Theranostics
13.3
Impact Factor
- Current Issue
- Advance Articles
- Volume 16; 2026
- Volume 15; 2025
- Volume 14; 2024
- Volume 13; 2023
- Volume 12; 2022
- Archive
- Cover Images
- Cover Suggestion
- Special Issues
Top
Introduction
Materials and methods
Results
Discussion
Conclusion
Supplementary Material
Acknowledgements
Authors'contributions
References
Introduction
Materials and methods
Results
Discussion
Conclusion
Supplementary Material
Acknowledgements
Authors'contributions
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2015; 5(12):1343-1362. doi:10.7150/thno.11685 This issue Cite
Research Paper
Blockade of Interplay between IL-17A and Endoplasmic Reticulum Stress Attenuates LPS-Induced Lung Injury
So Ri Kim1* ![]() , Hee Jung Kim1*, Dong Im Kim1, Kyung Bae Lee1, Hae Jin Park1, Jae Seok Jeong1, Seong Ho Cho2, Yong Chul Lee1
, Hee Jung Kim1*, Dong Im Kim1, Kyung Bae Lee1, Hae Jin Park1, Jae Seok Jeong1, Seong Ho Cho2, Yong Chul Lee1 ![]()
1. Department of Internal Medicine, Research Center for Pulmonary Disorders, Chonbuk National University Medical School, Research Institute of Clinical Medicine of Chonbuk National University-Biomedical Research Institute of Chonbuk National University Hospital, San 2-20 Geumam-dong, Deokjin-gu, Jeonju, 561-180, South Korea;
2. Division of Allergy-Immunology, Department of Internal Medicine, Morsani College of Medicine, University of South Florida, Tampa, FL, USA
* These authors contributed equally to this work.
Received 2015-1-25; Accepted 2015-8-30; Published 2015-10-7
Citation:
Kim SR, Kim HJ, Kim DI, Lee KB, Park HJ, Jeong JS, Cho SH, Lee YC. Blockade of Interplay between IL-17A and Endoplasmic Reticulum Stress Attenuates LPS-Induced Lung Injury. Theranostics 2015; 5(12):1343-1362. doi:10.7150/thno.11685. https://www.thno.org/v05p1343.htm
Other stylesAbstract

IL-17 is a cytokine mainly from IL-17-producing T cells, which are one of subsets of CD4+ T cells and play a role in adaptive immune system. Recent studies have demonstrated that IL-17A can act rapidly as an innate immune responder during infection before the onset of its classic adaptive immune response. This role of IL-17A in innate immune response is implicated in lipopolysaccharide (LPS)-induced lung inflammation. Very recently, we have reported that endoplasmic reticulum (ER) stress is involved in LPS-induced lung inflammation in vivo and in vitro. This study aimed to elucidate the role of IL-17A in LPS-induced lung injury, focusing on the link with ER stress. We treated a murine model of LPS-induced lung injury with IL-17A neutralizing antibody and 4-phenylbutyrate (4-PBA), a representative ER stress inhibitor. In addition, we evaluated the effects of IL-17A on ER stress in LPS-stimulated bronchial epithelial cells. Our results showed that inhibition of IL-17A decreased LPS-induced pulmonary neutrophilia, vascular leakage, nuclear translocation of nuclear factor-κB (NF-κB), infiltration of dendritic cells, increased expression of Toll-like receptor 4 (TLR4), activation of NLRP3 inflammasome, and increased ER stress in the lung. 4-PBA or TAK-242, a TLR4 inhibitor attenuated expression of IL-17A thereby improving LPS-induced lung inflammation. Intriguingly, we observed that stimulation with LPS increased expression of IL-17A in airway epithelial cells and co-stimulation with IL-17A further increased ER stress and NF-κB activation. This study indicates that the interrelationship between IL-17A and ER stress plays an important role in LPS-induced injury showing a positive feedback in airway epithelial cells and suggests that targeting their interaction can be a potential therapeutic approach to overcome one of severe refractory pulmonary disorders.
Keywords: IL-17A, ER stress, LPS, inflammation, epithelial cell, acute lung injury
Introduction
IL-17 is a cytokine mainly from IL-17-producing T cells (Th17 cells), one of subsets of CD4+ T cells, and plays a role in adaptive immune system [1]. In addition, it is well known to regulate innate immune responses of various cells, including macrophages, neutrophils, or epithelial cells in several pathologic processes [1]. In fact, genes induced by IL-17 encode antimicrobial proteins (β-defensins, cathelicidin, RegIII, lipocalin 2, salivary histatins), neutrophil-activating factors (G-CSF, CXC chemokines), and inducers of the acute phase response (IL-6) [2]. Especially, IL-17A, IL-17C, and IL-17F can directly target epithelial cells to induce various antimicrobial responses against extracellular pathogens and promote tissue remodeling [3]. These findings imply that IL-17 acts rapidly as an innate immune responder during infection before the onset of a classic role of IL-17 through adaptive T cell response. The indispensable roles of IL-17A and IL-17F in host defense are supported by various in vivo infection models [4, 5]. However, uncontrolled IL-17 responses can amplify inflammation and cause tissue damage in many inflammatory and infectious diseases [4, 6].
Although IL-17A is a signature cytokine from the Th17 cells and also commonly called IL-17, the IL-17 cytokine family consists of six members: IL-17A, IL-17B, IL-17C, IL-17D, IL-17E (IL-25), and IL-17F [4]. Their immune responses are initiated by binding of IL-17 cytokines to receptor complexes composed of heterodimers of several IL-17 receptor subunits: IL-17RA, IL-17RB, IL-17RC, IL-17RD, and IL-17RE [2]. Among the IL-17 family members, IL-17A and IL-17F share the highest amino acid sequence identity (50%), whereas the sequence of IL-17B, IL-17C, and IL-17E differ from those of IL-17A and IL-17F, suggesting that IL-17B, IL-17C, and IL-17E may form a distinct subclass [7, 8]. Despite the high structural homology, IL-17A and IL-17F function distinctly due to the differences in cellular sources, the induction of pro-inflammatory cytokines, and the distribution of their functional receptors [4]. In brief, recent accumulating evidence has indicated that IL-17A is involved in the induction of pro-inflammatory responses associated with development of certain diseases, and also plays important roles in the host defenses against bacterial and fungal infections, whereas IL-17F is mainly involved in mucosal host defense mechanisms [4]. Alternatively, IL-17E has known to be involved in promoting Th2 cell-type immune responses. The proposed functions of IL-17B, IL-17C, and IL-17D include pro-inflammatory cytokine induction and neutrophil recruitments, however, their roles remain largely unclear and these cytokines do not target the epithelial cells mainly. These characteristics make researchers more inclined towards IL-17A than other IL-17 family members in infectious diseases.
IL-17A is involved in neutrophil recruitment to the lung by both infectious and noninfectious agents [9, 10]. In fact, we have reported that IL-17A is one of the key players in eosinophilic as well as neutrophilic airway inflammation using animal models of asthma induced by toluene diisocyanate or ovalbumin [11, 12]. In addition, IL-17 has been reported to be necessary for lipopolysaccharide (LPS)-induced airway neutrophilia [13]. As well known, LPS fragments that coat the outer membrane of Gram-negative bacteria are ubiquitous in the environment and commonly involved in a number of infectious and inflammatory disorders [14]. As for the role of IL-17A, however, it is not fully understood in LPS-induced lung inflammation in vivo, especially in the innate immune responses.
Endoplasmic reticulum (ER) stress is defined as accumulation of unfolded or misfolded proteins in the ER and is implicated in several diseases, such as cancer, neurodegeneration, diabetes, and inflammatory conditions [15]. To cope with stress, cells activate a complex signal transduction pathway known as the unfolded protein response (UPR) that integrates information on the intensity and duration of the stress to adapt and recover ER homeostasis or to trigger apoptosis of irreversibly damaged cells [16]. The UPR signaling is made up of inositol-requiring 1α (IRE1α), double-stranded RNA-dependent protein kinase (PKR)-like ER kinase (PERK), and activating transcription factor (ATF)-6. When these protein sensors recognize the increased ER stress, they activate the UPR process such as increases of expression of glucose-regulated protein 78 (GRP78) and CCAAT/enhancer-binding protein-homologous protein (CHOP). Very recently, we have demonstrated that ER stress is involved in LPS-induced lung inflammation in vivo and expression of GRP78 and CHOP is up-regulated in LPS-stimulated airway epithelial cells in vitro [17]. Studies do indicate not only that ER stress amplifies inflammatory reactions [18] but also that several pro-inflammatory cytokines such as TNF-α and IFN-γ induce ER stress [19, 20], thus mounting and propagating inflammatory processes. Moreover, several reports have suggested that UPR stress sensors are evolutionarily related to regulators of innate immunity [18, 21, 22].
Considering the contribution of IL-17A and ER stress to infectious and inflammatory processes, the possible interaction between IL-17A and ER stress in LPS-induced lung inflammation is emerging as a pathogenic component of infectious pulmonary disorders. However, there is no available information on the relationship between IL-17A and ER stress in lung inflammation. In this study, we elucidated the role of IL-17A in LPS-induced lung inflammation, focusing on the link with ER stress in a mouse model of LPS-induced lung injury using an IL-17A neutralizing antibody and 4-phenylbutyrate (4-PBA), a representative ER stress inhibitor. In addition, we evaluated the effects of IL-17A on ER stress in LPS-stimulated bronchial epithelial cells.
Materials and methods
Animals and experimental protocol
Female C57BL/6 mice, 7 to 8 weeks of age and free of murine specific pathogens, were obtained from the Orient Bio Inc. (Seoungnam, Korea). They were housed throughout the experiments in a laminar flow cabinet and were maintained on standard laboratory chow ad libitum. All experimental animals used in this study were under a protocol approved by the Institutional Animal Care and Use Committee of the Chonbuk National University (Jeonju, Korea). Mice were treated once by intratracheal instillation with 50 μg of LPS (Sigma-Aldrich, St. Louis, MO, USA) in 50 μl saline (or with saline as a control) under anesthesia using inhaled isoflurane (Matrix, Orchard Park, NY, USA). For the induction of ER stress in saline-instilled mice, thapsigargin (20 μg/mouse, Sigma-Aldrich) was administered intraperitoneally once into saline-instilled mice at 8 hours before the sacrifice. In addition, recombinant human IL-17A (rhIL-17A)-treated mice were created by the intratracheal instillation of rhIL-17A (5 μg/mouse, R&D Systems, Minneapolis, MN, USA) at 24 hours before the sacrifice. Bronchoalveolar lavage (BAL) was performed at 48 hours after intratracheal instillation of LPS. At the time of lavage, the mice (5 or 6 mice in each group) were sacrificed with dislocation of cervical vertebrae. The chest cavity was exposed to allow for expansion, after which the trachea was carefully intubated. Prewarmed 0.9% NaCl solution was slowly instilled into the lung and withdrawn. The collected solutions were pooled and then kept at 4°C. A part of each pool was used for total cell counting. After centrifugation, the BAL supernatants were stored at -70oC until use. Cell pellets were resuspended with phosphate-buffered saline (PBS, Gibco-Invitrogen, Carlasbad, CA, USA) for cell differentials or immunofluorescence staining. Total cell numbers were counted with a Nucleocounter (Chemometec., Gydevang, Denmark). Smears of BAL cells were prepared by cytospin (Thermo Electron, Waltham, MA, USA), and stained with Diff-Quik solution (Dade Diagnostics of Puerto Rico Inc., Aguada, Puerto Rico) in order to examine cell differentials. At least 400 cells were counted on each of four different corners using a microscope.
Administration of 4-PBA, TAK-242, thapsigargin, rhIL-17A, and neutralizing antibodies for IL-17A, IL-1β, and TNF-α
4-PBA (200 mg/kg of body weight; Calbiochem, San Diego, CA, USA) dissolved in PBS was administered intraperitoneally 4 times to each animal, once at 1 hour before the administration of LPS and at 6, 18, and 24 hours after the administration of LPS. TAK-242 (200 μg/mouse, Calbiochem) dissolved in PBS was administered intravenously 2 times to each animal 1 hour and 24 hours after LPS instillation. Thapsigargin (20 μg/mouse, Sigma-Aldrich) dissolved in DMSO was administered once intratracheally to each animal 40 hours after LPS instillation. rhIL-17A (5 μg/mouse, R&D Systems) was administered once intratracheally to each animal 24 hours after the instillation of LPS. For IL-17A neutralization, murine anti-IL-17A antibody or isotype control monoclonal antibody (5 mg/kg of body weight/day; R&D Systems) was administered by intraperitoneal injection twice to each animal once at 6 hours before the instillation of LPS and the second time at 6 hours after the instillation of LPS. Murine anti-IL-1β monoclonal antibody or isotype control antibody (100 μg/mouse, eBioscience, San Diego, CA, USA) was administered intravenously two times to each animal at 1 hour before LPS instillation and at 6 hours after LPS instillation. Murine anti-TNF-α monoclonal antibody or isotype control antibody (10 μg/mouse, R&D Systems) was administered once intravenously to each animal at 12 hours before instillation of LPS.
Cell culture and treatment
Normal human bronchial epithelial (NHBE) cells were purchased from Lonza (Walkersville, MD, USA). The cells were cultured in bronchial epithelial basal medium (BEBM, Lonza) supplemented with bovine pituitary extract, gentamicin, amphotericin B, hydrocortisone, epidermal growth factor, epinephrine, insulin, triiodothyronine, transferrin, and retinoic acid, which were maintained in a humidified incubator of 5% CO2 atmosphere at 37°C. Cells were seeded in culture dishes and grown until 70% confluence. The medium was then replaced with a basal medium containing vehicle (phosphate buffered saline; PBS), 10 mmol/L 4-PBA (Calbiochem), or 1 μmol/L TAK-242 (Calbiochem) at 1 hour before LPS stimulation. The cells were pre-activated with 50 ng/ml of TNF-α and then stimulated with 100 μg/ml of LPS and/or 100 ng/ml of IL-17A (R&D systems) for 12 hours with 4-PBA or vehicle. Cells were harvested and extracted.
For the neutralization of IL-17A, 2 μg/ml of a human anti-IL-17A antibody or an isotype control monoclonal antibody (R&D Systems) was added to the complete growth medium at 1 hour before LPS treatment [23].
Immunofluorescence staining for GRP78, CHOP, IL-1β, TNF-α, and IL-17A
Paraffin-embedded lung tissue sections were deparaffinized and hydrated. After antigen retrieval, the tissues were permeabilized with 0.1% Triton X-100 (Sigma-Aldrich) in PBS. After blocking with 2% bovine serum albumin (BSA, Sigma-Aldrich) in PBS for 1 hour, they were probed with an anti-GRP78 antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA) and anti-CHOP antibody (Santa Cruz Biotechnology). The slides were then stained with Alexa Fluor 488 (green) or Alexa Fluor 546 (red) conjugated secondary antibodies (Invitrogen, Carlasbad, CA, USA) for detection of GRP78 and CHOP, respectively. For identification of nuclei, the fluorescent nucleic acid dye DAPI (Invitrogen) was applied for 10 minutes. Stained tissues were mounted on slides using fluorescent mounting medium (Golden Bridge International, Inc., Mukilteo, WA, USA), and then visualized using a confocal microscope (Zeiss LSM 510 Meta, Karl Zeiss, Jena, Germany) equipped with a C-Apochromat 63×/1.20W Korr UV-VIS-IR M27 water immersion objective.
For examination of BAL cells, total cells of each group were prepared at an equal number and smeared on slides. As for the examination of cultured NHBE cells, NHBE cells were seeded on coverslips in 3.5 cm dishes and cultured for 24 hours. After the LPS stimulation, cells were quickly washed with PBS on coverslips. Fixation, permeabilization, and blocking had been processed as described above. Cells were probed with antibodies against GRP78 (Santa Cruz Biotechnology), CHOP (Santa Cruz Biotechnology), IL-1β (Thermo Fisher Scientific Inc. Rockford, IL, USA), TNF-α (Thermo Fisher Scientific Inc.), of IL-17A (Santa Cruz Biotechnology), and the above methods of fluorescence staining and mounting were used. Confocal microscopic examination was performed under identical condition and magnification (BAL cells, × 630; NHBE cells, × 200/400).
RNA isolation and semi-quantitative RT-PCR
Levels of mRNA expression were analyzed by RT-PCR assay using total RNA isolated from lung tissues or NHBE cells by a rapid extraction method (TRI-Reagent) as previously described [24]. RNA was quantified by measuring absorption at 260 nm and was stored at -70ºC until use. One μg of total RNA was reverse-transcribed to cDNA using transcriptor first strand cDNA synthesis kit (Roche Diagnostics, Basel, Switzerland). The primers used were as follows: GRP78, sense: 5′-GAAAGGATGGTTAATGATGCTGAG-3′, antisense: 5′-GTCTTCAATGTCCGCATCCTG-3′; CHOP, sense: 5′-CATACACCACCACACCTGAAAG-3′, antisense: 5′-CCGTTTCCTAGTTCTTCCTTGC-3′; IL-17A, sense: 5′-CCCCTAGACTCAGGCTTCCT-3′, antisense: 5′-TCAGCTCCTTTCTGGGTTGT-3′; β-actin, sense: 5'-CAGATCATGTTTGAGACCTTC-3', antisense: 5'-ACTTCATGATGGAATTGAATG-3' and GAPDH, sense: 5′-GGCCTCCAAGGAGTAAGACC-3′, antisense: 5′-AGGGGTCTACATGGCAACTG-3′. PCR reactions were performed in a thermal cycler (MJ mini, Bio-Rad Laboratories, Hercules, CA, USA). The amplified PCR products were electrophoresed using 1.6% agarose gels stained with ethidium bromide. DNA bands were visualized using Gel Doc XR+ Imaging System (Bio-Rad Laboratories). The results were expressed as a relative ratio of the target intensity to intensity of β-actin or GAPDH. The relative ratio of the target intensity of SV mice or control group is arbitrarily presented as 1.
Western blot analysis
The proteins of lung tissues and NHBE cells were extracted in the presence of protease inhibitors. Protein concentrations were determined using Bradford reagent (Bio-Rad Laboratories). Proteins in the samples were separated by SDS-PAGE on denaturing gels of 8% to 20% at 120 V and transferred onto polyvinylidene difluoride membranes (Bio-Rad Laboratories) at 250 mA for 90 minutes by the wet transfer method. After blocking nonspecific binding sites with 3% nonfat milk in Tris-buffered saline Tween 20 (TBST; 20 mmol/L Tris pH 7.5, 500 mmol/L NaCl, 0.1% Tween 20) for 1 hour, and the blots were probed with an anti-IL-17A antibody (R&D systems), anti-GRP78 antibody (Cell Signaling Technologies, Beverly, MA, USA), anti-CHOP antibody (Santa Cruz Biotechnology), anti-phospho-eIF2α (Ser51) antibody (Cell Signaling Technologies), anti-eIF2α antibody (Cell Signaling Technologies), anti-TNF-α antibody (Thermo Fisher Scientific Inc.), anti-IL-1β (Thermo Fisher Scientific Inc.), anti-VEGF antibody (Santa Cruz Biotechnology), anti-KC antibody (R&D systems), anti-ICAM-1 antibody (Santa Cruz Biotechnology), anti-IL-4 antibody (Serotec Ltd, Oxford, UK), anti-NLRP3 antibody (AdipoGen, San Diego, CA, USA), anti-caspase-1 antibody (Santa Cruz Biotechnology), anti-IL-18 antibody (Santa Cruz Biotechnology), anti-caspase-3 antibody (Santa Cruz Biotechnology), anti-IκB-α antibody (Cell Signaling Technologies), anti-Toll-like receptor 4 (TLR4) antibody (Santa Cruz Biotechnology), or anti-actin antibody (Sigma-Aldrich). Anti-rabbit or anti-mouse horseradish peroxidase (HRP)-conjugated secondary antibodies (Cell Signaling Technologies) and anti-goat or anti-rat HRP-conjugated secondary antibodies (Santa Cruz Biotechnology) were used to detect binding of antibodies. The binding of the specific antibody was visualized by exposing to a photographic film or using Fuji film LAS-3000 (Fuji film, Tokyo, Japan) after treating with enhanced chemiluminescence system reagents (GE Healthcare, Little Chalfont, Buckinghamshire, UK). The film was scanned and the densitometric anlysis was performed using the quantification software (GS 800, Bio-Rad Laboratories; Multi Gauge program, Fuji film). For the quantification of specific bands, the same size square was drawn around each band to measure the density and then the value was adjusted by the density of the background near that band. The results of densitometric analysis were expressed as a relative ratio of the target protein to reference protein. The relative ratio of the target protein of control group is arbitrarily presented as 1.
Histology
At 48 hours after the instillation of LPS, mice were euthanized for histological assessment. The lung and trachea of mice were removed from the mice, and then for fixation, 10% (v/v) neutral buffered formalin was used. Specimens were dehydrated and embedded in paraffin. For histological examination, 4-μm sections of fixed embedded tissues were cut on a Leica model 2165 rotary microtome (Leica Microsystem Nussloch GmbH, Wetzlar, Germany), placed on glass slides, deparaffinized, and stained sequentially with H&E (Richard-Allan Scientific, Kalamazoo, MI, USA). Stained slides were analyzed with a light microscope (Axio Imager M1, Karl Zeiss, Goettingen, Germany) under identical conditions, including magnification (× 10), gain, camera position, and background illumination.
Micro-computed tomography (CT)
The lung of each mouse was scanned with a high resolution in vivo micro-CT system for small animal imaging (Skyscan 1076, Kontich, Belgium) at 48 hours after LPS or saline instillation. During the scan, mouse was lying on a computer-controlled rotation holder and scanned over a total rotation of 360 ° in rotation steps of 0.6 °. Microfocus X-ray tube was operated at a voltage of 48 kV and a current of 200 μA. The exposure time for each of one image was 0.3 seconds. Acquired images were reconstructed with NRecon program (Skyscan) by using Feldkamp back projection algorithm, resulting approximately 600 continuous scan images. Image analysis was performed with Data Viewer software version 1.4.3 (Skyscan), a 3D-rendering program package.
Measurement of plasma exudation
To assess lung permeability, we used Evans blue dye (EBD, Sigma-Aldrich) assay and measured the amount of total protein in BAL fluid. EBD solution (5 mg/ml) was injected into the tail vein at a concentration of 50 mg/kg. After 30 minutes, the animals were sacrificed and their chests were opened. Normal saline containing 5 mmol/L EDTA was perfused into the right ventricle until all venous fluid returned to the opened right atrium was clear. Lungs were carefully excised and weighed wet. EBD was extracted in 2 ml formamide kept in a water bath at 60°C for 3 hours, and the samples were centrifuged at 500 g for 10 minutes. The absorption of light at 595 nm was measured spectrophotometrically (Microplate reader, Bio-Rad Laboratories) with the supernatant. The dye extracted was quantified by interpolation against a standard curve of dye concentration in the range of 0.01-10 μg/ml and is expressed as ng of dye/mg of wet lung.
Measurement of myeloperoxidase (MPO) activity
MPO was extracted from each homogenized lung tissue sample by suspending the sample in 0.5% hexadecyltrimethylammonium bromide (Sigma-Aldrich) in 50 mmol/L potassium phosphate buffer, pH 6.0, before sonication in an ice bath for 20 seconds. The samples were freeze-thawed 2 times, after which sonication was repeated. Suspensions were then centrifuged at 12,000 g for 10 minutes, and the supernatant was assayed. MPO activity was determined by mixing 3 μL of supernatant with 97 μL of the above potassium phosphate buffer containing 0.167 mg/mL o-dianisidine dihydrochloride (Sigma-Aldrich) and 0.0005% hydrogen peroxide (Sigma-Aldrich). The change in absorbance at 450 nm was measured over 3 minutes. MPO activity was then derived from the observed change in absorbance per minute. The activity was expressed as units of MPO activity per milligram of protein, in which one unit of MPO activity was defined as the change in absorbance by one optical density unit per minute.
Measurement of dendritic cells (DCs) in lung
Dendritic cells were measured as described elsewhere [25]. Briefly, the lungs were excised and minced in RPMI-1640 medium (Gibco-Invitrogen) containing 25 mmol/L HEPES, 0.187% sodium bicarbobate, 1 mg/ml collagenase D (Sigma-Aldrich) and 0.002% DNase (Sigma-Aldrich). The suspensions were passed through a 40-μm cell strainer and centrifuged at 500 g for 5 minutes. Cells were resuspended in 15 ml red blood cell (RBC) lysing buffer consisting of 0.165 mol/L NH4Cl and 0.17 mol/L Tris (pH 7.65), and incubated for 10 minutes at room temperature. After the addition of above RPMI-1640 medium, cell numbers were counted with Nucleocounter (Chemometec.). Lung cells were washed in PBS containing 1% BSA (Sigma-Aldrich) and then stained with FITC-conjugated anti-CD11c (BD Pharmingen, San Diego, CA, USA), APC-conjugated anti-CD11b (BD Pharmingen), PE-conjugated anti-MHC II (BD Pharmingen), and PE-conjugated anti-CD8α (BD Pharmingen) for 30 minutes on ice. Lung cells were washed two times with PBS supplemented with 1% BSA and analyzed with a FACSCalibur apparatus (BD Biosciences, San Jose, CA, USA). Cells were gated on CD11b+and CD11c+ population (blue dot) and analyzed for MHC class II expression and CD8α. The classic quadrant gating, i.e., FSC/SSC properties was used for more specific identification of DCs to restrict high level MHC II expression and low level CD8α expression (MHCII+CD8α-) in CD11b+ and CD11c+ cell population. The number of DCs was calculated as percentage of each subset × number of cells counted in the single-cell suspensions of lung. Levels of MHC II+ cells in the CD11b+and CD11c+ population were expressed as the mean fluorescence intensity (MFI). The data were analyzed with a CellQuest Pro software (BD Biosciences) and presented as a representative dot-plot graph.
FACS analysis for cellular expression of nuclear factor-κB (NF-κB), TLR4, IL-1β, and TNF-α
Lung cells were stained for surface antigens to discriminate cellular phenotype as previously described [26-28]. The monoclonal antibodies conjugated to FITC, PE, or APC were used to detect surface expression of F4/80, CD11b, CD11c, LY6C/6G, CD326, or CD62E. Antibodies against CD11b (M1/70), CD11c (HL3), LY6C/6G (RB6-8C5), or CD62E (10E9.6) were purchased from BD Pharmingen. Anti-F4/80 (BM8) antibody and anti-CD326 (G8.8) antibody were from eBioscience. Cells were stained with the appropriate combinations of the antibodies for 30 minutes on ice and then washed with PBS. After centrifugation, cell pellets were fixed and permeabilized. Cells were probed with a biotinylated anti-TLR4 antibody (eBioscience), anti-p-NF-κB p65 antibody (Santa Cruz Biotechnology), anti-IL-1β antibody (Thermo Fisher Scientific Inc.), or anti-TNF-α antibody (Thermo Fisher Scientific Inc.). Probed cells with biotinylated antibodies were reacted with a FITC, PE, or APC conjugated streptavidin (BD Pharmingen) for immunofluorescence staining. Cells applied with non-labelled primary antibodies were stained with Alexa Fluor 488 (green), Alexa Fluor 546 (red), or Alexa Fluor 647 (blue) conjugated secondary antibodies (Invitrogen). Stained cells were measured by flow cytometry using a FACSCalibur apparatus (BD Biosciences) and the results were analyzed with a CellQuest Pro software (BD Biosciences). Lung cells were first gated on populations showing the FSC/SSC properties of each subset in the dot plot. Gates were further set F4/80+ and CD11b+ cells (macrophage), LY6C/6G+ and CD11b+ cells (neutrophil), CD11b+ and CD11c+ cells (DCs), or CD326+ and CD62E- cells (epithelial cells) according to the expression of surface marker. Levels of TLR4, p-NF-κB-p65, IL-1β, or TNF-α were analyzed on each cellular population. Fluorescence was determined by histogram analysis and expressed as MFI.
Cytosolic or nuclear protein extractions for analysis of transcription factors
Lungs were homogenized in 2 volume of buffer A (20 mmol/L HEPES, pH 7.9, 1.5 mmol/L MgCl2, 10 mmol/L KCl, 0.05% NP40, 0.5 mmol/L EDTA, 10% glycerol, 1 mmol/L Na3VO4, 0.5 mmol/L DTT, and 0.02% sodium deoxycholate) containing a protease inhibitor cocktail. The homogenates were centrifuged at 2,000 g for 10 minutes at 4°C. The supernatants were collected and incubated on ice for 10 minutes. After centrifugation at 12,000 g for 10 minutes at 4°C, the supernatants collected were used as cytosolic proteins for analysis of NF-κB p65. The pellets were washed twice in buffer A and solubilized with buffer B consisting of 20 mmol/L HEPES (pH 7.9), 150 mmol/L NaCl, 0.5 % NP40, 0.5 mmol/L EDTA, 20 % glycerol, 1 mmol/L Na3VO4, 10 mmol/L DTT, 0.02 % sodium deoxycholate, 1% SDS, and a protease inhibitor cocktail. The mixture was kept on ice for 30 minutes and centrifuged at 12,000 g for 10 minutes. The resulting supernatants were used as soluble nuclear proteins for analysis of XBP-1, ATF-4, ATF-6, NF-κB p65, and lamin B. The levels of these proteins were analyzed by Western blotting using antibodies against XBP-1 (Santa Cruz Biotechnology), ATF-4 (Santa Cruz Biotechnology), ATF-6 (Santa Cruz Biotechnology), NF-κB p65 (Upstate Biotech, Lake Placid, NY), or lamin B (Santa Cruz Biotechnology) as described above.
Measurement of histone deacetylase (HDAC) enzyme activity
HDAC activity was determined in crude nuclear extracts of lung tissues using a Fluor-de-Lys® fluorescent assay kit (HDAC assay kit; Biomol, Plymouth Meeting, PA, USA), as reported previously [29]. Fluorescence was measured by using a Multimode Microplate Reader System (EnSpire, Perkin Elmer, Waltham, MA, USA), with excitation at 360 nm and emission at 460 nm. The results are presented as arbitrary fluorescence units (AFU).
Statistical analysis
We used SPSS statistical software (version 16.0, SPSS, Chicago, IL). Data were expressed as mean ± SEM. Statistical comparisons were performed using one-way ANOVA followed by the Scheffe's test. Statistical differences between two groups were determined using the unpaired Student's t test. A value of P < 0.05 was considered statistically significant.
Results
Changes of ER stress and UPR in LPS-induced lung injury
At first, we analyzed the mRNA or protein levels of GRP78 and CHOP in BAL cells and lung tissues using RT-PCR, confocal microscopy, and Western blotting to evaluate whether ER stress is induced in LPS-instilled mice. Immunofluorescence intensities of GRP78 and CHOP in lung tissues from LPS-instilled mice injected with vehicle (LV mice) increased markedly 48 hours after LPS instillation especially in bronchiolar epithelium and inflammatory cells around bronchioles of lung tissues, compared with saline instilled mice injected with vehicle (SV mice) (Fig. 1A). All BAL cells from LV mice also showed significant increases in GRP78 and CHOP level, predominantly in the cytoplasm, compared to SV mice (Fig. 1B). Anti-IL-17A antibody or 4-PBA reduced the increases of GRP78 and CHOP expression in BAL cells and lung tissues of LV mice. Consistent with these observations, the mRNA and protein levels of GRP78 or CHOP in lung tissues (Figs. 1C-F) were significantly elevated in LV mice compared with SV mice. The LPS-induced increases of GRP78 and CHOP in lung tissues of LV mice were reduced by administration of anti-IL-17A antibody or 4-PBA. In addition, the inhibitory effects of anti-IL-17A antibody and 4-PBA on the expression of GRP78 and CHOP were stronger when they were administered together into LV mice (Figs. S1A-D).
Figure 1
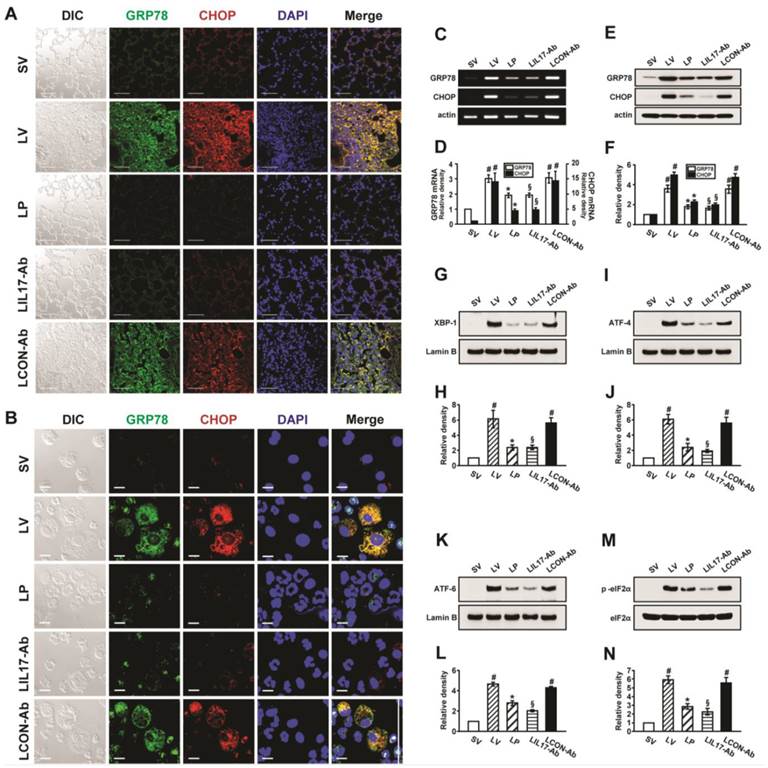
Effects of anti-IL-17A antibody or 4-PBA on ER stress markers and UPR-related proteins. (A and B) Representative confocal laser immunofluorescence photomicrographs of lung tissues (A) and BAL cells (B) from saline-instilled mice given injections of vehicle (SV), LPS-instilled mice given injections of vehicle (LV), LPS-instilled mice given intraperitoneal injections of 4-PBA of 200 mg/kg (LP), LPS-instilled mice given intravenous injections of anti-IL-17 antibody of 5 mg/kg (LIL17-Ab), or LPS-instilled mice given intravenous injections of isotype control monoclonal antibody (LCON-Ab). Sampling was performed at 48 hours after the instillation of LPS. Bars indicate 50 μm. DIC means 'Differential interference contrast'. (C and D) Representative RT-PCR and semi-quantative analyses for mRNA of GRP78 and CHOP. (E-N) Representative immunoblots of GRP78 (E), CHOP (E), XBP-1 (G), ATF-4 (I), ATF-6 (K) and p-eIF2α (M) in lung tissues and densitometric analyses of GRP78 (F), CHOP (F), XBP-1 (H), ATF-4 (J), ATF-6 (L), and p-eIF2α (M). Bars represent mean ± SEM from 5 or 6 mice/group. #P < 0.05 versus SV; *P < 0.05 versus LV; §P < 0.05 versus LCON-Ab.
To investigate effects of anti-IL-17A antibody or 4-PBA on UPR-related markers, we measured levels of X-box binding protein-1 (XBP-1), ATF-4, ATF-6, and phosphorylation of eukaryotic initiation factor 2α (eIF2α), in lung tissues of LV mice. Levels of ATF-4, ATF-6, and XBP-1 were increased in nuclear protein extracts from lung tissues of LV mice, compared with SV mice, but were substantially reduced by administration of anti-IL-17 antibody or 4-PBA (Figs. 1G-L). The increase in phospho-eIF2α in lung tissues from the LV mice was also reduced by anti-IL-17 antibody or 4-PBA (Figs. 1M, N), but total eIF2α protein levels did not change.
Effect of anti-IL-17A antibody or 4-PBA in cells and lung tissues of LPS-instilled mice
To determine the roles of IL-17A and ER stress in LPS-induced lung injury, we examined the effects of anti-IL-17A antibody and 4-PBA on various typical features of lung injury. Histological analyses showed that marked infiltration of inflammatory cells into the alveolar space, peribronchial wall thickening, and vascular congestion were increased markedly in the lung of LV mice (Fig. 2A; H&E). On micro-CT scan, LV mice showed bilateral diffuse ground glass opacities with the prominent bronchovascular bundle and bronchial wall thickenings in both lung fields, forming contrast to the findings of SV mice (Fig. 2A; micro-CT). These histologic and radiologic changes were dramatically reduced by administration of anti-IL-17A antibody or 4-PBA. The increased number of inflammatory cells in BAL fluids from LV mice was reduced by administration of anti-IL-17A antibody or 4-PBA (Fig. 2B). Supporting these observations, plasma exudation (Fig. 2C) and total protein levels in BAL fluids (Fig. 2D) were increased in LV mice compared with SV mice. Anti-IL-17A antibody or 4-PBA reduced the increase of plasma exudation.
MPO is a component of the neutrophil cytoplasm that is used to quantify neutrophils. Activity of MPO was increased substantially in lung tissues of LV mice, and was reduced by administration of anti-IL-17A antibody or 4-PBA (Fig. 2E).
Figure 2
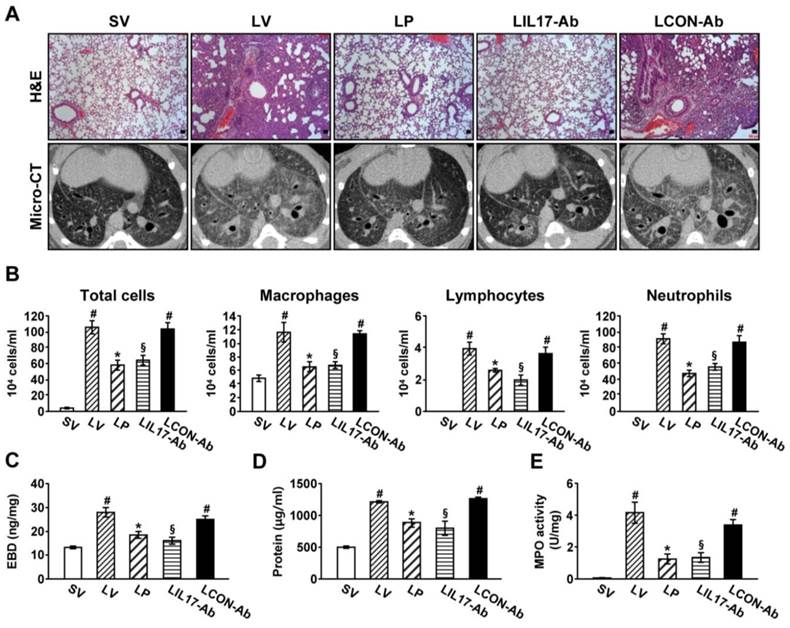
Lung injury and inflammation in LPS-instilled mice. (A) Representative H&E stained sections and micro-CT images of the lungs from SV, LV, LP, LIL17-Ab, and LCON-Ab. Sampling was performed at 48 hours after the instillation of LPS. Bars indicate 50 μm. (B) Cellular changes in BAL fluids from LPS-instilled mice. (C) EBD assay of lung tissues of LPS-inhaled mice. (D) Changes of total protein amounts in BAL fluids of LPS-instilled mice. (E) MPO activity in lung tissues. Bars represent mean ± SEM from 6 mice/group. #P < 0.05 versus SV; *P < 0.05 versus LV; §P < 0.05 versus LCON-Ab.
Inhibitory effects of anti-IL-17A antibody or 4-PBA on production of inflammatory mediators and activation of NLRP3 inflammasome
As IL-1β and TNF-α are representative pro-inflammatory mediators in various infectious disorders, we measured these levels in the lung of LPS-instilled mice. All BAL cells from LV mice showed significant increases in IL-1β and TNF-α expression compared to SV mice (Figs. 3A, B). Anti-IL-17A antibody or 4-PBA reduced the increased expression of IL-1β and TNF-α in BAL cells from LV mice. Supporting these observations, FACS analysis showed that the fluorescence intensity of IL-1β and TNF-α was significantly increased in sorted macrophages and neutrophils from lung cells of LV mice and these increases were dramatically reduced by administration of 4-PBA and anti-IL-17A antibody (Fig. S2). Consistent with these observations, the IL-1β and TNF-α level in lung tissues were also increased in LV mice compared with SV mice (Figs. 3C-F). The LPS-induced increases of IL-1β and TNF-α in lung tissues of LV mice were significantly reduced by administration of anti-IL-17A antibody or 4-PBA. As well known, IL-1β is one of the hallmarks indicating activation of NLRP3 inflammasome. Thus, to evaluate whether these findings are associated with activation of NLRP3 inflammasome in LPS-induced lung injury, we measured the protein levels of NLRP3, caspase-1, and IL-18 in lung tissues of LV mice (Figs. 3G-L). Interestingly, the protein levels were substantially increased in lung tissues of LV mice compared to those of SV mice and these increases were remarkably inhibited by administration of anti-IL-17A antibody or 4-PBA. In addition, LPS-induced increased levels of other inflammatory mediators (IL-4, ICAM-1, VEGF, IL-17, and keratinocyte-derived chemokine [KC]) in lung tissues of LV mice were significantly reduced by administration of anti-IL-17A antibody or 4-PBA (Figs. 4A-J). Specifically, IL-17A protein levels in lung tissue of LV mice were gradually increased up to 48 hours after LPS instillation and the increased levels of IL-17A were then maintained up to 72 hours (Figs. 4K, L). Interestingly, the anti-inflammatory effects of anti-IL-17A antibody or 4-PBA were further strengthened when they were injected together to LV mice (Figs. S1E-J).
Figure 3
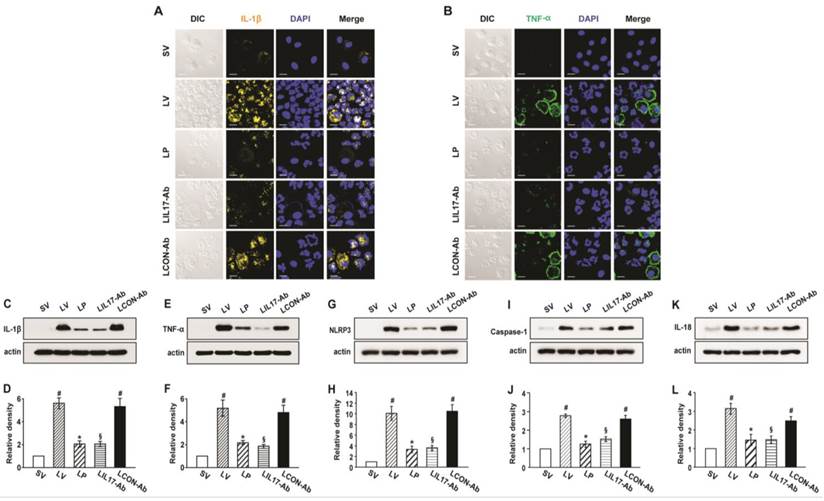
Levels of IL-1β, TNF-α, NLRP3, caspase-1, and IL-18 in lung tissues or BAL cells from LPS-instilled mice. (A and B) Representative confocal laser immunofluorescence photomicrographs showing location of IL-1β (A) and TNF-α (B) in BAL cells from SV, LV, LP, LIL17-Ab, and LCON-Ab mice. Sampling was performed at 48 hours after the instillation of LPS. Bars indicate 10 μm. (C-L) Representative immunoblots of IL-1β (C), TNF-α (E), NLRP3 (G), caspase-1 (I), and IL-18 (K) protein in lung tissues and densitometric analyses of IL-1β (D), TNF-α (F), NLRP3 (H), caspase-1 (J), and IL-18 (L) in lung tissues. Bars represent mean ± SEM from 6 mice/group. #P < 0.05 versus SV; *P < 0.05 versus LV; §P < 0.05 versus LCON-Ab.
Figure 4
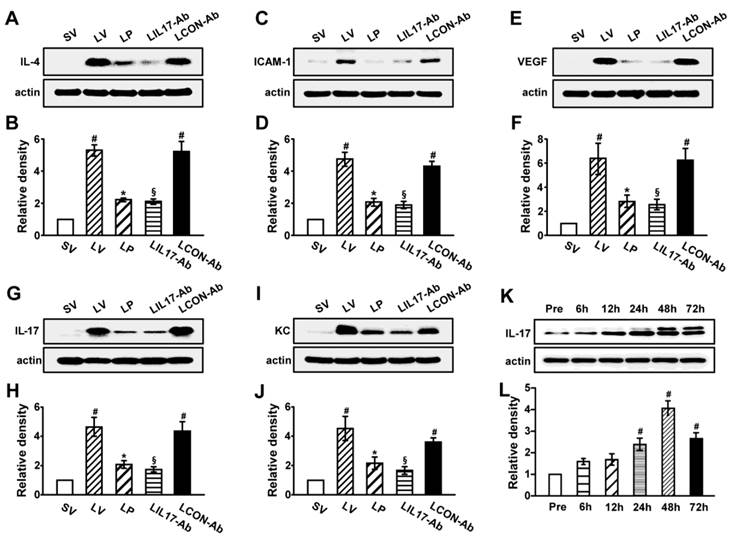
Levels of pro-inflammatory mediators and time-kinetics of IL-17A in lung tissues of LPS-instilled mice. Representative immunoblots of IL-4 (A), ICAM-1 (C), VEGF (E), IL-17 (G) and KC (I) in lung tissues and densitometric analysis of IL-4 (B), ICAM-1 (D), VEGF (F), IL-17 (H) and KC (J). Sampling was performed at 48 hours after the instillation of LPS. (K and L) Kinetics of IL-17A in lung tissues over time; One, 6, 12, 24, 48, and 72 hours are the time periods of the sampling after the instillation of LPS. Pre, 1 hour before the instillation of LPS. Bars represent mean ± SEM from 5 or 6 mice/group. #P < 0.05 versus SV or Pre; *P < 0.05 versus LV; §P < 0.05 versus LCON-Ab.
Changes of ER stress and UPR in lung tissues of rhIL-17A-instilled mice
To evaluate whether IL-17A can induce ER stress in lung without any other stimuli or stress, we measured the protein levels of GRP78, CHOP, and various UPR markers in lung tissues of mice treated with rhIL-17A intratracheally. GRP78 or CHOP levels in lung tissues were significantly elevated in saline-instilled mice administered with rhIL-17A (SrhIL-17 mice) compared with SV mice (Figs. S3A-D). Consistent with these observations, levels of XBP-1, ATF-4, and ATF-6α in the nuclear extracts of lung tissues and phospho-eIF2α were substantially increased in lung tissues of SrhIL-17 mice compared with SV mice (Figs. S3E-L).
Effects of rhIL-17A on ER stress-induced cell death in lung tissues of LV mice and LPS-stimulated NHBE cells
CHOP (GADD153) and caspase-3 are well known markers for the ER stress-associated cell death, thus we measured their levels in LPS-exposed lung tissues and NHBE cells to evaluate the role of IL-17A in ER stress-induced apoptosis. Western blot analyses revealed that the protein levels of CHOP and caspase-3 were increased by LPS stimulation in lung tissues and NHBE cells. The increases of CHOP and caspase-3 in the lung and epithelial cells were further enhanced by rhIL-17A treatment although it was not statistically significant (Fig. S4).
Thapsigargin-induced ER stress contributes to LPS-induced lung inflammation, plasma exudation, and the production of pro-inflammatory mediators
To confirm the contribution of ER stress to develop LPS-induced lung injury, we observed the effects of thapsigargin, a representative ER stress inducer, on LPS-induced typical features of lung injury. Thapsigargin increased the expression of GRP78 and CHOP in lung tissues of saline-instilled mice (Figs. S5A-D) as well as LPS-instilled mice (Fig. S6), although the increased extent was different between two groups of mice; LPS-instilled mice showed the exaggerated increases in levels of GRP78 and CHOP in lung tissues. The increases were dramatically reduced by administration of 4-PBA (Fig. S6). As for IL-17A production, thapsigargin also increased the expression of IL-17A protein in lung tissues of saline-instilled mice (Figs. S5E, F) showing that pure ER stress can induce IL-17A production without pathogen-stimulation such as LPS. The number of inflammatory cells including neutrophils in BAL fluids from LPS-instilled mice was significantly increased compared to those of control mice. The increased number of cells in BAL fluid was further elevated by administration of thapsigargin (Fig. S7A). Histological assessment showed that numerous inflammatory cells infiltrated into the lung of LPS-instilled mice treated with vehicle control (Fig. S7C) compared to saline-instilled mice treated with vehicle control (Fig. S7B). Interestingly, the LPS-instilled mice treated with thapsigargin (Fig. S7D) showed somewhat severer pathologic features than the LPS-instilled mice treated with vehicle control. Consistent with these pathologic findings, the plasma exudation and the expression of IL-17A, KC, IL-1β, and TNF-α in the lung tissues of LPS-instilled mice treated with thapsigargin were increased compared to those of LPS-instilled mice treated with vehicle control (Figs. S7F-N). In addition, all these parameters were substantially reduced by the treatment with 4-PBA. These findings suggest that pure ER stress is associated with the pathogenesis of LPS-induced lung injury.
Effects of blockade of IL-1β, TNF-α, or IL-17A on the induction of ER stress in the lung tissues of LPS-instilled mice
To investigate the direct effects of IL-1β, TNF-α, and IL-17A on ER stress, we used the neutralizing antibodies for each cytokine. Western blot analysis revealed that the protein levels of GRP78 and CHOP in the lung tissues of LV mice were greatly increased compared to the levels in SV mice (Fig. 5). The neutralization of IL-1β (Figs. 5A-D) or TNF-α (Figs. 5E-H) using their blocking antibodies substantially decreased the expression of GRP78 and CHOP in the lung tissues of LV mice. In addition, administration of anti-IL-17A antibody dramatically reduced the exaggerated increases in protein levels of GRP78 and CHOP by thapsigargin in the lung tissues of LV mice (Fig. S8).
Figure 5
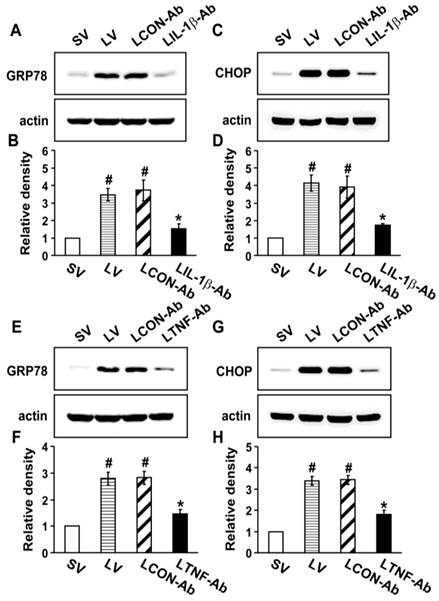
Effect of anti-IL-1β antibody or anti-TNF-α antibody on levels of GRP78 and CHOP in lung tissues of LPS-instilled mice. Representative immunoblots of GRP78 (A, E) and CHOP (C, G) lung tissues from SV, LV, LCON-Ab, LPS-instilled mice given intravenous injections of anti-IL-1β antibody of 100 μg/mouse (LIL-1β-Ab), or LPS-instilled mice given intravenous injections of anti-TNF-α antibody of 10 μg/mouse (LTNF-Ab) and densitometric analyses of GRP78 (B, F) and CHOP (D, H). Sampling was performed at 48 hours after the instillation of LPS. Bars represent mean ± SEM from 5 mice/group. #P < 0.05 versus SV; *P < 0.05 versus LCON-Ab.
Figure 6
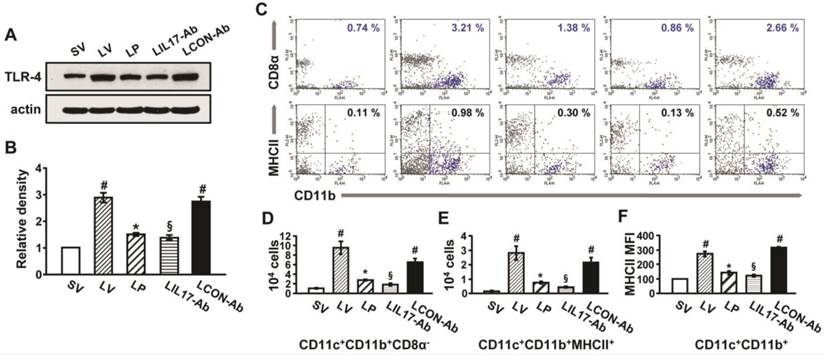
Effect of anti-IL-17A antibody or 4-PBA on levels of TLR4 and infiltration of CD11b+CD11c+MHC II+ cells in lung tissues of LPS-instilled mice. (A and B) Representative immunoblot of TLR4 in lung tissues (A) and densitometric analysis of TLR4 (B). (C) Dot plot analysis of CD11b+CD11c+MHC II+ cells that infiltrated lung tissues in SV, LV, LP, LIL17-Ab, and LCON-Ab mice. (D and E) Numbers of infiltrated DCs. (F) Mean fluorescence intensity for MHCII+ cells. Sampling was performed at 48 hours after the instillation of LPS. Bars represent mean±SEM from 6 mice/group. #P < 0.05 versus SV; *P < 0.05 versus LV; §P < 0.05 versus LCON-Ab.
Expression of TLR4 and infiltration of DCs in the lungs of LPS-instilled mice
To understand that LPS-stimulated TLR4 is connected to induce IL-17A production and ER stress, we analyzed the expression of TLR4 in lung tissues and lung cells. In addition, DCs that expressed TLR4 were counted in the lung tissues of LPS-instilled mice. As shown in Fig. 6, the level of TLR4 protein and numbers of DCs were greatly increased in the lung tissues of LV mice compared with SV mice, and these increases were reduced by administration of anti-IL-17A antibody or 4-PBA.
Effects of blockade of TLR4 pathway on LPS-induced IL-17A production and ER stress in murine lung tissues and NHBE cells
To determine the role of TLR4 signaling pathway in LPS-induced IL-17A and ER stress in the lung, we analyzed the effects of TAK-242, a TLR4 inhibitor, on the protein levels of IL-17A, GRP78, and CHOP in LPS-exposed lung tissues and NHBE cells. The levels of IL-17A were significantly elevated in the lung tissues of LV mice compared with SV mice (Figs. 7A, B). The LPS-induced increases of IL-17A were reduced by administration of TAK-242. Consistent with these observations, the increased levels of GRP78 or CHOP in the lung tissues of LV mice were dramatically reduced by administration of TAK-242 (Figs. 7C-F). In addition, these findings were consistent in LPS-stimulated NHBE cells (Figs. 7G-L). Moreover, we also found that the combination treatment with anti-IL-17A antibody and TAK-242 further inhibited the increased expression of GRP78, CHOP, IL-17A, IL-1β, and TNF-α in the lung tissues of LV mice than the treatment with a single agent, i.e., anti-IL-17A antibody or TAK-242 (Figs. S9A-J).
Effects of anti-IL-17A antibody, 4-PBA, or TAK-242 on nuclear translocation of NF-κB p65 and degradation of inhibitor-κBα (IκBα)
Since NF-κB is known to be a major transcriptional factor for inflammation, we measured the changes of NF-κB activity both in vivo and in vitro experimental systems. The level of NF-κB p65 in nuclear extracts of lung tissues of LV mice was significantly increased after the instillation of LPS compared with SV mice; this increase was reduced by administration of anti-IL-17A antibody, 4-PBA, or TAK-242 (Figs. 8A-D). At the same time, LPS-induced decrease of the level of IκBα protein in lung tissues was restored by anti-IL-17A antibody or 4-PBA (Figs. 8E, F). In LPS-stimulated NHBE cells, the nuclear translocation of NF-κB was significantly increased compared to control group and the increases were blocked by the pretreatment with TAK-242 (Figs. 8G, H). As expected, Western blot analyses also revealed that anti-IL-17A antibody, 4-PBA, or TAK-242 worked additively on the inhibition of nuclear translocation of NF-κB in the lung tissues of LV mice (Figs. S1K, L, S9K, L).
Expression of TLR4 and nuclear NF-κB in various lung cells of LPS-instilled mice
FACS analyses revealed that the immunofluorescence intensity for TLR4 in the isolated airway epithelial cells, macrophages, neutrophils, and DCs in the lungs of LV mice was significantly increased compared to the levels of SV mice (Figs. 9A-E). In addition, the levels of NF-κB intensity in nuclear extracts were consistent with the data of TLR4 in the cells (Figs. 9F-J). These increases were dramatically reduced by administration of 4-PBA or anti-IL-17A antibody in all cell groups isolated from the lung of LV mice.
Interaction between IL-17A and ER stress in LPS-stimulated NHBE cells
LPS-stimulated NHBE cells showed significant increases in immunofluorescence intensities of IL-17A, GRP78, and CHOP compared to control (Fig. 10A). Pre-treatment with anti-IL-17A antibody or 4-PBA reduced the LPS-induced increases in IL-17A, GRP78, and CHOP in NHBE cells. In addition, the mRNA levels of IL-17A in NHBE cells were significantly increased after LPS stimulation compared to control and the increase was markedly reduced by pre-treatment with 4-PBA (Figs. 10B, C). Meanwhile, the GRP78 level in NHBE cells was significantly increased after IL-17A stimulation compared to control, and co-stimulation with LPS further increased the GRP78 level induced by IL-17A (Figs. 10D, E). The increases in the protein level of GRP78 were substantially reduced by pre-treatment with 4-PBA.
Effects of IL-17A and ER stress on nuclear translocation of NF-κB in LPS-stimulated NHBE cells
The level of NF-κB p65 in nuclear extracts of NHBE cells was significantly increased after the stimulation with IL-17A compared with control; this increase was further enhanced by co-stimulation with LPS (Figs. 10F, G). Pre-treatment with 4-PBA markedly reduced the NF-κB p65 level in nuclear extracts of NHBE cells stimulated with IL-17A and/or LPS. Additionally, LPS-induced nuclear translocation of NF-κB p65 was blocked by the pre-treatment with anti-IL-17A antibody or 4-PBA (Figs. 10H, I).
Figure 7
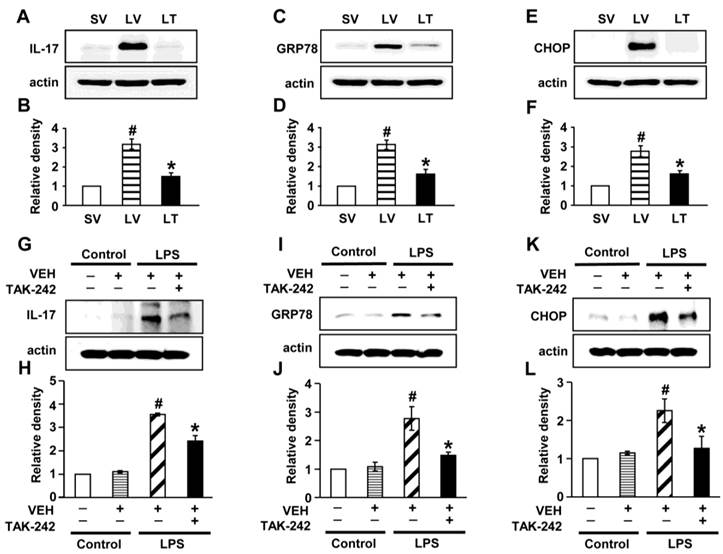
Levels of IL-17A, GRP78, and CHOP in lung tissues from LPS-instilled mice and LPS-stimulated NHBE cells. Representative immunoblots of IL-17A (A), GRP78 (C), and CHOP (E) in lung tissues from SV, LV, and LPS-instilled mice given intravenous injections of TAK-242 of 200 μg/mouse (LT) and densitometric analysis of IL-17A (B), GRP78 (D), and CHOP (F). Sampling was performed at 48 hours after the instillation of LPS. Bars represent mean ± SEM from 5 mice/group. #P < 0.05 versus SV; *P < 0.05 versus LV. Representative immunoblots of IL-17A (G), GRP78 (I), and CHOP (K) in NHBE cells and densitometric analyses of IL-17A (H), GRP78 (J), and CHOP (L). NHBE cells were pre-treated with 1 μmol/L TAK-242 or drug vehicle at 1 hour before LPS stimulation. Bars represent mean ± SEM from 3 independent experiments. #P < 0.05 versus Control; *P < 0.05 versus LPS-stimulated cells with drug vehicle only.
Figure 8
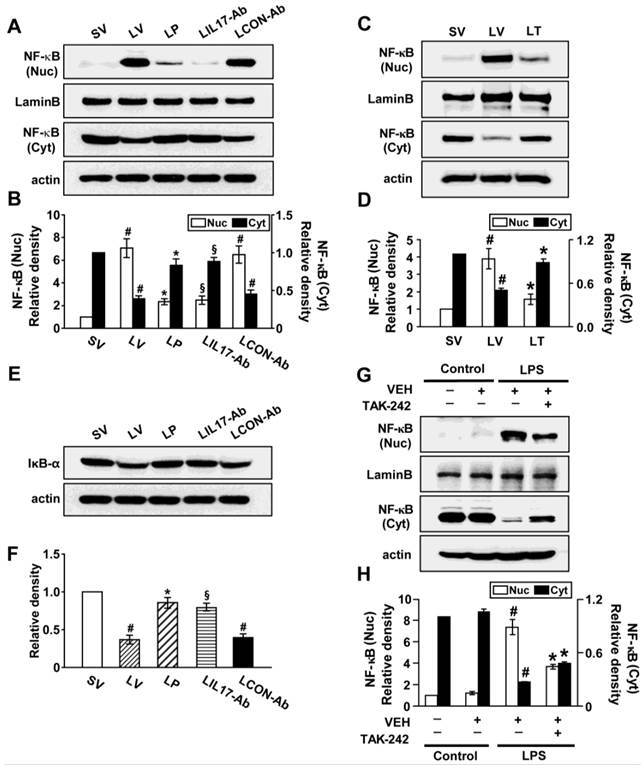
Nuclear translocation of NF-κB p65 and degradation of IκBα in lung tissues of LPS-instilled mice and LPS-stimulated NHBE cells. Representative immunoblots of NF-κB (A, C) and IκBα (E) in lung tissues of SV, LV, LP, LIL17-Ab, LCON-Ab, and LT mice and densitometric analyses of NF-κB p65 (B, D) and IκBα (F) levels. Sampling was performed at 48 hours after the instillation of LPS. Bars represent mean ± SEM from 6 mice/group. #P < 0.05 versus SV; *P < 0.05 versus LV; §P < 0.05 versus LCON-Ab. (G and H) Representative immunoblots of NF-κB (G) in NHBE cells and densitometric analyses of NF-κB. NHBE cells were pre-treated with 1 μmol/L TAK-242 or drug vehicle at 1 hour before LPS stimulation. Bars represent mean ± SEM from 3 independent experiments. #P < 0.05 versus Control; *P < 0.05 versus LPS-stimulated cells with drug vehicle only.
4-PBA does not affect HDAC activity significantly in the lung tissues of LPS-instilled mice
To evaluate off-target effects of 4-PBA in this study, we measured the changes of HDAC activity in the lung tissues of LPS-instilled mice after 4-PBA administration. LPS-instilled mice showed increased HDAC activity compared to those of control mice (Fig. S10). The increased HDAC activity was not affected significantly by 4-PBA administration in the lung tissues of LV mice. These findings suggest that 4-PBA works on the features of LPS-induced lung injury mainly through inhibition of ER stress not via inhibition of HDAC activity.
Figure 9
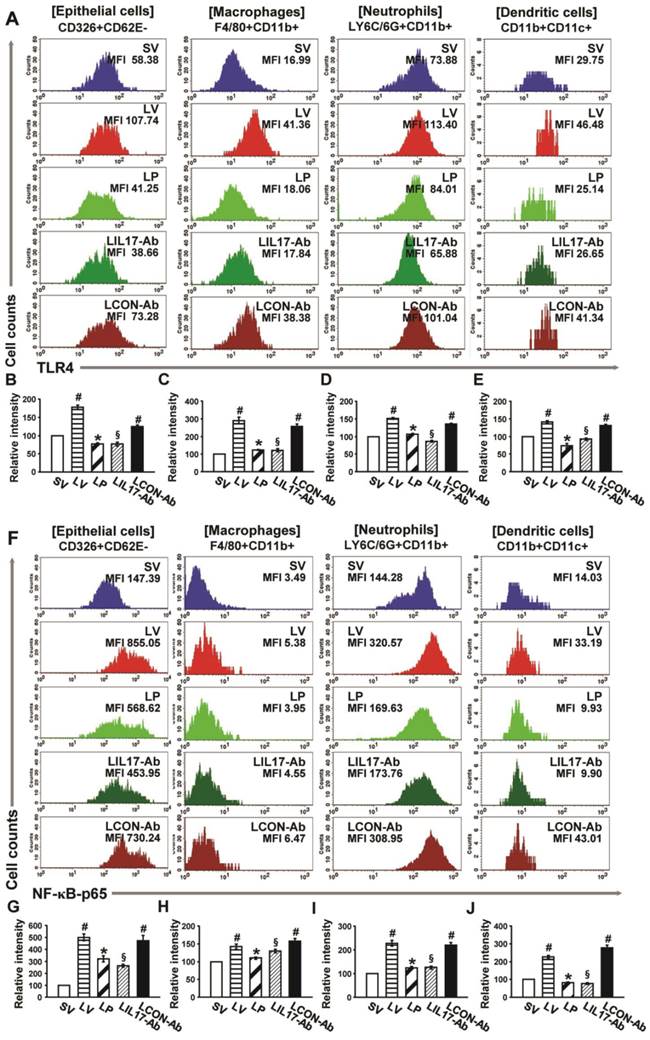
Effects of anti-IL-17A antibody and 4-PBA on the expression of TLR4 and NF-κB in airway epithelial cells, macrophages, neutrophils, and dendritic cells from LPS-instilled mice. (A) Representative histogram of the expression of TLR4 in various cells of lung from LV-instilled mice. (B-E) Fluorescence intensity of TLR4 in airway epithelial cells (B), macrophage (C), neutrophils (D), and dendritic cells (E) is presented as the ratio of the levels of TLR4 in each group relative to those in SV mice. (F) Representative histogram of the expression of NF-κB in nuclear extracts of various cells of the lung from LV-instilled mice. (G-J) Fluorescence intensity of NF-κB in nuclear extracts of airway epithelial cells (G), macrophage (H), neutrophils (I), and dendritic cells (J) is presented as the ratio of the levels of NF-κB in each group relative to those in SV mice. Sampling was performed at 48 hours after the instillation of LPS. Bars represent mean ± SEM from 5 mice/group. #P < 0.05 versus SV; *P < 0.05 versus LV; §P < 0.05 versus LCON-Ab.
Figure 10
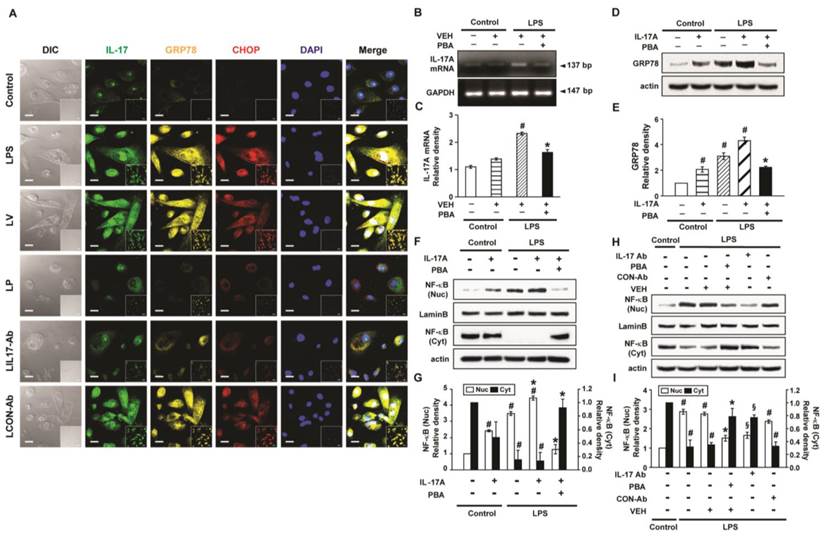
Levels of IL-17A, GRP78 and NF-κB p65 in LPS-stimulated NHBE cells. (A) Representative confocal laser immunofluorescence photomicrographs of NHBE cells with no stimulation (Control), LPS stimulation (LPS), pre-treatment of vehicle and LPS stimulation (LV), pre-treatment with 10 mmol/L 4-PBA for 1 hour and LPS stimulation (100 μg/ml) (LP), pre-treatment with 2 μg/ml of anti-IL-17 antibody and LPS stimulation (100 μg/ml) (LIL17-Ab), or pre-treatment with 2 μg/ml of isotype control monoclonal antibody and LPS stimulation (100 μg/ml) (LCON-Ab). Bars indicate 20 μm. (B and C) Representative RT-PCR of IL-17A mRNA and semi-quantitative analyses of IL-17A mRNA. (D and E) Representative immunoblots of GRP78 in NHBE cells and densitometric analyses of GRP78. (F-I) Representative immunoblots of NF-κB p65 in NHBE cells and densitometric analyses of NF-κB p65. Data are from 3 independent experiments. NHBE cells were stimulated with 100 μg/ml of LPS and/or 100 ng/ml of IL-17A. Pre-treatment with 10 mmol/L 4-PBA or 2 μg/ml of a human anti-IL-17A antibody (IL-17Ab) or an isotype control monoclonal antibody (CON-Ab) was performed at 1 hour before LPS stimulation. Cells were harvested at 12 hours after LPS stimulation. #P < 0.05 versus Control; *P < 0.05 versus LPS-stimulated cells with drug vehicle only; §P < 0.05 versus LPS-stimulated cells with isotype control antibody only.
Discussion
All the time, the lung is vulnerable to air environment. Therefore, this organ is often exposed to the risk of the damages directly or indirectly from various harmful agents. These noxious stimuli include exogenous components such as microbial and environmental antigens, as well as a plethora of host-derived danger signals. A common final pathway initiated by these exogenous and endogenous triggers is the development of acute lung injury (ALI) [30, 31]. Despite recent advances in mechanical ventilation and a better understanding of the inflammatory pathophysiology of ALI and acute respiratory distress syndrome (ARDS), many patients still progress to the state of respiratory failure, resulting in increased mortality [32].
As for the role of IL-17A in the pathogenesis of various inflammatory processes, there is still controversy between pro-inflammatory role and anti-inflammatory one. However, recent data have indicated that IL-17A is associated with development of various chronic inflammatory, allergic, and autoimmune disorders [4]. Moreover, several studies have reported that IL-17A is necessary for LPS-induced airway neutrophilia, is produced increasingly in patients with sepsis, induces the production of neutrophil-mobilizing cytokines such as IL-6 and IL-8 [33, 34], and is required for the augmented ozone-induced neutrophil influx in the lung [6]. In this study, we found that IL-17A was significantly increased in the lung from LPS-induced lung injury model of mice. Furthermore, substantial increases of airway inflammatory cell infiltrations including neutrophils with enhanced MPO activity, vascular leakages, and various pro-inflammatory mediators (IL-1β, TNF-α, KC, VEGF, ICAM-1, and IL-4) in the lung of LPS-instilled mice were observed. Interestingly, inhibition of IL-17A using a neutralizing anti-IL-17A antibody markedly reduced these typical manifestations of endotoxin-induced lung inflammation/injury including pulmonary neutrophilia and histologic and radiologic changes. These findings suggest that IL-17A is an important player in the development/maintenance of LPS-induced ALI/ARDS, providing additional evidence for pro-inflammatory role of IL-17A.
The ER stress is a potent evolutionarily conserved response to misfolded proteins resulted from cellular stress such as chronic inflammation. Moreover, ER stress pathway is associated with the pathogenesis of LPS-induced lung inflammation [35]. In addition, very recently, we have revealed that ER stress is involved in the LPS-induced acute lung injury in vivo and is enhanced in LPS-stimulated airway epithelial cells in vitro [17]. However, the investigation for the role of cytokines related to various inflammatory/infectious disorders in inducing ER stress is limited. Recent studies have shown that pro-inflammatory mediators such as TNF-α, IL-1β, and IFN-γ contribute to induction of ER stress in multiple cell types [20, 36]. In our current study, we have found that GRP78 and CHOP mRNA and protein levels as well as the levels of UPR-related markers were substantially increased in lung tissues of a murine model of LPS-induced lung injury. Very intriguingly, the increases of these protein levels were significantly reduced not only by the administration of ER stress inhibitor, 4-PBA but also by the treatment with a neutralizing anti-IL-17A antibody. As supporting data, we also showed that administration of rhIL-17A induced the significant increased expression of GRP78, CHOP, XBP-1, ATF-4, ATF-6, and p-eIF2α in the lung tissues of saline-instilled mice and that under LPS stimulation rhIL-17A contributed to somewhat of ER stress-induced cell death. These findings have implied that IL-17A is implicated with the induction of ER stress in LPS-induced lung injury. Additionally, the blockade of IL-1β or TNF-α which is the inflammatory mediator controlled by IL-17A also ameliorated the expression of GRP78 and CHOP in the lung tissues of LPS-instilled mice indicating that IL-1β and TNF-α team up with IL-17A to induce ER stress in the LPS-induced lung injury. Furthermore, treatment with 4-PBA substantially attenuated the pathophysiological features of LPS-induced lung inflammation/injury, including inflammatory cell recruitment and vascular leakage and reduced the nuclear translocation of NF-κB in lung tissues. Taken together, IL-17A can mediate LPS-induced lung injury through the enhancement of ER stress linked to NF-κB activation in mice.
NF-κB, a multiprotein complex, is involved in early cellular defense reactions in higher organisms and plays a pivotal role in immune and inflammatory responses [37]. In this study, treatment with 4-PBA or anti-IL-17A antibody blocked the nuclear translocation of NF-κB and degradation of IκBα, thereby reducing production of various pro-inflammatory mediators in the lungs of LPS-instilled mice. In fact, ER stress has been well known as a representative inducer or trigger of NF-κB activation and three canonical UPR pathways appear to be tightly interrelated with the NF-κB signaling pathway [38-41]. More specifically, among three pathways, PERK-eIF2α-mediated suppression of IκB translation can induce expression of inflammatory cytokines, allowing the excess of free NF-κB to enter the nucleus [41]. In addition to PERK, PKR can also phosphorylate eIF2α, leading to activation of various inflammatory responses. Furthermore, PKR is implicated in TLR signal transduction in response to bacterial cell wall components in which activated TLR2/TLR4 signaling induces PKR phosphorylation related to ER stress (i.e., eIF2α) and inflammatory responses [42]. Taken together, eIF2α seems to contribute to the induction of inflammatory responses more actively than other UPR pathways in the LPS-induced lung inflammation though it is very difficult to exclude the roles of other components of ER stress and UPR. Consequently, 4-PBA is expected to suppress the nuclear translocation of NF-κB in LPS-induced lung injury of mice. Naturally, 4-PBA has up to three pharmacological modes of action: it acts as an ammonia scavenger [43], a weak histone deacetylase (HDAC) inhibitor [44], and an ER stress inhibitor [45-47]. Considering that the action as an HDAC inhibitor has been reported in anti-inflammatory effects of certain disorders, we needed to determine which action mechanism attenuates LPS-induced inflammatory features. Our data revealed that HDAC activity was increased in the lung tissues of LPS-instilled mice. However, the increased HDAC activity was not affected by 4-PBA administration, suggesting that the inhibitory effects of 4-PBA on the features of LPS-induced lung injury including NF-κB activation was mainly through inhibition of ER stress. In addition, NF-κB is known as IL-17R signaling downstream and IL-17A can induce ER stress [48], thus the treatment with anti-IL-17 antibody appears to be able to suppress the nuclear translocation of NF-κB via both direct blockade of signaling pathway and indirect inhibition of ER stress.
Airway epithelial cells lie at the interface between the host and the environment and represent the first line of defense against microorganisms, toxic gases, and allergens [49]. In addition, the pulmonary epithelium is involved in inflammation-associated conditions such as ALI, ARDS, chronic obstructive pulmonary disease, and asthma, partly through the induction of inflammatory cytokines [50, 51]. Although airway epithelial cells are known as sources of some IL-17, i.e. IL-17C and IL-17E, there is little information that epithelial cells can produce and secrete IL-17A. The majority of reports have indicated that these epithelial cells are major targets and effectors of IL-17A. In fact, our results revealed that stimulation of human airway epithelial cells with exogenous IL-17A induced the activation of NF-κB pathway as well as increased expression of ER stress markers, GRP78 and CHOP. A neutralizing anti-IL-17A antibody significantly inhibited NF-κB nuclear translocation and increased expression of GRP78 protein in epithelial cells. Moreover, we unexpectedly found the expression of IL-17A protein as well as IL-17A mRNA under the stimulation with LPS in NHBE cells, which is a very surprising result, indicating that airway epithelial cells can also be an important cellular source of IL-17A production. In addition, increased expression of IL-17A mRNA and protein was reduced by treatment with a neutralizing anti-IL-17A antibody or 4-PBA. These findings suggest that IL-17A signaling can induce ER stress and NF-κB activation which is linked to the expression of various pro-inflammatory mediators in airway epithelial cells and that a positive feedback loop between IL-17A and ER stress exists in LPS-stimulated airway epithelial cells (Fig. 11).
Figure 11
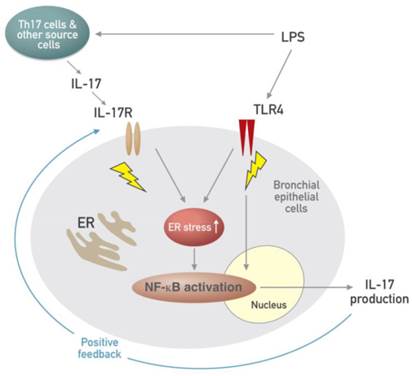
Schematic diagram for the role of the interplay between ER stress and IL-17A in LPS-induced lung injury.
LPS is a ligand for TLR4, one of the pattern recognition receptors (PRRs), and subsequent stimulation of the receptor activates NF-κB that plays a central role in regulation of many genes responsible for the production of inflammatory mediators. In addition, infection of airway epithelia or exposure to environmental stimuli up-regulates TLR4 expression and promotes its localization to the cell membrane, thus increasing endotoxin responsiveness [52, 53]. Another arm of PRRs, NLRP3 inflammasome is a major intracellular multi-protein and also plays as an important inflammatory pathway of the innate immune system. Accumulating evidence has indicated that agonists of TLRs such as LPS do not directly activate the NLRP3 inflammasome, however TLR ignition is a prerequisite for optimal activation of the NLRP3 inflammasome, resulting in production of relevant pro-inflammatory mediators such as IL-1β and IL-18 [54]. In addition, it has been recently reported that NLRP3 inflammasome is essential for the development of LPS-induced lung injury [55]. Consistent with these observations, expression of TLR4, NLRP3, caspase-1, IL-1β, and IL-18 was substantially increased in the lung tissues of LPS-instilled mice and these increases were dramatically inhibited by administration of anti-IL-17A antibody and 4-PBA. As expected, nuclear translocation of NF-κB was also significantly increased in the lung tissues of LPS-instilled mice and LPS-stimulated NHBE cells and LPS-induced nuclear translocation of NF-κB was effectively reduced by the administration of anti-IL-17A antibody, 4-PBA, or TAK-242. More clearly, the changes of the TLR4 expression and NF-κB activation induced by LPS instillation into mice were observed consistently in sorted each lung cell component including epithelial cells, macrophages, neutrophils, and DCs suggesting that IL-17A and ER stress modulates immune responses at least in part via the control of TLR4 and/or NLRP3 signaling pathways linked to NF-κB activation in LPS-induced lung inflammation.
Several studies have demonstrated that TLR4 signaling activation induces ER stress and they contribute to pro-inflammatory responses synergistically in various inflammatory and metabolic disorders [56-60]. In this current study, blockade of TLR4 with TAK-242 substantially reduced the induction of LPS-induced ER stress and IL-17A production in lung tissues of mice. More intriguingly, LPS-stimulated NHBE cells also revealed dramatic inhibition of the development of ER stress and IL-17A expression in case of pre-treatment with TAK-242. These data indicate that the link exists among TLR4 activation, ER stress, and IL-17A production in the lung, specifically in airway epithelium of LPS-induced lung injury. Moreover, we also found that combination treatment of anti-IL-17A antibody, 4-PBA, or TAK-242 showed more effective inhibition on the expression of ER stress markers and IL-17A and the nuclear translocation of NF-κB in lung tissues than the treatment with single each agent, suggesting that TLR4 pathway, ER stress, and IL-17A synergistically contribute to the pathogenesis of LPS-induced lung injury.
Recent studies have shown that epithelial cell activation is a key triggering event in the recognition of inhaled antigens that activates the local network of DCs and these DCs coordinate subsequent immune responses [61]. DCs play a key role in the pathogenesis of LPS-induced lung inflammation [62]. In keeping with these findings, infiltration of DCs into the lung was significantly increased in LPS-instilled mice, and the administration of the neutralizing anti-IL-17A antibody reduced the increases in the infiltrated DCs. These results suggest that inhibition of IL-17A using an anti-IL-17A antibody modulates recruitment/activation of DCs at least in part through inhibition of inflammatory cytokines including IL-17A produced from various lung cells including epithelial cells by LPS infection.
Conclusion
In conclusion, we have revealed an involvement of IL-17A in the development/maintenance of LPS-induced lung inflammation/injury. Our results have demonstrated that IL-17A might mediate the enhancement of ER stress and vice versa, with showing a positive feedback loop between IL-17A and ER stress exists in LPS-stimulated airway epithelial cells. Taken together, this study provides a new concept, an interrelationship between IL-17A and ER stress in the pathogenesis of LPS-induced lung injury, in which immune responses for LPS-containing Gram negative bacterial infection are modulated at least in part through the action of cytokines including IL-17A released from activated airway epithelial cells by the cooperation with ER stress and suggests their interaction can be a new therapeutic target for endotoxin-induced lung inflammatory disorders.
Supplementary Material
Figures S1-S10.
Acknowledgements
We thank Professor Mie-Jae Im for critical readings of the manuscript. This work was supported by the Korea Healthcare Technology R&D Project, Ministry for Health and Welfare, Republic of Korea; Grant A121931 (to Yong Chul Lee) and Grant A111992 (to So Ri Kim), by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT and future Planning (NRF-2014R1A2A1A01002823), and by the fund of Biomedical Research Institute, Chonbuk National University Hospital.
Authors'contributions
So Ri Kim designed research, interpreted data, and wrote the manuscript; Hee Jung Kim performed experiments and analyzed data; Jae Seok Jeong reviewed the manuscript; Seong Ho Cho reviewed and edited the manuscript; Yong Chul Lee designed research, interpreted data, and edited the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Miossec P, Korn T, Kuchroo V. Interleukin-17 and type 17 helper T cells. N Engl J Med. 2009;361:888-98
2. Gaffen S. Structure and signalling in the IL-17 receptor family. Nat Rev Immunol. 2009;9:556-67
3. Pappu R, Rutz S, Ouyang W. Regulation of epithelial immunity by IL-17 family cytokines. Trends Immunol. 2012;33:343-9
4. Iwakura Y, Ishigame H, Saijo S. et al. Functional specialization of interleukin-17 family members. Immunity. 2011;34:149-62
5. Cua DJ, Tato CM. Innate IL-17-producing cells: the sentinels of the immune system. Nat Rev Immunol. 2010;10:479-89
6. Kasahara DI, Kim HY, Williams AS. et al. Pulmonary inflammation induced by subacute ozone is augmented in adiponectin-deficient mice: role of IL-17A. J Immunol. 2012;188:4558-67
7. Gerhardt S, Abbott WM, Hargreaves D. et al. Structure of IL-17A in complex with a potent, fully human neutralizing antibody. J Mol Biol. 2009;394:905-21
8. Hymowitz SG, Filvaroff EH, Yin JP. et al. IL-17s adopt a cystine knot fold: Structure and activity of a novel cytokine, IL-17F, and implications for receptor binding. EMBO J. 2001;20:5332-41
9. Miyamoto M, Prause O, Sjo¨strand M. et al. Endogenous IL-17 as a mediator of neutrophil recruitment caused by endotoxin exposure in mouse airways. J Immunol. 2003;170:4665-72
10. Pichavant M, Goya S, Meyer EH. et al. Ozone exposure in a mouse model induces airway hyperreactivity that requires the presence of natural killer T cells and IL-17. J Exp Med. 2008;205:385-93
11. Kim SR, Lee KS, Park SJ. et al. PTEN down-regulates IL-17 expression in a murine model of toluene diisocyanate-induced airway disease. J Immunol. 2007;179:6820-9
12. Park SJ, Lee KS, Kim SR. et al. Peroxisome proliferator-activated receptor gamma agonist down-regulates IL-17 expression in a murine model of allergic airway inflammation. J Immunol. 2009;183:3259-67
13. Ferretti S, Bonneau O, Dubois GR. et al. IL-17, produced by lymphocytes and neutrophils, is necessary for lipopolysaccharide-induced airway neutrophilia: IL-15 as a possible trigger. J Immunol. 2003;170:2106-12
14. Singh J, Schwartz DA Endotoxin, the lung. Insight into the host-environment interaction. J Allergy Clin Immunol. 2005;115:330-3
15. Hosoi T, Ozawa K. Endoplasmic reticulum stress in disease: mechanisms and therapeutic opportunities. Clin Sci (Lond). 2009;118:19-29
16. Hetz C. The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat Rev Mol Cell Biol. 2012;13:89-102
17. Kim HJ, Jeong JS, Kim SR. et al. Inhibition of endoplasmic reticulum stress alleviates lipopolysaccharide-induced lung inflammation through modulation of NF-κB/HIF-1α signaling pathway. Sci Rep. 2013;3:1142
18. Martinon F, Glimcher LH. Regulation of innate immunity by signaling pathways emerging from the endoplasmic reticulum. Curr Opin Immunol. 2011;23:35-40
19. Watanabe Y, Suzuki O, Haruyama T. et al. Interferon-gamma induces reactive oxygen species and endoplasmic reticulum stress at the hepatic apoptosis. J Cell Biochem. 2003;89:244-53
20. Xue X, Piao JH, Nakajima A. et al. Tumor necrosis factor alpha (TNFalpha) induces the unfolded protein response (UPR) in a reactive oxygen species (ROS)-dependent fashion, and the UPR counteracts ROS accumulation by TNFalpha. J Biol Chem. 2005;280:33917-25
21. Richardson CE, Kooistra T, Kim DH. An essential role for XBP-1 in host protection against immune activation in C. elegans. Nature. 2010;463:1092-5
22. Sun J, Singh V, Kajino-Sakamoto R. et al. Neuronal GPCR controls innate immunity by regulating noncanonical unfolded protein response genes. Science. 2011;332:729-32
23. Jones CE, Chan K. Interleukin-17 stimulates the expression of interleukin-8, growth-related oncogene-α, and granulocyte-colony-stimulating factor by human airway epithelial cells. Am J Respir Cell Mol Biol. 2002;26:748-53
24. Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidium thiocyanate-phenol-chloroform extraction. Anal Biochem. 1987;162:156-9
25. Eppert BL, Motz GT, Wortham BW. et al. CCR7 deficiency leads to leukocyte activation and increased clearance in response to pulmonary Pseudomonas aeruginosa infection. Infect Immun. 2010;78:2099-107
26. Kirby AC, Raynes JG, Kaye PM. CD11b regulates recruitment of alveolar macrophages but not pulmonary dendritic cells after pneumococcal challenge. J Infect Dis. 2006;193:205-13
27. McCaskill J, Singhania R, Burgess M. et al. Efficient Biodistribution and Gene Silencing in the Lung epithelium via Intravenous Liposomal Delivery of siRNA. Mol Ther Nucleic Acids. 2013;2:e96
28. Mackay LS, Dodd S, Dougall IG. et al. Isolation and characterisation of human pulmonary microvascular endothelial cells from patients with severe emphysema. Respir Res. 2013;14:23
29. Myzak MC, Karplus PA, Chung FL. et al. A novel mechanism of hemoprotection by sulforaphane: inhibition of histone deacetylase. Cancer Res. 2004;64:5767-74
30. Bastarache JA, Ware LB, Bernard GR. The role of the coagulation cascade in the continuum of sepsis and acute lung injury and acute respiratory distress syndrome. Semin Respir Crit Care Med. 2006;27:365-76
31. Tolle LB, Standiford TJ. Danger-associated molecular patterns (DAMPs) in acute lung injury. J Pathol. 2013;229:145-56
32. Martin C, Papazian L, Payan MJ. et al. Pulmonary fibrosis correlates with outcome in adult respiratory distress syndrome. A study in mechanically ventilated patients. Chest. 1995;107:196-200
33. Fossiez F, Djossou O, Chomarat P. et al. T cell interleukin-17 induces stromal cells to produce pro-inflammatory and hematopoietic cytokines. J Exp Med. 1996;183:2593-603
34. Laan M, Cui ZH, Hoshino H. et al. Neutrophil recruitment by human IL-17 via C-X-C chemokinerelease in the airways. J Immunol. 1999;162:2347-52
35. Endo M, Oyadomari S, Suga M. et al. The ER stress pathway involving CHOP is activated in the lungs of LPS-treated mice. J Biochem. 2005;138:501-7
36. Hasnain SZ, Lourie R, Das I. et al. The interplay between endoplasmic reticulum stress and inflammation. Immunol Cell Biol. 2012;90:260-70
37. Barnes PJ, Karin M. Nuclear factor-kappaB: a pivotal transcription factor in chronic inflammatory diseases. N Engl J Med. 1997;336:1066-71
38. Chaudhari N, Talwar P, Parimisetty A. et al. A molecular web: endoplasmic reticulum stress, inflammation, and oxidative stress. Front Cell Neurosci. 2014;8:213
39. Yamazaki H, Hiramatsu N, Hayakawa K. et al. Activation of the Akt-NF-kappaB pathway by subtilase cytotoxin through the ATF6 branch of the unfolded protein response. J Immunol. 2009;183:1480-7
40. Hu P, Han Z, Couvillon AD. et al. Autocrine tumor necrosis factor alpha links endoplasmic reticulum stress to the membrane death receptor pathway through IRE1alpha-mediated NF-kappa B activation and downregulation of TRAF2 expression. Mol Cell Biol. 2006;26:3071-84
41. Jiang HY, Wek SA, McGrath BC. et al. Phosphorylation of the alpha subunit of eukaryotic initiation factor 2 is required for activation of NF-kappaB in response to diverse cellular stresses. Mol Cell Biol. 2003;23:5651-63
42. Cabanski M, Steinmüller M, Marsh LM. et al. PKR regulates TLR2/TLR4-dependent signaling in murine alveolar macrophages. Am J Respir Cell Mol Biol. 2008;38:26-31
43. Lichter-Konecki U, Diaz GA, Merritt JL 2nd. et al. Ammonia control in children with urea cycle disorders (UCDs); phase 2 comparison of sodium phenylbutyrate and glycerol phenylbutyrate. Mol Genet Metab. 2011;103:323-9
44. Miller AC, Cohen S, Stewart M. et al. Radioprotection by the histone deacetylase inhibitor phenylbutyrate. Radiat Environ Biophys. 2011;50:585-96
45. Basseri S, Lhotak S, Sharma AM. et al. The chemical chaperone 4-phenylbutyrate inhibits adipogenesis by modulating the unfolded protein response. J Lipid Res. 2009;50:2486-501
46. Xiao C, Giacca A, Lewis GF. Sodium phenylbutyrate, a drug with known capacity to reduce endoplasmic reticulum stress, partially alleviates lipid-induced insulin resistance and beta-cell dysfunction in humans. Diabetes. 2011;60:918-24
47. Yam GH, Gaplovska-Kysela K, Zuber C. et al. Sodium 4-phenylbutyrate acts as a chemical chaperone on misfolded myocilin to rescue cells from endoplasmic reticulum stress and apoptosis. Investig Oph-thalmol Vis Sci. 2007;48:1683-90
48. Awane M, Andres PG, Li DJ. et al. NF-kappa B-inducing kinase is a common mediator of IL-17-, TNF-alpha-, and IL-1 beta-induced chemokine promoter activation in intestinal epithelial cells. J Immunol. 1999;162:5337-44
49. Xiao C, Puddicombe SM, Field S. et al. Defective epithelial barrier function in asthma. J Allergy Clin Immunol. 2011;128:549-56
50. Borchers MT, Wesselkamper SC, Curull V. et al. Sustained CTL activation by murine pulmonary epithelial cells promotes the development of COPD-like disease. J Clin Invest. 2009;119:636-49
51. Kropski JA, Fremont RD, Calfee CS. et al. Clara cell protein (CC16), a marker of lung epithelial injury, is decreased in plasma and pulmonary edema fluid from patients with acute lung injury. Chest. 2009;135:1440-7
52. Pace E, Ferraro M, Siena L. et al. Cigarette smoke increases Toll-like receptor 4 and modifies lipopolysaccharide-mediated responses in airway epithelial cells. Immunology. 2008;124:401-11
53. Monick MM, Yarovinsky TO, Powers LS. et al. Respiratory syncytial virus up-regulates TLR4 and sensitizes airway epithelial cells to endotoxin. J Biol Chem. 2003;278:53035-44
54. Bauernfeind FG, Horvath G, Stutz A. et al. Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J Immunol. 2009;183:787-91
55. Grailer JJ, Canning BA, Kalbitz M. et al. Critical role for the NLRP3 inflammasome during acute lung injury. J Immunol. 2014;192:5974-83
56. Sodhi CP, Jia H, Yamaguchi Y. et al. Itestinal Epithelial TLR-4 Activation Is Required for the Development of Acute Lung Injury after Trauma/Hemorrhagic Shock via the Release of HMGB1 from the Gut. J Immunol. 2015;194:4931-9
57. Pierre N, Deldicque L, Barbé C. et al. Toll-like receptor 4 knockout mice are protected against endoplasmic reticulum stress induced by a high-fat diet. PLoS One. 2013;8:e65061
58. Martinon F, Chen X, Lee AH. et al. TLR activation of the transcription factor XBP1 regulates innate immune responses in macrophages. Nat Immunol. 2010;11:411-8
59. DeLay ML, Turner MJ, Klenk EI. et al. HLA-B27 misfolding and the unfolded protein response augment interleukin-23 production and are associated with Th17 activation in transgenic rats. Arthritis Rheum. 2009;60:2633-43
60. Mahadevan NR, Rodvold J, Sepulveda H. et al. Transmission of endoplasmic reticulum stress and pro-inflammation from tumor cells to myeloid cells. Proc Natl Acad Sci USA. 2011;108:6561-6
61. Lambrecht BN, Hammad H. Biology of lung dendritic cells at the origin of asthma. Immunity. 2009;31:412-24
62. Banchereau J, Briere F, Caux C. et al. Immunobiology of dendritic cells. Annu Rev Immunol. 2000;18:767-811
Author contact
![]() Corresponding authors: Yong Chul Lee, MD, PhD. Department of Internal Medicine, Chonbuk National University Medical School, san 2-20, Geumam-dong, Deokjin-gu, Jeonju, 561-180, South Korea. Phone: 82-63-250-1664; Fax: 82-63-259-3212; E-mail: leeycac.kr. So Ri Kim, MD, PhD. Department of Internal Medicine, Chonbuk National University Medical School, san 2-20, Geumam-dong, Deokjin-gu, Jeonju, 561-180, South Korea. Phone: 82-63-250-2475; Fax: 82-63-259-3212; E-mail: soriac.kr.
Corresponding authors: Yong Chul Lee, MD, PhD. Department of Internal Medicine, Chonbuk National University Medical School, san 2-20, Geumam-dong, Deokjin-gu, Jeonju, 561-180, South Korea. Phone: 82-63-250-1664; Fax: 82-63-259-3212; E-mail: leeycac.kr. So Ri Kim, MD, PhD. Department of Internal Medicine, Chonbuk National University Medical School, san 2-20, Geumam-dong, Deokjin-gu, Jeonju, 561-180, South Korea. Phone: 82-63-250-2475; Fax: 82-63-259-3212; E-mail: soriac.kr.
Citation styles
APA
Kim, S.R., Kim, H.J., Kim, D.I., Lee, K.B., Park, H.J., Jeong, J.S., Cho, S.H., Lee, Y.C. (2015). Blockade of Interplay between IL-17A and Endoplasmic Reticulum Stress Attenuates LPS-Induced Lung Injury. Theranostics, 5(12), 1343-1362. https://doi.org/10.7150/thno.11685.
ACS
Kim, S.R.; Kim, H.J.; Kim, D.I.; Lee, K.B.; Park, H.J.; Jeong, J.S.; Cho, S.H.; Lee, Y.C. Blockade of Interplay between IL-17A and Endoplasmic Reticulum Stress Attenuates LPS-Induced Lung Injury. Theranostics 2015, 5 (12), 1343-1362. DOI: 10.7150/thno.11685.
NLM
Kim SR, Kim HJ, Kim DI, Lee KB, Park HJ, Jeong JS, Cho SH, Lee YC. Blockade of Interplay between IL-17A and Endoplasmic Reticulum Stress Attenuates LPS-Induced Lung Injury. Theranostics 2015; 5(12):1343-1362. doi:10.7150/thno.11685. https://www.thno.org/v05p1343.htm
CSE
Kim SR, Kim HJ, Kim DI, Lee KB, Park HJ, Jeong JS, Cho SH, Lee YC. 2015. Blockade of Interplay between IL-17A and Endoplasmic Reticulum Stress Attenuates LPS-Induced Lung Injury. Theranostics. 5(12):1343-1362.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) License. See http://ivyspring.com/terms for full terms and conditions.