major elements of the graft copolymer structure (courtesy of Terence O'Loughlin, M.D., MGH-CMIR); B - the comparison of immune response to DTPAGd conjugated either to a protein (BSA-DTPAGd, open symbols) or to PGC (closed symbols) as determined by ELISA of blood plasma (in mice, n=3) using ovalbumin conjugated to DTPAGd for detecting the formation of antibodies. The numbers in parenthesis indicate the time interval before Gd injection and ELISA test. Adapted from [18].
Figure 3
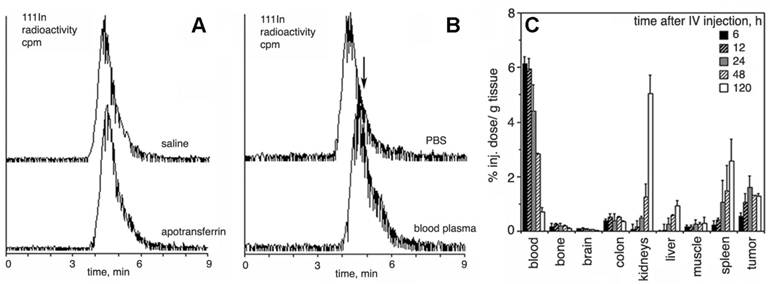
Resistance to transchelation, degradation in blood, and biodistribution in vivo. A- size exclusion HPLC profiles (scintillation radioactivity detector) of 111In-labeled, Gd-saturated sample of PGC-DTPA incubated in saline or PBS/2 mg/ml apo-transferrin for 48 h; B- size exclusion HPLC of 111In-labeled, Gd-saturated sample of PGC-DTPA incubated in PBS or whole blood for 48 h. The arrow shows the shift of the peak migration and the formation of a shoulder indicating biodegradation. C - residual blood-corrected biodistribution of PGC-DTPA (111In-labeled, Gd-saturated) in R3230 AC implanted Fisher rats (n=4 for each time point). Adapted from [17].
4. Applications in imaging
4.1. Quantitative MR and radioisotope imaging of blood volume and dynamics
Most pathological processes involve changes, at some level, in vascular structure or function. In some cases (atherosclerosis, stroke, and heart disease), these changes cause the disease and in other cases (diabetes, cancer, and inflammation) these changes promote and exacerbate the spread/pathology of the disease. Thus high resolution morphological and functional vascular imaging is the center stone of radiological methods for the diagnosis and staging of multiple diseases, as well as in treatment planning and evaluation (such as determining the metastatic potential of tumors or the amount of salvageable brain area after stroke).
Gadolinium labeled PGC (PGC-DTPAGd) enables high-quality, high resolution images of blood vessel morphology [18], in rodents and rabbits [27] that can be obtained at various magnetic field strengths (1.5-9.4T), Fig. 4. After injecting PGC-DTPAGd, we were able to track arteriovenous circulation on the obtained images by using angiographic MR pulse sequences. Both the arteries and veins of small diameter (0.25-1 mm) became readily discernible for 3 h after the injection. We achieved a level of contrast equivalent to that achieved by the low mass contrast agent DTPAGd, but at a much lower dose of Gd (20 µmol PGC-DTPAGd versus 100-200 µmol DTPAGd). With the introduction of high field MRI (9.4T) much higher signal-to-noise images could be obtained by injecting approximately 0.15 mmol PGC-DTPAGd/kg (Fig 4). We further demonstrated the feasibility of PGC-DTPAGd assisted detection of slow gastrointestinal bleeding [28]. In the above model study we showed that at a dose of 35 μmol/kg PGC-DTPAGd we could clearly delineate small volumes of blood in the gastrointestinal (GI) tract (stomach and small intestine), whereas the same volume of Gd-free blood was not detectable. These results demonstrate that due to the long blood half life of PGC, blood pool imaging can potentially be helpful in detecting venous as well as arterial bleeding and in imaging intermittent GI bleeding over time.
The advantages of DTPAGd-labeled PGC for determining blood flow and blood volume in animal models of disease have been assessed in several studies [29, 30]. Unlike radiochemical CAs, the signal changes generated by MR blood pool agents do not directly reflect their concentration since MR signal changes do not arise due to CA itself but instead are the consequence of CA effects on relaxation properties of surrounding proton spins. Because water exchange occurs between blood and the extravascular space, protons of water molecules carry magnetization out of the blood and into the extravascular space, rendering inaccuracies in the calculation of hemodynamic parameters. In an effort to offset these inaccuracies, most studies of blood flow and volume make a priori assumptions about the proton exchange rate (that it is either zero or rapid) for the organ or system under investigation. The use of a strictly intravascular CA such as PGC-DTPAGd allowed estimating the accuracy and precision of blood volume measurements [31]. When assuming fast exchange of protons between blood compartments, the true blood volume was generally overestimated and when assuming slow or no exchange the opposite was true (an underestimation occurred). As for the use of PGC-GdDTPA in measuring blood volume, we found that the best approach was to assume contributions from fast and slow proton exchange to be equal while carefully selecting imaging parameters. Such considerations were found to be of upmost importance in the accurate assessment of blood volume in an animal model of tumor neovascularization (see section 4.2).
Figure 4
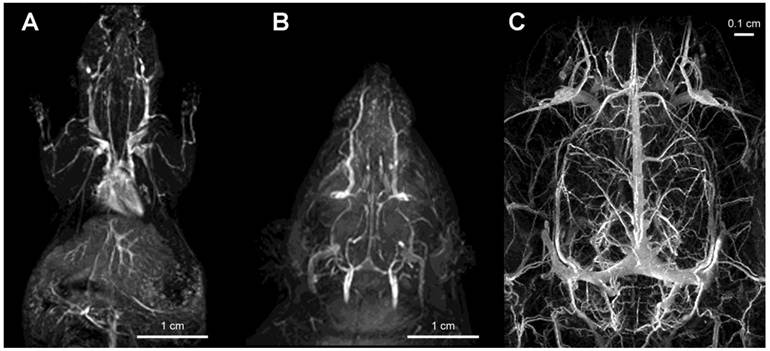
Imaging of vascular supply using PGC-DTPAGd. A- maximum intensity pixel (MIP) projection image of blood circulation in a mouse obtained using gradient-echo pulse sequence (1.5T, GRASS, TR/TE 60/8, FA 60, 2NEX, 8 cm field of view) after injecting PGC-DTPAGd at 25 µmol Gd/kg; B- a MIP image showing circulation in the head of a rat (imaging parameters similar to that of A); C - an improvement in imaging detail of rat cerebral circulation at 9.4T results from increased dose of PGC-DTPAGd (150 µmol Gd/kg) due to the lower molar relaxivity of Gd3+ at high magnetic fields. Adapted from [18, 103].
Based on the above findings we assessed transvascular water exchange in an acute stroke model using PGC-DTPAGd [32]. Because low molecular weight DTPAGd permeates the compromised brain vessels, its presence in the extravascular space is widely used for detecting this pathology in stroke. However early brain ischemia may not be detectible using conventional DTPAGd-enhanced MR angiography [33] [34]. We measured the early movement of water molecules out of the vasculature in mice with middle cerebral artery occlusion by acquiring MR images after the injection of PGC-DTPAGd. We found that water exchange across the vessel walls increased early (within 1h) after the middle cerebral artery occlusion and apparent diffusion coefficient (ADC) values in the same brain regions decreased over time consistent with the progression of edema due to increased intracellular volume.
Subsequently we tested a similar PGC compound obtained by chelating the paramagnetic metal dysprosium with PGC-DTPA. We found that a blood volume-dependent tissue signal (generated by T2- or T2*-weighted pulse sequences) can be monitored continuously, without interfering with arterial spin labeling technique, thereby allowing simultaneous blood volume and cerebral blood flow measurements (CAPTIVE imaging) [35]. Due to a very low r1 of dysprosium PGC-DTPADy does not interfere with the ASL induced signal, and thus affords an advantage over imaging with DTPAGd. CAPTIVE allowed high spatial resolution imaging of blood flow and volume changes occurring after middle cerebral artery occlusion in rats and these hemodynamic changes could be correlated with ischemia induced tissue damage. In a separate study [36] the CAPTIVE method was used successfully to measure hemodynamic changes during hemorrhagic hypotension.
Collectively, our results with PGC as a carrier for MR imaging CAs suggest that steady-state imaging with long-circulating agents potentially has important advantages for a number of MR clinical applications. These should be of greatest utility in cases such as stroke and GI bleeding when timely diagnosis is key to positive patient outcome.
We have also developed PGC-DTPA for blood pool imaging in nuclear medicine applications. We hypothesized that the availability of a long-circulating synthetic molecule that could be easily labeled with gamma-emitting isotopes would obviate the need to use human blood components as carriers for isotopes and therefore would be a substantial improvement in patients' and operators' safety. Our major expectation was that PGC-DTPA could be formulated into a kit form which needs only the addition of pertechnetate-99m solution to obtain a [99mTc]-labeled injection-ready preparation for imaging left ventricular ejection fraction in oncology patients undergoing potentially cardiotoxic treatments [37].
To obtain a labeling kit, we combined PGC-DTPA with a buffering solution in the presence of Sn(II) to enable reductive conversion of pertechnetate to a Tc oxocomplex [37]. After testing the kit in vitro and in three animal species [37] (representative SPECT/CT image in rats Fig. 5A) we tested PGC-DTPA[99mTc] in a phase I clinical trail that showed promising results. Twenty mCi (70 MBq) of the compound were injected I.V. and serial anterior and posterior whole body images were collected immediately and after a delay [38] (Fig. 5B). The compound showed the expected distribution and the heart and major blood vessels were clearly distinguishable up to 24 h post injection. The blood half life of the compound was 20.6 ± 2.3 hr. In four of the volunteers 22.1% ± 2.5% of the compound had been excreted through the kidneys 24 hr after iv administration, while the other two volunteers showed a greater distribution of the agent to the liver and spleen presumably due to DTPA labeling instability and not due to immunogenicity. A combination of the PGC long blood half-life with the advantages of a simple labeling protocol make this synthetic copolymer kit a potential substitute for blood products in blood pool imaging under the conditions when other blood pool imaging modalities fail to produce adequate results.
4.2. Imaging of tumor neovascularity
Neovascularization is one of the key characteristics of tumor growth [39]. Although neovascularity of a solid tumor does not necessarily imply malignancy, extensive sprouting of capillaries in malignant tumors has been shown to correlate with metastatic potential [39]. Non-invasive detection of altered tumor vessels is potentially important in biopsy planning, staging and in treatment evaluation (since decreased vascularized margins can be indicative of tumor regression). We initially used PGC-DTPAGd-assisted MR imaging for detecting the increase of the diameter of host vessels, e.g. epigastric vein, as an early sign of tumor progression in a rat model of mammary adenocarcinoma [40]. Six to 8 days after tumor implantation, vessels at the tumor/host interface became clearly visible by MRI. At later stages of tumor development, we observed formation of tumor blood vessels with abnormal morphology (Fig. 6A-C). Time-delayed imaging of the same animals (48 h post IV administration) revealed the enhanced leakage of PGC from immature and permeable tumor blood vessels (Fig. 6D-F) Therefore, PGC-DTPAGd-generated vessel enhancement of MR signal is effective in tracking the developing vascular tumor architecture non-invasively in vivo. The method is sensitive in the whole volume of the tumor at even early time points and has the capacity to be instrumental in detecting areas of enhanced vascular permeability.
We further tested whether PGC-DTPAGd distribution reflects tumor and interstitial relative blood volume fractions in a model of rat glioma [41]. We used consecutive I.V. injections of PGC-DTPAGd and DTPAGd where the first injection selectively saturates the blood signal so that the second I.V. injection of DTPAGd alters the MR signal of the tumor interstitium due only to its escape through abnormally permeable vessels of the tumor. Therefore, a simple subtraction of MR images obtained using the two contrast agents allowed mapping of relative vascular (VVF) and relative interstitial volume fraction (IVF) of the tumor within the same imaging slice, assuming very slow water exchange between compartments (which is valid given the use of specific pulse sequences). We were able to map patterns of intratumoral and intertumoral heterogeneity of both volume fractions at submillimeter pixel resolution and these patterns agreed with tissue histology data.
Figure 5
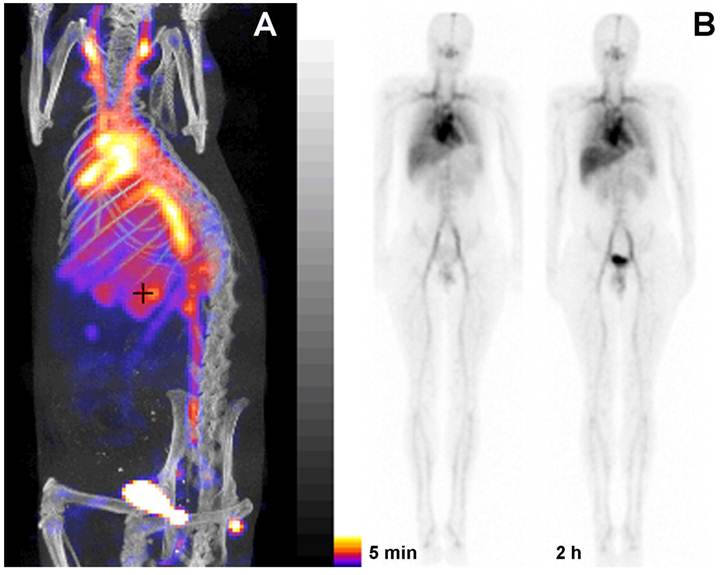
PGC as a blood pool imaging agent. A- a fusion SPECT/CT image of a rat 30 min after the injection of 99mTc-labeled PGC (2 mg PGC-DTPA/kg, 1 mCi 99mTc/rat). The image was obtained using NanoSPECT/CT (Bioscan Inc.); B- an image obtained in a healthy volunteer during Phase I clinical trials. Serial anterior whole-body images are shown taken immediately (30 min) and 2 h after the injection of 25 µg PGC-[99mTc]DTPA/kg (total dose - 20 mCi 99mTc). Note the distribution in the blood pool and low uptake in the lung, spleen and liver. Adapted from [38].
Figure 6
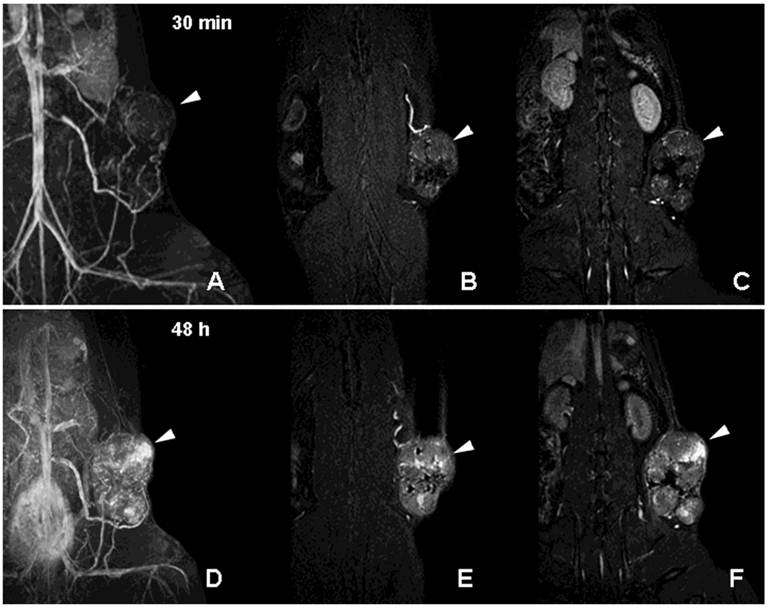
MR imaging of tumor neovascularity and interstitial leakage in a rat model of adenocarcinoma. A-C- a MIP projection (A) and two non-consecutive MR imaging slices obtained 30 min after I.V injection of PGC-DTPA(Gd) in rats with R3230 adenocarcinoma. D-F - MR images obtained in the same animal 48 h after the injection of PGC-DTPAGd. Note accumulation in the tumor at 48 h (the location of the tumor is shown by arrowheads). For MR imaging conditions see Figure 4A, legend. Adapted from [40].
The use of PGC-DTPAGd was further refined for measurement of absolute blood and interstitial volumes in a murine model of prostate cancer [42]. The measurements of blood and interstitial volumes calculated from Gd-based methods could be skewed when a prior assumptions of either large or negligible water exchange between the inter and extravascular compartment were made [31]. The is primarily due to the fact that some tumors (for example rat brain 9L gliosarcoma model) have significantly different water exchange rates than the surrounding normal tissue. In this study, we modeled a range of water exchange rates in order to establish a more precise method for quantification of absolute blood and interstitial volume in mice bearing slow growing MDA PCa 2b prostate tumors. Results showed that unlike more aggressive tumors, MDA PCa 2b tumors have very similar intra-/extravascular water exchange rates and blood volumes for tumor and non-tumor compartments. This raises the intriguing possibility that a dual tracer method using a large molecular weight, strictly intravascular CA such as PGC-DTPAGd in combination with DTPAGd may allow differentiation of aggressive versus slow-growing tumors. In a model of MCF-7 mammary adenocarcinoma overexpressing VEGF, PGC-GdDTPA revealed significantly higher vascular volume fractions in VEGF expressing tumors (8.9 ± 2.1) compared to control tumors (1.7 ± 0.5, p<0.003). VEGF overexpressing tumors had 1.6 fold higher microvascular density as assessed by histology [43]. Overall, the ability to separately measure relative vascular and extravascular tissue volume fractions in the same study may provide new algorithms for assessing tumor aggressiveness and response to treatment. In addition, because the size and make up of the tumor insterstitial volume will affect the ultimate concentration and distribution of chemotherapeutics that extravasate from the blood pool and reach the tumor, the knowledge of the IVF will be important in treatment planning and assessment of tumoral response to therapy.
In view of the latter, PGC-DTPAGd is especially useful for monitoring anti-angiogenesis. Since our studies in a tumor model overexpressing VEGF demonstrated a pro-angiogenic increase of tumor blood volume, we tested whether PGC-DTPAGd can also be used to detect anti-angiogenic effects of the experimental VEGF type 2 receptor tyrosine kinase inhibitor (VEGFR2-TKI) [44]. By using a steady-state blood pool imaging technique we were able to detect a very early decrease in tumor blood volume (Vb) after treating mice with two doses of VEGFR2-TKI, which in turn resulted in a decrease of vascular density and tumor cell apoptosis (Fig. 7A,B). By measuring the dependence of Vb measurement on flip angle using a gradient echo pulse sequence, we determined that in ectopic and orthotopic colon adenocarcinomas, a Vb change from 2.4% to 1.1% (at high flip angle of 80o [45]) was detectable only in tumors as opposed to the normal muscle blood volumes, which were not decreased by VEGFR2-TKI treatment (Fig. 7C). Alongside with Vb, water exchange across the blood vessel wall, reflected by the slope of Vb dependence on flip angle, was also decreased in treated tumors but not in the muscle (Fig. 7C).
Stable synthetic peptides derived from type 1 repeats of thrombospondin (TSP1) are a second class of molecules that we have tested for antiproliferative effects in a rat model of glioma [46]. After treating animals with TSP-1 derived peptides, we observed suppression of glioma growth that coincided with apparent inhibition of de-novo blood vessel formation, i.e. according to our PGC-DTPAGd enhanced MR imaging, TSP-1 derived peptides did not induce anti-angiogenesis but instead resulted in angiostasis. More recent MR studies have used both fluorescence and PGC-DTPAGd assisted MRI to show a reduction in functional de-novo vessel formation occurring after adoptive transfer of GFP/Noggin-expressing human endothelial cells in immunocompromised mice [47]. The effect of Noggin (a potent antagonist of bone morphogenetic protein signaling) on tumor proliferation and metastases awaits further study, but should be readily assessed with the use of PGC-DTPAGd -based perfusion imaging methods.
Figure 7
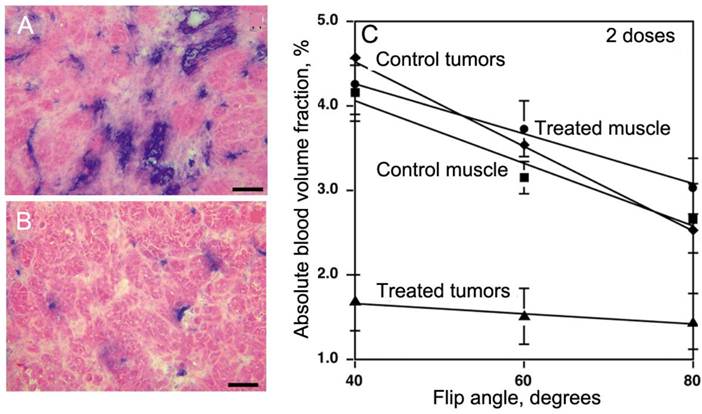
PGC-DTPAGd in imaging of anti-angiogenesis. A,B- histology of MV522 tumors prior to (A) and after treating animals with VEGFR2 tyrosine kinase inhibitor (TKI). Anti-CD31 staining (blue) reflects changes in vascular diameter and density. C - The dependence of absolute tissue blood volume fraction (Vb), measured after the injection of 50 µmol Gd/kg of PGC-DTPAGd from flip angle of MR SPGR pulse sequence that reflects changes of Vb due to TKI treatment. The slopes reflect changes in water exchange rates across the blood vessel wall. Adapted from [44].
4.3. Imaging the tumor microenvironment
Some of the properties of the tumor environment can potentially be imaged by using probes that respond to altered expression of hydrolases due to tumor-mediated recruitment of stromal cells in response to rapid extracellular matrix remodeling, locally elevated angiogenesis, and apoptosis. Specifically, protease expression and resultant extracellular matrix remodeling are regulated by complex interactions between extracellular matrix components and cells populating the tumor, including mesenchymal and hematopoetic stromal cells [48].
This has been initially demonstrated by conjugating a near-infrared fluorescent (NIRF) dye, Cy5.5 to PGC instead of GdDTPA (Fig. 8A).
Figure 8
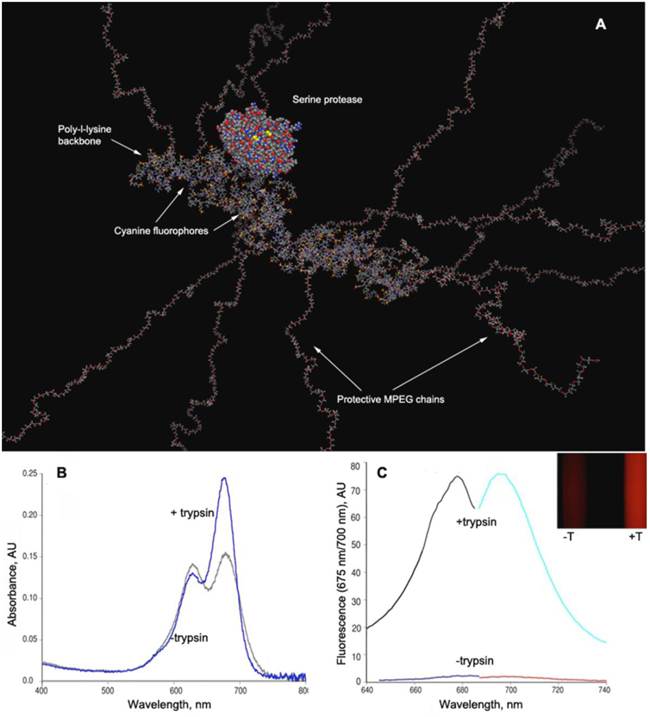
PGC-based protease-sensing optical imaging probe. A - a computer-generated model (Molecular operating environment, MOE) of a Cy5.5 cynaine dye-conjugated PGC showing 12 MPEG protective chains linked to a PL backbone (degree of polymerization - n=50) with the rest of N-ε-amino groups modified by cyanine dye. A molecule of trypsin (a model serine protease, m.w. 22 kD) is shown for comparison. The modeling was performed assuming a tetramolecular water hydration layer which is not shown (Laboratory of Molecular Imaging Probes, University of Massachusetts Medical School); B - change of Cy5.5 absorbance spectra before and after the trypsinolysis of N-ε-cyanine modified PGC; C - the increase of Cy5.5 fluorescence excitation and emission values after treating PGC-Cy5.5 with trypsin which translates into an increase of fluorescence intensity detectable with imaging after the trypsinolysis (inset, , -T - in the absence and +T in the presence of trypsin). Adapted from [49].
NIRF dyes are advantageous for in vivo optical imaging due to higher transmittance of the tissues in the near-infrared range of the spectrum (700-850 nm) as opposed to visible light, and lower emission of inherent biomolecules, thus allowing imaging of deeper tissue. In its native state, the PGC-Cy5.5 protease sensing probe has Cy5.5 molecules attached to the PL in close physical proximity and in a quenched state thereby preventing the dyes from emitting fluorescence upon light activation. After injection into tumor bearing animals, the probe accumulates in tumors due to high vascular permeability, and is subsequently exposed to high levels of tumor associated proteases. The poly-L-lysine backbone of the probe is readily cleaved by serine proteases, which apparently can gain access to the backbone poly-l-lysine due to relatively small diameters of the protein molecule (Fig. 8A), thereby freeing the Cy5.5 and rendering PGC fluorescent, due to degradation of the copolymer and physical separation of the quenched fluorophores (Fig. 8B,C).
In initial tests, this protease-sensing approach resulted in tumor associated fluorescence which was 12 times higher than surrounding tissue and that allowed detection of tumor xenographts in vivo [49, 50]. A family of cysteine proteases (possibly cathepsins B, H, or L) were determined to be the activating enzyme in this study although other proteases have since been shown to be detectable using specifically designed PGC based probes in a number of experimental models of cancer including those expressing cathepsin D [51, 52], matrix metalloproteinase [53-55], urokinase plasminogen-activator [56, 57] and caspase-1 [58]. The strength of this approach is that PGC attachment affords the accumulation of the probe in the tumor, while the cleaved products are sequestered in cells and tissues, thus allowing the amplification of signal over an extended time span (>24 hrs). Subsequent studies showed that the probe was taken up in some tumor cells but was mainly present in stromal cells recruited to the site of tumor formation (fibroblasts, monocytes/ macrophages, and dendritic cells) [59].
High molecular weight activatable probes should have a number of advantages in detection of tumors accessible to clinical optical imaging applications (i.e., those within 7-10 mm of the skin surface such as breast, prostate, and colon cancers), especially given the demonstrated sensitivity of these probes (detecting subnanomole levels of tumor-associated enzymes). These probes may prove valuable in differential diagnosis and staging of cancers. For example, cathespsin B-PGC probes allowed visualization of differential cathespin B expression in a well-differentiated human breast cancer (BT20) and a highly invasive metastatic human breast cancer (DU4475) in mice. Both types of breast cancers activated the NIRF probe so that tumors became readily detectable. However, in tumors of equal size, there was a 1.5-fold higher fluorescence signal in the highly invasive breast cancer which correlated with a higher cathepsin-B protein content [60] (Fig. 9). Furthermore, these probes should have utility in assessing therapeutic response as indicated in a study that proved feasibility of PGC-based metalloproteinase-3 sensing probe for detecting the effect of oncolytic adenovirus in tumors [55].
Figure 9
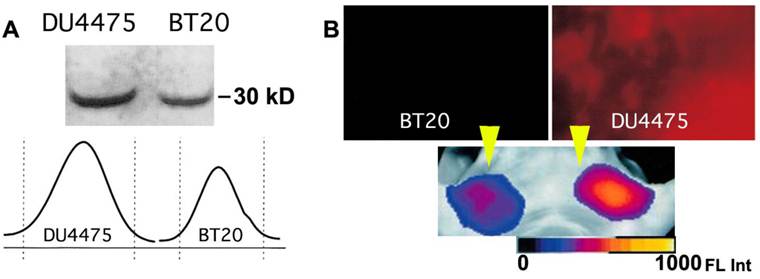
Imaging of PGC-Cy5.5 breakedown in mammary adenocarcinoma tumors. A- the immunoblot demonstrating a higher level of cathepsin B expression in DU4475 than in BT20 tumors, with the band integration profiles shown below the image of a blot. Reproduced with a permission from [60]; B - ex vivo planar imaging of Cy5.5 fluorescence in adenocarcinoma samples and the corresponding in vivo image of orthotopic tumors implanted in the same animal bilaterally (shown by yellow arrowheads). The animals were injected with 2 nmol of Cy5.5 conjugated to PGC 24 h prior to optical imaging.
4.4. Imaging markers of inflammation, atherosclerosis and diabetes
Diseases with different etiologies often share common pathological phenotypes due to activation of the body's innate defense system which is triggered by nearly all external and internal insults. For example, vascular changes such as increased vascular permeability accompany many diseases as the body attempts to increase blood flow to sites that will require efficient crossing of endothelial barriers by the cells of the immune system. Thus the PGC-based imaging agents described thus far may have utility in detecting a wide range of diseases because of their extravasation that is highly atypical in normal tissues. When used to detect E. coli induced infections in rats, Gupta et al [61] found that PGC-DTPAGd yielded higher relative signal than [111In] labeled IgG due to the lower accumulation of PGC in normal muscle tissue, and PGC-DTPAGd yielded similar signal ratios when compared to [111In]-labeled white blood cells and [99mTc]-labeled chemotactic peptides. Increased vascular permeability in infected tissue was regarded as the main reason for increased probe concentration in these areas. Similar permeability changes were detectable with high-field MRI in the pancreatic vasculature of type 1 diabetic rats [62], suggesting that MR imaging may be useful in detecting very early changes in pancreatic vasculature that are believed to precede the onset of full disease.
PGC-based NIRF probes designed to target inflammation specific molecules have also been evaluated in animal models of osteo- and rheumatoid arthritis. The disease is characterized by early inflammatory responses in which the release of matrix-degrading enzymes (e.g., matrix metalloproteinases (MMPs), cysteine proteases and cathepsin B), into the synovial fluid cause the destruction of arthritic joints due to the degeneration of proteoglycan and type II collagen. These events occur before morphological changes in joint structure can be detected. In two separate studies, cathepsin B activated NIR probe (PGC-Cy5.5) injected 24 hr prior to imaging resulted in a 3-fold increased fluorescence signal intensity in the affected joints of animals with experimentally induced arthritis as compared to control animals [63, 64]. In arthritic animals treated with the anti-inflammatory drug methotrexate (35 mg of MTX/kg 48 hours prior to probe injection), a significantly lower fluorescent signal was observed as compared with untreated arthritic animals [63]. Although X-ray and MRI have established utility in assessing the progression of arthritis, these imaging methods detect changes only in more advanced disease. Thus NIR imaging of protease activated probes may offer an advantage in detecting the earliest changes in the disease, when anti-inflammatory therapy has the greatest chance of limiting joint damage. In a more recent study, an altered form of the probe was used in which poly-D-lysine replaced poly-L-lysine and a cathepsin K sensitive peptide linker (GHPG-GPQGKC) was used for attachment of Cy5.5. This configuration resulted in a probe that was activatable only by cathepsin K [65], and was subsequently shown to detect osteoclast-derived cathepsin K mediated bone degradation, which is largely responsible for bone resorption.
Atherosclerosis is another highly prevalent disease that is now considered to be largely linked to vascular wall inflammation, and that may be detectable in its early stages with long circulating protease sensitive PGC based probes. An early pathological sign of the disease, the fatty streak, is an inflammatory lesion which is caused by the accumulation of monocyte-derived macrophages and T lymphocytes [66]. The final (and often fatal) sequelea of the disease, plaque rupture, is also associated with inflammation and has been correlated with the presence of highly activated macrophages [67, 68] which release proteolytic enzymes that hasten plaque erosion and rupture. Experimentally induced atherosclerotic lesions in mice were successfully imaged using fluorescence-mediated tomography (FMT) after the injection of PGC-based “activatable” near-infrared fluorescence (NIRF) probes for cathepsin B [69] and matrix metalloproteinases (MMP2 and MMP9) [70]. Histological analysis showed co-localization of the probes with the targeted enzymes and with activated macrophages. In addition, the probe mediated signal increase could be diminished after treatment with protease inhibitors [70]. MMP sensitive probes have also been used to detect inflammatory responses occurring after myocardial infarction in mice [71].
5. Applications in drug delivery
5.1. Synthesis and applications of PGC-covalently linked drugs
Developments in synthetic chemistry and nanotechnology as well as improvements in the ability to re-engineer intrinsic biomolecules, have supplied the field with a number of promising drug delivery systems. Ideally, these systems should be capable of protecting the active therapeutic as it travels to its organ of destination and then releasing the payload in full so that the drug concentration at the site of action is equivalent to the administered dose. When administered directly into the circulation, the drug-carrier complex should ideally exhibit low binding to serum proteins and be resistant to immediate uptake by the reticuloendothelial system of the liver and spleen, or to rapid elimination by the kidneys. Passage of the drug through the tight junctions of the vascular endothelial barrier and into the interstitium is another necessary step for delivery of a therapeutic molecule to its cellular destination. Ultimately, these characteristics are conferred by the biochemical properties of the drug (such as its molecular weight and charge), and can also be influenced by the disease process itself. For example, many high molecular weight drug conjugates are known to accumulate in tumors to a greater extent than their free drug counterparts. This phenomenon termed “enhanced permeability and retention (EPR)” [72, 73] occurs because abnormal (i.e. 'leaky') vascular structure in tumors are highly permeable to high molecular weight drug conjugates.
Methods for encapsulating therapeutic compounds (such as the use of liposomes or polymeric micelles) and for targeting these compounds to specific tissues and cells (such as attachment of antibodies) have greatly widened the rage of compounds that can now be considered for development as therapeutics [74].
The development of protected graft copolymers has made available a new and potentially very flexible method for drug delivery. The potential for use of PGC based compounds as drug delivery agents is related not only to their ability to carry therapeutic molecules in a non-specific manner (“passive targeting”) through the vascular system and into the interstitium, but to their ability to be modified so as to selectively target molecules in the extracellular space (such as adhesion molecules or enzymes) or on the cell surface (i.e. “active targeting”). We have found that PGCs have a slow rate of biodegradation, are biocompatible (show low immunogenicity) and are eventually eliminated from the body after cleavage into small fragments. Additional advantages of PGCs include: 1) they accumulate at sites of high vascular permeability, 2) they are large enough to escape glomerular clearance (> 4nm in diameter) even when bound to drug, 3) they have a very slow RES clearance rate, and 4) they can potentially decrease immunogenicity of the drug to which they are bound.
Somewhat surprisingly, the capacity of PGC to 'passively' accumulate in tumors was found to be as high as that of an antibody targeted drug conjugate [75]. Using radionuclide and fluorescent labeled derivatives of PGC and BR96 (a tumor-specific chimeric monoclonal antibody [76]) we found that PGC slowly accumulated in LX-1 human small cell lung carcinoma xenografts (Fig 10A) within a 48 h period (to 5.2 ±1.7% of the injected dose per gram of tissue), while the antibody accumulation peaked at 24 h and was only slightly higher at 6.0± 3.2% ID/g (Fig.10B). Tumoral tissue distribution studies showed that roughly 43.9% of the PGC was associated with the tumor cell fraction while the remainder had accumulated in the interstitium. The large accumulation of PGC in tumor cells is likely due to its extravasation from leaky tumor vascular and subsequent retention in the tissue which serves as a depot for prolonged fluid phase endocytotic uptake by the cells. This hypothesis is supported by in vitro studies showing that the copolymer showed negligible binding to the cell surface as compared to the antibody which showed high affinity binding (Kd of 4.5 x 10-7 M) followed by active vesicular internalization. In a subsequent study by Choi and colleagues [77], this approach for achieving high concentrations of PGC bound drugs in tumors was shown to have therapeutic applicability when used to accumulate chlorine e6 (Ce6) in tumors. Ce6 is a porphyrin-based molecule that can be excited by light to emit fluorescence and simultaneously produce cytotoxic singlet oxygen, which causes local tissue damage. When used to treat xenografted HT1080 fibrosarcoma tumors in mice, a single dose of PGC-Ce6 followed by phototherapy was sufficient to decrease the tumor proliferation rate by one half.
In order to render PGC molecules more applicable for the diagnosis and treatment of diseases that are not characterized by leaky vasculature, PGC can be targeted to specific tissues and cells by conjugation to antibodies and other targeting moieties. PGC probes were successfully targeted to cultured human endothelial cells by conjugating the F(ab')2 fragment of monoclonal anti-human E- selectin monoclonal antibody (H18/7 [78]) to PGC modified by the addition of either a sulfhydryl-reactive terminal vinylsulfonate group or a terminal carboxyl group [79] (Fig. 11A). The two conjugation strategies resulted in compounds with similar antibody/PGC ratios and apparent hydrodynamic radii. When added to human cultures, 125I labeled F(ab')2-PCG showed a binding affinity to HUVEC cells expressing high levels of E-selectin (after interleukin 1β mediated activation) that was 25 to 30 fold larger than the non-specific uptake observed for non-stimulated HUVEC cells (Fig. 11B,C). Furthermore the binding yield of the conjugate (expressed as a per cent of the total added antibody fragment) was almost 25% higher than for free F(ab')2 indicating that conjugates with the higher F(ab')2 content were preferentially bound to the cell surface due to the increased avidity effect. This targeted PGC probe should have indications for in vivo imaging of a host of diseases in which E-selectin expression is upregulated in association with inflammation and angiogenesis.
Figure 10
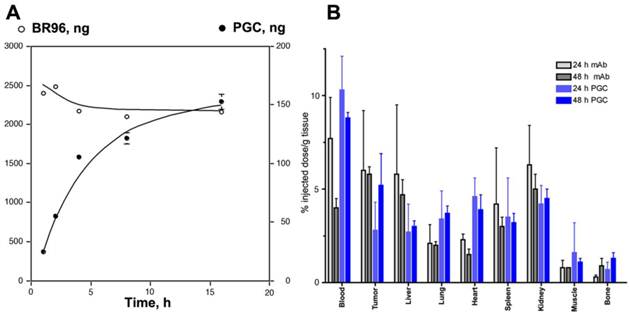
Targeted antibody and non-targeted PGC accumulation in the same LX-1 non-small cell lung carcinoma model. A - a time course of 111In-labeled monoclonal internalizing antibody BR96 and PGC uptake in LX-1 cells; B - biodistribution of 111In-PGC (blue bars) and 111In-BR96 (grey bars) in LX-1 xenografts after intravenous injection in LX-1 bearing mice at 24h (n=4 animals/time point). Adapted from [75].
Figure 11
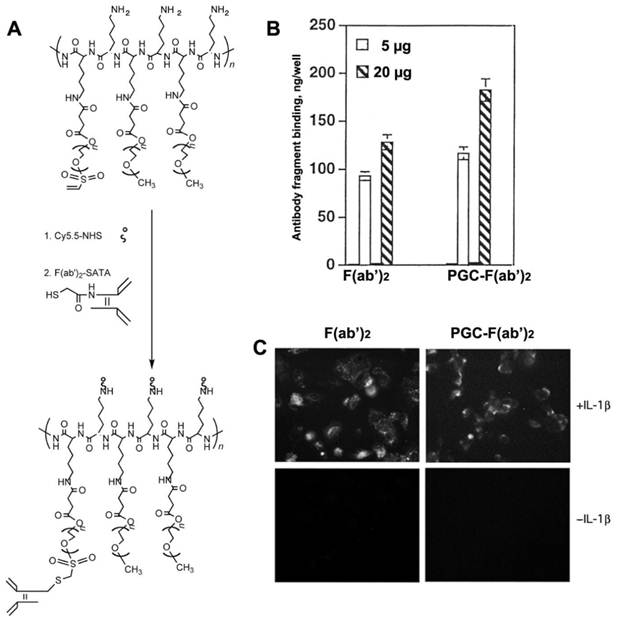
Human endothelial cell activation-dependent uptake of anti-E-selectin conjugated PGC. A - synthesis of anti-E selectin targeted PGC using SATA-modified F(ab')2 antibody fragment conjugated to heterobifunctional PEG-linkers carrying vinyl sulfone groups; B - comparative uptake of non-conjugated F(ab')2 antibody fragments and PGC- F(ab')2 conjugate; C - fluorescence microscopy of IL-1β activated and non-activated cells demonstrating the uptake of both Cy5.5-labeled antibody fragment and PGC- F(ab')2 in LX-1 cells. Adapted from [79].
5.2. PGC- cisplatin complex and anti-proliferative activity
Our initial attempt at modifying PGCs for use in drug delivery entailed the synthesis of an adduct of the antineoplastic drug cis-diamminedichloroplatinum(II) (cDDP) and PGC [80]. cDDP and other antineoplastic drugs containing platinum are effective cancer treatments due to their ability to cross-link DNA and thus inhibit division/ proliferation of malignant cells [81]. The motivation in creating a modified form of cDDP was to increase the bioavalibility of the drug after I.V. administration which is otherwise very low due to cDDP's rapid clearance by the kidney and irrevesible high affinity binding to plasma protiens [82, 83]. We also hypothesized that binding to PGC would decrease the non-specific toxicity of the drug. Since Pt(II) is known to form complexes with carboxyl residues of polycarboxylic acids, we hypothesized that succinyl residues attached to the backbone PGC with succinylated amino groups will similarly bind the aquated platinum(II)-diammine complex. We found that simple co-incubation of cDDP (12 mg/ml in DMF) with a water solution of succinylated PGC (20 mg/ml in water) at 37 °C resulted in an adduct of PGC-cDDP containing 4.3% of platinum by weight [80].
In vitro studies showed a 50% inhibition of human BT-20 adenocarcinoma cell proliferation at a PGC-cDDP dose of 0.9 ± 0.2 μM as compared to 0.3 ± 0.1 μM for free cDDP. Because only free platinum complexes are effective in inhibitiing cell division, this result suggest that cDDP could dissociate from the PGC complex in vitro. Indium-111 labeled PGC-cDDP was used to study the kinetics and biodistribution of the compound in vivo. We determined that due to a long blood half-life of 14 h, PGC-cDDP accumulated in rat mammary tumors (13762NF and R3230) at a dose that was 5-fold higher (2.5 to 3.5% of the injected dose/ gram of tissue) than a control adduct of cDDP with a short half life. In vivo imaging using radiolabeled PGC-cDDP showed high regional activity in tumors, whereas the control adduct accumulated mainly in the kidneys. This pattern of distribution suggested that accumulation in tissue results from extravasation of cDDP and release of the drug in the tumor. We also found that 80% of the PGC-cDDP adduct was eliminated from the circulation 48 h after I.V. administration, and exhibited a nearly 5-times lower concentration in kidney.
In a following study [17] we assessed the therapeutic efficacy of PGC-cDDP in a cDDP-sensitive F9 murine teratoma model. We injected identical doses (7.5 mg) of free or PGC-linked cDDP/kg every 3 d. Non-treated mice showed a 17-fold increase in tumor volume over 30 d after implantation. While animals treated with free cDDP showed reduction in tumor volume, they also showed an average 24% weight loss over the course of 6 d (2 treatments) with 100% mortality. All animals treated with PGC-cDDP survived six consecutive treatments over 15 d and showed on average a 10% reduction in tumor volume, with 25% of the animals showing a complete regression of the tumors. The collective results from these studies demonstrate that that PGC-succinate can serve as a high-capacity carrier for sustained release of cDDP in vivo. The drug conjugate shows less systemic toxicity than the free drug while retaining antiproliferative effectiveness.
5.3. Non-covalent complexes of drugs and PGC
The obvious advantages of the non-covalent association of drugs with drug delivery carriers is in scalability, in the ease of formulating drugs-containing compositions and in the lack of the need to chemically modify the drug molecules. The latter is a critical advantage in developing drug delivery formulations containing physiologically active peptides and proteins, many of which are capable of inactivation during chemical modification. The development of new formulations and devices for administration of such peptides, proteins, as well as low molecular weight drugs is driven by the need to achieve targeted and highly specific physiological effects. With respect to peptides and proteins, many of them are unstable in the gastro-intestinal tract and therefore may need to be stabilized and protected or otherwise delivered via systemic circulation. In addition, peptides and proteins that have low molecular masses tend to have short biological half-lives due to their efficient removal from systemic circulation via kidneys. A fraction of these peptides and proteins can also be removed via reticuloendothelial uptake due to recognition by monocyte/macrophages and sinusoidal endothelial cells exacerbated by opsonization of drug carriers by the complement system. As a rule, many peptides and proteins lose their activity in vivo due to proteolysis (peptide bond cleavage). Several drug delivery strategies can be useful to circumvent these undesirable effects and thus increase peptide and protein delivery in vivo. First, a pump can be employed for continuous systemic drug infusion. This strategy is proven efficient in clinical practice but may be impractical or disadvantageous for outpatients because it restricts mobility, lowers quality of life and potentially results in I.V. line infections. Second, peptides and proteins can be included in an implantable pump with a membrane allowing diffusion of the drug at a desirable release rate. Due to limited volume of these capsules, peptides and proteins are often used in a concentrated formulation, which leads to aggregation resulting in loss of solubility and potential loss of specific activity. In most cases, the drug is released into the extracellular space and is carried by lymphatics. Overall concentration of peptide or protein may be affected by local lymph node activity and the efficacy of lymph node drainage at the implantation site. There is also a likelihood of host reaction to capsule material. Third, the drug release system can be made biodegradable as a result of encapsulation or inclusion into degradable drug delivery vehicles or carriers, e.g. polymeric matrices, particles or membrane vesicles (liposomes). These delivery systems are usually either implantable or injectable. Implantable drug delivery systems are often placed under the epidermis where the components of the system are usually slowly degraded as a result of biological activity of surrounding cells (i.e. as a result of the release of enzymes degrading chemical bonds that hold these implants together).
The potential problems associated with the use of delivery compositions containing non-covalently bound drugs is in their potentially low thermodynamic stability in solutions that necessitates formulating drug delivery compositions shortly before use. This disadvantage is not shared by PGC-based carriers that can be subjected to lyophilization together with the drug load due to their cryoprotective properties [84].
5.3.1. PGC with hydrophobic cores (HC-PGC) as drug carriers
Most peptides are purified after synthesis using reversed phase HPLC because of their affinity for alkyl groups covalently linked to the resin. Compared to resin with diameter of several microns, PGC has diameter of 10 to 50 nm. Despite these small diameters, PGCs can be modified to contain a high density of long-chain fatty acid groups covalently linked to the PL backbone. Specifically, PGC can be synthesized to have a hydrophobic core consisting of stearic acid residues covalently linked to PGC (Fig. 12A). Such structure has certain similarity to an HPLC carrier matrix that is covalently linked to octadecane groups. However, unlike a silanol-grafted HPLC matrix, the bonds in PGC are peptide and there are also MPEG protective chains covalently linked to PL. We have previously shown that such “hydrophobic core” (HC)-modified PGC is capable of reversibly binding peptides/proteins that are then capable of partitioning into hydrophobic cores. Alternatively, they can have affinity for fatty acids and thus linked to the MPEG-grafted PL backbone. One such example, i.e. a computer-generated model of leptin non-covalently associated with aliphatic chains of HC-PGC, is shown in Fig. 12B. The peptide formulation with HC-PGC is accomplished by simple co-lyophilization of peptide with PGC followed by reconstitution in the injection buffer. Peptide formulations prepared in this manner demonstrate an extended blood circulation time after subcutaneous injection.
In general, when mixed with an appropriate peptide that binds to HC, the resulting HC-PGC formulation is expected to have an affinity-based peptide release mechanism. Under normal physiological conditions in vivo, one would expect such peptide-HC-PGC complexes to dissociate due to a depletion of free peptide pool (which depends on kinetic and thermodynamic stability of the peptide complex) which can accelerate in the presence of fatty-acid binding proteins (e.g. serum albumin). However, due to steric hindrance imposed by the drug carrier-linked MPEG chains [85], which slows down the process of peptide displacement from the complex, such competition is likely a less significant factor in peptide-HC-PGC complex dissociation than in the case where MPEG chains were absent. Hydrophilic MPEG chains are also the major reason why HC-PGC can exist as a colloidal solution in water. The overall hydrodynamic diameter of HC-PGC is 20 nm, which circumvents the rapid kidney elimination but at the same time, enables trafficking from subcutaneous sites to the blood, predominantly via the lymphatic system.
Stabilization by PGC is particularly useful for peptides that rapidly degrade due to proteolysis. Most proteolytic enzymes in the blood have a Michaelis constant (Km) in the range of several micromolar and most peptide receptor effective thermodynamic constants (Kd) are in the nanomolar range. Therefore, for PGC delivery of peptides to be successful, the complex of peptide-HC-PGC must have a Kd below the enzyme Km but above the receptor Kd (preferably <1uM). In the past we tested several highly potent small peptides, e.g. glucagon-like peptides (GLP-1 [19, 86-88], GLP-2), atrial natriuretic peptide (ANP) and leptin as potential cargo for loading HC-PGC. Below are some of the examples of our efforts directed at improving the stability and bioavailability of the above peptides in vivo.
5.3.2. Formulation and HC-PGC-mediated delivery of atrial natriuretic peptide (ANP) and leptin
Atrial natriuretic peptide (ANP) and brain natriuretic peptide (BNP) are hormones produced by the heart that bind to natriuretic peptide receptor A (NPR-A) in target organs (blood vessels, kidney, adrenal, and heart) and generate intracellular cGMP [89]. They act through many parallel mechanisms to promote cardiac output and function, i.e., they antagonize the renin-angiotensin-aldosterone system [90-92], inhibit endothelin secretion, reduce systemic and renal sympathetic activity, increase natriuretic, diuretic and vasorelaxant activity, and act directly on the kidney by dilating the afferent arteriole and constricting the efferent arteriole [93, 94]. Thus natriuretic peptides (NP) can be viewed as inherent biological molecules which are capable of producing most of the therapeutic results that are produced by first line medications in use today for the treatment of heart failure (HF). In addition, NPs may offer additional therapeutic benefits in treating patients with HF because they have the unique ability to inhibit TGFβ induced Smad translocation into the nucleus [95] and thus suppress myocardial fibrosis and hypertrophy or remodeling [96, 97] which would otherwise lead to further heart deterioration in patients with heart disease. Currently used treatments for HF directly inhibit cardiac scarring and remodeling and thus patients remain at a greater risk of future cardiac events and generally poor long-term outcome. Several studies have shown benefits of ANP and BNP in preventing cardiac remodeling in ischemic injury. Patients who received an infusion of ANP or BNP during or for 2-3 days after reperfusion procedures have shown decreased infarct size, fewer reperfusion injuries, and better outcomes (lower incidence of readmission to hospital) than controls [98].
Advances in formulation chemistry over the last several years have lead to increased clinical use of NPs. ANP has a blood half-life of only 2.4 min and, as a result, has to be administered as an I.V. infusion. BNP also has a short half-life (12.1 min), and has been approved for the treatment of HF (also as an infusion). Unfortunately, bolus administration of large amounts of these peptides leads to hypotension and this effect persist (10% incidence) even if lower amounts are injected (0.025 µg/kg/min of carperitide) [95]. Therefore, the development of a formulation of these peptides that has a longer blood half-life would potentially allow patients to self-administer the drug outside the hospital setting. Our goal has been the development of a long-acting formulation of NP that can be used by patients who have received reperfusion treatment to minimize future cardiac events and also to improve the long term outcome for those who have had mild myocardial infarction without receiving reperfusion treatment. We envision that the use of a PGC drug delivery system will allow the development of injectable NPs (ANP and BNP) for use in patients with both mild and severe myocardial infarction, thus expanding the benefits of NP therapy to the majority of patients suffering from coronary heart disease.
Leptin is a 16 kDa protein hormone that plays a key role in regulating energy intake and energy expenditure, including appetite and metabolism. Leptin is one of the most important adipose-derived hormones. Leptin binds to the ventromedial nucleus of the hypothalamus, known as the "appetite center." A small group of humans possess homozygous mutations for the leptin gene, which leads to a constant desire for food, resulting in severe obesity [99]. This condition can be successfully treated by the administration of recombinant human leptin [100, 101].
Our initial formulation of the HC-PGC complex with ANP and leptin was obtained by simply combining HC-PGC and the corresponding peptide in isotonic buffered saline. This particular HC-PGC had a 20kD PL backbone and a high (55% of the N-ε-amino groups) modified with 5kD MPEG, while the remaining amino groups were modified by acylation with stearic acid N-hydroxysuccinimide ester.
After 1 h, the mixture was separated using a membrane of 100 kD cutoff that efficiently retained the HC-PGC complex. The free and the HC-PGC bound ANP and leptin complexes were released from HC-PGC by treating them with 50% acetonitrile. The free and bound ANP and leptin were then analyzed by quantitative HPLC which enables estimation of the effective Kd value (Fig. 13A). We obtained a Kd value of 1.1 uM and determined that each 350kD HC-PGC carrier had on average 9 ANP molecules (mol. mass 3kD) bound to it. Leptin formed a more stable complex with HC-PGC (Fig. 13A). It should be noted, that estimation of effective Kd values reflecting the affinity of leptin and ANP to HC-PGC shows that the affinity correlates with GRAVY index (hydropathicity of leptin is greater than ANP), while the capacity of HC-PGC carrier inversely correlates with peptide mass (ANP<leptin).
The pharmacokinetic profile of the PGC-ANP formulation was determined in mice at various time points using ELISA of blood serum after intravenous administration of either free ANP or HC-PGC-ANP. Data were corrected for endogenous background ANP. It should be noted that mice have a higher metabolic rate than humans and the conversion factor (mouse to human) in dosing is 12:1 to 9:1 [102]. Hydrophobic core-PGC bound ANP had an extended circulation time compared to non-conjugated peptide and the extension of circulation was a function of the ratio of peptide to PGC (Fig. 13B). It is clear that reversible association of peptides with HC-PGC enables sustained release after systemic administration in mice (2µg peptide/animal, n=4, group) with a resultant large increase of area under the curve (AUC).
Figure 12
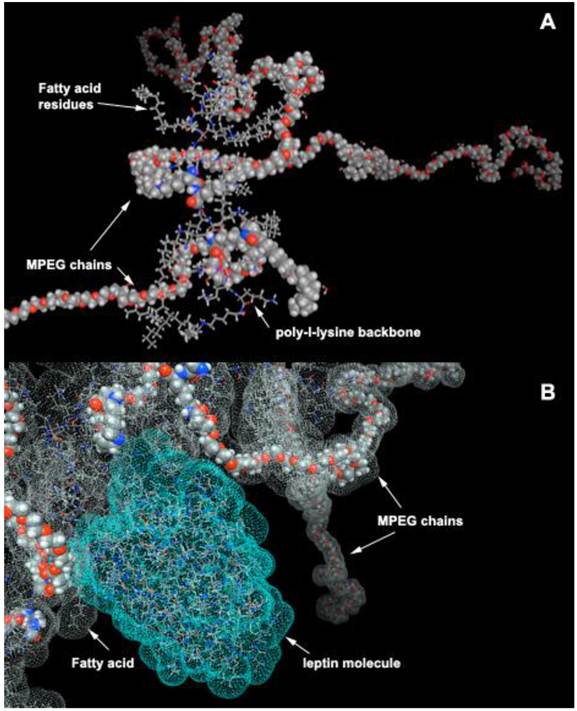
HC-PGC as a drug carrier. A- a computer generated model of HC-PGC fragment with MPEG protective chains (shown space filled) and hydrophobic elements represented by fatty acid residues; B - a computer simulated representation of a leptin molecule binding to the hydrophobic “core”. Interacting surfaces are shown in green and grey for leptin and HC-PGC, respectively. The modeling is performed in MOE (Laboratory of Molecular Imaging Probes, Univesity of Massachusetts Medical School).
Figure 13
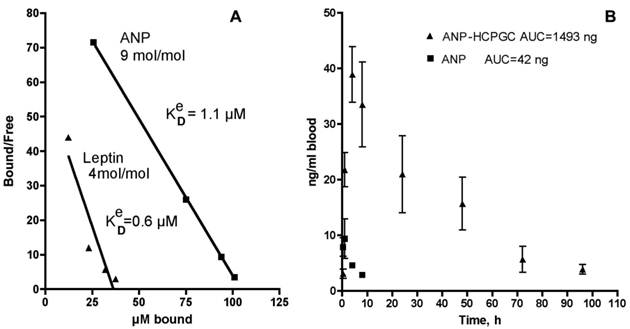
Polypeptide binding to HC-PGC and the release of HC-PGC bound cargo after the I.V. administration. A - The binding of atrial natriuretic peptide (ANP) and leptin to HC-PGC. The affinity to HC-PGC correlates with GRAVY index (hydropathicity, leptin>ANP) and the capacity of HC-PGC carrier inversely correlates with peptide mass (ANP<leptin). B - reversible association of ANP with HC-PGC enables sustained release after intravenous administration in mice (2µg peptide/animal, n=4, group) with a resultant strong increase of AUC compared to native ANP (Data provided by Pharmain Corp. Seattle WA).
6. Conclusions and perspectives
As summarized in this review, the development of MPEG-protected polyamino acids has resulted in one of the most successful graft-copolymers to date for use as a carrier of potential diagnostic and therapeutic agents. Much of the potential of PGC lies in its versatility and ease of chemical modification and thus adaptation for delivering both convalently and non-covalently linked cargo. PGC can be engineered as a covalent amphiphile that is capabe of binding to hydrophobic residues of small proteins and peptides resulting in a “hydrophobic core” (HC)-modified PGC formulation that acts as an affinity-based peptide release mechanism. PGCs can also be made with zinc bridges for anchoring zinc-binding proteins. Alternately, non-covalent attachment can be designed for carriers containing physiologically active peptides and proteins, many of which are rendered inactive upon chemical modification. PGC-based carries can be synthesized relatively inexpensively and on a large scale bases for commercial applications. Most importantly, these formulations are expected to remain stable in solutions or in lyophilized form for extended periods of time.
PGCs offer a plurality of advantages in comparison to other carries designed using PEGylation technology, e.g., multiple available linking sites for drug and adaptor molecules, long circulation times without compromised biodegradability, and no toxicity or immunogenicity. The safety and biocampatability profile of PGC has in fact resulted in its being one of the few 100% synthetic non-proteinaceous macromolecules that has suceeded in passing the initial safety phase of clinical trials. This safety profile, along with the capability of MPEGs to protect the inner drug cargo from uptake and degradation, forms the bases of its outstanding potential for use in the development of a wide range of future therapeutic and diagnostic agents.
PGCs conjugated to contrast agents such as paramagnetic gadolinium are capable of long circulation times after injection into the blood stream and as such offer advantages in angiographhic imaging applications used for the assesment of disease related vascular pathologies. These should be of greatest utility in cases such as stroke and GI bleeding when timely diagnosis is key to positive patient outcome and will also be beneficial in chronic diseases such as cancer where assessment of tumor vascularity following therapeutic intervention is critical for further treatment planning. In addition, because of their extravasation that is highly atypical in normal tissues, PGC-based therapeutic agents can accumulate in diseased tissue in sufficient quantities to be of therapeutic benefit. On the same bases, accumulation of enzyme sensing PGC-based imaging agents allows for amplification of diagnostic signal and thus higher sensitivity detection of pathological changes occurring at the cellular and tissue level. Furthermore, PGC technology applied to near-infrared fluorophores as optical imaging probes is also anticipated to have a number of advantages in detection of tumors accessible to clinical optical imaging applications (i.e., those within 7-10 mm of the skin surface such as breast, prostate, and colon cancers), especially given the demonstrated sensitivity of these probes (detecting subnanomole levels of tumor-associated enzymes). These probes may prove invaluable in the differential diagnosis and staging of cancers. A future, but as yet untested use of PGC-based carriers is in non-viral delivery of genes and other nucleotides such as for the delivery of siRNA. As a non-modified copolymer, PGC carries multiple positive charges and can potentially form polyionic complexes with polyanions, e.g. plasmid DNA.
In conclusion, the chemical versatility, safety, cost-effectiveness and tissue permeability and accessibility profile of PGC-based compounds offers an exciting pathway for future development of highly sensitive and targeted diagnostics and therapeutics.
Acknowledgements
We are grateful for contributions to PGC applications development to current and former members of the Laboratory of Molecular Imaging Probes (University of Massachusetts Medical School) and Center for Molecular imaging Research (Massachusetts General Hospital). This work was supported by NIH grants: R01CA074424-02, R44AA019584 and R44DK069727.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Toncheva V, Wolfert MA, Dash PR, Oupicky D, Ulbrich K, Seymour LW. et al. Novel vectors for gene delivery formed by self-assembly of DNA with poly(L-lysine) grafted with hydrophilic polymers. Biochim Biophys Acta. 1998;1380:354-68
2. Choi YH, Liu F, Kim JS, Choi YK, Park JS, Kim SW. Polyethylene glycol-grafted poly-L-lysine as polymeric gene carrier. J Control Release. 1998;54:39-48
3. Bogdanov A, Kayne L, Weissleder R. Graft copolymers as carriers for systemic delivery of expression vectors. In: (ed.) Controlled Release Society I. The 25th Int. Symp. Controlled Rel. Bioact. Mater. Las Vegas, NV: Controlled Release Society, Inc. 1998:91-2
4. Read ML, Dash PR, Clark A, Howard KA, Oupicky D, Toncheva V. et al. Physicochemical and biological characterisation of an antisense oligonucleotide targeted against the bcl-2 mRNA complexed with cationic-hydrophilic copolymers. Eur J Pharm Sci. 2000;10:169-77
5. Choi YR, Chae SY, Ahn CH, Lee M, Oh S, Byun Y. et al. Development of polymeric gene delivery carriers: PEGylated copolymers of L-lysine and L-phenylalanine. J Drug Target. 2007;15:391-8
6. Bogdanov AJ, Tsai E, Bogdanova A, Papisov M, Brady T, Weissleder R. Novel agent for contrast-enhanced MR angiography and targeting. Radiology. 1992;185:128
7. Xun W, Wang HY, Li ZY, Cheng SX, Zhang XZ, Zhuo RX. Self-assembled micelles of novel graft amphiphilic copolymers for drug controlled release. Colloids Surf B Biointerfaces. 2011;85:86-91
8. Licciardi M, Cavallaro G, Di Stefano M, Pitarresi G, Fiorica C, Giammona G. New self-assembling polyaspartylhydrazide copolymer micelles for anticancer drug delivery. Int J Pharm. 2010;396:219-28
9. Kopecek J, Kopeckova P, Minko T, Lu Z. HPMA copolymer-anticancer drug conjugates: design, activity, and mechanism of action. Eur J Pharm Biopharm. 2000;50:61-81
10. Katchalski E, Sela M, Silman H, Berger A. Polyamino acids as protein models. In: (ed.) Neurath H. The Proteins. New York: Academic Press Inc. 1964:405-581
11. Nathan A, Zalipsky S, Ertel SI, Agathos SN, Yarmush ML, Kohn J. Copolymers of lysine and polyethylene glycol: a new family of functionalized drug carriers. Bioconj Chem. 1993;4:54-62
12. Wolfert MA, Schacht EH, Toncheva V, Ulbrich K, Nazarova O, Seymour LW. Characterization of vectors for gene therapy formed by self-assembly of DNA with synthetic block co-polymers. Human Gene Ther. 1996;7:2123-33
13. Trubetskoy VS, Gazelle GS, Wolf GL, Torchilin VP. Block-copolymer of polyethylene glycol and polylysine as a carrier of organic iodine: design of long-circulating particulate contrast medium for X-ray computed tomography. J Drug Target. 1997;4:381-8
14. Shiraishi K, Kawano K, Minowa T, Maitani Y, Yokoyama M. Preparation and in vivo imaging of PEG-poly(L-lysine)-based polymeric micelle MRI contrast agents. J Control Release. 2009;136:14-20
15. Chou PY, Scheraga HA. Calorimetric measurement of enthalpy change in the isothermal helix--coil transition of poly-L-lysine in aqueous solution. Biopolymers. 1971;10:657-80
16. Bogdanov AJr, Weissleder R, Brady T. Protected angiopolymers and their use in blood pool imaging. In: (ed.) VP T. Handbook of Targeted Delivery of Imaging Agents. Boca Raton, FL: CRC Press. 1995:502-22
17. Bogdanov A Jr, Wright SC, Marecos EM, Bogdanova A, Martin C, Petherick P. et al. A long-circulating co-polymer in "passive targeting" to solid tumors. J Drug Target. 1997;4:321-30
18. Bogdanov AA, Weissleder R, Frank HW, Bogdanova AV, Nossif N, Schaffer BK. et al. A New Macromolecule as a contrast agent for MR-angiography - preparation, properties, and animal studies. Radiology. 1993;187:701-6
19. Castillo G, Reichstetter S, Bolotin E. Extending residence time and stability of peptides by Protected Graft Copolymer (PGC) excipient: GLP-1 example. Pharm Res. 2011;29:306-18
20. Bogdanov AA, Lewin M, Weissleder R. Approaches and agents for imaging the vascular system. Adv Drug Deliv Rev. 1999;37:279-93
21. Baxter AB, Melnikoff S, Stites DP, Brasch RC. AUR Memorial Award 1991. Immunogenicity of gadolinium-based contrast agents for magnetic resonance imaging. Induction and characterization of antibodies in animals. Invest Radiol. 1991;26:1035-40
22. Smyth H, Carpenter C, Shaffer C. The toxicity of high molecular weight poly (ethylene glycols): chronic and parenteral administration. J Am Pharm Assoc. 1947;36:157-60
23. Abuchowski A, van Es T, Palczuk NC, Davis FF. Alteration of immunological properties of bovine serum albumin by covalent attachment of poly(ethylene glycol). J Biol Chem. 1977;252:3578-81
24. Nucci M, Schorr R, Abuchowski A. The therapeutic value of poly(ethylene glycol)-modified proteins. Adv Drug Delivery Revs. 1993;6:133-51
25. Katre N. The conjugation of proteins with polyethylne glycol and other polymers. Altering properties of proteins to enhance their therapeutic potential. Adv Drug Delivery Revs. 1993;10:91-114
26. Bogdanov AJ, Weissleder R, Brady T. Long-circulating blood pool imaging agents. Adv Drug Delivery Revs. 1995;16:335-48
27. Frank H, Weissleder R, Bogdanov AAJr, Brady TJ. Detection of pulmonary emboli by using MR angiography with MPEG-PL-GdDTPA: an experimental study in rabbits. Amer J Roentgenol. 1994;162:1041-6
28. Gupta H, Weissleder R, Bogdanov AAJr, Brady TJ. Experimental gastrointestinal hemorrhage: detection with contrast-enhanced MR imaging and scintigraphy. Radiology. 1995;196:239-44
29. Donahue KM, Weisskoff RM, Burstein D. Water diffusion and exchange as they influence contrast enhancement. J Magn Reson Imaging. 1997;7:102-10
30. Kim YR, Savellano MD, Weissleder R, Bogdanov A Jr. Steady-state and dynamic contrast MR imaging of human prostate cancer xenograft tumors: a comparative study. Technol Cancer Res Treat. 2002;1:489-95
31. Donahue KM, Weisskoff RM, Chesler DA, Kwong KK, Bogdanov AAJr, Mandeville JB. et al. Improving MR quantification of regional blood volume with intravascular T1 contrast agents: accuracy, precision, and water exchange. Magn Reson Med. 1996;36:858-67
32. Kim YR, Tejima E, Huang S, Atochin DN, Dai G, Lo EH. et al. In vivo quantification of transvascular water exchange during the acute phase of permanent stroke. Magn Reson Med. 2008;60:813-21
33. Jiang Q, Ewing JR, Ding GL, Zhang L, Zhang ZG, Li L. et al. Quantitative evaluation of BBB permeability after embolic stroke in rat using MRI. J Cereb Blood Flow Metab. 2005;25:583-92
34. Gotoh O, Asano T, Koide T, Takakura K. Ischemic brain edema following occlusion of the middle cerebral artery in the rat. I: The time courses of the brain water, sodium and potassium contents and blood-brain barrier permeability to 125I-albumin. Stroke. 1985;16:101-9
35. Zaharchuk G, Bogdanov AAJr, Marota JJ, Shimizu-Sasamata M, Weisskoff RM, Kwong KK. et al. Continuous assessment of perfusion by tagging including volume and water extraction (CAPTIVE): a steady-state contrast agent technique for measuring blood flow, relative blood volume fraction, and the water extraction fraction. Magn Reson Med. 1998;40:666-78
36. Zaharchuk G, Mandeville JB, Bogdanov AAJr, Weissleder R, Rosen BR, Marota JJ. Cerebrovascular dynamics of autoregulation and hypoperfusion. An MRI study of CBF and changes in total and microvascular cerebral blood volume during hemorrhagic hypotension. Stroke. 1999;30:2197-204
37. Bogdanov AAJr, Callahan RJ, Wilkinson RA, Martin C, Cameron JA, Fischman AJ. et al. Synthetic copolymer kit for radionuclide blood-pool imaging. J Nucl Med. 1994;35:1880-6
38. Callahan RJ, Bogdanov A Jr, Fischman AJ, Brady TJ, Weissleder R. Preclinical evaluation and phase I clinical trial of a 99mTc-labeled synthetic polymer used in blood pool imaging. Amer J Roentgenol. 1998;171:137-43
39. Weidner N, Folkman J, Pozza F, Bevilacqua P, Allred E, Moore D. et al. Tumor angiogenesis: a new significant andindependent prognostic indicator in early-stage breast carcinoma. Jour Natl Cancer Inst. 1992;84:1875-87
40. Bogdanov AJr, Weissleder R, Tsai E, Schaffer B, Bogdanova A, Nossiff N. et al. Tumor neovascularity imaging by contrast-enhanced MR angiography. New York, NY: 12th Annual Scientific Meeting of the Society of Magnetic Resonance in Medicine. 1993:577
41. Weissleder R, Cheng HC, Marecos E, Kwong K, Bogdanov A Jr. Non-invasive in vivo mapping of tumour vascular and interstitial volume fractions. Eur J Cancer. 1998;34:1448-54
42. Kim YR, Savellano MD, Savellano DH, Weissleder R, Bogdanov A Jr. Measurement of tumor interstitial volume fraction: method and implication for drug delivery. Magn Reson Med. 2004;52:485-94
43. Lewin M, Bredow S, Sergeyev N, Marecos E, Bogdanov A Jr, Weissleder R. In vivo assessment of vascular endothelial growth factor-induced angiogenesis. Int J Cancer. 1999;83:798-802
44. Kim YR, Yudina A, Figueiredo J, Reichardt W, Hu-Lowe D, Petrovsky A. et al. Detection of early antiangiogenic effects in human colon adenocarcinoma xenografts: in vivo changes of tumor blood volume in response to experimental VEGFR tyrosine kinase inhibitor. Cancer Res. 2005;65:9253-60
45. Kim YR, Rebro KJ, Schmainda KM. Water exchange and inflow affect the accuracy of T1-GRE blood volume measurements: implications for the evaluation of tumor angiogenesis. Magn Reson Med. 2002;47:1110-20
46. Bogdanov A Jr, Marecos E, Cheng HC, Chandrasekaran L, Krutzsch HC, Roberts DD. et al. Treatment of experimental brain tumors with trombospondin-1 derived peptides: an in vivo imaging study. Neoplasia. 1999;1:438-45
47. Kang HW, Walvick R, Bogdanov A Jr. In vitro and In vivo imaging of antivasculogenesis induced by Noggin protein expression in human venous endothelial cells. FASEB J. 2009;23:4126-34
48. Stetler-Stevenson WG, Aznavoorian S, Liotta LA. Tumor cell interactions with the extracellular matrix during invasion and metastasis. Ann Rev Cell Biol. 1993;9:541-73
49. Weissleder R, Tung CH, Mahmood U, Bogdanov A Jr. In vivo imaging of tumors with protease-activated near-infrared fluorescent probes. Nat Biotechnol. 1999;17:375-8
50. Mahmood U, Tung CH, Bogdanov A Jr, Weissleder R. Near-infrared optical imaging of protease activity for tumor detection. Radiology. 1999;213:866-70
51. Tung C, Mahmood U, Bredow S, Bogdanov A, Weissleder R. A cathepsin D sensitive near infrared fluorescence probe for in vivo imaging of enzyme activity. Bioconj Chem. 1999;10:892-6
52. Marten K, Bremer C, Khazaie K, Sameni M, Sloane B, Tung CH. et al. Detection of dysplastic intestinal adenomas using enzyme-sensing molecular beacons in mice. Gastroenterology. 2002;122:406-14
53. Bremer C, Tung CH, Weissleder R. In vivo molecular target assessment of matrix metalloproteinase inhibition. Nat Med. 2001;7:743-8
54. Pham W, Choi Y, Weissleder R, Tung CH. Developing a peptide-based near-infrared molecular probe for protease sensing. Bioconj. Chem. 2004;15:1403-7
55. Lamfers ML, Gianni D, Tung CH, Idema S, Schagen FH, Carette JE. et al. Tissue inhibitor of metalloproteinase-3 expression from an oncolytic adenovirus inhibits matrix metalloproteinase activity in vivo without affecting antitumor efficacy in malignant glioma. Cancer Res. 2005;65:9398-405
56. Law B, Curino A, Bugge TH, Weissleder R, Tung CH. Design, synthesis, and characterization of urokinase plasminogen-activator-sensitive near-infrared reporter. Chem & Biol. 2004;11:99-106
57. Hsiao JK, Law B, Weissleder R, Tung CH. In-vivo imaging of tumor associated urokinase-type plasminogen activator activity. J Biomed Opt. 2006;11:34013
58. Messerli SM, Prabhakar S, Tang Y, Shah K, Cortes ML, Murthy V. et al. A novel method for imaging apoptosis using a caspase-1 near-infrared fluorescent probe. Neoplasia. 2004;6:95-105
59. Bogdanov AJ, Lin C, Simonova M, Matuszewski L, Weissleder R. Cellular activation of the self-quenched fluorescent reporter probe in tumor microenvironment. Neoplasia. 2002;4:228-36
60. Bremer C, Tung CH, Bogdanov A Jr, Weissleder R. Imaging of differential protease expression in breast cancers for detection of aggressive tumor phenotypes. Radiology. 2002;222:814-8
61. Gupta H, Wilkinson RA, Bogdanov AAJr, Callahan RJ, Weissleder R. Inflammation: imaging with methoxy poly(ethylene glycol)-poly-L-lysine-DTPA, a long-circulating graft copolymer. Radiology. 1995;197:665-9
62. Medarova Z, Castillo G, Dai G, Bolotin E, Bogdanov A, Moore A. Noninvasive magnetic resonance imaging of microvascular changes in type 1 diabetes. Diabetes. 2007;56:2677-82
63. Wunder A, Tung CH, Muller-Ladner U, Weissleder R, Mahmood U. In vivo imaging of protease activity in arthritis: a novel approach for monitoring treatment response. Arthritis Rheum. 2004;50:2459-65
64. Lai WF, Chang CH, Tang Y, Bronson R, Tung CH. Early diagnosis of osteoarthritis using cathepsin B sensitive near-infrared fluorescent probes. Osteoarthritis Cartilage. 2004;12:239-44
65. Kozloff KM, Quinti L, Patntirapong S, Hauschka PV, Tung CH, Weissleder R. et al. Non-invasive optical detection of cathepsin K-mediated fluorescence reveals osteoclast activity in vitro and in vivo. Bone. 2009;44:190-8
66. Naruko T, Ueda M, Haze K, van der Wal AC, van der Loos CM, Itoh A. et al. Neutrophil infiltration of culprit lesions in acute coronary syndromes. Circulation. 2002;106:2894-900
67. Falk E. Why do plaques rupture? Circulation. 1992;86:11130-42
68. Schroeder AP, Falk E. Pathophysiology and inflammatory aspects of plaque rupture. Cardiol Clin. 1996;14:211-20
69. Chen J, Tung CH, Mahmood U, Ntziachristos V, Gyurko R, Fishman MC. et al. In vivo imaging of proteolytic activity in atherosclerosis. Circulation. 2002;105:2766-71
70. Deguchi JO, Aikawa M, Tung CH, Aikawa E, Kim DE, Ntziachristos V. et al. Inflammation in atherosclerosis: visualizing matrix metalloproteinase action in macrophages in vivo. Circulation. 2006;114:55-62
71. Chen J, Tung CH, Allport JR, Chen S, Weissleder R, Huang PL. Near-infrared fluorescent imaging of matrix metalloproteinase activity after myocardial infarction. Circulation. 2005;111:1800-5
72. Matsumura Y, Oda T, Maeda H. General mechanism of intratumor accumulation of macromolecules: advantage of macromolecular therapeutics. Gan To Kagaku Ryoho. 1987;14:821-9
73. Maeda H. The enhanced permeability and retention (EPR) effect in tumor vasculature: the key role of tumor-selective macromolecular drug targeting. Adv Enzyme Regul. 2001;41:189-207
74. Torchilin VP. Passive and active drug targeting: drug delivery to tumors as an example. Handb Exp Pharmacol. 2010:3-53
75. Marecos E, Weissleder R, Bogdanov A Jr. Antibody-mediated versus nontargeted delivery in a human small cell lung carcinoma model. Bioconj Chem. 1998;9:184-91
76. Trail PA, Willner D, Lasch SJ, Henderson AJ, Hofstead S, Casazza AM. et al. Cure of xenografted human carcinomas by BR96-doxorubicin immunoconjugates. Science. 1993;261:212-5
77. Choi Y, Weissleder R, Tung CH. Selective antitumor effect of novel protease-mediated photodynamic agent. Cancer Res. 2006;66:7225-9
78. Bevilacqua MP, Pober JS, Mendrick DL, Cotran RS, Gimbrone MA Jr. Identification of an inducible endothelial-leukocyte adhesion molecule. Proc Natl Acad Sci USA. 1987;84:9238-42
79. Kang HW, Weissleder R, Bogdanov A Jr. Targeting of MPEG-protected polyamino acid carrier to human E-selectin in vitro. Amino Acids. 2002;23:301-8
80. Bogdanov AAJr, Martin C, Bogdanova AV, Brady TJ, Weissleder R. An adduct of cis-diamminedichloroplatinum(II) and poly(ethylene glycol)poly(L-lysine)-succinate: synthesis and cytotoxic properties. Bioconjug Chem. 1996;7:144-9
81. Pinto AL, Lippard SJ. Binding of the antitumor drug cis-diamminedichloroplatinum(II) (cisplatin) to DNA. [Review] [71 refs]. Biochimica et Biophysica Acta. 1985;780:167-80
82. Sherman SE, Lippard SJ. Structural Aspects of Platinum Anticancer Drug Interactions with DNA. Cem Rev. 1987;87:1153-81
83. Muggia FM, Rozencweig M, Penta J. Clinical implications of cisplatin pharmacology. Recent Results Cancer Res. 1980;74:132-8
84. Reichstetter S, Castillo GM, Lai M, Nishimoto-Ashfield A, Banerjee A, Bogdanov A. et al. Protected Graft Copolymer (PGC) basal formulation of insulin as potentially safer alternative to Lantus(R) (Insulin-Glargine): A streptozotocin-induced, diabetic sprague dawley rats study. Pharm Res. 2011 doi:10.1007/s11095-011-0646-8
85. Torchilin V, Omelyanenko V, Papisov M, Bogdanov AJ, Trubetskoy V, Herron J. et al. Poly(ethylene glycol) on the liposome surface: on the mechanism of polymer-coated liposome longevity. Biochimi Biophys Acta. 1994;1195:11-20
86. Reichstetter S, Castillo G, Perfetti R, Leahy M, Bolotin E. Long acting GLP-1 for the treatment of type 2 diabetes. Diabetes. 2007;56:A144-A
87. Reichstetter S, Castillo G, Medarova Z, Moore A, Perfetti R, Leahy M. et al. Long acting GLP-1 for the treatment of type I diabetes. Diabetes. 2007;56:A73-A
88. Castillo G, Reichstetter S, Medarova Z, Moore A, Bolotin E. PGC-GLP-1: Pharmacokinetics in rodents. Diabetes. 2007;56:A554-A
89. Koller KJ, Lowe DG, Bennett GL, Minamino N, Kangawa K, Matsuo H. et al. Selective activation of the B natriuretic peptide receptor by C-type natriuretic peptide (CNP). Science. 1991;252:120-3
90. Harris PJ, Thomas D, Morgan TO. Atrial natriuretic peptide inhibits angiotensin-stimulated proximal tubular sodium and water reabsorption. Nature. 1987;326:697-8
91. Gambaryan S, Wagner C, Smolenski A, Walter U, Poller W, Haase W. et al. Endogenous or overexpressed cGMP-dependent protein kinases inhibit cAMP-dependent renin release from rat isolated perfused kidney, microdissected glomeruli, and isolated juxtaglomerular cells. Proc Natl Acad Sci U S A. 1998;95:9003-8
92. Cherradi N, Brandenburger Y, Rossier MF, Vallotton MB, Stocco DM, Capponi AM. Atrial natriuretic peptide inhibits calcium-induced steroidogenic acute regulatory protein gene transcription in adrenal glomerulosa cells. Mol Endocrinol. 1998;12:962-72
93. Marin-Grez M, Fleming JT, Steinhausen M. Atrial natriuretic peptide causes pre-glomerular vasodilatation and post-glomerular vasoconstriction in rat kidney. Nature. 1986;324:473-6
94. Kishimoto I, Dubois SK, Garbers DL. The heart communicates with the kidney exclusively through the guanylyl cyclase-A receptor: acute handling of sodium and water in response to volume expansion. Proc Natl Acad Sci U S A. 1996;93:6215-9
95. Li P, Wang D, Lucas J, Oparil S, Xing D, Cao X. et al. Atrial natriuretic peptide inhibits transforming growth factor beta-induced Smad signaling and myofibroblast transformation in mouse cardiac fibroblasts. Circ Res. 2008;102:185-92
96. Lopez MJ, Wong SK, Kishimoto I, Dubois S, Mach V, Friesen J. et al. Salt-resistant hypertension in mice lacking the guanylyl cyclase-A receptor for atrial natriuretic peptide. Nature. 1995;378:65-8
97. Oliver PM, Fox JE, Kim R, Rockman HA, Kim HS, Reddick RL. et al. Hypertension, cardiac hypertrophy, and sudden death in mice lacking natriuretic peptide receptor A. Proc Natl Acad Sci U S A. 1997;94:14730-5
98. Kitakaze M, Asakura M, Kim J, Shintani Y, Asanuma H, Hamasaki T. et al. Human atrial natriuretic peptide and nicorandil as adjuncts to reperfusion treatment for acute myocardial infarction (J-WIND): two randomised trials. Lancet. 2007;370:1483-93
99. Hamilton BS, Paglia D, Kwan AY, Deitel M. Increased obese mRNA expression in omental fat cells from massively obese humans. Nat Med. 1995;1:953-6
100. Heymsfield SB, Greenberg AS, Fujioka K, Dixon RM, Kushner R, Hunt T. et al. Recombinant leptin for weight loss in obese and lean adults: a randomized, controlled, dose-escalation trial. JAMA. 1999;282:1568-75
101. Paz-Filho G, Wong ML, Licinio J. Ten years of leptin replacement therapy. Obes Rev. 2011;12:e315-23
102. Freireich EJ, Gehan EA, Rall DP, Schmidt LH, Skipper HE. Quantitative comparison of toxicity of anticancer agents in mouse, rat, hamster, dog, monkey, and man. Cancer Chemother Rep. 1966;50:219-44
103. KIm Y, Dai G, Bogdanov A, Rosen BR. Novel quantification method for measuring vessel size selective blood volume and transvascular water exchange rate: implication of visualizing neurovascular unit. ISMRM 13th scientific meeting Miami FL: ISMRM Berkeley. 2005
Author contact
![]() Corresponding author: E-mail: Alexei.Bogdanovedu. The Laboratory of Molecular Imaging Probes, S6-434, Department of Radiology, University of Massachusetts Medical School, 55 Lake Ave North, Worcester MA 01655. Tel. 508-856-5571. FAX 508-856-1860
Corresponding author: E-mail: Alexei.Bogdanovedu. The Laboratory of Molecular Imaging Probes, S6-434, Department of Radiology, University of Massachusetts Medical School, 55 Lake Ave North, Worcester MA 01655. Tel. 508-856-5571. FAX 508-856-1860
Citation styles
APA
Bogdanov Jr, A.A., Mazzanti, M., Castillo, G., Bolotin, E. (2012). Protected Graft Copolymer (PGC) in Imaging and Therapy: A Platform for the Delivery of Covalently and Non-Covalently Bound Drugs. Theranostics, 2(6), 553-576. https://doi.org/10.7150/thno.4070.
ACS
Bogdanov Jr, A.A.; Mazzanti, M.; Castillo, G.; Bolotin, E. Protected Graft Copolymer (PGC) in Imaging and Therapy: A Platform for the Delivery of Covalently and Non-Covalently Bound Drugs. Theranostics 2012, 2 (6), 553-576. DOI: 10.7150/thno.4070.
NLM
Bogdanov Jr AA, Mazzanti M, Castillo G, Bolotin E. Protected Graft Copolymer (PGC) in Imaging and Therapy: A Platform for the Delivery of Covalently and Non-Covalently Bound Drugs. Theranostics 2012; 2(6):553-576. doi:10.7150/thno.4070. https://www.thno.org/v02p0553.htm
CSE
Bogdanov Jr AA, Mazzanti M, Castillo G, Bolotin E. 2012. Protected Graft Copolymer (PGC) in Imaging and Therapy: A Platform for the Delivery of Covalently and Non-Covalently Bound Drugs. Theranostics. 2(6):553-576.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) License. See http://ivyspring.com/terms for full terms and conditions.