Impact Factor
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Challenges in CTC research
Trends in CTC research and...
Concluding remarks
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2013; 3(6):377-394. doi:10.7150/thno.5195 This issue Cite
Review
Detecting Circulating Tumor Cells: Current Challenges and New Trends
Bin Hong1, Youli Zu2 ![]()
1. TeloVISION, LLC, 1281 Win Hentschel Blvd. West Lafayette, IN 47906, USA;
2. Department of Pathology and Genomic Medicine, The Methodist Hospital, 6565 Fannin, MS205, Houston, TX 77030, USA.
Received 2012-9-10; Accepted 2012-11-1; Published 2013-4-23
Abstract
Circulating tumor cells (CTCs) in the blood stream play a critical role in establishing metastases. The clinical value of CTCs as a biomarker for early cancer detection, diagnosis, prognosis, prediction, stratification, and pharmacodynamics have been widely explored in recent years. However, the clinical utility of current CTC tests is limited mainly due to methodological constraints. In this review, the pros and cons of the reported CTC assays are comprehensively discussed. In addition, the potential of tumor cell-derived materials as new targets for CTC detection, including circulating tumor microemboli, cell fragments, and circulating DNA, is evaluated. Finally, emerging approaches for CTC detection, including telomerase-based or aptamer-based assays and cell functional analysis, are also assessed. Expectantly, a thorough review of the current knowledge and technology of CTC detection will assist the scientific community in the development of more efficient CTC assay systems.
Keywords: CTC: Circulating tumor cell, CTM: Circulating tumor microemboli, CTMat: Circulating tumor materials, CTDNA: Circulating tumor DNA, POCT: Point-of-care test.
Introduction
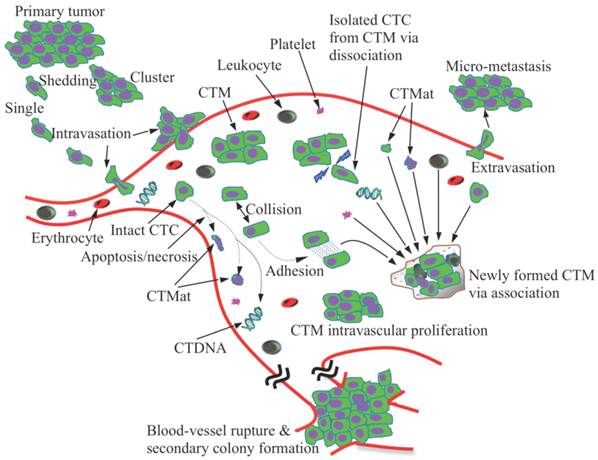
Cancer is the second leading cause of death worldwide [1]. Greater than 90% of deaths in cancer patients are attributed to metastasis [2]. Cancer is considered a localized disease in its early stage; however, it has often become systemic by the time a patient becomes symptomatic and the disease is detected by the currently available imaging modalities such as traditional radiography (X-ray), magnetic resonance imaging (MRI), computed tomography (CT), positron emission tomography (PET), or ultrasound [3]. There is growing evidence that cancer cells are shed from the primary tumor into the circulation prior to the presentation of clinical symptoms [4]. These circulating tumor cells (CTCs) may finally colonize at distant sites and form metastases (Figure 1).
Schematic illustration of metastasis development via CTCs/CTM. Tumor cells and cell clusters are shed from the primary tumor and intravasate into the circulation, which might involve the process of epithelial-to-mesenchymal transition. The majority of the CTCs are, however, killed via apoptosis and necrosis, releasing debris, cell fragments and intracellular substances (CTMat and CTDNA). CTM, the even rarer species than CTCs in blood, undergo a dynamic life. Tumor cells can dissociate from CTM when subjected to shear force and/or frequent collisions in blood; they can also attach to other tumor or blood cells upon collision due to increased adhesion. The microenvironment established within CTM is unique, protecting the tumor cells inside from damage. CTM are, therefore, believed to be more aggressive than individual CTCs as they proliferate in the vessel and eventually rupture the vessel. Conversely, CTCs have to extravasate in order to form metastasis.
The presence of CTCs was first reported approximately 140 years ago [5]. However, it was not a widespread topic in cancer research until recently. Because CTCs are ultra-rare events, with numbers as low as one CTC in 106-107 leukocytes of the peripheral blood of cancer patients, enrichment and investigation of CTCs have been extremely difficult. It was often akin to pinpointing a needle in a haystack until, in 2004, the CellSearch System (Veridex, Raritan, NJ) was introduced, which is the only medical device currently cleared by the Food and Drug Administration (FDA) for CTC selection and enumeration. However, researchers are still facing various challenges, including the methodological constraints imposed by the CellSearch instrument, physics, and statistics [6], and the translational issues [7], thereby limiting the clinical implementation of CTC tests and accurate interpretation of the test results. Requirement of a multi-step cell preparation and isolation process in the current CTC detection method may lead to loss and damage of tumor cells, and have an adverse impact on the assay accuracy.
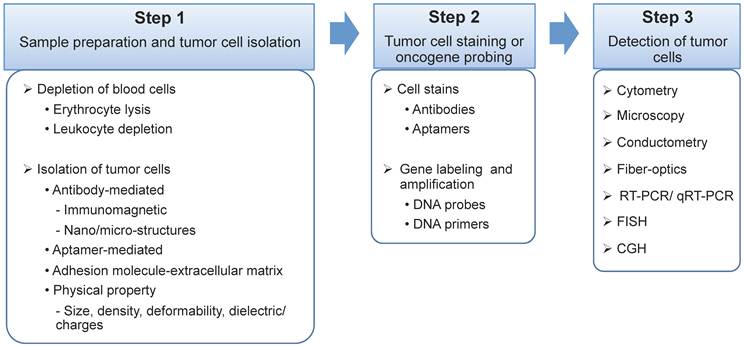
The majority of CTC detection methods are designed as bench-top instruments, such as flow cytometers [8-10], the CellSearch system [11], high-definition fluorescence scanning microscopy [12], fiber-optic array scanning technology (FAST) [13, 14], isolation by size of epithelial tumor cells (ISET) [15, 16], and laser scanning cytometers [17, 18]. Some methods combine bench-top instruments with an additional assay system, such as the processes of Ficoll [19], OncoQuick [20], and RT-PCR [21, 22]. Interestingly, CTC microdevices have undertaken a different approach by providing miniature structure [23-29], microfluidic reaction kinetics [24-26, 28, 29] and integrated processes [23, 24, 26]. When compared to bench-top devices, the CTC microdevices demonstrated superior sensitivity [23, 25-28], improved cell recovery [23-25, 29], high purity [24], enhanced enrichment [23, 24, 27, 28], and low cost [23, 24, 26]. More importantly, CTC microdevices are ideal for point-of-care testing [25, 30, 31]. Since CTCs are mainly characterized and identified by their morphology and immunostaining pattern, their heterogeneity is a major obstacle for CTC detection. The CTCs derived from different types of tissues significantly distinguish from each other with different size, shape, and immunophenotyping profile. However, there is broad morphological and immunophenotypical variation within CTCs derived from the same tissue of origin. During epithelial to mesenchymal transition, the expression of epithelial markers on CTCs, such as epithelial cell adhesion molecule (EpCAM) and cytokeratin (CK), may be down-regulated and become undetectable [2, 11]. Therefore, accurate detection of CTCs based on morphological and immunophenotypical profiling is still challenged. Additionally, CTCs may be damaged and fragmented, in vivo and/or in vitro, due to multi-step cell preparation processes, causing inaccurate detection and misinterpretation. In addition to the presence of significant heterogeneity, as the biology of CTCs evolves, additional challenges, as well as opportunities, are expected to present. It is also important to note that simple enumeration of CTCs will not contribute significantly to the development of improved or more personalized cancer treatments. Instead, the contributions of CTCs will stem more from obtaining a better understanding of this cell population through complete characterization and functional analysis. From a technical standpoint, almost all CTC assays have three major steps: 1) blood sample preparation and tumor cell separation; 2) cell staining by antibodies or gene probing by DNA probes; and 3) CTC detection (Figure 2).
Schematic illustration of the three major steps of a CTC assay. Step 1. Sample preparation and tumor cell isolation: blood samples may be pre-treated to remove the majority of the erythrocytes and/or leukocytes, and therefore, rare CTCs can be detected at a higher frequency with low background interference. The isolation of CTCs from blood cells can be based on various ligand-receptor interactions, such as antibody-antigen, aptamer-receptor, or adhesion molecule-extracellular matrix, and unique physical properties of cancer cells, including cell size, density, deformability, and dielectric/charges. Step 2. Tumor cell staining or oncogene probing: CTCs can be labeled by antibodies or aptamers, or probed by DNA primers. Step 3. CTC detection via cytometry, microscopy, conductometry, fiber-optics, reverse transcription polymerase chain reaction (RT-PCR), quantitative RT-PCR (qRT-PCR), fluorescence in situ hybridization (FISH), and comparative genomic hybridization (CGH).
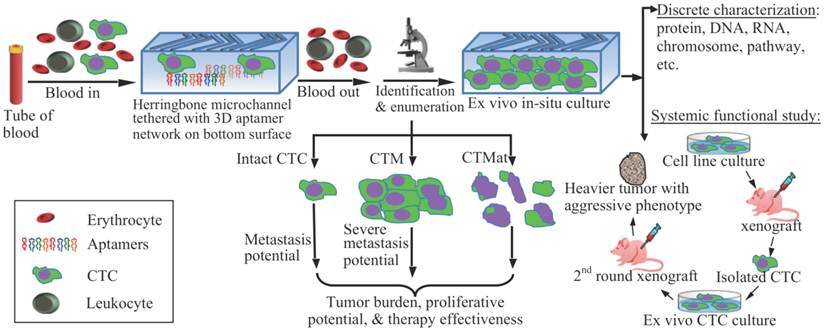
Although the last step of the CTC detection approach can be rapidly completed, the cell preparation and antibody staining are labor- and time-consuming. Many research and review articles have recently appeared covering the technical advances, research progress, and clinical demonstrations related to CTCs [7, 32-38]. This article will not exhaustively review all of the existing literature. Instead, it will focus on analyzing several outstanding pitfalls in the current CTC technologies and clinical research, followed by, as an important contribution of this article, a summary of the emerging approaches and powerful tools that have represented the latest trend in the field to better characterize CTCs (Figure 3). The topics to be discussed in this article are as follows: 1) circulating tumor microemboli (CTM), which are tumor cell clusters/aggregates and associated with high metastatic potential; 2) circulating tumor materials (CTMat), which are debris and fragments of dead tumor cells and may possess antigenic or genetic characteristics of intact cells, and, therefore, present independent prognostic value from that of intact and viable CTCs; 3) telomerase-targeted CTC detection, a method that may target the entire CTC subpopulation; 4) aptamer-based CTC detection, which could specifically identify and capture the CTCs without knowing the binding sites on them; 5) functional studies on live CTCs to assess their specific role in metastasis development; and 6) mathematical modeling, which is beneficial to CTC research because it can analyze data for precision and accuracy, and display hidden boundaries to expose outsiders.
Schematic representation of CTC enrichment, detection, and characterization. After blood collection, the whole blood is processed by a microdevice, for example, a herringbone-chip tethered with tumor-specific recognition molecules, e.g., aptamers, to isolate the CTCs from the majority of normal blood cells. Enriched CTCs are then identified, enumerated, and characterized via cytopathology, molecular biology, and other examinations. By means of morphology, tumor cells could be categorized into intact CTCs, circulating tumor microemboli (CTM), and circulating tumor materials (CTMat). Each of them may present particular metastatic potential as independent indicators for prediction. Captured CTCs could be cultured ex vivo, followed by additional systemic functional studies. For instance, cultured CTCs are injected into nude mice subcutaneously for a xenograft; and a heavier tumor is found than that induced by the parental cell line.
Challenges in CTC research
CTCs as a disease marker
As CTC research expanded rapidly in recent years, data have been accumulated to demonstrate the potential clinical values of CTCs as early detection, diagnostic, prognostic, predictive, surrogate, stratification, and pharmacodynamic biomarkers [11, 39-42]. However, the role of CTCs as a disease marker may be unique in different clinical conditions and should be carefully interpreted. A good example is the comparison between the prognostic and predictive biomarkers. Both biomarkers employ progression-free survival and overall survival for data interpretation; however, the prognostic biomarker is independent of specific drug treatment or therapy, and used for the determination of outcomes before treatment, while the predictive biomarker is related to a particular treatment to predict the response [43]. The definitions of all types of biomarkers [44, 45] are summarized in Table 1.
Definition/ intended use for commonly used biomarkers [44, 45].
| Type of biomarker | Definition/ intended use |
|---|---|
| Prognostic marker | Used to assess the survival probabilities of patients or detect an aggressive phenotype and determine how a disease will behave |
| Predictive marker | Used to predict whether a drug or other therapies will be effective, or to monitor the effectiveness of treatment |
| Stratification marker | A marker used to predict the likely response to a drug prior to treatment by classifying individuals as responsive or nonresponsive |
| Clinical endpoint | A characteristic or variable that reflects how a patient feels, functions, or survives. Clinical endpoints are distinct measurements or analyses of disease characteristics observed in a study or a clinical trial that reflect the effect of a therapeutic intervention. |
| Surrogate marker or surrogate endpoint | A biomarker that is intended to substitute for a clinical endpoint. A surrogate endpoint is expected to predict clinical benefit (or harm or lack of benefit or harm) based on epidemiologic, therapeutic, pathophysiologic, or other scientific evidence. |
| Efficacy marker or outcome marker | Correlated with the desired effect of a treatment, but does not have as much validation as a surrogate endpoint |
| Diagnostic marker | Indicates the presence or likelihood of a particular disease in patients or in animal models |
| Early detection marker | Used for screening patients to detect cancer or other progressive diseases early |
| Pharmacodynamic marker | Provides information to determine highest dose in clinical trials, or time-associated measure of a pharmacologic response |
| Kinetic marker | An in vivo measurement of flux through the key pathways that drive disease processes and therapeutic response |
| Toxicity marker or safety marker | Indicates potentially adverse effects in in vitro, preclinical, or clinical studies |
| Distal marker | A marker that reflects disease progression |
| Screening marker | A marker discriminating the healthy state from an early disease state, preferably prior to symptoms |
| Target marker | Shows that a drug interacts with a particular molecular target or receptor |
| Bridging marker or translational marker | Used to measure and compare the same endpoint in preclinical (animal) and clinical studies |
| Authentic marker | Reveals the activity of a pathway that is integrally involved in disease activity or therapeutic action |
| Routine marker | A marker that is analyzed in laboratories with well-established methods, such as in diagnostic clinical chemistry |
| Mechanism marker | Provides evidence that a drug affects a desired pathway |
| Known valid marker | A marker that is measured in an analytical test system with well-established performance characteristics and for which there is widespread agreement in the medical or scientific community about the physiologic, toxicologic, pharmacologic, or clinical significance of results; test required |
| Probable valid marker | A marker that is measured in an analytical test system with well-established performance characteristics and for which there is a scientific framework or body of evidence that appears to elucidate the physiologic, toxicologic, pharmacologic, or clinical significance of results; test recommended |
| Exploratory marker | A marker that is measured in the exploratory research without well-established performance characteristics and for which there is limited initial information to support the physiologic, toxicologic, pharmacologic, or clinical significance of results; information only |
Inconsistent results are increasingly reported among the various CTC assay methods, specifically pertaining to results for the CTC detection rate, patient positivity rate, and the correlation between the presence of CTCs and survival rate [36, 46, 47]. In addition to heterogeneity of CTCs, a number of technical factors may also contribute to the discordance, including the methodological discrepancies and flaws, lack of reference standard, spectrum and selection bias, operator variability and bias, sample size, blurred clinical impact with known clinical/pathologic data, use of diverse capture antibodies from different sources, lack of awareness of the pre-analytical phase, oversimplification of the cytopathology process, use of dichotomous decision criteria, etc. [48-51]. To minimize the inconsistencies, a standard protocol is indispensable for all CTC assays [52], although the CellSearch system is the only one currently cleared by the FDA.
Sample size and sampling probability
It is generally hypothesized that blood samples withdrawn from a vein statistically represent the whole blood of the entire body. However, it may not be true for the detection of CTCs, since they are so rare that the frequency of CTCs obtained may not reflect the entire cell population. Often, in most clinical studies, only a small aliquot of sample is processed; for instance, 7.5 mL aliquot vs. 5 L of total blood in an adult human. It is therefore important to consider: 1) whether the frequency of the CTC population is large enough to deem the sample size as infinite; 2) whether cell loss during processing significantly affects the probability of acquiring CTCs; 3) whether the frequency of the CTC population measured in an aliquot is a statistical representative of the entire sample; and 4) whether the results of one assay correlate with the assays that use a different amount of blood for the sample [53].
The most effective way to increase the CTC detection rate in whole blood would be to increase the sample volume within the clinically allowable range. A study has compared the numbers of CTCs in 7.5 mL and 30 mL of blood from the same patient, using the CellSearch System, for a group of 15 patients with colorectal liver metastases [54]. In 7.5 mL of blood, the median number of CTCs was 1 with a range of 0 to 4, while in 30 mL of blood, the median number was 2 with a range of 0 to 9. CTCs were found in 13% and 47% of the patient samples with 7.5 mL and 30 mL blood, respectively. Analyzing 30 mL of blood resulted in 20% more patients having detectable CTCs. A different study also recognized that the volume of blood sample was a limiting factor for the sensitivity of flow cytometry-based CTC assays [8]. Based on a mathematical model using the Poisson distribution, the probability of collecting ≥1 CTCs in one aliquot of 7.5 mL blood from a patient with 500 CTCs in vivo is 50%, and the probability increases with the number of aliquots to 78%, 90% and 95% for 2, 3, and 4 tubes of blood, respectively [55]. It is estimated that, if the frequency of CTCs is 10-6, at least 15,705,214 nucleated cell events must be acquired by a flow cytometer to pinpoint 10 cells of interest with 95% confidence [56]. It is then a corresponding 13 mL of blood, based on the fact that 1 mL blood has roughly 1.2 million events at medium acquisition rate (~30 µl/min) after lysing the majority of the erythrocytes and gating out debris and dead cells. Twenty mL of whole blood would have to be assessed if the cell event was elevated for lower frequency at 1 CTC in 107 leukocytes [57].
Notably, it has been reported that the CellSearch system might undercount the number of CTCs. Nagrath et al. have demonstrated that the average CTC number per mL of whole blood is approximately 79-155 in various cancers [28]. Similarly, in contrast to the FDA-cleared CellSearch Epithelial method, an investigative CellSearch Profile approach (for research use only) could detect an approximately 30-fold higher number of the median CTC counts by using the same paired blood samples [58]. These findings indicate that the actual CTC numbers in patients' blood could be at least 30-100 fold higher than that currently reported by the only FDA-cleared CellSearch system. Theoretically, as more sensitive and accurate detection methods are developed, the CTC counts might be evaluated using smaller blood volumes (1 mL of whole blood instead of 7.5 mL), and assays with shorter turnaround time. Small sample amounts and rapid turnaround time are both prerequisites when developing microdevices for point-of-care applications.
Point-of-care testing and clinical resistance
Point-of-care testing continues to play an ever-increasing role in patient diagnosis and management due to its convenience, rapid results, and low cost [59]. However, barriers remain to the development of a CTC-based point-of-care test (POCT). First, the clinical significance of screening CTCs needs to be confirmed, although growing evidence has demonstrated that the presence of CTCs in the peripheral blood of cancer patients is an important prognostic factor and the number of the detected CTCs post-treatment is predictive of response to therapy [46, 60-62].
The second factor that needs to be addressed is the simplicity of the CTC assay as a POCT [63]. CellSearch, for instance, may not be an ideal type of POCT, unless the CTC assignment process could be implemented more simply and automatically. CellSearch employs a multi-step cell preparation approach and a manual image screening process to identify intact or damaged CTCs. The interpretation of individual CTCs is performed by a trained operator based on the established selection guideline [11, 64]. The morphology identification of CTCs is not as easy as reading the result from a classic POCT, such as a pregnancy test, by showing black and white lines, or a diabetes test, by displaying a digital number. With this test, it is possible that a degree of subjectivity can occur during the CTC assignment process. The inter-operator variability of the CellSearch test has been assessed by interpreting 509 blinded images, as well as the inter-laboratory variation from 14 independent laboratories [65]. Results showed that the inter-operator variability was the primary cause of the poor inter-laboratory coefficient of variation (CV; 45% ~ 64%), while the inconsistency in CTC assignment, especially when samples contained a large number of apoptotic cells [66], was the leading contributor for inter-operator variability. The presence of apoptotic cells is therefore problematic, and may cause bias. In addition, CTCs have been shown as doublets, clusters, and with irregular shapes, including elongated and multinucleated forms [11, 12]. CTC fragments were also frequently observed [11, 12] as the result of in vivo and/or in vitro cell destruction, probably due to the multi-step preparation [4]. With the large volume of blood to be processed in a CTC assay, the quantity of cell fragments would be substantial because of the degradation of both CTCs and normal blood cells. In addition, expanding the selection criteria to include widely varying cell size, nuclear size, and nuclear-cytoplasmic ratio were could further complicate the assignment process [11]. Hence, identification of CTCs by their morphology can be challenging. To minimize cell misinterpretation, anatomic pathologists may have to participate in the process, helping to differentiate CTCs from cell debris/fragments, apoptotic/damaged cells, and tumor-like cells [12, 67-69]. Considering the heterogeneity of the CTC population, and the co-captured white blood cells, immunofluorescence is still the gold standard for CTC counting. Even though CTC isolation can be miniaturized and contained within microdevices, a bulky fluorescence microscope for CTC counting and classification is still needed [12, 70]. These would be important in preventing false-positive conclusions when the measured CTC number is close to the threshold. However, a pathologist typically analyzes samples and confirms results in an office separated from the laboratory, or, sometimes at a satellite site. Therefore, inclusion of an anatomic pathologist in the process would preclude the realization of a POCT. So far, many CTC detection platforms share the same variables for CTC identification. The resolution of the cell images needs to be upgraded [67] and the screening process is ideally automated by an intelligent computer program [64]. The latest advances confirm this trend [12, 70], and a universal standard may soon be developed [52].
Thirdly, as a diagnostic tool, clinical acceptance is mandatory. In 2007, the American Society of Clinical Oncology (ASCO) [71] decided not to recommend the CellSearch CTC test for the diagnosis of metastatic breast cancer or patient management due to its unclear clinical value. In 2009, the National Academy of Clinical Biochemistry (NACB) [72], the academy of the American Association for Clinical Chemistry (AACC), declined to recommend various CTC assays in routine clinical practice for screening, detecting, and monitoring prostate cancer metastasis until more evidence was accumulated in further validation studies. In 2010, the American Joint Committee on Cancer (AJCC) [73] reinforced ASCO's guideline that routine use of CTCs for diagnostic or management purposes is not readily accepted because the benefit of using CTC tests for managing metastatic breast cancer patients is not clear. The Breast Cancer Task Force, therefore, did not incorporate CTCs into the TNM (i.e., Tumor, Lymph Node and Metastasis) staging system, especially for M1 stage sub-classification. Thus, CTC testing is still investigational and should be utilized with caution, and only in specific clinical setting(s) [46]. To prepare for future widespread clinical acceptance and use, a comprehensive guideline for all phases of CTC technology development was recently published by the Foundation for the National Institutes of Health (FNIH) Biomarkers Consortium [52]. These guidelines suggest a framework for development and clinical validation, procedure standardization, context of use, biomarker validation and qualification, and, importantly, the strategies and processes which have been specifically designed for the manipulation of rare CTCs.
Lastly, the turnaround time for a typical POCT is normally within a 1-hour window, but a 30-minute window is preferred to accomplish a truly rapid diagnosis followed by an immediate clinical management decision [74, 75]. It seems none of the reported CTC detection systems have fulfilled this requirement, including the CTC microdevices.
Microdevices
Microdevices have attracted plenty of attention with their unique merits on providing rapid, low-cost, simple, and automated immunoassays [76, 77]. In recent years, microdevices, such as the micro sinusoidal chip [24, 30], microfilter [23, 31, 78, 79], MagSweeper [27, 78, 80], micropost CTC-chip [25, 28], microvortex herringbone-chip [25], micropillar chip [26], serpentine chaotic nanopillar chip [29], micro crescent chip [81, 82], micro column wall chip [83], micro GEDI (geometrically enhanced differential immunocapture) chip [84, 85], micro pinched flow chip [86], micro double spiral chip [87], micro eDAR (ensemble-decision aliquot ranking) cytometer [88], and microsieve chip [69] have been used to enrich and enumerate rare cells. The physical dimensions and assay characteristics of these microdevices are summarized in Tables 2-3. Most microdevices have a planar structure comparable to a glass slide and the capability of processing 1 mL of whole blood in ~ 20 to 60 minutes, not including preparation time for the microdevice, the sample, and the actual detection process. The MagSweeper, microfilter, micro double spiral chip, and microsieve chip, however, present a significantly higher processing capacity or shorter processing time. Human blood has been used for spiking tumor cells in analytical validation studies for all of the microdevices, except the micro sinusoidal chip and the micropillar chip; human patient samples were also tested on nine of 14 microdevices during a pilot trial for clinical validation. Normally, assay preparation consists of microdevice precondition, sample pre-treatment (e.g., dilution and/or plasma removal), and cell manipulation (e.g., fixation, permeablization, fluorescence staining, and rinsing), and takes 30 minutes or longer. The sinusoidal microchip is an exception because it employs conductometric, label-free cell counting. Preparation time for this device is less because the process does not require fluorescence staining and microscopic enumeration, although immunofluorescence or histologic staining is highly recommended for tumor cell identification [12]. To compare the assay performance, the CellSearch system serves as a control in Tables 2-3. It demonstrated 27% to 70% sensitivity and 89% to 100% specificity [11, 39, 89, 90]. Utilizing the CellSearch system as a presumptive standard, the microfilter [31], the nanopillar chip [29], the micro GEDI chip [84], and the micro eDAR cytometer [88] demonstrated 96%, 75%, 94%, and 100% sensitivity, and 16%, 22%, 0%, and 0% specificity, respectively. The significant discordance on specificity may indicate a lack of a solid reference standard since the CellSearch system is known for relatively low analytical sensitivity [28, 40, 58]. The clinical studies using other microdevices have assumed that metastatic cancer patients should have CTCs in the peripheral blood at the time of blood draw and healthy subjects should not. It may not be wise to rely on such an assumption, especially in the absence of specific validation studies. Three types of enrichment principles have been employed by these microdevices (Tables 2-3): antibody/aptamer based, size only based, and both size and deformability based. Most microdevices, despite employing only one principle, have still demonstrated better CTC recovery and enrichment rates than the CellSearch system, and a capability in maintaining cell viability, which is important for additional CTC characterization and potential function analysis. However, depending on the type of microdevice used, there is a notable trade-off between the sample processing time and the purity of CTCs. Immuno-detection-based microdevices demonstrate greater purity than the CellSearch system, but have a longer sample processing time; however, they both use epithelial marker antibodies and therefore experience similar losses of CTCs. In contrast, size exclusion-based microdevices provide a significantly shorter processing time than the CellSearch system but fail to enrich CTCs at a higher purity. Without the use of antibodies in size-based separation, the aforementioned method of cell loss is avoided, but depletion of CTCs smaller than the size cutoff and/or failure to separate CTCs from blood cells of similar sizes produces another manner of cell loss. Adding a second enriching parameter by means of cell deformability, in addition to cell size, has improved the purity.
Comparison of CTC microdevices: physical dimension and assay performance characteristics.
| Microdevice | Year of invention | Principle of enrichment | Physical dimension (cm) | Sample: species, volume, spiked and/or clinical sample from patients | Sample processing timea (min) | Assay preparation timeb (min) | Total turnaround timec (min) |
|---|---|---|---|---|---|---|---|
| CellSearch (control) | 2004 | Antibody based | AutoPrep System: 173L× 69W× 69H; Analyzer II: 73L× 63W× 41H | Hu, WB; 7.5 mL; spiked & clinical | < 60 | > 30 | ~90 |
| Micropost chip | 2007 | Antibody based | 6.6L × 2.5W | Hu, WB; 2.7 mL; spiked & clinical | 80~160 | > 165 | > 387~609 |
| Microfilter | 2007 | Cell size | 1L × 1W | Hu, WB; 7.5 mL; spiked & clinical | < 2 | > 140 | > 142 |
| Microsinusoidal chip | 2008 | Antibody/aptamer based | 5L × 2Wd | Rb, WB; 1 mL; spiked | ~30 | > 35 | > 260 |
| MagSweeper | 2008 | Antibody based | 0.6D | Hu, WB; <9 mL; spiked & clinical | 60 | > 60 | > 120 |
| Micropillar chip | 2009 | Antibody based | na | Mo, WB; 1 mL; spiked | 60 | > 30 | > 480 |
| Microcrescent chip | 2009 | Cell size and deformability | 2L × 2We | Hu, WB; 1-3 mL; spiked & clinical | >86 | > 350 | > 575f |
| Microwall chip | 2009 | Cell size and deformability | 6L × 3W | Hu, WB; 1 mL; spiked | 60 | na | > 450 |
| MicroGEDI chip | 2009 | Antibody based | 2L × 1Wg | Hu, WB; 1 mL; spiked & clinical | 60 | > 100 | > 550 |
| Microvortex chip | 2010 | Antibody based | 7.6L × 2.5W | Hu, WB; ~4 mL; spiked & clinical | 96~160 | > 120 | > 300~420 |
| Nanopillar chip | 2010 | Antibody based | 5L × 2.5W | Hu, WB; 1 mL; spiked & clinical | 60 | > 100 | > 550 |
| Micropinching chip | 2011 | Cell size | na | Hu, WB; 1 mL; spiked | 50 | > 95 | > 208 |
| Microspiral chip | 2012 | Cell size | 4L × 3Wd | Hu, WB; 1 mL; spiked | 3h | na | na |
| MicroeDAR cytometer | 2012 | Antibody based | 5L × 3.5Wg | Hu, WB; 1 mL; spiked & clinical | 20 | > 80 | > 230 |
| Microsieve chip | 2012 | Cell size | 0.75D | Hu, WB; 1 mL; spiked & clinical | 3 | 90i | 113i |
Hu, Human; Rb, Rabbit; Mo, Mouse; WB, Whole blood; L, Length; W, Width; H, Height; D, Diameter; na, not available
a Time to flow a sample through and interact with the microdevice
b Time to prime the microdevice, pre-treat the sample before added to the microdevice, and post-treat the captured tumor cells in or outside the microdevice for identification, which was underestimated by adding the time for each step described in the assay protocol. The time to image and count CTCs was excluded, except the microsieve chip, for lack of information.
c Total consuming time for the entire assay of processing a fixed volume of 7.5 mL blood by combining the sample processing time and the assay preparation time. Sample processing time was proportionally extrapolated, while assay preparation time was kept constant.
d Estimated from the schematic of the chip
e Estimated by the information from the commercial product
f Time by simultaneous processing using multiple microdevices
g Estimated from the real photo of the chip
h Estimated by the flow rate of 20 mL/hour
I Time including the operation time for CTC imaging and enumeration.
Comparison of CTC microdevices: physical dimension and assay performance characteristics (continued).
| Microdevice | Clinical sensitivityj | Clinical specificityk | Viability | Recovery | Purity | Enrichment factorl | References |
|---|---|---|---|---|---|---|---|
| CellSearch (control) | 27%, 32%, 70% | 89%, 99.7%, 93%, 100% | na | 42%, 85% | 0.1%, 1.4% | 4 × 104 | 11, 31, 39, 67, 89, 90, 158 |
| Micropost chip | 64%m, 99%m | 100%m | ~99% | > 60% | 9%, 50% | 106 | 25, 28 |
| Microfilter | 96%n | 16%n | 85% | 86-90% | na | 107 | 23, 31, 78, 79 |
| Microsinusoidal chip | na | na | na | 90-97% | 100% | 2.5 × 108 | 24, 30 |
| MagSweeper | 100%m | na | 94% | > 50% | 51-100% | 108 | 27, 78, 80 |
| Micropillar chip | na | na | na | 71% | na | na | 26 |
| Microcrescent chip | na | na | na | 80% | 83-89% | 4.9 × 109 | 81, 82 |
| Microwall chip | na | na | na | na | na | na | 83 |
| MicroGEDI chip | 94%n | 0%n | na | 80-100% | 62-74% | 109 | 84, 85 |
| Microvortex chip | 93%m | na | 95% | 92% | 14% | na | 25 |
| Nanopillar chip | 75%n | 22%n | na | > 95% | na | na | 29 |
| Micropinching chip | na | na | > 90% | 80-90% | na | 3.25 × 105 p | 86 |
| Microspiral chip | na | na | na | 89% | na | 19 | 87 |
| MicroeDAR cytometer | 100%n | 0%n | na | 93% | 10-50% | na | 88 |
| Microsieve chip | na | na | na | > 80% | < 1%q | na | 69 |
na, not available
j The ratio of true test positive to overall actual positive as defined by the health condition of the human subject or provided by the presumptive gold standard, CellSearch.
k The ratio of true test negative to overall actual negative as defined by the health condition of the human subject or provided by the presumptive gold standard, CellSearch.
l A measure of the enrichment capability of a given microdevice
m The agreement between the test results and the hypothesis that the blood samples from known metastatic cancer patients are true positives, while samples from healthy subjects are true negatives. The assumption has not been validated by an independent method or CellSearch.
n The agreement between the microdevice and the CellSearch system, assuming CellSearch test is the gold standard for CTC enumeration, thereby providing true positives and negatives.
p Only feasible by sequential processing using two devices
q Estimated from the patient data
On the other hand, the turnaround time of microdevices is somewhat disappointing. The median of the total time consumed to perform the entire assay (Tables 2-3), if extrapolated for processing a fixed volume of 7.5 mL of blood, is approximately 6 hours (360 min), which does not include the time to image and count each single CTC. Therefore, it may be challenging to complete multiple CTC assays, from loading samples to generating results, in a typical eight-hour shift as worked by most clinical laboratory technicians. Moreover, a long turnaround time may also have an adverse impact on viability of CTCs and stability of cell immunophenotyping and genotyping profiles, if CTCs are not fixed or permanently stabilized upfront. Notably, recent studies have revealed that the actual CTC numbers in patients' blood could be, at least, 30-100 fold higher than that currently reported by the only FDA-cleared CellSearch system [28, 58]. Thus, as more sensitive microdevices are developed, the CTC counts could be evaluated with 1 mL of whole blood, instead of 7.5 mL, and completed within 2-3 hours.
Trends in CTC research and development
Detection of circulating tumor microemboli
The role of circulating tumor microemboli (CTM) in metastasis development has been emphasized in recent years in both animal and clinical studies. Occurring in blood or lymphatic vessels, CTM are composed of at least two tumor cells, and occasionally, normal blood cells. The generation of CTM is believed to result from collective migration of tumor cells [91] and intravasation of tumor cell clusters via a leaky vessel in the primary tumor [91, 92]. The inhibition of VEGF-A could also play a role in this process [93]. Circulating tumor microemboli have been observed in the peripheral blood of metastatic lung [25, 37, 94], liver [95], colorectal [96], renal [36, 93], breast [97] and prostate [25, 98] cancer patients, as well as in the lymphatic vessels of breast cancer patients and an animal model of lymphatic metastasis [99, 100]. The morphology of CTM is highly variable [11, 25, 94, 96]; irregular shapes, such as clusters, circles, and strands, are often seen [94]. It is likely that CTM form as a result of the increased adhesive characteristic of tumor cells in circulation [94]. With greater adhesiveness, possibly due, in part, to sticky DNA molecules released by dead cells, CTCs might accumulate and form CTM upon collision at low-shear blood flow conditions (Figure 1). However, further study is needed to confirm this process.
Increased evidence has suggested that the presence of CTM could be an indication of higher metastatic potential [95, 96, 98, 101]. In one study, CTM or CTCs were intravenously injected and a higher tendency of metastasis was shown for CTM than CTCs [101]. Additionally, more metastatic foci were observed by inoculation with larger sized CTM than an equal number of smaller CTM. Another study also suggested that the presence of CTM might be a more relevant prognostic factor for malignancy than that of CTCs alone [95]. In the setting of liver cancer, patients with one CTM or four or more CTCs displayed significantly shorter survival than those without CTM, or with less than four CTCs [95].
The possible explanation for the increased metastatic potential of CTM, when compared to CTCs, is that: 1) CTMs are more easily trapped in the narrow vasculature than CTCs [101]; and 2) CTM present a favorable microenvironment for tumor cell survival [94]. After arrested in capillaries, CTM microenvironment and derived growth factors (e.g., EGF and VEGF) could stimulate the proliferation of tumor cells and inhibit the apoptosis [102]. When CTM grow to a certain size and rupture capillary walls, metastasis is formed locally. Hence, extravasation may not be necessary in CTM-mediated metastasis [36]. If a cancer stem cell resides in the CTM, the resulting metastatic tumor will grow faster and become more malignant [103]. As a comparison, a solitary CTC has to extravasate the capillary wall and form the necessary microenvironment to survive and grow, and accordingly, endure a much longer latency period before it is detected. In addition, the majority of CTCs do not survive for long periods in the circulation, probably due to anoikis [94, 104]. Conversely, CTM maintain the cell-cell contact, which provides a favorable microenvironment for tumor cells to crosstalk and survive. This configuration leaves the innermost cells protected from the immunological assault by lymphocytes and natural-killer cells [94]. A study recently found that there were no apoptotic tumor cells within the CTM of small cell lung cancer (SCLC) or non-small cell lung cancer (NSCLC) patients [94, 104]. The production of autocrine pro-migratory factors and matrix proteases, the enzymes required for collective migration, by CTM might also contribute to their high metastatic potential [101].
The current knowledge for CTM is incomplete due to their extremely low abundance; sensitive and specific detection of CTM could, therefore, expedite the investigative process. Several methods have been used for CTM collection/detection, such as flow cytometry [9], immunomagnetic isolation (CellSearch) [37, 94], high-definition fluorescence scanning microscopy [12], size-based filtration (ISET) [37, 94, 104], the microsieve chip [69], and the microvortex herringbone-chip [25]. Both CellSearch and ISET were able to detect CTM in blood samples from SCLC patients, although more CTM, especially large sized, were found by ISET; while for NSCLC patients, CTM were detected by ISET, but not by CellSearch [94]. The causes for limited or unsuccessful CellSearch detection might be that: 1) CTM lack sufficient expression of EpCAM and/or CKs for immunomagnetic enrichment and identification [37, 69, 94, 105]; 2) large CTM are not effectively attracted by magnetic forces [94]; 3) dissociation of the CTM occurs during prolonged manipulation (Figure 1) [106]; or 4) tumor cells within CTM are surrounded by blood cells [69, 105] and, thus, could not be detected or recognized. When compared to CellSearch, ISET minimizes the manipulation of the blood sample and excludes the use of epithelial markers for enrichment [37]. As a result, the enrichment of CTM by ISET seems more reliable; while the burden of CTM measured by CellSearch could be underestimated [12]. Among the existing technologies, flow cytometry is a readily accessible platform for the detection and enumeration of CTM; it distinguishes cell clusters from CTCs by size and fluorescence intensity. However, large and variable-sized CTM may not be detectable and often block the flow by forming clumps, and sorting out the acquired CTM can be a challenge. After successful CTM enrichment, cytopathological and/or cytochemical examination is highly recommended for accurate identification, especially when considering the complexity and heterogeneity of CTM. Identifying the cell composition and microenvironmental effect, as well as gene and protein expression profiles, would be critical to further understand the metastatic potential of CTM [107, 108].
Detection of circulating tumor materials
CTCs are shed from solid tumors at a daily rate of 3.2 to 4.1x106 per gram of tissue, based on a rat model study [109]; however, half of these CTCs perish within 2.4 hours [110]. This elimination process initiates apoptotic signaling and causes membrane perforation of the CTCs, resulting in leakage of intracellular components, e.g., electrolytes, small molecules, DNA, and chromatin [4]. Extracellular substances, e.g. cytotoxic anti-cancer drugs used in chemotherapy, can also trigger the cell death via necrosis. In addition to in vivo immunological, apoptotic, and necrotic processes, surviving CTCs may be further stressed and damaged in vitro during the blood draw and subsequent sample processing, by temperature shock, fluidic turbulence, shear force, surface tension, and other harsh conditions. As a result of these destructive processes, damaged cells, fragmented cells, cellular debris, microparticles, and clump-like aggregates are created. Because CTCs are continuously shed and destroyed, such circulating tumor materials (CTMat) can accumulate and present in greater numbers than intact, damaged, and fragmented CTCs in whole blood samples [4, 111]. In 18 blood samples from prostate cancer patients, the intact CTCs, damaged CTCs, and CTC fragments were found to comprise 0% to 22% (mean 4%; smallest sub-population), 1% to 100% (mean 34%), and 0% to 93% (mean 62%; largest sub-population) of the total CTC cell population, respectively [4].
Although the prognostic and predictive roles of intact and viable CTCs have been the focus of most research efforts, the potential of CTMat as an independent biomarker for disease monitoring and prognosis should not be neglected [64, 66]. In a 2010 study, CTC fragments (large) and microparticles (small) were found to predict overall survival similar to intact CTCs (P < 0.001) [64]. Use of CTMat, instead of intact CTCs, could provide flexibility in prognosis due to their high abundance, which is particularly important to patients who have no intact CTCs detectable in the blood. Additionally, the use of CTMat may allow for less stringent target identification, easier enumeration, and fully automated image processing [64]. It is therefore speculated that the numbers of intact CTCs and CTMats, as well as their ratio to one another, may provide an important tool for the assessment of tumor burden, the proliferative capability of tumor cells, and therapeutic efficacy [4, 66].
Circulating tumor DNA (CTDNA) is one critical component of CTMat [112-114]. The release of CTDNA into a patient's blood is thought to derive mainly from the apoptotic and necrotic cells in solid tumors and only modestly from the disintegrated CTCs [4, 112, 115, 116]. Similar to the shedding of CTCs, CTDNA is fed into the blood circulation continuously and the yield is estimated to be 3.3% of tumor DNA per 100 gram of solid tumor per day [117]. The level of CTDNA in patients' blood has been demonstrated correlated with the malignancy status and the therapy response [118, 119]. A study using personalized analysis of rearranged ends (PARE) reported that the amount of CTDNA in plasma decreased immediately after removal of the primary tumor, followed by an increase in subsequent weeks, and then another decrease after chemotherapy and removal of a metastatic tumor in one colorectal patient [113, 114]. It was also found that the plasma CTDNA level was correlated with the tumor stage as demonstrated by the significant difference between localized and metastasized prostate cancer [112].
Telomerase activity and CTC detection
Epithelial markers, such as CK 8, 18, 19, and EpCAM, are the commonly used biomarkers for CTC identification, although they only identify tissue origin and not biological behavior (benign vs. malignant). It is known that tumor cells could decrease or lose epithelial marker expression during metastasis/dissemination, causing significant heterogeneity [37, 94]. Therefore, the CTCs detected by epithelial markers may not represent the actual or entire CTC population. Telomerase has been found activated in the majority of cancer types and is known to be associated with malignant properties [120]. Seventy-nine% of patients with localized prostate cancer [121], 100% with stage IV ovarian cancer [122], 84% with stage IV breast cancer [123], 73% with stage IIIB/IV NSCLC [124], 72% with stage C or D (Dukes classification) colon cancer [124], and 90% with metastatic bladder cancer [125] have demonstrated telomerase activity in blood samples, but no activity was demonstrated in the samples of healthy volunteers [121-125]. Since elevated telomerase activity is one of the hallmarks for stem cell identification/characterization [126], the abovementioned results could be also related to circulating cancer stem cells. Although assessing telomerase activity may have potential as a method to more accurately detect entire CTC populations, the whole blood sample has to be lysed in order to measure the enzyme activity, thereby destroying all intact CTCs, and precluding enumeration and any other follow-up analysis. This constitutes a significant downside to this method.
Aptamer technology in CTC Detection
Antibody-based immuno-identification has been the mainstream for CTC detection. This strategy, however, is limited by the availability and specificity of the antibodies [30]. Aptamers, single-stranded RNA or DNA molecules, have demonstrated potential as an alternative identification method [30, 127, 128]. The molecular weight of the aptamer is small, about 8 to 15 kDa, leading to rapid tumor penetration and blood clearance. Aptamers also provide high stability, resistance to harsh conditions (pH, urea, organic solvents, chaotropic salts, and detergents), reversible denaturation, negligible toxicity and immunogenicity, and oriented surface immobilization. These qualities are typically not found in antibodies. Remarkably, aptamers can be developed directly against the binding targets, ranging from small compounds to large cell membrane and/or transmembrane proteins, without prior knowledge of these target molecules.
The technique of Systematic Evolution of Ligands by Exponential Enrichment (i.e., SELEX) has been employed to facilitate the automation of in vitro aptamer production [129]. When compared to antibodies, the manufacture of aptamers is relatively less expensive and much faster [127, 128]. This is mainly due to their small size, simple structure, and because they do not require the use of animal cells. In numerous studies, aptamers have been created to target cancer cells via extracellular membrane proteins, such as prostate-specific membrane antigen (PSMA), nucleolin, human epidermal growth factor receptor 3 (HER-3), RET, tenascin-C, and muc1 [130]. The synthesized aptamers against PSMA were developed with nanomolar affinity, which is equivalent to that of an antibody, affording them great potential for cancer imaging or therapeutics [131]. Differential Cell-SELEX technology could be employed to produce aptamers to target CTCs. The method isolates a group of aptamers from a synthesized oligonucleotide library that specifically targets differentially expressed rare cell surface proteins without the need for previous information for the proteins. It is postulated that one aptamer binds to one specific surface antigen on the rare cell, while multiple diversified aptamers recognize the fingerprint of a cell phenotype with greater binding avidity, allowing the aptamers to distinguish cell type (e.g., CTC vs. normal cell), cancer type, malignant status, metastatic potential (e.g., epithelial type CTC vs. mesenchymal type CTC), and proliferation capability [127]. Therefore, multivalent CTC enrichment and analysis using aptamers specific to the molecular signature could elucidate the intrinsic heterogeneity of CTCs, and eventually realize the separation of the subpopulation that best represents the metastasis. Moreover, aptamer probes and antibodies could simultaneously bind to the same biomarker molecule by targeting different epitopes.
Perhaps the incorporation of aptamer technology into microdevices has the most potential for the development of a fast, easy, and accurate method for cell isolation, enrichment and related purposes such as detection, enumeration, and characterization (Figure 3). An aptamer-based, cell-affinity chromatography microdevice has been developed that is capable of isolating tumor cells from a mixture of control cells [132], and simultaneously sorting and enriching multiple distinct cells into independent fractions [133]. The enrichment factor of the microdevice was determined to be ~130 times. The sorted cells were able to grow as well as the cultured cells at 96% purity. A micro-sinusoidal chip tethered with PSMA-specific aptamers also displayed high performance on the detection and enumeration of rare LNCaP cells in whole blood [30]. Results demonstrated a 90% recovery and ~100% purity and detection efficiency. A specially structured aptamer also displayed the potential for rapid and efficient isolation of CTCs [134]. A microdevice immobilized with long DNA aptamers was developed and each aptamer was prepared by the process of rolling circle amplification [135] and composed of multiple binding sites. A number of these multivalent aptamers were able to capture rare cells via an improved avidity and efficiency. Many of these long DNA aptamers subsequently created a three dimensional network, enabling rapid cell capture under high flow rates.
CTC functional study
It is important to note that research on CTCs has not been limited to detection or enumeration. In many instances, when viable and proliferative CTCs have been successfully isolated, scientists have attempted to investigate their functions in in vivo models (Figure 3). Viable CTCs have been isolated from murine blood, cultured, and implanted into other mice (Figure 3) [78, 80]. Results showed that isolated CTCs displayed a greater capability of colony formation than the parental cultured cells. Moreover, as expected, xenografts generated from isolated CTCs were larger and heavier, and metastasized faster than the original cell line. If CTCs are more aggressive than their parental cell line, a derivative hypothesis could be that CTCs have stem or progenitor cell-like properties.
Because often only a limited number of CTCs are captured in most CTC assays, it is important to expand the isolated CTCs ex vivo and, ideally, in situ in order to perform subsequent functional analyses. A plastic version of the micropost CTC-chip has been developed to capture viable CTCs and culture them directly on the chip [26]. Viable CTCs were found to expand after 24 hr culture using this chip. Cell clusters were observed after five days and, after 12 days, the clusters had expanded and cells were found on top of the microposts. Moreover, the cultured CTCs were found >99% viable.
Chemotherapeutic efficacy is often monitored by radiological imaging and/or the level of the serum tumor markers, in which it could take months to recognize a change [136, 137]. The count of CTCs appears to respond more rapidly to the treatment, in as little as a few weeks [138, 139]. Beyond an in vitro number count, an ex vivo functional study on patient-derived CTCs might provide for an immediate treatment decision regarding drug resistance [84]. To reveal the real-time, individualized drug-target interaction, live CTCs, freshly removed from the patient, can be the target for the anti-cancer drug candidates. Lately, the activity of docetaxel and paclitaxel was preliminarily studied on CTCs isolated from castrate-resistant prostate cancer patients using a GEDI microdevice [84]. The interaction between taxane drugs and CTCs was monitored at the molecular level via microtubule bundling. Some patients failed docetaxel, and consistently, their CTCs showed no activity when exposed ex vivo to docetaxel. Other patients displayed drug-dependent efficacy during chemotherapy, and correspondingly, their CTCs demonstrated aberrant mitotic arrest or apoptotic nuclei in response to paclitaxel, but not to docetaxel.
Analyzing the proteins directly secreted by viable CTCs is an emerging approach to profile valuable tumor markers that may be relevant to metastasis. The epithelial immunospot (EPISPOT) technique has been utilized to immunospot the released marker proteins in culture media [140-143]. It was found that, by using EPISPOT, 100% of metastatic breast and 83.3% of prostate cancer patients had cells present in blood that secreted tumor antigens mucin 1 and prostate-specific antigen, respectively. These cells were not detected in healthy donors or patients with benign disease. The markers that potentially affect metastasis, such as cathepsin D protease, fibroblast growth factor 2, and cytokeratin-19, were also within the scope of secretion by the tumor cells [141, 142].
Mathematical Modeling
Variation in CTC detection methodologies and clinical data management has created discordance across studies [46, 47, 60] and generated significant disagreement amongst both scientists and clinicians on how to interpret the data and apply it clinically [71]. Mathematical modeling could be a useful tool in differentiating rational results from those that are not, and especially for ruling out data that is outside of the inherent boundaries in rare event measurement [11]; supplementary materials. Three commonly asked boundaries have been demonstrated by mathematical modeling: 1) what is the lowest number of CTCs that would need to be in a 7.5 mL blood sample to allow CellSearch system to detect one CTC at 85% recovery? The minimum is 1.2 ± 0.4 based on the binomial distribution analysis and lower than the limit will cause the failure; 2) what is the theoretical CV for CTC quantification? On the basis of a Poisson distribution, it decreases with the increase of the CTC counts. Therefore, 50%, 24%, 12%, 6%, and 3% are the estimated CVs for acquiring 4, 18, 71, 286, and 1142 CTCs, respectively; 3) what is the range of the CTC numbers that might have actually been in that tube of blood, if one obtains 5 CTCs in the clinical study? It is between 2 and 12, with 95% confidence. This range narrows downward when the confidence level decreases.
Mathematical models could also reveal the methodological constraints for the CellSearch system [55] and flow cytometry [56, 57]. When the CellSearch system is used for a patient with 500 CTCs in circulation, the probability of collecting ≥1 CTCs in one aliquot of 7.5 mL blood is 50%, while the probability of detecting 1 CTC is only 18% considering the statistical contribution from each step of the process [55]. As for flow cytometry-based CTC acquisition, the correlation between the sample size (i.e., number of cell events) and detection precision (i.e., CV) is inverse; the smaller the CV, the more events are to be collected. For instance, a 40% CV requires 6.3 x 106 events (approximately 1 mL blood); while for a reduction to a 10% CV, 109 events needs to be acquired from approximately 200 mL blood [57].
Dichotomization is a commonly used method in CTC analysis, which is based on a single cutoff; for instance, 5 CTCs for metastatic breast cancer [90, 144, 145]. The method categorizes cancer patients into two response groups, with favorable or unfavorable prognosis [11, 146-148]. While the use of dichotomization is straightforward, it has its limitations, including loss of statistical power, loss of nonlinearity, generation of observer bias, origination of discordance across studies, and creation of false prognostic factors [149-153]. Analysis with standard categorical survival analysis and spline regression models may resolve the problems [153]. A multivariable spline Cox regression model has been used for the interpretation of the relationship between the number of CTCs and the prognosis of metastatic breast cancer. Four categories, 0, 1-4, 5-20, and >20, were applied to illustrate the relationship. Results showed that, with an increasing number of CTCs, the risks of cancer progression (hazard ratio of progression-free survival) and death (hazard ratio of overall survival) increased continuously. A linear relationship was demonstrated with a small number of CTCs (< 5), however, the increase in both risks was then tapered after the number reached 5. When more than 20 CTCs were detected, the risk of progression became slightly changed, while the risk of death was almost constant. Notably, by using more sensitive detection approaches, recent studies demonstrated the presence of at least a 30-100 fold higher number of CTCs, in a variety of cancer patients, than that currently reported by the CellSearch system [28, 58]. Thus, technology advancing the mathematical modeling of CTCs may need to be refined according to the sensitivity of detection methods and volume of blood samples used for assays.
Concluding remarks
The road for CTC exploration is long and rough. What we have experienced thus far has been inspiring; however, we want to make sure we do not move at such a rapid pace that something is overlooked. The rare cells that have been identified via EpCAM and/or CK may be better called circulating epithelial cells due to the existence of non-tumorous epithelial cells in blood [12, 84, 154]. More aggressive cell subpopulations might have been missed or ignored because they were EpCAM-, CK- or CD45+/CK+/EpCAM+ [155-157]. The blood volume of 7.5 mL seems widely accepted along with the use of the CellSearch system, although additional aliquots of blood, which is clinically allowed, could make significant improvements in sensitivity and sampling accuracy. Notably, as shown in Figure 2, the sensitivity and specificity depend on all three major steps of the CTC assay. Introduction of new technology in each step will certainly enhance the power of the CTC assay. Translation of CTC research findings from the laboratory to clinical practice has been slow. Eight years ago, CellSearch system was cleared by FDA for CTC based cancer diagnosis. Until now CellSearch system is still the only one on the market and has not been demanded to the extent that it could be. The main reason well accepted is the unclear benefit of using CTC in treatment decision-making, and so to clarify the concern, extended validation has been recommended since 2007 [71]. However, aforementioned methodological constraints, inherent in the CellSearch sytem [28, 40, 58], cannot be neglected. A detection system with inadequate cell recovery, analytical sensitivity, and sampling accuracy may continue providing mixed results, and cumulatively, further stress the limited utility.
Opportunities frequently come with challenges. Increasing knowledge of CTCs and their tumor cell-associated and/or -derived materials, such as CTM and CTMat (including CTDNA), will likely lead to exciting discoveries and provide novel insight into tumor burden, therapy efficacy, metastatic potential, proliferative capability, and heterogeneity. Measuring elevated telomerase activity might be a promising avenue to target the majority of tumor cell populations in blood. The advances with aptamers and microdevices have succeeded, through a variety of routes, in minimizing the technical constraints. The application of mathematical models could smartly direct the rare event handling and data management. Successful ex-vivo CTC culture methods will allow for subsequent functional studies that are poised as the next important step in CTC research. There are also numerous opportunities to evaluate CTCs systemically and systematically using proteomics and genomics. Most importantly, however, is to move the science forward and beyond simple enumeration, and to view and respect CTCs as a contributing, functional system.
Conflict of Interest
The authors have declared that no conflict of interests exists.
Acknowledgements
This project was supported by NIH grants (to Y.Z.) R01CA151955, R33CA173382, and 5P50CA126752 and Golden Key Alumni Professional Development Grant (to B.H.).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61(2):69-90
2. Wicha MS, Hayes DF. Circulating tumor cells: not all detected cells are bad and not all bad cells are detected. J Clin Oncol. 2011;29(12):1508-1511
3. Fass L. Imaging and cancer: a review. Mol Oncol. 2008;2(2):115-52
4. Rao GC, Larson C, Repollet M, Rutner H, Terstappen LW, O'hara SM, Gross S. Analysis of circulating tumor cells, fragments, and debris. United States Patent 7863012. 2011
5. Asworth TR. A case of cancer in which cells similar to those in tumors were seen in the blood after death. Aust Med J. 1869;14:146-149
6. Leary JF. Rare event detection and sorting of rare cells. Durack G, Robinson JP, ed. Emerging tools for cell analysis: advances in optical measurement technology, Wilmington, DE: Wiley-Liss. 2000:49-72
7. Attard G, de Bono JS. Utilizing circulating tumor cells: challenges and pitfalls. Curr Opin Genet Dev. 2011;21(1):50-58
8. Racila E, Euhus D, Weiss AJ, Rao C, McConnell J, Terstappen LW, Uhr JW. Detection and characterization of carcinoma cells in the blood. Proc Natl Acad Sci USA. 1998;95(8):4589-4594
9. Takao M, Takeda K. Enumeration, characterization, and collection of intact circulating tumor cells by cross contamination-free flow cytometry. Cytometry A. 2011;79(2):107-117
10. Hong B, Mace M, Crowder R, Coleman T, He W, Szczepanski F, Feczko J, Lyzak J. Metastatic breast cancer detection and therapy monitoring using folate-targeting flow cytometry. J Clin Oncol. 2011;29(Suppl 27):23
11. Allard WJ, Matera J, Miller MC, Repollet M, Connelly MC, Rao C, Tibbe AG, Uhr JW, Terstappen LW. Tumor cells circulate in the peripheral blood of all major carcinomas but not in healthy subjects or patients with nonmalignant diseases. Clin Cancer Res. 2004;10(20):6897-6904
12. Marrinucci D, Bethel K, Kolatkar A, Luttgen MS, Malchiodi M, Baehring F, Voigt K, Lazar D, Nieva J, Bazhenova L, Ko AH, Korn WM, Schram E, Coward M, Yang X, Metzner T, Lamy R, Honnatti M, Yoshioka C, Kunken J, Petrova Y, Sok D, Nelson D, Kuhn P. Fluid biopsy in patients with metastatic prostate, pancreatic and breast cancers. Phys Biol. 2012;9(1):016003
13. Krivacic RT, Ladanyi A, Curry DN, Hsieh HB, Kuhn P, Bergsrud DE, Kepros JF, Barbera T, Ho MY, Chen LB, Lerner RA, Bruce RH. A rare-cell detector for cancer. Proc Natl Acad Sci USA. 2004;101(29):10501-10504
14. Hsieh HB, Marrinucci D, Bethel K, Curry DN, Humphrey M, Krivacic RT, Kroener J, Kroener L, Ladanyi A, Lazarus N, Kuhn P, Bruce RH, Nieva J. High speed detection of circulating tumor cells. Biosens Bioelectron. 2006;21(10):1893-1899
15. Pinzani P, Salvadori B, Simi L, Bianchi S, Distante V, Cataliotti L, Pazzagli M, Orlando C. Isolation by size of epithelial tumor cells in peripheral blood of patients with breast cancer: correlation with real-time reverse transcriptase-polymerase chain reaction results and feasibility of molecular analysis by laser microdissection. Hum Pathol. 2006;37(6):711-718
16. Wong NS, Kahn HJ, Zhang L, Oldfield S, Yang LY, Marks A, Trudeau ME. Prognostic significance of circulating tumour cells enumerated after filtration enrichment in early and metastatic breast cancer patients. Breast Cancer Res Treat. 2006;99(1):63-69
17. Rolle A, Günzel R, Pachmann U, Willen B, Höffken K, Pachmann K. Increase in number of circulating disseminated epithelial cells after surgery for non-small cell lung cancer monitored by MAINTRAC ® is a predictor for relapse: A preliminary report. World J Surg Oncol. 2005;3(1):18
18. Pachmann K, Camara O, Kavallaris A, Krauspe S, Malarski N, Gajda M, Kroll T, Jörke C, Hammer U, Altendorf-Hofmann A, Rabenstein C, Pachmann U, Runnebaum I, Höffken K. Monitoring the response of circulating epithelial tumor cells to adjuvant chemotherapy in breast cancer allows detection of patients at risk of early relapse. J Clin Oncol. 2008;26(8):1208-1215
19. Rosenberg R, Gertler R, Friederichs J, Fuehrer K, Dahm M, Phelps R, Thorban S, Nekarda H, Siewert JR. Comparison of two density gradient centrifugation systems for the enrichment of disseminated tumor cells in blood. Cytometry. 2002;49(4):150-158
20. Gertler R, Rosenberg R, Fuehrer K, Dahm M, Nekarda H, Siewert JR. Detection of circulating tumor cells in blood using an optimized density gradient centrifugation. Recent Results Cancer Res. 2003;162:149-155
21. Bossolasco P, Ricci C, Farina G, Soligo D, Pedretti D, Scanni A, Deliliers GL. Detection of micrometastatic cells in breast cancer by RT-pCR for the mammaglobin gene. Cancer Detect Prev. 2002;26(1):60-63
22. Zieglschmid V, Hollmann C, Böcher O. Detection of disseminated tumor cells in peripheral blood. Crit Rev Clin Lab Sci. 2005;42(2):155-196
23. Zheng S, Lin H, Liu JQ, Balic M, Datar R, Cote RJ, Tai YC. Membrane microfilter device for selective capture, electrolysis and genomic analysis of human circulating tumor cells. J Chromatogr A. 2007;1162(2):154-161
24. Adams AA, Okagbare PI, Feng J, Hupert ML, Patterson D, Göttert J, McCarley RL, Nikitopoulos D, Murphy MC, Soper SA. Highly efficient circulating tumor cell isolation from whole blood and label-free enumeration using polymer-based microfluidics with an integrated conductivity sensor. J Am Chem Soc. 2008;130(27):8633-8641
25. Stott SL, Hsu CH, Tsukrov DI, Yu M, Miyamoto DT, Waltman BA, Rothenberg SM, Shah AM, Smas ME, Korir GK, Floyd FP Jr, Gilman AJ, Lord JB, Winokur D, Springer S, Irimia D, Nagrath S, Sequist LV, Lee RJ, Isselbacher KJ, Maheswaran S, Haber DA, Toner M. Isolation of circulating tumor cells using a microvortex-generating herringbone-chip. Proc Natl Acad Sci USA. 2010;107(43):18392-18397
26. Helzer KT, Barnes HE, Day L, Harvey J, Billings PR, Forsyth A. Circulating tumor cells are transcriptionally similar to the primary tumor in a murine prostate model. Cancer Res. 2009;69(19):7860-7866
27. Talasaz AH, Powell AA, Huber DE, Berbee JG, Roh KH, Yu W, Xiao W, Davis MM, Pease RF, Mindrinos MN, Jeffrey SS, Davis RW. Isolating highly enriched populations of circulating epithelial cells and other rare cells from blood using a magnetic sweeper device. Proc Natl Acad Sci USA. 2009;106(10):3970-3975
28. Nagrath S, Sequist LV, Maheswaran S, Bell DW, Irimia D, Ulkus L, Smith MR, Kwak EL, Digumarthy S, Muzikansky A, Ryan P, Balis UJ, Tompkins RG, Haber DA, Toner M. Isolation of rare circulating tumous cells in cancer patients by microchip technology. Nature. 2007;450(7173):1235-1239
29. Wang S, Liu K, Liu J, Yu ZT, Xu X, Zhao L, Lee T, Lee EK, Reiss J, Lee YK, Chung LW, Huang J, Rettig M, Seligson D, Duraiswamy KN, Shen CK, Tseng HR. Highly efficient capture of circulating tumor cells by using nanostructured silicon substrates with integrated chaotic micromixers. Angew Chem Int Ed Engl. 2011;50(13):3084-3088
30. Dharmasiri U, Balamurugan S, Adams AA, Okagbare PI, Obubuafo A, Soper SA. Highly efficient capture and enumeration of low abundance prostate cancer cells using prostate-specific membrane antigen aptamers immobilized to a polymeric microfluidic device. Electrophoresis. 2009;30(18):3289-3300
31. Lin HK, Zheng S, Williams AJ, Balic M, Groshen S, Scher HI, Fleisher M, Stadler W, Datar RH, Tai YC, Cote RJ. Portable filter-based microdevice for detection and characterization of circulating tumor cells. Clin Cancer Res. 2010;16(20):5011-5018
32. Sun YF, Yang XR, Zhou J, Qiu SJ, Fan J, Xu Y. Circulating tumor cells: advances in detection methods, biological issues, and clinical relevance. J Cancer Res Clin Oncol. 2011;137(8):1151-73
33. Swaby RF, Cristofanilli M. Circulating tumor cells in breast cancer: a tool whose time has come of age. BMC Med. 2011;9:43
34. Yu M, Stott S, Toner M, Maheswaran S, Haber DA. Circulating tumor cells: approaches to isolation and characterization. J Cell Biol. 2011;192(3):373-82
35. Ghadially R. The role of stem and circulating cells in cancer metastasis. J Surg Oncol. 2011;103(6):555-7
36. Paterlini-Brechot P, Benali NL. Circulating tumor cells (CTC) detection: clinical impact and future directions. Cancer Lett. 2007;253(2):180-204
37. Hou JM, Krebs M, Ward T, Morris K, Sloane R, Blackhall F, Dive C. Circulating tumor cells, enumeration and beyond. Cancers. 2010;2(2):1236-1250
38. Maheswaran S, Haber DA. Circulating tumor cells: a window into cancer biology and metastasis. Curr Opin Genet Dev. 2010;20(1):96-99
39. Cohen SJ, Punt CJ, Iannotti N, Saidman BH, Sabbath KD, Gabrail NY, Picus J, Morse M, Mitchell E, Miller MC, Doyle GV, Tissing H, Terstappen LW, Meropol NJ. Relationship of circulating tumor cells to tumor response, progression-free survival, and overall survival in patients with metastatic colorectal cancer. J Clin Oncol. 2008;26(19):3213-3221
40. Tanaka F, Yoneda K, Kondo N, Hashimoto M, Takuwa T, Matsumoto S, Okumura Y, Rahman S, Tsubota N, Tsujimura T, Kuribayashi K, Fukuoka K, Nakano T, Hasegawa S. Circulating tumor cell as a diagnostic marker in primary lung cancer. Clin Cancer Res. 2009;15(22):6980-6986
41. Devriese LA, Voest EE, Beijnen JH, Schellens JH. Circulating tumor cells as pharmacodynamic biomarker in early clinical oncological trials. Cancer Treat Rev. 2011;37(8):579-589
42. Mostert B, Sleijfer S, Foekens JA, Gratama JW. Circulating tumor cells (CTCs): detection methods and their clinical relevance in breast cancer. Cancer Treat Rev. 2009;35(5):463-474
43. Oldenhuis CN, Oosting SF, Gietema JA, de Vries EG. Prognostic versus predictive value of biomarkers in oncology. Eur J Cancer. 2008;44(7):946-953
44. DeCaprio AP. Introduction to toxicologic biomarkers. DeCaprio AP, ed. Toxicologic Biomarkers, New York: Taylor and Francis Group. 2006:6-8
45. Saijo N. Critical comments for roles of biomarkers in the diagnosis and treatment of cancer. Cancer Treat Rev. 2011;38(1):63-7
46. Nagaiah G, Abraham J. Circulating tumor cells in the management of breast cancer. Clin Breast Cancer. 2010;10(3):209-216
47. Alunni-Fabbroni M, Sandri MT. Circulating tumour cells in clinical practice: Methods of detection and possible characterization. Methods. 2010;50(4):289-297
48. Gion M. Serum tumour markers: from quality control to total quality management. Breast. 2000;9(6):306-311
49. Sturgeon C. Limitations of assay techniques for tumor markers. In: (ed.) Diamandis EP, Fritsche HA, Lilja H, Chan DW, Schwartz MK. Tumor markers: physiology, pathobiology, technology, and clinical applications. Washington, DC: AACC Press. 2002:65-82
50. Gion M, Daidone MG. Circulating biomarkers from tumour bulk to tumour machinery: promises and pitfalls. Eur J Cancer. 2004;40(17):2613-2622
51. Knottnerus JA, van Weel C, Muris JW. Evaluation of diagnostic procedures. BMJ. 2002;324:477-480
52. Parkinson DR, Dracopoli N, Gumbs Petty B, Compton C, Cristofanilli M, Deisseroth A, Hayes DF, Kapke G, Kumar P, Lee JS, Liu MC, McCormack R, Mikulski S, Nagahara L, Pantel K, Pearson-White S, Punnoose EA, Roadcap LT, Schade AE, Scher HI, Sigman CC, Kelloff GJ. Considerations in the development of circulating tumor cell technology for clinical use. J Transl Med. 2012;10(1):138
53. Leary JF. Strategies for rare cell detection and isolation. Darzynkiewicz Z, Robinson JP, Crissman HA, ed. Methods in Cell Biology: Flow Cytometry, San Diego: Academic Press. 1994:331-358
54. Lalmahomed ZS, Kraan J, Gratama JW, Mostert B, Sleijfer S, Verhoef C. Circulating tumor cells and sample size: the more, the better. J Clin Oncol. 2010;28(17):e288-e289
55. Tibbe AG, Miller MC, Terstappen LW. Statistical considerations for enumeration of circulating tumor cells. Cytometry A. 2007;71(3):154-162
56. Rosenblatt JI, Hokanson JA, McLaughlin SR, Leary JF. Theoretical basis for sampling statistics useful for detecting and isolating rare cells using flow cytometry and cell sorting. Cytometry. 1997;27(3):233-238
57. Allan AL, Keeney M. Circulating tumor cell analysis: technical and statistical considerations for application to the clinic. J Oncol. 2010;2010:426218
58. Flores LM, Kindelberger DW, Ligon AH, Capelletti M, Fiorentino M, Loda M, Cibas ES, Jänne PA, Krop IE. Improving the yield of circulating tumour cells facilitates molecular characterisation and recognition of discordant HER2 amplification in breast cancer. Br J Cancer. 2010;102(10):1495-502
59. Cramb R. Guideline for point-of-care testing. In: Price CP, John A, Hicks J (ed) Point-of-Care Testing, Washington, DC: AACC Press. 2004:155
60. Amadori A, Rossi E, Zamarchi R, Carli P, Pastorelli D, Jirillo A. Circulating and disseminated tumor cells in the clinical management of breast cancer patients: unanswered questions. Oncology. 2009;76(6):375-386
61. Dotan E, Cohen SJ, Alpaugh KR, Meropol NJ. Circulating tumor cells: evolving evidence and future challenges. Oncologist. 2009;14(11):1070-1082
62. Lianidou ES, Mavroudis D, Sotiropoulou G, Agelaki S, Pantel K. What's new on circulating tumor cells? A meeting report. Breast Cancer Res. 2010;12(4):307
63. Drenck N. Point of care testing in Critical Care Medicine: the clinician's view. Clin Chim Acta. 2001;307(1-2):3-7
64. Coumans FA, Doggen CJ, Attard G, de Bono JS, Terstappen LW. All circulating EpCAM+CK+CD45- objects predict overall survival in castration-resistant prostate cancer. Ann Oncol. 2010;21(9):1851-1857
65. Kraan J, Sleijfer S, Strijbos MH, Ignatiadis M, Peeters D, Pierga JY, Farace F, Riethdorf S, Fehm T, Zorzino L, Tibbe AG, Maestro M, Gisbert-Criado R, Denton G, de Bono JS, Dive C, Foekens JA, Gratama JW. External quality assurance of circulating tumor cell enumeration using the CellSearch® system: A feasibility study. Cytometry B Clin Cytom. 2011;80(2):112-118
66. Rossi E, Basso U, Celadin R, Zilio F, Pucciarelli S, Aieta M, Barile C, Sava T, Bonciarelli G, Tumolo S, Ghiotto C, Magro C, Jirillo A, Indraccolo S, Amadori A, Zamarchi R. M30 neoepitope expression in epithelial cancer: quantification of apoptosis in circulating tumor cells by CellSearch analysis. Clin Cancer Res. 2010;16(21):5233-43
67. Fischer AH. Circulating tumor cells: seeing is believing. Arch Pathol Lab Med. 2009;133(9):1367-1369
68. Titus K. Sizing up a role for circulating tumor cells. CAP today. 2011;25:1-38
69. Lim LS, Hu M, Huang MC, Cheong WC, Gan AT, Looi XL, Leong SM, Koay ES, Li MH. Microsieve lab-chip device for rapid enumeration and fluorescence in situ hybridization of circulating tumor cells. Lab Chip. 2012;12(21):4388-96
70. Ligthart ST, Coumans FA, Attard G, Cassidy AM, de Bono JS, Terstappen LW. Unbiased and automated identification of a circulating tumour cell definition that associates with overall survival. PLoS One. 2011;6(11):e27419
71. Harris L, Fritsche H, Mennel R, Norton L, Ravdin P, Taube S, Somerfield MR, Hayes DF, Bast RC Jr. American Society of Clinical Oncology 2007 update of recommendations for the use of tumor markers in breast cancer. J Clin Oncol. 2007;25(33):5287-5312
72. Lilja H, Babaian R, Dowell B, Klee GG, Rittenhouse H, Semjonow A, Sibley P, Sokoll L, Stephan C. Tumor markers in prostate cancer. In: (ed.) Sturgeon CM, Diamandis E. Laboratory medicine practice guidelines: Use of tumor markers in testicular, prostate, colorectal, breast, and ovarian cancers. Washington, DC: The National Academy of Clinical Biochemistry, The American Association for Clinical Chemistry (AACC). 2009:25
73. Edge SB, Byrd DR, Compton CC, Fritz AG, Greene FL, Trotti A. AJCC Cancer Staging Handbook: From the AJCC Cancer Staging Manual, 7th ed. New York, USA: Springer. 2010
74. Kilgore ML, Steindel SJ, Smith JA. Evaluating stat testing options in an academic health center: therapeutic turnaround time and staff satisfaction. Clin Chem. 1998;44(8 Pt 1):1597-603
75. Louie RF, Tang Z, Shelby DG, Kost GJ. Point-of-Care Testing: Millennium Technology for Critical Care. Lab Medicine. 2000;31:402-408
76. Bange A, Halsall HB, Heineman WR. Microfluidic immunosensor systems. Biosens Bioelectron. 2005;20(12):2488-2503
77. Chen J, Li J, Sun Y. Microfluidic approaches for cancer cell detection, characterization, and separation. Lab Chip. 2012;12(10):1753-67
78. Kaiser J. Medicine. Cancer's circulation problem. Science. 2010;327(5969):1072-1074
79. Zheng S, Lin HK, Lu B, Williams A, Datar R, Cote RJ, Tai YC. 3D microfilter device for viable circulating tumor cell (CTC) enrichment from blood. Biomed. Microdevices. 2011;13(1):203-213
80. Ameri K, Luong R, Zhang H, Powell AA, Montgomery KD, Espinosa I, Bouley DM, Harris AL, Jeffrey SS. Circulating tumour cells demonstrate an altered response to hypoxia and an aggressive phenotype. Br J Cancer. 2010;102(3):561-569
81. Tan SJ, Lakshmi RL, Chen P, Lim WT, Yobas L, Lim CT. Versatile label free biochip for the detection of circulating tumor cells from peripheral blood in cancer patients. Biosens Bioelectron. 2010;26(4):1701-5
82. Tan SJ, Yobas L, Lee GY, Ong CN, Lim CT. Microdevice for the isolation and enumeration of cancer cells from blood. Biomed Microdevices. 2009;11(4):883-92
83. Mohamed H, Murray M, Turner JN, Caggana M. Isolation of tumor cells using size and deformation. J Chromatogr A. 2009;1216(47):8289-95
84. Kirby BJ, Jodari M, Loftus MS, Gakhar G, Pratt ED, Chanel-Vos C, Gleghorn JP, Santana SM, Liu H, Smith JP, Navarro VN, Tagawa ST, Bander NH, Nanus DM, Giannakakou P. Functional characterization of circulating tumor cells with a prostate-cancer-specific microfluidic device. PLoS One. 2012;7(4):e35976
85. Gleghorn JP, Pratt ED, Denning D, Liu H, Bander NH, Tagawa ST, Nanus DM, Giannakakou PA, Kirby BJ. Capture of circulating tumor cells from whole blood of prostate cancer patients using geometrically enhanced differential immunocapture (GEDI) and a prostate-specific antibody. Lab Chip. 2010;10(1):27-9
86. Bhagat AA, Hou HW, Li LD, Lim CT, Han J. Pinched flow coupled shear-modulated inertial microfluidics for high-throughput rare blood cell separation. Lab Chip. 2011;11(11):1870-8
87. Sun J, Li M, Liu C, Zhang Y, Liu D, Liu W, Hu G, Jiang X. Double spiral microchannel for label-free tumor cell separation and enrichment. Lab Chip. 2012;12(20):3952-60
88. Schiro PG, Zhao M, Kuo JS, Koehler KM, Sabath DE, Chiu DT. Sensitive and high-throughput isolation of rare cells from peripheral blood with ensemble-decision aliquot ranking. Angew Chem Int Ed Engl. 2012;51(19):4618-22
89. Beveridge R. Circulating tumor cells in the management of metastatic breast cancer patients. Commun Oncol. 2007;4(2):79-82
90. De Giorgi U, Valero V, Rohren E, Dawood S, Ueno NT, Miller MC, Doyle GV, Jackson S, Andreopoulou E, Handy BC, Reuben JM, Fritsche HA, Macapinlac HA, Hortobagyi GN, Cristofanilli M. Circulating tumor cells and [18F]fluorodeoxyglucose positron emission tomography/computed tomography for outcome prediction in metastatic breast cancer. J Clin Oncol. 2009;27(20):3303-3311
91. Friedl P, Wolf K. Tumour-cell invasion and migration: diversity and escape mechanisms. Nat Rev Cancer. 2003;3(5):362-374
92. Christiansen JJ, Rajasekaran AK. Reassessing epithelial to mesenchymal transition as a prerequisite for carcinoma invasion and metastasis. Cancer Res. 2006;66(17):8319-8326
93. Kats-Ugurlu G, Roodink I, de Weijert M, Tiemessen D, Maass C, Verrijp K, van der Laak J, de Waal R, Mulders P, Oosterwijk E, Leenders W. Circulating tumour tissue fragments in patients with pulmonary metastasis of clear cell renal cell carcinoma. J Pathol. 2009;219(3):287-93
94. Hou JM, Krebs M, Ward T, Sloane R, Priest L, Hughes A, Clack G, Ranson M, Blackhall F, Dive C. Circulating tumor cells as a window on metastasis biology in lung cancer. Am J Pathol. 2011;178(3):989-996
95. Vona G, Estepa L, Béroud C, Damotte D, Capron F, Nalpas B, Mineur A, Franco D, Lacour B, Pol S, Bréchot C, Paterlini-Bréchot P. Impact of cytomorphological detection of circulating tumor cells in patients with liver cancer. Hepatology. 2004;39(3):792-797
96. Molnar B, Ladanyi A, Tanko L, Sréter L, Tulassay Z. Circulating tumor cell clusters in the peripheral blood of colorectal cancer patients. Clin Cancer Res. 2001;7(12):4080-4085
97. Witzig TE, Bossy B, Kimlinger T, Roche PC, Ingle JN, Grant C, Donohue J, Suman VJ, Harrington D, Torre-Bueno J, Bauer KD. Detection of circulating cytokeratin-positive cells in the blood of breast cancer patients using immunomagnetic enrichment and digital microscopy. Clin Cancer Res. 2002;8(5):1085-91
98. Brandt B, Junker R, Griwatz C, Heidl S, Brinkmann O, Semjonow A, Assmann G, Zänker KS. Isolation of prostate-derived single cells and cell clusters from human peripheral blood. Cancer Res. 1996;56(20):4556-4561
99. Carr I. Experimental lymphatic metastasis. J Microsc. 1983;131(Pt 2):211-220
100. Madhavan M, Srinivas P, Abraham E, Ahmed I, Mathew A, Vijayalekshmi NR, Balaram P. Cadherins as predictive markers of nodal metastasis in breast cancer. Mod Pathol. 2001;14(5):423-427
101. Liotta LA, Saidel MG, Kleinerman J. The significance of hematogenous tumor cell clumps in the metastatic process. Cancer Res. 1976;36(3):889-94
102. Zhang X, Nie D, Chakrabarty S. Growth factors in tumor microenvironment. Front Biosci. 2010;15:151-165
103. Theodoropoulos PA, Polioudaki H, Agelaki S, Kallergi G, Saridaki Z, Mavroudis D, Georgoulias V. Circulating tumor cells with a putative stem cell phenotype in peripheral blood of patients with breast cancer. Cancer Lett. 2010;288(1):99-106
104. Hou JM, Krebs MG, Lancashire L, Sloane R, Backen A, Swain RK, Priest LJ, Greystoke A, Zhou C, Morris K, Ward T, Blackhall FH, Dive C. Clinical significance and molecular characteristics of circulating tumor cells and circulating tumor microemboli in patients with small-cell lung cancer. J Clin Oncol. 2012;30(5):525-532
105. Deng G, Herrler M, Burgess D, Manna E, Krag D, Burke JF. Enrichment with anti-cytokeratin alone or combined with anti-EpCAM antibodies significantly increases the sensitivity for circulating tumor cell detection in metastatic breast cancer patients. Breast Cancer Res. 2008;10(4):R69
106. Hori H, Fujimori T, Fujii S, Ichikawa K, Ohkura Y, Tomita S, Ono Y, Imura J, Kuroda Y. Evaluation of tumor cell dissociation as a predictive marker of lymph node metastasis in submucosal invasive colorectal carcinoma. Dis Colon Rectum. 2005;48(5):938-45
107. Fehm T, Sagalowsky A, Clifford E, Beitsch P, Saboorian H, Euhus D, Meng S, Morrison L, Tucker T, Lane N, Ghadimi BM, Heselmeyer-Haddad K, Ried T, Rao C, Uhr J. Cytogenetic evidence that circulating epithelial cells in patients with carcinoma are malignant. Clin Cancer Res. 2002;8(7):2073-2084
108. Fehm T, Solomayer EF, Meng S, Tucker T, Lane N, Wang J, Gebauer G. Methods for isolating circulating epithelial cells and criteria for their classification as carcinoma cells. Cytotherapy. 2005;7(2):171-185
109. Butler TP, Gullino PM. Quantitation of cell shedding into efferent blood of mammary adenocarcinoma. Cancer Res. 1975;35(3):512-516
110. Meng S, Tripathy D, Frenkel EP, Shete S, Naftalis EZ, Huth JF, Beitsch PD, Leitch M, Hoover S, Euhus D, Haley B, Morrison L, Fleming TP, Herlyn D, Terstappen LW, Fehm T, Tucker TF, Lane N, Wang J, Uhr JW. Circulating tumor cells in patients with breast cancer dormancy. Clin Cancer Res. 2004;10(24):8152-8162
111. Moreno JG, O'Hara SM, Gross S, Doyle G, Fritsche H, Gomella LG, Terstappen LW. Changes in circulating carcinoma cells in patients with metastatic prostate cancer correlate with disease status. Urology. 2001;58(3):386-392
112. Schwarzenbach H, Alix-Panabières C, Müller I, Letang N, Vendrell JP, Rebillard X, Pantel K. Cell-free tumor DNA in blood plasma as a marker for circulating tumor cells in prostate cancer. Clin Cancer Res. 2009;15(3):1032-1038
113. Leary RJ, Kinde I, Diehl F, Schmidt K, Clouser C, Duncan C, Antipova A, Lee C, McKernan K, De La Vega FM, Kinzler KW, Vogelstein B, Diaz LA Jr, Velculescu VE. Development of personalized tumor biomarkers using massively parallel sequencing. Sci Transl Med. 2010;2(20):20ra14
114. Kaiser J. Medicine. Keeping tabs on tumor DNA. Science. 2010;327(5969):1074
115. Fleischhacker M, Schmidt B. Circulating nucleic acids (CNAs) and cancer-a survey. Biochim Biophys Acta. 2007;1775(1):181-232
116. Jung K, Fleischhacker M, Rabien A. Cell-free DNA in the blood as a solid tumor biomarker--a critical appraisal of the literature. Clin Chim Acta. 2010;411(21-22):1611-24
117. Diehl F, Li M, Dressman D, He Y, Shen D, Szabo S, Diaz LA Jr, Goodman SN, David KA, Juhl H, Kinzler KW, Vogelstein B. Detection and quantification of mutations in the plasma of patients with colorectal tumors. Proc Natl Acad Sci USA. 2005;102(45):16368-16373
118. Ziegler A, Zangemeister-Wittke U, Stahel RA. Circulating DNA: a new diagnostic gold mine? Cancer Treat Rev. 2002;28(5):255-71
119. Schwarzenbach H, Hoon DS, Pantel K. Cell-free nucleic acids as biomarkers in cancer patients. Nat Rev Cancer. 2011;11(6):426-37
120. Blackburn EH. Telomere states and cell fates. Nature. 2000;408(6808):53-6
121. Fizazi K, Morat L, Chauveinc L, Prapotnich D, De Crevoisier R, Escudier B, Cathelineau X, Rozet F, Vallancien G, Sabatier L, Soria JC. High detection rate of circulating tumor cells in blood of patients with prostate cancer using telomerase activity. Ann Oncol. 2007;18(3):518-21
122. Sapi E, Okpokwasili NI, Rutherford T. Detection of telomerase-positive circulating epithelial cells in ovarian cancer patients. Cancer Detect Prev. 2002;26(2):158-67
123. Soria JC, Gauthier LR, Raymond E, Granotier C, Morat L, Armand JP, Boussin FD, Sabatier L. Molecular detection of telomerase-positive circulating epithelial cells in metastatic breast cancer patients. Clin Cancer Res. 1999;5(5):971-5
124. Gauthier LR, Granotier C, Soria JC, Faivre S, Boige V, Raymond E, Boussin FD. Detection of circulating carcinoma cells by telomerase activity. Br J Cancer. 2001;84(5):631-5
125. Soria JC, Morat L, Durdux C, Housset M, Cortez A, Blaise R, Sabatier L. The molecular detection of circulating tumor cells in bladder cancer using telomerase activity. J Urol. 2002;167(1):352-6
126. Maurelli R, Zambruno G, Guerra L, Abbruzzese C, Dimri G, Gellini M, Bondanza S, Dellambra E. Inactivation of p16INK4a (inhibitor of cyclin-dependent kinase 4A) immortalizes primary human keratinocytes by maintaining cells in the stem cell compartment. FASEB J. 2006;20(9):1516-8
127. Cerchia L, de Franciscis V. Targeting cancer cells with nucleic acid aptamers. Trends Biotechnol. 2010;28(10):517-525
128. Xu Y, Yang X, Wang E. Review: Aptamers in microfluidic chips. Anal Chim Acta. 2010;683(1):12-20
129. Tuerk C, Gold L. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science. 1990;249(4968):505-10
130. Vorhies JS, Nemunaitis JJ. Nucleic acid aptamers for targeting of shRNA-based cancer therapeutics. Biologics. 2007;1(4):367-376
131. Lupold SE, Hicke BJ, Lin Y, Coffey DS. Identification and characterization of nuclease-stabilized RNA molecules that bind human prostate cancer cells via the prostate-specific membrane antigen. Cancer Res. 2002;62(14):4029-4033
132. Phillips JA, Xu Y, Xia Z, Fan ZH, Tan W. Enrichment of cancer cells using aptamers immobilized on a microfluidic channel. Anal Chem. 2009;81(3):1033-1039
133. Xu Y, Phillips JA, Yan J, Li Q, Fan ZH, Tan W. Aptamer-based microfluidic device for enrichment, sorting, and detection of multiple cancer cells. Anal Chem. 2009;81(17):7436-7442
134. Cui CH, Zhao W, Guo D, Bose S, Philips J, Karnik R, Karp JM. Capture and detection of circulating tumour cells using three- dimensional, multivalent DNA aptamer network. Molecular Med TRI-CON 2011 (Poster Presentation), San Francisco, CA. 2011
135. Zhao W, Ali MM, Brook MA, Li Y. Rolling circle amplification: applications in nanotechnology and biodetection with functional nucleic acids. Angew Chem Int Ed Engl. 2008;47(34):6330-7
136. Boss DS, Olmos RV, Sinaasappel M, Beijnen JH, Schellens JH. Application of PET/CT in the development of novel anticancer drugs. Oncologist. 2008;13(1):25-38
137. Mohler JL, Armstrong AJ, Bahnson RR, Boston B, Busby JE, D'Amico AV, Eastham JA, Enke CA, Farrington T, Higano CS, Horwitz EM, Kantoff PW, Kawachi MH, Kuettel M, Lee RJ, Macvicar GR, Malcolm AW, Miller D, Plimack ER, Pow-Sang JM, Roach M 3rd, Rohren E, Rosenfeld S, Srinivas S, Strope SA, Tward J, Twardowski P, Walsh PC, Ho M, Shead DA. Prostate Cancer, Version 3.2012 Featured Updates to the NCCN Guidelines. J Natl Compr Canc Netw. 2012;10(9):1081-1087
138. Hayes DF, Smerage J. Is There a Role for Circulating Tumor Cells in the Management of Breast Cancer? Clin Cancer Res. 2008;14:3646
139. Saad A, Abraham J. Role of tumor markers and circulating tumors cells in the management of breast cancer. Oncology. 2008;22(7):726-31
140. Czerkinsky CC, Nilsson LA, Nygren H, Ouchterlony O, Tarkowski A. A solid-phase enzyme-linked immunospot (ELISPOT) assay for enumeration of specific antibody-secreting cells. J Immunol Methods. 1983;65(1-2):109-121
141. Alix-Panabières C, Brouillet JP, Fabbro M, Yssel H, Rousset T, Maudelonde T, Choquet-Kastylevsky G, Vendrell JP. Characterization and enumeration of cells secreting tumor markers in the peripheral blood of breast cancer patients. J Immunol Methods. 2005;299(1-2):177-188
142. Alix-Panabières C, Vendrell JP, Pellé O, Rebillard X, Riethdorf S, Müller V, Fabbro M, Pantel K. Detection and characterization of putative metastatic precursor cells in cancer patients. Clin Chem. 2007;53(3):537-539
143. Alix-Panabières C, Riethdorf S, Pantel K. Circulating tumor cells and bone marrow micrometastasis. Clin Cancer Res. 2008;14(16):5013-5021
144. Cristofanilli M, Hayes DF, Budd GT, Ellis MJ, Stopeck A, Reuben JM, Doyle GV, Matera J, Allard WJ, Miller MC, Fritsche HA, Hortobagyi GN, Terstappen LW. Circulating tumor cells: a novel prognostic factor for newly diagnosed metastatic breast cancer. J Clin Oncol. 2005;23(7):1420-1430
145. Nolé F, Munzone E, Zorzino L, Minchella I, Salvatici M, Botteri E, Medici M, Verri E, Adamoli L, Rotmensz N, Goldhirsch A, Sandri MT. Variation of circulating tumor cell levels during treatment of metastatic breast cancer: prognostic and therapeutic implications. Ann Oncol. 2008;19(5):891-897
146. Cristofanilli M, Budd GT, Ellis MJ, Stopeck A, Matera J, Miller MC, Reuben JM, Doyle GV, Allard WJ, Terstappen LW, Hayes DF. Circulating tumor cells, disease progression, and survival in metastatic breast cancer. N Engl J Med. 2004;351(8):781-791
147. Budd GT, Cristofanilli M, Ellis MJ, Stopeck A, Borden E, Miller MC, Matera J, Repollet M, Doyle GV, Terstappen LW, Hayes DF. Circulating tumor cells versus imaging-predicting overall survival in metastatic breast cancer. Clin Cancer Res. 2006;12(21):6403-6409
148. Hayes DF, Cristofanilli M, Budd GT, Ellis MJ, Stopeck A, Miller MC, Matera J, Allard WJ, Doyle GV, Terstappen LW. Circulating tumor cells at each follow-up time point during therapy of metastatic breast cancer patients predict progression-free and overall survival. Clin Cancer Res. 2006;12(14 Pt 1):4218-4224
149. Altman DG, Lausen B, Sauerbrei W, Schumacher M. Dangers of using optimal cutpoints in the evaluation of prognostic factors. J Natl Cancer Inst. 1994;86(11):829-835
150. Faraggi D, Simon R. A simulation study of cross-validation for selecting an optimal cutpoint in univariate survival analysis. Stat Med. 1996;15(20):2203-2213
151. Austin PC, Brunner LJ. Inflation of the type I error rate when a continuous confounding variable is categorized in logistic regression analyses. Stat Med. 2004;23(7):1159-1178
152. Royston P, Altman DG, Sauerbrei W. Dichotomizing continuous predictors in multiple regression: a bad idea. Stat Med. 2006;25(1):127-141
153. Botteri E, Sandri MT, Bagnardi V, Munzone E, Zorzino L, Rotmensz N, Casadio C, Cassatella MC, Esposito A, Curigliano G, Salvatici M, Verri E, Adamoli L, Goldhirsch A, Nolè F. Modeling the relationship between circulating tumour cells number and prognosis of metastatic breast cancer. Breast Cancer Res Treat. 2010;122(1):211-217
154. CellSearch. Circulating Tumor Cell Epithelial Kit (IVD), CellSearch® Instructions for Use (IFU), No. e631500023. CellSearch. 2012
155. Lustberg MB, Balasubramanian P, Miller B, Garcia Villa A, Carothers S, Michael B, Mrozek E, Ramaswamy B, Layman RM, Wesolowski R, Shapiro CL, Chalmers JJ. EpCAM-negative cancer-associated circulating cells in blood samples of women with triple-negative breast cancer. J Clin Oncol. 2011;29(Suppl):11559
156. Pecot CV, Bischoff FZ, Lin YG, Jaladurgam P, Merritt WM, Pircher TJ, Mikolajczyk S, Mayer JA, Wong K, Pham T, Bottsford-Miller JN, Stone RL, Celestino J, Nick AM, Eng C, Sood A. Clinical relevance of cytokeratin-negative circulating tumor cells. J Clin Oncol. 2011;29(Suppl):21101
157. Pecot CV, Bischoff FZ, Mayer JA, Wong KL, Pham T, Bottsford-Miller J, Stone RL, Lin YG, Jaladurgam P, Roh JW, Goodman BW, Merritt WM, Pircher TJ, Mikolajczyk SD, Nick AM, Celestino J, Eng C, Ellis LM, Deavers MT, Sood AK. A novel platform for detection of CK+ and CK- CTCs. Cancer Discov. 2011;1(7):580-6
158. Eifler RL, Lind J, Falkenhagen D, Weber V, Fischer MB, Zeillinger R. Enrichment of circulating tumor cells from a large blood volume using leukapheresis and elutriation: proof of concept. Cytometry B Clin Cytom. 2011;80(2):100-111
Author contact
![]() Corresponding author: Bin Hong, Ph.D. TeloVISION, LLC, 1281 Win Hentschel Blvd. West Lafayette, IN 47906. (502) 445-6362. infocom or Youli Zu, MD., PhD. Department of Pathology and Genomic Medicine, The Methodist Hospital, 6565 Fannin, MS205, Houston, TX 77030. Tel 713-441-4460 Fax 713-441-1565. yzuorg.
Corresponding author: Bin Hong, Ph.D. TeloVISION, LLC, 1281 Win Hentschel Blvd. West Lafayette, IN 47906. (502) 445-6362. infocom or Youli Zu, MD., PhD. Department of Pathology and Genomic Medicine, The Methodist Hospital, 6565 Fannin, MS205, Houston, TX 77030. Tel 713-441-4460 Fax 713-441-1565. yzuorg.