Impact Factor
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Conclusion
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2016; 6(1):142-154. doi:10.7150/thno.13130 This issue Cite
Research Paper
Anti-tumor Efficiency of Lipid-coated Cisplatin Nanoparticles Co-loaded with MicroRNA-375
Tan Yang1, Pengxuan Zhao1, Zhao Rong1, Bin Li1, Huiying Xue1, Jia You1, Chuanchuan He1, Weijie Li2, Xingxing He2, Robert J. Lee3, Xiang Ma1, ![]() , Guangya Xiang1,
, Guangya Xiang1, ![]()
1. School of Pharmacy, Tongji Medical College, Huazhong University of Science and Technology, Wuhan 430030, P. R. China
2. Tongji Hospital, Huazhong University of Science and Technology, Wuhan 430030, P. R. China
3. Division of Pharmaceutics, College of Pharmacy, the Ohio State University, Columbus, Ohio, USA
Received 2015-7-3; Accepted 2015-9-30; Published 2016-1-1
Abstract

One of the major challenges in the hepatocellular carcinoma (HCC) treatment is its insensitivity to chemotherapeutic drugs. Here, we report the development of novel lipid-coated cisplatin nanoparticles co-loaded with microRNA-375 (NPC/miR-375) as a potential treatment for chemotherapy insensitive HCC. The NPC/miR-375 was fabricated by mixing two reverse microemulsions containing KCl solution and a highly soluble cis-diaminedihydroplatinum (II) coated with a cationic lipid layer. Subsequently, the miR-375 was incorporated into the lipid-coated cisplatin nanoparticles. The NPC/miR375 nanoparticles were expected to further decrease cell proliferation and to enhance the anti-tumor effect of cisplatin in chemotherapy resistant HCC cells. In vitro analysis of intracellular trafficking revealed that NPC/miR-375 were able to escape from the late endosomes instead of lysosomes thus avoiding degradation of the miR-375 in lysosomes. Importantly, NPC/miR-375 enhanced apoptosis and induced cell cycle arrest in HCC cells in vitro. In the double oncogenes Akt/Ras-induced primary HCC mouse model, multiple doses of NPC/miR-375 significantly inhibited tumor growth and delayed the tumor relapse. Our results indicate that cisplatin nanoparticles co-loaded with miR-375 represent a potential therapeutic agent for chemotherapy-insensitive HCC.
Keywords: Hepatocellular Carcinoma, Co-delivery, miR-375, Cisplatin, Nanoparticles
Introduction
Liver cancer is a lethal disease that caused the death of more than 745,000 people worldwide in 2012 [1]. Most (85% to 90%) of primary liver cancers occurring worldwide are hepatocellular carcinomas (HCC) [2]. Therapeutic options for unresectable HCC have been very limited and ineffective so far, until the discovery of new therapeutic agents such as sorafenib that target the molecular pathways involved in hepatocarcinogenesis [3, 4]. Sorafenib is currently the FDA-approved mono therapeutic drug for the treatment of patients with advanced HCC [5]. However, sorafenib was beneficial only in about 30% of advanced HCC patients increasing the survival term for only 2-3 months [6]. Due to limited efficacy and high price of sorafenib, chemotherapy is still an important modality in the treatment paradigm for HCC. Standard chemotherapeutic agents, including doxorubicin, cisplatin, and 5-fluorouracil, are used in the clinic either individually or in combination. None of these agents or combinations has been shown to have significant survival benefit in large randomized trials, although there are many reports of partial responses in a small number of patients [7]. The long-term survival of patients with advanced HCC is poor, with a 1-year survival rate of 17 to 44 % and a 3-year survival rate of 8 to 17% [8]. The lack of survival benefits of treatment with conventional drugs as well as sorafenib makes it imperative to develop novel therapeutic strategies with new agents directed against novel therapeutic targets.
One of the major challenges in drug development for HCC treatment resides in the fact that HCC is a highly heterogeneous disease at the molecular level. It is, therefore, conceivable that only certain HCC subsets would benefit from the use of targeted therapies [5]. In the past decades, studies have focused on investigating the genes and proteins underlying the development of HCC subsets [9]. A large number of microRNAs (miRNA) whose expression was down- or up-regulated in human HCC specimens has been identified [10-12]. Functional studies further supported their critical roles in hepatocarcinogenesis by their ability to modulate cell proliferation, survival, and invasive properties [12]. Thus, targeting miRNAs for therapeutic purposes would simultaneously affect multiple genes and signaling cascades by possibly regulating the growth of HCCs with heterogeneous molecular features [13]. Several studies performed in vitro and xenograft models have provided evidence that modulating miRNA expression in HCCs has a significant therapeutic potential [14-16]. One of these miRNAs, miR 375, showed tumor suppressor activity in liver cancer and in hepatocellular carcinoma by targeting Hippo-signaling effector Yes-associated protein (YAP) and astrocyte elevated gene-1 (AEG-1), respectively [16, 17]. Elevated YAP and AEG-1 levels and increased nuclear localization have been reported in multiple cancerous tissues [18, 19]. In a conditional transgenic mouse model, overexpressed YAP caused a dramatic increase in liver size eventually leading to tumorigenesis [20]. We have previously reported that up-regulation of miR-375 significantly suppressed the growth of HCC both in vitro and in vivo [16]. Despite these promising results, delivery of miRNAs into cells has been the major challenge for miRNA-based therapeutics so far [21]
Nanocarrier-based miRNA anticancer therapies, either alone or in combination with conventional chemotherapeutic drugs, are currently under development with the goal of improving survival and clinical outcome [22, 23]. Nanocarriers were frequently selected for miRNA delivery due to their low toxicity, clinical potential and the ease of production [22, 23]. Currently, there is growing interest in developing nanocarriers to simultaneously deliver miRNA or siRNA with chemotherapeutic drugs to produce synergetic effects by targeting different therapeutic mechanisms [23-26]. The combination delivery systems of miRNA or siRNA with chemotherapeutic drugs partially reverse drug resistance of cancer cells and hence enhance the efficacy of chemotherapeutic drugs to eventually improve overall treatment outcomes [27, 28].
Recently, lipid-coated nanoparticles of cisplatin (NPC) with high drug loading capability were investigated and showed excellent safety and anti-tumor efficacy in mice [29]. Platinum agents are among the most widely used cytotoxic agents for cancer therapy. One of these agents, cisplatin, causes DNA damage as its primary mechanism of cellular cytotoxicity [30]. However, several cellular pathways are also activated in response to the genotoxic stress including phosphorylation and activation of YAP that helps the cells tolerate the DNA damage [31]. We hypothesized that nanoparticles carrying a combination of YAP- targeting miR375 with cisplatin could promote cellular uptake of both therapeutic agents. This would enable efficient endosomal escape in cancer cells and overcome cisplatin-induced resistance by targeting molecules such as YAP or AEG-1 at the molecular level. In this study, we used the lipid-coated nanoparticles to simultaneously deliver cisplatin and miR375 for enhancing the cisplatin-associated chemosensitivity in hepatocarcinogenesus. We evaluated the inhibitory effects of miR-375 in a primary HCC mouse model induced by two oncogenes [32]. Our results showed that NPC co-loaded with miR-375, NPC/miR375 exhibited significant repression capability in HCC cells in vitro and abrogated the progression of HCC in a primary mouse model.
Materials and Methods
Materials
Cisplatin (purity > 95%) was obtained from the Sino-Platinum Metals (Guiyang, Guizhou, China). FA-PEG-CHEMS was synthesized in our laboratory as previously reported [25]. 1,2-dioleoyl-3-trimethylammonium-propane (chloride salt) (DOTAP) and 1,2-dioleoyl-sn-glycerol-3-phosphate (DOPA) were purchased from Avanti Polar Lipids Inc. (Alabaster, AL, USA). Monomethoxy polyethylene glycol 2000-distearoyl phosphatidylethanolamine (mPEG-DSPE2000) was obtained from Lipoid GmbH (Ludwigshafen, Germany). Cholesterol (CHOL) was purchased from Acros Organics (Geel, Belgium). 1-Hexanol was purchased from Alfa Aesar (Ward Hill, MA, USA). Igepal® CO-520, triton™ X-100, cyclo-hexane, and silver nitrate were obtained from Sigma-Aldrich (St. Louis, MO, USA). All solvents and reagents were of analytical or HPLC grade and used without further purification.
DMEM was purchased from Sigma-Aldrich Corp. (St. Louis, MO, USA). Fetal bovine serum (FBS) was purchased from Zhejiang Tianhang Biological Technology Co., Ltd. (Hangzhou, China). HCC cell line HepG2 was obtained from the China Center for Type Culture Collection at Wuhan University (Wuhan, China). Balb/C nude mice (20 g, 6 - 8 weeks old) and FVB mice (16 g, 6 weeks old) were obtained from Beijing Huafukang Bioscience Technology Co., Ltd. (Beijing, China). The miR-375 sequences for mice were CCCCGCGACGAGCCCCUCGCACAAACCGGACCUGAGCGUUUUGUUCGUUCGGCUCGCGUGAGGC; the sequences in mice and humans were the same.
Cell culture
HepG2 and Hep3B cells were cultured with high glucose DMEM supplemented with penicillin, streptomycin, and 10% FBS in 37°C and 5% CO2 incubators. M-plasmocin (Invivogen, San Diego, CA, USA) at a concentration of 2.5 μg/mL was used to prevent the possible mycoplasma infections.
Animals
Balb/C nude and FVB mice were kept in filter-topped cages with standard rodent chow, water available ad libitum, and a 12 h light/dark cycle. The experiment protocol was approved by the Committee on Ethical Animal Experiment at Huazhong University of Science and Technology.
Synthesis of cis-[Pt(NH3)2(H2O)2](NO3)2 precursor
Cis-[Pt (NH3)2 (H2O)2] (NO3)2 was prepared according to the literature [29]. In brief, AgNO3 (66.2 mg, 0.39 mmol) was added to 1 mL suspension of cisplatin in double distilled H2O (cisplatin, 60 mg, 0.20 mmol) to make a mixture. Then the mixture was heated for 3 h at 60°C and then stirred overnight in a flask under dark with aluminum foil package. Next, the mixture was centrifuged at 12,000g for 15 min to remove the AgCl precipitate. The solution was then filtered through a 0.2 μm syringe filter. The concentration of cis-[Pt(NH3)2(H2O)2](NO3)2 was measured using inductively coupled plasma mass spectrometry (ICP-MS, Agilent 7500, CA, USA) and adjusted to 200 mM.
Preparation of NPC/miR-375
The synthesis of NPC/miR-375 nanoparticles is illustrated in Figure 1a. Firstly, 100 μL of 200 mM cis-[Pt (NH3)2 (H2O)2] (NO3)2 was dispersed in a solution composed of a mixture of 6 mL cyclohexane/Igepal CO-520 (71:29, V:V) and 2 mL cyclohexane/triton-X100/hexanol (75:15:10, V:V:V) to generate a well-dispersed, water-in-oil reverse micro-emulsion. DOPA (100 μL, 20 mM) was then added to the cisplatin-containing micro-emulsion and the mixture was stirred for 20 min. Another emulsion containing KCl was prepared by adding 100 μL of 800 mM KCl in water into a separate 8.0 mL same reverse micro-emulsion. Then the two emulsions were mixed and the reaction proceeded for 30 min. Subsequently, 16.0 mL ethanol was added to the micro-emulsion, and the mixture was centrifuged at 12,000 g for at least 15 min to remove the cyclohexane and surfactants. After being extensively washed with ethanol 2-3 times, the pellets were re-dispersed in 3.0 mL chloroform to obtain Lipid-Pt-Cl nanoparticles.
Preparation and characterization of NPC/miR-375 nanoparticles: (a) Schematic illustration of NPC/miR-375 nanoparticles preparation. (b) The structure of NPC/miR-375 nanoparticles imaged using TEM. NPC/miR-375 were negatively stained with 2% phosphotungstic acid. (c) The structure of NPC/miR-375 nanoparticles imaged using atomic force microscope. (d) Determination of diameter distribution of NPC/miR-375 nanoparticles using dynamic light scattering (DLS). (e) A representative agarose gel electrophoresis image illustrating miR-375 loading efficiency at ratios from 20:1 to 200:1 (w/w).
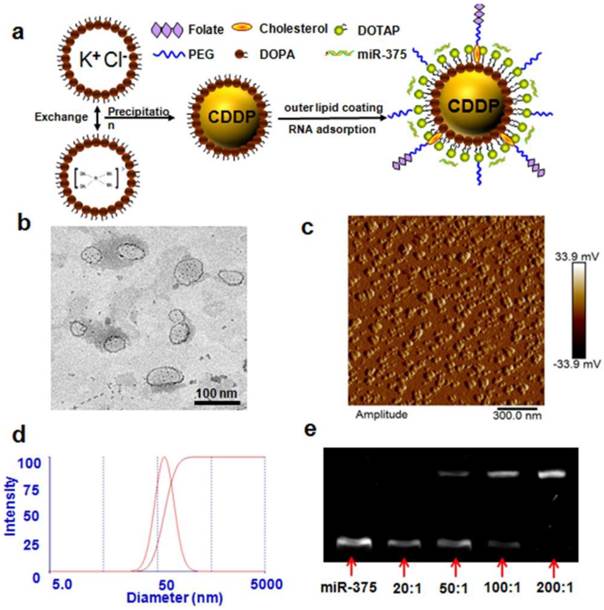
To prepare Nano-Particles of Cisplatin (NPC), 28 mg DOTAP, 21.3 mg cholesterol, 12.6 mg mPEG-DSPE and 2.1 mg folate-PEG-CHEMS (molar ratio 40:55:4.5:0.5) and 1.0 mL Lipid-Pt-Cl nanoparticle core were dispersed in 5.0 mL chloroform. After chloroform had been removed by rotary evaporation, residual lipids were dispersed in 2.0 mL of ddH2O to generate NPC.
To prepare NPC/miR-375, NPC and miR 375 were mixed at w/w (weight NPC/weight siRNA) ratio of more than 200:1 in RNase free H2O by adding a stock solution of NPC into a miR-375 solution. The samples were vortexed 2-3 min and then incubated at room temperature for 30 min to ensure formation of NPC/miR-375 nanoparticles.
Characterization of NPC/miR-375
Particle size and zeta potential of NPC/miR-375 were measured by Zeta PALS (Zeta Potential Analyzer, Brookhaven Instruments Corporation, Austin, TX) according to the manufacturer's instructions. All measurements were carried out at room temperature. Each parameter was measured 3 times; average values and standard deviations were calculated.
The drug-loading capacity and platinum content were measured using ICP-MS. Samples were digested in 70% HNO3 and diluted in water to a final acid content of 2%. A standard curve was derived from a series of dilutions of cisplatin solutions and the platinum concentration was determined according to the standard curve. The shapes of the NPC/miR-375 particles were determined on a transmission electron microscope (JEOL 100CX II TEM, Japan). NPC/miR-375 were negatively stained with 2% phosphotungstic acid. Atomic force microscope (AFM, Nanoscope III A, Digital Instruments, Brucker, CA, USA) was used to characterize the nanoparticles as well. Briefly, 5 μL of the solutions was deposited on freshly cleaved mica. After 5 min incubation, the mica surface was rinsed with 3 drops of ddH2O for 4 times and dried under a flow of dry nitrogen. The samples of the condensates were imaged with a tapping mode AFM. AFM images were flattened using Nanoscope 6.14 software.
Agarose gel electrophoresis
Agarose gel electrophoresis was used to evaluate miRNA loading in NPC. The miRNA or NPC/miR-375 (the dose of miRNA was 120 pmol) was applied to a 2% (w/v) agarose gel in TAE buffer containing Goldview staining reagent (Solarbio, China). Free miRNA will migrate in the gels, but NPC/miR-375 will be stuck in the loading wells. Images were obtained using a UV transilluminator and a digital imaging system (Life Science Technologies, USA).
Uptake of NPC/miR-375
To analyze uptake of NPC/miR-375 in vitro, HepG2 cells were seeded on a 24-well plate at 1 × 105 cells per well and cultured for 24 h. Fluorescently labeled NPC/miR-375 (equivalent to 5 μM cisplatin and 50 nM miRNA/well) and NPC (equivalent to 5 μM cisplatin) were then added to the medium for 12 h. For measuring uptake of miR-375, cells were washed with PBS 3 times and then trypsinized, collected and examined by FACS (BD Accuri C6 Flow Cytometer, San Jose, CA, USA). To determine the uptake of cisplatin, cells were collected and digested in 70% HNO3, and then diluted in water to a final acid content of 2%. Then the solutions were used for the detection of cisplatin by ICP-MS. For platinum concentration determination in mouse tissue, tissues were isolated from the drug-treated mice after sacrifice. Then the tissues were digested by 70% HNO3 and diluted in water to a final acid content of 2% for ICP-MS analysis.
Intracellular trafficking examination
To investigate NPC/miR-375 uptake, HepG2 cells were seeded in 96-well plates and incubated with NPC/miR-375 (50 nM) for different amounts of time. The cells were then rinsed with PBS for three times and fixed in 4% PFA for 15 min. Next, the cells were permeated with 0.1% Triton X-100 for 3 min. After blocking with 0.5% goat serum in PBS for 0.5 h, the cells were incubated with different primary antibodies against the protein markers of interest in goat serum for 1.5 h at 37°C. The cells were rinsed with PBS 3 times and then incubated with an AlexaFluor-488 labeled secondary antibody (Invitrogen Alexa Fluor 488 Goat Anti-Rabbit IgG (H+L)) at 7.5 μg/mL in PBS for 45 min at 37°C. The cells were imaged using a fluorescence microscope (Olympus SZX12, Japan). The primary antibodies used included rabbit Rab9 (CST #5118, cell signaling) and rabbit LAMP1 (CST #9091, cell signaling). Concentrations of these antibodies used were 0.5% (LAMP1) and 2% (Rab9). MiR-375 was labeled with Cy3, and the nucleus was counterstained with DAPI.
Cytotoxicity
The cytotoxic effects of NPC/miR-375 on HepG2 and Hep3B cells were measured using Cell Counting Kit-8 kit (CCK-8 kit, Dojindo Laboratories, Kumamoto, Japan). The cells were seeded onto 96-well plates at a density of 1 × 104 cells/well. After 24 h, cells were treated with free cisplatin, NPC or NPC/miR-375. After incubation for an additional 24 h, the portions of viable cells were measured using CCK-8 kits according to the user's manual. Cell viability within each group was expressed as a percentage of the viability of untreated control cells.
Cell cycle and apoptosis analyses
For cell cycle analysis by FACS, HepG2 cells (4 × 105 per well) were seeded in DMEM with 10% FBS on 6-well plates and allowed to attach overnight. The medium was then changed to fresh DMEM with 10% FBS and cells were treated with PBS (control), free cisplatin (5 μM), NPC (containing 5 μM cisplatin), or NPC/miR-375 (containing 5 μM cisplatin and 50 nM miR 375). Cells were harvested by trypsinization, collected and fixed in 70% ethanol at 4°C overnight. After being washed and resuspended in 200 μL phosphate-buffered saline, cells were treated with 5 μL RNase (20 mg/mL) at 37°C for 30 min and stained with 20 μL propidium iodide (500μg/mL, KeyGen Biotech Co. Ltd., Nanjing, China) at 4°C for 30 min. For apoptosis analysis, HepG2 cells (4 × 105 per well) were seeded in DMEM with 10% FBS on 6-well plates and allowed to attach overnight. The medium was then changed to fresh DMEM with 10% FBS and cells were treated with PBS (control), free cisplatin, NPC (containing 5 μM cisplatin), or NPC/miR-375 (containing 5 μM cisplatin and 50 nM miR 375) for 12 h. The cells were harvested by trypsinization and were stained using an Annexin V-FITC Apoptosis Detection Kit (KeyGen Biotech) according to the manufacturer's protocol. Stained cells were immediately analyzed on a BD Accuri C6 Flow Cytometer.
Western blotting
HepG2 cells were incubated in CelLytic M Cell Lysis Reagent (Sigma-Aldrich, MA, USA) for 30 min on ice. The supernatant was collected after centrifugation at 12,000 rpm (Eppendorf 5415D). Cell lysates were separated on a 10% polyacrylamide gel and transferred to a PVDF membrane. The membrane was blocked for 1 h in 5% skim milk and then incubated with monoclonal antibody against cyclin A2 (1:1000, Abcam, Cambridge, UK ), cyclin B1 (1:1000, Abcam), cyclin D1 (1:1000, Abcam), Bax (1:1000, Cell Signaling, Danvers, MA, USA), Bcl-2 (1:1000, Cell Signaling), caspase 3 (1:1000, Cell Signaling), or tubulin (1:1000, Abcam) overnight. The membrane was washed in TBST (PBS with 0.1% Tween-20) three times and then incubated for one hour with a secondary antibody. Then the membrane was washed four times and developed by an enhanced chemiluminescence system according to the manufacturer's instructions (Perkin Elmer, Waltham, MA, USA).
Akt and Ras-induced primary HCC mouse model
To determine the tumor-suppression activity of NPC/miR-375 in primary HCC, we generated an Akt/Ras double overexpression HCC mouse model. Wild-type FVB/N mice were used at 6 to 8 weeks of age. The hyperactive transposase construct pCMV/SB, Ras gene construct pCaggs-RasV12, and AKT gene construct myr-AKT/pT3EF1α were provided by Dr. Xin Chen at the University of California at San Francisco (UCSF, CA, USA). All plasmids were purified using the GenElute Endotoxin-free plasmid Maxiprep Kit (Sigma, USA) before administration into mice.
Hydrodynamic injections were performed as reported previously [32, 33] . In brief, FVB/N mice were injected with 5 μg myr-AKT/pT3EF1α, 5 μg NRasV12/pT2-CAGGS, or 0.4 μg pCMV/SB in 2 mL saline via tail vein in 5 - 10 s. Upon injection, the Ras and Akt plasmid, along with transponsase plasmid were taken up by hepatocytes and integrated into their chromosomes. The continuous overexpression of Ras and Akt induced the formation of tumors in the liver. The tumors were monitored by bimanual palpation for ballooning and stiffness of livers, and confirmed by dissection before drug administration.
In vivo/ex vivo tissue imaging and cisplatin distribution in mouse tissue
A xenograft mouse model was generated by subcutaneous injection of HepG2 cells (2.5 × 106 cells per mouse) in the right front flank of female Balb/C nude mice (6 - 8 weeks, 20 g). For in vivo imaging, the tumor bearing mice (n = 3) were injected via tail vein a single dose of cisplatin (1.0 mg/kg) or NPC/miR-375 (170 μg/kg miRNA, 1.0 mg/kg cisplatin) when the tumors reached ~500 mm3. The mice were imaged in a small animal imaging system (Brucker In-vivo Fx Pro, CA, USA) by X-ray and fluorescence at 1, 3, 8, and 12 h after injection. Subsequently, the mice were sacrificed and organs were collected for ex vivo tissue imaging.
For determination of cisplatin concentration in tissues, mice were given a single dose of cisplatin (1.0 mg/kg) or NPC/miR-375 (170 μg/kg siRNA, 1.0 mg/kg cisplatin) and then sacrificed at 1, 3 and 12 h following injection. Collected tissue samples were digested by concentrated nitric acid overnight at room temperature and processed according to the procedure reported previously [28]. The concentration of the platinum was measured using ICP-MS as described above.
Anti-tumor efficacy study in vivo
In vivo anti-tumor efficacy of NPC/miR-375 was evaluated in both the Akt/Ras primary HCC mouse model and xenograft tumor bearing Balb/c nude mice. Upon HCC formation in the liver, mice were randomly distributed into 4 groups (PBS, free cisplatin, NPC and NPC/miR-375). The mice (n = 3 - 6) were given intravenous injections of different formulations via the tail vein at the dose of 1.0 mg/kg cisplatin and 170 μg/kg miRNA once a week for a total of 4 doses. Mice were sacrificed 4 weeks after the first injection. Then body weight, liver weight, tumor number, and the largest tumor size were recorded. The tumor size was calculated by equation V = length × width2 × 0.52. All data analysis was performed using GraphPad Prism (version 5.01).
Microscopy
Cells and tissue section slides were examined on an Olympus SZX12 fluorescence microscope equipped with a digital camera and connected to a computer running MacroFire 2.0 camera software (Optronics, Goleta, CA, USA). Pictures were taken at equal exposure time for each sample.
Statistical analysis
Comparison of two groups was performed using Student's t-test (SPSS Software, Chicago, IL). Multiple groups were compared by one-way ANOVA with Dunnett's post-test. A value of p < 0.05 was considered significant and p < 0.01 was considered highly significant.
Results
Preparation and characterization of NPC/miR-375 nanoparticles
NPC and NPC/miR-375 were synthesized as described in the Methods section and were characterized by TEM, AFM and DLS. The average particle sizes of NPC and NPC/miR-375 were 88.2 and 97.6 nm, respectively as measured by DLS (Table 1). This indicated that miRNA binding to NPC increased the diameter of NPC by about 10 nm. The PDI of NPC and NPC/miR-375 were 0.112 and 0.141, respectively showing the narrow distributions. The zeta potentials of NPC and NPC/miR-375 were 37.9 and 16.8 mV, respectively (Table 1). The reduced zeta potential of NPC/miR-375 was due to the incorporation of the miRNA with a negative potential. The loading efficiencies of cisplatin in NPC and NPC/miR-375 were 97.7% and 94.1%, respectively (Table 1). TEM images showed that the NPC/miR-375 nanoparticles were dispersed in the solution and particle shape was uniform (Figure 1b). The average particle size of the NPC/miR-375 determined by TEM was about 80 nm. A similar result was obtained by AFM (Figure 1c). DLS determination also indicated that the particle size had a normal distribution (Figure 1d). The weight ratio for the best loading efficiency of miRNA was carefully optimized using the agarose gel electrophoresis analysis. The miRNA was fully entrapped in the loading wells with NPC when the ratio of NPC and miRNA was 200:1 (w/w) (Figure 1e) suggesting maximum loading of the miRNA into the NPC at this ratio.
Physicochemical properties of NPC and NPC/miR-375
| Nano-particles | Diameter (nm) | PDI | Zeta (mV) | EE of cisplatin (%) |
|---|---|---|---|---|
| NPC | 88.2 ± 10.1 | 0.112 ± 0.015 | 37.9 ± 2.9 | 97.7 ± 11.2 |
| NPC/miR-375 | 97.6 ± 10.6 | 0.141 ± 0.035 | 16.8 ± 4.1 | 94.1 ± 13.7 |
Note: each value represents the mean ± SD (n=3).
Abbreviations: EE, encapsulation efficiency of cisplatin; PDI, polydispersity index
Intracellular trafficking and cellular uptake of NPC/miR-375 in HepG2 cells
One of the critical points for miRNA delivery into the cells is to avoid its degradation during the intracellular trafficking. We therefore checked the release and trafficking of miR-375 delivered by NPC/miR-375 in HepG2 cells. One hour after treatment, most of the NPC/miR-375 (red) were adhered to cell membranes and were entrapped in the late endosomes. At three hours, a majority of the NPC/miR-375 remained in late endosomes, whereas at six hours, a fraction of the NPC/miR-375 appeared to have escaped from the late endosomes. After 12 hours, most of NPC/miR-375 escaped from the late endosomes (Figure 2a). Notably, no localization of the NPC/miR-375 was observed in lysosomes (Figure 2b). These observations indicate that NPC/miR-375 can deliver the miR-375 and facilitate its escape from late endosomes into the cytoplasm. The absence of lysosome localization suggests that degradation of miRNA in the lysosomes was potentially avoided.
Intracellular trafficking and cellular uptake of NPC/miR-375 nanoparticles by HepG2 cells: MiR-375 was labeled by Cy3 (red) and nuclei were counterstained with DAPI (blue). The cellular organelles were identified by immunostaining with markers Rab9 (late endosomes) (a) and LAMP1 (lysosomes) (b) that were stained as green. The pictures were taken by confocal microscopy using the same exposure time with a magnification of × 400. (c) Cisplatin content in HepG2 cells treated with cisplatin or NPC/miR-375 determined by ICP-MS. (d) MiR-375 uptake by HepG2 cells determined by FACS. (e) Quantitative analysis of the expression of miR-375 by qRT-PCR in HepG2 cells.
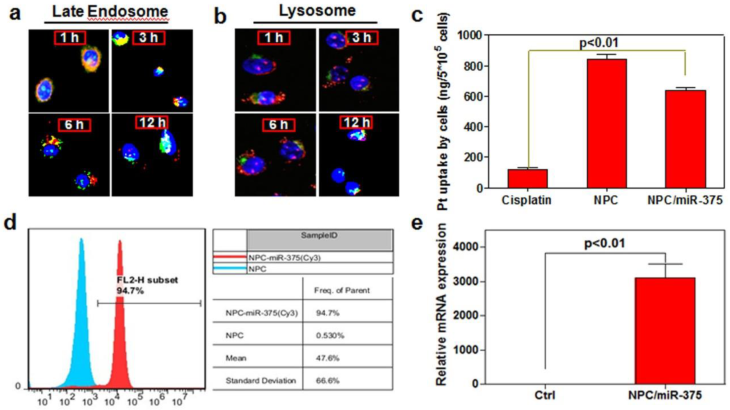
To determine whether NPC/miR-375 nanoparticles can be taken up by HCC cells, we treated HepG2 cells with cisplatin, NPC or NPC/miR-375 at a cisplatin concentration of 5 μM. The concentration of the platinum determined by ICP-MS was 112, 823, and 614 ng/5×105 cells, respectively, in cisplatin, NPC and NPC/miR-375 treated HepG2 cells (Figure 2c). Flow cytometry analysis showed that NPC/miR-375 was taken up by the HepG2 cells. Cy3 labeled miRNA was detected in 94.7% of HepG2 cells treated with NPC/miR-375 (Figure 2d). TeqMan RT-PCR analysis showed that NPC/miR-375 enormously increased the expression of miR-375 (Figure 2e). These data suggest that NPC/miR-375 can efficiently deliver both miR-375 and cisplatin to the HepG2 cells.
Cytotoxicity of NPC/miR-375 in HepG2 cells and Hep3B cells
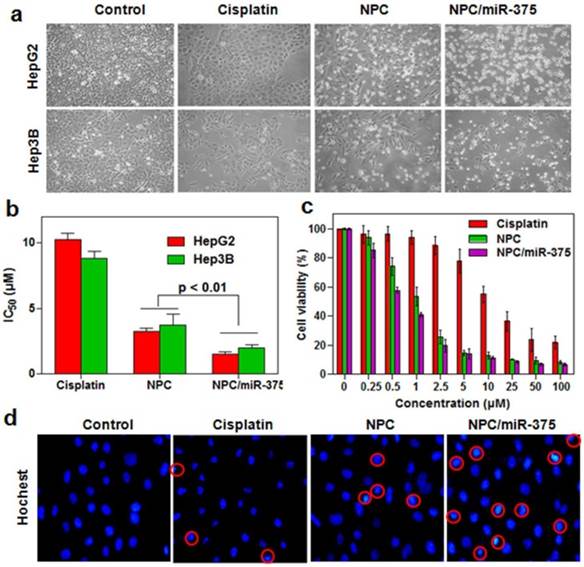
To determine whether the NPC/miR-375 were able to suppress the growth of HCC cells, we measured cytotoxicity following treatment of HepG2 and Hep3B cells with NPC/miR-375. Free cisplatin significantly inhibited the growth of HepG2 cells after 12 hours of treatment, whereas NPC were found to be highly cytotoxic (Figure 3a). Based on morphology, NPC/miR-375 treatment also led to death or apoptosis of most HepG2 and Hep3B cells (Figure 3a). Consistent with this observation, the IC50 of cisplatin, NPC and NPC/miR-375 were 10.4, 3.1 and 0.98 μM for HepG2 and 8.8, 3.9 and 1.1 μM for Hep3B cells (Figure 3b). The cytotoxic effects of cisplatin, NPC and NPC/miR-375 showed an excellent dose-effect relationship, suggesting that the killing effects of these drugs were dose dependent (Figure 3c). We further incubated HepG2 cells with DAPI and then examined the morphology of nuclei. NPC/miR-375-treated HepG2 cells showed a significant percentage of condensed and brighter nuclei, suggesting a high level of apoptosis in these cells (Figure 3d). These data clearly suggest that NPC/miR-375 has enhanced cytotoxic effect for HepG2 cells in vitro.
In vitro anti-tumor activity of NPC/miR-375 in HepG2 cells: (a) Morphology of HepG2 and Hep3B cells treated with NPC/miR-375containing 5 μM cisplatin and 100 nM miR-375 for 12 h. The pictures were taken under an inverted light microscope (× 100), (b) IC50 of cisplatin, NPC and NPC/miR-375 in HepG2 and Hep3B cells. HepG2 and Hep3B cells were treated with NPC/miR-375 containing 5 μM cisplatin and 100 nM miR-375 for 24 h, (c) Viabilities of HepG2 cells treated with different dose levels of cisplatin, NPC and NPC/miR-375 for 24 h, (d) DAPI staining of HepG2 cells treated with drugs. Data are shown as mean ± SEM of 3 independent experiments.
NPC/miR-375 induces cell apoptosis and cell cycle arrest in HepG2 cells
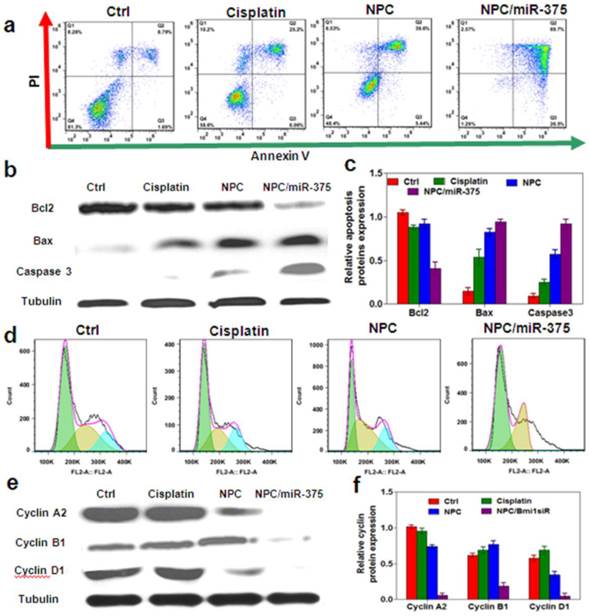
Cisplatin was known to induce apoptosis and necrosis [34]. MiR-375 can also induce apoptosis and block cell cycle progression in cancer cells. To further define the role of miR-375 on growth inhibition, we treated HepG2 cells with low dose cisplatin and an equivalent dose of NPC or NPC/miR-375. The results showed that 5 μM cisplatin-induced apoptosis in 31% HepG2 cells compared with 8.8% of untreated control cells (Figure 4a). NPC treatment induced apoptosis in 45% HepG2 cells, whereas NPC/miR-375 induced apoptosis in 97% HepG2 cells (Figure 6a). These results strongly suggest that miR-375 promotes apoptosis of the HepG2 cells. To confirm this observation, we determined the expression levels of apoptosis-related genes. NPC/miR-375 treatment group significantly up-regulated the expression of apoptosis proteins Bax and caspase 3 (Figure 4b), suggesting miR-375 promoted apoptosis (Figure 4b and 4c). The apoptosis blocker Bcl2 was down-regulated by 60% compared with the control group (Figure 4b and 4c). We also examined the effect of NPC/miR-375 on cell cycle progression in HepG2 cells. FACS analysis showed that NPC/miR-375-treated HepG2 cells were arrested in G1 phase (Figure 4d). Furthermore, Western blotting revealed that G1 phase-related cyclin D1 was down-regulated, consistent with the observation of accumulation of HepG2 cells in the G1 phase (Figure 4e and 4f). Besides, G2 phase-related cyclins B1 and A1 were repressed as well, suggesting that the cells were blocked in G1 phase and unable to progress to S phase (Figure 4e and 4f). Taken together, these results clearly indicate that the improved anti-tumor effect of NPC/miR-375 compared with NPC is due to apoptosis and cell cycle arrest induced by miR-375.
Anti-tumor mechanisms of NPC/miR-375 in vitro: (a) Apoptosis of HepG2 cells treated with NPC/miR-375 by FACS analysis. Quadrants from lower left to upper left (counter clockwise) represent healthy, early apoptotic, late apoptotic, and necrotic cells respectively. (b) Western blotting of Bcl2, Bax and Caspase3 in HepG2cells treated with PBS, cisplatin, NPC and NPC/miR-375 for 24 h (tubulin was used as a loading control). (c) Quantitative analysis of the western blotting bands. (d) Cell cycle profiles of HepG2 cells incubated with PBS, cisplatin, NPC, and NPC/miR-375. Cells were fixed by 70% ethanol and labeled with propidium iodide and analyzed for fluorescent DNA content using FACS. (e) Western blotting of cyclin A2, cyclin B1 and cyclin D1 in HepG2 cells treated with PBS, cisplatin, NPC and NPC/miR-375 (tubulin was used as a loading control). (f) Quantitative analysis of the western blotting bands. Data are expressed as mean ± SEM of 3 independent experiments.
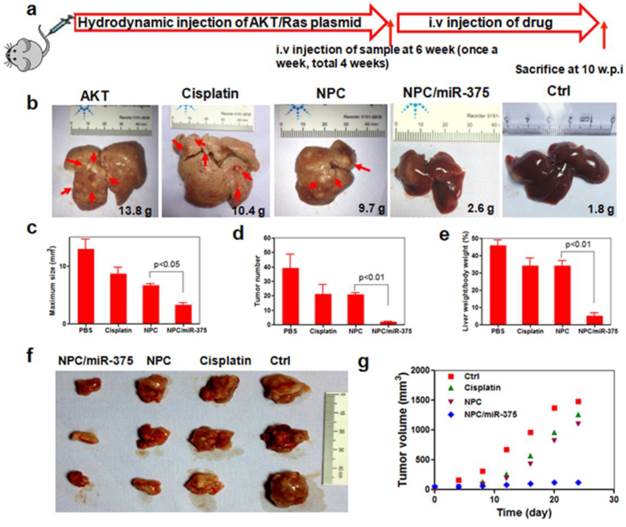
Anti-tumor effects of NPC/miR-375 in an Akt-Ras induced primary HCC mouse model and a HepG2 xenograft tumor model: (a) Overall diagram of study design. Akt and Ras plasmids were hydrodynamically injected into 6 weeks old FVB mice via the tail vein. The mice received an injection of PBS, free cisplatin, NPC, and NPC/miR-375 (once a week, total 4 weeks) starting from 6 weeks after plasmid injection when tumors were formed. Mice were sacrificed and livers were excised at 4 weeks after drug administration. (b) The ex vivo liver images of mice treated with PBS, free cisplatin, NPC, NPC/miR-375 and control. The average liver weights for each group (n = 5) are shown in the images. (c) Maximal tumor sizes in the liver of HCC mice. (d) HCC nodule numbers of mice after treatment with drugs or vehicle. (e) Liver weight/body weight of mice after sacrifice. The results are displayed as mean ± SEM of 5 animals per group. (f) Excised tumors at the end point of the experiment (day 24th after the drug injection). (g) Growth curves of xenograft tumors treated with PBS, free cisplatin, NPC, and NPC/miR-375 in 4 doses given by tail vein injection ( 170 μg/kg miR-375, 1 mg/kg cisplatin) (n = 5).
In vivo fluorescence imaging and biodistribution study of NPC/miR-375
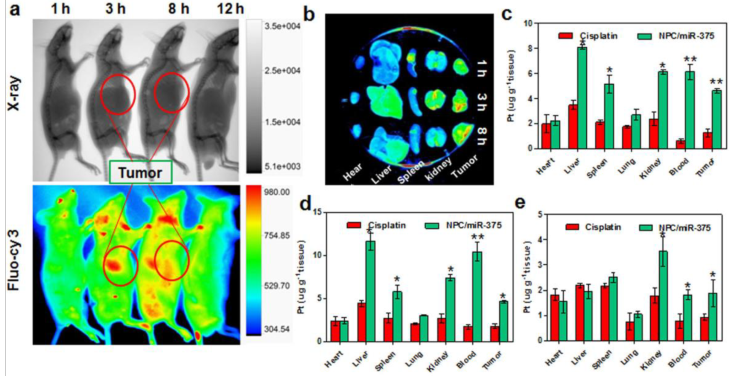
We determined the distribution of NPC/miR-375 in tumor-bearing nude mice. The mice received a single injection of NPC/miR-375 at a dose of 170 μg/kg miR-375 and 1.0 mg/kg cisplatin. Using in vivo imaging system, we observed that NPC/miR-375 started to enter into the tumor at one hour, and the concentration reached the peak at three hours after injection (red circle) (Figure 5a and 5b). Also, NPC/miR-375 particles showed accumulation in the liver, kidney and tumor (Figure 5a and 5b). Later images showed that miR-375 levels in the tumor and liver were decreased at eight hours. Observation of isolated organs at one, three, and eight hours from injected mice corroborated the results of in vivo imaging (Figure 5b). We then determined cisplatin distribution following NPC/miR-375 injections. At one and three hours after injection, concentrations of platinum at each time point delivered by NPC/miR-375 were significantly higher than that of free cisplatin, suggesting preferential tumor localization of NPC/miR-375. At 12 hours, a high concentration of cisplatin remained in blood, indicating that NPC/miR-375 has long circulation time. Although cisplatin showed a higher concentration in the spleen, lung and kidney at three hours, it was rapidly cleared from these organs as indicated by the data obtained at 12 hours (Figure 5c, 5d and 5e). These results collectively demonstrate that NPC/miR-375 can efficiently deliver both cisplatin and miR-375 into tumor tissues.
MiR-375 and cisplatin distributions following administration of NPC/miR-375 in xenograft nude mice: Tumor-bearing nude mice were injected via tail vein with a single dose of cisplatin (1.0 mg/kg), NPC/miR-375 (170 μg/kg siRNA, 1.0 mg/kg cisplatin) (n = 3) when tumors were ~500 mm3. (a) X-ray and bioluminescence imaging of mice injected with NPC/miR-375 via the tail vein. (b) Ex-vivo X-ray and bioluminescence imaging of tissues excised from the animals 1, 3 and 8 h after tail vein injection of NPC/miR-375. (c and d) Tissue distributions of Pt at 1, 3 and 12 h after intravenous injection of cisplatin and NPC/miR-375. Data are expressed as mean ± SEM (n = 5).
NPC/miR-375 suppresses tumor growth in a primary HCC mouse model and a HepG2 xenograft tumor model
NPC/miR-375 was evaluated in a primary HCC mouse model. To mimic the etiology of human HCC, which requires both lipogenic and oncogenic signaling, an Akt/Ras-induced primary HCC mouse model was created by hydrodynamic injection of a plasmid carrying these genes. In this model, the Akt gene promotes lipogenesis and Ras drives oncogenesis in the liver. Tumors are formed six weeks after injecting both oncogenes AKT and Ras. At that point, PBS, cisplatin, NPC, or NPC/miR-375 were administrated in four doses by tail injections. Four weeks after the first injection, mice were sacrificed and tumors were examined (Figure 6a). PBS injected mice had many HCC nodules distributed on the surface of the liver (Figure 6b). Compared with the PBS group, both cisplatin and NPC groups showed tumor inhibition (Figure 6b). More importantly, administration of NPC/miR-375 led to significant inhibition of tumor growth (Figure 6b). The liver weights of NPC/miR-375-treated mice were significantly smaller than those from the NPC or cisplatin-treated mice (Figure 6b). Moreover, the number of tumor nodules in NPC/miR-375-treated mice was much lower than those of the NPC-treated (21 nodules) and cisplatin-treated mice (21 nodules), indicating that most of the tumors were eliminated at early stages (Figure 6d). Furthermore, maximal tumor sizes in NPC/miR-375 treated mice were considerably lower than those from other groups (Figure 6c). Notably, one mouse treated with NPC/miR-375 showed a liver that appeared normal (Figure 6b). The liver /body weight ratio also indicated the tumor growth was largely inhibited by NPC/miR-375 (Figure 6e). These data suggested that co-delivery of cisplatin and miR-375 by NPC produced significantly enhanced tumor inhibition. The anti-tumor effect of both compounds was further evaluated in a HepG2 xenograft tumor model in mice that produced similar results (Figure 6f and 6g).
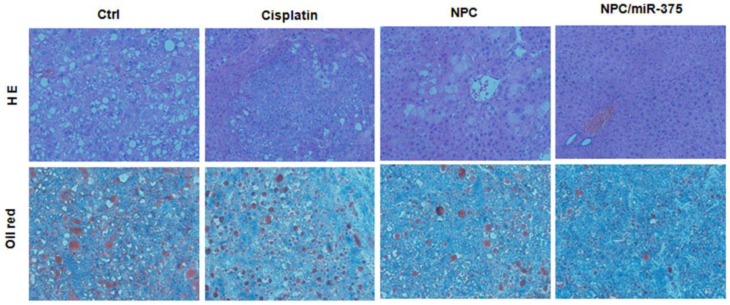
We subsequently performed the histological examination to determine tumor morphology. For PBS and cisplatin-treated mice, the liver showed apparent lipid droplet accumulation that was induced by the Akt oncogene (Figure 7, HE and oil red staining). Treatment of mice with NPC/miR-375 notably reduced lipid accumulation in the liver.
Histological analysis of livers from primary HCC mice treated with cisplatin, NPC or NPC/miR-375.
Discussion
MiRNAs are small noncoding RNAs that play significant roles in cancers. Due to its imperfect complementarity for targeting mRNA transcripts, a single miRNA can regulate multiple target genes and thus functions as a molecular 'rheostat' [35]. Alterations in miRNA expression can modulate key cellular processes involved in tumorigenesis. Evaluation of changes in miRNA profile can provide insight into the basic mechanisms of tumor formation and progression [36]. Chemo-resistance is one of the key causal factors in cancer death. Emerging evidence suggests that miRNAs have critical roles in the regulation of chemo-sensitivity in cancers. Therefore, miRNAs have potential roles in the diagnosis and treatment of cancers. Our previous study showed that up-regulation of miR-375 can significantly inhibit the growth of HCC both in vitro and in vivo [16]. In liver cancer, miR-375 has been reported to be down-regulated [17]. However, despite its important roles in carcinogenesis, the therapeutic potential of miR-375 has not been adequately evaluated in a translational research setting.
A variety of carriers has been designed for miRNA delivery and has shown high delivery efficiency over a broad range of cell types. There is growing interest in fabricating multi-agent co-delivery nanocarriers that can simultaneously incorporate and deliver multiple types of therapeutic agents including miRNA or siRNA to target sites [24]. In our previous study, we explored the feasibility of delivering siRNA together with doxorubicin (Dox) by liposomes for cancer therapy in a xenograft mouse tumor model [25]. Although this study demonstrated the efficacy of co-delivered siRNA/Dox by folate-targeted liposomes, it failed to show similar efficacy in the primary cancer model. In the current study, we selected miR-375 and cisplatin as a combination for the treatment of HCC. The rationale was based on the fact that the targeted effectors or genes of miR-375, such as YAP and ASG-1, are associated with cisplatin-based chemoresistance. We reasoned that the co-delivery of miR-375 together with a chemotherapeutic drug using nanocarriers might potentially produce synergetic effects and improve overall treatment outcomes for HCC.
In this study, NPC/miR-375 were successfully synthesized using the method originally developed by Guo et al [37]. The novel cisplatin nanoformulation method provided a good example of using an unmodified poorly soluble drug for the assembly of NPs. We further extended the application of this formulation to gene delivery. We took advantage of the positive charge feature of DOTAP that absorbed miR-375 and incorporated miR-375 into NPC thereby co-delivering miR-375 and cisplatin. It has already been demonstrated that this novel mono-delivering cisplatin NP exhibit enhanced anticancer activity compared to free cisplatin [37]. However, despite the encouraging antitumor efficacy of the mono-delivering cisplatin NP, drug resistance eventually contributes to treatment failure. Thus, our efforts focused on combining cisplatin NP with another therapeutic mechanism. We selected miR-375 to inhibit YAP or other genes which are induced by mono-cisplatin and subsequently reverse tumor cell autonomous resistance. The role of miR-375 in HCC has previously been investigated. We reported that miR-375 was significantly down-regulated in HCC tissues and cell lines [16]. Overexpression of miR-375 in liver cancer cells decreased cell proliferation, clonogenicity, migration/invasion and simultaneously induced G1 arrest and apoptosis [16]. Apparently, mono gene therapy or chemotherapy alone has not been very promising for HCC. We, therefore, developed NPC as a carrier for co-delivering miR-375 to augment the cisplatin NP efficacy. As shown in this study, NPC/miR-375 system was able to efficiently deliver cisplatin into tumors and release miR-375 from endosomes instead of lysosomes thus guaranteeing efficient cytoplasmic delivery of the miRNA. Our results clearly showed that miR-375 delivered by NPC produced a synergetic effect with cisplatin significantly inhibiting tumor growth in a relevant mouse model of HCC.
Lack of an appropriate mouse model has been an obstacle for the evaluation of anti-cancer drugs, often leading to the failure of drug candidates in clinical trials. Xenograft mouse models have long been used for in vivo evaluation of anti-tumor drugs. However, xenograft tumor implants are different from the primary tumor in many aspects. For example, xenograft tumor cells grow in an environment that lacks supporting niches, including oxygen supply, neovascularization, and protection from fibroblast or mesenchymal cells. Especially, nude or NOD/SCID mice used for xenograft tumor models are immune-deficient, whereas tumors in humans are still subject to immune surveillance. These deficiencies lead to many false positive results in anti-tumor drug evaluations. The primary HCC mouse model used in this study fully mimics the etiology and morphology of human HCC. In this model, oncogenes Akt and Ras cooperate to initiate the HCC by driving lipogenesis and uncontrolled mitosis, respectively, which are two major biochemical alterations during the hepatic carcinogenesis. Therefore, the robust anti-tumor effects of NPC/miR-375 exhibited in this mouse model are far more convincing in terms of potential therapeutic utility than the data obtained in xenograft mouse models.
Our study also confirmed the significance of up-regulation of miR-375 for overcoming chemoresistance in HCC cells. Despite strong apoptotic signals observed in vitro as well as in the cisplatin- and NPC-treated mice, strong growth signals were also seen in these mice. This suggests that although cisplatin induced apoptosis in HCC, there were still many cisplatin insensitive tumor cells that were proliferating. Both in vitro and in vivo results clearly showed synergistic effects o NPC/miR-375. This was possibly due to the elimination of chemotherapy insensitive HCC cells by miR-375. Abolishing chemoresistance in HCC cells reduced the possibility of re-growth of tumor cells after the end of treatments.
To our knowledge, this is the first report on the therapeutic value of miR-375 in a primary tumor model when it is co-delivered with cisplatin in lipid-coated nanoparticles. We have provided evidence that nanoparticles co-delivering a chemotherapy drug and a microRNA modulating specific molecular targets in hepatocellular carcinogenes represent a potential therapeutic agent for chemotherapy insensitive HCC.
Conclusion
The co-delivery nano vehicles described here can be used for therapeutic delivery of a broad spectrum of siRNAs or miRNAs. NPC/miR-375 represents a promising approach to overcome drug resistance in cancer therapy and warrants further studies. We envision that this co-delivery system can be generalized to other miRNAs and other cancer types.
Supplementary Material
Supplementary Figures 1-2.
Acknowledgements
This work was supported by Natural Science Foundation of China grants (No.81301235 and No.81370058), Natural Science Foundation of Hubei Province (No. 2014CFB405), and Independent Innovation Foundation of HUST (2014YGYL013), Specialized Research Fund for the Doctoral Program of Higher Education of China (20120142120095), the Fundamental Research Funds for the Central Universities of HUST (2014TS090).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA: a cancer journal for clinicians. 2015;65:87-108
2. El-Serag HB, Rudolph KL. Hepatocellular carcinoma: epidemiology and molecular carcinogenesis. Gastroenterology. 2007;132:2557-76
3. Cervello M, Bachvarov D, Lampiasi N, Cusimano A, Azzolina A, McCubrey JA. et al. Molecular mechanisms of sorafenib action in liver cancer cells. Cell cycle. 2012;11:2843-55
4. Llovet JM, Bruix J. Molecular targeted therapies in hepatocellular carcinoma. Hepatology. 2008;48:1312-27
5. Spangenberg HC, Thimme R, Blum HE. Targeted therapy for hepatocellular carcinoma. Nature reviews Gastroenterology & hepatology. 2009;6:423-32
6. Siegel AB, Olsen SK, Magun A, Brown RS Jr. Sorafenib: where do we go from here? Hepatology. 2010;52:360-9
7. Ikeda M, Okusaka T, Ueno H, Takezako Y, Morizane C. A phase II trial of continuous infusion of 5-fluorouracil, mitoxantrone, and cisplatin for metastatic hepatocellular carcinoma. Cancer. 2005;103:756-62
8. Schwartz JD, Beutler AS. Therapy for unresectable hepatocellular carcinoma: review of the randomized clinical trials-II: systemic and local non-embolization-based therapies in unresectable and advanced hepatocellular carcinoma. Anti-cancer drugs. 2004;15:439-52
9. Aravalli RN, Cressman EN, Steer CJ. Cellular and molecular mechanisms of hepatocellular carcinoma: an update. Archives of toxicology. 2013;87:227-47
10. Gailhouste L, Ochiya T. Cancer-related microRNAs and their role as tumor suppressors and oncogenes in hepatocellular carcinoma. Histology and histopathology. 2013;28:437-51
11. Giordano S, Columbano A. MicroRNAs: new tools for diagnosis, prognosis, and therapy in hepatocellular carcinoma? Hepatology. 2013;57:840-7
12. Wong CM, Kai AK, Tsang FH, Ng IO. Regulation of hepatocarcinogenesis by microRNAs. Frontiers in bioscience. 2013;5:49-60
13. Drakaki A, Hatziapostolou M, Iliopoulos D. Therapeutically targeting microRNAs in liver cancer. Curr Pharm Des. 2013;19:1180-91
14. Kota J, Chivukula RR, O'Donnell KA, Wentzel EA, Montgomery CL, Hwang HW. et al. Therapeutic microRNA delivery suppresses tumorigenesis in a murine liver cancer model. Cell. 2009;137:1005-17
15. Park JK, Kogure T, Nuovo GJ, Jiang J, He L, Kim JH. et al. miR-221 silencing blocks hepatocellular carcinoma and promotes survival. Cancer research. 2011;71:7608-16
16. He XX, Chang Y, Meng FY, Wang MY, Xie QH, Tang F. et al. MicroRNA-375 targets AEG-1 in hepatocellular carcinoma and suppresses liver cancer cell growth in vitro and in vivo. Oncogene. 2012;31:3357-69
17. Liu AM, Poon RT, Luk JM. MicroRNA-375 targets Hippo-signaling effector YAP in liver cancer and inhibits tumor properties. Biochem Biophys Res Commun. 2010;394:623-7
18. Zhao B, Wei X, Li W, Udan RS, Yang Q, Kim J. et al. Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genes & development. 2007;21:2747-61
19. Brown DM, Ruoslahti E. Metadherin, a cell surface protein in breast tumors that mediates lung metastasis. Cancer Cell. 2004;5:365-74
20. Camargo FD, Gokhale S, Johnnidis JB, Fu D, Bell GW, Jaenisch R. et al. YAP1 increases organ size and expands undifferentiated progenitor cells. Current biology: CB. 2007;17:2054-60
21. Gandellini P, Profumo V, Folini M, Zaffaroni N. MicroRNAs as new therapeutic targets and tools in cancer. Expert Opinion on Therapeutic Targets. 2011;15:265-79
22. Deng X, Cao M, Zhang J, Hu K, Yin Z, Zhou Z. et al. Hyaluronic acid-chitosan nanoparticles for co-delivery of MiR-34a and doxorubicin in therapy against triple negative breast cancer. Biomaterials. 2014;35:4333-44
23. Chen Y, Zhu X, Zhang X, Liu B, Huang L. Nanoparticles modified with tumor-targeting scFv deliver siRNA and miRNA for cancer therapy. Molecular therapy: the journal of the American Society of Gene Therapy. 2010;18:1650-6
24. Chen AM, Zhang M, Wei D, Stueber D, Taratula O, Minko T. et al. Co-delivery of doxorubicin and Bcl-2 siRNA by mesoporous silica nanoparticles enhances the efficacy of chemotherapy in multidrug-resistant cancer cells. Small. 2009;5:2673-7
25. Yang T, Li B, Qi S, Liu Y, Gai Y, Ye P. et al. Co-delivery of doxorubicin and Bmi1 siRNA by folate receptor targeted liposomes exhibits enhanced anti-tumor effects in vitro and in vivo. Theranostics. 2014;4:1096-111
26. Kim C, Shah BP, Subramaniam P, Lee KB. Synergistic induction of apoptosis in brain cancer cells by targeted codelivery of siRNA and anticancer drugs. Molecular pharmaceutics. 2011;8:1955-61
27. Creixell M, Peppas NA. Co-delivery of siRNA and therapeutic agents using nanocarriers to overcome cancer resistance. Nano Today. 2012;7:367-79
28. He C, Liu D, Lin W. Self-assembled nanoscale coordination polymers carrying siRNAs and cisplatin for effective treatment of resistant ovarian cancer. Biomaterials. 2015;36:124-33
29. Guo S, Wang Y, Miao L, Xu Z, Lin CM, Zhang Y. et al. Lipid-coated Cisplatin nanoparticles induce neighboring effect and exhibit enhanced anticancer efficacy. ACS Nano. 2013;7:9896-904
30. Wang D, Lippard SJ. Cellular processing of platinum anticancer drugs. Nat Rev Drug Discov. 2005;4:307-20
31. Lee KK, Yonehara S. Identification of mechanism that couples multisite phosphorylation of Yes-associated protein (YAP) with transcriptional coactivation and regulation of apoptosis. J Biol Chem. 2012;287:9568-78
32. Ho C, Wang C, Mattu S, Destefanis G, Ladu S, Delogu S. et al. AKT (v-akt murine thymoma viral oncogene homolog 1) and N-Ras (neuroblastoma ras viral oncogene homolog) coactivation in the mouse liver promotes rapid carcinogenesis by way of mTOR (mammalian target of rapamycin complex 1), FOXM1 (forkhead box M1)/SKP2, and c-Myc pathways. Hepatology. 2012;55:833-45
33. Chen X, Calvisi DF. Hydrodynamic transfection for generation of novel mouse models for liver cancer research. The American journal of pathology. 2014;184:912-23
34. Fuertes MA, Castilla J, Alonso C, Perez JM. Cisplatin biochemical mechanism of action: from cytotoxicity to induction of cell death through interconnections between apoptotic and necrotic pathways. Curr Med Chem. 2003;10:257-66
35. Baek D, Villen J, Shin C, Camargo FD, Gygi SP, Bartel DP. The impact of microRNAs on protein output. Nature. 2008;455:64-71
36. Braconi C, Patel T. MicroRNA expression profiling: a molecular tool for defining the phenotype of hepatocellular tumors. Hepatology. 2008;47:1807-9
37. Guo S, Miao L, Wang Y, Huang L. Unmodified drug used as a material to construct nanoparticles: delivery of cisplatin for enhanced anti-cancer therapy. Journal of Controlled Release. 2014;174:137-42
Author contact
![]() Corresponding authors: Guangya Xiang (gyxiang1968com) and Xiang Ma ( xiangmaedu.cn), School of Pharmacy, Tongji Medical College, Huazhong University of Science and Technology, 13 Hangkong Rd., Wuhan 430030, P. R. China, Tel.: +86-27-8369-2793; Fax: +86-27-8369-2762.
Corresponding authors: Guangya Xiang (gyxiang1968com) and Xiang Ma ( xiangmaedu.cn), School of Pharmacy, Tongji Medical College, Huazhong University of Science and Technology, 13 Hangkong Rd., Wuhan 430030, P. R. China, Tel.: +86-27-8369-2793; Fax: +86-27-8369-2762.