Impact Factor
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and Methods
RESULTS
Discussion
Conclusion
Supplementary Material
Abbreviations
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2014; 4(6):565-578. doi:10.7150/thno.7581 This issue Cite
Research Paper
Anti-Angiogenesis Therapy in the Vx2 Rabbit Cancer Model with a Lipase-cleavable Sn 2 Taxane Phospholipid Prodrug using αvβ3-Targeted Theranostic Nanoparticles
Dipanjan Pan1*, Anne H. Schmieder1*, Kezheng Wang1,2, Xiaoxia Yang1, Angana Senpan1, Grace Cui1, Kendall Killgore3, Benjamin Kim1, John S. Allen1, Huiying Zhang1, Shelton D. Caruthers1, Baozhong Shen2, Samuel A. Wickline1, Gregory M. Lanza1 ![]()
1. Department of Medicine, Washington University School of Medicine, St Louis, MO 63108, USA.
2. Radiology Department and Molecular Imaging Center, 4th Affiliated Hospital, Harbin Medical University, Harbin, Heilongjiang 150001, China.
3. Kereos, Inc., Center for Emerging Technology, St. Louis, MO 63108, USA.
* Equal first author co-contributors.
Received 2013-9-3; Accepted 2014-1-27; Published 2014-3-11
Abstract
In nanomedicine, the hydrophobic nature of paclitaxel has favored its incorporation into many nanoparticle formulations for anti-cancer chemotherapy. At lower doses taxanes are reported to elicit anti-angiogenic responses. In the present study, the facile synthesis, development and characterization of a new lipase-labile docetaxel prodrug is reported and shown to be an effective anti-angiogenic agent in vitro and in vivo. The Sn 2 phosphatidylcholine prodrug was stably incorporated into the lipid membrane of αvβ3-integrin targeted perfluorocarbon (PFC) nanoparticles (αvβ3-Dxtl-PD NP) and did not appreciably release during dissolution against PBS buffer or plasma over three days. Overnight exposure of αvβ3-Dxtl-PD NP to plasma spiked with phospholipase enzyme failed to liberate the taxane from the membrane until the nanoparticle integrity was compromised with alcohol. The bioactivity and efficacy of αvβ3-Dxtl-PD NP in endothelial cell culture was as effective as Taxol® or free docetaxel in methanol at equimolar doses over 96 hours. The anti-angiogenesis effectiveness of αvβ3-Dxtl-PD NP was demonstrated in the Vx2 rabbit model using MR imaging of angiogenesis with the same αvβ3-PFC nanoparticle platform. Nontargeted Dxtl-PD NP had a similar MR anti-angiogenesis response as the integrin-targeted agent, but microscopically measured decreases in tumor cell proliferation and increased apoptosis were detected only for the targeted drug. Equivalent dosages of Abraxane® given over the same treatment schedule had no effect on angiogenesis when compared to control rabbits receiving saline only. These data demonstrate that αvβ3-Dxtl-PD NP can reduce MR detectable angiogenesis and slow tumor progression in the Vx2 model, whereas equivalent systemic treatment with free taxane had no benefit.
Keywords: lipase-labile docetaxel prodrug, perfluorocarbon, nanoparticles
Introduction
Paclitaxel is a cytoskeletal drug that interacts with tubulin to corrupt mitotic spindle assembly, chromosome segregation, and cell division by inhibiting microtubule disassembly. As a consequence, chromosomes are unable to achieve proper metaphase configuration and mitotic progression is blocked. The inability to complete cell division promotes apoptosis or reversion to a quiescent G0-phase of the cell cycle. The rarity of paclitaxel, initially isolated from the Pacific yew, led to synthetic chemical pathway development from which emerged docetaxel, a close chemical analogue of the index compound with similar biological properties. Taxanes have found clinical applications as a cytotoxic cancer chemotherapeutic [1] as well as a stent-delivered inhibitor of restenosis following angioplasty. [2] In nanomedicine, the hydrophobic nature of paclitaxel has favored its incorporation into many nanoparticle formulations, including into the phospholipid outer membrane of perfluorocarbon nanoparticles. [3]
Perfluorocarbon (PFC) nanoparticles have been targeted to a myriad of epitopes for diagnostic imaging and site-specific drug delivery for applications in atherosclerosis, restenosis, cancer and rheumatoid arthritis. [4-7] Early comparisons of hydrophobic drugs, including doxorubicin, paclitaxel, and fumagillin, revealed that fumagillin, a potent anti-angiogenic mycotoxin, was retained best in dissolution studies and was effective in vivo. [8, 9] Unfortunately, fumagillin is not readily available in large quantities, and it is photochemically unstable. [10-12] Taxanes are reported to elicit potent anti-angiogenic effects, particularly at low doses, and the compounds are readily available and chemically stable. [13]
Preliminary studies in the Vx2 model showed that paclitaxel incorporated into the outer lipid membrane of PFC nanoparticles was not effective for anti-angiogenesis whereas fumagillin was. [14] (Additional file 1: Supplemental Figure 1) In vivo pharmacokinetic studies revealed that both fumagillin and paclitaxel included in the PFC nanoparticle lipid membrane were significantly lost in circulation en route to the endothelial target cells, with the premature release of the taxane occurring quicker and to a greater extent than the more hydrophobic fumagillin.
We have reported effective in vivo delivery of hydrophobic drugs entrapped in the phospholipid surfactant through a mechanism, we described as “contact facilitated drug delivery” (CFDD). [9] Tethering of the nanoparticle to the target cell surface facilitated the interaction and hemifusion of the two lipid membranes, which affords the passive transfer of the drug and phospholipids from the nanoparticle surface to the outer leaflet of the target cell membrane. The drug is then translocated to the inner leaflet through an ATP dependent mechanism. [15, 16] CFDD eliminates the need for particle internalization with subsequent endosomal drug payload escape or extracellular particle release with diffusion into the cell. We hypothesized that a phospholipid prodrug approach that couples the active pharmaceutical ingredient (API) through the Sn 2 acyl position (i.e., stereospecific hydroxyl group of the second carbon of glycerol) would provide a stable membrane complex in the nanoparticle during circulatory transit to the target site. Subsequent transfer of the monolayer components into the target cell membrane would allow cell surface or cytosolic phospholipases to enzymatically cleave the Sn 2 ester and release the drug, allowing it to diffuse into the cytosol for effect. (Figure 1)
Schematic representation of the concept of the lipase-cleavable Sn 2 phosphatidylcholine docetaxel prodrug: The lipophilic docetaxel prodrug is incorporated into the phospholipid surfactant, where the drug enters the protective hydrophobic acyl layer. The active pharmaceutical ingredient (API) is protected from premature dissolution into serum or serum lipase liberation during circulation to the target. The API is delivered to the target cell through a “contact facilitated drug delivery” (CFDD) mechanism wherein the prodrug exchanges passively into the outer target cell leaflet. The prodrug transfers through an ATP dependent mechanism into the inner cell membrane leaflet, which is contiguous with the internal cell membranes (except the mitochondria). The prodrug undergoes a region-selective enzymatic cleavage at the Sn 2 site by phospholipases, such as phospholipase A-2 (PLA-2), but up to 60 endothelial lipases could functionally participate in drug liberation into the cytosol.
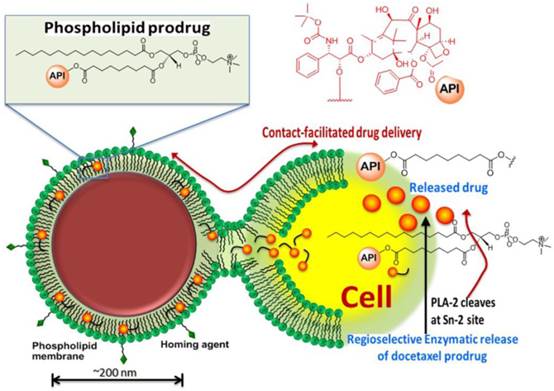
The objectives of the present work were: 1) to develop and characterize an Sn 2 lipase-labile prodrug of docetaxel (Dxtl-PD); 2) to demonstrate the stability of the taxane prodrug in the PFC nanoparticles in the presence of serum and excess lipase, 3) to demonstrate the anti-neovascular efficacy of the agent in the rabbit Vx2 cancer model using magnetic resonance (MR) angiogenesis molecular imaging, 4) to demonstrate the efficacy advantage of the integrin-targeted Dxtl-PD nanoparticle compared with the molar equivalent dose of Abraxane® given systemically, 5) to microscopically characterize the impact of targeted Dxtl-PD on intratumoral apoptosis and cell proliferation.
Materials and Methods
Sn 2 lipase-labile taxane phosphatidylcholine prodrug synthesis
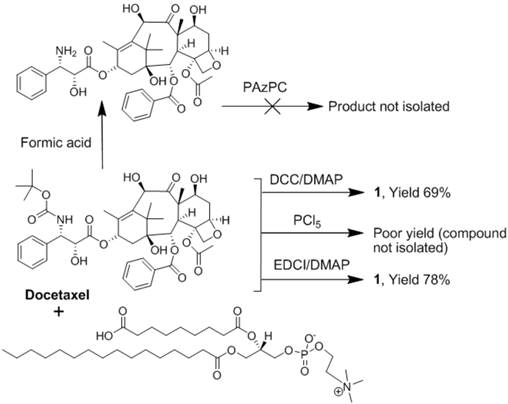
Docetaxel (AvaChem Scientific), a clinically relevant broad-spectrum anticancer agent, was utilized in the present study for site-specific anti-angiogenesis therapy. Docetaxel has a low solubility in water (<0.1 μg/ml) and from the synthetic stand point, chemistries involving esterification of the 2'-hydroxyl group, which is the most reactive of the hydroxy groups, is feasible. Any conventional method of protecting a hydroxy group, such as esterification, can be utilized to protect this hydroxy group at the 2' position, while leaving the hydroxy groups at the 7 and 10 positions free for reaction. For the synthesis of docetaxel prodrug, several coupling methods were attempted that included dicyclohexyl carbodiimide (DCC) / dimethylaminopyridine (DMAP), phosphorous pentachloride (PCl5), and 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide/dimethylaminopyridine (EDCI/DMAP). A summary of those results are consolidated into Figure 2. Among these reagents, EDCI/DMAP was the most efficient approach and produced the desired compound in 78% yield. (Figure 3) The compound was isolated and purified by preparative thin layer chromatography (prep-TLC) and the structure was confirmed by NMR and ESI MS spectroscopy. Paclitaxel prodrug (Kereos, Inc., St. Louis, MO) was prepared analogously as illustrated and described in the Additional file 1: Supplemental Figure 2.
Summary of the attempted synthetic approaches to docetaxel phosphatidylcholine prodrug.
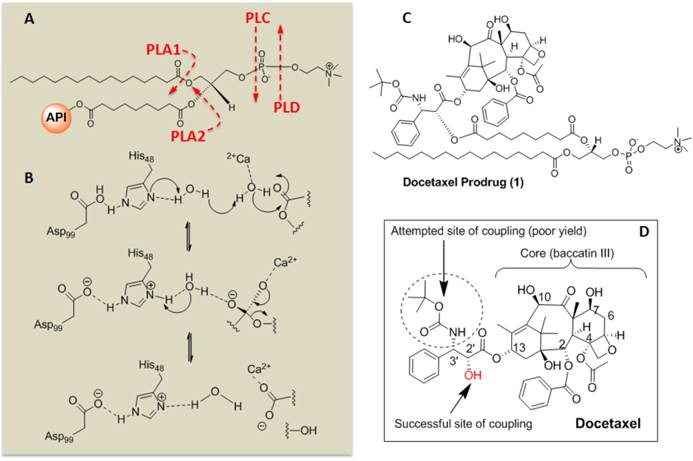
Sn 2 Docetaxel Prodrug. (A) Cleavage sites of phospholipase enzymes, PLA2 regiospecifically recognizes the Sn 2 acyl bond of phospholipids; (B) predicted mechanistic pathways of hydrolysis catalyzed by PLA2; (C) structure of the docetaxel prodrug synthesized in this work; (D) structure of parent molecule docetaxel and depicting different sites for functionalization and activity. PLA2 = phospholipase A2, PLA1=phospholipase A1.
Of note, specific 2'-modification of docetaxel was drawn from analogy based on the scientific literature when 1H-NMR spectroscopic studies to confirm the conjugation at the 2' position failed to resolve the overlapping resonating peaks from the neighboring protons of the tetracyclic 17-carbon (heptadecane) skeleton. [17] Further, the initial approach to synthesize Dxtl-PD involved coupling chemistries through deprotected 3'-NH2 group. (Figure 2) Docetaxel was subjected to hydrolytic deprotection in presence of formic acid to produce 3'-amine deprotected docetaxel. However, the attempted conjugation of 1-palmitoyl-2-azelaoyl-sn-glycero-3-phosphocholine (PAz-PC) through the free amine group resulted in a complex mixture of products and the desired compound was never isolated.
ανβ3-integrin antagonist homing ligand
The ανβ3-integrin antagonist was a quinalone nonpeptide developed by Bristol-Myers Squibb Medical Imaging (US patent 6,511,648 and related patents) and initially reported and characterized as the 111In-DOTA conjugate RP478 and cyan 5.5 homologue TA145 [18]. PFC nanoparticles present ~300 ligands/particle with an IC50 of 50 pM for the Mn2+-activated αvβ3-integrin (unpublished data from Kereos, Inc., St. Louis, MO, USA). Targeting of the lipid-nanoparticles with this peptidomimetic was shown to be specific for neovascular endothelial cells expressing the αvβ3-integrin, PECAM, but not Tie-2 (a maturity biomarker of stabilized endothelium). [19]
Perfluorocarbon nanoparticle synthesis incorporating docetaxel-prodrug
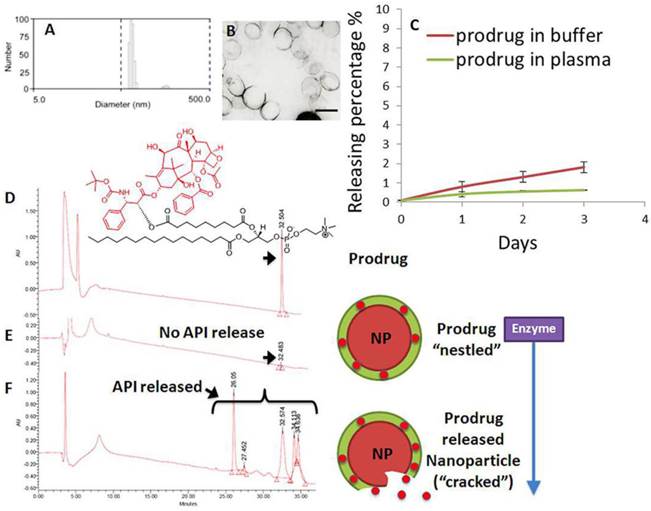
Phospholipid-encapsulated perfluorocarbon (PFC) nanoparticles (NP) were prepared as a microfluidized suspension of 20% (v/v) perfluoroctylbromide (Exfluor Inc., Round Rock, TX), 2.0% (w/v) of a surfactant co-mixture, and 1.7% (w/v) glycerin in pH 6.5 carbonate buffer. The surfactant co-mixture of nanoparticles included: ~ 97.6 mole% lecithin, 0.15 mole% of αvβ3-ligand conjugated lipid and 2 mole% (3.75 wt%) of the taxane prodrug. An αvβ3-integrin antagonist coupled to phosphatidylethanolamine-PEG2000 was used for homing angiogenesis. For MR imaging, the surfactant excluded the taxane prodrug but included 20 mole% gadolinium-DOTA-amide-caproyl-phosphoethanolamine at the expense of equimolar substitution of phosphatidylcholine in the surfactant commixture (i.e., αvβ3-Gd-DOTA NP). Nontargeted (NT) nanoparticles excluded the homing ligands. The surfactant components for each formulation were combined with the perfluorocarbon, buffer, and glycerin with pH adjusted to 6.5, and the mixtures were homogenized at 20,000 psi for 4 minutes. The nanoparticles were preserved under inert gas in sterile sealed vials until use. Upon completion of the synthesis, nanoparticles were thoroughly characterized by physical-chemical techniques e.g. dynamic light scattering (DLS), electrophoretic potential (zeta) and transmission electron microscopy (TEM). A typical preparation of αvβ3-Dxtl-PD NP resulted in: particle size, 214±20 nm; zeta potential, (ζ) -23±12 mv; and polydispersity index, 0.15±0.5. (Figure 4a,b)
Characterization of docetaxel prodrug (Dxtl-PD) loaded PFC nanoparticles (NP). (A) Number-averaged hydrodynamic diameter distribution of αvβ3-Dxtl-PD in hydrated state; (B) transmission electron microscopy (TEM) image of the integrin targeted nanoparticles (scale bar: 200 nm); (C) dissolution of Dxtl-PD over 3 days when incubated in PBS with albumin (red) or human plasma (green); HPLC traces of Dxtl-PD (D), Dxtl-PD-NP treated with PLA2 (E) and Dxtl-PD-NP “cracked” with isopropanol and exposed to PLA2. PLA2=phospholipase A2.
Comparison of docetaxel, docetaxel-prodrug, paclitaxel, and paclitaxel-prodrug on endothelial cell proliferation in vitro
Mouse vascular endothelial cells (2F2B) were plated at 2500 cells/well in media with Angiotensin II (1nM) for overnight culture at 37°C, 5% CO2. Cells were treated with paclitaxel, docetaxel, paclitaxel prodrug, or docetaxel prodrug at 0.5 µg/ml, 1 µg/ml, 5 µg/ml, 10 µg/ml, 50 µg/ml and 100 µg/ml for 1 hour with mild agitation. The treatments were removed; cells were washed three times with PBS; fresh media was added; and the plates were returned back to the incubator. Cell proliferation was assayed at 24, 48, and 72 hours. At each time point, 20 µl of Alamar Blue was added to each well and incubated for 4 hours. The supernatants were transferred to a new replicate plate for spectroscopic analysis. The original wells were replenished with fresh media and all plates were returned to the incubator until the next time point. The reaction was monitored using a fluorescence plate reader (λex 570nm, λem 585nm) where the resulting signal is proportional to the number of viable cells present. Signal intensity from each sample was normalized to signal from the positive saline control. Each treatment was replicated three times.
In vitro dissolution stability of docetaxel-prodrug in PFC nanoparticles
The stability of the Dxtl-PD incorporated at 2 mole% (3.75 wt%) into the surfactant (360mg) of PFC nanoparticles was evaluated by dissolution in plasma and buffer over three days in triplicate. Drug-laden nanoparticles were secured within dialysis tubing (MWCO=60K, Spectrum Laboratories, Inc.), which were washed externally to remove possible surface contamination, and then incubated in 3.5 ml of human plasma at 37°C for 3 days with constant slow rotation. The dialysis tube samples were transferred to 3.5 ml of fresh plasma daily and 200µl of the 3.5 ml dialysate was injected into a reversed-phase HPLC system (Waters 600S controller, 626 delivery pump, 717 auto sampler, and 2487 Dual λ absorbance detector) through a 0.2 µm syringe filter for analysis of prodrug release. Dxtl-PD was detected at a wavelength 227 nm. A Grace Vydac C18 column, 4.6 x 250 mm, was used with a mobile phase consisting of: (A) 80% acetonitrile, 20% methanol and (B) 100% 0.2 M ammonium acetate, pH 4.5 using gradient conditions (from 100% A to 40% A 60% B within 30 min then returning to 100% A in 5 min for an additional 5 min (run time 40 min; drug retention time ~33 min). Flow rate was 1 ml/min.
To further assess the potential susceptibility of Dxtl-PD in PFC nanoparticles to premature lipase liberation, drug-laden nanoparticles (250 µl) were incubated for 24 hours with 3.5 ml fresh plasma or fresh whole blood preserved with EDTA each spiked with 250 µl 0.9 % NaCl, 15-30 mM CaCl2 and 0.278 mg phospholipase A2. This was referenced to an equimolar amount of Dxtl or Dxtl-PD in DMSO diluted in 3.5ml of PBS pH 7.4. The samples were continuously agitated on a nutator to prevent settling. As a positive control, 250 µl 2-propanol was added to the lipase-spiked plasma containing Dxtl-PD in PFC nanoparticles to “crack” the emulsion's integrity, exposing the lipase-labile prodrug to the enzyme-rich plasma milieu. The plasma-enzyme-NP mixture (250 µl) was combined with 250 µl Cleanascite™ for lipid removal and clarification (Biotech Support Group, Inc., Monmouth Junction, NJ) at 25°C for 15min. The mixture was centrifuged at 5000 rpm (30 min) and the supernatant (200 µl) was analyzed by HPLC.
To determine the potential exchange of Dxtl-PD from the nanoparticle surfactant to erythrocyte membranes, 2-propranolol (200 µl) and water (200 µl) were added to the erythrocyte aliquot (100 µl) and the mixture was centrifuged at 1000 rpm for 10 min. The RBC fraction was separated from the pelleting PFC nanoparticles (specific gravity of 2). The RBC fraction was analyzed in triplicate for the Dxtl-PD and its major metabolites by HPLC.
Docetaxel-prodrug testing in vitro: cytotoxicity and metabolic assays
Mouse endothelial cells (2F2B; ATCC® CRL2168™, Manassas, VA) were seeded on a 96 well plate (2000-5000 cells/well) in Dulbecco's Modified Eagle's Medium (DMEM) with 4.5 g/L glucose; L-glutamine; and 10% heat-inactivated fetal bovine serum plus 10 µM angiotensin II (Sigma Chemical Co, St. Louis, MO) and incubated at 37ºC with 5% CO2. After 24 hours, cells were incubated for 1 hour with 1) αvβ3-Dxtl-PD NPs, 2) equimolar equivalent of Taxol® (0.3 µM), 3) αvβ3-No drug NPs, or 4) saline. After incubation the wells were washed three times with PBS and returned to the incubator. At 96 hours after drug exposure, cell proliferation was measured using the MTT assay for cytotoxicity, which measures the activity of cellular enzymes that reduce the tetrazolium dye, MTT, to the insoluble formazan (Invitrogen; Carlsbad, CA, USA). Each treatment was replicated 6 times.
In a confirmatory experiment using human endothelial cells (HUVEC; Lonza Walkersville, MD, USA), cell metabolic activity was assessed using a Vybrant Cell Metabolic Assay Kit (Invitrogen). The experiment was conducted as described above with 6 replicates/treatment and measurements assessed at 48, 72, and 96 hours. The treatments applied were: αvβ3-Dxtl-PD NPs, free Dxtl in methanol (0.3 µM) and αvβ3-No drug NPs. This assay is based on the reduction of non-fluorescent C12-resazurin to red-fluorescent C12-resorufin in metabolically active cells, which is attributed to different oxido-reductase enzyme systems that use NAD(P)H as the primary electron donor. The redox reaction was monitored using a fluorescence plate reader (λex 570nm, λem 587nm) where the resulting signal is proportional to the number of viable cells present. Signal intensity from each sample was normalized to signal from the positive control (αvβ3-targeted no drug nanoparticles).
Docetaxel-prodrug evaluation in rabbit Vx2 tumor angiogenesis model
All animal studies were conducted in accordance with a protocol approved by the Washington University Animal Studies Committee.
Surgical preparation
The anti-angiogenesis effects of Dxtl-PD were evaluated in the Vx2 rabbit tumor model using MR angiogenesis imaging and histology. Male New Zealand White rabbits (~2 kg) were anesthetized with intramuscular ketamine and xylazine. One hind leg was shaved, prepped under sterile conditions, and infiltrated with Marcaine. A 2-3 mm3 rabbit Vx2 tumor fragment (squamous cell carcinoma, National Cancer Institute Tumor Repository, Frederick, MD, USA) was implanted at a depth of approximately 0.5 cm through a small incision into the popliteal fossa. Anatomical planes were closed and secured with a single absorbable suture. The skin was sealed with Dermabond skin glue. Animals were recovered by reversing the effect of ketamine and xylazine with yohimbine.
Experimental Design
On days 9, 12 and 15 post tumor implantation, rabbits were treated with either: αvβ3-Dxtl-PD NP, non-targeted Dxtl-PD NPs, or αvβ3-targeted no drug NPs at a 2ml/kg i.v. dose. On day 17, rabbits were imaged in a clinical 3T MR scanner (Philips Achieva) with an 8-element SENSE coil using a high-resolution, T1-weighted, fat suppressed, 3D gradient echo sequence (TR/TE = 42/5.7ms, 45o flip angle, 2 NSA, 0.25 x 0.25 x 0.5 mm resolution). Images were obtained before and 3 hours after i.v. administration of αvβ3-Gd-DOTA NP (1ml/kg) to compare the influence of the three treatments on angiogenesis. At each time point, rabbits were anesthetized with 1% to 2% Isoflurane in oxygen to effect. In a second cohort of rabbits (n=4/group), the anti-angiogenesis effect of serially administered Abraxane® at the same drug dose and days of treatment described above were compared to an analogously treated saline control. MRI molecular imaging of tumor volumes and angiogenesis as well as image post processing were performed as described.
Image analysis
In each experiment, the signal intensities of the MR images were analyzed with MATLAB software (The Math Works, Natick, MA, USA). Dynamic T1-weighted signal intensities were normalized to a Gd-DTPA doped water reference standard included within each imaging field-of-view. For each animal, a region-of-interest (ROI) was manually placed around the tumor edge on each baseline slice, and the standard deviation of the average tumor signal at baseline was calculated. Voxels on post-injection MRI images were defined as enhanced if the signal intensity exceeded the mean tumor signal at baseline by more than three standard deviations. The tumor ROI was automatically partitioned into a rim and core region using an automated erosion method developed in MATLAB. The core region was set to approximately half of the ROI volume. The volume of enhancement was calculated, as well as the relative distribution of enhancing voxels in the periphery versus the core of the tumor. High-resolution 3D tumor reconstructions were created in MATLAB to map the spatial distribution of enhancing neovasculature in each treatment group. The overall 3D structure of the tumor was displayed as a mesh surface plot using isosurface rendering and a smoothing filter. A surface plot of the enhancing voxels was reconstructed similarly, and overlaid onto the tumor volume.
Histology
Immediately after MR imaging, animals were euthanized, and tumors were resected, weighed, and frozen in optimum cutting temperature (OCT) compound. Tumors (n=4/treatment group) were sectioned and stained for cell proliferation (proliferating cell nuclear antigen, PCNA) antibody; Abcam, Cambridge, MA, USA) and apoptosis (TUNEL assay; Promega, Madison, WI, USA) to corroborate MR results. Microscopic images (40x) were obtained with an Olympus BX61 microscope, Color-view CCD Camera and analyzed with ImageJ software using manual placement of regions of interest on each microscopic image (National Institute of Mental Health, Bethesda, Maryland, USA.).
Statistical Analysis
Data were analyzed using analysis of variance (SAS Inc.; Cary, NC, USA) (p < 0.05). Data are presented as the mean ± standard error of the mean unless otherwise stated.
RESULTS
Stability of docetaxel-prodrug in perfluorocarbon nanoparticles during dissolution
The retention of the Dxtl-PD in the PFC nanoparticle during in vitro dissolution over 3 days in PBS with albumin or human plasma was excellent with < 6% of prodrug released, indicating that the drug was sufficiently membrane stable. (Figure 4c) Phospholipases A2 (PLA2s) EC 3.1.1.4, commonly found in mammalian tissues, specifically recognizes the Sn 2 acyl bond of phospholipids and regioselectively hydrolyzes the bond, releasing free fatty acid and lysophospholipids. The catalytic mechanism of PLA2 is presumably initiated by a His-48/Asp-99/calcium complex within the active site in the presence of calcium or other small cations like cobalt and nickel. A simplistic mechanism of catalytic action of PLA2 on Dxtl-PD has been proposed in Figure 3. Figure 4d illustrates the HPLC reference retention time of Dxtl-PD (~32min). In the presence of excess lipase, Dxtl-PD nanoparticles had nearly negligible release of the drug (Figure 4e). Incubation of the Dxtl-PD nanoparticles in whole blood under slow continuous agitation revealed that the prodrug was not passively transferred to erythrocytes by transient contact (Additional file 1: Supplemental Figure 3). Supplemental Figure 4 shows the relative retention times of free docetaxel, Dxtl-PD, and the metabolism of Dxtl-PD in methanol added to phospholipase enriched plasma. As expected, docetaxel retention was somewhat less than the HPLC retention of Dxtl-PD. When Dxtl-PD was added to lipase rich plasma, docetaxel was liberated and differentiated from the parent prodrug. Regioselective hydrolysis of the prodrug by lipase in vitro required the addition of 2-propranol to “crack” the nanoparticle and expose the membrane components to the enzyme. The appearance of multiple peaks around 30 minutes showed that the Dxtl-PD was released and hydrolyzed from the prodrug backbone. (Figure 4f) While the 32.5 minutes corresponded to intact Dxtl-PD, the peak at 26.5 minutes in the presence of enzyme was confirmed by MS to be a hydrolyzed product, baccatin III [m/z 524 (M+Na)], (Figure 3). The cleavage of the side chain at C-13 of the taxane ring is known in the literature and also results in the formation of baccatin III in rats and is formed as a minor species in humans. [20, 21]
Collectively, these results suggest that Dxtl-PD is snuggled into the hydrophobic membrane and protected from hydrolysis and lipase activation in plasma until the nanoparticle is destabilized, such as with alcohol. Over the relatively brief time course of systemic targeting, αvβ3-Dxtl-PD NP retention of drug is anticipated to be very high, as opposed to earlier results with free paclitaxel dissolved into the surfactant, with negligible passive transference to co-circulating erythrocytes.
Bioequivalence of docetaxel, paclitaxel, docetaxel prodrug, and paclitaxel-prodrug
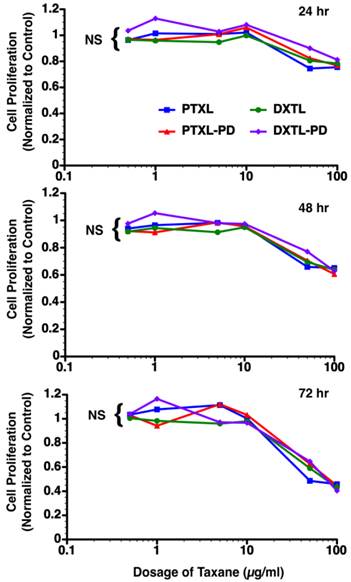
The bioequivalence of paclitaxel, docetaxel, paclitaxel prodrug, or docetaxel prodrug at 0.5 µg/ml, 1 µg/ml, 5 µg/ml, 10 µg/ml, 50 µg/ml and 100 µg/ml was studied in angiotensin II stimulated mouse vascular endothelial cells (2F2B). Monitoring of the proliferative response of these endothelial cells to the varying doses of drugs at 24, 48 and 72 hours post exposure reveal no significant difference in anti-endothelial cell proliferation. (Figure 5)
Mouse vascular endothelial cells (2F2B) stimulated with angiotensin II (1nM) were treated with paclitaxel, docetaxel, paclitaxel prodrug, or docetaxel prodrug at 0.5 µg/ml, 1 µg/ml, 5 µg/ml, 10 µg/ml, 50 µg/ml and 100 µg/ml for 1 hour. Cultures were monitored for proliferation at 24, 48, and 72 hours. No difference (p= NS) in biopotency of the different taxane forms was detected.
In vitro cytotoxicity and metabolic assays
The cytotoxicity of Dxtl-PD incorporated into PFC NPs was compared to Taxol® in vitro using 2F2B endothelial cell cultures stimulated with angiotensin II to up-regulate activated αvβ3-integrin expression. αvβ3-Dxtl-PD NPs decreased (p<0.05) cell proliferation 34% at 96 hours compared to the saline control, showing that the drug retained bioactivity through the prodrug synthesis, and that the compound was effectively transferred from the particle, into the cell membranes from where it was liberated in adequate concentrations to induce cell cytotoxicity. (Figure 6a) The αvβ3-no-drug NPs had no effect on cell proliferation. At the equivalent low dose of taxane, the Taxol® treatment decreased cell proliferation less, 8%, which was more similar to the controls but not significantly less than the αvβ3-Dxtl-PD NP. The slightly greater effectiveness of the Dxtl-PD is related to the number of drug laden nanoparticles bound to the endothelial cells through the integrin receptor within the brief incubation window versus the amount of free paclitaxel able to permeate the cell. (Figure 6a)
A) Cell proliferation via MTT assay comparing docetaxel prodrug targeted to angiotensin II activated integrin receptor in 2F2B endothelial cells versus Taxol® and no drug targeted particles. At 96 hours, the prodrug was significantly (p < 0.05) more effective than the control and at least equivalent to Taxol®. B) Cell proliferation via Vybrant Cell Metabolic Assay comparing docetaxel prodrug in HUVEC endothelial cells versus docetaxel in methanol and no drug targeted particles. Marked and equivalent reductions in cell proliferation were appreciated at 48, 72, and 96 hours with free drug and the integrin targeted Dxtl-PD NPs. * p<0.05.
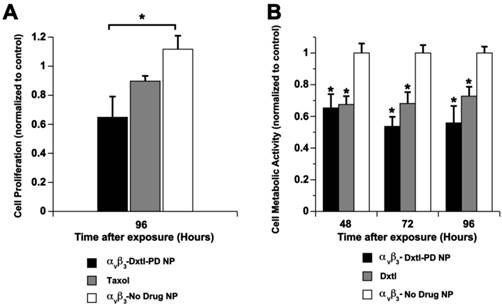
In a follow up experiment using angiotensin II stimulated HUVEC cells, cell metabolic activity responses to nanoparticle delivered docetaxel were assessed at earlier time points: 48, 72, and 96 hours. (Figure 6b) In this experiment native docetaxel in methanol added to the medium was compared to its integrin targeted lipase-labile prodrug analog on an equimolar drug basis. The treatments applied were: αvβ3-Dxtl-PD NPs, free Dxtl, and αvβ3-no drug NPs. As shown in Figure 6b, the nanoparticle delivered docetaxel prodrug or parent compound equivalently decreased (p<0.05) cell metabolic activity at 48 hours when compared with the targeted no-drug nanoparticle control. The marked reductions in cell metabolic activity persisted for both drug forms at 72 and 96 hours. These results substantiate the comparable bioequivalence of the native docetaxel and targeted Sn 2 lipase-labile prodrug in angiotensin II activated endothelial culture. These data further suggest that the intracellular enzymatic hydrolysis requirement to achieve efficacy of Dxtl-PD was not limiting in the activated HUVEC cells.
Docetaxel prodrug evaluation in rabbit Vx2 tumor angiogenesis model
In a previously unpublished pilot study, the effectiveness of paclitaxel incorporated into the membrane of αvβ3-targeted PFC nanoparticles was evaluated in the Vx2 tumor model (Additional file 1: Supplemental Figure 1). The results showed that serial doses of the agent produced neither anti-neovascular nor anti-tumor benefit when evaluated by MR molecular imaging with αvβ3-paramagnetic PFC nanoparticles. Studies simultaneously tracking the perfluorocarbon core, paclitaxel, and homing ligand in whole blood from rats showed that the particles were intact but the drug was rapidly and prematurely loss from the lipid membrane (Kereos, St. Louis, unpublished data).
In the present study, the efficacy of the docetaxel Sn 2 prodrug incorporated into the lipid membrane of αvβ3-targeted PFC nanoparticles (αvβ3-Dxtl-PD NP) was compared in the Vx2 rabbit tumor model to non-targeted Dxtl-PD NPs (NT-Dxtl-PD NP) and αvβ3-No Drug NPs. Animals were serially treated on days 9, 12, and 15 days post implant. On day 17, rabbits were imaged in a clinical 3T MR scanner (Philips Achieva) using a high-resolution, T1-weighted, fat suppressed, 3D gradient echo sequence at baseline and 3 hours after i.v. administration of αvβ3-Gd-DOTA nanoparticles (1ml/kg), previously characterized and demonstrated in vivo.
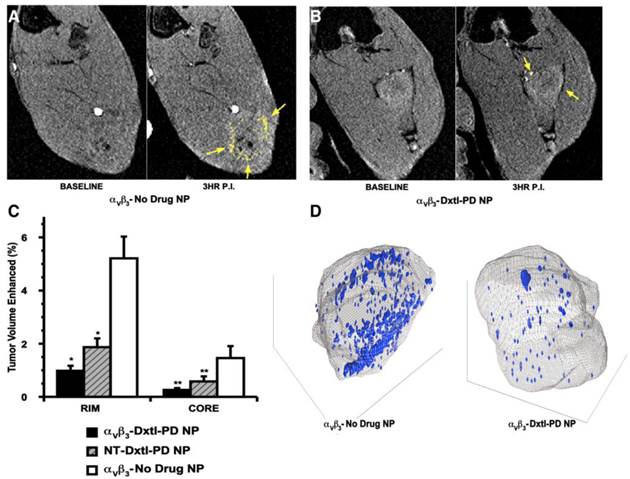
As previously reported, angiogenesis contrast in the control animals was concentrated in the tumor periphery with minimal signal enhancement localized to the core, except for the rare lobulated tumor with an internal segment of neovascular-rich capsule. (Figure 7a,c,d) The peripheral distributions of contrast-enhanced voxels (i.e., 3 standard deviations greater than baseline) in the control were typically confluent and asymmetrically distributed, presumably reflecting the most active regions of tumor growth at the time of imaging. αvβ3-Dxtl-PD NPs substantially decreased (p<0.05) MR detectable angiogenesis in the Vx2 tumor model after serial treatment, as opposed to the negative results achieved previously with the targeted particles incorporating native paclitaxel. (Figure 7b,c,d) The magnitude of neovascular suppression achieved with NT-Dxtl-PD NP was similar to the results obtained with αvβ3-Dxtl-PD NP, suggesting that the passive entrapment of the frequently dosed NT-Dxtl-PD NP within the irregular neovascular bed provided enough sustained particle-endothelial cell contact to afford prodrug transfer, at least to the extent detectable by MR molecular imaging. We have previously shown with dynamic MR imaging in the Vx2 rabbit tumor [24] and mouse melanoma [25] models, that passive entrapment of nontargeted paramagnetic PFC nanoparticles within the tumor peripheral neovasculature is detectable with approximately half the level of contrast signal observed with αvβ3-targeted paramagnetic particles at two hours. No differences in tumor volume between any treatment group were measured. (Additional file 1: Supplemental Figure 5)
A) T1w MR images of Vx 2 tumor obtained at baseline and 3 hours after αvβ3-Gd-DOTA NP in animals receiving αvβ3-no drug NP showing peripheral tumor angiogenesis contrast enhancement false colored in yellow. B) The same image sequence as panel A in Vx2 tumor rabbits following αvβ3-Dxtl-PD NP. C) Histogram illustrating marked angiogenesis in the control rabbits rim (tumor peripheral 50%) versus those receiving Dtxl-PD with or without targeting. Note minimal contrast signal with the tumor core. Marked decrease in contrast in nontargeted (NT) animal group suggests that the dosage or frequency of treatment was in excess of the therapeutic need with passive entrapment providing adequate particle-to-endothelium contact to afford significant anti-angiogenesis. D) Representative 3D neovascular maps of Vx2 tumor in control and targeted Dxtl-PD treatment groups. Note asymmetric distribution of confluent neovascular regions in the control rabbit was not appreciated in the treated animal. Blue voxels equate to αvβ3-Gd-DOTA NP contrast enhancement exceeding 3 standard deviations over baseline. * p<0.05, ** p<0.01.
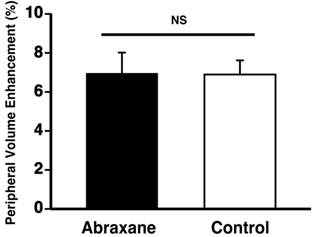
To verify that the neovascular effect of the Dxtl-PD NP, appreciated by MR molecular imaging, was not attributable to the systemic premature release of taxanes in one or both groups, a separate cohort of rabbits implanted with the Vx2 tumor were treated serially with Abraxane® given at an equimolar dose and compared to a saline control. MR molecular imaging of angiogenesis with αvβ3-Gd-DOTA nanoparticles on day 17 revealed no anti-angiogenic effectiveness in the Abraxane® treated rabbits versus the controls. (Figure 8) In fact, the magnitudes of neovascular contrast measured in each group were nearly identical, as were the tumor volumes (Additional file 1: Supplemental Figure 6). These results demonstrate that the retention and delivery method of Dxtl-PD in the nanoparticle was essential to elicit the MR measureable change in tumor neovasculature.
Vx 2 tumor angiogenesis following serial dosages of Abraxane® (equivalent to treatment regimen used in Figure 7) versus control rabbits receiving saline as assessed by MR molecular imaging with αvβ3-Gd-DOTA NP. No effect of the free taxane drug was detected, which was equivalent to the results previously obtained with free Taxol® in the preliminary data (Additional file 1: Supplemental Figure 1).
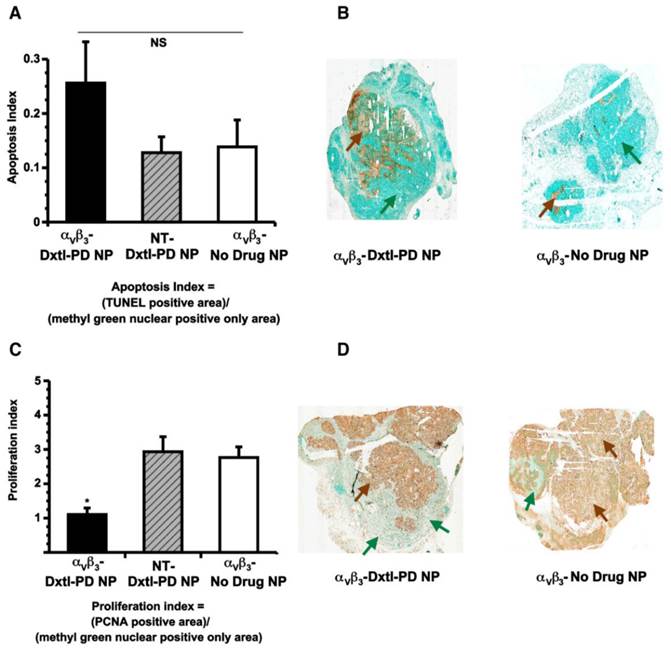
Histological assessments of tumor showed that αvβ3-Dxtl-PD NP decreased (p<0.05) tumor cell proliferation, as assessed by PNCA staining of the lesion. (Figure 9c,d) The same trend was measured for tumor apoptosis. (Figure 9a,b) In each case, the patterns of staining appeared as patchy confluencies, distributed variably among animal tumors. These data suggest that targeted delivery of Dxtl-PD was biologically more effective than NT Dxtl-PD treatment, which is consistent with previously reported dynamic imaging results discussed above. [24] [25] Further, while MR angiogenesis imaging is able to robustly track the progression of Vx2 tumor neovasculature around 12 days post implantation or longer, MR sensitivity may permit only assessment of the macroscopic changes of neovasculature and may not adequately resolve microscopic differences in neovasculature which may still impart significant tumor biology impact. Changes in microvasculature counts following theranostic anti-angiogenesis treatment were correlated previously with MR molecular imaging of angiogenesis in atherosclerotic rabbits[26], However, given the extensive heterogeneity of angiogenesis presentation in the tumor, replication here would require complete serial sectioning and quantification of neo/microvasculature to compare the extent of microscopic angiogenesis persisting with and without integrin-targeting, and was precluded in this project.
A) Histogram showing a trend (nonsignificant (NS), p = 0.2) toward increased apoptosis among Vx 2 tumor rabbits receiving αvβ3-Dxtl-PD NP versus controls. B) Illustrative sections (40x) of Vx 2 tumor following TUNEL staining (brown arrows) using a methyl green nuclear positive reference for tumor area (green arrows). C). Histogram showing a significant (p < 0.05) decrease in proliferation index among Vx2 tumor rabbits receiving αvβ3-Dxtl-PD NP versus controls. D) Representative sections (40x) of Vx2 tumor following PCNA staining (brown arrows) using a methyl green nuclear positive reference for tumor area (green arrows). * p < 0.05.
Discussion
The facile synthesis, development and characterization of a new Sn 2 lipase-labile docetaxel prodrug is reported and shown to be an effective anti-angiogenic agent in vitro and in vivo. The Sn 2 phosphatidylcholine drug stably incorporated into the nanoparticle lipid membrane and did not appreciably release during dissolution against PBS buffer or plasma over three days. Further, overnight exposure of αvβ3-Dxtl-PD NP to plasma spiked with phospholipase enzyme failed to liberate the taxane from the membrane, unless the nanoparticle itself was first “cracked” with alcohol. The bioactivity and efficacy of Dxtl-PD was demonstrated in endothelial cell culture and found to be equivalent to docetaxel, paclitaxel, and paclitaxel prodrug. The effectiveness of αvβ3-Dxtl-PD NP was at least as effective as Taxol® at equimolar doses over 96 hours, with 4-fold numerical advantage noted for the prodrug over the free compound. At earlier time points, in vitro cell metabolism studies demonstrated that free docetaxel had equivalent efficacy to αvβ3-Dxtl-PD NP, indicating that requisite intracellular lipase liberation of docetaxel did not limit prodrug effectiveness at 48 hours, and this result persisted through 96 hours. The effectiveness of αvβ3-Dxtl-PD NP compared with the negative control was demonstrated in the Vx2 rabbit model using MR molecular imaging of angiogenesis. Nontargeted Dxtl-PD nanoparticles had a similar anti-angiogenic response as measured with MR molecular imaging, indicating that passive entrapment of the particles in the tumor neo-/microvasculature was adequate to decrease MR detectable neovasculature. However, histology of these tumors revealed that αvβ3-Dxtl-PD NP impacted tumor progression and apoptosis, which was not appreciated for NT Dxtl-PD NP or the control groups. Abraxane® given at an equimolar drug dosage and frequency as the αvβ3-Dxtl-PD NP had no effect on angiogenesis when compared to control rabbits receiving saline only, confirming that the effectiveness of this low taxane exposure was dependent on nanoparticle delivery and concentration at the tumor.
Dxtl-PD was not incorporated into the αvβ3-Gd-DOTA nanoparticles to support simultaneous imaging and therapy serially as previously reported in other models. [22] In a previous time course study characterizing MR angiogenesis mapping in the Vx2 tumor on days 9, 12, and 15 post tumor implantation, MR molecular imaging was unable to delineate and differentiate a coherent neovascular pattern or increased integrated MR contrast signal between animals receiving αvβ3-Gd-DOTA PFC NPs or NT-Gd-DOTA PFC NPs on day 9. Although angiogenesis is well known to occur robustly at a microscopic level as tumors grow beyond 2 to 3 mm, the insensitivity of MRI, even using a strong molecular imaging agent such as αvβ3-Gd-DOTA PFC NP, was unable to differentiate specific neovascular targeting from passive nontargeted entrapment in the Vx2 tumor neo/microvasculature on day 9 post implantation. [23] However, on days 12 and 15 of that study, clear differences in neovasculature signal were measured between the two groups and a very close correspondence between the neovasculature pattern determined on day 12 and day 15 in the same animal was illustrated. [23] Consequently, the pursuit of theranostic MR molecular imaging approach beginning on day 8 post-implantation as a baseline imaging reference, as demonstrated in other experimental models[22], was precluded and a companion diagnostic design with serial treatments followed by terminal imaging measurement was adopted.
The direct targeting and delivery of low dose docetaxel to nascent endothelial cells is a distinctly different mechanism of achieving anti-angiogenesis treatment than the vascular endothelial growth factor (VEGF) inhibitors approved to date. Certainly, tumor angiogenesis is a complex process with a dominant role attributed to VEGF. Bevacizumab, anti-VEGF-A, was initially intended to block angiogenesis and vascular permeability by adsorbing VEGF-A in the tumor milieu. However, VEGF-A is produced not only by the tumor but also by surrounding stromal cells, such that monotherapy with the drug was unsuccessful. Ultimately, bevacizumab was approved as the first inhibitor for combination use in metastatic colorectal cancer [27], then later for non-squamous, non-small cell lung cancer [28], renal cell carcinoma [29], and glioblastoma multiforme [30]. Several other VEGF-directed drugs have followed which competitively inhibit the intracellular ATP at the catalytic binding site of tyrosine kinases, including sunitinib (Pfizer), pazopanib (Glaxo-Smith-Kline), sorafenib (Nexavar), vandetanib (Astrazeneca), carbozantinib (Exelixis), axitinib (Inlyta), and linifanib (Abbott). Tyrosine kinase inhibitors (TKI) are designed to blockade the downstream signaling pathways that promote tumor growth and metastasis when stimulated by angiogenic factors. The third class of current anti-angiogenesis inhibitors impact mTOR, mammalian target of rapamycin, which, under upstream control via the PI3K/AKT pathway, influences cell growth by modulating metabolism, protein synthesis, and autophagy. Rapamycin and its analogs, temsirolimus (CCI-779) and everolimus (RAD001), are allosteric inhibitors of mTORC1. In general, these agents have had limited effectiveness because they are specifically directed to mTORC1, and a second mTOR (mTORC2) is able to exert negative feedback to AKT/PKB that can antagonize the anti-tumor effects of the rapalogs. Resistance to all of these drugs may result from negative feedback, invocation of alternative angiogenic pathways that circumvent the blockade, selection pressure for tumor or stem cells able survive the hypoxic environments, induction of cryoprotective tumor cell autophagy or dormancy, or tumor extravasation to better environs. The nanomedicine approach with αvβ3-Dxtl-PD NP is independent of how the neovasculature expansion is stimulated to occur, and the endothelium, which is a normal replicating cell, as opposed to the epigenetically morphing tumor cells, can be anticipated to respond uniformly to a directly targeted nanosystem approach regardless of the tumor type. As with other anti-angiogenesis therapies, αvβ3-Dxtl-PD NP treatment will best be used as an adjuvant combined with effective chemotherapy rather than standalone monotherapy. Further, MR molecular imaging, which was used in this study to assess anti-angiogenesis response, is a robust technique amenable to identifying patient populations most likely to have the greatest treatment benefit-to-risk ratio and longitudinally monitor for recurrence, but it may lack the sensitivity required to detect biologically relevant microscopic levels of neovascularity that may indicate a complete therapeutic response.
In the present study, the concept and efficacy of using docetaxel as an Sn 2 lipase labile prodrug was studied and demonstrated in vitro and in vivo. In contradistinction to the original failed pilot studies pursued with αvβ3-paclitaxel (free) NP, the use of Dxtl-PD NPs resulted in significant anti-angiogenesis. Further, Abraxane® given in equivalent doses following the same treatment regimen was ineffective, emphasizing the importance of the targeted drug delivery paradigm. Whereas some effective nanomedicine applications are intended to be improved excipients, providing more extended drug release in circulation, αvβ3-Dxtl-PD NP is an anti-angiogenesis agent that concentrates a small amount of systemically administered taxane directly on the target for a highly potent effect.
Conclusion
The synthesis of the Sn 2 docetaxel prodrug was accomplished, which produced a lipophilic prodrug incorporated into the phospholipid surfactant of PFC nanoparticles. In endothelial cell culture, the biopotency of paclitaxel, docetaxel, paclitaxel prodrug and docetaxel prodrug were equivalent. In vitro experiments confirm that the drug was stable in the particle membrane in plasma and whole blood, even in the presence of excess phospholipase. Prodrug-laden particles were bound to target cells, the docetaxel prodrug was delivered and equivalently bioactive to the parent drug in the same nanosystem. In the Vx2 cancer model, αvβ3-Dxtl-PD NP elicited strong anti-angiogenesis effects as assessed by MR neovascular molecular imaging and histopathology, but acute changes in tumor volumes were not appreciated. In contradistinction to current anti-angiogenic treatments, for which the intent to blockade or modulate tumor production of VEGF can be circumvented or ameliorated by countering biochemical mechanisms in the epigenetically morphing tumor cells, αvβ3-Dxtl-PD NP should deliver uniform anti-angiogenic results regardless of the molecular mechanism of neovessel stimulation and may be an effective adjuvant to chemotherapies in aggressive cancers dependent on an expanding rich neovasculature.
Supplementary Material
Additional File 1Supplemental Figure 1-Figure 6.
Abbreviations
PFC: perfluorocarbon
CFDD: contact-facilitated drug delivery
API: active pharmaceutical ingredient
Dxtl-PD: docetaxel prodrug
MR(I): magnetic resonance (imaging)
DCC: dicyclohexyl carbodiimide
DMAP: dimethylaminopyridine
PCl5: phosphorous pentachloride
EDCI: 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide
NMR: nuclear magnetic resonance
ESI MS: electron spin ionization mass spectrometry
PAz-PC: 1-palmitoyl-2-azelaoyl PC, Azelaoyl PC or 1-O-hexadecanoyl-2-O-(9-carboxyoctanoyl)-sn-glyceryl-3-phosphocholine
Gd-DOTA: gadolinium 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid
Gd-DTPA: gadolinium diethylenetriaminepentaacetic
TLC: thin layer chromatography
NP: nanoparticle
DLS: dynamic light scattering
Zeta: zeta / electrophoretic potential
TEM: transmission electron microscopy
Sn 2: glycerophospholipids use "sn" notation, stereospecific numbering. By convention the hydroxyl group of the second carbon of glycerol (Sn 2) is on the left on a Fischer projection.
PECAM: platelet endothelial cell adhesion molecule
Tie-2: endothelium-specific receptor tyrosine kinase
MWCO: molecular weight cut-off
HPLC: high performance liquid chromatography
PEG: polyethylene glycol
DMEM: Dulbecco's Modified Eagle's Medium
MTT: colorimetric assay that measures the reduction of yellow 3-(4,5-dimethythiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) by mitochondrial succinate dehydrogenase
DMSO: dimethyl sulfoxide
NAD(P)H: nicotinamide adenine dinucleotide phosphate (reduced)
SENSE: sensitivity encoding for fast MRI
ROI: region-of-interest
OCT: optimal cutting temperature
PCNA: proliferating cell nuclear antigen
HUVEC: human vascular endothelial cell
PBS: phosphate buffered saline
PLA2: phospholipase A2
VEGF: vascular endothelial growth factor
TKI: Tyrosine kinase inhibitors
PI3K: Phosphatidylinositol 3-kinase
AKT: Protein Kinase B
mTOR: mammalian target of rapamycin
mTORC1: mammalian target of rapamycin complex 1
mTORC2: mammalian target of rapamycin complex 2
Acknowledgements
The financial support from the AHA under grant number 0835426N (DP), from NIH under grant numbers HL073646 (SAW), HL112518, HL113392, CA154737, CA136398, NS073457, and from the DOD CA100623 is greatly appreciated. Additional support was derived from the International Cooperation and Exchanges Program of the National Ministry of Science and Technology (2009DFB30040) (BS), the National Natural Science Foundation of China (81130028, 30970807,30570527, 31210103913,81225010) (BS), the Key Grant Project of Heilongjiang Province (GA12C302) and the Ph.D. Programs Foundation of Ministry of Education of China (201123071100203) (BS), the National Natural Science Foundation for Young Scholars of China (81101086) (KW), China Postdoctoral Science Foundation (20100471020) (KW), China Postdoctoral Special Science Foundation (2012T50375) (KW) and Medical Scientific Research Foundation of Heilongjiang Province Health Department (2010-156) (KW).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Jones SE, Erban J, Overmoyer B, Budd GT, Hutchins L, Lower E. et al. Randomized phase III study of docetaxel compared with paclitaxel in metastatic breast cancer. J Clin Oncol. 2005;23:5542-51
2. Stone GW, Ellis SG, Cox DA, Hermiller J, O'Shaughnessy C, Mann JT. et al. A polymer-based, paclitaxel-eluting stent in patients with coronary artery disease. N Engl J Med. 2004;350:221-31
3. Zhang Z, Mei L, Feng SS. Paclitaxel drug delivery systems. Expert Opin Drug Deliv. 2013;10:325-40
4. Zhou HF, Hu G, Wickline SA, Lanza GM, Pham CT. Synergistic effect of antiangiogenic nanotherapy combined with methotrexate in the treatment of experimental inflammatory arthritis. Nanomedicine (Lond). 2010;5:1065-74
5. Lanza GM, Winter PM, Caruthers SD, Hughes MS, Hu G, Schmieder AH. et al. Theragnostics for tumor and plaque angiogenesis with perfluorocarbon nanoemulsions. Angiogenesis. 2010;13:189-202
6. Lanza GM, Caruthers SD, Winter PM, Hughes MS, Schmieder AH, Hu G. et al. Angiogenesis imaging with vascular-constrained particles: the why and how. Eur J Nucl Med Mol Imaging. 2010;37(Suppl 1):S114-26
7. Zhou HF, Chan HW, Wickline SA, Lanza GM, Pham CT. Alpha v beta 3-targeted nanotherapy suppresses inflammatory arthritis in mice. FASEB J. 2009;23:2978-85
8. Schmieder AH, Caruthers SD, Zhang H, Williams TA, Robertson JD, Wickline SA. et al. Three-dimensional MR mapping of angiogenesis with alpha 5 beta 1(alpha v beta 3)-targeted theranostic nanoparticles in the MDA-MB-435 xenograft mouse model. FASEB J. 2008;22:4179-89
9. Lanza GM, Yu X, Winter PM, Abendschein DR, Karukstis KK, Scott MJ. et al. Targeted antiproliferative drug delivery to vascular smooth muscle cells with a magnetic resonance imaging nanoparticle contrast agent: implications for rational therapy of restenosis. Circulation. 2002;106:2842-7
10. Ingber D, Fujita T, Kishimoto S, Sudo K, Kanamaru T, Brem H. et al. Synthetic analogues of fumagillin that inhibit angiogenesis and suppress tumour growth. Nature. 1990;348:555-7
11. Garrett ER, Eble TE. Studies on the stability of fumagillin. I. Photolytic degradation in alcohol solution. J Am Pharm Assoc Am Pharm Assoc (Baltim). 1954;43:385-90
12. Garrett ER. Studies on the stability of fumagillin. III. Thermal degradation in the presence and absence of air. J Am Pharm Assoc Am Pharm Assoc (Baltim). 1954;43:539-43
13. Wang J, Lou P, Lesniewski R, Henkin J. Paclitaxel at ultra low concentrations inhibits angiogenesis without affecting cellular microtubule assembly. Anti-Cancer Drugs. 2003;14:13-9
14. Winter PM, Schmieder AH, Caruthers SD, Keene JL, Zhang H, Wickline SA. et al. Minute dosages of alpha v beta 3-targeted fumagillin nanoparticles impair Vx-2 tumor angiogenesis and development in rabbits. FASEB J. 2008;22:2758-67
15. Partlow K, Lanza G, Wickline S. Exploiting lipid raft transport with membrane targeted nanoparticles: A strategy for cytosolic drug delivery. Biomaterials. 2008;29:3367-75
16. Soman N, Baldwin S, Hu G, Marsh J, Lanza G, Heuser J. et al. Molecularly targeted nanocarriers deliver the cytolytic peptide melittin specifically to tumor cells in mice, reducing tumor growth. J Clin Invest. 2009;119(c):2830-42
17. Jubo Liu, Payam Zahedi, Faquan Zeng, Allen C. Nano-sized assemblies of a PEG-docetaxel conjugate as a formulation strategy for docetaxel. J Pharm Sci. 2008;97:3274-90
18. Meoli DF, Sadeghi MM, Krassilnikova S, Bourke BN, Giordano FJ, Dione DP. et al. Noninvasive imaging of myocardial angiogenesis following experimental myocardial infarction. J Clin Invest. 2004;113:1684-91
19. Pan D, Pramanik M, Senpan A, Allen JS, Zhang H, Wickline SA. et al. Molecular photoacoustic imaging of angiogenesis with integrin-targeted gold nanobeacons. FASEB J. 2011;25:875-82
20. Cresteil T, Monsarrat B, Dubois J, Sonnier M, Alvinerie P, Gueritte F. Regioselective metabolism of taxoids by human CYP3A4 and 2C8: structure-activity relationship. Drug Metab Dispos. 2002;30:438-45
21. Hendrikx. JJMA, Dubbelman. A-C, Rosing. H, Schinkel. AH, Schellens. JHM, Beijnen. JH. Quantification of docetaxel and its metabolites in human plasma by liquid chromatography / tandem mass spectrometry. Rapid Commun Mass Spectrom. 2013;27:1-10
22. Winter P, Caruthers S, Zhang H, Williams T, Wickline S, Lanza G. Antiangiogenic synergism of integrin-targeted fumagillin nanoparticles and atorvastatin in atherosclerosis. J Am Coll Cardiol Img. 2008;1:624-34
23. Schmieder AH, Winter PM, Williams TA, Allen JS, Hu G, Zhang H. et al. Molecular MR imaging of neovascular progression in the Vx2 tumor with alpha v beta 3-targeted paramagnetic nanoparticles. Radiology. 2013;268:470-80
24. Winter PM, Caruthers SD, Kassner A, Harris TD, Chinen LK, Allen JS. et al. Molecular imaging of angiogenesis in nascent Vx-2 rabbit tumors using a novel alpha v beta 3-targeted nanoparticle and 1.5 tesla magnetic resonance imaging. Cancer Res. 2003;63:5838-43
25. Schmieder AH, Winter PM, Caruthers SD, Harris TD, Williams TA, Allen JS. et al. Molecular MR imaging of melanoma angiogenesis with alpha v beta 3-targeted paramagnetic nanoparticles. Magn Reson Med. 2005;53:621-7
26. Winter P, Neubauer A, Caruthers S, Harris T, Robertson J, Williams T. et al. Endothelial alpha v beta 3 -Integrin targeted fumagillin nanoparticles inhibit angiogenesis in atherosclerosis. Arterioscler Thromb Vasc Biol. 2006;26:2103- 9
27. Hurwitz H, Fehrenbacher L, Novotny W, Cartwright T, Hainsworth J, Heim W. et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med. 2004;350:2335-42
28. Sandler A, Gray R, Perry MC, Brahmer J, Schiller JH, Dowlati A. et al. Paclitaxel-carboplatin alone or with bevacizumab for non-small-cell lung cancer. N Engl J Med. 2006;355:2542-50
29. Escudier B, Pluzanska A, Koralewski P, Ravaud A, Bracarda S, Szczylik C. et al. Bevacizumab plus interferon alfa-2a for treatment of metastatic renal cell carcinoma: a randomised, double-blind phase III trial. Lancet. 2007;370:2103-11
30. Friedman HS, Prados MD, Wen PY, Mikkelsen T, Schiff D, Abrey LE. et al. Bevacizumab alone and in combination with irinotecan in recurrent glioblastoma. J Clin Oncol. 2009;27:4733-40
Author contact
![]() Corresponding author: Gregory M. Lanza, MD PhD. Division of Cardiology, Campus Box 8215, 660 Euclid Ave, Washington University School of Medicine, St. Louis, MO 63108. Tel: 314-454-8813, Fax: 314-454-5265, Email: greg.lanzacom.
Corresponding author: Gregory M. Lanza, MD PhD. Division of Cardiology, Campus Box 8215, 660 Euclid Ave, Washington University School of Medicine, St. Louis, MO 63108. Tel: 314-454-8813, Fax: 314-454-5265, Email: greg.lanzacom.