Theranostics
13.3
Impact Factor
 Global reach, higher impact
Global reach, higher impact
Theranostics 2016; 6(1):104-117. doi:10.7150/thno.13580 This issue Cite
Research Paper
In Vivo Stabilization of a Gastrin-Releasing Peptide Receptor Antagonist Enhances PET Imaging and Radionuclide Therapy of Prostate Cancer in Preclinical Studies
Kristell L.S. Chatalic1,2,3, ![]() , Mark Konijnenberg1, Julie Nonnekens1,4, Erik de Blois1, Sander Hoeben3, Corrina de Ridder3, Luc Brunel5, Jean-Alain Fehrentz5, Jean Martinez5, Dik C. van Gent4, Berthold A. Nock6, Theodosia Maina6, Wytske M. van Weerden3, Marion de Jong1,2
, Mark Konijnenberg1, Julie Nonnekens1,4, Erik de Blois1, Sander Hoeben3, Corrina de Ridder3, Luc Brunel5, Jean-Alain Fehrentz5, Jean Martinez5, Dik C. van Gent4, Berthold A. Nock6, Theodosia Maina6, Wytske M. van Weerden3, Marion de Jong1,2
1. Department of Nuclear Medicine, Erasmus MC, Rotterdam, The Netherlands
2. Department of Radiology, Erasmus MC, Rotterdam, The Netherlands
3. Department of Urology, Erasmus MC, Rotterdam, The Netherlands
4. Department of Genetics, Erasmus MC, Rotterdam, The Netherlands
5. IBMM, UMR 5247, CNRS, Université Montpellier, ENSCM, France
6. Molecular Radiopharmacy, INRASTES, NCSR Demokritos, Athens, Greece
Received 2015-8-17; Accepted 2015-9-25; Published 2016-1-1
Citation:
Chatalic KLS, Konijnenberg M, Nonnekens J, de Blois E, Hoeben S, de Ridder C, Brunel L, Fehrentz JA, Martinez J, van Gent DC, Nock BA, Maina T, van Weerden WM, de Jong M. In Vivo Stabilization of a Gastrin-Releasing Peptide Receptor Antagonist Enhances PET Imaging and Radionuclide Therapy of Prostate Cancer in Preclinical Studies. Theranostics 2016; 6(1):104-117. doi:10.7150/thno.13580. https://www.thno.org/v06p0104.htm
Other stylesAbstract

A single tool for early detection, accurate staging, and personalized treatment of prostate cancer (PCa) would be a major breakthrough in the field of PCa. Gastrin-releasing peptide receptor (GRPR) targeting peptides are promising probes for a theranostic approach for PCa overexpressing GRPR. However, the successful application of small peptides in a theranostic approach is often hampered by their fast in vivo degradation by proteolytic enzymes, such as neutral endopeptidase (NEP). Here we show for the first time that co-injection of a NEP inhibitor (phosphoramidon (PA)) can lead to an impressive enhancement of diagnostic sensitivity and therapeutic efficacy of the theranostic 68Ga-/177Lu-JMV4168 GRPR-antagonist. Co-injection of PA (300 µg) led to stabilization of 177Lu-JMV4168 in murine peripheral blood. In PC-3 tumor-bearing mice, PA co-injection led to a two-fold increase in tumor uptake of 68Ga-/177Lu-JMV4168, 1 h after injection. In positron emission tomography (PET) imaging with 68Ga-JMV4168, PA co-injection substantially enhanced PC-3 tumor signal intensity. Radionuclide therapy with 177Lu-JMV4168 resulted in significant regression of PC-3 tumor size. Radionuclide therapy efficacy was confirmed by production of DNA double strand breaks, decreased cell proliferation and increased apoptosis. Increased survival rates were observed in mice treated with 177Lu-JMV4168 plus PA as compared to those without PA. This data shows that co-injection of the enzyme inhibitor PA greatly enhances the theranostic potential of GRPR-radioantagonists for future application in PCa patients.
Keywords: GRPR, neutral endopeptidase, enzyme inhibition, phosphoramidon, theranostics, PET imaging, radionuclide therapy, prostate cancer.
Introduction
Prostate cancer (PCa) is the most frequently diagnosed cancer and the second leading cause of cancer-related death among men in the United States [1]. The five-year relative survival rate for localized and regional PCa is 100%, but decreases to 29% when the cancer has spread to distant sites [2]. Prognosis and treatment options are strongly related to disease stage and grade at time of diagnosis. Early diagnosed and locally confined tumors can be treated by radical prostatectomy and/or local radiation therapy, whereas for advanced, disseminated PCa curative treatment is not available [2]. This underlines the importance of early detection and accurate staging for the successful management of PCa patients.
Currently, local staging is mostly based on ultrasound, biopsies and level of serum prostate-specific antigen. Recent advances in molecular imaging techniques have highlighted the potential of PET imaging, providing a sensitive and non-invasive detection method for PCa lesions. Metabolic PET radiotracers, such as 18F-fluoro-2-deoxy-D-glucose (FDG) or 18F-fluoro-choline have been used to detect recurrent metastatic PCa with high metabolic rates, but are suboptimal for the detection of slow-growing primary tumors.
An alternative strategy focuses on the development of receptor-directed PET imaging agents. PET radiotracers targeting prostate-specific membrane antigen (PSMA) and gastrin-releasing peptide receptor (GRPR) have shown promising results for detection of primary and metastasized PCa in early clinical studies [3-6]. Besides their use for diagnostic imaging, these targeting agents may also be used as therapeutic agents for tumor-targeted radionuclide therapy of PCa. PSMA-targeted agents labeled with the β-emitting radioisotope 177Lu are currently evaluated in clinical studies [7, 8]. The powerful combination of diagnosis and therapy by one single probe in a “theranostic” strategy could effectively contribute to a more personalized management of PCa patients, whereby imaging can guide the choice of the most suitable therapeutic option for each patient.
The aim of the present study was to develop a theranostic agent based on a GRPR-antagonist with application prospects in the personalized treatment of PCa patients.
GRPRs are overexpressed in the majority of primary prostate tumors and in a subset of patients with lymph node and/or bone metastasis [9, 10], making it an interesting target for radionuclide therapy. GRPR-antagonists are promising molecular probes, owing to their superior tumor-targeting and pharmacokinetic properties compared to agonists [11]. GRPR agonists were also shown to induce side effects in patients, owing to their physiological activity [12, 13]. The potential of the 68Ga-labeled GRPR-antagonist, JMV4168, for PET imaging of prostate cancer was reported previously in preclinical studies [14].
However, delivery of radiopeptides to the tumor will be challenged by the action of proteolytic enzymes present in blood, vasculature walls, liver, lungs, kidneys and gastrointestinal tract. In vivo degradation of radiopeptides by proteolytic enzymes may hamper their targeting properties, thereby limiting their successful application as theranostic probes. One of the key proteolytic enzymes is neutral endopeptidase 24.11 (NEP, EC 3.4.24.11, neprilysin, CD10, MME), a cell surface metallopeptidase responsible for catalytic inactivation of many neuropeptide substrates, including bombesin-like peptides [15-17]. NEP is abundantly expressed in the human body, including vasculature walls, major organs and tissues, potentially hampering radiopeptide-based imaging and therapy by cleaving radiopeptides on the amino side of hydrophobic amino acids. The released inactive radiometabolites will not be able to interact with the tumor-associated target-receptors, and as a result, diagnostic sensitivity and therapeutic efficacy will be severely compromised. In previous studies we have shown that co-injection of a single enzyme inhibitor, phosphoramidon (PA), could stabilize radiopeptides in vivo by inhibition of NEP. This strategy led to notable enhancement of tumor uptake of radiopeptides, whereas uptake in most non-target healthy tissues was not affected [18]. We hypothesize that this strategy would be particularly interesting to enhance the therapeutic efficacy of radionuclide therapy as well, by delivering a higher cytotoxic radiation dose to cancer cells at similar dose exposure of healthy tissues.
The aim of this study was to investigate the effect of in vivo stabilization by PA on diagnostic sensitivity and therapeutic efficacy of the GRPR-targeted theranostic agent 68Ga/177Lu-JMV4168 in nude mice with subcutaneous (sc) human prostate tumors.
Materials and Methods
Peptide, reagents, cell line and mice
JMV4168 (DOTA-βAla-βAla-[H-D-Phe-Gln-Trp-Ala-Val-Gly-His-Sta-Leu-NH2], Figure 1) was synthesized as described previously [19]. Chemicals were purchased from Sigma-Aldrich, unless otherwise stated. Phosphoramidon (PA) was purchased from Peptides International Inc. 177LuCl3 was purchased from IDB Holland and no-carrier added (n.c.a.) ItG 177LuCl3 was obtained from ITG Isotope Technologies Garching GmbH. 175Lu was obtained from Merck as 1 g/L standard solution in nitric acid. The human PCa cell line PC-3 was obtained from the American Type Culture Collection (CRL 1435) and cell culture reagents from Life Technologies. Cells were cultured in Ham's F-12K (Kaighn's) Medium supplemented with 10% fetal bovine serum, penicillin (100 units/mL), and streptomycin (100 µg/mL). Cells were grown in tissue culture flasks at 37°C in a humidified atmosphere containing 5% CO2. Male nude BALB/c mice (8 weeks old) were obtained from Janvier. All animal experiments were approved by the Animal Experiments Committee under the Dutch Experiments on Animal Act and adhered to the European Convention for Protection of Vertebrate Animals used for Experimental Purposes (Directive 86/609/EEC).
Labeling of JMV4168 with 68Ga, 177Lu and 175Lu
Elution of 68Ga from a 68Ga/68Ge generator (IGG-100, Eckert & Ziegler AG) was performed using fractionated elution with 0.1 M HCl (Rotem Industries Ltd). For PET imaging and biodistribution studies, JMV4168 (1-2 nmol) was mixed with 68Ga eluate (200 µL), sodium acetate (0.5 M, 50 µL) and ethanol (30 µL). The reaction mixture was heated for 10 min at 95°C. After reaction, ethylenediaminetetraacetic acid (EDTA, 4 mM) was added to complex free 68Ga, and the reaction mixture was filtered (0.02 µm WhatmanTM filter, GE Healthcare) to remove 68Ga-hydroxides [20].
JMV4168 was labeled with carrier-added 177LuCl3 (IDB Holland) with a specific activity (ratio between amount of bound radioactivity and total molar quantity of peptide) of 125 MBq/nmol for in vivo stability studies and 60 MBq/nmol for biodistribution studies. Labeling was performed in 20 mM sodium acetate, for 15 min at 80°C. Radioprotectants (gentisic acid, ascorbic acid and methionine, 3.5 mM) were added to prevent radiolysis.
To obtain higher specific activity (i.e. 250 MBq/nmol) for in vivo therapy studies, JMV4168 was labeled with n.c.a. 177LuCl3 (ITG Munich) as the presence of 176Lu in carrier-added 177LuCl3 limits the maximum achievable specific activity to 125 MBq/nmol. Labeling was performed in 50 mM sodium acetate for 15 min at 80°C with radioprotectants. An excess of diethylenetriaminepentaacetic acid (DTPA, 4 mM) was added to complex free 177LuCl3 after reaction.
For control experiments, JMV4168 was labeled with the stable isotope 175Lu. JMV4168 was incubated with a 2-fold molar excess 175Lu in 80 mM sodium acetate, for 15 min at 80°C.
Vehicle for animal injection
To allow for injection into mice, the radiolabeled peptide was diluted in a vehicle. For biodistribution studies, vehicle consisted of 5% (v/v) ethanol, 0.05% (w/v) bovine serum albumin (BSA) in phosphate-buffered saline (PBS), pH 7.4, containing a mixture of 0.5 mM radioprotectants. For therapy studies with higher activity concentration, vehicle consisted of 5% (v/v) ethanol, 0.05% (w/v) BSA in PBS, pH 7.4, containing 5 mM radioprotectants.
Quality control
Labeling efficiency was assessed by instant thin layer chromatography (iTLC) using silica gel coated paper (Varian Medical Systems, Inc.) and 0.1 M citrate buffer pH 5 as eluent. Colloid formation was determined by iTLC using silica gel-coated paper and 1 M NH4OAc:methanol (1:3) as eluent. Radiochemical purity of labeled peptides was analyzed by RP-HPLC on a Breeze system (Waters). A C-18 column (Symmetry Shield, 4.6 mm x 250 mm; particle size 5 μm, Waters) was used at a flow rate of 1 mL/min with the following buffer system: buffer A, 0.1% v/v trifluoroacetic acid in water; buffer B, methanol; with a gradient as follows: 100% buffer A (0-5 min), 60% buffer B (5-5.01 min), 80% buffer B (5.01-20 min), 100% buffer B (20.01-25 min), 100% buffer A (25.01-30 min). The radioactivity of the eluate was monitored using an in-line NaI radiodetector, digital multichannel analyzer and dedicated software (MetorX B.V.). Elution profiles were analyzed using Empower 3 software (Waters).
In vivo stability studies
Non-tumor bearing mice were injected intraperitoneally (ip) with 177Lu-JMV4168 (25 MBq, 200 pmol) in vehicle, or in vehicle containing PA (300 μg). Blood was drawn 15 min after injection and collected in a polypropylene tube coated with EDTA containing PA (30 μg), and 50 nmol of radioprotectants on ice. Plasma proteins were precipitated by adding 50% ethanol, centrifuged for 10 min at 4°C and the extract was analyzed by radio-HPLC. HPLC fractions were collected every minute and counted in a γ-counter to quantify intact radiopeptide and degradation products.
Figure 1
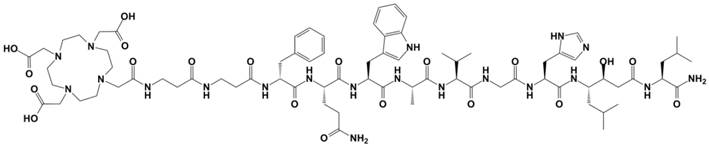
Chemical structure of JMV4168 (DOTA-βAla-βAla-[H-D-Phe-Gln-Trp-Ala-Val-Gly-His-Sta-Leu-NH2])
Prostate cancer xenograft model
Mice were injected subcutaneously on the right shoulder with a PC-3 cell suspension (3 x 106 cells, 200 µL, 67% RPMI, 33% Matrigel [BD Bioscience]). Three to four weeks after inoculation, tumor size averaged 200 mm3 and mice were used for biodistribution, PET imaging and therapy studies.
Biodistribution studies
68Ga-JMV4168 (1 MBq, 200 pmol) was injected intravenously (iv) with vehicle (control) or with PA (300 µg) in vehicle. Mice were euthanized 1 h or 2 h after injection. 177Lu-JMV4168 (0.7 MBq, 200 pmol) was injected iv or ip with or without PA (300 µg) in vehicle. Mice were sacrificed at 1 h, 4 h or 24 h after injection for 177Lu-JMV4168 in controls, and 1 h or 4 h for 177Lu-JMV4168 with PA. Blood, tumor, and relevant organs and tissues were collected, weighed and counted in a γ-counter (WIZARD, PerkinElmer) with a counting time of 60 sec per sample, isotope-specific energy window and an error not exceeding 5%. The percentage of the injected dose per gram (%ID/g) was determined for each tissue sample.
Dosimetry
Biodistribution data of 177Lu-JMV4168 in controls (iv and ip) were used to estimate the absorbed radiation doses to the organs with physiological uptake and tumor. A single-exponential expression was fitted to the activity data at 1, 4 and 24 h, and the cumulative activity concentration in each organ was calculated by analytic integration of the fitted expression. Absorbed radiation doses of 177Lu-JMV4168 with PA were estimated assuming equivalent clearance as without PA from 4 to 24 h.
Absorbed radiation doses to the organs were estimated according to the MIRD-scheme [21] with the following equation: 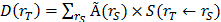 , with
, with 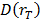 the absorbed dose to a target organ
the absorbed dose to a target organ  ,
, 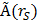 the time-integrated activity in a source organ
the time-integrated activity in a source organ 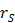 and
and 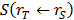 the absorbed dose rate per unit activity of 177Lu. The S-values were obtained for a standardized 25 g mouse from the RADAR realistic animal models [22]. The biodistribution data are measured in activity concentration and hence the time-integrated activity concentration is obtained and this is multiplied with the source organ mass, as used in the phantom for the S-value calculation.
the absorbed dose rate per unit activity of 177Lu. The S-values were obtained for a standardized 25 g mouse from the RADAR realistic animal models [22]. The biodistribution data are measured in activity concentration and hence the time-integrated activity concentration is obtained and this is multiplied with the source organ mass, as used in the phantom for the S-value calculation.
The Linear Quadratic (LQ) model was used to predict cell survival for PC-3 radiation response (α = 0.13 /Gy, β = 0.024 /Gy2, doubling time of 11 days, sub-lethal repair half-life Tμ = 1 h, clonogenic cell density; 107/g) [23]. Radiation sensitivity was estimated by external 137Cs gamma irradiation (data not shown), resulting in LQ model fit (Pearson R2 = 0.99) with α= 0.13 ± 0.02 /Gy, β = 0.025 ± 0.005 /Gy2 (α/β = 5.5 Gy). Doubling time was estimated by fitting an exponential growth expression into PC-3 tumor growth curve in our model. Tumor control as a function of absorbed dose D over time T was expressed by Poisson statistics expressing the probability for survival S of 1 or more clonogenic cells from the original number of clonogenic cells Nclonogens by:
, with the survival
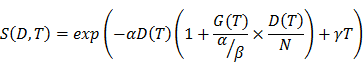
with γ the tumor growth constant and N the number of therapy cycles used.
The dose prolongation factor G(T) can be approximated by the value integrated to infinity for single exponential dose build-up with effective half-life Teff: 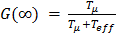 [24]. A Tumor Control Probability of > 90% was considered as being successful.
[24]. A Tumor Control Probability of > 90% was considered as being successful.
Small-animal PET
Mice were injected iv with 2-8 MBq of 68Ga-JMV4168 (200 pmol) in vehicle or vehicle containing PA (300 µg). Mice were scanned 2 h after injection, on a heated bed under isoflurane/O2 anesthesia, in a small animal PET scanner (Inveon; Siemens Preclinical Solutions). PET emission scans were acquired for 45 min, followed by a transmission scan (402 s). Scans were reconstructed using Inveon Acquisition Workplace software (version 1.5; Siemens Preclinical Solutions), using a 3-dimensional ordered-subset expectation maximization/maximization a posteriori algorithm with the following parameters: matrix, 256 x 256 x 161; pixel size, 0.40 x 0.40 x 0.796 mm; and β-value, 1.0, with uniform variance and OPMAP. Images were rescaled to account for differences in injected dose.
Therapy studies
Mice received four ip injections in 2-days intervals of either PBS, 177Lu-JMV4168 (50 MBq, 200 pmol) in vehicle or 177Lu-JMV4168 (50 MBq, 200 pmol) in vehicle containing PA (300 µg). To exclude a possible anti-tumor effect of radioprotectants, GRPR-antagonist or PA, a separate experiment was performed: mice received four ip injections of PBS, 175Lu-JMV4168 in vehicle, or 175Lu-JMV4168 in vehicle containing PA (300 µg). Tumor size was followed twice weekly by caliper measurements, endpoints were tumor size exceeding 1.5 cm3, or survival of 94 days after the first injection.
GRPR expression, histology and molecular characteristics
In order to study the molecular response of tumor cells to therapy, 2 mice of each group were sacrificed 8 days after start of therapy, and tumor was dissected. At the endpoint of the experiment, tumor, kidney and pancreas from sacrificed animals of each group were dissected. For each organ half was frozen in liquid nitrogen and half was fixed in 10% neutral buffered formalin at 4°C. After fixation and routine dehydration, tissue samples were embedded in paraffin.
GRPR expression after radionuclide therapy treatment was assessed on frozen tumor sections (10 µm) using autoradiography as described earlier [14]. In brief, frozen tissue sections were incubated with 177Lu-JMV4168 (10-10 M) for 1 h in binding buffer containing 1% w/v BSA. Unlabeled JMV4168 was used for blocking (10-6 M). Radioactivity was measured using a phosphor imager system (Cyclone, Packard A431201, PerkinElmer) and data obtained on different phosphor screens was normalized against a positive control slide (PC-295 xenograft tumor [25]).
Histology was performed on paraffin-embedded kidney and pancreas sections (4 µm). Sections were stained with hematoxylin and eosin for microscopic examination using standard protocols. Immunofluorescence and TUNEL staining were performed on paraffin-embedded tumor sections (4 µm) of mice sacrificed 8 days after therapy.
For immunofluorescence staining, tumor sections were deparaffinized, rehydrated and target antigen retrieval was performed using DAKO Antigen Retrieval buffer (pH 6.9) for 15 min at 100oC. Cells were permeabilized in PBS containing 0.1% Triton X-100 for 10 min and incubated in blocking buffer [PBS, 0.1% Triton X-100, 2% BSA]. Primary antibodies [anti-GEMININ (10802-1-AP, Proteintech Group, 1/400 dilution), anti-53BP1 (NB100-304, Novus Biologicals, dilution 1/1000), or anti-γH2AX (05-636, Merck-Millipore, dilution 1/500)] were diluted in blocking buffer and incubated for 90 min at room temperature (RT). Secondary antibodies (Alexa Fluor 594 or 488, Life Technologies; dilution 1/1000) were diluted in blocking buffer and incubated for 60 min at RT.
For apoptotic cell detection TUNEL staining was performed. Tumor sections were stained using an in situ cell death detection kit (11684795910, Roche). Briefly, cells were deparaffinized, rehydrated and incubated in PBS containing 0.5% Triton X-100 and proteinase K (20 μg/ml) for 15 min at RT. Cells were incubated in PBS containing 0.1% Triton X-100 and 0.1% sodium citrate for 2 min on ice, followed by TUNEL labeling for 1 h at 37oC. Imaging was performed using a DM4000 fluorescent microscope (Leica).
γH2AX and 53BP1 imaging was performed using a TCS SP5 confocal microscope (Leica) and GEMININ imaging using a DM4000 fluorescent microscope (Leica).
53BP1/γH2AX foci quantification was performed with ImageJ software using automated threshold settings. GEMININ and TUNEL positive cells were counted manually and compared to the total number of DAPI-positive cells. Approximately 1000 (53BP1/γH2AX) or 400 (GEMININ/TUNEL) DAPI-positive cells from different microscope fields per condition were evaluated.
Statistical Analysis
Statistical analysis was performed using GraphPad Prism version 5.01. Biodistribution data are represented as the mean %ID/g ± SD, with group sizes of 3-4 mice. Statistical analysis of biodistribution data was performed using a 2-way ANOVA with a Tukey's multiple comparisons test, and the level of significance was set at P < 0.05.
Mean tumor size with 95% confidence interval were displayed for the period that all mice of one group were still in the experiments. Mice were sacrificed when tumor exceeded 1.5 cm3, causing the mean size to drop, which made the comparison of the means not fair after that period. Survival analysis is displayed up to day 94, corresponding to a maximum follow up time.
Statistical analysis of mean tumor size of all groups was performed up to day 24, when all animals were still in the experiment. A 2-way ANOVA with repeated measures was used to compare the three groups, with a Tukey's multiple comparison test. Tumor size of the two treated groups was compared up to day 66, with a two-tailed paired t-test. Statistical analysis of survival data was performed using a Mantel-Cox Chi square test, with a level of significance set at P < 0.05, corrected to 0.0167 for multiple comparisons.
Tumor growth was analyzed by performing a non-linear regression fit of an exponential growth curve to the control group. The Aikake information criterion was used to determine whether the growth was exponential or showed a Gompertzian type of growth. The tumor growth data of the treated animals were fitted by an initial exponential growth curve till the onset time of shrinkage and an exponential regrowth curve. Extrapolations of the growth curves for animals that were taken out of the experiment due to too large tumor sizes enabled establishment of a mean growth curve.
Results
Radiolabeling of JMV4168 with 68Ga and 177Lu
68Ga-JMV4168 was obtained with a specific activity of 50 MBq/nmol. 177Lu-JMV4168 could be obtained with a specific activity of up to 125 MBq/nmol using carrier added 177Lu/176Lu and 250 MBq/nmol using n.c.a. 177Lu. Labeling efficiency for 68Ga-JMV4168 and 177Lu-JMV4168 exceeded 97% as determined by iTLC. Radiopeptides were not purified and radiochemical purity of preparations used in in vivo experiments exceeded 96% for 68Ga-JMV4168 and 94% for 177Lu-JMV4168, as determined by RP-HPLC. An excess of EDTA/DTPA was added to complex free 68Ga/177Lu and limit accumulation in bone. Protection of 177Lu-JMV4168 with radioprotectants (methionine, gentisic acid and ascorbic acid) was required to preserve the peptide from radiolysis [26]. Radioprotectants were added in the labeling reaction, as well as in the formulation for mouse injection. Radio-HPLC elution profiles of 68Ga- and 177Lu-JMV4168 are shown in Figure 2. 68Ga-JMV4168 and 177Lu-JMV4168 had retention times of 9.99 min and 12.57 min, respectively.
Figure 2
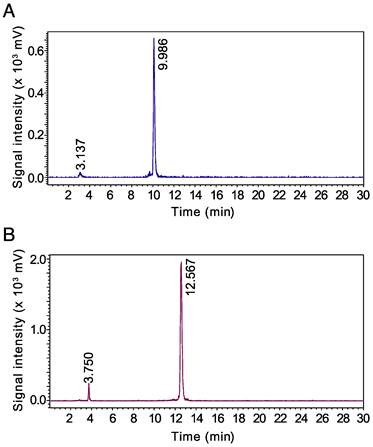
Radio-HPLC chromatograms of 68Ga-JMV4168 (A) and 177Lu-JMV4168 (B).
PA stabilizes 177Lu-JMV4168 in murine peripheral blood
In vivo stability studies resulted in 48% intact 177Lu-JMV4168 in peripheral blood after 15 min, and an increase to 84% intact radiopeptide upon co-injection of PA.
PA enhances tumor uptake of 68Ga- and 177Lu-JMV4168 in PC-3 xenograft mice
The results of the biodistribution studies of 68Ga- and 177Lu-JMV4168 are summarized in Figure 3. Biodistribution patterns of 68Ga- and 177Lu-JMV4168 at 1 h after injection were comparable (p > 0.05 for all organs). Tumor uptake (in %ID/g ± SD) at 1 h after iv injection increased from 9.9 ± 1.3 to 19.1 ± 2.6 (68Ga-JMV4168, p < 0.0001) and from 9.0 ± 3.9 to 17.2 ± 1.7 (177Lu-JMV4168, p < 0.0001) upon co-injection of PA. Uptake in the GRPR-positive pancreas was increased at 1 h after iv injection for 68Ga-JMV4168 (p < 0.0001), but not for 177Lu-JMV4168, upon co-injection of PA. No significant increase in uptake in non-target tissue was observed, resulting in high tumor-to-background ratios. A 3-fold decrease in pancreatic uptake was observed for 68Ga-JMV4168 from 1 h to 2 h after injection, while tumor uptake remained stable, leading to increased tumor/pancreas ratio over time. Biodistribution patterns of 177Lu-JMV4168 after iv or ip injection were fairly comparable. Increased uptake in pancreas was observed at 1 h after ip injection in comparison with iv injection (p < 0.0001), but no differences were observed at later time points. Tumor uptake values (in %ID/g ± SD) at 1 h, 4 h, and 24 h after injection were 9.0 ± 3.9, 8.3 ± 1.7, and 4.2 ± 1.0 after iv, versus 8.9 ± 2.4, 7.2 ± 2.1, and 4.5 ± 0.7 after ip injection, respectively. Tumor uptake at 4 h after injection increased to 12.9 ± 3.5 (iv), and 14.5 ± 3.7 (ip) upon co-administration of PA.
Dosimetry of 177Lu-JMV4168 in PC-3 xenograft mice
Single-exponential curves could be fitted to the biodistribution data; the tumor showed comparable clearance half-lives for the two types of injection (20.8 ± 8.5 h (iv) and 24.4 ± 8.3 h (ip)). Estimated absorbed radiation doses (in Gy) from 50 MBq 177Lu-JMV4168 in tumor, kidneys and pancreas were 10, 3.5, 2.5 Gy, and 11, 7.3, 4.4 Gy after iv and ip injections, respectively. Absorbed doses for tumor, kidneys and pancreas for 177Lu-JMV4168 with PA were 16, 3.7, 3.1 and 20, 7.6, 4.8 Gy after iv and ip injections, respectively, assuming equivalent clearance from 4 to 24 h as without PA. No significant differences were observed for absorbed radiation doses of normal organs for 177Lu-JMV4168 and 177Lu-JMV4168 with PA.
Based on the absorbed doses in PC-3 tumors, it was predicted that a single ip dose of 367 MBq (81 Gy) 177Lu-JMV4168 would have a 95% probability to elicit full tumor control. However, a maximum of 50 MBq (200 pmol) could be administered per injection (specific activity of 250 MBq/nmol). For 177Lu-JMV4168 alone, 8 cycles of therapy (8 x 50 MBq, ip) with 2-days intervals would be needed to reach 98% TCP. Upon co-administration of 177Lu-JMV4168 and PA, 4 cycles of therapy (4 x 50 MBq, ip) with 2-days intervals would be needed to reach 93% TCP (Figure 4).
Figure 3
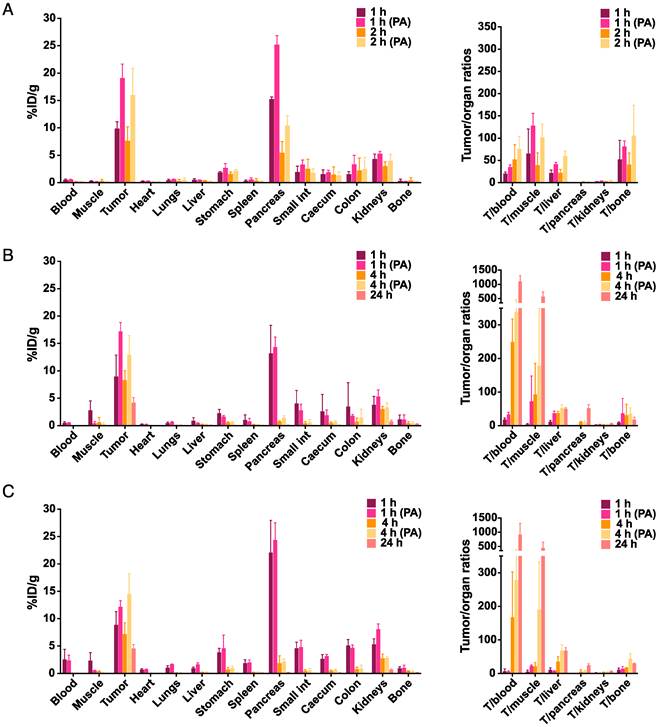
Effect of phosphoramidon (PA) on the biodistribution of 68Ga-JMV4168 (200 pmol, 1 MBq) administered intravenously (iv) at 1 h and 2 h after injection (A) and 177Lu-JMV4168 (200 pmol, 0.7 MBq) administered intravenously (B) or intraperitoneally (C) at 1 h, 4 h and 24 h after injection (n=3-4 per group). Biodistribution of 68Ga-JMV4168 and 177Lu-JMV4168 at 1 h after injection were comparable. Tumor uptake of 177Lu-JMV4168 injected intraperitoneal and intravenously were comparable. Tumor uptake and tumor-to-background ratios were increased in the presence of PA.
Figure 4
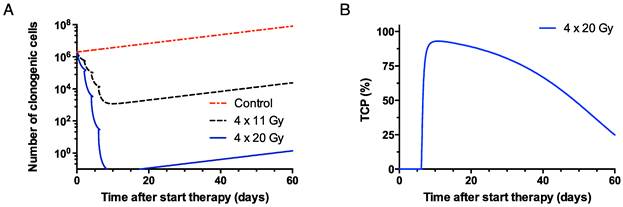
Theoretical survival curve of clonogenic cells in PC-3 xenograft (200 mg, 2 x 106 clonogenic cells at the start) after therapy with 4 injections (2-days intervals) of 50 MBq of 177Lu-JMV4168 (4 x 11 Gy) and 177Lu-JMV4168 + phosphoramidon (4 x 20 Gy) (A). Tumor control probability (TCP) curve for the therapy with phosphoramidon is shown, reaching a 93% TCP after 9 days (B).
PA enhances sensitivity of small-animal PET imaging of 68Ga-JMV4168 in PC-3 xenograft mice
PET images obtained at 2 h after iv injection of 68Ga-JMV4168 are presented in Figure 5, showing clear, high-contrast visualization of PC-3 tumors. Visualization of PC-3 tumors was substantially improved by co-injection of PA, as shown by the increased signal intensity in the tumors.
Figure 5
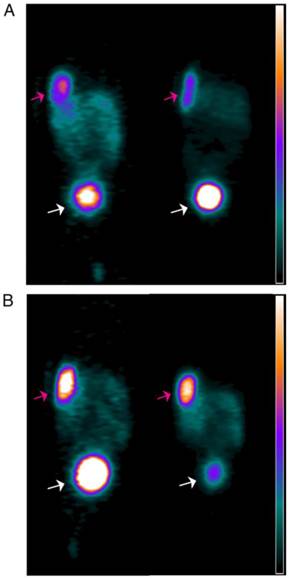
PET images of duplicate mice bearing subcutaneous PC-3 xenografts near the shoulder (pink arrow) injected with 68Ga-JMV4168 (A), or 68Ga-JMV4168 with phosphoramidon (B) at 2 h after injection, illustrating the higher tumor uptake in the presence of phosphoramidon. Excretion of the radiolabeled peptide in the bladder (white arrow) can be observed. Scale bar; 0 to 105 kBq/mL.
PA augments therapeutic efficacy of 177Lu-JMV4168 in PC-3 xenograft mice
Therapy studies were conducted in PC-3 tumor-bearing mice receiving four cycles of PBS, 177Lu-JMV4168, or 177Lu-JMV4168 plus PA (Figures 6 and 7). The 177Lu-JMV4168-treated mice showed significant reduction in tumor size as compared to control (PBS) mice. At day 24 after start of therapy, mean tumor volumes (in mm3 ±SD) were 1181 ± 402, 146 ± 35 and 145 ± 45 for PBS, 177Lu-JMV4168 and 177Lu-JMV4168 plus PA, respectively. Tumor regrowth was observed for both treated groups, with mean tumor volumes (in mm3 ± SD) of 770 ± 456 and 381 ± 264 at day 66 for 177Lu-JMV4168 and 177Lu-JMV4168 plus PA, respectively. Consequently, mice in both treated groups had an increased survival as compared to the control group (P < 0.0001) and mice treated with 177Lu-JMV4168 plus PA had an increased survival as compared to mice treated with 177Lu-JMV4168 alone (P = 0.0157). Median survival increased from 35 to 79 days upon treatment with 177Lu-JMV4168, whereas the median survival in mice treated with 177Lu-JMV4168 plus PA was > 94 days, which was the endpoint of the experiment. Animal body weight remained stable during the course of the experiment and no apparent toxicity was observed (Figure 7).
No significant differences in tumor growth and survival were observed for the groups treated with PBS, 175Lu-JMV4168 or 175Lu-JMV4168 plus PA, showing that vehicle, 175Lu-JMV4168 and PA had no effect on tumor growth (Figure 6).
177Lu-JMV4168 radionuclide therapy results in DNA damage, apoptosis and decreased proliferation and does not affect GRPR expression of PC-3 tumors
Histological analysis of tumors, kidneys and pancreas dissected at the endpoint of the experiment showed overall healthy tissue, with some necrotic areas in the tumors (data not shown). Staining of molecular characteristics (DNA damage, replication, apoptosis and GRPR expression) of tumors from mice sacrificed 8 days after the last injection is presented in Figures 8, 9 and Figure 10.
Tumors from the groups treated with 177Lu-JMV4168 and 177Lu-JMV4168 plus PA showed an increased number of DNA double strand breaks (DSBs) as shown by phosphorylated histone 2AX (γH2AX) and p53-binding protein 1 (53BP1) foci formation in the tumor cell nuclei. Co-injection with PA increased the amount of produced DSBs. Furthermore, decreased numbers of cells expressing GEMININ (replicating cells) and increased numbers of TUNEL-positive cells (apoptotic cells) were observed after 177Lu-JMV4168 treatment. GRPR binding of 177Lu-JMV4168 on frozen tumor sections was preserved in PC-3 tumors from all groups.
Figure 6
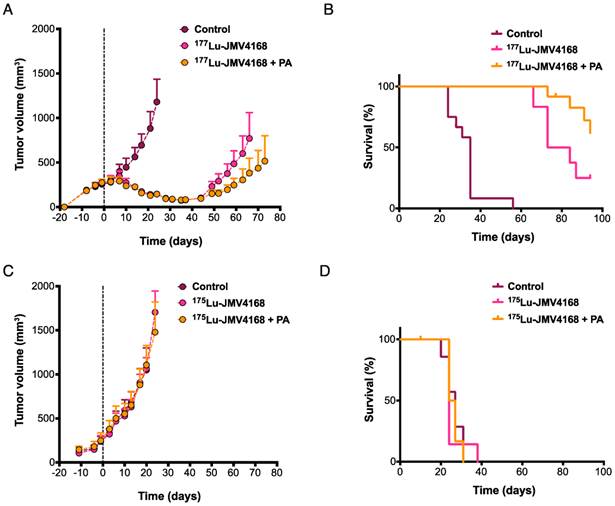
Effect of phosphoramidon (PA) on therapeutic efficacy of 177Lu-JMV4168 to treat subcutaneous PC-3 tumors. Mice were treated with four injections of 177Lu-JMV4168 (50 MBq, 200 pmol, n=12 per group) or 175Lu-JMV4168 (200 pmol, n=7 per group) on days 0, 2, 4 and 6, with or without PA. Mean tumor volume with 95% confidence interval (A, C) are displayed for the duration that no data point was missing. Kaplan-Meier survival curves (B, D) are displayed for the event of tumor volume exceeding 1500 mm3. All growth curves of the control mice could be fitted by an exponential growth curve, none should evidence for Gompertzian type of growth. The mean tumor volume doubling time was 11.2 ± 1.2 days. Regrowth of tumors in the treated animals was found after a kick-off time of 32 ± 4 days (177Lu-JMV4168) and 35 ± 6 days (177Lu-JMV4168 + PA) and showed comparable regrowth doubling times to the initial and control growth curves. No significant differences in tumor growth and survival were observed for the groups treated with PBS, 175Lu-JMV4168 or 175Lu-JMV4168 with PA.
Figure 7
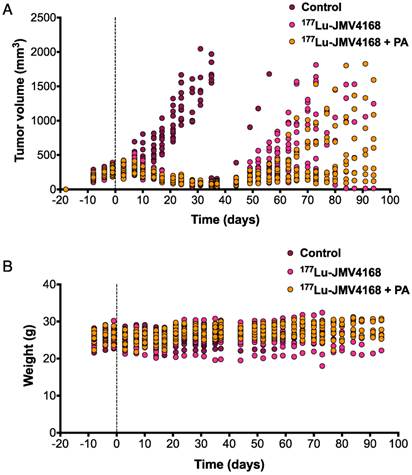
Therapeutic efficacy of 177Lu-JMV4168 to treat subcutaneous PC-3 tumors. Tumor size (A) and weight (B) follow up of mice treated with four injections of 177Lu-JMV4168 (50 MBq, 200 pmol) on days 0, 2, 4 and 6, with or without PA (n=12 per group).
Figure 8
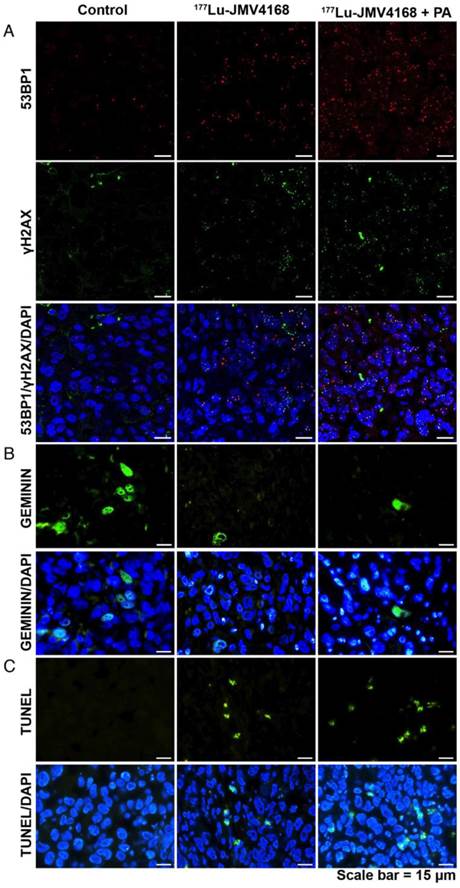
Representative pictures of 53BP1 and γH2AX staining (A), GEMININ staining (B) and TUNEL assay (C) on PC-3 paraffin-embedded tissues, 8 days after radionuclide therapy (control, 177Lu-JMV4168 and 177Lu-JMV4168 with phosphoramidon (PA)). Scale bar = 15 μm.
Discussion
In the search for an improved personalized cancer management, the field of theranostics is emerging, applying imaging diagnostics into the choice of the most suitable therapy for each patient. Bombesin analogs have been widely used for molecular imaging [27] and radionuclide therapy [28-30] of PCa. The field was initially focused on bombesin agonists until a change in paradigm was introduced with somatostatin receptor (SSTr) antagonists, which despite their poor internalization rate have shown more favorable pharmacokinetics and higher tumor uptake than agonists [31-33]. Similarly to SSTr antagonists, GRPR antagonists have shown superior tumor-targeting and pharmacokinetic properties as compared to agonists [11], with promising characteristics for imaging [3, 4] and therapeutic applications [34]. Moreover, the use of GRPR antagonists should prevent side effects such as nausea, hot flush and sweating, which were previously observed in patients injected with a bombesin receptor agonist [12, 13]. In a previous study performed by Dumont et al. [34] the potential of GRPR-targeted radionuclide therapy using a GRPR antagonist, 177Lu-RM2, and enhancement of therapeutic efficacy in combination with rapamycin were reported. GRPR-antagonists can be used as theranostic agents for PCa, but rapid in vivo degradation of these peptides by proteolytic enzymes may strongly hamper their targeting properties. Strategies to improve in vivo stability of radiolabeled peptides include various structural modifications, such as key amino acid substitutions, reduction/methylation of biodegradable bonds or cyclization, but these modifications can cause undesired changes in pharmacokinetics and/or impair receptor affinity [35-37]. The proteolytic enzyme NEP, which is abundant in the human body [38-40], cleaves peptides on the amino side of hydrophobic amino acids, thereby inactivating a broad range of neuropeptides, including bombesin-like peptides [15-17]. Recently we have shown that co-injection of the NEP inhibitor PA [41] could stabilize radiolabeled bombesin, minigastrin and somatostatin analogs in vivo, leading to enhanced tumor uptake [18, 42].
In this preclinical study, we have brought this in vivo stabilization concept to the next level, showing that co-injection of PA can contribute to an impressive enhancement in diagnostic sensitivity and therapeutic efficacy of a GRPR-targeted theranostic agent. We labeled the GRPR-antagonist JMV4168 with 68Ga for PET imaging and 177Lu for radionuclide therapy. All in vivo studies were performed with 200 pmol JMV4168, which appeared to be the optimal peptide dose in previous experiments (data not shown). The dose of 300 µg of PA was chosen based on previous experience [18]. Optimization of PA-dose for this 68Ga-/177Lu-JMV4168 theranostic pair has not been conducted, as the major purpose of the present study has been to show the PA-effect in therapy in a qualitative “proof-of-principle” approach. 177Lu-labeling of JMV4168 with very high specific activities (250 MBq/nmol) could be achieved using n.c.a. 177Lu. In vivo stability studies in mice showed excellent stabilization of 177Lu-JMV4168 in peripheral blood upon co-injection of PA.
Figure 9
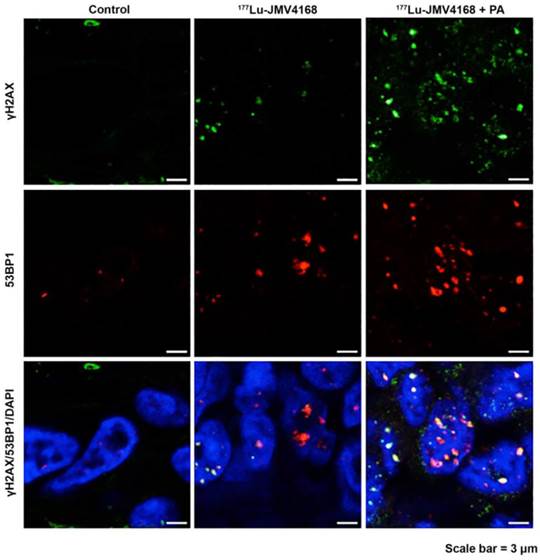
Representative pictures of γH2AX and 53BP1 foci staining on PC-3 paraffin-embedded tissues 8 days after radionuclide therapy (control, 177Lu-JMV4168 and 177Lu-JMV4168 with phosphoramidon (PA)). Scale bar = 3 μm.
Figure 10
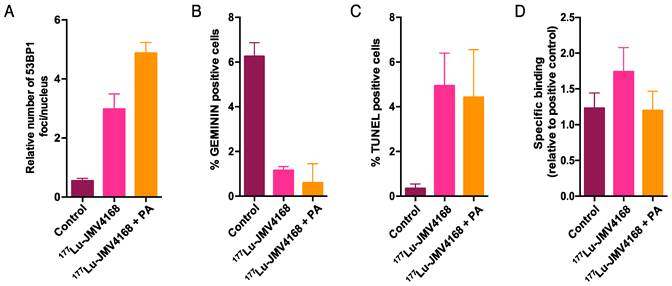
Quantification of number of 53BP1 foci per nucleus (A), and number of cells scored positive for GEMININ (B) or TUNEL (C) on PC-3 paraffin-embedded tissue, 8 days after radionuclide therapy. (D) GRPR binding of 177Lu-JMV4168 on PC-3 frozen sections after radionuclide therapy (endpoint).
Biodistribution of 68Ga-JMV4168 and 177Lu-JMV4168 was compared in nude mice with sc PC-3 human tumors. The similar biodistribution patterns of 68Ga-JMV4168 and 177Lu-JMV4168 at 1 h after injection indicate that 68Ga-JMV4168 could serve as a surrogate to 177Lu-JMV4168 for PET imaging-based patient selection and follow up of therapy outcome. Tumor uptake of 177Lu-JMV4168 was similar to tumor uptake of 177Lu-RM2 reported by Dumont et al. [34]. Tumor uptake of 68Ga- and 177Lu-JMV4168 was doubled by co-injection of PA, while most non-target background organs and blood levels were not enhanced, leading to higher tumor-to-background ratios. Although uptake of 68Ga-JMV4168 in the GRPR-positive pancreas was also increased by co-injection of PA at 1 h after injection, rapid washout from the pancreas was observed between 1 h and 2 h after injection, in agreement with previous findings [14]. These increased tumor uptake values were visualized on PET images acquired at 2 h after injection, showing higher signal intensity in the PC-3 tumors in mice that received co-injection of PA, with unaltered low intensity in the pancreas area. This suggests that PA could increase sensitivity of detection of small PCa lesions in patients.
Dosimetry of 177Lu-JMV4168 was evaluated to predict therapy outcome and to optimize the design of therapy studies. Administration of 177Lu-JMV4168 via the ip route was preferred to reduce injection failure rate and radiation exposure rate to researchers. Based on dosimetry calculations, therapy studies were conducted in nude mice with sc PC-3 tumors with 4 cycles of 50 MBq of 177Lu-JMV4168 (ip) with 2-days intervals. This schedule resulted in an impressive decrease and stabilization of tumor size as compared to that in untreated mice. More strikingly, co-administration of PA to 177Lu-JMV4168 resulted in increased survival. Several reports have suggested a role of NEP in PCa progression [43-45]. Minimal effect of NEP inhibition on PC-3 tumor growth was expected, as previous studies have shown lack of NEP expression in PC-3 cells [45-47]. Consistently, our results revealed no effect of PA on PC-3 xenograft tumor growth. However, we would recommend testing the effect of PA for each new model being investigated in similar studies.
In order to confirm the efficacy of radionuclide therapy at the molecular level, we evaluated the occurrence of DSBs, proliferation and apoptosis in PC-3 tumors. The β-particles emitted by the radionuclide 177Lu induce several types of DNA damage, among which DSBs are the most genotoxic; because unrepaired DSBs can trigger cell cycle arrest, cell death (apoptosis) or chromosomal aberrations. DSBs produced by ionization radiation activate a cascade of events, including protein modifications (such as γH2AX) and accumulation of several DNA damage response proteins (one of these proteins is the key DSB repair regulator 53BP1) [48]. After DSB induction, γH2AX and 53BP1 can be visualized as nuclear foci at the DSBs providing a measure of DSB induction.
PC-3 tumors dissected 8 days after the last therapeutic injection revealed increased number of DSBs, as shown by the increase in γH2AX and 53BP1 foci for the groups treated with 177Lu-JMV4168 and 177Lu-JMV4168 with PA. Furthermore, an increased production of DSBs was observed in mice that received co-administration of PA, which was consistent with the higher 177Lu-JMV4168 tumor uptake and the improved therapeutic outcome.
In both treated groups, we also observed decreased replication, as shown by the reduction in the number of cell expressing the cell cycle marker GEMININ. In line with these results, TUNEL staining showed an increased level of apoptosis after treatment. No significant difference in the number of GEMININ- and TUNEL-positive cells was observed between both treated groups, unlike DNA damage data. This could be due to different kinetics of DNA damage repair, proliferation and apoptosis.
Altogether, these parameters confirm tissue damage and subsequent tumor volume reduction induced by the radionuclide treatment with 177Lu-JMV4168. Importantly, GRPR expression was maintained on re-growing PC-3 tumors after radionuclide therapy, excluding the possibility of a selection of GRPR-negative cells, and allowing repeated treatment with the same probe.
In conclusion, this study highlights the potential of the GRPR-targeted peptide-based theranostic approach for imaging and treatment of PCa. Besides treatment of PCa patients, this approach could be applied to a wider range of GRPR-expressing tumors, such as breast tumors [49]. Moreover, it shows how co-injection of the NEP inhibitor PA can greatly enhance diagnostic sensitivity and therapeutic efficacy. We believe that this strategy may contribute to an improved management of PCa patients and warrants translation into a clinical setting. For that purpose, clinically certified NEP inhibitors should be preferred to facilitate translation.
Acknowledgements
This study was supported by the Erasmus MC grant “Novel Radio-Antagonists for PET/MRI Imaging and Therapy of Prostate Cancer”, the Dutch Cancer Society (Ride for the Roses Cancer Research Grant) and the Netherlands Organization for Scientific Research (ZON-MW grant 40-42600-98-018). The authors thank Cecile Beerens for her technical assistance with staining tumor samples and Jan de Swart for his technical assistance during PET experiments. We thank Sebastian Marx (ITG Munich) for providing n.c.a. 177Lu. This research was carried out within the animal molecular imaging facility (AMIE) and optical imaging center (OIC) of the Erasmus MC. This research is implemented in the Molecular Medicine graduate school (MolMed) of the Erasmus MC.
Competing Interests
Marion de Jong has stock ownership with the radiopharmaceutical company Advanced Accelerator Applications (AAA). No other potential conflict of interest relevant to this article was reported.
References
1. Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA Cancer J Clin. 2012;62:10-29
2. Siegel R, DeSantis C, Virgo K. et al. Cancer treatment and survivorship statistics, 2012. CA Cancer J Clin. 2012;62:220-41
3. Roivainen A, Kahkonen E, Luoto P. et al. Plasma pharmacokinetics, whole-body distribution, metabolism, and radiation dosimetry of 68Ga bombesin antagonist BAY 86-7548 in healthy men. J Nucl Med. 2013;54:867-72
4. Kahkonen E, Jambor I, Kemppainen J. et al. In vivo imaging of prostate cancer using [68Ga]-labeled bombesin analog BAY86-7548. Clin Cancer Res. 2013;19:5434-43
5. Afshar-Oromieh A, Malcher A, Eder M. et al. PET imaging with a [68Ga]gallium-labelled PSMA ligand for the diagnosis of prostate cancer: biodistribution in humans and first evaluation of tumour lesions. Eur J Nucl Med Mol Imaging. 2013;40:486-95
6. Afshar-Oromieh A, Zechmann CM, Malcher A. et al. Comparison of PET imaging with a (68)Ga-labelled PSMA ligand and (18)F-choline-based PET/CT for the diagnosis of recurrent prostate cancer. Eur J Nucl Med Mol Imaging. 2014;41:11-20
7. Tagawa ST, Milowsky MI, Morris M. et al. Phase II study of Lutetium-177-labeled anti-prostate-specific membrane antigen monoclonal antibody J591 for metastatic castration-resistant prostate cancer. Clin Cancer Res. 2013;19:5182-91
8. Vallabhajosula S, Nikolopoulou A, Jhanwar YS. et al. Radioimmunotherapy of Metastatic Prostate Cancer with 177Lu-DOTA-huJ591 Anti Prostate Specific Membrane Antigen Specific Monoclonal Antibody. Curr Radiopharm. 2015 [Epub ahead of print]
9. Ananias HJ, van den Heuvel MC, Helfrich W. et al. Expression of the gastrin-releasing peptide receptor, the prostate stem cell antigen and the prostate-specific membrane antigen in lymph node and bone metastases of prostate cancer. Prostate. 2009;69:1101-8
10. Markwalder R, Reubi JC. Gastrin-releasing peptide receptors in the human prostate: relation to neoplastic transformation. Cancer Res. 1999;59:1152-9
11. Cescato R, Maina T, Nock B. et al. Bombesin receptor antagonists may be preferable to agonists for tumor targeting. J Nucl Med. 2008;49:318-26
12. Basso N, Lezoche E, Speranza V. Studies with bombesin in man. World J Surg. 1979;3:579-85
13. Bodei L, Ferrari M, Nunn A. et al. Lu-177-AMBA Bombesin analogue in hormone refractory prostate cancer patients: A phase I escalation study with single-cycle administrations. Eur J Nucl Med Mol Imaging. 2007;34:S221-S
14. Chatalic KL, Franssen GM, van Weerden WM. et al. Preclinical comparison of Al18F- and 68Ga-labeled gastrin-releasing peptide receptor antagonists for PET imaging of prostate cancer. J Nucl Med. 2014;55:2050-6
15. Shipp MA, Tarr GE, Chen CY. et al. CD10/neutral endopeptidase 24.11 hydrolyzes bombesin-like peptides and regulates the growth of small cell carcinomas of the lung. Proc Natl Acad Sci U S A. 1991;88:10662-6
16. Erdos EG, Skidgel RA. Neutral endopeptidase 24.11 (enkephalinase) and related regulators of peptide hormones. Faseb j. 1989;3:145-51
17. Roques BP, Noble F, Dauge V. et al. Neutral endopeptidase 24.11: structure, inhibition, and experimental and clinical pharmacology. Pharmacol Rev. 1993;45:87-146
18. Nock BA, Maina T, Krenning EP. et al. "To serve and protect": enzyme inhibitors as radiopeptide escorts promote tumor targeting. J Nucl Med. 2014;55:121-7
19. Marsouvanidis PJ, Nock BA, Hajjaj B. et al. Gastrin releasing peptide receptor-directed radioligands based on a bombesin antagonist: synthesis, (111)in-labeling, and preclinical profile. J Med Chem. 2013;56:2374-84
20. Ali M, Hsieh W, Smyth D. et al. A simple and rapid method to quantify (68)Ga-hydroxides in a (68)Ga-DOTATATE formulation. Intern Med J. 2014;44:36-7
21. Bolch WE, Eckerman KF, Sgouros G. et al. MIRD pamphlet No. 21: a generalized schema for radiopharmaceutical dosimetry-standardization of nomenclature. J Nucl Med. 2009;50:477-84
22. Keenan MA, Stabin MG, Segars WP. et al. RADAR realistic animal model series for dose assessment. J Nucl Med. 2010;51:471-6
23. Konijnenberg M, Breeman W, de Blois E. et al. Therapeutic application of CCK2R-targeting PP-F11: influence of particle range, activity and peptide amount. EJNMMI Research. 2014;4:1-15
24. O'Donoghue JA, Bardies M, Wheldon TE. Relationships between tumor size and curability for uniformly targeted therapy with beta-emitting radionuclides. J Nucl Med. 1995;36:1902-9
25. de Visser M, van Weerden WM, de Ridder CM. et al. Androgen-dependent expression of the gastrin-releasing peptide receptor in human prostate tumor xenografts. J Nucl Med. 2007;48:88-93
26. de Blois E, Chan HS, Konijnenberg M. et al. Effectiveness of quenchers to reduce radiolysis of (111)In- or (177)Lu-labelled methionine-containing regulatory peptides. Maintaining radiochemical purity as measured by HPLC. Curr Top Med Chem. 2012;12:2677-85
27. Sancho V, Di Florio A, Moody TW. et al. Bombesin receptor-mediated imaging and cytotoxicity: review and current status. Curr Drug Deliv. 2011;8:79-134
28. Lantry LE, Cappelletti E, Maddalena ME. et al. 177Lu-AMBA: Synthesis and characterization of a selective 177Lu-labeled GRP-R agonist for systemic radiotherapy of prostate cancer. J Nucl Med. 2006;47:1144-52
29. Johnson CV, Shelton T, Smith CJ. et al. Evaluation of combined (177)Lu-DOTA-8-AOC-BBN (7-14)NH(2) GRP receptor-targeted radiotherapy and chemotherapy in PC-3 human prostate tumor cell xenografted SCID mice. Cancer Biother Radiopharm. 2006;21:155-66
30. Wild D, Frischknecht M, Zhang H. et al. Alpha- versus beta-particle radiopeptide therapy in a human prostate cancer model (213Bi-DOTA-PESIN and 213Bi-AMBA versus 177Lu-DOTA-PESIN). Cancer Res. 2011;71:1009-18
31. Ginj M, Zhang H, Waser B. et al. Radiolabeled somatostatin receptor antagonists are preferable to agonists for in vivo peptide receptor targeting of tumors. Proc Natl Acad Sci U S A. 2006;103:16436-41
32. Cescato R, Waser B, Fani M. et al. Evaluation of 177Lu-DOTA-sst2 antagonist versus 177Lu-DOTA-sst2 agonist binding in human cancers in vitro. J Nucl Med. 2011;52:1886-90
33. Wild D, Fani M, Behe M. et al. First clinical evidence that imaging with somatostatin receptor antagonists is feasible. J Nucl Med. 2011;52:1412-7
34. Dumont RA, Tamma M, Braun F. et al. Targeted radiotherapy of prostate cancer with a gastrin-releasing peptide receptor antagonist is effective as monotherapy and in combination with rapamycin. J Nucl Med. 2013;54:762-9
35. Pernot M, Vanderesse R, Frochot C. et al. Stability of peptides and therapeutic success in cancer. Expert Opin Drug Metab Toxicol. 2011;7:793-802
36. Adessi C, Soto C. Converting a peptide into a drug: strategies to improve stability and bioavailability. Curr Med Chem. 2002;9:963-78
37. Vlieghe P, Lisowski V, Martinez J. et al. Synthetic therapeutic peptides: science and market. Drug Discov Today. 2010;15:40-56
38. Shipp MA, Richardson NE, Sayre PH. et al. Molecular cloning of the common acute lymphoblastic leukemia antigen (CALLA) identifies a type II integral membrane protein. Proc Natl Acad Sci U S A. 1988;85:4819-23
39. Erdos EG, Schulz WW, Gafford JT. et al. Neutral metalloendopeptidase in human male genital tract. Comparison to angiotensin I-converting enzyme. Lab Invest. 1985;52:437-47
40. Sato Y, Itoh F, Hinoda Y. et al. Expression of CD10/neutral endopeptidase in normal and malignant tissues of the human stomach and colon. J Gastroenterol. 1996;31:12-7
41. Oefner C, D'Arcy A, Hennig M. et al. Structure of human neutral endopeptidase (Neprilysin) complexed with phosphoramidon. J Mol Biol. 2000;296:341-9
42. Marsouvanidis PJ, Melis M, de Blois E. et al. In vivo enzyme inhibition improves the targeting of [177Lu]DOTA-GRP(13-27) in GRPR-positive tumors in mice. Cancer Biother Radiopharm. 2014;29:359-67
43. Osman I, Dai J, Mikhail M. et al. Loss of neutral endopeptidase and activation of protein kinase B (Akt) is associated with prostate cancer progression. Cancer. 2006;107:2628-36
44. Albrecht M, Doroszewicz J, Gillen S. et al. Proliferation of prostate cancer cells and activity of neutral endopeptidase is regulated by bombesin and IL-1beta with IL-1beta acting as a modulator of cellular differentiation. Prostate. 2004;58:82-94
45. Dawson LA, Maitland NJ, Turner AJ. et al. Stromal-epithelial interactions influence prostate cancer cell invasion by altering the balance of metallopeptidase expression. Br J Cancer. 2004;90:1577-82
46. Usmani BA, Harden B, Maitland NJ. et al. Differential expression of neutral endopeptidase-24.11 (neprilysin) and endothelin-converting enzyme in human prostate cancer cell lines. Clin Sci (Lond). 2002;103(Suppl 48):314s-7s
47. Ho ME, Quek SI, True LD. et al. Prostate cancer cell phenotypes based on AGR2 and CD10 expression. Mod Pathol. 2013;26:849-59
48. van Gent DC, van der Burg M. Non-homologous end-joining, a sticky affair. Oncogene. 2007;26:7731-40
49. Dalm SU, Martens JW, Sieuwerts AM. et al. In vitro and in vivo application of radiolabeled gastrin-releasing Peptide receptor ligands in breast cancer. J Nucl Med. 2015;56:752-7
Author contact
![]() Corresponding author: Kristell Chatalic, MSc, PhD student, Erasmus MC, Department of Nuclear Medicine, Na-620, Postbus 2040, 3000 CA Rotterdam, The Netherlands. E-mail: k.chatalicnl; Phone: +31(0)107041033
Corresponding author: Kristell Chatalic, MSc, PhD student, Erasmus MC, Department of Nuclear Medicine, Na-620, Postbus 2040, 3000 CA Rotterdam, The Netherlands. E-mail: k.chatalicnl; Phone: +31(0)107041033
Citation styles
APA
Chatalic, K.L.S., Konijnenberg, M., Nonnekens, J., de Blois, E., Hoeben, S., de Ridder, C., Brunel, L., Fehrentz, J.A., Martinez, J., van Gent, D.C., Nock, B.A., Maina, T., van Weerden, W.M., de Jong, M. (2016). In Vivo Stabilization of a Gastrin-Releasing Peptide Receptor Antagonist Enhances PET Imaging and Radionuclide Therapy of Prostate Cancer in Preclinical Studies. Theranostics, 6(1), 104-117. https://doi.org/10.7150/thno.13580.
ACS
Chatalic, K.L.S.; Konijnenberg, M.; Nonnekens, J.; de Blois, E.; Hoeben, S.; de Ridder, C.; Brunel, L.; Fehrentz, J.A.; Martinez, J.; van Gent, D.C.; Nock, B.A.; Maina, T.; van Weerden, W.M.; de Jong, M. In Vivo Stabilization of a Gastrin-Releasing Peptide Receptor Antagonist Enhances PET Imaging and Radionuclide Therapy of Prostate Cancer in Preclinical Studies. Theranostics 2016, 6 (1), 104-117. DOI: 10.7150/thno.13580.
NLM
Chatalic KLS, Konijnenberg M, Nonnekens J, de Blois E, Hoeben S, de Ridder C, Brunel L, Fehrentz JA, Martinez J, van Gent DC, Nock BA, Maina T, van Weerden WM, de Jong M. In Vivo Stabilization of a Gastrin-Releasing Peptide Receptor Antagonist Enhances PET Imaging and Radionuclide Therapy of Prostate Cancer in Preclinical Studies. Theranostics 2016; 6(1):104-117. doi:10.7150/thno.13580. https://www.thno.org/v06p0104.htm
CSE
Chatalic KLS, Konijnenberg M, Nonnekens J, de Blois E, Hoeben S, de Ridder C, Brunel L, Fehrentz JA, Martinez J, van Gent DC, Nock BA, Maina T, van Weerden WM, de Jong M. 2016. In Vivo Stabilization of a Gastrin-Releasing Peptide Receptor Antagonist Enhances PET Imaging and Radionuclide Therapy of Prostate Cancer in Preclinical Studies. Theranostics. 6(1):104-117.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) License. See http://ivyspring.com/terms for full terms and conditions.