Theranostics
13.3
Impact Factor
- Current Issue
- Advance Articles
- Volume 16; 2026
- Volume 15; 2025
- Volume 14; 2024
- Volume 13; 2023
- Volume 12; 2022
- Archive
- Cover Images
- Cover Suggestion
- Special Issues
Top
1. Introduction
2. Nuclei represent primary...
3. Targeting cancer cell...
4. Targeting AuNPs to the ER
5. Targeting AuNPs to...
6. Perspectives: Limitations and...
7. Summary and highlights
Abbreviations
Supplementary Material
Author Contributions
References
1. Introduction
2. Nuclei represent primary...
3. Targeting cancer cell...
4. Targeting AuNPs to the ER
5. Targeting AuNPs to...
6. Perspectives: Limitations and...
7. Summary and highlights
Abbreviations
Supplementary Material
Author Contributions
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2015; 5(4):357-370. doi:10.7150/thno.10657 This issue Cite
Review
Off to the Organelles - Killing Cancer Cells with Targeted Gold Nanoparticles
Mohamed Kodiha1, Yi Meng Wang1, Eliza Hutter2, Dusica Maysinger2, Ursula Stochaj1 ![]()
1. Department of Physiology, McGill University, Montreal, Canada;
2. Department of Pharmacology & Therapeutics, McGill University, Montreal, Canada.
Received 2014-9-27; Accepted 2014-12-16; Published 2015-1-21
Citation:
Kodiha M, Wang YM, Hutter E, Maysinger D, Stochaj U. Off to the Organelles - Killing Cancer Cells with Targeted Gold Nanoparticles. Theranostics 2015; 5(4):357-370. doi:10.7150/thno.10657. https://www.thno.org/v05p0357.htm
Other stylesAbstract

Gold nanoparticles (AuNPs) are excellent tools for cancer cell imaging and basic research. However, they have yet to reach their full potential in the clinic. At present, we are only beginning to understand the molecular mechanisms that underlie the biological effects of AuNPs, including the structural and functional changes of cancer cells. This knowledge is critical for two aspects of nanomedicine. First, it will define the AuNP-induced events at the subcellular and molecular level, thereby possibly identifying new targets for cancer treatment. Second, it could provide new strategies to improve AuNP-dependent cancer diagnosis and treatment.
Our review summarizes the impact of AuNPs on selected subcellular organelles that are relevant to cancer therapy. We focus on the nucleus, its subcompartments, and mitochondria, because they are intimately linked to cancer cell survival, growth, proliferation and death. While non-targeted AuNPs can damage tumor cells, concentrating AuNPs in particular subcellular locations will likely improve tumor cell killing. Thus, it will increase cancer cell damage by photothermal ablation, mechanical injury or localized drug delivery. This concept is promising, but AuNPs have to overcome multiple hurdles to perform these tasks. AuNP size, morphology and surface modification are critical parameters for their delivery to organelles. Recent strategies explored all of these variables, and surface functionalization has become crucial to concentrate AuNPs in subcellular compartments.
Here, we highlight the use of AuNPs to damage cancer cells and their organelles. We discuss current limitations of AuNP-based cancer research and conclude with future directions for AuNP-dependent cancer treatment.
Keywords: Gold nanoparticles, AuNPs, cancer cell imaging
This review provides an update on the therapeutic potential of gold nanoparticles (AuNPs) for oncology. To this end, we introduce AuNPs as therapeutic tools, summarize the current strategies that target AuNPs to specific cell compartments and discuss how this targeting impacts cancer cell killing. For subcellular targeting, our focus is on nuclei and mitochondria, since both organelles are intimately linked to cancer cell survival, growth and proliferation and therefore primary targets for anti-cancer agents [1, 2]. We conclude by highlighting unsolved questions and potential roadblocks in the field.
1. Introduction
Nanotechnology is in the spotlight of therapeutic innovation [3], and AuNPs are particularly promising tools to improve cancer treatment [4]. Due to their unique optical properties, non-toxic nature, relatively simple preparation and functionalization, AuNPs are excellent candidates for many biological applications, such as imaging, drug delivery and photothermal therapy. These applications commonly take advantage of the particles' strong light scattering, intense absorption, and electromagnetic field enhancement that result from localized surface plasmon resonance [5, 6].
AuNPs can be produced in large quantities with defined shapes and sizes. The most common approaches synthesize AuNPs in situ through chemical reduction of gold salts and seed-mediated growth [7], which enlarges the particles step by step. This method is ideal to control AuNP size and shape [8-10] and used to produce large spherical, semi-spherical, rod-like, branched or other particle shapes [7]. AuNP surfaces are amenable to covalent and non-covalent surface modifications; this property is crucial for cellular and subcellular targeting. As the physico-chemical characterization of AuNPs and their detection have been reviewed by others [11-15], it will not be discussed here.
The development of AuNP-based strategies for the eradication of cancer cells is important, because effective therapies are frequently not available for rapidly progressing cancers [16]. So far, many of the studies on AuNPs suggest that cancer cells are especially vulnerable to these particles. Thus, AuNP-based treatment can destroy cancer cells, with minimal injury to healthy cells [17].
The therapeutic value of AuNPs is based on (i) their distinctive physical properties and (ii) their ability to interact with tumors and damage cancer cells. Thus, the enhanced permeability and retention (EPR) characteristics of many, but not all, tumors facilitate AuNP infiltration into the tumor [18]. Due to this passive targeting, AuNPs (~6-200 nm) access the tumor tissue, where they accumulate in the extracellular matrix before entering the cells [19]. Following their association with tumor cells, AuNPs promote unique ways of killing (Fig. 1). They can destroy cancer cells by photothermal ablation, as exemplified by AuroShell [20, 21], through mechanical damage, or as drug delivery systems for anticancer agents, such as tumor necrosis factor [21, 22] or doxorubicin [23, 24].
What are the benefits of subcellular AuNP targeting?
While AuNPs are relevant for different clinical applications, further improvements of AuNP-based strategies are expected to optimize the therapeutic outcomes. One such improvement is based on the concept that AuNP targeting to specific organelles maximizes the impact on tumor cells. To this end, AuNPs are being developed that accumulate in subcellular compartments where they destroy intrinsic cancer cell functions that are essential for tumor survival. Once in their proper intracellular location, AuNPs can enhance cancer cell destruction by different means. This includes the confined delivery of anti-cancer agents [25], localized subcellular mechanical damage, and improved efficiency of photothermal ablation due to high local AuNP concentrations [26, 27]. Such controlled AuNP action will not only increase cancer cell killing, but also diminish toxic side effects, because it reduces the necessary amounts of AuNPs and drug-load. Candidate compounds for nanoparticle-dependent subcellular delivery are doxorubicin [23], platinum-based drugs [28] and paclitaxel [29]. These anticancer agents interfere with nuclear and mitochondrial functions, respectively [30-33] and have been used to functionalize AuNPs [23, 34-37]. Aside from drugs, AuNPs can also deliver oligonucleotides to alter gene expression or splicing ([38] and references therein).
Figure 1
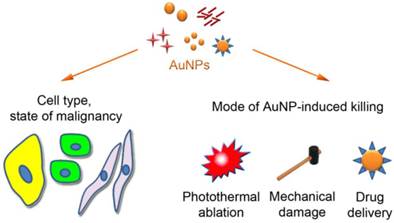
Impact of AuNPs on cancer cells. Size, morphology, functional groups on the AuNP surface and the cell type determine the subcellular distribution of AuNPs. AuNPs can cause tumor cell death by photothermal ablation, mechanical damage, and increase in the localized drug concentration. These events can be combined to enhance their killing efficiency.
What are the bottlenecks for AuNP targeting to specific subcellular compartments?
Once accumulated in tumor tissue, AuNPs have to overcome multiple obstacles before they can concentrate in the desired cell compartment: (i) cell surface binding, (ii) cellular uptake, (iii) escape from lysosomes/endosomes, and (iv) association with a particular subcellular location, such as nuclei or mitochondria (Fig. 2; [39]). The first three steps are general features that regulate the intracellular destination of all AuNPs. These steps have been reviewed extensively [13, 39-42]; we will only briefly summarize them here and then provide a more detailed discussion of AuNP targeting to nuclei, mitochondria and the ER.
Figure 2
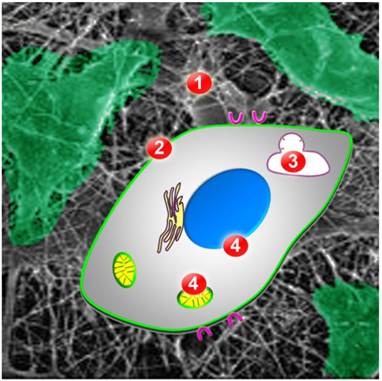
Obstacles AuNPs have to overcome for successful targeting to intracellular organelles or compartments. Once AuNPs are in the extracellular matrix of the tumor (ECM, barrier 1), they have to bind to the cancer cell surface. Cellular uptake requires translocation across the plasma membrane (barrier 2), by endocytosis or other mechanisms. Inside the cell, AuNPs have to escape from endosomes or lysosomes (barrier 3) to subsequently associate with the desired organelle or cell compartment (barrier 4). Possible final destinations are the nucleus (blue) or mitochondria (yellow).
Binding to the cancer cell surface, internalization and escape from endosomes/lysosomes
AuNP uptake, subcellular distribution and toxicity are determined by particle size, morphology and surface modification. Although AuNPs of different shapes (spherical, shells, rods, diamonds) or sizes (1-100 nm) accumulate in various cancer cells, their uptake kinetics and toxicity may vary profoundly (Fig. 1, reviewed by [13]). Besides shape and size, AuNP-based bio-nano interactions are further modulated by their functionalization [11, 43, 44]. In general, positive charges on the AuNP surface stimulate cellular uptake, possibly due to electrostatic interactions with the cell surface [45]. Positive charges can also improve AuNP transport to the nucleus, because nuclear localization sequences (NLSs) of many proteins are enriched for basic amino acid residues.
Particle uptake not only depends on AuNP properties, but also relies on the cell type. When grown in culture, cancer and non-tumorigenic cells differ significantly in this respect. Notably, tumor cells are often more vulnerable to AuNPs [46, 47]. Nevertheless, there is variability even among cancer cells; for example, AuNP uptake differs in hepatocellular carcinoma (HepG2) and cervix carcinoma cells (HeLa) [48]. Despite such cell-type specific differences, nanoparticle binding to cancer cells can be enhanced by exploiting tumor-related changes in plasma membrane composition (Additional File 1: Table S1 and associated references [41-91]). To this end, particle surfaces have been functionalized with ligands that improve docking at the tumor cell membrane. Ligands that promote high avidity binding to cancer cells include EGF (epidermal growth factor; associates with EGFR) or peptides containing the RGD motif (arginine-glycine-aspartic acid; recognized by some integrin family members) [32, 92-94]. Nucleolin, transferrin or antibodies against Her2 and EGFR [55, 95-97] can also enhance nanoparticle binding to cancer cells.
Once bound to the cell surface, AuNPs enter the cell; in most cases this occurs by an energy-dependent process, for which endocytosis is the main route (reviewed in [13, 14]). Following cellular uptake, AuNPs initially locate to endosomes and/or lysosomes, where the organellar membrane integrity determines AuNP retention (reviewed in [14]). However, proper functionalization of AuNPs can stimulate their endosomal/lysosomal escape or promote uptake by non-endocytotic pathways ([13, 14] and references therein).
2. Nuclei represent primary targets for cancer therapy
As nuclei mastermind essential aspects of tumor cell biology, they have become important targets in cancer therapy in general, and AuNP-dependent interventions in particular. The nucleus controls cell growth, proliferation and apoptosis, and many anti-cancer drugs obliterate these functions. At the same time, nuclear homeostasis is often altered in cancer cells, which display changes in nuclear size, shape, envelope, lamina and chromatin organization, nucleolar function or nucleocytoplasmic trafficking [98-104]. For example, the concentration of nuclear transport carriers is frequently increased in transformed cells or tumor samples, and this correlates with augmented signal-mediated nuclear import and export [105]. As compared to their normal counterparts, transformed cells often transport larger AuNPs, and AuNP nuclear translocation is more efficient [106-108].
Nuclear interactions of non-targeted AuNPs
Even in the absence of subcellular targeting signals, certain AuNP types associate with the nucleus and its subcompartments. In some ─but not all─ cases this nuclear localization is accompanied by organelle damage. As such, AuNPs (3.7nm average diameter) were modified with both 3-mercaptopropionic acid and polyethylene glycol (PEG) and conjugated to FITC [58]. Although the particles accumulated in HeLa cell nuclei after a 24-hour incubation period, cell viability still amounted to 85% of the control samples. Even after 72 hours and with up to10µM of AuNPs, cell survival was maintained at 70%, suggesting low cytotoxicity in HeLa cells [58].
On the other hand, Au55 gold clusters (1.4nm) entered the cell nucleus where they interacted with DNA and elicited toxic effects [109]. Comparison of healthy and tumor cell lines revealed maximum toxicity for the metastatic melanoma cell lines MV3 and BLM. The interaction between Au55 gold clusters and nuclear DNA is likely the underlying cause of this toxicity.
Our recent studies examined AuNPs of different sizes and morphologies. In breast cancer cells, nuclear membranes, nuclear laminae and nucleolar functions were compromised by small spherical AuNPs or gold nanoflowers, but not by large spherical AuNPs ([46], Fig. 3). The damage inflicted by non-spherical gold nanoflowers is particularly interesting; despite their large size (40-120nm), they entered the nucleus and destroyed nuclear homeostasis in breast cancer cells, but not in normal breast cells.
Figure 3
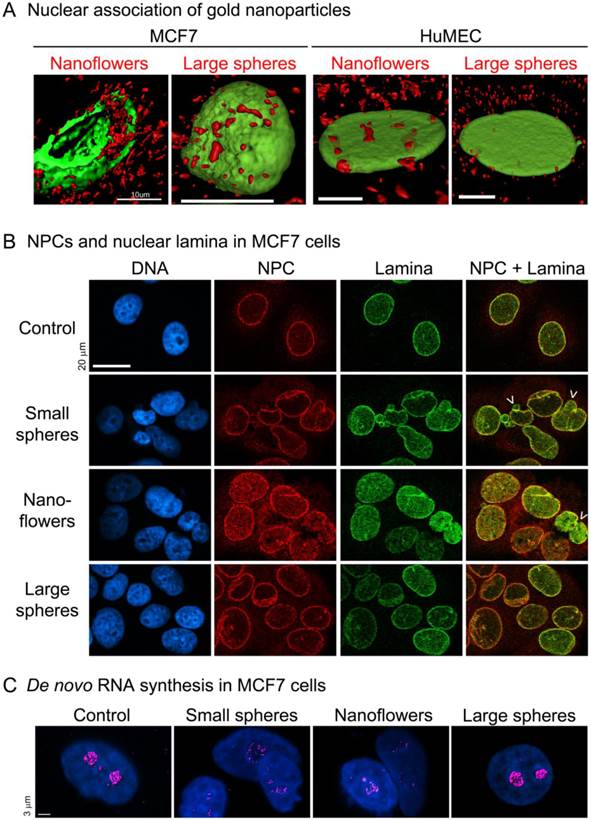
(A) Gold nanoflowers and large gold nanospheres (red) associate with nuclei of different breast cells, i.e. MCF7 and non-tumorigenic human mammary epithelial cells (HuMEC). Note that gold nanoflowers can be detected in the nuclear interior of MCF7 cells, where they disrupt the nuclear lamina (green). Scale bars are 10µm. (B) Small gold nanospheres (15.6nm diameter) and gold nanoflowers (40-120nm), but not large gold nanospheres (60nm), alter the nuclear organization in MCF7 cells. In particular, nuclear pore complexes (NPC, red) and the nuclear lamina (Lamin A, green) show severe changes. Arrows mark some of the nuclei with altered morphology; scale bar is 20µm. (C) Small gold nanospheres and gold nanoflowers inhibit de novo RNA synthesis (magenta) in the nucleolus. Scale bar is 3µm. Adapted from [46] with permission.
Targeting AuNPs to the nucleus
Which AuNP size fits the nucleus? The optimal AuNP properties for nuclear targeting depend on the desired impact of the particle. If AuNPs are destined for the nuclear interior, the size of the transport channel of the nuclear pore complex (NPC) has to be considered. Accordingly, spherical particles of 9nm or less in diameter may cross the NPC by diffusion, whereas particles up to 39nm in diameter can be delivered to the nucleus if they carry a nuclear transport signal [110]. These are not fixed values, as the nuclear transport machinery is often altered in cancer cells (see above). In the context of cancer therapy, three different scenarios can apply to nuclear AuNPs: (i) they are transported into the nuclear interior or (ii) clog the NPC, or (iii) release cytotoxic drugs in the vicinity of nuclei.
Nuclear import of AuNPs. The ground-breaking work by Carl Feldherr built the foundation for AuNP targeting to the nucleus. His laboratory coated AuNPs with conjugates that contained serum albumin and the nuclear localization sequences (NLSs) derived from SV40 T-antigen or nucleoplasmin. Upon injection into the cytoplasm, these functionalized AuNPs translocated across the nuclear pore complex (NPC) and accumulated inside the nucleus [111, 112].
Feldheim's group went beyond these studies and defined the different stages of AuNP nuclear localization in intact cultured cells [39, 59]. These studies showed that aside from the initial binding, cellular uptake and release from endosomes/lysosomes (see above), targeting to the nucleus and passage across the nuclear envelope (NE) are critical steps that limit the delivery to the nucleus. While AuNPs destined for the nucleus show cell-type specific differences in cellular uptake, escape from endosomes/lysosomes and nuclear targeting [113], they also share common features. For example, uptake of NLS-modified AuNPs is temperature-dependent in cultured cells, consistent with endocytosis [59, 113]. Furthermore, increasing the number of NLS-peptides on the particle surface enhances AuNP nuclear localization and toxicity [59].
Various strategies have been employed to target gold nanoparticles to the nucleus. So far, there is no protocol that has been universally successful for different tumor cells. Therefore, we discuss specific examples for AuNPs that were functionalized with NLSs, cell-penetrating peptides, peptide combinations, oligonucleotides or other moieties (summarized in Additional File 1: Table S1). Peptide sequences are depicted in the one-letter code.
The success of multiple peptide modifications was demonstrated for AuNPs modified with SV40-NLS or an NLS derived from adenovirus fiber proteins; these particles did not enter the nucleus of HepG2 cells. However, upon further addition of the adenovirus receptor-mediated endocytosis sequence, AuNPs localized to nuclei [39]. Notably, when AuNPs carried the NLS and the receptor-mediated endocytosis peptide as separate entities, nuclear targeting was enhanced as compared to a longer peptide that combines both sequences. If combined into one peptide, the two sequence elements may be less accessible to cellular binding partners.
In other studies, AuNPs stabilized with the non-natural amino acid tiopronin [94] were functionalized with GRKKRRQRRR, a peptide derived from the HIV-1 protein Tat [61]. After a 1-hour treatment, Tat-modified particles were located in the nuclear interior of hTERT-BJ1 human fibroblasts. By contrast, AuNPs carrying tiopronin alone accumulated in cytoplasmic vacuoles or at the mitochondrial periphery [61]. Upon a 24-hour incubation period with up to 10µM AuNPs (core size 2.8nm), toxicity was ≤20% for both types of particles.
To improve cellular uptake and nuclear accumulation of 30nm AuNPs, particles were decorated with peptides that contained the sequence CALNN. Addition of mixed peptides containing CALNN and CALNN plus eight arginine residues (CALNNR8) promoted AuNP accumulation in nuclei of HeLa cells [52]. After incubation with 0.32nM AuNPs for 24 hours, cell death could reach 95%.
In further experiments, 16 or 14nm PEGylated AuNPs were modified with different CALLN-derived peptides. Functionalization with CALLN-fusions containing the cell penetrating peptide of TAT (CALLN-AGRKKRRQRRR) or Pntn (CALLN-GRQIKIWFQNRRMKWKK) stimulated cell entry, whereas an NLS (CALLN-GGFSTSLRARKA) was added for nuclear targeting [66, 114]. In this scenario, AuNPs modified simultaneously with NLS, TAT and Pntn-containing peptides led to higher intracellular gold content when compared to AuNPs carrying one peptide species only. Furthermore, simultaneous coating with NLS, TAT and Pntn promoted AuNP nuclear localization.
Surprisingly, if AuNPs carry a positive surface charge, nuclear targeting can even be accomplished with peptides that have no sequence similarity to a basic NLS ([64], Fig. 4).
Nucleolin is a multifunctional protein that resides in the nucleolus and other cell compartments, such as the plasma membrane. For some cancer cells nucleolin abundance is high in the plasma membrane, where the protein can serve as a docking site for AuNPs. The aptamer AS1411 binds nucleolin and was tested in phase I and II clinical trials for advanced solid tumors and acute myeloid leukemia [21]. When gold nanostars were functionalized with AS1411, they entered HeLa cells and concentrated in the vicinity of nuclei [55]. This uptake required both nucleolin on the cell surface and the aptamer. AS1411-gold nanostar localization close to the nucleus correlated with changes in nuclear morphology, suggesting mechanical damage to the nuclear envelope. Irradiation liberated the aptamer from AuNPs, which in turn activated caspases 3 and 7 and increased cell death. Collectively, the study supports the model that the aptamer release close to the nucleus enhanced damage and the ensuing cell death.
Figure 4
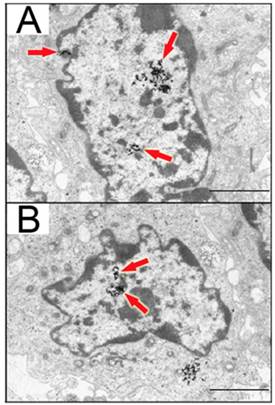
Detection of 13nm AuNPs modified with CIPGNVG-PEG-NH3+ in 1BR3G cells (transformed human skin fibroblasts). Cells were incubated for 3 hours with functionalized AuNPs, and particles were visualized by transmission electron microscopy. Some of the AuNPs were present in the nuclear interior, as indicated by the red arrows. Panels A and B depict two different nuclei. Scale bars are 2µm. Adapted from Ojea-Jiménez et al. [64] with permission.
The examples above suggest that targeting to the nucleus can amplify organelle-specific insults. This idea is validated by NLS-modified AuNPs that increased DNA damage and interfered with cell division, in particular cytokinesis [56]. Specifically, 30nm PEGylated AuNPs decorated with RGD- and NLS-peptides accumulated in nuclei and triggered apoptosis in 20% of the cancer cells [56].
Aside from targeting to the nuclear interior, AuNPs can also be used to obstruct the NPC. Thus, NLS-modified AuNPs (~39 or 32nm in size) blocked nuclear transport in HeLa cells and induced autophagic cell death [115]. Interestingly, this was cell type specific, because it was not observed in SiHa cells, another cervix carcinoma cell line.
Delivery of nucleic acids. One of the applications of nuclear AuNPs is the modulation of gene expression; this can be achieved by knockdown or changes in splicing [60, 116-119]. An example of this approach is the use of 13nm AuNPs that carried oligonucleotides to adjust the alternative splicing of mRNAs for pro-survival factors in different experimental systems [119]. As such, the particles were detected in nuclei of cultured cells and led to tumor shrinkage in a xenograft model based on LoVo cells (human colon carcinoma).
In MCF7 breast cancer cells, spherical ultrasmall (6 and 2nm) tiopronin-coated AuNPs reside in the cytoplasm and enter the nucleus after a 24-hour incubation period [60]. When conjugated to a fluorescent FITC-tag, only 2nm particles were detected in MCF7 nuclei. FITC-labeling increased the size of 6nm AuNPs to 10nm, which may have limited the passage through NPCs. To induce cell death, an oligonucleotide was added to 2nm tiopronin-AuNPs. This oligonucleotide generated triplex structures with the P2 promoter of c-MYC. As a result, expression of the proto-oncogene was downregulated and cell viability was reduced to 70%. When compared to the triplex-forming oligonucleotide alone, AuNP-attached oligonucleotides increased c-MYC silencing and MCF7 cell death [60].
Interestingly, AuNPs functionalized with dexamethasone also delivered plasmid DNA to nuclei [76], suggesting that steroid hormones can be used for AuNP nuclear targeting. These studies were performed in cultured cells and experimental animals; they are discussed in section 5.
Delivery of anti-cancer drugs. AuNPs that locate to the nuclear periphery can provide transport vehicles for anti-cancer drugs. One such delivery system carried doxorubicin and was tested in HeLa (cervix carcinoma), A549 (lung carcinoma) and NIH3T3-L1 cells (fibroblasts with pre-adipose characteristics). To this end, 25nm PEGylated spherical AuNPs were functionalized with cell penetrating peptides, such as TAT [24]. TAT-AuNPs were taken up in all cells tested, whereas other peptides displayed cell type-specific differences. Doxorubicin-loaded AuNPs were especially cytotoxic to HeLa and A549 cells, but NIH3T3-L1 cells were less affected. Cell death was attributed to the release of doxorubicin close to the nucleus, where AuNPs concentrated.
3. Targeting cancer cell mitochondria with AuNPs
Mitochondria are the major sites of cellular energy production, and their dysfunction is associated with a wide range of diseases and pathophysiologies, including cancer [120]. Changes in bioenergetics are a hallmark of many tumors, but mitochondria are also key regulators of apoptotic cell death. These properties make mitochondria prime targets for cancer treatment [121, 122]. AuNPs often impair mitochondrial functions and thereby induce cell death. However, this does not always involve stable physical contact between AuNPs and the organelle. Here, we focus on examples that report AuNP association with mitochondria. The emphasis is on studies that were designed to deliver AuNPs specifically to mitochondria.
Which AuNP size fits mitochondria? The conceptual differences for AuNP targeting to nuclei and mitochondria are based on the distinct organelle borders to the cytoplasm. Unlike the nucleus, mitochondria do not contain large pores that provide easy access for AuNPs. Both the outer and the inner mitochondrial membranes present barriers to AuNPs that are destined for the mitochondrial matrix. In heart cells, 3nm particles, but not 6nm AuNPs, translocated across the outer mitochondrial membrane [86]. In the inner mitochondrial membrane, protein import channels provide openings of <2nm ([123], reviewed in [124]). This restricts AuNP entry into the matrix of intact mitochondria. Accordingly, mitochondrial targeting sequences, which interact with the mitochondrial protein import apparatus, have only limited value for AuNP delivery. However, AuNP translocation across the mitochondrial membranes is not obligatory to damage the organelle, and other strategies have been successful. Common effects of AuNPs on mitochondria are morphological changes, loss of membrane potential and production of reactive oxygen species.
Mitochondrial delivery of AuNPs. An alternative to peptide-derived AuNP targeting was liposomes that fuse with mitochondrial membranes [87]. AuNPs (8-12nm) were delivered by encapsulation in Mito-porter; this envelope contains fusogenic lipids and octa-arginine surface modifications. Octa-arginine served multiple functions; it stimulated particle uptake by micropinocytosis, escape into the cytosol, and binding to mitochondria. Once the particles bound to mitochondria, Mito-porter lipids mediated membrane fusion, and AuNPs were released into mitochondria.
Another approach was based on AuNP functionalization with cetyltrimethylammonium bromide (CTAB) [88]. This compound damages lipid bilayers and can facilitate the permeation of membranes, leading to the mitochondrial association of gold nanorods. CTAB stimulated lysosomal escape in some cell lines, likely because it compromised lysosomal membrane integrity. Among lung carcinoma (A549), normal bronchial epithelial (16HBE) and primary adult stem cells, the nanoparticles affected A549 cells to the greatest extent [47]. Such CTAB-gold nanorods accumulated in mitochondria (Fig. 5) and caused cell death through a collapse of the mitochondrial membrane potential and the generation of oxidative stress.
Figure 5
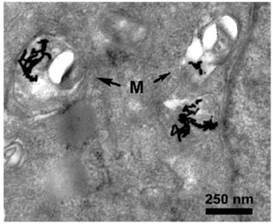
A549 cells were incubated for 24 hours with gold nanorods. Swelling and rounding was observed for a fraction of the mitochondria. Moreover, some cristae were lost and vacuoles appeared in mitochondria. Gold nanorods associated as aggregates with mitochondria (M), as indicated by the arrows. The transmission electron micrograph was adapted from [47] with permission.
The biological properties of mitochondria can be exploited for AuNP targeting with triphenylphosphonium. Driven by the mitochondrial membrane potential, this lipophilic cation can increase the concentration of various agents in the mitochondrial matrix (reviewed in [125]). Triphenylphosphonium targeted gold nanoclusters to HeLa cell mitochondria [89]. However, under the conditions tested, the cytotoxicity of these gold nanoclusters was low, as ≥80% of the cells remained viable.
Pre-photoactivation seemed to enhance the deleterious effects of AuNPs on cancer cells [90]. Thus, AuNPs were more toxic, if photoexcited before addition to pancreatic cancer cells (1.4E7 cells). Their heightened toxicity correlated with increased mitochondrial damage, such as swelling and loss of mitochondrial membrane potential. Pre-photocativation also promoted AuNP location to mitochondria and the production of reactive oxygen species.
A pro-apoptotic peptide containing the sequence (KLAKLAK)2 targeted 13.5nm AuNPs to HeLa cell mitochondria [33]. These peptide-functionalized AuNPs associated with mitochondria and induced their swelling, loss of membrane potential and ultimately cell death. Peptide-modified AuNPs were more toxic than their non-functionalized counterparts or the pro-apoptotic peptide alone.
Taken together, nuclei and mitochondria have been successfully targeted by AuNPs. The general principles that route AuNPs to the nucleus or mitochondria have been identified (Fig. 6), but mechanistic details are frequently unknown.
Figure 6
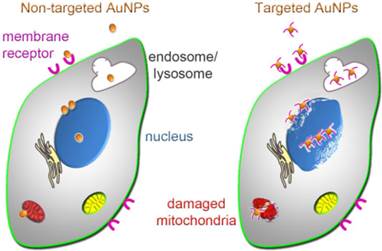
Uptake and subcellular targeting of AuNPs. Non-targeted (left) and targeted (right) AuNPs bind to the plasma membrane; this may involve receptors on the cell surface. Upon internalization, AuNPs initially concentrate in endosomes or lysosomes. After escape from these membrane-bound compartments, AuNPs associate with nuclei or mitochondria, where they can cause irreversible damage that culminates in cancer cell death. Cellular injury is enhanced if AuNPs are targeted to nuclei or mitochondria (right); this subcellular targeting increases cancer cell killing.
4. Targeting AuNPs to the ER
Unlike AuNP delivery to nuclei or mitochondria, strategies for ER targeting are less well developed. However, given the importance of the ER for many tumor types and the links between mitochondria and the ER [126], AuNPs located in the ER could play a significant role in cancer therapy. AuNPs have been detected in this membrane system; for example, non-functionalized 13nm spherical AuNPs colocalized with the ER and Golgi apparatus in B16F10 melanoma cells [127]. In HeLa cells, 18nm AuNPs associated with cytoplasmic vesicles that were presumably part of the ER [128]. In Kupffer cells of the liver, 13nm PEGylated AuNPs located to lysosomes and the smooth ER [129], and AuNPs derivatized with CALNNR8 peptides were trapped in the ER of HeLa cells [52]. Simultaneous association with nuclei and the ER has also been reported for K562 cells (human, chronic myelogenous leukemia) [130]. Notably, these particles induced ER stress, which likely contributed to their toxicity.
5. Targeting AuNPs to subcellular organelles in vivo
As compared to cultured cells or spheroids, AuNP targeting to cell organelles in vivo faces extra hurdles. These additional challenges include accumulation in healthy organs, tissue-specific barriers, clearance by the reticuloendothelial system and insufficient concentration at the tumor site. Once accumulated in the tumor, AuNPs will follow the same steps depicted in Fig. 1. As such, after cellular uptake, particles have to escape lysosomes/endosomes and locate to their final subcellular destination.
To overcome these obstacles, several variables can be changed, including size, surface modification, dosage and route of entry. Accumulation at the tumor sites in vivo is a prerequisite for subcellular targeting, and this topic has been discussed in previous reviews [16, 18]. At present, only a limited number of studies have attempted to optimize AuNPs to reach a subcellular destination in vivo. Here, we discuss several studies that show organelle targeting with AuNPs both in cultured cells and in experimental animals.
Targeting nuclei in vitro and in vivo. Huang et al. [85] compared the performance of AuNPs in three different model systems. Spherical tiopronin-coated AuNPs of 2, 6 or 15nm size were analyzed in MCF7 cells, grown in monolayers or spheroids. These particles were also examined in xenografts of Balb/c nude mice. In all of these model systems, cellular uptake was size-dependent and more efficient for smaller AuNPs. Moreover, particle size determined the subcellular AuNP distribution (see Additional File 1: Table S1). Small AuNPs of 2 and 6nm located to the nucleus and cytoplasm, whereas 15nm particles were restricted to the cytoplasm. Following intravenous injection, all AuNPs were cleared from the blood; this was most efficient for 15nm particles. By contrast, tumors accumulated preferentially 2nm AuNPs, but 2nm particles also concentrated in the kidney and lung. AuNPs of 15nm size associated predominantly with non-tumor tissue, with preference for the liver and spleen. Collectively, these results suggest that the subcellular distribution of tiopronin-coated AuNPs is similar in MCF7 cell monolayers, spheroids and xenografts.
Chen et al. [76] examined AuNP nuclear targeting in vitro in Hep3B (hepatocellular carcinoma) and 293T cells (human embryonic kidney cells containing SV40 T-antigen); the experiments were extended to Hep3B cell-derived xenografts. To deliver DNA efficiently to the cell nucleus, AuNPs were coated with different molecules in a sandwich-like fashion. The sandwich included plasmid DNA, polyethylenimine (PEI) and dexamethasone, a synthetic agonist of the glucocorticoid receptor. PEI enhanced endosomal escape, while dexamethasone served several purposes. First, it improved uptake, which reached 82.5% in cultured HepB3 cells. Second, dexamethasone stimulated AuNP nuclear targeting through its association with the glucocorticoid receptor. Specifically, functionalized AuNPs (carrying dexamethasone, DNA and PEI) form complexes with glucocorticoid receptor in the cytoplasm and then translocate to the nucleus. With this approach, cultured cells were transfected effectively, while cytotoxicity was low.
Building on these in vitro studies, functionalized AuNPs were also used for gene delivery in vivo (Balb/c nude mice bearing HepB3-derived tumors). DNA encoding tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) was introduced into the animals using AuNPs of ~55nm size. Tumors produced the highest levels of TRAIL when the DNA was delivered by an AuNP sandwich that contained both PEI and dexamethasone. Concomitant with efficient DNA delivery, tumor growth was inhibited. Thus, functionalized AuNPs induced TRAIL synthesis in vivo and thereby interfered with tumor growth [76], while TRAIL expression was low in non-tumor tissues. This study demonstrated AuNP nuclear targeting in cultured cells. Although not shown experimentally in vivo, results suggest that nuclear targeting also occurred in experimental animals. Thus, AuNP functionalization with dexamethasone can be useful to target tumors that synthesize glucocorticoid receptor.
Targeting mitochondria in vitro and in vivo. The preferred route of ATP production in many forms of cancer is anaerobic glycolysis; this pathway relies on the enzyme hexokinase which synthesizes glucose-6-phosphate. Hexokinase 2 is especially important among the four enzyme isoforms, because it is highly abundant in aggressive tumor cells and predominantly associated with the outer mitochondrial membrane. Since hexokinase 2 interaction with the outer mitochondrial membrane stimulates glycolytic energy production and reduces apoptosis, the enzyme has become a therapeutic target for cancer therapy. Hexokinase 2 is inhibited by 3-bromopyruvate, a toxic compound with off-target effects. To improve 3-bromopyruvate specificity and diminish toxicity, AuNPs were developed for mitochondrial delivery [91]. PEGylated spherical AuNPs (2.9 - 4.3nm) were modified with TPP, thereby generating T-AuNPs which are characterized by positively charged and lipophilic moieties on the surface. These properties improved T-AuNP uptake and mitochondrial association in PC3 cells. Results were similar when T-AuNPs were further modified with 3-bromopyruvate (T-3-BP-AuNP). After 4 hours, T-3-BP-AuNPs bound to the outer mitochondrial membrane where hexokinase 2 resides. These AuNPs accumulated in the mitochondrial matrix at 12 hours. T-3-BP-AuNPs reduced PC3 and DU145 (prostate carcinoma) cell viability, but had little effect on human mesenchymal stem cells. The impact on cancer cells was attributed to the loss of mitochondrial functions and glycolysis. Overall, AuNPs modified with 3-BP were more toxic to cancer than normal cells. At the same time, AuNPs targeted to mitochondria (T-AuNPs) were more harmful than their non-targeted counterparts (NT-AuNPs). The toxicity of AuNPs was further enhanced by a 1min laser irradiation at 660nm. To evaluate in vivo effects, the biodistribution and pharmacokinetics of T-AuNP and NT-AuNP were assessed in male Sprague Dawley rats. Clearance from the plasma was faster for T-AuNPs than NT-AuNPs. Both types of particles accumulated in the liver and spleen, but their subcellular localization was not determined [91].
6. Perspectives: Limitations and future directions
The promise of AuNP-dependent cancer therapy is emphasized by numerous publications and clinical trials [21]. At present, several hurdles limit the rapid improvement of AuNP-based treatments. For example, the approaches for AuNP targeting to subcellular locations differ widely. Since AuNP size, morphology, functionalization, concentration and the cell types analyzed vary significantly, it is difficult to compare results from different laboratories. Moreover, although essential to improve AuNP-based intervention, the biological mechanisms underlying AuNP-induced cell killing are often not defined. Nevertheless, some general conclusions can be drawn for the diverse strategies that lead to AuNP association with subcellular organelles (Table 1).
Our review discusses the potential benefits of targeting AuNPs to specific cell organelles, and we present the barriers AuNPs have to overcome to reach their intracellular destination. Aside from the restrictions these barriers impose, AuNP-mediated cancer cell killing could be limited by export via exocytosis (reviewed in [131]). However, such particle loss can be reduced with appropriate surface functionalization [132]. At the organismal level, it will further be important to design strategies that control AuNP clearance. Clearance is mostly achieved through the hepatobiliary system and the kidney, and it is modulated by AuNP surface modification [133]. As the immune system contributes to nanoparticle clearance [134], clearance rates likely differ among patients. Although these variations challenge the universal application for cancer therapy, they may nevertheless offer the opportunity to produce AuNPs for personalized medicine.
Cell type dependent differences in uptake and subsequent intracellular targeting complicate the optimization of AuNPs for cancer therapy. On the other hand, if systematically examined, these differences could be explored to develop AuNPs that are selective for specific types of cancer. Moreover, AuNP targeting to tumor cell organelles could be enhanced by exploiting the differences between normal and cancer cells. For example, nuclear transport is more efficient in proliferating cancer cells as compared to their non-tumorigenic counterparts (see above). This is -in part- caused by the overexpression of importin-α and importin-β family members and other soluble factors or nucleoporins that support signal-mediated nuclear import [135-137]. To date, AuNP nuclear targeting relies predominantly on NLSs that bind importin-α (SV40 T-antigen, nucleoplasmin) or importin-β1 (TAT, [138]). In the future, nuclear delivery could be improved through AuNP functionalization that stimulates NPC binding. Some nucleoporins are more abundant in cancer cells [139], and therefore provide potential AuNP docking sites at the nuclear pore.
With respect to mitochondria, functionalizing small spherical or rod-shaped AuNPs with mitochondrial targeting sequences may be suitable to damage organellar functions or even clog mitochondrial protein import sites, analogous to what has been observed for NPCs.
While targeting to subcellular organelles is of particular interest to nanomedicine, the major bottleneck after cellular uptake is AuNP escape from endosomes/lysosomes. Recent studies suggest additional strategies to enhance this escape. Besides low energy laser irradiation [140], particle surface modifications that increase the “proton sponge effect” could promote endosome swelling and AuNP release [141]. Moreover, AuNPs that undergo pH-dependent aggregation or enhance pH-dependent lipid rupture could be useful for the liberation from endosomes. Release from endosomes is critical for AuNPs to reach nuclei; however, it may not be mandatory for mitochondria. For instance, fusogenic lipids as described for the Mito-porter could circumvent the need for endosomal escape. On the other hand, it is conceivable that the direct delivery of material from endosomes to mitochondria [142] will be exploited to bring AuNPs to mitochondria.
So far, cell culture models have dominated the characterization of AuNP bio-nano interactions. More recently, AuNP-induced cell damage and death have been assessed in tumor cell spheroids [85, 143], a model system that mimics multiple aspects of tumor tissues in vivo. For example, AuNP penetration across multiple cell layers was studied in spheroids derived from U87 glioblastoma cells [143]. The progress in spheroid production sets the stage to explore this model further and examine AuNP targeting to nuclei and mitochondria. To date, there are only few studies that examine the subcellular distribution of AuNPs in vivo. While in vitro analyses located AuNPs in nuclei or mitochondria, it is in most cases not clear whether these AuNPs are targeted to the same tumor cell organelles in experimental animals. Clearly, this information has to be provided in the future to fully assess the value of AuNP subcellular targeting in vivo.
Table 1
Advantages and limitations of current approaches for AuNP delivery to subcellular organelles.
| Approach | Subcellular organelle | Advantages | Limitations |
|---|---|---|---|
| Exploitation of AuNP physical properties for organellar delivery without specific targeting moieties: size (e.g. small or large particles, nanoclusters), shape (spheres, rods, flowers, urchins), charge | nonspecific subcellular distribution, often concentrated in endosomes/lysosomes, particles may escape endosomes/lysosomes and associate with nuclei and/or mitochondria | easy to prepare, low cost, fast clearance may limit toxicity, PEGylated small AuNPs often damage cancer cells, positive charges frequently enhance uptake | uptake not specific to cancer cells, fast clearance may limit accumulation in tumor, effect of positive charges cell-type dependent, low endosomal/lysosomal escape, low concentration in subcellular organelles, high toxicity, thus damage to non-tumor tissues |
| Targeting to cell surface: RGD transferrin, EGF, antibodies or aptamers that bind cell surface components | nonspecific subcellular distribution, often concentrated in endosomes/lysosomes, particles may escape endosomes/lysosomes and associate with nuclei and/or mitochondria | enhanced targeting to cancer cells, thereby improved uptake by tumor cells | endosomal escape and subcellular delivery may require additional modifications; cell surface receptor signatures for efficient tumor targeting not available for all cancer types |
| Improved cellular uptake: cell penetrating (CPP) and other peptides; e.g. CALNN, CALNNR8, TAT, Pntn, lysosomal sorting peptides | nucleus and other subcellular compartments | may enhance nuclear targeting through increase of cellular uptake; some CPPs also function as nuclear localization signal | some peptides inefficient for endosomal/lysosomal escape and nuclear targeting; nuclear localization can depend on cell type |
| Nuclear localization signals (NLSs): biological and synthetic signals; linear and cyclic peptides: SV40-NLS, adenoviral NLS, cyclic [KW]5 | enriched in nucleus | positive charges of NLS enhance cellular uptake; specific and efficient nuclear targeting; frequently low toxicity in vitro; useful for drug delivery to nucleus | may need additional modifications to improve tumor targeting in vivo and to facilitate endosomal/lysosomal escape |
| Combination of peptides with different functions: e.g. CPP+NLS, RGD+NLS | enriched in nucleus | improved tumor targeting and cellular uptake; useful to exploit cell surface receptors | functionalization with multiple peptides; specific ratio of peptides may be required |
| Other molecules: CTAB, tiopronin, cysteamine, thioglucose, dexamethasone | can lead to enrichment in nucleus | may stimulate cellular uptake, endosomal/lysosomal escape or both; this can enhance nuclear association | some modifications highly toxic (e.g. CTAB); except for dexamethasone, nuclear targeting not efficient; targeting may be cell type specific (e.g. nuclear accumulation by dexamethasone relies on glucocorticoid receptor) |
| Different types of functionalization: octa-arginine, CTAB, TPP, chitosan, polyvinylpyrrolidone | enriched in mitochondria | can stimulate cellular uptake and/or endosomal/lysosomal escape; this can enhance mitochondrial association | frequently toxic (e.g. TPP); may require permeabilization of plasma membrane (e.g. polyvinylpyrrolidone) |
As the model systems to evaluate AuNP performance are becoming more diverse, so are AuNPs. As such, AuNPs can assemble into chains, plasmonic vesicles [144, 145] or other structures that provide multiple functions at the same time, including drug delivery, enhanced photothermal ablation and particle tracking. Large complex AuNPs that are able to disassemble into smaller AuNP units [145, 146] could help overcome the different roadblocks that limit AuNP subcellular targeting (Fig. 2). Such complex AuNPs, when disassembled, may have the additional benefit of rapid renal or hepatobiliary clearance.
Taken together, AuNPs offer unique opportunities to translate the insights of basic research into clinical applications. Given the success of AuNPs for photothermal ablation, mechanical injury and targeted drug delivery, future strategies that combine these effects for AuNP-dependent cancer cell killing are particularly promising.
7. Summary and highlights
Recent studies have begun to reveal how AuNPs impinge on the structural/functional organization of cancer and normal cells. This knowledge is critical for two aspects of nanomedicine. First, it will help define the AuNP-induced events at the subcellular level. This will set the stage for the identification of new molecular targets for cancer therapy. Second, it will direct the design of AuNPs with physico-chemical properties that overcome the current limitations these particles face in basic research, diagnosis and therapy. Thus, optimization of AuNP surfaces for cell and organelle-specific delivery is anticipated to enhance the efficiency of cancer cell killing, while minimizing the impact on non-tumor tissues. Based on their essential role for cancer cell survival, nuclei and mitochondria are prime targets for this approach.
Abbreviations
AuNP: gold nanoparticle
CALNN: cys-ala-leu-asp-asp
CTAB: cetyltrimethylammonium bromide
EGF: epidermal growth factor
EGFR: EGF receptor
EPR: enhanced permeability and retention
LSP: localized surface plasmon resonance
MTS: mitochondrial targeting sequence
NIR: near infrared radiation
NLS: nuclear localization sequence
PEG: polyethylene glycol, surface coating of AuNPs that reduces particle aggregation, non-specific interactions with biomolecules and uptake by the reticuloendothelial system
PEI: polyethylenimine
Pntn: Penetratin, peptide sequence derived from Drosophila protein Antennapedia that promotes entry into the cell
TAT: HIV-1-trans-activating protein
TPP: triphenylphosphonium
TRAIL: tumor necrosis factor-related apoptosis-inducing ligand
Supplementary Material
Additional File 1Table S1: Impact of AuNP size, morphology and functionalization on cellular uptake, subcellular localization and cell survival.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Hanahan D, Weinberg Robert A. Hallmarks of cancer: The next generation. Cell. 2011;144:646-74
2. Ubah OC, Wallace HM. Cancer therapy: Targeting mitochondria and other sub-cellular organelles. Curr Pharm Des. 2014;20:201-22
3. Wright J. Nanotechnology: Deliver on a promise. Nature. 2014;509:S58-S9
4. Ghosh P, Han G, De M, Kim CK, Rotello VM. Gold nanoparticles in delivery applications. Adv Drug Del Rev. 2008;60:1307-15
5. Hutter E, Maysinger D. Gold nanoparticles and quantum dots for bioimaging. Microsc Res Tech. 2011;74:592-604
6. Hutter E, Maysinger D. Gold-nanoparticle-based biosensors for detection of enzyme activity. Trends Pharmacol Sci. 2013;34:497-507
7. Zhao P, Li N, Astruc D. State of the art in gold nanoparticle synthesis. Coord Chem Rev. 2013;257:638-65
8. Jana NR, Gearheart L, Murphy CJ. Wet chemical synthesis of high aspect ratio cylindrical gold nanorods. J Phys Chem B. 2001;105:4065-7
9. Brown KR, Natan MJ. Hydroxylamine seeding of colloidal au nanoparticles in solution and on surfaces. Langmuir. 1998;14:726-8
10. Nikoobakht B, El-Sayed MA. Preparation and growth mechanism of gold nanorods (NRs) using seed-mediated growth method. Chem Mater. 2003;15:1957-62
11. Sapsford KE, Algar WR, Berti L, Gemmill KB, Casey BJ, Oh E. et al. Functionalizing nanoparticles with biological molecules: Developing chemistries that facilitate nanotechnology. Chem Rev. 2013;113:1904-2074
12. Huefner A, Septiadi D, Wilts BD, Patel II, Kuan W-L, Fragniere A. et al. Gold nanoparticles explore cells: Cellular uptake and their use as intracellular probes. Methods. 2014;68:354-63
13. Dykman LA, Khlebtsov NG. Uptake of engineered gold nanoparticles into mammalian cells. Chem Rev. 2014;114:1258-88
14. Lévy R, Shaheen U, Cesbron Y, Sée V. Gold nanoparticles delivery in mammalian live cells: A critical review. Nano Reviews. 2010;1:10.3402 /nano.v1i0.4889
15. Jeremic B, Aguerri AR, Filipovic N. Radiosensitization by gold nanoparticles. Clin Translat Oncol. 2013;15:593-601
16. Jain S, Hirst DG, O'Sullivan JM. Gold nanoparticles as novel agents for cancer therapy. Br J Radiol. 2012;85:101-13
17. Ahmad MZ, Akhter S, Rahman Z, Akhter S, Anwar M, Mallik N. et al. Nanometric gold in cancer nanotechnology: Current status and future prospect. J Pharm Pharmacol. 2013;65:634-51
18. Dreaden EC, Austin LA, Mackey MA, El-Sayed MA. Size matters: Gold nanoparticles in targeted cancer drug delivery. Ther Deliv. 2012;3:457-78
19. Holback H, Yeo Y. Intratumoral drug delivery with nanoparticulate carriers. Pharm Res. 2011;28:1819-30
20. Leung J, Wu S, Chou K, Signorell R. Investigation of sub-100 nm gold nanoparticles for laser-induced thermotherapy of cancer. Nanomaterials. 2013;3:86-106
21. 2014. Clinical trials: NIH. http://clinicaltrials.gov/
22. Libutti SK, Paciotti GF, Byrnes AA, Alexander HR, Gannon WE, Walker M. et al. Phase I and pharmacokinetic studies of CYT-6091, a novel PEGylated colloidal gold-rhTNF nanomedicine. Clin Cancer Res. 2010;16:6139-49
23. Alexander CM, Hamner KL, Maye MM, Dabrowiak JC. Multifunctional DNA-gold nanoparticles for targeted doxorubicin delivery. Bioconjug Chem. 2014
24. Park H, Tsutsumi H, Mihara H. Cell-selective intracellular drug delivery using doxorubicin and α-helical peptides conjugated to gold nanoparticles. Biomaterials. 2014;35:3480-7
25. Sakhrani NM, Padh H. Organelle targeting: Third level of drug targeting. Drug Des Devel Ther. 2013;7:585-99
26. Yuan H, Fales AM, Vo-Dinh T. Tat peptide-functionalized gold nanostars: Enhanced intracellular delivery and efficient nir photothermal therapy using ultralow irradiance. J Am Chem Soc. 2012;134:11358-61
27. Kong T, Zeng J, Wang X, Yang X, Yang J, McQuarrie S. et al. Enhancement of radiation cytotoxicity in breast-cancer cells by localized attachment of gold nanoparticles. Small. 2008;4:1537-43
28. Brown SD, Nativo P, Smith J-A, Stirling D, Edwards PR, Venugopal B. et al. Gold nanoparticles for the improved anticancer drug delivery of the active component of oxaliplatin. J Am Chem Soc. 2010;132:4678-84
29. Seidman AD, Conlin AK, Bach A, Moynahan ME, Lake D, Forero A. et al. Randomized phase II trial of weekly vs. every 2 weeks vs. every 3 weeks nanoparticle albumin-bound paclitaxel with bevacizumab as first-line chemotherapy for metastatic breast cancer. Clin Breast Cancer. 2013;13:239-46.e1
30. Jaggi AS, Singh N. Mechanisms in cancer-chemotherapeutic drugs-induced peripheral neuropathy. Toxicology. 2012;291:1-9
31. Vyas D, Laput G, Vyas AK. Chemotherapy-enhanced inflammation may lead to the failure of therapy and metastasis. Onco Targets Ther. 2014;7:1015-23
32. Mackey MA, Saira F, Mahmoud MA, El-Sayed MA. Inducing cancer cell death by targeting its nucleus: Solid gold nanospheres versus hollow gold nanocages. Bioconjug Chem. 2013;24:897-906
33. Chen W-H, Chen J-X, Cheng H, Chen C-S, Yang J, Xu X-D. et al. A new anti-cancer strategy of damaging mitochondria by pro-apoptotic peptide functionalized gold nanoparticles. Chem Commun (Camb). 2013;49:6403-5
34. Wang F, Wang Y-C, Dou S, Xiong M-H, Sun T-M, Wang J. Doxorubicin-tethered responsive gold nanoparticles facilitate intracellular drug delivery for overcoming multidrug resistance in cancer cells. ACS Nano. 2011;5:3679-92
35. Gibson JD, Khanal BP, Zubarev ER. Paclitaxel-functionalized gold nanoparticles. J Am Chem Soc. 2007;129:11653-61
36. Kumar A, Huo S, Zhang X, Liu J, Tan A, Li S. et al. Neuropilin-1-targeted gold nanoparticles enhance therapeutic efficacy of platinum(IV) drug for prostate cancer treatment. ACS Nano. 2014;8:4205-20
37. Chen H, Zhang X, Dai S, Ma Y, Cui S, Achilefu S. et al. Multifunctional gold nanostar conjugates for tumor imaging and combined photothermal and chemo-therapy. Theranostics. 2013;3:633-49
38. Almeida JPM, Figueroa ER, Drezek RA. Gold nanoparticle mediated cancer immunotherapy. Nanomed Nanotechnol Biol Med. 2014;10:503-14
39. Tkachenko AG, Xie H, Coleman D, Glomm W, Ryan J, Anderson MF. et al. Multifunctional gold nanoparticle-peptide complexes for nuclear targeting. J Am Chem Soc. 2003;125:4700-1
40. Akhter S, Ahmad MZ, Ahmad FJ, Storm G, Kok RJ. Gold nanoparticles in theranostic oncology: Current state-of-the-art. Expert Opin Drug Deliv. 2012;9:1225-43
41. Murphy CJ, Gole AM, Stone JW, Sisco PN, Alkilany AM, Goldsmith EC. et al. Gold nanoparticles in biology: Beyond toxicity to cellular imaging. Acc Chem Res. 2008;41:1721-30
42. Dreaden EC, El-Sayed MA. Detecting and destroying cancer cells in more than one way with noble metals and different confinement properties on the nanoscale. Acc Chem Res. 2012;45:1854-65
43. Verma A, Stellacci F. Effect of surface properties on nanoparticle-cell interactions. Small. 2010;6:12-21
44. Llevot A, Astruc D. Applications of vectorized gold nanoparticles to the diagnosis and therapy of cancer. Chem Soc Rev. 2012;41:242-57
45. Cho EC, Xie J, Wurm PA, Xia Y. Understanding the role of surface charges in cellular adsorption versus internalization by selectively removing gold nanoparticles on the cell surface with a I2/KI etchant. Nano Lett. 2009;9:1080-4
46. Kodiha M, Hutter E, Boridy S, Juhas M, Maysinger D, Stochaj U. Gold nanoparticles induce nuclear damage in breast cancer cells which is further amplified by hyperthermia. Cell Mol Life Sci. 2014;71:4259-73
47. Wang L, Liu Y, Li W, Jiang X, Ji Y, Wu X. et al. Selective targeting of gold nanorods at the mitochondria of cancer cells: Implications for cancer therapy. Nano Lett. 2010;11:772-80
48. Tkachenko AG, Xie H, Liu YL, Coleman D, Ryan J, Glomm WR. et al. Cellular trajectories of peptide-modified gold particle complexes: Comparison of nuclear localization signals and peptide transduction domains. Bioconjug Chem. 2004;15:482-90
49. Chithrani BD, Ghazani AA, Chan WCW. Determining the size and shape dependence of gold nanoparticle uptake into mammalian cells. Nano Lett. 2006;6:662-8
50. Chithrani BD, Chan WCW. Elucidating the mechanism of cellular uptake and removal of protein-coated gold nanoparticles of different sizes and shapes. Nano Lett. 2007;7:1542-50
51. Mandal D, Maran A, Yaszemski M, Bolander M, Sarkar G. Cellular uptake of gold nanoparticles directly cross-linked with carrier peptides by osteosarcoma cells. J Mater Sci Mater Med. 2009;20:347-50
52. Sun L, Liu D, Wang Z. Functional gold nanoparticle-peptide complexes as cell-targeting agents. Langmuir. 2008;24:10293-7
53. Dekiwadia CD, Lawrie AC, Fecondo JV. Peptide-mediated cell penetration and targeted delivery of gold nanoparticles into lysosomes. J Pept Sci. 2012;18:527-34
54. Song J, Zhou J, Duan H. Self-assembled plasmonic vesicles of sers-encoded amphiphilic gold nanoparticles for cancer cell targeting and traceable intracellular drug delivery. J Am Chem Soc. 2012;134:13458-69
55. Dam DHM, Lee JH, Sisco PN, Co DT, Zhang M, Wasielewski MR. et al. Direct observation of nanoparticle-cancer cell nucleus interactions. ACS Nano. 2012;6:3318-26
56. Kang B, Mackey MA, El-Sayed MA. Nuclear targeting of gold nanoparticles in cancer cells induces DNA damage, causing cytokinesis arrest and apoptosis. J Am Chem Soc. 2010;132:1517-9
57. Shah NB, Dong J, Bischof JC. Cellular uptake and nanoscale localization of gold nanoparticles in cancer using label-free confocal raman microscopy. Mol Pharm. 2010;8:176-84
58. Gu Y-J, Cheng J, Lin C-C, Lam YW, Cheng SH, Wong W-T. Nuclear penetration of surface functionalized gold nanoparticles. Toxicol Appl Pharmacol. 2009;237:196-204
59. Ryan JA, Overton KW, Speight ME, Oldenburg CM, Loo L, Robarge W. et al. Cellular uptake of gold nanoparticles passivated with bsa-sv40 large t antigen conjugates. Anal Chem. 2007;79:9150-9
60. Huo S, Jin S, Ma X, Xue X, Yang K, Kumar A. et al. Ultrasmall gold nanoparticles as carriers for nucleus-based gene therapy due to size-dependent nuclear entry. ACS Nano. 2014;8:5852-62
61. de la Fuente JM, Berry CC. Tat peptide as an efficient molecule to translocate gold nanoparticles into the cell nucleus. Bioconjug Chem. 2005;16:1176-80
62. Berry CC, de la Fuente JM, Mullin M, Chu SW, Curtis AS. Nuclear localization of HIV-1 tat functionalized gold nanoparticles. IEEE Trans Nanobioscience. 2007;6:262-9
63. Arvizo RR, Miranda OR, Thompson MA, Pabelick CM, Bhattacharya R, Robertson JD. et al. Effect of nanoparticle surface charge at the plasma membrane and beyond. Nano Lett. 2010;10:2543-8
64. Ojea-Jimenez I, Garcia-Fernandez L, Lorenzo J, Puntes VF. Facile preparation of cationic gold nanoparticle-bioconjugates for cell penetration and nuclear targeting. ACS Nano. 2012;6:7692-702
65. Chithrani BD, Stewart J, Allen C, Jaffray DA. Intracellular uptake, transport, and processing of nanostructures in cancer cells. Nanomed Nanotechnol Biol Med. 2009;5:118-27
66. Nativo P, Prior IA, Brust M. Uptake and intracellular fate of surface-modified gold nanoparticles. ACS Nano. 2008;2:1639-44
67. Oyelere AK, Chen PC, Huang X, El-Sayed IH, El-Sayed MA. Peptide-conjugated gold nanorods for nuclear targeting. Bioconjug Chem. 2007;18:1490-7
68. Yang C, Uertz J, Yohan D, Chithrani BD. Peptide modified gold nanoparticles for improved cellular uptake, nuclear transport, and intracellular retention. Nanoscale. 2014;6:12026-33
69. Connor EE, Mwamuka J, Gole A, Murphy CJ, Wyatt MD. Gold nanoparticles are taken up by human cells but do not cause acute cytotoxicity. Small. 2005;1:325-7
70. Dixit V, Van den Bossche J, Sherman DM, Thompson DH, Andres RP. Synthesis and grafting of thioctic acid-PEG-folate conjugates onto au nanoparticles for selective targeting of folate receptor-positive tumor cells. Bioconjug Chem. 2006;17:603-9
71. Huff TB, Hansen MN, Zhao Y, Cheng J-X, Wei A. Controlling the cellular uptake of gold nanorods. Langmuir. 2007;23:1596-9
72. Huff TB, Tong L, Zhao Y, Hansen MN, Cheng J-X, Wei A. Hyperthermic effects of gold nanorods on tumor cells. Nanomedicine. 2007;2:125-32
73. Bhattacharya R, Patra CR, Earl A, Wang S, Katarya A, Lu L. et al. Attaching folic acid on gold nanoparticles using noncovalent interaction via different polyethylene glycol backbones and targeting of cancer cells. Nanomed Nanotechnol Biol Med. 2007;3:224-38
74. Shirazi AN, Tiwari RK, Oh D, Sullivan B, McCaffrey K, Mandal D. et al. Surface decorated gold nanoparticles by linear and cyclic peptides as molecular transporters. Mol Pharm. 2013;10:10.1021 /mp400199e
75. Shukla R, Bansal V, Chaudhary M, Basu A, Bhonde RR, Sastry M. Biocompatibility of gold nanoparticles and their endocytotic fate inside the cellular compartment: A microscopic overview. Langmuir. 2005;21:10644-54
76. Chen Z, Zhang L, He Y, Li Y. Sandwich-type Au-PEI/DNA/PEI-Dexa nanocomplex for nucleus-targeted gene delivery in vitro and in vivo. ACS Appl Mater Interfaces. 2014;6:14196-206
77. Han G, You C-C, Kim B-j, Turingan RS, Forbes NS, Martin CT. et al. Light-regulated release of DNA and its delivery to nuclei by means of photolabile gold nanoparticles. Angew Chem Int Ed Engl. 2006;45:3165-9
78. El-Sayed IH, Huang X, El-Sayed MA. Surface plasmon resonance scattering and absorption of anti-egfr antibody conjugated gold nanoparticles in cancer diagnostics: Applications in oral cancer. Nano Lett. 2005;5:829-34
79. Huang X, El-Sayed IH, Qian W, El-Sayed MA. Cancer cell imaging and photothermal therapy in the near-infrared region by using gold nanorods. J Am Chem Soc. 2006;128:2115-20
80. Hauck TS, Ghazani AA, Chan WCW. Assessing the effect of surface chemistry on gold nanorod uptake, toxicity, and gene expression in mammalian cells. Small. 2008;4:153-9
81. Oh E, Delehanty JB, Sapsford KE, Susumu K, Goswami R, Blanco-Canosa JB. et al. Cellular uptake and fate of PEGylated gold nanoparticles is dependent on both cell-penetration peptides and particle size. ACS Nano. 2011;5:6434-48
82. Liu X, Huang N, Li H, Jin Q, Ji J. Surface and size effects on cell interaction of gold nanoparticles with both phagocytic and nonphagocytic cells. Langmuir. 2013;29:9138-48
83. Coradeghini R, Gioria S, García CP, Nativo P, Franchini F, Gilliland D. et al. Size-dependent toxicity and cell interaction mechanisms of gold nanoparticles on mouse fibroblasts. Toxicol Lett. 2013;217:205-16
84. Pan Y, Neuss S, Leifert A, Fischler M, Wen F, Simon U. et al. Size-dependent cytotoxicity of gold nanoparticles. Small. 2007;3:1941-9
85. Huang K, Ma H, Liu J, Huo S, Kumar A, Wei T. et al. Size-dependent localization and penetration of ultrasmall gold nanoparticles in cancer cells, multicellular spheroids, and tumors in vivo. ACS Nano. 2012;6:4483-93
86. Salnikov V, Lukyanenko YO, Frederick CA, Lederer WJ, Lukyanenko V. Probing the outer mitochondrial membrane in cardiac mitochondria with nanoparticles. Biophys J. 2007;92:1058-71
87. Yamada Y, Akita H, Kamiya H, Kogure K, Yamamoto T, Shinohara Y. et al. Mito-porter: A liposome-based carrier system for delivery of macromolecules into mitochondria via membrane fusion. Biochim Biophys Acta. 2008;1778:423-32
88. Wang L, Jiang X, Ji Y, Bai R, Zhao Y, Wu X. et al. Surface chemistry of gold nanorods: Origin of cell membrane damage and cytotoxicity. Nanoscale. 2013;5:8384-91
89. Zhuang Q, Jia H, Du L, Li Y, Chen Z, Huang S. et al. Targeted surface-functionalized gold nanoclusters for mitochondrial imaging. Biosensors Bioelectron. 2014;55:76-82
90. Mocan L, Ilie I, Tabaran FA, Dana B, Zaharie F, Zdrehus C. et al. Surface plasmon resonance-induced photoactivation of gold nanoparticles as mitochondria-targeted therapeutic agents for pancreatic cancer. Expert Opin Ther Targets. 2013;17:1383-93
91. Marrache S, Dhar S. The energy blocker inside the power house: Mitochondria targeted delivery of 3-bromopyruvate. Chem Science. 2015 doi:10.1039/C4SC01963
92. Cheng Y, Meyers JD, Agnes RS, Doane TL, Kenney ME, Broome A-M. et al. Addressing brain tumors with targeted gold nanoparticles: A new gold standard for hydrophobic drug delivery? Small. 2011;7:2301-6
93. Scarì G, Porta F, Fascio U, Avvakumova S, Dal Santo V, De Simone M. et al. Gold nanoparticles capped by a gc-containing peptide functionalized with an rgd motif for integrin targeting. Bioconjug Chem. 2012;23:340-9
94. de la Fuente JM, Berry CC, Riehle MO, Curtis ASG. Nanoparticle targeting at cells. Langmuir. 2006;22:3286-93
95. Sykes EA, Chen J, Zheng G, Chan WCW. Investigating the impact of nanoparticle size on active and passive tumor targeting efficiency. ACS Nano. 2014;8:5696-706
96. Eghtedari M, Liopo AV, Copland JA, Oraevsky AA, Motamedi M. Engineering of hetero-functional gold nanorods for the in vivo molecular targeting of breast cancer cells. Nano Lett. 2008;9:287-91
97. El-Sayed IH, Huang X, El-Sayed MA. Selective laser photo-thermal therapy of epithelial carcinoma using anti-EGFR antibody conjugated gold nanoparticles. Cancer Lett. 2006;239:129-35
98. de las Heras JI, Batrakou DG, Schirmer EC. Cancer biology and the nuclear envelope: A convoluted relationship. Semin Cancer Biol. 2013;23:125-37
99. Jevtić P, Edens LJ, Vuković LD, Levy DL. Sizing and shaping the nucleus: Mechanisms and significance. Curr Opin Cell Biol. 2014;28:16-27
100. Matchett KB, McFarlane S, Hamilton SE, Eltuhamy YS, Davidson MA, Murray JT. et al. Ran GTPase in nuclear envelope formation and cancer metastasis. Adv Exp Med Biol. 2014;773:323-51
101. Miyamoto Y, Loveland KL, Yoneda Y. Nuclear importin alpha and its physiological importance. Commun Integr Biol. 2012;5:220-2
102. Quin JE, Devlin JR, Cameron D, Hannan KM, Pearson RB, Hannan RD. Targeting the nucleolus for cancer intervention. Biochim Biophys Acta. 2014;1842:802-16
103. Ruggero D. Revisiting the nucleolus: From marker to dynamic integrator of cancer signaling. Sci Signal. 2012;5:pe38
104. Mor A, White MA, Fontoura BMA. Nuclear trafficking in health and disease. Curr Opin Cell Biol. 2014;28:28-35
105. Kuusisto HV, Wagstaff KM, Alvisi G, Roth DM, Jans DA. Global enhancement of nuclear localization-dependent nuclear transport in transformed cells. FASEB J. 2012;26:1181-93
106. Feldherr CM, Akin D. The permeability of the nuclear envelope in dividing and nondividing cell cultures. J Cell Biol. 1990;111:1-8
107. Feldherr CM, Akin D. Signal-mediated nuclear transport in proliferating and growth-arrested BALB/c 3T3 cells. J Cell Biol. 1991;115:933-9
108. Feldherr CM, Akin D. Regulation of nuclear transport in proliferating and quiescent cells. Exp Cell Res. 1993;205:179-86
109. Tsoli M, Kuhn H, Brandau W, Esche H, Schmid G. Cellular uptake and toxicity of Au55 clusters. Small. 2005;1:841-4
110. Panté N, Kann M. Nuclear pore complex is able to transport macromolecules with diameters of ∼39 nm. Mol Biol Cell. 2002;13:425-34
111. Feldherr CM, Akin D, Cohen RJ. Regulation of functional nuclear pore size in fibroblasts. J Cell Sci. 2001;114:4621-7
112. Feldherr CM, Akin D. The location of the transport gate in the nuclear pore complex. J Cell Sci. 1997;110:3065-70
113. Novak JP, Nickerson C, Franzen S, Feldheim DL. Purification of molecularly bridged metal nanoparticle arrays by centrifugation and size exclusion chromatography. Anal Chem. 2001;73:5758-61
114. Krpetić Ž, Saleemi S, Prior IA, Sée V, Qureshi R, Brust M. Negotiation of intracellular membrane barriers by TAT-modified gold nanoparticles. ACS Nano. 2011;5:5195-201
115. Tsai TL, Hou CC, Wang HC, Yang ZS, Yeh CS, Shieh DB. et al. Nucleocytoplasmic transport blockage by SV40 peptide-modified gold nanoparticles induces cellular autophagy. Int J Nanomedicine. 2012;7:5215-34
116. Huschka R, Barhoumi A, Liu Q, Roth JA, Ji L, Halas NJ. Gene silencing by gold nanoshell-mediated delivery and laser-triggered release of antisense oligonucleotide and siRNA. ACS Nano. 2012;6:7681-91
117. Ryou S-M, Park M, Kim J-M, Jeon CO, Yun C-H, Han SH. et al. Inhibition of xenograft tumor growth in mice by gold nanoparticle-assisted delivery of short hairpin RNAs against Mcl-1L. J Biotechnol. 2011;156:89-94
118. Kim J-H, Jang HH, Ryou S-M, Kim S, Bae J, Lee K. et al. A functionalized gold nanoparticles-assisted universal carrier for antisense DNA. Chem Commun (Camb). 2010;46:4151-3
119. Kim D-W, Kim J-H, Park M, Yeom J-H, Go H, Kim S. et al. Modulation of biological processes in the nucleus by delivery of DNA oligonucleotides conjugated with gold nanoparticles. Biomaterials. 2011;32:2593-604
120. Wallace DC. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: A dawn for evolutionary medicine. Annu Rev Genet. 2005;39:359-407
121. Pathania D, Millard M, Neamati N. Opportunities in discovery and delivery of anticancer drugs targeting mitochondria and cancer cell metabolism. Adv Drug Del Rev. 2009;61:1250-75
122. Wen S, Zhu D, Huang P. Targeting cancer cell mitochondria as a therapeutic approach. Future Med Chem. 2012;5:53-67
123. Rehling P, Model K, Brandner K, Kovermann P, Sickmann A, Meyer HE. et al. Protein insertion into the mitochondrial inner membrane by a twin-pore translocase. Science. 2003;299:1747-51
124. Szabo I, Zoratti M. Mitochondrial channels: Ion fluxes and more. Physiol Rev. 2014;94:519-608
125. Smith RA, Hartley RC, Murphy MP. Mitochondria-targeted small molecule therapeutics and probes. Antioxid Redox Signal. 2011;15:3021-38
126. Clarke Hanna J, Chambers Joseph E, Liniker E, Marciniak Stefan J. Endoplasmic reticulum stress in malignancy. Cancer Cell. 2014;25:563-73
127. Chang M-Y, Shiau A-L, Chen Y-H, Chang C-J, Chen HHW, Wu C-L. Increased apoptotic potential and dose-enhancing effect of gold nanoparticles in combination with single-dose clinical electron beams on tumor-bearing mice. Cancer Sci. 2008;99:1479-84
128. Khan JA, Pillai B, Das TK, Singh Y, Maiti S. Molecular effects of uptake of gold nanoparticles in HeLa cells. ChemBioChem. 2007;8:1237-40
129. Cho W-S, Cho M, Jeong J, Choi M, Cho H-Y, Han BS. et al. Acute toxicity and pharmacokinetics of 13 nm-sized PEG-coated gold nanoparticles. Toxicol Appl Pharmacol. 2009;236:16-24
130. Tsai Y-Y, Huang Y-H, Chao Y-L, Hu K-Y, Chin L-T, Chou S-H. et al. Identification of the nanogold particle-induced endoplasmic reticulum stress by omic techniques and systems biology analysis. ACS Nano. 2011;5:9354-69
131. Kim CS, Le NDB, Xing Y, Yan B, Tonga GY, Kim C. et al. The role of surface functionality in nanoparticle exocytosis. Adv Healthc Mater. 2014;3:1200-2
132. Oh N, Park J-H. Surface chemistry of gold nanoparticles mediates their exocytosis in macrophages. ACS Nano. 2014;8:6232-41
133. Simpson CA, Salleng KJ, Cliffel DE, Feldheim DL. In vivo toxicity, biodistribution, and clearance of glutathione-coated gold nanoparticles. Nanomedicine. 2013;9:257-63
134. Jones SW, Roberts RA, Robbins GR, Perry JL, Kai MP, Chen K. et al. Nanoparticle clearance is governed by Th1/Th2 immunity and strain background. J Clin Invest. 2013;123:3061-73
135. van der Watt PJ, Stowell CL, Leaner VD. The nuclear import receptor kpnb1 and its potential as an anticancer therapeutic target. Crit Rev Eukaryot Gene Expr. 2013;23:1-10
136. Christiansen A, Dyrskjøt L. The functional role of the novel biomarker karyopherin α 2 (KPNA2) in cancer. Cancer Lett. 2013;331:18-23
137. Fan H, Lu Y, Qin H, Zhou Y, Gu Y, Zhou J. et al. High ran level is correlated with poor prognosis in patients with colorectal cancer. Int J Clin Oncol. 2013;18:856-63
138. Truant R, Cullen BR. The arginine-rich domains present in human immunodeficiency virus type 1 Tat and Rev function as direct importin β-dependent nuclear localization signals. Mol Cell Biol. 1999;19:1210-7
139. Ori A, Banterle N, Iskar M, Andrés-Pons A, Escher C, Khanh Bui H. et al. Cell type-specific nuclear pores: A case in point for context-dependent stoichiometry of molecular machines. Mol Syst Biol. 2013;9:648
140. Krpetic Z, Nativo P, See V, Prior IA, Brust M, Volk M. Inflicting controlled nonthermal damage to subcellular structures by laser-activated gold nanoparticles. Nano Lett. 2010;10:4549-54
141. Neuberg P, Kichler A. Recent developments in nucleic acid delivery with polyethylenimines. Adv Genet. 2014;88:263-88
142. Sheftel AD, Zhang AS, Brown C, Shirihai OS, Ponka P. Direct interorganellar transfer of iron from endosome to mitochondrion. Blood. 2007;110:125-32
143. Huo S, Ma H, Huang K, Liu J, Wei T, Jin S. et al. Superior penetration and retention behavior of 50 nm gold nanoparticles in tumors. Cancer Res. 2013;73:319-30
144. Lin M, Guo C, Li J, Zhou D, Liu K, Zhang X. et al. Polypyrrole-coated chainlike gold nanoparticle architectures with the 808 nm photothermal transduction efficiency up to 70%. ACS Appl Mater Interfaces. 2014;6:5860-8
145. Song J, Fang Z, Wang C, Zhou J, Duan B, Pu L. et al. Photolabile plasmonic vesicles assembled from amphiphilic gold nanoparticles for remote-controlled traceable drug delivery. Nanoscale. 2013;5:5816-24
146. Kanaras AG, Wang Z, Brust M, Cosstick R, Bates AD. Enzymatic disassembly of DNA-gold nanostructures. Small. 2007;3:590-4
Author contact
![]() Corresponding author: Ursula Stochaj, Ph.D. Department of Physiology, McGill University, 3655 Promenade Sir William Osler, Montreal, QC, Canada, H3G 1Y6. Phone: 514-398-2949 Fax: 514-398-7452 E-mail: ursula.stochajca.
Corresponding author: Ursula Stochaj, Ph.D. Department of Physiology, McGill University, 3655 Promenade Sir William Osler, Montreal, QC, Canada, H3G 1Y6. Phone: 514-398-2949 Fax: 514-398-7452 E-mail: ursula.stochajca.
Citation styles
APA
Kodiha, M., Wang, Y.M., Hutter, E., Maysinger, D., Stochaj, U. (2015). Off to the Organelles - Killing Cancer Cells with Targeted Gold Nanoparticles. Theranostics, 5(4), 357-370. https://doi.org/10.7150/thno.10657.
ACS
Kodiha, M.; Wang, Y.M.; Hutter, E.; Maysinger, D.; Stochaj, U. Off to the Organelles - Killing Cancer Cells with Targeted Gold Nanoparticles. Theranostics 2015, 5 (4), 357-370. DOI: 10.7150/thno.10657.
NLM
Kodiha M, Wang YM, Hutter E, Maysinger D, Stochaj U. Off to the Organelles - Killing Cancer Cells with Targeted Gold Nanoparticles. Theranostics 2015; 5(4):357-370. doi:10.7150/thno.10657. https://www.thno.org/v05p0357.htm
CSE
Kodiha M, Wang YM, Hutter E, Maysinger D, Stochaj U. 2015. Off to the Organelles - Killing Cancer Cells with Targeted Gold Nanoparticles. Theranostics. 5(4):357-370.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) License. See http://ivyspring.com/terms for full terms and conditions.