Impact Factor
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Pathophysiology challenges for...
Aptamer-mediated drug delivery...
Free Aptamer as molecularly...
Aptamer-drug delivery system
Aptamer-nanoparticle-drug...
Aptamer-mediated nucleic acid...
Aptamer-mediated co-delivery...
Current progress in...
Aptamer-Guided Theranostic...
Future perspectives
Conclusions
Abbreviations
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2015; 5(1):23-42. doi:10.7150/thno.10202 This issue Cite
Review
Nucleic Acid Aptamer-Guided Cancer Therapeutics and Diagnostics: the Next Generation of Cancer Medicine
Dongxi Xiang1*, Sarah Shigdar1*, Greg Qiao2, Tao Wang1, Abbas Z. Kouzani3, Shu-Feng Zhou4, Lingxue Kong5, Yong Li6, Chunwen Pu7 ![]() , Wei Duan1
, Wei Duan1 ![]()
1. School of Medicine, Deakin University, Pigdons Road, Waurn Ponds, Victoria 3216, Australia
2. Department of Chemical and Biomolecular Engineering, Melbourne School of Engineering, The University of Melbourne, Parkville, Victoria 3010, Australia
3. School of Engineering, Deakin University, Pigdons Road, Waurn Ponds, Victoria 3216, Australia
4. Department of Pharmaceutical Sciences, College of Pharmacy, University of South Florida, Tampa, FL 33612, USA
5. Institute for Frontier Materials, Deakin University, Waurn Ponds, Victoria, Australia
6. Cancer Care Centre, St George Hospital and St George Clinical School, University of New South Wales (UNSW), Kensington, NSW2052, Australia
7. The Affiliated Zhongshan Hospital of Dalian University, 6 Jiefang Road, Dalian, Liaoning, The People's Republic of China, 116001
* These authors contributed equally.
Received 2014-7-28; Accepted 2014-9-1; Published 2015-1-1
Abstract

Conventional anticancer therapies, such as chemo- and/or radio-therapy are often unable to completely eradicate cancers due to abnormal tumor microenvironment, as well as increased drug/radiation resistance. More effective therapeutic strategies for overcoming these obstacles are urgently in demand. Aptamers, as chemical antibodies that bind to targets with high affinity and specificity, are a promising new and novel agent for both cancer diagnostic and therapeutic applications. Aptamer-based cancer cell targeting facilitates the development of active targeting in which aptamer-mediated drug delivery could provide promising anticancer outcomes. This review is to update the current progress of aptamer-based cancer diagnosis and aptamer-mediated active targeting for cancer therapy in vivo, exploring the potential of this novel form of targeted cancer therapy.
Keywords: aptamer, cancer therapy, active targeting, cancer stem cell, chemoresistance, cancer theranostic
Introduction
The success of cancer chemotherapy depends on enough delivery of therapeutic drugs to tumors while sparing normal tissues. Inadequate amounts of drug reaching cancer cells after initial treatment is responsible for an increase in drug resistance and tumor recurrence, which are due to an outgrowth of residual tumor cells [1]. Factors such as abnormality in tumor microenvironment and tumor cell heterogeneity contribute to the failure of cancer treatment. The abnormal tumor physiology, such as vasculature changes, introduces a set of delivery challenges that limit drug transport to original and metastatic tumor sites, and in return promotes systematic drug resistance [2]. Aptamers are short chemically synthesised single-stranded RNA or DNA oligonucleotides that specifically bind to molecular targets with high affinity and specificity. They can be further chemically modified to acquire desirable attributes for clinical applications [3]. A wide range of diagnostic agents or chemotherapeutic agents can be linked to aptamers, via physical or chemical conjugation, to enhance cancer diagnosis and treatment efficacies. This review will provide an overview of obstacles that therapeutic agents face and discuss the latest advances in aptamer-based cancer diagnostics and therapy.
Pathophysiology challenges for drug transport to solid tumors
During the course of pharmacological treatment of cancer, delivery and metabolism of a chemotherapeutic agent is a complex journey starting from the site of administration and ending where the tumor cell is located. The five main factors involved in this journey are discussed in the following sections [4, 5].
Abnormal blood and lymphatic vessels
Efficient drug delivery to solid tumors relies on good blood circulation in the tumor, followed by drug transportation through vascular walls [4, 5]. In the normal angiogenesis system, there is a balance between new blood vessel growth and shrinking of the old one under the influence of antiangiogenic factors [6, 7]. In contrast, tumor angiogenesis is characterised by uncontrolled overproduction of new blood vessels that are inhomogeneous in size and density, thus hampering fluid perfusion and drug transportation and resulting in further drug resistance, tumor mutagenesis, metastases and/or recurrence [8-10]. The lymphatic system functions to return interstitial fluid to the blood circulation and to clear macromolecules as well as detached tumor cells [11, 12]. The impaired lymphatic drainage is a prominent characteristic of solid tumors and this, coupled with a leaky tumor vasculature, results in an enhanced accumulation and retention of molecules with a molecular mass of 40 kDa or higher in the tumor, an important phenomenon known as the enhanced permeability and retention (EPR) effect [8, 13, 14]. The compressed lymphatic vessels lose their function of returning interstitial fluid, resulting in fluid retention and elevation of interstitial fluid pressure (IFP), generating another barrier for drug transport [8, 15].
Elevated interstitial fluid pressure (IFP)
Interstitial fluid is the extracellular solution that baths and surrounds most tissues, excluding the extravasation fluid from the lymph and blood vessels [16]. The interstitial fluid mediates the exchange of oxygen, nutrients and waste products between cells and their surroundings through the interstitial space among capillaries, blood vessels and the lymphatic system [17]. IFP is the hydrostatic and colloid osmotic pressures exerted by the free interstitial fluid. In contrast to normal interstitium, the IFP is often elevated in solid tumors partly because of low lymphatic drainage and leaky tumor vessels [16-19]. The elevated IFP is a major obstacle for anticancer drug transport [20]. Many factors can lead to an increased IFP, including individual or combined effects of hyper-permeable blood vessels and a defective lymphatic system [21-23]. Chemotherapeutic drugs are transported and distributed through the tumor interstitial space via diffusion and convection, for small and large molecule transport, respectively [24-26]. Drug diffusion depends on diffusivity and concentration gradients, while convection of large molecules relies on hydraulic conductivity and pressure differences [27]. High molecular weight drugs mainly travel to the interstitial space by convection, but the uniform IFP and negligible fluid pressure gradients weaken the convection power, leaving diffusion largely responsible for drug transport in solid tumors [28, 29]. Increased IFP decreases vascular and capillary functions and limits drug exposure, distribution and uptake by tumor cells, resulting in the failure of anticancer therapy [4, 30].
Dense interstitial structure and extracellular matrix
Extracellular matrices in healthy tissue serve many functions, including maintaining cellular homeostasis, sequestering tissues or cellular growth factors from one to another, and eliciting protease activities for regulating cellular functions [31]. The abnormal extracellular matrices in the tumor are largely composed of structural macromolecules (collagen) and space-filling proteins (polysaccharides) that make the tumor interstitium a highly viscous space [31, 32]. This viscoelastic and tortuous area forms a major barrier for effective drug transport [30]. Components such as collagen, glycosaminoglycan and fibroblasts are responsible for the lower hydraulic conductivity and increase of interstitium tension, resulting in a poorer convection and increased flow resistance [4, 5]. In addition, the dense interstitial matrices mediate solid stress and compress blood and lymphatic vessels, leading to further restrictions on drug distribution.
High tumor cell density
High cell density is also responsible for the increase in IFP, compression of blood vessels, and extracellular matrix that lead to a dense and tortuous tumor microenvironment [9, 33]. In contrast, low cell density along with larger interstitial spaces facilitates more effective drug transport to their targets.
Tumor acidic environment
The leaky tumor vessel is inadequate to supply enough nutrients, resulting in a reduction in the transport of oxygen to tumor cells [34, 35]. Together with hypoxia, the nutrient-deprived niche provides a physiological-associated pressure stimulating the development of malignant tumor cells as well as resistance to chemotherapy [36-39]. The low pH (pH 5.7-7.4) in the tumor can naturally induce cell death, but cells that live in such conditions are hard to be killed, even with drug treatment, contributing to a failure of cancer therapies and disease progression [36-39].
Aptamer-mediated drug delivery for targeted cancer therapy
Aptamer
Aptamers, nucleic acid-based ligands, are small single-stranded DNA or RNA oligonucleotides that are produced in vitro via a process known as systematic evolution of ligands by exponential enrichment (SELEX) [40, 41] (Fig. 1). Isolation of aptamers specific for the target of interest can be achieved via SELEX from a large pool (> 1 x 1014) of single-stranded oligonucleotides with random sequences [42, 43]. After the incubation of the random aptamer pool with the target followed by the removal of non-binding aptamers, the bound aptamer species are recovered. These recovered nucleic acid sequences are amplified with PCR (in the case of DNA aptamer) or RT-PCR (in the case of RNA aptamers). An enriched pool of potential binders generated from PCR amplification is used in the subsequent selection rounds (typically 6-10 rounds). At the last round of SELEX, the resulting highly enriched pool of aptamers are cloned or subjected to next generation sequencing followed by further characterization and engineering of individual cloned aptamers (Fig. 1). In addition to selection against a purified or highly enriched target molecule, one can perform SELEX using live cells and even in a whole mouse to isolate cell- or organ-specific aptamers against a specific type of cells or an organ/tissue [44, 45]. Therefore, the power of SELEX enables one to generate specific aptamers against an inorganic or organic molecule, a protein, a cell and even an organ of interest.
Isolation of aptamer and aptamer-target cell interaction. (A) Schematic illustration of RNA aptamer generation by SELEX. Negative control cells or proteins are incubated with a large pool (> 1 x 1014) of single-stranded RNA oligonucleotides with random sequences, followed by removal of bound RNAs. The unbound sequences are then subjected to positive selection by incubating with specific targets of cells or proteins. The bound aptamers are eluted and amplified by RT-PCR, followed by new SELEX rounds. After the last round of selection (typically 6-10 round), the remaining RNA aptamers are converted to DNA and subjected to next generation sequencing or conventional cloning followed by characterization and further engineering. (B) Schematic diagram depicting the aptamer-target interactions. Aptamers bind to targets at the cell surface through shape complementarity involving non-covalent bonds.
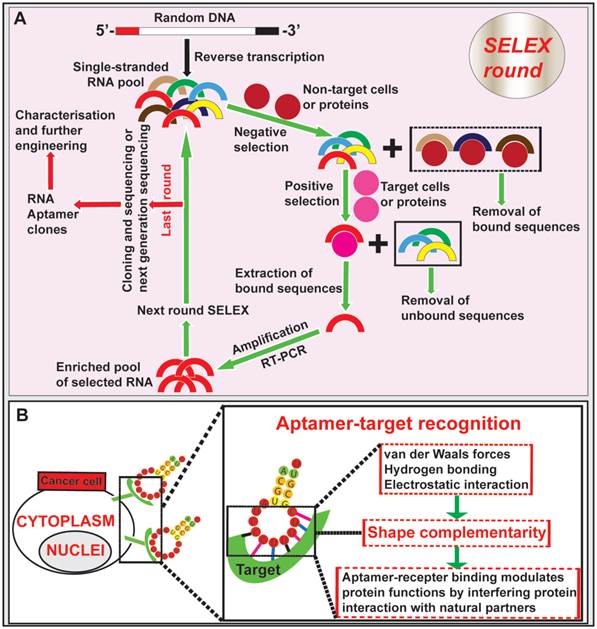
Aptamers fold into tertiary conformations and bind to their targets through shape complementarity at the aptamer-target interface [46]. An aptamer binds to a protein can modulate protein functions by interfering with protein interaction with natural partners. Besides to binding to small organic and inorganic molecules, aptamers have the unique ability to recognize and bind to large targets, such as proteins, whole cells or even organs [47-50]. Similar to antibodies, aptamers gain entrance to target cells via receptor-mediated endocytosis upon binding to cell surface ligands [51, 52]. In addition, the in vitro generation of aptamers via SELEX confers a low-cost advantage over the long and arduous development process of antibodies [51, 53-55]. One important advantage of aptamers over antibodies is that, once selected, they can be chemically synthesised instead of being produced in animals or cultured mammalian cells, thus simplifying the production of therapeutic grade materials, which represents a key advantage for commercial development [56, 57]. Importantly, aptamers can penetrate into tumor cores much more efficiently than antibodies due to their ~20-25-fold smaller sizes compared with full sized monoclonal antibodies [58-60]. Given that the nucleic acid aptamers function in vivo through the blood plasma, several limitations of aptamers should be considered. Being polynucleotides, nucleic acid aptamers are naturally susceptible to enzymes degradation by exo- and/or endo-nucleases, leading to a reduced in vivo circulatory half-life. This drawback can be alleviated by introducing backbone or side chain chemical modifications to aptamers, incorporating unnatural nucleotide bases (locked and unlocked nucleic acids) and capping the aptamer ends, thus minimizing the susceptibility to endonuclease and exonuclease attack [61-63]. Short blood residence time is another challenge with in vivo aptamer applications, which is due to fast removal of aptamer from the circulation by renal filtration as most aptamers have a size smaller than the renal filtration threshold of 40 kDa [63]. To achieve desired serum half-life, aptamers can be engineered by conjugation with a terminal polyethylene glycol (PEG), although this may compromise the extent of tumor penetration [62, 64]. It is worth noting that post-SELEX modifications following the selection of aptamers may alter the 3-D structure of the aptamers, leading to the lost or altered binding affinity and specificity. Such risks can be prevented by using random aptamer pools containing modified nucleotides during the SELEX selection [63, 64]. In addition, the ability of aptamers to interact with cells may decrease due to repulsion of nucleic acids by negatively-charged cell membranes [49]. This can be refuted by increasing the binding affinity and specificity of aptamers toward their cell surface receptors to trigger receptor-mediated endocytosis [61].
Aptamer-guided active targeting system: overcoming pathophysiology barriers
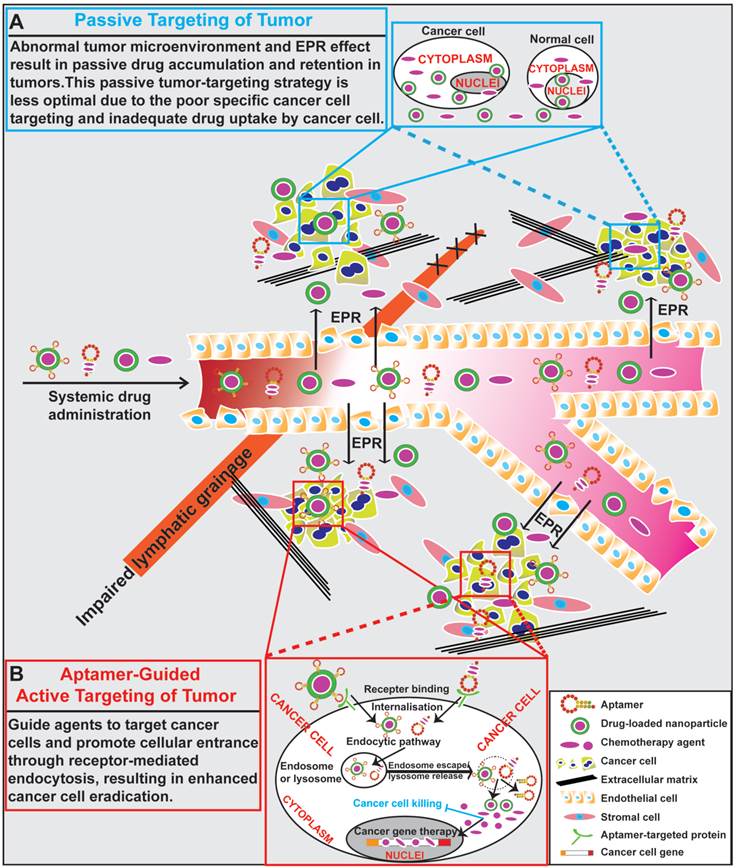
The ultimate aim of anticancer research is to improve the rates of treatment response and overall survival in patients with cancer. To achieve this goal, a tumor targeting system that delivers therapeutic agents selectively to cancer cells is necessary [65]. Passive targeting based on EPR utilises the fundamental differences in the organisation of vasculatures between normal tissues and a tumor to achieve preferential accumulation of nano-sized anti-tumor agents in tumors [66]. Based on the EPR effect, a passive tumor-targeting strategy has improved patient compliance and reduced adverse effects over the conventional free drug approach [67]. However, there have been heated debate on the existence of EPR and whether EPR plays a role in enhanced delivery of nano therapeutics in at least some types of tumors [66, 68]. Moreover, the clinical efficacy of nano-sized drugs guided by a passive targeting system is still far from optimal due to poor tumor penetration and specific drug uptake by cancer cells [67, 69]. To further improve treatment efficacy, an “active targeting” approach has been attracted huge attentions. The function of active targeting is to guide therapeutic agents with the aid of targeting ligands to tumor cells and promote their subsequent cellular entry through receptor-mediated endocytosis [70-72]. Development of novel molecular probes that bind to specific tumor biomarkers as actively guiding devices represents one of the major advances in the next generation of tumor therapeutics.
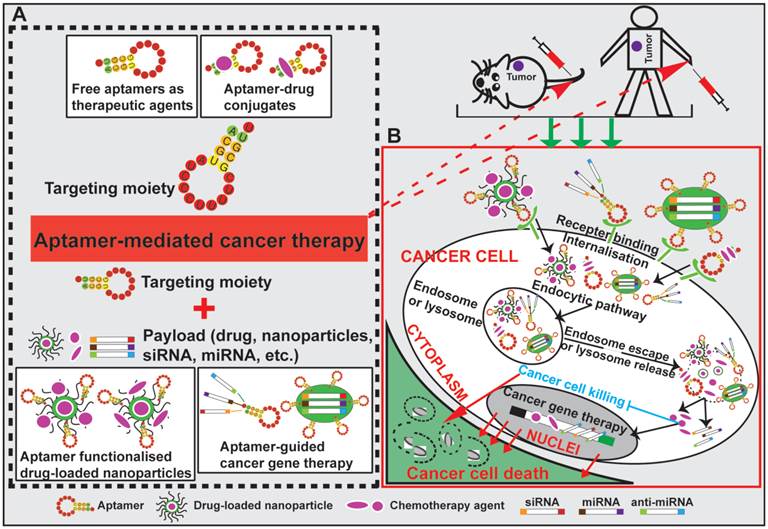
The goal of surface modification of drug carriers by tumor specific recognition molecules, such as antibodies, peptides and aptamers, is to enhance specific drug accumulation, internalization and retention in tumors via specific ligand-mediated interactions thus increasing the therapeutic index [73, 74]. Targeting specificity and drug delivering capacity are two important aspects that need to be considered in the development of an active targeting system [70]. Antibodies are the most commonly used target ligands due to their high specificity and wide availability [51, 52]. Recently, aptamers have emerged as an attractive alternative to antibodies as a ligand for active targeting of tumors (Fig. 2). Actively targeted nano-sized therapeutic is envisioned as a promising complementary strategy to passive targeting via EPR to further augment the efficiency of cancer nanomedicines. In this regard, effective delivery of drugs to tumors can be achieved via active targeting utilizing tumor-specific aptamers binding to their targets present on the surface of tumor cells. For instance, epithelial cell adhesion molecule (EpCAM) is expressed at low levels in normal epithelium, but is overexpressed (up to 1000-fold) in various solid tumors, including colon, hepatic and breast carcinomas, which relates to poor prognosis [49, 75]. Aptamer-guided active targeting enables the increased delivery of therapeutic agents to tumors as well as a reduction in toxicity and side effects by minimizing the exposure of normal tissues to the therapeutic agent. As an active targeting ligand, antibodies suffer from immunogenicity as even humanised antibodies may elicit immune responses in patients [57, 76, 77]. In contrast, being nucleic acids, aptamers are generally non-immunogenic or low-immunogenic [78, 79]. The chemical synthesis of aptamers confers additional advantages to aptamers, such as low batch-to-batch variation, and they are easier to scale up production for large scale manufacturing with minimal risk of contamination of microorganisms and endotoxins [76, 80, 81]. Therefore, aptamers are being intensively investigated as specific targeting moieties for cancer therapy. Aptamer-mediated active targeting for cancer therapy can be categorised into four groups: free aptamer as molecularly targeted agents, aptamer-drug conjugates, aptamer-nanoparticle-drug delivery system and aptamer-mediated cancer nucleic acid therapy (Fig. 3).
General scheme of drug delivery to tumors. (A) Schematic illustration of drug delivery via passive targeting. Abnormal tumor microenvironment and EPR effect result in passive drug accumulation and retention in tumors. This passive tumor-targeting strategy exhibits limitations of poor cancer cell targeting and inadequate drug uptake. (B) Schematic illustration of aptamer-guided active targeting of tumor. Aptamers undergo endocytosis after binding to cell surface targets, along with aptamer-conjugated therapeutic agents or drug-encapsulated nanoparticles. Following the entrance and escape from endosomes and/or lysosomes, the released therapeutic agents or small RNAs engage their cytoplasmic or nucleic targets to eliminate cancer cells.
Schematic illustration of aptamer-mediated cancer therapy. Four approaches are presented: free aptamer as chemotherapeutic agents, aptamer-drug conjugates, aptamer-nanoparticle-drug delivery system and aptamer-guided cancer gene therapy. After specific binding of aptamers to their cell surface target, aptamer-conjugated therapeutic agents (including exogenous therapeutic nucleotides) or drug-encapsulated nanoparticles are internalised into cancer cells via receptor-mediated endocytosis. Subsequent escape from endosomes and/or lysosomes enables the released therapeutic agents to eradicate cancer cell.
Free Aptamer as molecularly targeted therapeutics
DNA aptamer targeting vimentin for tumor therapy
To study the anti-tumor activity of a DNA aptamer, Zamay and colleagues selected a DNA aptamer NAS-24 (80-nt in length) that targets vimentin and investigated its efficacy in inducing apoptosis of mouse ascites adenocarcinoma cells in vivo [82]. They utilized arabinogalactan (AG), a natural polysaccharide with low toxicity (LD50 > 5g/kg) and high biocompatibility, to enhance in vivo delivery of anti-vimentin aptamers [83, 84]. In vivo anti-tumor efficacy of AG-NAS-24 self-forming complex was evaluated using an Ehrlich ascites adenocarcinoma mouse model in which one million adenocarcinoma cells were transplanted into the abdominal cavity of imprinting control region male mice. Starting from day 4 post cell inoculation, mice were treated with AG-NAS-24 complex (1.6µg/kg), free AG or aptamer daily via intraperitoneal injection for 5 days. On day nine, following the 5-day treatment, there was a 5-fold higher suppression of carcinoma growth in mice treated with AG-NAS-24 compared with those treated with free aptamer, free AG or saline. Aptamer delivered by AG caused ~2-fold more apoptosis in ascites tumor cells than that treated by saline or AG alone [82]. After intracellular delivery of AG-NAS-24 complex, aptamers may promote vimentin degradation which causes morphological changes and apoptosis in cancer cells with higher vimentin expression [82]. This study illustrates that intracellular delivery of NAS-24 aptamer could target vimentin and initiate signalling events leading to cellular apoptosis.
Aptamer-mediated tumor cell apoptosis and suppression of tumor progression
Myeloid-derived suppressor cells (MDSC) and tumor-associated macrophages (TAM) can enhance tumor angiogenesis, invasion, and limit the immunotherapeutic function of T-cells, resulting in further tumor aggression and metastasis [85-87]. In order to selectively target MDSCs and TAM activities for inhibiting the tumor progression, Roth et al. generated an RNA aptamer, cl.42 with 79 nucleotides, that specifically binds to IL-4 receptor-a (IL4Ra) with a Kd =14 nM, which is a critical molecule for MDSC function [85]. In the IL4Ra+/+ or IL4Ra-/- 4T1 breast cancer-bearing mice, cl.42 aptamer or a control aptamer (16 pmol/L/g) were administrated intravenously 3 times a week. In addition to a reduction of MDSCs and TAM expression in vivo, tumor progression was significantly inhibited in the IL4Ra+/+ tumor-bearing mice treated with the cl.42 aptamer. In contrast, no discernable anti-tumor effect was observed in mice bearing IL4Ra-/- 4T1 tumors or those treated with the control aptamer. To better understand the effect of the anti-IL4Ra aptamer on MDSCs, Roth et al. isolated immortalized mesenchymal stem cells (MSC2) and MDSCs from IL4Ra+/+ tumor-bearing mice. Cell viability of MDSCs was analysed after treatment with cl.42 aptamer or control aptamer for 1-4 days. Compared to untreated or control-aptamer treated groups, the cl.42 aptamer treatment resulted in a 2-fold decrease in viability on day 1, and a 3-3-fold higher increase in apoptosis in MSC2 cells. More importantly, only the IL4Ra-specific aptamer (150 nM), but not the control irrelevant aptamer, triggers MDSC apoptosis and drastically reduced phosphoSTAT6 (pSTAT6) signalling which is known to play crucial roles in MDSC activation. This study suggests that aptamer-triggered apoptosis in MDSCs via blocking of the IL4Ra-STAT6 signalling pathway could be a promising strategy to arrest immune escape in cancer treatment. Recent studies which utilize free aptamers as molecularly targeted agents are summarized in Table 1.
Application of free aptamers as cancer-targeted therapeutics
| Aptamer | Target | Cancer | References |
|---|---|---|---|
| DNA aptamer | DNA-binding proteins | Ovarian cancer and breast cancer | Mern et al. 2010 [88, 89] |
| DNA aptamer | MCF-10TA1 cell | Breast cancer | Tan et al. 2011 [90] |
| DNA aptamer | VEGF165 protein | Human hepatocellular carcinoma | Yung et al. 2013[91] |
| DNA aptamer | AGE product | Melanoma | Yamagishi et al. 2014 [92] |
| RNA aptamer | CEA protein | Colorectal cancer | Lee et al. 2012 [93] |
| RNA aptamer | MDSC and TAM | Multiple carcinomas (colon, mammary, fibrosarcoma, melanoma) | Serafini et al. 2012 [85] |
AGE, advanced glycation end; CEA, carcinoembryonic antigen; MDSC, myeloid-derived suppressor cells; TAM, tumor-associated macrophage; VEGF, vascular endothelial growth factor.
Aptamer-drug delivery system
Conventional therapeutic drugs often lead to severe adverse effects. Conjugating chemotherapeutic drugs to tumor-targeting aptamers can increase the drugs delivery to tumor cells while minimizing the exposure of non-target sites to the chemotherapy agents [94]. Doxorubicin (DOX) is one of the most potential anticancer agents ever developed and has been used for treatment of a wide range of liquid and solid cancers, including acute lymphoblastic leukemia and malignant lymphomas, breast, ovarian, prostate, and bronchogenic carcinomas [95]. DOX can intercalate into genomic DNA, resulting in the disruption of DNA replication and apoptotic cell death [96]. However, its efficacy is impeded by dose-limiting cardiotoxicity, inspiring intense effort in transforming this free drug into a new and targeted DOX-delivery system [97]. Aptamers are known to form tertiary structures with short double stranded regions through intra-molecular base pairing [98], which is available for the intercalation of DOX to form a physical complex. In order to investigate the anticancer efficacy of aptamer-DOX conjugates, Tan's group developed two DNA aptamers sgc8c and TLS11a-GC that specifically bind to CCRF-CEM cells (T-cell acute lymphoblastic leukemia, T-cell ALL) and LH86 cells (a human hepatocellular carcinoma cell line), respectively [52, 99]. After intercalating DOX to the TLS11a-GC aptamer, they evaluated whether the aptamer-DOX conjugate could specifically kill their targets with a low toxicity towards non-target cells. Cell viability tests demonstrated that the aptamer-DOX conjugate exhibited high therapeutic potency similar to free DOX, but prevented the nonspecific uptake of membrane-permeable DOX to non-targeted cells. After the establishment of an in vivo liver cancer model, the tumor bearing NOD Cg-Prkdc (SCID) IL2 mice were treated with DOX and TLS11a-GC-DOX complex (2 mg/kg) via a tail vein injection on days 1, 2, 3, 4, 5, 9, 10 and killed on day 11. The TLS11a-GC-DOX complex had a more efficient anti-tumor effect (2-fold more decrease in volume) compared to free DOX, which indicated that TLS11a-GC-DOX conjugates increased the local DOX concentration in the tumors with the aid of a targeting aptamer. This aptamer-drug intercalation approach thus represents a promising tactic for targeted drug delivery to cancer cells. Recent examples of developing aptamer-drug conjugates as targeted therapeutics for cancer are summarized in Table 2.
Application of aptamer-drug conjugations as cancer-targeted therapeutics
| Aptamer | Target | Drug | Cancer | Research Group |
|---|---|---|---|---|
| DNA aptamer | CCRF-CEM cell | DOX | T-cell ALL | Tan et al. 2009 [100] |
| DNA aptamer | LH86 cell | DOX | Human hepatocellular carcinoma | Tan et al. 2012 [52] |
| DNA aptamer | MUC1 protein | DOX | Lung cancer and breast cancer | Yang et al. 2012 [101] |
| DNA aptamer | HER2 protein | DOX | Breast cancer | Yang et al. 2012 [102] |
| RNA aptamer | EpCAM protein | DOX | Retinoblastoma | Krishnakumar et al. 2012 [103] |
| RNA aptamer | EGFR protein | GEM | Pancreatic cancer | White et al. 2012 [104] |
ALL: acute lymphoblastic leukemia; CCRF-CEM, T-acute lymphoblastic leukemia cell line; GEM, gemcitabine; LH86, human hepatocellular carcinoma cell line.
Aptamer-nanoparticle-drug delivery system
The past decade has witnessed promising advances in the synthesis and characterization of various nano-materials, which have been optimized for anti-cancer drug delivery vehicles [105]. The hybrid aptamer-nanoparticle system significantly enhanced cancer-specific cytotoxicity both in vitro and in vivo [106, 107] (Fig. 4). Aptamer-guided nano therapeutics aid in circumventing pathophysiology barriers to enhance both the uptake and retention of drugs by tumor cells, leading to improved therapeutic efficacy [70, 108]. There are four major merits when nanoparticles are surface functionalized by aptamers for targeted cancer drug delivery [109]: 1) the specific recognition of aptamers to targets enables a targeted detection or binding to cancer cells; 2) the straightforward synthesis and chemical modification of aptamers can boost the translation of aptamers into clinical practice; 3) the biocompatible nanoparticle-loading ability enhances the intensity of analytical signal, leading to effective cell recognition, drug delivery and anticancer treatment, and 4) aptamers linking to the surface of nanoparticles facilitates internalisation and cellular uptake of drugs with the aid of aptamer-guided active targeting. Aptamers have been extensively used to functionalize nanomaterials for targeted drug delivery, which is briefly presented below.
Aptamer-guided nano-drugs and their applications. (A) Schematic illustration of aptamer functionalised and drug-encapsulated nanoparticles. (B) Representative results from studies by Farokhzad et al. on A10 RNA aptamer functionalised Dtxl-encapsulated PLGA-PEG nanoparticles (Dtxl-NP-Apt) and anticancer efficacy in vivo. Even a single intratumoral administration of Dtxl-NP-Apt is significantly more efficient in tumor reduction as evident from the ~6.5-fold smaller tumor volume than that in mice treated with the controls (saline, NP, or Dtxl) (a and b) and improved survival (109 days vs. 73 days in Dtxl-NP group) (c and d). Reproduced from Ref. [113], Copyright (2006), with permission from National Academy of Sciences, U.S.A..
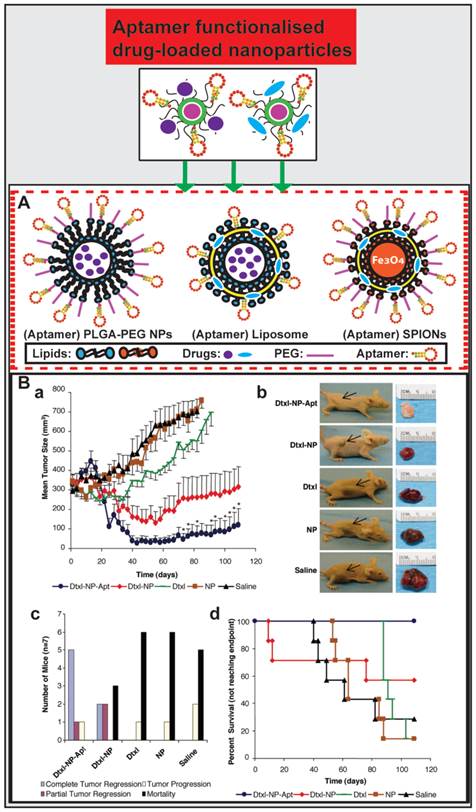
PLGA-PEG nanoparticles
Biodegradable lipid-PLGA-PEG nanoparticles take advantage of polymeric nanoparticles and have emerged as a robust drug delivery platform. They are composed of PLGA as a hydrophobic core for encapsulating drugs and a PEG shell which can prolong circulation half-life in vivo and link targeting ligands [110, 111]. PLGA-PEG nanoparticles conjugated with aptamers function via both active and passive targeting for anticancer drug delivery [26].
Farokhzad and co-workers encapsulated docetaxel (Dtxl) with PLGA-PEG nanoparticles that were functionalized with a 2-fluoropyrimidine modified RNA aptamer (A10) that binds to the prostate-specific membrane antigen (PSMA) [112, 113]. These Dtxl-encapsulated bioconjugates (Dtxl-NP-Apt) effectively targeted PSMA-expressing LNCaP prostate cancer cells and were taken up by these cells leading to significant enhancement of cellular toxicity in vitro compared with that of control nanoparticles lacking PSMA aptamers (P < 0.004) and control cells that do not express PSMA proteins. After the establishment of an LNCaP xenograft model in BALB/c nude mice, they treated the mice with Dtxl-NP-Apt and other controls at a reported tolerated dose of 40 mg/kg Dtxl [114]. Surprisingly, even a single intratumoral administration of Dtxl-NP-Apt could achieve significant efficacy in tumor reduction (a 6.5-fold higher reduction in tumor volume than any of control treatment groups of saline, NP, or Dtxl), better survival time (109 days vs. 73 days in Dtxl-NP group) and decreased toxicity as compared with non-targeted NPs and controls (Fig. 4B). This study demonstrates the bioconjugation between PLGA-PEG nanoparticle and aptamer constitutes a promising strategy for future targeted cancer therapy.
AS1411, a 26-nucleotide DNA aptamer which specifically binds to the external domain of nucleolin, has been extensively used for glioma-targeted therapy [115], as nucleolin is a nuclear matrix protein over-expressed on the cell surface of several types of cancers, including lung cancer, glioblastoma (C6 cells) and pancreatic cancer. Chen et al. developed AS1411 aptamer functionalised PLGA-PEG nanoparticles encapsulated with Paclitaxel (PTX) (Apt-PTX-NPs) and evaluated the aptamer-guided anti-glioma efficacy of PTX [116]. Following the same dose administration, this AS1411-PTX-NPs showed a 2.3-fold and 5.4-fold increase in circulation half-life and AUC compared to free Taxol, respectively. Mice or rats bearing glioma xenografts were intravenously administered with Taxol, PTX-NPs, Apt-PTX-NPs (equivalent to PTX of 3 mg/kg) every 2 days for seven consecutive injections. Until the 20th day on which the animals were humanely killed, Apt-PTX-NPs treated mice exhibited a 1.7-fold higher tumor inhibition than those treated with PTX-NP, while the prolonged survival of tumor-bearing rats received Apt-PTX-NPs was observed compared with those treated with PTX-NP (P < 0.05) and Taxol (P < 0.01).
Liposomes
Liposomes have been successfully utilised as a very versatile drug delivery carrier. Various sizes and formulation methods of liposomes contribute to a wide range of options for drug delivery in tumor treatment [117, 118]. The liposome surface can easily be decorated with PEG, which can prolong the in vivo half-life in systemic circulation, resulting in the accumulation of chemotherapeutic agents at tumor sites [119, 120]. In addition, the outer lipid bilayer allows further incorporation of targeting molecules, such as antibodies and aptamers capable of delivering drugs by active and/or passive targeting [118, 121, 122].
Lu et al. developed AS1411 aptamer-functionalized liposomes encapsulating DOX (Apt-DOX-Lip) for targeted treatment of breast cancer [123]. Enhancement of cell internalization and cytotoxicity afforded by Apt-DOX-Lip was observed in MCF-7 cells in vitro compared to controls (P < 0.01). In an in vivo model, nude mice bearing MCF-7 tumors were treated with Apt-DOX-Lip or Ctrl- DOX -Lip (25 µg DOX equivalent per tumor via intratumoral injection) every 4 days until day 24. Apt-Dox-Lip significantly inhibited MCF-7 tumor growth compared with the Ctrl-Dox-Lip group (P = 0.004). Intratumoral treatment of mice with Apt-Dox-Lip caused an earlier onset of tumor inhibition on day 8 as compared with that of day 16 in Ctrl-Dox-Lip treated mice. Apt-DOX-Lip exhibited superior tumor penetration compared with the control treatment in MCF-7 xenograft models (~1000 µm vs. ~ 500 µm), contributing to the enhancement of anti-tumor efficacy. This study concluded that aptamer-functionalized drug-loaded liposomes could be a potential strategy for targeted drug delivery in anticancer treatment.
Superparamagnetic iron oxide NPs
One of the most promising and biocompatible magnetic nanoparticles is the superparamagnetic iron oxide nanoparticles (SPIONs), which possess several favourable features that are desirable for diagnostic and therapeutic applications. They can be easily degraded in humans, leading to a short body residence and lower toxicity in vivo [124]. The surface of SPIONs is also available for modification with polymers, metal elements and various targeting ligands for applications in nanomedicine [124, 125].
Farokhzad's group formulated a multifunctional bioconjugate composed of A10 RNA aptamer modified SPIONs and DOX (TCL-SPION-Apt) for detection and therapeutic delivery of chemotherapeutic agents to prostate cancer cells (LNCaP and PC3 cells) [126]. The cellular uptake of TCL-SPION-Apt was observed in LNCaP cells as early as 3 h after dosing followed by a time-dependent increase, while there was no obvious intracellular uptake of TCL-SPION-Apt in PC3 cells that do not express PMSA, nor was with non-targeted TCL-SPIONs. The TCL-SPION-Apt bioconjugates selectively delivered DOX to their targeted cells without significant loss in cytotoxicity. In addition, Abnous's group developed an Epirubicin (Epi)-encapsulated versatile aptamer-SPION bioconjugate (Epi-Apt-SPION) for cancer imaging and therapy in vitro and in vivo [127]. Using an in vitro cytotoxic assay and flow cytometric analysis, the Epi-Apt-SPION complex was found to be capable of specifically and effectively delivering Epi to C26 cells (colon carcinoma cells) but not to non-targeted CHO-K1 cells (Chinese hamster ovary cells). Epi-Apt-SPION complex is able to accumulate at the tumor and display a high MRI signal one hour post-injection. Furthermore, enrichment of Epi-Apt-SPION at the tumor resulted in more tumor volume reduction (~2-fold and ~3.3-fold, respectively) compared with that of free Epi- and saline-treated groups.
In addition, aptamers can also be conjugated with other nanostructures for targeted and high-payload cancer therapy, such as single-walled carbon nanotubes, bovine serum albumin-based NPs, dendrimers and mesoporous silica nanoparticles [128-132] (Table 3). However, more studies on potential toxicity and in vivo fate of the nano-carriers are needed before successful translation of aptamer-nano-drugs from bench to bedside.
Application of aptamer-functionalised drug-encapsulated NPs as cancer-targeted therapeutics
| Aptamer | Target | Payload | Nanoparticle / carrier | Cancer | Research Group |
|---|---|---|---|---|---|
| RNA aptamer | PSMA | Dtxl, DOX, miRNA, CUR, GEM, TMPyP4, PTX | PLA-PEG-COOH NPs, SPION, Atelocollagen Liposomes, PLGA NPs, MSNs, Carbon nanotubes, AuNPs, PLGA-PEG NPs | Prostate cancer, Breast cancer, Liver cancer, Murine cancer, C6 glioma | Farokhzad et al. 2004, 2006, 2008, 2012 [112, 113, 126, 133] Wang et al. 2014 [134] Lu et al. 2013 [123] Kumar et al. 2012, 2013 [135-137] Cai et al. 2014 [138] Zhang et al. 2014 [139] Qu et al. 2012 [131, 140] Huang et al. 2014 [141] Chen et al. 2011 [116] |
| DNA aptamer | MUC1 | PTX, Epi, DOX | PLGA NPs, SPION, MSNs | Breast cancer, Colon cancer | Yang et al. 2011 [142] Abnous et al. 2013 [127] Santos-Oliveira et al. 2013 [132] |
| DNA aptamer | CCRF-CEM | DOX | Nanoassembly system, DNA nanotrains | T-cell ALL | Tan et al. 2013 [143, 144] |
ALL: acute lymphoblastic leukemia; AuMP, mesoporous silica-encapsulated Au nanorod; CUR, curcumin; Dtxl, docetaxel; DOX, doxorubicin; EGFR, epidermal growth factor receptor; Epi, Epirubicin; GEM, gemcitabine; MSN, Mesoporous silica nanoparticle; PSMA, prostate-specific membrane antigen; PTX, paclitaxel; SPION, super paramagnetic iron oxide nanoparticle.
Aptamer-mediated nucleic acid therapy for cancer
Aptamer-guided nucleic acid therapy
In contrast to antibodies that can be easily inactivated due to the increased acidity in cellular organelles, aptamers generated with chemical modifications maintain stability after endocytosis [85, 88, 89, 91, 93, 145]. Aptamers have been exploited as guiding moieties for both drug delivery and nucleic acid transport vehicles such as small interfering RNA (siRNA), micro RNA (miRNA) and short hairpin RNA (shRNA) [61, 146-148] (Fig. 5). Aptamers can mediate nucleic acid therapy by increasing nucleic acid targeting efficacy for oncogenes and supressing overexpressed signalling molecules or regulatory nucleic acids in cancer [112, 149, 150]. Intense effort has been devoted to explore aptamers as an effective nucleic acid therapy modality for cancer therapeutics.
Schematic illustration of aptamer-guided cancer nucleic acid therapy. Exogenous therapeutic small RNAs (siRNA, miRNA and anti-miRNA) can be directly conjugated with aptamers or encapsulated within nanoparticles functionalized with an aptamer. As targeted-delivery modalities, aptamers bind to cell surface targets on cancer cells, followed by internalisation via receptor-mediated endocytosis. Through unknown mechanisms, small RNAs escape endosomes and engage cytoplasmic RNAi/miRNA protein machinery. The mature siRNA/shRNA/miRNA interact with their cytoplasmic or nucleic target sequences, leading to mRNA degradation, translation promotion or repression or modulation of gene expression.
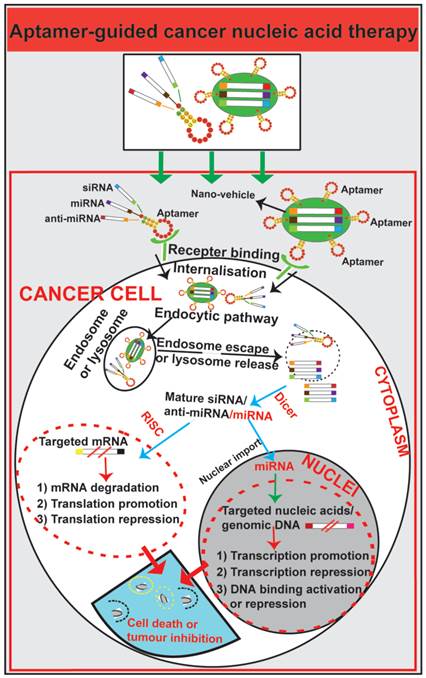
Aptamer-siRNA chimera for targeted cancer nucleic acid therapy
Small interfering RNA (siRNA) can be integrated into RNA-induced silencing complex (RISC), contributing to the degradation of target mRNA by RNA interference (RNAi) [151]. Despite the potential of efficiently inhibiting nucleic acid expression of unlimited targets, the insufficient dose of siRNA delivered to target tissues in vivo hampers the translation of this strategy from bench to bedside. Generating a chimera by linking siRNA with an aptamer, aptamer-guided siRNA delivery via receptor-mediated endocytosis serves as a promising approach for cancer-targeted RNAi therapy. After transportation to the cytoplasm by a yet to be fully elucidated route or mechanism, the therapeutic siRNA can be recognized and processed by Dicer to produce the mature siRNA followed by the target mRNA degradation [152].
In order to clarify the importance of memory T cells in mediating protective immunity against cancer, Berezhnoy et al. used a siRNA to downregulate intracellular mediators of CD8+ T cell effector differentiation [153]. To reduce the undesirable immunosuppressive effects that could result from the downregulation of siRNA in unactivated cells, they conjugated the siRNA targeting the mTORC1 component raptor to an oligonucleotide aptamer specific for 4-1BB (Kd = ~40 nM), a co-stimulatory molecule that is expressed on CD8+ T cells. To build up a B16/F10 melanoma tumor models, C57Bl/6 mice were firstly immunized with 106 irradiated B16 melanoma cells (GVAX), and one day later mice were treated with aptamer-siRNA conjugates (0.25 nmol/mouse) administered i.v. or with 1.5 μg of rapamycin injected i.p. three times daily. On day 50, the mice were xenotransplanted s.c. with 106 B16/F10 tumor cells. In this study, systemic administration of the 4-1BB aptamer-raptor siRNA to mice downregulated mTOR complex 1 (mTORC1) activity in CD8+ T cells, leading to the enhancement of antigen-activated CD8+ T cell function and boosted vaccine-induced protective immunity in tumor-bearing mice. Mice treated only with rapamycin, but not with 4-1BB aptamer-conjugated siRNA, failed to reject a subsequent tumor challenge. This study indicated that aptamer-targeted siRNA downregulation of mTORC1 in CD8+ T cells can enhance their differentiation into memory T cells and promote antitumor immunity more effectively than pharmacologic inhibitors like rapamycin. In addition, Lai et al. utilised two nucleolin aptamer-siRNA chimeras (aptNCL-SLUGsiR and aptNCL-NRP1siR) to evaluate the synergistic effect in blocking key signalling pathways involved in tumor invasion and angiogenesis [154]. After establishment of an in vivo lung cancer model in NOD-SCID mice by subcutaneously inoculating 2 × 106 CL1-5 cells, Lai and co-workers administered single chimera (2 mM of aptNCL-ControlsiR, aptNCL-SLUGsiR, or aptNCL-NRP1siR) and combined chimera (1 mM of each chimera) (aptNCL-SLUGsiR and aptNCLNRP1siR) in 50 µL PBS intratumorly three times per week until day 42. The tumor growth rate in the chimera-treated groups (aptNCL-SLUGsiR, aptNCL-NRP1siR, and combined treatment) was found to be 3-fold (with single chimera treatment) to 4-fold (with combined chimera treatment) lower than the group treated with saline. Furthermore, the combined-chimera treatment synergistically suppressed tumor invasion (1.2-fold suppression vs. control chimera) even at half-dose administration. These data indicate that aptamer-siRNA chimera holds promise for targeted cancer nucleic acid therapy.
Aptamer-miRNA active targeting system
MicroRNA (miRNA), a small non-coding RNA with 20-22 nucleotides, regulates transcriptional and post-transcriptional nucleic acid expression and modulates normal cell functions such as proliferation, apoptosis and tumor destruction [155, 156]. The manipulation of miRNA function constitutes a promising approach for anti-cancer therapy [157, 158]. However, interaction with target nucleic acids requires the specific delivery of miRNAs to their targets to minimize side effects [157]. After conjugation of a miRNA with an aptamer, the miRNA can be specifically delivered to their targets expressing proteins recognized by the aptamer. Following internalisation into cancer cells via ATP-dependent endocytosis, miRNA is processed by Dicer followed by the loading of such miRNA duplexes into an Argonaute protein in the miRNA-induced silencing complex. Esposito et al. engineered a multifunctional aptamer-miRNA conjugate by linking a tumor suppressor let-7g miRNA to the GL21.T aptamer specifically recognising the oncogenic receptor tyrosine kinase Axl [159]. The results indicated that the functional let-7g miRNA was delivered by this conjugate to Axl-expressing A549 lung cancer cells and let-7g target nucleic acids could be effectively silenced both in vitro and in vivo (1.6 nmol/mouse, three injections a week for 3 weeks), contributing to the prevention of tumor cell proliferation and migration in vitro and the tumor growth inhibition in a lung adenocarcinoma model.
Aptamer-mediated shRNA delivery for cancer therapy
As an alternative approach to RNAi, shRNA can be produced by various DNA constructs such as DNA plasmid, linear template, or packaged viral vectors [160, 161]. Being produced in host cells, shRNA elicits a more durable gene silencing compared to siRNA that are normally degraded around 48 h in vivo [162]. However, the requirement of using gene transfection or viral vectors hinders the shRNA application in clinic. To explore vector-free RNAi using shRNA, Ni et al. explored a chimera linking RNA A10-3 aptamer (56 nucleotides) targeting PSMA to a shRNA (50 nucleotides) against DNA-activated protein kinase catalytic polypeptide (DNAPK), a target protein for radiosensitisation [148]. After incubating LNCaP cells with aptamer-shRNA chimera (400 nM) for 2 days, the chimera significantly reduced levels of BRCA2, DNAPK and ATM mRNA. The in vivo efficacy of such treatment was evaluated in a xenograft LNCaP mouse model in which the mice were treated with radiation plus two intratumoral injections of 200 pmol A10-3 aptamer-shRNA. The time for reaching quadruple tumor volume was dramatically extended from one week (as in mice treated with control) to ~10 weeks. This aptamer-guided delivery of shRNA resulted in an effective knockdown of DNAPK and significantly improved tumor response to ionizing radiation.
Aptamer-mediated co-delivery system
Compared to a single agent delivery system, co-delivery of more therapeutic agents, such as chemotherapeutic agents and small RNAs, represents a more powerful approach to cancer treatment [163-166] (Fig. 6). Such a combinatorial treatment strategy may 1) provide synergistic or addictive effects beyond one single drug therapy; 2) circumvent multiple drug resistance of cancer cells by targeted delivery of anticancer agents intracellularly; and 3) afford lower doses of individual drugs thus minimising systematic side effects of individual agents. A physical conjugate of DOX and A10 RNA aptamer was bridged to Dtxl-encapsulated PLGA-PEG NPs to generate a targeted co-delivery of both hydrophobic and hydrophilic drugs to tumors [106]. Both Dtxl and DOX were selectively delivered by LNCaP cells with high PSMA expression while limited DOX was internalised to the control PSMA-negative PC3 cells. In addition, anti-proliferation assays indicated that relative cell viability of LNCaP cells treated by NP (Dtxl)-Apt (DOX) was 54%, compared with that of 58 %, 86%, and 100%, respectively, for cells treated with NP (Dtxl)-Apt, NP-Apt (DOX), and NP-Apt. These results suggest that the co-delivery of DOX-loaded aptamer with Dtxl-encapsulated polymeric NPs exhibit more efficient killing of target cancer cells compared with the same amount of individual drugs.
Schematic illustration of co-delivery of therapeutic drugs and therapeutic small RNA using free aptamer or aptamer-conjugated nanoparticles. The choice of drugs and small RNA is based on rational design and subject to extensive in vivo testing. Therapeutic agents can be directly conjugated with aptamers without further encapsulated into a nanoparticle, while aptamers can either be grafted on to the surface of nanoparticles as a targeting agent, and/or conjugated with a drug.
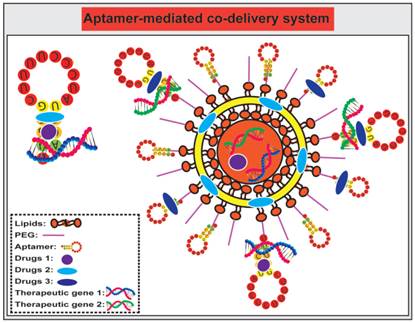
The combination of chemotherapeutic agents with small RNAs, i.e., siRNA, miRNA, anti-miRNA and shRNAs, has emerged as a potential strategy for achieving synergistic efficacies for targeted cancer therapy [167]. For example, a co-drug delivery system that combines PTX and the chimera comprising of MUC1 aptamer and let-7i miRNA specifically delivered let-7i miRNA and PTX to target ovarian cancer cells for circumventing PTX-induced chemoresistance [168]. Furthermore, Kim and colleagues synthesized PSMA aptamer-conjugated polyplexes (APs) that selectively targeted co-delivered shRNA against the anti-apoptotic gene Bcl-xL and DOX to prostate cancer cells by aptamer-mediated interaction [169]. This system efficiently activated intrinsic apoptosis of cancer cells and resulted in a 17-fold improvement in IC50 compared with the simple mixture of shRNA/Lipofectamine with DOX. These results suggest that aptamer-mediated co-delivery of anticancer drugs with shRNA could broaden the therapeutic window for selective eradication of cancer cells.
Current progress in aptamer-based diagnostics
Clinical diagnostic imaging needs to use specific probes, except for nonspecific contrast agents, to interrogate a biologic target. These specific imaging probes either target a physiologic process or cellular surface biomarkers in which the biologic targeting ligand requires high affinity, sensitivity, and specificity [170]. Aptamers possess several key advantages over proteins. The smaller size enables aptamers to achieve the enhanced tumor-to-blood and/or tumor-to-normal tissue ratio due to robust penetration into the core of the tumor, maintaining a significantly high concentration at the tumor for a clinically meaningful period and effective systemic clearance via renal filtration. Such superior signal-to-background ratio makes aptamers better imaging ligands than antibodies for effective clinical cancer diagnosis and tracking [91, 170-174]. Aptamers conjugated with diagnostic agents such as fluorescent molecules, magnetic resonance imaging (MRI) contrast enhancing agents and radioisotopes for positron emission tomography (PET) and single-photon emission computed tomography (SPECT) have been actively exploited for detecting both primary tumor and metastases (Fig. 7) [170, 175].
Application of aptamers in cancer diagnostics and therapeutics. (A) Schematic illustration of aptamer-based cancer diagnostic and therapeutic applications. (B) Representative results from the study carried out by Li et al. where microPET/CT images of 64Cu-DOTA-AS1411 or 64Cu-CB-TE2A-AS1411 at multiple time points shown that 64Cu-CB-TE2A-AS1411 was stable in tumor site and was cleared rapidly from the blood, liver, and kidneys 1 h post-injection, resulting in a much higher tumor-to-liver ratio (0.56) and the tumor-to-kidney ratio (1.57) than that of 0.12 and 0.21 for that for 64Cu-DOTA-AS1411, respectively. a) microPET image slices obtained at different time points for 64Cu-DOTA-AS1411; b) and c) Quantitative results from PET imaging for the liver, kidney, tumor, and muscle uptake of 64Cu-DOTA-AS1411 and 64Cu-CB-TE2A-AS1411; d) microPET image slices obtained at different time points for 64Cu-CB-TE2A-AS1411; e) Overview of microCT (left), microPET (middle), and microPET/CT fusion images (right) 1 h post-injection of 64Cu-CB-TE2A-AS1411. Reproduced from Ref. [180], Copyright (2014), with permission from Elsevier. (C) Representative results from the study carried out by Charlton and co-workers where in vivo imaging of inflammation sites in a rat reverse passive Arthus reaction model by (99mTc) aptamer NX21909 indicated that the aptamer achieved a significantly higher target-to-background ratio (4.3 ± 0.6 in 2 hours) in less time than IgG (3.1 ± 0.1 at 3 hours). Reproduced from Ref. [181], Copyright (1997), with permission from Elsevier.
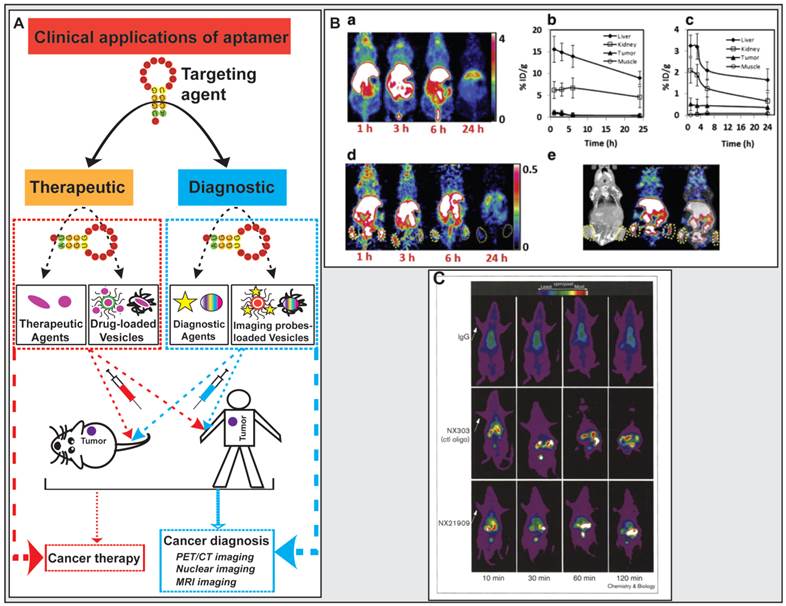
PET/CT imaging
Aptamer-based molecular probes have been used in PET/CT imaging. 64Cu represents an attractive radiotracer that has broadly applied for in vivo PET imaging due to its decay characteristics and easy attachment to ligands with high affinity [176, 177]. A chelator CB-TE2A, 4,11-bis(carboxymethyl)-1,4,8,11-tetraazabicyclo[6.6.2] hexadecane, has been known to display good kinetic stability, enhanced accumulation in tumor and reduced liver or kidney uptake, which would be a potentially effective chelator for aptamers to conjugate radiotracers for in vivo imaging [178, 179]. During their study of novel aptamer-mediated imaging agents for early detection of lung cancer, Li and colleagues labelled AS1411 with 64Cu using various chelators (DOTA and CB-TE2A), and assessed the in vivo uptake characteristics of the modified aptamer for microPET/CT imaging [180]. Using a mouse H460 tumor xenograft model, 7.4 MBq (200 μCi) of 64Cu-DOTA-AS1411 or 64Cu-CB-TE2A-AS1411 was administrated to mice via tail vein injection followed by microPET/CT imaging at multiple time points (Fig. 7). The microCT image confirmed the presence of tumors. After the injection of 64Cu-CB-TE2A-AS1411, two tumors were clearly detected by microPET imaging, with the highest tumor-to-background contrast reached at 1 h. In contrast, two tumors in mice injected with 64Cu-DOTA-AS1411 were not discernable due to the high liver uptake and slow blood clearance, which were confirmed by the time-activity curves of tumor to organ ratios (liver, kidney and muscle). 64Cu-CB-TE2A-AS1411 was stable in the tumor site but was cleared rapidly from blood, liver, and kidney 1 h post injection, resulting in a much higher tumor-to-liver ratio (0.56) and tumor-to-kidney ratio (1.57) than that of 0.12 and 0.21 for that for 64Cu-DOTA-AS1411, respectively (Fig. 7).
Single-photon emission computerized tomography (SPECT) imaging
One earlier successful aptamer-based imaging system was to use a radiolabeled (Technetium, 99mTc) aptamer NX21909 specifically targeting human neutrophil elastase in identifying inflammation sites in a rat model of reverse passive Arthus reaction [181]. After systemic administration, the aptamer achieved a significantly higher target-to-background ratio of 4.3 ± 0.6 in 2 hours than that of IgG, 3.1 ± 0.1 in 3 hours. The difference in tumor-over-background values is due to the rapid clearance of the aptamer signal from the blood than the IgG probe. These results provide a good example of great potential of aptamers in diagnostic imaging.
MRI imaging
In addition to their potential as excellent radio-tracers for nuclear medicine, aptamers also achieve a higher target-to-background ratio in a shorter period of time when conjugated with agents used in other imaging modalities, e.g. gadolinium-containing compounds and superparamagnetic iron oxide nanoparticles (SPIONs) [180, 182]. This is illustrated by aptamer-based contrast agents targeting vascular endothelial growth factor (VEGF) in magnetic resonance imaging (MRI) [182]. Due to its role in development of tumor angiogenesis and metastasis, VEGF165 is an ideal target for the generation of a tumor imaging probe [183]. Radioisotope-labelled antibodies as sensitive imaging agents, such as 124I-HuMV833 (humanized anti-VEGF-A mouse monoclonal antibody) and 89Zr-bevacizumab (VEGF recombinant humanized monoclonal antibody), have been used for PET imaging-based assessment of cancer treatment outcomes [184, 185]. However, the in vivo imaging application of antibodies has been limited by their large size, immunogenicity and relatively poor tumor penetration [186]. To address these limitations, You et al. developed a tumor targeted MRI probe by chemically crosslinking ultrasmall superparamagnetic iron oxide (USPIO) nanoparticles (~20 nm) with an aptamer specifically targeting VEGF165 (VEGF165-aptamer-USPIO). By examining T2-weighted imaging (T2WI) contrast enhancement, they verified the specific enhancement of MRI signal by VEGF165-aptamer-USPIO in mice bearing liver cancer xenografts [182]. Three hours after intravenous administration of VEGF165-aptamer-USPIO probe to mice, the signal-to-noise ratio of the tumor was statistically lower than that of pre-injection of the agent (P < 0.05), this negative enhancement phenomenon disappeared 6 h after the probe injection (Fig. 7C). In contrast, the signal-to-noise ratio of the tumor xenografts remained similar before and after the same amount of control USPIO was administrated. The results suggest that the VEGF165-aptamer-mediated USPIO probe can accumulate in the tumor, demonstrating promise for future in vivo diagnostic applications.
Aptamer-Guided Theranostic Nanomedicine
Theranostic refers to a strategy that integrates disease diagnosis and therapy within a single system. It is an interdisciplinary field involving chemistry, materials science, nanotechnology, molecular cell biology, pharmaceutical science, molecular imaging and medicine [187, 188]. The application of theranostics in oncology is anticipated to facilitate the selection of a safe and efficacious dose, recognition of toxic effects at the early stages of therapy, objective response monitoring and follow-up therapy planning [187, 189]. The simple form of theranostics consists of a single entity system in which a therapeutic agent is labelled with a radioisotope as exemplified by radio-labelled therapeutic monoclonal antibody, e.g. application of 111In-evacizumab and 124I-girentuximab in whole body biodistribution studies to quantify target accessibility [190]. However, recent work focus on a more sophisticated multi-component system in which targeting, imaging, diagnostic, and therapeutic functions are assembled in smartly engineered nanocarrier. Such theranostic nanoparticles capitalise on the advantages of nanoparticles, including high surface area, high payload capacity, prolonged circulation half-life and increased delivery to tumors [191, 192]. The commonly used tumor targeting agents are antibodies. However, the large size and other limitations associated with antibodies demand the development of alternative targeting moiety for theranostics. Aptamer-functionalised nanoparticles emerge as promising targeting agents for theranostic applications due to the sensitive tracking and imaging of aptamers in vivo and the ability to facilitate drug transport across physiological barriers [62, 193, 194].
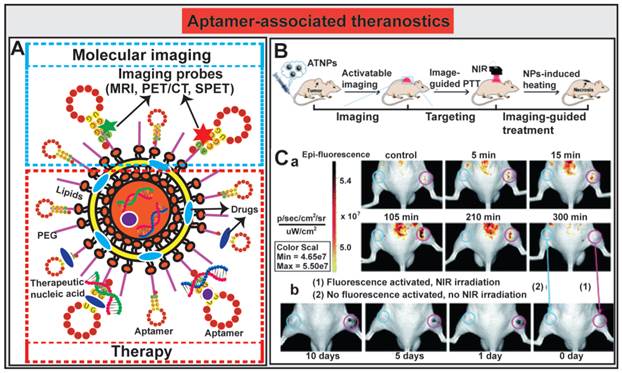
The innovative approaches to aptamer-guided theranostics are exemplified by an Au@Ag/Au nanoparticles assembled with activatable aptamer probes (AAPs) developed by Jia and co-workers [193]. The therapeutic arm of the system consists of a photothermal therapy (PTT) entity composed of Au@Ag/Au nanoparticles capable of large absorption in the near-infrared (NIR) window followed by efficacious hyperthermia; while the diagnostic arm of the system is made of AAPs that target human A549 lung cancer cells. The fluorescence of the labeled activatable aptamer is quenched in sites where there are no tumors by a fluorescence-labeled DNA (F-cDNA) co-grafted onto the same nanoparticles. Once the aptamer-Au@Ag/Au nanoparticle binds to tumor cells and interacts with the target protein on A549 cells, the quencher fluorophore on the suppressor F-cDNA moves away from the aptamer-Au@Ag/Au nanoparticle, leading to the activation of the fluorescence from the A549-targeting aptamer. The subsequent tumor-specific fluorescence imaging allows the precise positioning of the NIR light towards the tumor to facilitate in vivo guided photothermal therapy (Fig. 8B). The authors demonstrated the utility of this novel theranostic therapy in an A549 mouse xenograft model. The tumor bearing mice were administered i.v. with 300 μL of the aptamer-nanoparticles (1.1 × 1010 particles per mL). Mice without tumor or non-targeted tumor-bearing mice had no or minimal activated fluorescence from the aptamer, while prominent fluorescence signals accumulated rapidly in A549 tumor sites 5 min after administration and reached to the peak at around 15 min (Fig. 8C). The relative highest-contrast tumor imaging signals appeared at around 105 min. The subsequent efficacy of inducing tumor necrosis was achieved after exposing the tumor to a 980 nm laser light. Prominent necrosis was observed in A549 tumors treated with irradiation, and the tumor growth was inhibited at necrotic sites (Fig. 8C). In light of the broad application of Au@Ag/Au NPs and the short development circle for aptamers, aptamer-mediated imaging-guided PTT strategy might be explored to a flexible platform for real-time detection and treatment of a wide range of solid cancers. The incorporation of chemotherapy agents into this system will further facilitate the transformation of this platform to a synergic anticancer strategy by combination of aptamer-guided chemotherapy and PTT.
Aptamer-guided theranostic nanomedicine. (A) Schematic representation of aptamer-associated theranostic nanomedicine. (B) Schematic illustration of the novel activatable theranostic nanoprobe (ATNP) strategy for aptamer-mediated imaging-guided photothermal therapy (PTT) in vivo. (C) In vivo aptamer-related activatable fluorescence imaging (a) and imaging-guided site-specific NIR PTT (b) of SPCAi (cyan circles) and A549 (pink circles) tumor following ATNP administration. Reproduced from Ref. [193], Copyright (2014), with permission from The Royal Society of Chemistry.
Future perspectives
Tumor heterogeneity: another obstacle for anticancer treatment
In addition to killing cancer cells, current chemotherapy or radiotherapy can also damage normal cells. Such intensive therapies can also alter the cancer cell ecosystem in which variant or drug-resistant cancer cells emerge. The altered cancer ecosystem can be explained by the evolving cancer models. The conventional stochastic cancer model suggests that each cell within a tumor has equal potential to propagate and maintain a tumor but it is hard to prospectively identify the tumorigenic subset [195-197]. Recent studies suggest that cancer can be considered as a hierarchical organisation [198], which is the basis of the cancer stem cell (CSC) model. In the CSC model, only a small population of cancer cells possesses the ability to generate a tumor. Generally, CSCs, as 'roots of cancer', are defined by their abilities to form new tumors that histologically resembles the original tumor when xenotransplanted into immunodeficient mice. However, their descendant cells lose their self-renewal potential through either asymmetric or symmetric division and proliferate extensively [199-202]. The CSC model provides a plausible account for poorly understood clinical phenomena, such as drug resistance to chemical- and/or radiotherapy, as CSCs can resume growth and lead to a new relapse after primary therapy [198]. However, a new concept - the dynamic CSC model - has emerged to indicate that the CSC phenotype is much more fluid than previously predicted and can be regulated by external signals [203]. It suggests that not only can CSCs self-renewal create to differentiated cells, but the de-differentiation of non-CSCs to CSCs can also occur and thus revert to CSC [1, 204, 205]. The observation of inter-conversion between CSCs and their offspring suggests the necessity to evolve current anticancer strategies to target both types of cancer cells with self-renewal potential and the bulk differentiated bulk cancer cells [206].
Aptamer-mediated chemotherapy for eradicating CSCs
Based on current studies, CSCs are attractive targets for anticancer therapies. Limitations, such as only a small population of CSC, overlapping of CSC-correlated surface markers and signal transduction circuits (e.g. Wnt and Notch pathway) with normal stem cells, hamper the effective targeting of CSCs [207]. However, cell surface markers and/or signal transduction routes that underline tumor clonogenicity and tumor resistance could still provide promising therapeutic targets for developing effective anticancer drugs [208]. One of the challenges facing effective anti-CSC therapy is the elevated expression of drug efflux pumps and intracellular detoxification capacities [206, 209-214]. Active targeting of CSCs might hold the answer for circumventing chemoresistance [215]. Emerging data indicate that sensitizing CSCs to current chemotherapy using a CSC-targeting strategy can eliminate CSCs and minimize cancer recurrence [216]. For example, McAuliffe et al. have shown that targeting CSC-associated Notch pathway with a γ-secretase inhibitor and Notch3 siRNA knockdown can increase tumor sensitivity to platinum treatment in both cancer cell lines and patient-derived xenograft ovarian cancer models [217]. In addition, following the isolation of CSCs from non-small-cell lung carcinoma (NSCLC) sphere cells and blocking the stem cell factor (SCF) c-kit signalling that modulates self-renewal and proliferation of lung CSCs, the anti-SCF antibody conjugated cisplatin could target both non-CSCs and CSCs, enhancing the anti-tumor efficacy in human NSCLC [218]. Instead of developing new drugs, these studies explored a potential effective strategy that utilises a CSC-targeting moiety to deliver existing chemotherapeutic agents to eradicate both non-CSCs and CSCs. Most recently, RNA aptamers for targeting CSC markers (EpCAM and CD133) have been isolated [49, 60, 219]. Upon binding to cancer cells expressing these CSC markers, these CSC-targeting aptamers efficiently internalised via receptor-mediated endocytosis, an important mechanism capable of bypassing ATP-binding cassette transporters (ABC transporters) that are responsible for multiple drug resistance in CSC [60, 100]. Indeed, by conjugating a cytotoxic agent to CSC-targeting RNA aptamers, we have been able to transform a classic chemotherapy agent into an effective CSC killer, circumventing drug resistance in CSCs and eradicating both non-CSC and CSC in vitro and in vivo (our unpublished data).
Conclusions
Aptamers as chemical antibodies possess attractive physicochemical and cell biological attributes for clinical applications both in cancer diagnosis and targeted cancer therapy. In particular, aptamers have been exploited as active targeting moieties for targeted delivery of therapeutic agents for cancer therapy. Through either direct physical conjugation or chemical conjugation onto nanoparticles, aptamers can guide drugs to tumor sites efficiently with superior tumor penetration than antibodies. The co-delivery of classic anticancer reagents coupled with single or multiple small RNA therapeutics may offer more efficacious cancer treatment regimens than either of these anticancer tactics alone. Further engineering of aptamers with shorter sequences but extended blood residence time and superior tumor penetration will facilitate the development of aptamer-guided cancer therapeutics with significantly improved clinical outcome and better quality of life for patients with cancer.
Abbreviations
2'-OMe, 2'-O-methoxy; ABC transporter, ATP-binding cassette transporter; AG, arabinogalactan; Apt, aptamer; AAP, activatable aptamer probe; CEA, carcinoembryonic antigen; Control, Ctrl; CSC, cancer stem cell; CT, computed tomography; DNAPK, DNA-activated protein kinase catalytic polypeptide; DOX, doxorubicin; Dtxl, docetaxel; EGFR, epidermal growth factor receptor; EpCAM, epithelial cell adhesion molecule; Epi, Epirubicin; EPR, enhanced permeability and retention; F-cDNA, fluorescence-labeled DNA. IFP, interstitial fluid pressure; IL4Ra, IL-4 receptor-a; Lip, liposome; MDSC, Myeloid-derived suppressor cells; miRNA, micro RNA; MRI, magnetic resonance imaging; MSC, mesenchymal stem cell; MSN, Mesoporous silica nanoparticle; mTORC1, mTOR complex 1; NIR, near-infrared. NP, nanoparticle; NSCLC, non-small-cell lung carcinoma; PEG, Polyethylene glycol; PET, positron emission tomography; PLGA, Poly(Lactide-co-Glycolide); PSMA, prostate-specific membrane antigen; pSTAT6, phosphoSTAT6; PTX, Paclitaxel; PTT, photothermal therapy. RISC, RNA-induced silencing complex; RNAi, RNA interference; SELEX, systematic evolution of ligands by exponential enrichment; shRNA, short hairpin RNA; siRNA, small interfering RNA; SNR, signal-to-noise ratio; SPECT, single-photon emission computed tomography; SPION, superparamagnetic iron oxide nanoparticle; T2WI, T2-weighted imaging; TAM, tumor-associated macrophage; USPIO, ultrasmall superparamagnetic iron oxide; VEGF, vascular endothelial growth factor.
Acknowledgements
This work was supported by an Australian Research Council grant DP140100002 (G.Q. and W. Duan), grants from CASS Foundation grant and Australia-India Strategic Research Fund (W.D.) and a Deakin University Postgraduate Research Scholarship (D. X.).
Competing Interests
The authors have declared that no competing interest exists.
References
1. O'Connor ML, Xiang D, Shigdar S. et al. Cancer stem cells: A contentious hypothesis now moving forward. Cancer Lett. 2014;344:180-7
2. Easwaran H, Tsai HC, Baylin SB. Cancer epigenetics: tumor heterogeneity, plasticity of stem-like states, and drug resistance. Mol Cell. 2014;54:716-27
3. Shigdar S, Macdonald J, O'Connor M. et al. Aptamers as theranostic agents: modifications, serum stability and functionalisation. Sensors. 2013;13:13624-37
4. Chauhan VP, Stylianopoulos T, Boucher Y. et al. Delivery of molecular and nanoscale medicine to tumors: transport barriers and strategies. Annu Rev Chem Biomol Eng. 2011;2:281-98
5. Jain RK. Normalizing tumor microenvironment to treat cancer: bench to bedside to biomarkers. J Clin Oncol. 2013;31:2205-18
6. Straw JA, Hart MM, Klubes P. et al. Distribution of anticancer agents in spontaneous animal tumors. I. Regional blood flow and methotrexate distribution in canine lymphosarcoma. J Natl Cancer Inst. 1974;52:1327-31
7. Straw JA, Klubes P, Hart MM. Distribution of anticancer agents in spontaneous animal tumors. II. Distribution of gallium in canine lymphosarcoma. J Natl Cancer Inst. 1975;55:199-202
8. Stacker SA, Williams SP, Karnezis T. et al. Lymphangiogenesis and lymphatic vessel remodelling in cancer. Nat Rev Cancer. 2014;14:159-72
9. Fong EL, Santoro M, Farach-Carson MC. et al. Tissue Engineering Perfusable Cancer Models. Curr Opin Chem Eng. 2014;3:112-17
10. Correa de Sampaio P, Auslaender D, Krubasik D. et al. A heterogeneous in vitro three dimensional model of tumor-stroma interactions regulating sprouting angiogenesis. PLoS One. 2012;7:e30753
11. Ruddle NH. Lymphatic vessels and tertiary lymphoid organs. J Clin Invest. 2014;124:953-9
12. Karaman S, Detmar M. Mechanisms of lymphatic metastasis. J Clin Invest. 2014;124:922-8
13. Fang J, Nakamura H, Maeda H. The EPR effect: Unique features of tumor blood vessels for drug delivery, factors involved, and limitations and augmentation of the effect. Adv Drug Deliv Rev. 2011;63:136-51
14. Taurin S, Nehoff H, Greish K. Anticancer nanomedicine and tumor vascular permeability; Where is the missing link? J Control Release. 2012;164:265-75
15. Hagendoorn J, Tong R, Fukumura D. et al. Onset of abnormal blood and lymphatic vessel function and interstitial hypertension in early stages of carcinogenesis. Cancer Res. 2006;66:3360-4
16. Welter M, Rieger H. Interstitial fluid flow and drug delivery in vascularized tumors: a computational model. PLoS One. 2013;8:e70395
17. Heldin C-H, Rubin K, Pietras K. et al. High interstitial fluid pressure - an obstacle in cancer therapy. Nat Rev Cancer. 2004;4:806-13
18. Ariffin AB, Forde PF, Jahangeer S. et al. Releasing pressure in tumors: what do we know so far and where do we go from here? A review. Cancer Res. 2014;74:2655-62
19. Stylianopoulos T, Martin JD, Snuderl M. et al. Coevolution of solid stress and interstitial fluid pressure in tumors during progression: implications for vascular collapse. Cancer Res. 2013;73:3833-41
20. Heldin CH, Rubin K, Pietras K. et al. High interstitial fluid pressure - an obstacle in cancer therapy. Nat Rev Cancer. 2004;4:806-13
21. Jain RK, Martin JD, Stylianopoulos T. The role of mechanical forces in tumor growth and therapy. Annu Rev Biomed Eng. 2014;16:321-46
22. Nichols JW, Bae YH. EPR: Evidence and fallacy. J Control Release. 2014;190C:451-64
23. Fuso Nerini I, Morosi L, Zucchetti M. et al. Intratumor Heterogeneity and Its Impact on Drug Distribution and Sensitivity. Clin Pharmacol Ther. 2014;96:224-38
24. Liu C, Krishnan, Xu XY. Towards an integrated systems-based modelling framework for drug transport and its effect on tumor cells. J Biol Eng. 2014;8:3
25. Ramanujan S, Pluen A, McKee TD. et al. Diffusion and convection in collagen gels: implications for transport in the tumor interstitium. Biophys J. 2002;83:1650-60
26. Jang SH, Wientjes MG, Lu D. et al. Drug delivery and transport to solid tumors. Pharm Res. 2003;20:1337-50
27. Yang X, Liu X, Liu Z. et al. Near-infrared light-triggered, targeted drug delivery to cancer cells by aptamer gated nanovehicles. Adv Mater. 2012;24:2890-5
28. Matsuo M, Matsumoto S, Mitchell JB. et al. Magnetic Resonance Imaging of the Tumor Microenvironment in Radiotherapy: Perfusion, Hypoxia, and Metabolism. Semin Radiat Oncol. 2014;24:210-17
29. Kharaishvili G, Simkova D, Bouchalova K. et al. The role of cancer-associated fibroblasts, solid stress and other microenvironmental factors in tumor progression and therapy resistance. Cancer Cell Int. 2014;14:41
30. Sheth RA, Hesketh R, Kong DS. et al. Barriers to drug delivery in interventional oncology. J Vasc Interv Radiol. 2013;24:1201-7
31. Fan D, Creemers EE, Kassiri Z. Matrix as an interstitial transport system. Circ Res. 2014;114:889-902
32. Goertz DE, Yu JL, Kerbel RS. et al. High-frequency Doppler ultrasound monitors the effects of antivascular therapy on tumor blood flow. Cancer Res. 2002;62:6371-5
33. Senger DR, Galli SJ, Dvorak AM. et al. Tumor cells secrete a vascular permeability factor that promotes accumulation of ascites fluid. Science. 1983;219:983-5
34. Azzi S, Hebda JK, Gavard J. Vascular permeability and drug delivery in cancers. Front Oncol. 2013;3:211
35. Chen P, Bonaldo P. Role of macrophage polarization in tumor angiogenesis and vessel normalization: implications for new anticancer therapies. Int Rev Cell Mol Biol. 2013;301:1-35
36. He X, Li J, An S. et al. pH-sensitive drug-delivery systems for tumor targeting. Ther Deliv. 2013;4:1499-510
37. Riemann A, Schneider B, Gundel D. et al. Acidic priming enhances metastatic potential of cancer cells. Pflugers Arch. 2014 doi: 10.1007/s00424-014-1458-6. [Epub ahead of print]
38. Rotin D, Robinson B, Tannock IF. Influence of hypoxia and an acidic environment on the metabolism and viability of cultured cells: potential implications for cell death in tumors. Cancer Res. 1986;46:2821-6
39. Wike-Hooley JL, Haveman J, Reinhold HS. The relevance of tumor pH to the treatment of malignant disease. Radiother Oncol. 1984;2:343-66
40. Ellington AD, Szostak JW. In vitro selection of RNA molecules that bind specific ligands. Nature. 1990;346:818-22
41. Shigdar S, Qian C, Lv L. et al. The use of sensitive chemical antibodies for diagnosis: detection of low levels of epcam in breast cancer. PLoS One. 2013;8:e57613
42. Cerchia L, de Franciscis V. Targeting cancer cells with nucleic acid aptamers. Trends Biotechnol. 2010;28:517-25
43. Keefe AD, Szostak JW. Functional proteins from a random-sequence library. Nature. 2001;410:715-8
44. Mi J, Liu Y, Rabbani ZN. et al. In vivo selection of tumor-targeting RNA motifs. Nat Chem Biol. 2010;6:22-4
45. Cheng C, Chen YH, Lennox KA. et al. In vivo SELEX for Identification of Brain-penetrating Aptamers. Mol Ther Nucleic Acids. 2013;2:e67
46. Colas P. Combinatorial protein reagents to manipulate protein function. Curr Opin Chem Biol. 2000;4:54-9
47. Doggrell SA. Pegaptanib: the first antiangiogenic agent approved for neovascular macular degeneration. Expert Opin Pharmacother. 2005;6:1421-3
48. Hicke BJ, Stephens AW. Escort aptamers: a delivery service for diagnosis and therapy. J Clin Invest. 2000;106:923-8
49. Shigdar S, Lin J, Yu Y. et al. RNA aptamer against a cancer stem cell marker epithelial cell adhesion molecule. Cancer Sci. 2011;102:991-8
50. Proske D, Blank M, Buhmann R. et al. Aptamers--basic research, drug development, and clinical applications. Appl Microbiol Biotechnol. 2005;69:367-74
51. Bruno JG. A review of therapeutic aptamer conjugates with emphasis on new approaches. Pharmaceuticals (Basel). 2013;6:340-57
52. Meng L, Yang L, Zhao X. et al. Targeted delivery of chemotherapy agents using a liver cancer-specific aptamer. PLoS One. 2012;7:e33434
53. Hedden L, O'Reilly S, Lohrisch C. et al. Assessing the real-world cost-effectiveness of adjuvant trastuzumab in HER-2/neu positive breast cancer. Oncologist. 2012;17:164-71
54. Jeyakumar A, Younis T. Trastuzumab for HER2-Positive Metastatic Breast Cancer: Clinical and Economic Considerations. Clin Med Insights Oncol. 2012;6:179-87
55. Phillips JA, Lopez-Colon D, Zhu Z. et al. Applications of aptamers in cancer cell biology. Anal Chim Acta. 2008;621:101-8
56. Jayasena SD. Aptamers: an emerging class of molecules that rival antibodies in diagnostics. Clin Chem. 1999;45:1628-50
57. Harding FA, Stickler MM, Razo J. et al. The immunogenicity of humanized and fully human antibodies: residual immunogenicity resides in the CDR regions. MAbs. 2010;2:256-65
58. White RR, Sullenger BA, Rusconi CP. Developing aptamers into therapeutics. J Clin Invest. 2000;106:929-34
59. Drolet DW, Nelson J, Tucker CE. et al. Pharmacokinetics and safety of an anti-vascular endothelial growth factor aptamer (NX1838) following injection into the vitreous humor of rhesus monkeys. Pharm Res. 2000;17:1503-10
60. Shigdar S, Qiao L, Zhou S-F. et al. RNA aptamers targeting cancer stem cell marker CD133. Cancer Lett. 2013;330:84-95
61. Shigdar S, Ward AC, De A. et al. Clinical applications of aptamers and nucleic acid therapeutics in haematological malignancies. Br J Haematol. 2011;155:3-13
62. Shigdar S, Macdonald J, O'Connor M. et al. Aptamers as theranostic agents: modifications, serum stability and functionalisation. Sensors (Basel). 2013;13:13624-37
63. Keefe AD, Pai S, Ellington A. Aptamers as therapeutics. Nat Rev Drug Discov. 2010;9:537-50
64. Keefe AD, Cload ST. SELEX with modified nucleotides. Curr Opin Chem Biol. 2008;12:448-56
65. Davis ME, Chen ZG, Shin DM. Nanoparticle therapeutics: an emerging treatment modality for cancer. Nat Rev Drug Discov. 2008;7:771-82
66. Prabhakar U, Maeda H, Jain RK. et al. Challenges and key considerations of the enhanced permeability and retention effect for nanomedicine drug delivery in oncology. Cancer Res. 2013;73:2412-7
67. Zhong Y, Meng F, Deng C. et al. Ligand-Directed Active Tumor-Targeting Polymeric Nanoparticles for Cancer Chemotherapy. Biomacromolecules. 2014
68. Hollis CP, Weiss HL, Leggas M. et al. Biodistribution and bioimaging studies of hybrid paclitaxel nanocrystals: lessons learned of the EPR effect and image-guided drug delivery. J Control Release. 2013;172:12-21
69. Petros RA, DeSimone JM. Strategies in the design of nanoparticles for therapeutic applications. Nat Rev Drug Discov. 2010;9:615-27
70. Bertrand N, Wu J, Xu X. et al. Cancer nanotechnology: the impact of passive and active targeting in the era of modern cancer biology. Adv Drug Deliv Rev. 2014;66:2-25
71. Allen TM. Ligand-targeted therapeutics in anticancer therapy. Nat Rev Cancer. 2002;2:750-63
72. Modery-Pawlowski CL, Gupta AS. Heteromultivalent ligand-decoration for actively targeted nanomedicine. Biomaterials. 2014;35:2568-79
73. Hirsjarvi S, Passirani C, Benoit J-P. Passive and active tumor targeting with nanocarriers. Curr Drug Discov Technol. 2011;8:188-96
74. Danhier F, Feron O, Preat V. To exploit the tumor microenvironment: Passive and active tumor targeting of nanocarriers for anti-cancer drug delivery. J Control Release. 2010;148:135-46
75. Patriarca C, Macchi RM, Marschner AK. et al. Epithelial cell adhesion molecule expression (CD326) in cancer: a short review. Cancer Treat Rev. 2012;38:68-75
76. Schrama D, Reisfeld RA, Becker JC. Antibody targeted drugs as cancer therapeutics. Nat Rev Drug Discov. 2006;5:147-59
77. Chari RVJ, Miller ML, Widdison WC. Antibody-drug conjugates: an emerging concept in cancer therapy. Angew Chem Int Ed Engl. 2014;53:3796-827
78. Bouchard PR, Hutabarat RM, Thompson KM. Discovery and development of therapeutic aptamers. Annu Rev Pharmacol Toxicol. 2010;50:237-57
79. Dassie JP, Liu XY, Thomas GS. et al. Systemic administration of optimized aptamer-siRNA chimeras promotes regression of PSMA-expressing tumors. Nat Biotechnol. 2009;27:839-49
80. Radom F, Jurek PM, Mazurek MP. et al. Aptamers: molecules of great potential. Biotechnol Adv. 2013;31:1260-74
81. Panowksi S, Bhakta S, Raab H. et al. Site-specific antibody drug conjugates for cancer therapy. MAbs. 2014;6:34-45
82. Zamay TN, Kolovskaya OS, Glazyrin YE. et al. DNA-aptamer targeting vimentin for tumor therapy in vivo. Nucleic Acid Ther. 2014;24:160-70
83. Lehr C-M, Gabor F. Lectins and glycoconjugates in drug delivery and targeting. Adv Drug Deliv Rev. 2004;56:419-20
84. Prescott JH, Enriquez P, Jung C. et al. Larch arabinogalactan for hepatic drug delivery: isolation and characterization of a 9 kDa arabinogalactan fragment. Carbohydr Res. 1995;278:113-28
85. Roth F, De La Fuente AC, Vella JL. et al. Aptamer-mediated blockade of IL4Ralpha triggers apoptosis of MDSCs and limits tumor progression. Cancer Res. 2012;72:1373-83
86. Yang L, DeBusk LM, Fukuda K. et al. Expansion of myeloid immune suppressor Gr+CD11b+ cells in tumor-bearing host directly promotes tumor angiogenesis. Cancer Cell. 2004;6:409-21
87. van Kempen LC, Coussens LM. MMP9 potentiates pulmonary metastasis formation. Cancer Cell. 2002;2:251-2
88. Mern DS, Hasskarl J, Burwinkel B. Inhibition of Id proteins by a peptide aptamer induces cell-cycle arrest and apoptosis in ovarian cancer cells. Br J Cancer. 2010;103:1237-44
89. Mern DS, Hoppe-Seyler K, Hoppe-Seyler F. et al. Targeting Id1 and Id3 by a specific peptide aptamer induces E-box promoter activity, cell cycle arrest, and apoptosis in breast cancer cells. Breast Cancer Res Treat. 2010;124:623-33
90. Zhang K, Sefah K, Tang L. et al. A novel aptamer developed for breast cancer cell internalization. ChemMedChem. 2012;7:79-84
91. Kaur H, Li JJ, Bay B-H. et al. Investigating the antiproliferative activity of high affinity DNA aptamer on cancer cells. PLoS One. 2013;8:e50964
92. Ojima A, Matsui T, Maeda S. et al. DNA aptamer raised against advanced glycation end products inhibits melanoma growth in nude mice. Lab Invest. 2014;94:422-9
93. Lee YJ, Han SR, Kim NY. et al. An RNA aptamer that binds carcinoembryonic antigen inhibits hepatic metastasis of colon cancer cells in mice. Gastroenterology. 2012;143:155-65.e8
94. Huang Y-F, Sefah K, Bamrungsap S. et al. Selective photothermal therapy for mixed cancer cells using aptamer-conjugated nanorods. Langmuir. 2008;24:11860-5
95. Tacar O, Sriamornsak P, Dass CR. Doxorubicin: an update on anticancer molecular action, toxicity and novel drug delivery systems. J Pharm Pharmacol. 2013;65:157-70
96. Agudelo D, Bourassa P, Berube G. et al. Intercalation of antitumor drug doxorubicin and its analogue by DNA duplex: Structural features and biological implications. Int J Biol Macromol. 2014;66:144-50
97. Minotti G, Menna P, Salvatorelli E. et al. Anthracyclines: molecular advances and pharmacologic developments in antitumor activity and cardiotoxicity. Pharmacol Rev. 2004;56:185-229
98. Sussman D, Nix JC, Wilson C. The structural basis for molecular recognition by the vitamin B 12 RNA aptamer. Nat Struct Biol. 2000;7:53-7
99. Zhu G, Meng L, Ye M. et al. Self-assembled aptamer-based drug carriers for bispecific cytotoxicity to cancer cells. Chem Asian J. 2012;7:1630-6
100. Huang YF, Shangguan D, Liu H. et al. Molecular assembly of an aptamer-drug conjugate for targeted drug delivery to tumor cells. Chembiochem. 2009;10:862-8
101. Hu Y, Duan J, Zhan Q. et al. Novel MUC1 aptamer selectively delivers cytotoxic agent to cancer cells in vitro. PLoS One. 2012;7:e31970
102. Liu Z, Duan JH, Song YM. et al. Novel HER2 aptamer selectively delivers cytotoxic drug to HER2-positive breast cancer cells in vitro. J Transl Med. 2012;10:148
103. Subramanian N, Raghunathan V, Kanwar JR. et al. Target-specific delivery of doxorubicin to retinoblastoma using epithelial cell adhesion molecule aptamer. Mol Vis. 2012;18:2783-95
104. Ray P, Cheek MA, Sharaf ML. et al. Aptamer-mediated delivery of chemotherapy to pancreatic cancer cells. Nucleic Acid Ther. 2012;22:295-305
105. Orilall MC, Wiesner U. Block copolymer based composition and morphology control in nanostructured hybrid materials for energy conversion and storage: solar cells, batteries, and fuel cells. Chem Soc Rev. 2011;40:520-35
106. Zhang L, Radovic-Moreno AF, Alexis F. et al. Co-delivery of hydrophobic and hydrophilic drugs from nanoparticle-aptamer bioconjugates. ChemMedChem. 2007;2:1268-71
107. Dhar S, Gu FX, Langer R. et al. Targeted delivery of cisplatin to prostate cancer cells by aptamer functionalized Pt(IV) prodrug-PLGA-PEG nanoparticles. Proc Natl Acad Sci U S A. 2008;105:17356-61
108. Xiao Z, Farokhzad OC. Aptamer-functionalized nanoparticles for medical applications: challenges and opportunities. ACS Nano. 2012;6:3670-6
109. Farokhzad OC, Karp JM, Langer R. Nanoparticle-aptamer bioconjugates for cancer targeting. Expert Opin Drug Deliv. 2006;3:311-24
110. Chan JM, Zhang L, Yuet KP. et al. PLGA-lecithin-PEG core-shell nanoparticles for controlled drug delivery. Biomaterials. 2009;30:1627-34
111. Zheng Y, Yu B, Weecharangsan W. et al. Transferrin-conjugated lipid-coated PLGA nanoparticles for targeted delivery of aromatase inhibitor 7alpha-APTADD to breast cancer cells. Int J Pharm. 2010;390:234-41
112. Farokhzad OC, Jon S, Khademhosseini A. et al. Nanoparticle-aptamer bioconjugates: a new approach for targeting prostate cancer cells. Cancer Res. 2004;64:7668-72
113. Farokhzad OC, Cheng J, Teply BA. et al. Targeted nanoparticle-aptamer bioconjugates for cancer chemotherapy in vivo. Proc Natl Acad Sci U S A. 2006;103:6315-20
114. Vanhoefer U, Cao S, Harstrick A. et al. Comparative antitumor efficacy of docetaxel and paclitaxel in nude mice bearing human tumor xenografts that overexpress the multidrug resistance protein (MRP). Ann Oncol. 1997;8:1221-8
115. Bates PJ, Laber DA, Miller DM. et al. Discovery and development of the G-rich oligonucleotide AS1411 as a novel treatment for cancer. Exp Mol Pathol. 2009;86:151-64
116. Guo J, Gao X, Su L. et al. Aptamer-functionalized PEG-PLGA nanoparticles for enhanced anti-glioma drug delivery. Biomaterials. 2011;32:8010-20
117. Dicheva BM, Koning GA. Targeted thermosensitive liposomes: an attractive novel approach for increased drug delivery to solid tumors. Expert Opin Drug Deliv. 2014;11:83-100
118. Deshpande PP, Biswas S, Torchilin VP. Current trends in the use of liposomes for tumor targeting. Nanomedicine (Lond). 2013;8:1509-28
119. Yang T, Cui F-D, Choi M-K. et al. Enhanced solubility and stability of PEGylated liposomal paclitaxel: in vitro and in vivo evaluation. Int J Pharm. 2007;338:317-26
120. Er Y, Barnes TJ, Fornasiero D. et al. The encapsulation and release of guanosine from PEGylated liposomes. J Liposome Res. 2009;19:29-36
121. Malam Y, Loizidou M, Seifalian AM. Liposomes and nanoparticles: nanosized vehicles for drug delivery in cancer. Trends Pharmacol Sci. 2009;30:592-9
122. Kang H, O'Donoghue MB, Liu H. et al. A liposome-based nanostructure for aptamer directed delivery. Chem Commun (Camb). 2010;46:249-51
123. Xing H, Tang L, Yang X. et al. Selective Delivery of an Anticancer Drug with Aptamer-Functionalized Liposomes to Breast Cancer Cells and in vitro and in vivo. J Mater Chem B Mater Biol Med. 2013;1:5288-97
124. Wang YX, Hussain SM, Krestin GP. Superparamagnetic iron oxide contrast agents: physicochemical characteristics and applications in MR imaging. Eur Radiol. 2001;11:2319-31
125. Bourrinet P, Bengele HH, Bonnemain B. et al. Preclinical safety and pharmacokinetic profile of ferumoxtran-10, an ultrasmall superparamagnetic iron oxide magnetic resonance contrast agent. Invest Radiol. 2006;41:313-24
126. Wang AZ, Bagalkot V, Vasilliou CC. et al. Superparamagnetic iron oxide nanoparticle-aptamer bioconjugates for combined prostate cancer imaging and therapy. ChemMedChem. 2008;3:1311-5
127. Jalalian SH, Taghdisi SM, Shahidi Hamedani N. et al. Epirubicin loaded super paramagnetic iron oxide nanoparticle-aptamer bioconjugate for combined colon cancer therapy and imaging in vivo. Eur J Pharm Sci. 2013;50:191-7
128. Meng L, Zhang X, Lu Q. et al. Single walled carbon nanotubes as drug delivery vehicles: targeting doxorubicin to tumors. Biomaterials. 2012;33:1689-98
129. Wang G, Uludag H. Recent developments in nanoparticle-based drug delivery and targeting systems with emphasis on protein-based nanoparticles. Expert Opin Drug Deliv. 2008;5:499-515
130. Lee IH, An S, Yu MK. et al. Targeted chemoimmunotherapy using drug-loaded aptamer-dendrimer bioconjugates. J Control Release. 2011;155:435-41
131. Li Z, Liu Z, Yin M. et al. Aptamer-capped multifunctional mesoporous strontium hydroxyapatite nanovehicle for cancer-cell-responsive drug delivery and imaging. Biomacromolecules. 2012;13:4257-63
132. Sa LTM, Simmons S, Missailidis S. et al. Aptamer-based nanoparticles for cancer targeting. J Drug Target. 2013;21:427-34
133. Xiao Z, Levy-Nissenbaum E, Alexis F. et al. Engineering of targeted nanoparticles for cancer therapy using internalizing aptamers isolated by cell-uptake selection. ACS Nano. 2012;6:696-704
134. Hao Z, Fan W, Hao J. et al. Efficient delivery of micro RNA to bone-metastatic prostate tumors by using aptamer-conjugated atelocollagen in vitro and in vivo. Drug Deliv. 2014:1-8
135. Aravind A, Jeyamohan P, Nair R. et al. AS1411 aptamer tagged PLGA-lecithin-PEG nanoparticles for tumor cell targeting and drug delivery. Biotechnol Bioeng. 2012;109:2920-31
136. Aravind A, Nair R, Raveendran S. et al. Aptamer conjugated paclitaxel and magnetic fluid loaded fluorescently tagged PLGA nanoparticles for targeted cancer therapy. J Magn Magn Mater. 2013;344:116-23
137. Sivakumar B, Aswathy RG, Nagaoka Y. et al. Aptamer conjugated theragnostic multifunctional magnetic nanoparticles as a nanoplatform for pancreatic cancer therapy. RSC Adv. 2013;3:20579-98
138. Zhang B, Luo Z, Liu J. et al. Cytochrome c end-capped mesoporous silica nanoparticles as redox-responsive drug delivery vehicles for liver tumor-targeted triplex therapy in vitro and in vivo. J Control Release. 2014;192C:192-201
139. Zhang H, Hou L, Jiao X. et al. In vitro and in vivo evaluation of antitumor drug -loaded aptamer targeted single-walled carbon nanotubes system. Curr Pharm Biotechnol. 2014 doi: 10.2174/1389201015666140408123710. [Epub ahead of print]
140. Yang X, Liu X, Liu Z. et al. Near-infrared light-triggered, targeted drug delivery to cancer cells by aptamer gated nanovehicles. Adv Mater. 2012;24:2890-5
141. Shiao YS, Chiu HH, Wu PH. et al. Aptamer-Functionalized Gold Nanoparticles As Photoresponsive Nanoplatform for Co-Drug Delivery. ACS Appl Mater Interfaces. 2014: doi:10.1021/am5026243. [Epub ahead of print].
142. Yu C, Hu Y, Duan J. et al. Novel aptamer-nanoparticle bioconjugates enhances delivery of anticancer drug to MUC1-positive cancer cells in vitro. PLoS One. 2011;6:e24077
143. Wu C, Han D, Chen T. et al. Building a multifunctional aptamer-based DNA nanoassembly for targeted cancer therapy. J Am Chem Soc. 2013;135:18644-50
144. Zhu G, Zheng J, Song E. et al. Self-assembled, aptamer-tethered DNA nanotrains for targeted transport of molecular drugs in cancer theranostics. Proc Natl Acad Sci U S A. 2013;110:7998-8003
145. Zhao Z, Xu L, Shi X. et al. Recognition of subtype non-small cell lung cancer by DNA aptamers selected from living cells. Analyst. 2009;134:1808-14
146. Thiel KW, Giangrande PH. Intracellular delivery of RNA-based therapeutics using aptamers. Ther Deliv. 2010;1:849-61
147. Cerchia L, Esposito CL, Camorani S. et al. Coupling Aptamers to Short Interfering RNAs as Therapeutics. Pharmaceuticals. 2011;4:1434-49
148. Ni X, Zhang Y, Ribas J. et al. Prostate-targeted radiosensitization via aptamer-shRNA chimeras in human tumor xenografts. J Clin Invest. 2011;121:2383-90
149. Chu TC, Marks JW 3rd, Lavery LA. et al. Aptamer:toxin conjugates that specifically target prostate tumor cells. Cancer Res. 2006;66:5989-92
150. McNamara JO 2nd, Andrechek ER, Wang Y. et al. Cell type-specific delivery of siRNAs with aptamer-siRNA chimeras. Nat Biotechnol. 2006;24:1005-15
151. Bumcrot D, Manoharan M, Koteliansky V. et al. RNAi therapeutics: a potential new class of pharmaceutical drugs. Nat Chem Biol. 2006;2:711-9
152. Zhou J, Bobbin ML, Burnett JC. et al. Current progress of RNA aptamer-based therapeutics. Front Genet. 2012;3:234
153. Berezhnoy A, Castro I, Levay A. et al. Aptamer-targeted inhibition of mTOR in T cells enhances antitumor immunity. J Clin Invest. 2014;124:188-97
154. Lai W-Y, Wang W-Y, Chang Y-C. et al. Synergistic inhibition of lung cancer cell invasion, tumor growth and angiogenesis using aptamer-siRNA chimeras. Biomaterials. 2014;35:2905-14
155. Iorio MV, Croce CM. MicroRNAs in cancer: small molecules with a huge impact. J Clin Oncol. 2009;27:5848-56
156. Zamore PD, Haley B. Ribo-gnome: the big world of small RNAs. Science. 2005;309:1519-24
157. Iorio MV, Croce CM. MicroRNA dysregulation in cancer: diagnostics, monitoring and therapeutics. A comprehensive review. EMBO Mol Med. 2012;4:143-59
158. Garofalo M, Condorelli GL, Croce CM. et al. MicroRNAs as regulators of death receptors signaling. Cell Death Differ. 2010;17:200-8
159. Esposito CL, Cerchia L, Catuogno S. et al. Multifunctional Aptamer-miRNA Conjugates for Targeted Cancer Therapy. Mol Ther. 2014;22:1151-63
160. Yu J-Y, DeRuiter SL, Turner DL. RNA interference by expression of short-interfering RNAs and hairpin RNAs in mammalian cells. Proc Natl Acad Sci U S A. 2002;99:6047-52
161. Wang Z, Rao DD, Senzer N. et al. RNA interference and cancer therapy. Pharm Res. 2011;28:2983-95
162. Rao DD, Vorhies JS, Senzer N. et al. siRNA vs. shRNA: similarities and differences. Adv Drug Deliv Rev. 2009;61:746-59
163. Frei E 3rd. Combination cancer therapy: Presidential address. Cancer Res. 1972;32:2593-607
164. Walsh C. Molecular mechanisms that confer antibacterial drug resistance. Nature. 2000;406:775-81
165. Hongrapipat J, Kopeckova P, Liu J. et al. Combination chemotherapy and photodynamic therapy with fab' fragment targeted HPMA copolymer conjugates in human ovarian carcinoma cells. Mol Pharm. 2008;5:696-709
166. Wiradharma N, Tong YW, Yang Y-Y. Self-assembled oligopeptide nanostructures for co-delivery of drug and gene with synergistic therapeutic effect. Biomaterials. 2009;30:3100-9
167. Yadav S, van Vlerken LE, Little SR. et al. Evaluations of combination MDR-1 gene silencing and paclitaxel administration in biodegradable polymeric nanoparticle formulations to overcome multidrug resistance in cancer cells. Cancer Chemother Pharmacol. 2009;63:711-22
168. Liu N, Zhou C, Zhao J. et al. Reversal of paclitaxel resistance in epithelial ovarian carcinoma cells by a MUC1 aptamer-let-7i chimera. Cancer Invest. 2012;30:577-82
169. Kim E, Jung Y, Choi H. et al. Prostate cancer cell death produced by the co-delivery of Bcl-xL shRNA and doxorubicin using an aptamer-conjugated polyplex. Biomaterials. 2010;31:4592-9
170. Wang AZ, Farokhzad OC. Current progress of aptamer-based molecular imaging. J Nucl Med. 2014;55:353-6
171. Mishra G, Giri D, Li MS. et al. Role of loop entropy in the force induced melting of DNA hairpin. J Chem Phys. 2011;135:035102
172. Turney K, Drake TJ, Smith JE. et al. Functionalized nanoparticles for liquid atmospheric pressure matrix-assisted laser desorption/ionization peptide analysis. Rapid Commun Mass Spectrom. 2004;18:2367-74
173. Fang X, Sen A, Vicens M. et al. Synthetic DNA aptamers to detect protein molecular variants in a high-throughput fluorescence quenching assay. Chembiochem. 2003;4:829-34
174. Scaglioni PP, Pandolfi PP. Medicine: taking apart a cancer protein. Nature. 2003;426:512-3
175. Khan H, Missailidis S. Aptamers in oncology: a diagnostic perspective. Gene Ther Mol Bio. 2008;12:111-28
176. Wadas TJ, Wong EH, Weisman GR. et al. Copper chelation chemistry and its role in copper radiopharmaceuticals. Curr Pharm Des. 2007;13:3-16
177. Li WP, Lewis JS, Kim J. et al. DOTA-D-Tyr(1)-octreotate: a somatostatin analogue for labeling with metal and halogen radionuclides for cancer imaging and therapy. Bioconjug Chem. 2002;13:721-8
178. Boswell CA, Sun X, Niu W. et al. Comparative in vivo stability of copper-64-labeled cross-bridged and conventional tetraazamacrocyclic complexes. J Med Chem. 2004;47:1465-74
179. Wu Y, Zhang X, Xiong Z. et al. microPET imaging of glioma integrin {alpha}v{beta}3 expression using (64)Cu-labeled tetrameric RGD peptide. J Nucl Med. 2005;46:1707-18
180. Li J, Zheng H, Bates PJ. et al. Aptamer imaging with Cu-64 labeled AS1411: preliminary assessment in lung cancer. Nucl Med Biol. 2014;41:179-85
181. Charlton J, Sennello J, Smith D. In vivo imaging of inflammation using an aptamer inhibitor of human neutrophil elastase. Chem Biol. 1997;4:809-16
182. You XG, Tu R, Peng ML. et al. Molecular magnetic resonance probe targeting VEGF165: preparation and in vitro and in vivo evaluation. Contrast Media Mol Imaging. 2014 doi: 10.1002/cmmi.1584. [Epub ahead of print]
183. Sauter ER, Nesbit M, Watson JC. et al. Vascular endothelial growth factor is a marker of tumor invasion and metastasis in squamous cell carcinomas of the head and neck. Clin Cancer Res. 1999;5:775-82
184. Jayson GC, Zweit J, Jackson A. et al. Molecular imaging and biological evaluation of HuMV833 anti-VEGF antibody: implications for trial design of antiangiogenic antibodies. J Natl Cancer Inst. 2002;94:1484-93
185. Nagengast WB, de Korte MA, Oude Munnink TH. et al. 89Zr-bevacizumab PET of early antiangiogenic tumor response to treatment with HSP90 inhibitor NVP-AUY922. J Nucl Med. 2010;51:761-7
186. Anderson CR, Rychak JJ, Backer M. et al. scVEGF microbubble ultrasound contrast agents: a novel probe for ultrasound molecular imaging of tumor angiogenesis. Invest Radiol. 2010;45:579-85
187. Chen XS. Introducing Theranostics Journal - From the Editor-in-Chief. Theranostics. 2011;1:1-2
188. Casciaro S. Theranostic applications: Non-ionizing cellular and molecular imaging through innovative nanosystems for early diagnosis and therapy. World J Radiol. 2011;3:249-55
189. Palekar-Shanbhag P, Jog SV, Chogale MM. et al. Theranostics for cancer therapy. Curr Drug Deliv. 2013;10:357-62
190. Fleuren ED, Versleijen-Jonkers YM, Heskamp S. et al. Theranostic applications of antibodies in oncology. Mol Oncol. 2014;8:799-812
191. Chen F, Hong H, Zhang Y. et al. In vivo tumor targeting and image-guided drug delivery with antibody-conjugated, radiolabeled mesoporous silica nanoparticles. ACS Nano. 2013;7:9027-39
192. Wang J, Tian S, Petros RA. et al. The complex role of multivalency in nanoparticles targeting the transferrin receptor for cancer therapies. J Am Chem Soc. 2010;132:11306-13
193. Shi H, Ye X, He X. et al. Au@Ag/Au nanoparticles assembled with activatable aptamer probes as smart "nano-doctors" for image-guided cancer thermotherapy. Nanoscale. 2014;6:8754-61
194. Fan Z, Senapati D, Singh AK. et al. Theranostic magnetic core-plasmonic shell star shape nanoparticle for the isolation of targeted rare tumor cells from whole blood, fluorescence imaging, and photothermal destruction of cancer. Mol Pharm. 2013;10:857-66
195. Nowell PC. The clonal evolution of tumor cell populations. Science. 1976;194:23-8
196. Visvader JE, Lindeman GJ. Cancer stem cells in solid tumors: accumulating evidence and unresolved questions. Nat Rev Cancer. 2008;8:755-68
197. Nguyen-Khac F, Lesty C, Eclache V. et al. Chromosomal abnormalities in transformed Ph-negative myeloproliferative neoplasms are associated to the transformation subtype and independent of JAK2 and the TET2 mutations. Genes Chromosomes Cancer. 2010;49:919-27
198. Shackleton M, Quintana E, Fearon ER. et al. Heterogeneity in cancer: cancer stem cells versus clonal evolution. Cell. 2009;138:822-9
199. Lee HE, Kim JH, Kim YJ. et al. An increase in cancer stem cell population after primary systemic therapy is a poor prognostic factor in breast cancer. Br J Cancer. 2011;104:1730-8
200. Beck B, Driessens G, Goossens S. et al. A vascular niche and a VEGF-Nrp1 loop regulate the initiation and stemness of skin tumors. Nature. 2011;478:399-403
201. Nguyen LV, Vanner R, Dirks P. et al. Cancer stem cells: an evolving concept. Nat Rev Cancer. 2012;12:133-43
202. Jiang W, Peng J, Zhang Y. et al. The Implications of Cancer Stem Cells for Cancer Therapy. Int J Mol Sci. 2012;13:16636-57
203. Vermeulen L, de Sousa e Melo F, Richel DJ. et al. The developing cancer stem-cell model: clinical challenges and opportunities. Lancet Oncol. 2012;13:e83-9
204. Chaffer CL, Brueckmann I, Scheel C. et al. Normal and neoplastic nonstem cells can spontaneously convert to a stem-like state. Proc Natl Acad Sci U S A. 2011;108:7950-5
205. Scheel C, Eaton EN, Li SH-J. et al. Paracrine and autocrine signals induce and maintain mesenchymal and stem cell states in the breast. Cell. 2011;145:926-40
206. Vermeulen L, de Sousa e Melo F, Richel DJ. et al. The developing cancer stem-cell model: clinical challenges and opportunities. Lancet Oncol. 2012;13:e83-e89
207. Yang YM, Chang JW. Current status and issues in cancer stem cell study. Cancer Invest. 2008;26:741-55
208. de Sousa EM, Vermeulen L, Richel D. et al. Targeting Wnt signaling in colon cancer stem cells. Clin Cancer Res. 2011;17:647-53
209. Aulmann S, Waldburger N, Penzel R. et al. Reduction of CD44(+)/CD24(-) breast cancer cells by conventional cytotoxic chemotherapy. Hum Pathol. 2010;41:574-81
210. Resetkova E, Reis-Filho JS, Jain RK. et al. Prognostic impact of ALDH1 in breast cancer: a story of stem cells and tumor microenvironment. Breast Cancer Res Treat. 2010;123:97-108
211. Fillmore CM, Kuperwasser C. Human breast cancer cell lines contain stem-like cells that self-renew, give rise to phenotypically diverse progeny and survive chemotherapy. Breast Cancer Res. 2008;10:R25
212. Ghods AJ, Irvin D, Liu G. et al. Spheres isolated from 9L gliosarcoma rat cell line possess chemoresistant and aggressive cancer stem-like cells. Stem Cells. 2007;25:1645-53
213. Francipane MG, Chandler J, Lagasse E. Cancer Stem Cells: A Moving Target. Curr Pathobiol Rep. 2013;1:111-18
214. Williams K, Motiani K, Giridhar PV. et al. CD44 integrates signaling in normal stem cell, cancer stem cell and (pre)metastatic niches. Exp Biol Med (Maywood). 2013;238:324-38
215. Koren A, Motaln H, Cufer T. Lung cancer stem cells: a biological and clinical perspective. Cell Oncol (Dordr). 2013;36:265-75
216. Singh SR. Gastric cancer stem cells: A novel therapeutic target. Cancer Lett. 2013;338:110-9
217. McAuliffe SM, Morgan SL, Wyant GA. et al. Targeting Notch, a key pathway for ovarian cancer stem cells, sensitizes tumors to platinum therapy. Proc Natl Acad Sci U S A. 2012;109:E2939-48
218. Levina V, Marrangoni A, Wang T. et al. Elimination of human lung cancer stem cells through targeting of the stem cell factor-c-kit autocrine signaling loop. Cancer Res. 2010;70:338-46
219. Li L, Xiang D, Shigdar S. et al. Epithelial cell adhesion molecule aptamer functionalized PLGA-lecithin-curcumin-PEG nanoparticles for targeted drug delivery to human colorectal adenocarcinoma cells. Int J Nanomedicine. 2014;9:1083-96
Author contact
![]() Corresponding author: The Affiliated Zhongshan Hospital of Dalian University, Dalian 116001, China, Tel.: +86 - 411 62893015; Fax: +86 - 411 62893555; E-mail: dahaixiaowencom (C. Pu). Or School of Medicine, Faculty of Health, Deakin University, Waurn Ponds, Victoria 3216, Australia. Tel.: +61 - 3 5227 1149; Fax: +61 - 3 5227 2945; E-mail: wduanedu.au (W. Duan).
Corresponding author: The Affiliated Zhongshan Hospital of Dalian University, Dalian 116001, China, Tel.: +86 - 411 62893015; Fax: +86 - 411 62893555; E-mail: dahaixiaowencom (C. Pu). Or School of Medicine, Faculty of Health, Deakin University, Waurn Ponds, Victoria 3216, Australia. Tel.: +61 - 3 5227 1149; Fax: +61 - 3 5227 2945; E-mail: wduanedu.au (W. Duan).