Impact Factor
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Methods
Results and Discussion
Conclusions
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2016; 6(2):192-203. doi:10.7150/thno.13657 This issue Cite
Research Paper
Mono-arginine Cholesterol-based Small Lipid Nanoparticles as a Systemic siRNA Delivery Platform for Effective Cancer Therapy
Jinju Lee1, Phei Er Saw2, Vipul Gujrati2, Yonghyun Lee2, Hyungjun Kim2, Sukmo Kang2, Minsuk Choi2, Jae-Il Kim1, Sangyong Jon2, ![]()
1. † School of Life Science, Gwangju Institute of Science and Technology, 123 Cheomdangwagi-ro, Gwangju 500-712, Republic of Korea.
2. ‡ KAIST Institute for the BioCentury, Department of Biological Sciences, Korea Advanced Institute of Science and technology (KAIST), 291 Daehak-ro, Daejeon 305-701, Republic of Korea
Received 2015-8-25; Accepted 2015-10-1; Published 2016-1-1
Abstract

Although efforts have been made to develop a platform carrier for the delivery of RNAi therapeutics, systemic delivery of siRNA has shown only limited success in cancer therapy. Cationic lipid-based nanoparticles have been widely used for this purpose, but their toxicity and undesired liver uptake after systemic injection owing to their cationic surfaces have hampered further clinical translation. This study describes the development of neutral, small lipid nanoparticles (SLNPs) made of a nontoxic cationic cholesterol derivative, as a suitable carrier of systemic siRNA to treat cancers. The cationic cholesterol derivative, mono arginine-cholesterol (MA-Chol), was synthesized by directly attaching an arginine moiety to cholesterol via a cleavable ester bond. siRNA-loaded SLNPs (siRNA@SLNPs) were prepared using MA-Chol and a neutral helper lipid, dioleoyl phosphatidylethanolamine (DOPE), as major components and a small amount of PEGylated phospholipid mixed with siRNA. The resulting nanoparticles were less than ~50 nm in diameter with neutral zeta potential and much lower toxicity than typical cationic cholesterol (DC-Chol)-based lipid nanoparticles. SLNPs loaded with siRNA against kinesin spindle protein (siKSP@SLNPs) exhibited a high level of target gene knockdown in various cancer cell lines, as shown by measurement of KSP mRNA and cell death assays. Furthermore, systemic injection of siKSP@SLNPs into prostate tumor-bearing mice resulted in preferential accumulation of the delivered siRNA at the tumor site and significant inhibition of tumor growth, with little apparent toxicity, as shown by body weight measurements. These results suggest that these SLNPs may provide a systemic delivery platform for RNAi-based cancer therapy.
Keywords: cationic lipids · cancer therapy · drug delivery · nanoparticles · RNAi · siRNA
Introduction
RNAi-based therapy has attracted great attention as the next generation of agents to treat various hard-to-cure diseases.[1, 2] RNAi therapy involves the delivery of small interfering RNA (siRNA) to the cytosol of cells, knocking down the expression of these genes.[3, 4] Several types of siRNA-based therapeutics are currently undergoing preclinical and clinical testing.[5-7] The key determinant for the success of RNAi-based therapy is a suitable platform to deliver siRNA to target cells of interest.
Cationic lipid-based nanocarriers are the most widely studied non-viral vehicles for RNAi therapy, as cationic lipids can easily form complexes with negatively charged siRNA via electrostatic interactions.[8-11] To date, several cationic lipid-based nanoparticles have demonstrated high potential as delivery platforms for therapeutic siRNA in hepatic diseases,[12-14] as these nanoparticles tend to preferentially localize to the liver upon systemic administration.[15-17] In contrast, the systemic delivery of siRNA to treat solid tumors has proven more difficult.[18, 19] Nanoparticles smaller than ~50 nm in diameter and with a neutral surface charge may be suitable nanocarriers for cancer therapy upon systemic injection, as they can achieve deeper penetration within tumor tissues after extravasation than larger sized nanoparticles.[20-22] Most of the cationic lipids developed to date, however, generate relatively large-sized lipid nanoparticles (> 100 nm in diameter) with highly cationic surfaces. These properties result in their being directed mainly to the liver rather than to the tumor site, limiting their clinical application in cancer therapy.[23, 24] In addition, the toxicity associated with the cationic lipids per se has limited the clinical use of these nanoparticles.[22, 24-27]
Neutral lipid-based liposomes have recently emerged as a delivery platform for siRNA and have shown potent therapeutic effects in in vivo cancer models without appreciable toxicity.[23, 28] These liposomes, however, range in size from ~80 to 150 nm, which may limit their ability to penetrate tumor tissue.[20, 21] In the context, here we report neutrally-charged, less toxic, small lipid nanoparticles (SLNPs) containing a new cationic cholesterol derivative as a key lipid component that may be a delivery platform suitable for RNAi-based cancer therapy. This study describes the design and synthesis of such a cholesterol derivative and the preparation and characterization of SLNP containing a therapeutic siRNA (siKSP@SLNPs) targeting kinesin spindle protein (KSP), a protein that has a critical function in mitosis. Blockage of this protein has been found to result in cell cycle arrest at mitosis and ultimately cell death.[29-31] The gene silencing efficacy of siKSP@SLNPs was evaluated in various cancer cell lines in vitro and in a human prostate tumor model in vivo.
Methods
Materials
Cholesterol (99%), Boc-Arg(Pbf)-OH (98%), 4-(dimethylamino) pyridine (DMAP), 1,3-dicyclohexyl-carbodiimide (DCC), trifluoroacetic acid (TFA), and the Sepharose CL-4B column were purchased from Sigma-Aldrich (St. Louis, MO). Lipofectamine 2000 was purchased from Invitrogen (Carlsbad, CA). Dioleoylphosphatidylethanolamine (DOPE), PEG1000-DSPE (ammonium salt), 3β-[N-(N`,N`-dimethylaminoethane) carbamoyl] cholesterol (DC-Chol), and a mini handheld extruder set were obtained from Avanti Polar Lipids (Alabaster, AL). All other organic reagents were of analytical grade and were purchased from Sigma Chemical (St. Louis, MO). siRNAs labeled with TAMRA or Cy5.5 and negative control siRNA were synthesized by ST Pharm Co. Ltd. (Seoul, Republic of Korea). The following siRNAs with the indicated sequences were used for this study: KSP-specific siRNA, 5'-CUG AAG ACC UGA AGA CAA UdTdT (sense) and 5'-AUU GUC UUC AGG UCU UCA GdTdT-3' (antisense); and negative control siRNA, 5'-AUG AAC GUG AAU UGC UCA AdTdT-3' (sense) and '5'- UUG AGC AAU UCA CGU UCA UdTdT-3' (antisense).
Synthesis of monoarginine-cholesterol (MA-Chol)
A solution of dicyclohexylcarbodiimide (DCC, 215.6 mg, 1.045 mmol) in anhydrous trichloromethane (CHCl3, 5 mL) was added dropwise while stirring to a solution of cholesterol (477 mg, 1.235 mmol), Boc-Arg(Pbf)-OH (500 mg, 0.95 mmol) and 4-dimethylaminopyridine (DMAP, 11.6 mg, 0.095 mmol) in anhydrous CHCl3 (20 mL). The solution was cooled in an ice bath to 0˚C and maintained for a further 5 min at 0˚C. The ice was removed and the solution was stirred at room temperature overnight. The white precipitate, dicyclohexylurea (DCU), was removed by filtration. The filtrate was washed twice with chloroform (250 mL) and HCl (250 mL). The organic solution was dried over magnesium sulfate to remove residual water and the remaining filtrate was evaporated, purified by column chromatography and recrystallized to obtain Boc-Arg(Pbf)-Chol. Trifluoroacetic acid (7 mL) was added dropwise to a solution of Boc-Arg(Pbf)-Chol in anhydrous CHCl3 (15 mL) at room temperature for 3 h to remove the Pbf and Boc groups. The reaction solution was evaporated and purified by column chromatography. The product was concentrated and recrystallized to obtain MA-Chol conjugate with a yield of 60% and further characterized by 1H-NMR (400 MHz, Jeol, Tokyo).
Estimation on degradability of MA-Chol
Degradability of MA-Chol (10 mg/mL) under physiological conditions was assessed by thin layer chromatography (TLC) upon incubation in PBS (pH 7.4) containing 10% FBS at 37˚C for predetermined times (0, 1, 6, 12, 24, and 48 h). At each time, 100 µL of aliquot was taken from the solution, vigorously mixed with 400 µL of chloroform to extract organic compounds, and further centrifuged for 5 min. The chloroform layer was collected, evaporated to concentrate the solvent, and developed for TLC using a solvent of chloroform : methanol = 15 : 1 v/v. The resulting TLC was stained with p-anisaldehyde solution and further heated for detection.
Preparation of small lipid nanoparticles (SLNPs)
Liposomes were prepared by a thin-film hydration method, as described previously.[32] Briefly, all lipids, including MA-Chol (1.95 µmol), DOPE (1.95 µmol) and PEG1000-DSPE (0.1 µmol), were added to a glass vial, dried under vacuum, and lyophilized overnight to remove residual chloroform. To optimize liposomes, MA-Chol, DOPE, and PEG1000-DSPE were mixed at the indicated ratios, keeping the total lipid amounts fixed at 4 µmol. The lipid film was rehydrated by adding 1 mL of HEPES-buffered 5% glucose (HBG) containing 3 nmol siRNA, resulting in a final lipid concentration of 4 mM (N/P ratio = 33.3). The solution was sonicated briefly, incubated for 4 h at room temperature with intermittent mixing and extruded at least 10 times through a stack of two polycarbonate membranes (100 nm size) using a hand held extruder (Avanti Polar Lipid, AL). The liposomes were then sterilized by passage through a 0.22 µm sterile filter.
Physicochemical characterization of SLNPs
The particle size and zeta potential of SLNPs (1 mg/mL in 5% HBG buffer, pH 7.4) were determined by dynamic light scattering (DLS) using a Zetasizer nano range (Malvern, Worcestershire, UK) at ambient temperature. Each sample was analyzed in triplicate. The size and morphology of liposomes were further characterized by transmission electron microscopy (JEOL-in situ TEM). Samples were stained with 1% uranyl acetate for negative staining.
To evaluate the siRNA encapsulation efficiency, as-prepared SLNPs were loaded onto a Sepharose CL-4B column, which was prewashed with HEPES-buffered saline (10 mM, pH 7.4). The column was eluted with the same buffer into fifteen 1 mL fractions, and the siRNA contents of each fraction were analyzed using OliGreen (Invitrogen) according to the manufacturer's protocol. Briefly, 100 µL of each fraction were mixed with 10 µL of 5% Triton-X 100 containing OliGreen, and the fluorescence intensity was assessed at UV excitation and emission wavelengths of 480 and 520 nm, respectively, to measure the amount of free siRNA. Liposome fractions were eluted in fractions 2-3 whereas free siRNA was eluted in fractions 6-10. The entrapment efficiency of siRNA was calculated as the area under the curve of each peak relative to the total area under the curve. The dose dependency of siRNA encapsulation efficiency was evaluated in SLNPs and DC-Chol/DOPE liposomes using 3 nmol of siRNA and lipid concentrations decreasing from 4 to 0.2 µmol/mL, with encapsulation efficiency analyzed as described above.
The stability of siRNA@SLNPs and the naked siRNA was evaluated in PBS containing 30% FBS at 37˚C for predetermined time (0.5, 1, 3, 6, 12, and 24 h). At each time, 3 units of RNase inhibitor (Invitrogen) was added to stop the enzymatic degradation and each sample was treated with 0.5% Triton-X 100. Aliquots containing 1 µg siRNA were loaded onto 12% agarose gels containing Redsafe (iNtRON Biotechnology Inc., Seongnam, Korea). Next, the integrity of siRNA@SLNPs upon incubation in PBS containing 10% FBS at 37˚C was further evaluated with respect to size and zeta potential at predetermined time points (0, 1, 3, 6, 12, and 24 h).
Cell culture
All cell lines were purchased form Korean Cell Line Bank (KCLB; Seoul, Korea). PC-3 human prostate cancer cells were maintained in Ham's F12-K medium supplemented with 10% FBS (GIBCO), 100 U/mL penicillin, and 100 µg/mL streptomycin. SK-OV-3 and AGS cells were cultured in RPMI-1640 medium containing 10% FBS, 100 U/mL penicillin, and 100 µg/mL streptomycin. U87MG cells were maintained in MEM medium with 10% FBS, 100 U/mL penicillin, and 100 µg/mL streptomycin. All cells were incubated at 37˚C in a humidified atmosphere containing 5% CO2.
In vitro cytotoxicity
Cytotoxicity of SLNPs was determined by CCK-8 cytotoxicity assays (Sigma). Briefly, PC-3 cells were seeded in 96-well plates at 5 x 103 cells/ well in Ham's F-12K and incubated overnight. Total 4 µmol lipid of MA-Chol/DOPE, at ratio of 1:1 containing 2.5 mol% PEG was encapsulated with negative control siRNA (siNC, 3 nmol) to a final lipid concentration of 4 mM. The cells were treated with the liposomes at the indicated concentration for 4 h, washed and further incubated in fresh medium for 48 h. The cells incubated in fresh medium containing 10% v/v of the CCK-8 proliferation reagent for 2 h at 37˚C, and the absorbance at 450 nm was measured using a microplate reader (VERASmaxTM, Molecular Devices). The viability of PC-3 cells treated with DC-Chol/DOPE liposomes containing negative control siRNA (siNC) was assessed by the same procedure.
Assessment of gene silencing in vitro
The cytotoxicity of siKSP@SLNPs was assessed in four cancer cell lines: PC-3, SK-OV-3, AGS, and U87MG. Cells were seeded into 96-well plates at 5 x 103 cells/ well, incubated overnight, and transfected for 4 h with free siKSP, siKSP@SLNPs, negative control siRNA (siNC)@SLNPs, or siKSP@LF, with final concentrations of siRNA of 100 nM. The cells were washed with PBS and incubated in fresh medium for 48 h. Cell viability was analyzed by the CCK-8 cytotoxicity assay.
Quantitative (real-time) reverse transcription polymerase chain reactions (qRT-PCR) were performed to optimize the transfection efficiency of liposomes and to evaluate target mRNA knockdown. Briefly, cells were incubated for 48 h with different molar formulations of liposomes (MA-Chol/DOPE ratios and mol% of PEG1000-DSPE) or lipofectamine containing siKSP at a final concentration of 100 nM. Total RNA was isolated using RNA purification kits (Hybrid-RTM, Gene All) according to the manufacturer's instructions, and 1 µg aliquots of RNA were reverse-transcribed to first strand cDNA using the ImProm-II Reverse Transcription System (Promega) as described by the manufacturer. A 2 µL aliquot of cDNA was subjected to qRT-PCR targeting KSP and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) using SYBR Premix (Kapa Systems) and a Rotor Gene Q System (Qiagen). The relative gene expression was quantified using the ΔΔCt method, and the expression levels for target transcripts were reported as fold changes in KSP expression relative to untreated controls and normalized to the endogenous reference, GAPDH mRNA. The KSP primers were 5'-GGC GTC GCA GCC AAA TTC GTC-3' (forward) and 5'-TGC CAG TTT GGC CAT ACG CA-3' (reverse); and the GAPDH primers were 5'-CGT CTT CAC CAC CAT GGA GA-3' (forward) and 5'-CGG CCA TCA CGC CAC AGT TT-3' (reverse).
Protein expression was analyzed by western blotting. PC-3 cells were seeded into six-well plates and incubated overnight. The cells were transfected with siKSP@SLNPs, at a final siRNA concentration of 100 nM, for 4 h in serum-free medium. The transfection medium was replaced with complete medium, followed by incubation for an additional 48 h. The cells were detached and lysed using protein extraction solution (PRO-PREPTM, iNtRON Biotechnology), according to the manufacturer's instructions. Total protein concentration in all samples was determined by the Bradford method, and equal amounts of total protein lysates (50 µg) were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) on 8% gels. The proteins were electrotransferred to polyvinylidene difluoride (PVDF) membranes at 30 V overnight at 4˚C. The blots were blocked with 5% skim milk in Tris-buffered saline containing Tween-20 (TBST) for 2 h at ambient temperature and incubated with primary antibodies against KSP (mouse anti-EG5 monoclonal antibody; Abcam) and GAPDH (mouse monoclonal; Santa Cruz Biotechnology) in 5% skim milk in TBST overnight at 4˚C. The membranes were washed, incubated with HRP-conjugated secondary antibodies for 2 h at ambient temperature, and specific bands were visualized by western blotting detection reagents (Bio Imaging Analyzer, BIO-RAD).
Imaging of intracellular uptake
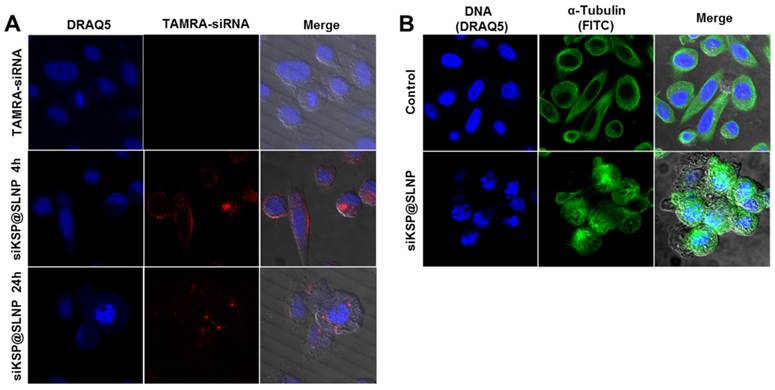
Cellular uptake of SLNPs was assessed by confocal fluorescence microscopy. PC-3 and U87MG cells were grown in Ham's F-12K and MEM media, respectively, supplemented with 10% FBS and antibiotics to ~70% confluence on coverslips in 12-well dishes. The medium was replaced with serum-free medium containing TAMRA-labeled siKSP@SLNPs or free TAMRA-labeled siKSP, both containing 100 nM siRNA. After incubation for 4 h at 37˚C, the cells were washed three times with PBS, followed by the addition of fresh medium containing 10% FBS and antibiotics. At specific time points (4 and 24 h), the cells were stained with DRAQ5 (Cell Signaling technology) before fixation with 4% (w/v) paraformaldehyde (PFA). Coverslips with fixed cells were mounted onto glass slides with fluorescence mounting medium (Dako). All samples were examined by confocal microscopy (LSM 510, Carl Zeiss).
The mechanism of cell death was evaluated by monitoring the monoastral spindle arrangement by confocal microscopy after 48 h of treatment with siKSP(100 nM)@SLNPs. Briefly, PC-3 cells in Ham's F-12K medium containing 10% FBS and antibiotics were grown on 0.1% gelatin-coated coverslips in 6-well dishes. When they reached approximately 70% confluence, the cells were treated with siKSP@SLNPs in serum-free medium for 4 h at 37˚C, followed by replacement with fresh medium containing 10% FBS and antibiotics and incubation for a further 48 h. The nuclei were stained with DRAQ5, and the cells were washed three times with PBS and fixed with 4% PFA. The cells were permeabilized by incubation in 0.25% Triton X-100 in PBS for 10 min and washed three times with PBS. The slides were incubated with anti-tubulin primary antibody (mouse monoclonal; Santa Cruz Biotechnology) and FITC-labeled anti-mouse secondary antibody (Santa Cruz Biotechnology) and examined by confocal microscopy.
In vivo biodistribution and antitumor effects
Xenograft studies were performed using 6-8 week-old female Balb/c nude mice (Orient Bio Inc.). All in vivo experiments was performed according to established guidelines and under the supervision of the Institutional Animal Care Committee (2012-BS25). PC-3 cells were injected subcutaneously into the flanks of nude mice at 5 x 106 cells/ mouse. To assess biodistribution, Cy5.5-siRNA(1.5 mg/kg)@SLNPs or equivalent free Cy5.5-siRNA was injected into the tail vein of three mice/group. After 24 h, in vivo fluorescence imaging of whole bodies and tissues of mice was performed using an IVIS 100 imaging system (PerkinElmer, Waltham, MA).
Antitumor efficacy was investigated when the tumor volume reached approximately 150-170 mm3. Twenty mice were randomly divided into four groups of five mice each, followed by intravenous injection into the tail vein of (1) saline, (2) siNC(0.5 mg/kg)@SLNPs (negative control siRNA), (3) siKSP(0.5 mg/kg)@SLNPs (low dose siKSP), or (4) siKSP(1 mg/kg)@SLNPs (high dose siKSP) every other day for a total of six injections. Tumor volume was calculated using the formula: Volume (mm3) = (a × b2)/2, where a and b are the major and minor axes of the tumor, respectively. Tumor volumes and body weights were monitored throughout the treatment. Tumor growth inhibition (TGI) on the final day was calculated as TGI = 100% × (TvolControl - TvolTreatment)/TvolControl, where Tvol = final tumor volume - initial tumor volume.[33] Twenty-four hours after the final injection, the mice were sacrificed and tumors were collected. KSP mRNA level was assessed by qRT-PCR and protein expression by western blotting, as above. Apoptotic cells in tumor sections were detected by TUNEL assay (Bio Vision) according to the manufacturer's protocol.
Hemolysis assay
SLNPs (1.34 mg/mL) or DC-Chol/DOPE-based lipid nanoparticles (1.34 mg/mL) was placed on horse blood agar plates and then further incubated at 37˚C overnight. The next day, plates were examined for formation of a clear zone (i.e., reflecting lysis of red blood cells) surrounding regions plated with either SLNPs or DC-Chol/DOPE-based lipid nanoparticles.
Results and Discussion
Design and synthesis of a new cationic cholesterol derivative
Cholesterol is an essential component in the preparation of lipid-based nanoparticles, such as liposomes and lipoplexes for siRNA delivery, making these nanoparticles mechanically stable.[9, 34-37] Thus, a cholesterol derivative with cationic charge may function as not only a stabilizer but also a complexation agent of siRNA. Although DC-Chol, a typical cationic cholesterol derivative, can efficiently form complexes with siRNA,[38-40] the in vivo use of DC-Chol has been hampered by its relatively high level of cytotoxicity. Therefore, we designed a new cationic cholesterol derivative composed of one arginine and one cholesterol molecule, in which the carboxylic acid moiety of the amino acid arginine is directly connected to a hydroxyl group of cholesterol through a cleavable ester bond (Figure 1A); this molecule has been designated MA-Chol (monoarginine-cholesterol). In the first step of synthesis, a carboxylic acid in the protected form of arginine [t-Boc-Arg(Pbf)-OH] was converted to DCC-activated arginine and subsequently allowed to react with a hydroxyl group of cholesterol to yield an ester product. Following deprotection of the arginine, MA-Chol was produced, with an overall yield of 60%. The 1H-NMR spectrum of MA-Chol indicates its successful synthesis (Supplementary Information: Figure S1). MA-Chol has several favorable attributes as a complexing agent for siRNA. First, unlike typical cationic lipids such as DOTAP and DC-Chol, MA-Chol has two positive charges under physiological conditions, on the amino group and the guanidinium side chain of arginine, both of which may be able to participate in forming complexes with siRNA.[41] Second, the guanidinium side chain of arginine can effectively bind to phosphodiester moieties of oligonucleotides, further facilitating the formation of complexes with siRNA.[41, 42] Lastly, as arginine is linked to cholesterol via a cleavable ester bond, hydrolysis of MA-Chol yields two nontoxic endogenous compounds, arginine and cholesterol. We examined the degradability of MA-Chol under physiological conditions by using TLC. Upon incubation of MA-Chol in PBS (pH 7.4) containing 10% FBS at 37˚C, cholesterol, an expected degradation product, started to be detected after 1 h on TLC; the production of cholesterol increased and became more apparent with time (Supplementary Information: Figure S2), indicating that the ester bond in MA-Chol was cleaved to yield cholesterol and arginine. Consequently, MA-Chol is expected to exhibit much lower cytotoxicity than other cationic lipids, such as DC-Chol, in which the cationic head group is linked to cholesterol via a non-cleavable bond.
Synthesis of MA-Chol and formation of a small lipid nanoparticle (SLNP). (A) Synthetic scheme for the synthesis of MA-Chol. Cholesterol was conjugated to an arginine by esterification using DMAP and DCC. After purification on a silica gel column and subsequent deprotection using TFA, pure MA-Chol was obtained. (B) Schematic illustration of a siRNA@SLNP prepared using MA-Chol:DOPE (1:1) and 2.5 mol% PEG1000-DSPE. (C) Representative TEM image of siRNA@SLNPs and a magnified image of a nanoparticle (inset). (D) DLS measurement of the hydrodynamic size of siRNA@SLNPs, showing a narrow size distribution.
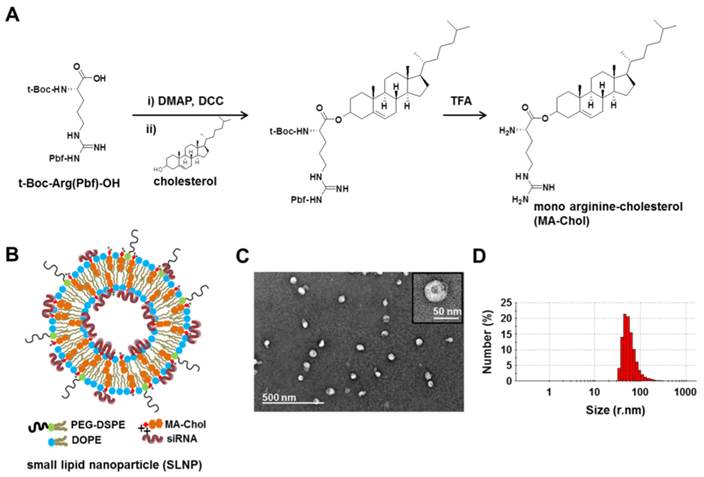
Preparation and characterization of small lipid nanoparticles (SLNPs)
Dioleoyl phosphatidylethanolamine (DOPE) is a neutral helper lipid that is frequently used for the preparation of cationic lipid nanoparticles, as it can destabilize the endosomal membrane upon endocytosis.[22, 43] Thus, DOPE was chosen as another key lipid component in preparation of SLNPs. siRNA-loaded SLNPs (siRNA@SLNPs) were prepared by a thin lipid film-rehydration method,[44] at a MA-Chol:DOPE molar ratio of 1:1 and 2.5 mol% PEGylated 1,2-distearoyl-sn-glycero-3-phosphoethanolamine (PEG1000-DSPE) as an essential component for in vivo applications, in which MA-Chol functions as both a liposomal stabilizer and complexation agent of siRNA (Figure 1B). Transmission electron microscopy (TEM) showed that the size of siRNA@SLNPs was ~45 ± 8.2 nm (mean ± s.d.; n = 30 particles) (Figure 1C), with a magnified image (Figure 1C, inset) showing that these particles have a spherical morphology with an empty inner core, similar to liposomes. The hydrodynamic size of siRNA@SLNPs, as measured by dynamic light scattering (DLS), was 52 ± 2 nm, far smaller than conventional lipid nanoparticles (~100-150 nm) (Figure 1D). In the absence of siRNA, SLNPs exhibited a slightly positive surface charge of 7.8 ± 1.5 mV, whereas the zeta potential of siRNA@SLNPs was decreased to ~0, suggesting that they became neutral lipid nanoparticles after siRNA loading. We speculate that the interactions of MA-Chol with siRNA are very tight owing to the two positive charges so as to effectively condense and compact the complex, thereby minimizing exposure of the cationic cholesterol on the surface of SLNPs and resulting in much smaller-sized nanoparticles (<50 nm) than other cationic lipids-based liposomes. Gel permeation chromatography showed that the encapsulation efficiency of siRNA was nearly 100% at MA-Chol:DOPE molar ratios of 1:1 and 1:2, whereas an unmodified normal cholesterol/DOPE formulation at the same molar ratios failed to encapsulate any siRNA (Supplementary Information: Figure S3). Furthermore, the colloidal stability of siRNA@SLNPs over time upon incubation in PBS containing 10% FBS at 37˚C was evaluated with respect to size and zeta potential. At 6 h post incubation, there was little change in the size as well as zeta potential of siRNA@SLNPs, indicating relatively high stability of the complex; however, after 24 h the size of SLNPs became appreciably increased and the zeta potential changed to more positive charge (Supplementary Information: Table S1).
The enzymatic stability of siRNA@SLNPs was also examined in the presence of 30% fetal bovine serum (FBS) at 37˚C. While naked siRNA showed rapid degradation within 3 h, siRNA encapsulated within SLNPs remained 75% intact for up to 6 h and 40% of siRNA was stable for up to 24 h (Supplementary Information: Figure S4).
Optimization and in vitro efficacy of siRNA-loaded SLNPs
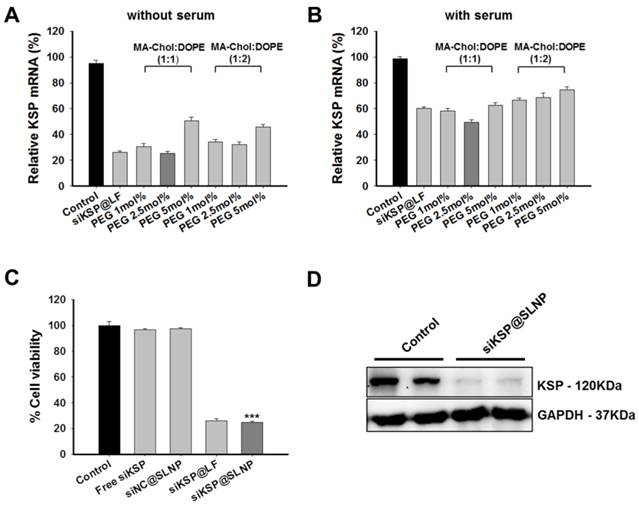
To optimize the composition of SLNPs for siRNA delivery, two sets of siRNA@SLNPs were prepared at MA-Chol:DOPE molar ratios of 1:1 and 1:2 and with different percentages of PEG1000-DSPE (1, 2.5, and 5 mol%), because PEG density on a nanoparticle surface affects its circulation half-life and biodistribution.[45, 46] A siRNA against kinesin spindle protein (siKSP) was chosen because KSP is overexpressed in various cancer cells and the inhibition of KSP function leads to cell cycle arrest, resulting in apoptosis and cell death.[29-31] The gene silencing efficacy of the two sets of siKSP(100 nM)@SLNPs was assessed in the human PC-3 prostate cancer cell line, both in the presence and absence of serum. Both siKSP@SLNPs formulations exhibited high levels of target gene silencing, comparable to the efficacy of a corresponding Lipofectamine system (siKSP@LF). In the absence of serum, siKSP@SLNPs with a 1:1 molar ratio of MA-Chol to DOPE and containing 2.5 mol% of PEG1000-DSPE exhibited the highest gene silencing efficacy (~75%), being slightly more effective than the formulation with a 1:2 molar ratio of MA-Chol to DOPE and 2.5 mol% of PEG1000-DSPE (~68%) (Figure 2A). As expected, in the presence of serum, the efficacy of gene knockdown decreased appreciably for all formulations; however, siKSP@SLNPs with a 1:1 molar ratio of MA-Chol:DOPE and 2.5 mol% of PEG1000-DSPE still showed the highest efficacy (~51%) and was therefore chosen as the optimal formulation (Figure 2B). The abilities of this formulation of siKSP@SLNPs to induce cell death and to knockdown KSP protein were also evaluated. siKSP(100 nM)@SLNPs induced substantially higher levels of cancer cell death than either free siKSP or the negative control, non-specific control siRNA-loaded SLNPs (siNC@SLNPs), suggesting that the target specific silencing of KSP mRNA led to cell death (Figure 2C). The cytotoxic effect of siKSP@SLNPs was of a similar level to that of siKSP@LF. Although Lipofectamine 2000 is known to be a powerful transfection reagent in vitro, it is not a suitable delivery carrier of siRNA for systemic RNAi therapy in vivo.[47] Western blotting showed that siKSP@SLNPs significantly reduced the production of KSP protein, consistent with its reduction of KSP mRNA (Figure 2D).
Optimization of liposome composition and in vitro efficacy of siKSP@SLNPs in PC-3 cells. (A, B) KSP gene silencing by SLNPs formed at various molar ratios of MA-Chol to DOPE and mol% PEG1000-DSPE in the absence (A) or presence (B) of serum. KSP mRNA levels were evaluated to determine the optimal molar ratio of MA-Chol to DOPE (1:1 or 1:2) at varying PEG1000-DSPE concentrations (1, 2.5, and 5 mol%). Results are shown as mean ± s.e. (n=6). (C) Cells were transfected for 4 h with siKSP@SLNPs comprised of MA-Chol/DOPE (1:1) and cytotoxicity was analyzed 48 h later. siKSP@SLNPs exhibited significantly greater cytotoxicity than control formulations. Data represent mean ± s.e. (n=9). ***p < 0.001. (D) Western blot analysis following that transfection of cells with siKSP@SLNPs resulted in substantial knockdown of KSP protein expression in PC-3 cells. Results in duplicate samples are shown.
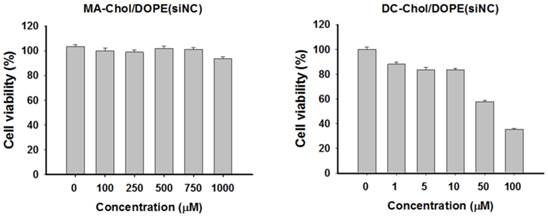
Despite cationic lipid derivatives such as DOTAP and DC-Chol having a high ability to form complexes and despite their high transfection efficiency, these lipids may be cytotoxic. We expected that MA-Chol would have much lower levels of toxicity because it could be degraded to produce two nontoxic endogenous compounds, arginine and cholesterol. Evaluation of the cytotoxicity in PC-3 cells of siNC@SLNPs with the optimal formulation (MA-Chol:DOPE of 1:1, 2.5 mol% PEG1000-DSPE) showed that this formulation had no appreciable cytotoxicity even at concentrations as high as 1 mM (Figure 3). In contrast, siNC-loaded DC-Chol/DOPE (1:1) began to show appreciable cytotoxicity even at much lower concentration of ~50 µM. Interestingly, although MA-Chol is a cationic cholesterol derivative like DC-Chol and has an additional positive charge compared to DC-Chol, it turned out that the former was far less toxic than the latter. Moreover, it should be noted that there was only a slight difference in the complex-forming ability and encapsulation efficiency for siRNA between SLNPs and DC-Chol/DOPE-based lipid nanoparticles (Supplementary Information: Figure S5). Taken together, these gene silencing and cytotoxicity results clearly suggest that SLNPs may have potential as a carrier of therapeutic siRNA.
Cytotoxicity of SLNPs and DC-Chol/DOPE liposomes with negative control siRNA. PC-3 cells were incubated for 4 h with liposomes of MA-Chol:DOPE (1:1) and DC-Chol:DOPE (1:1) encapsulating the same amount of negative control siRNA and assayed 48 h later. MA-Chol:DOPE liposomes did not show any notable toxicity, whereas DC-Chol:DOPE exhibited dose-related cytotoxicity, even at low lipid concentrations. The results represent mean ± s.e. (n=9).
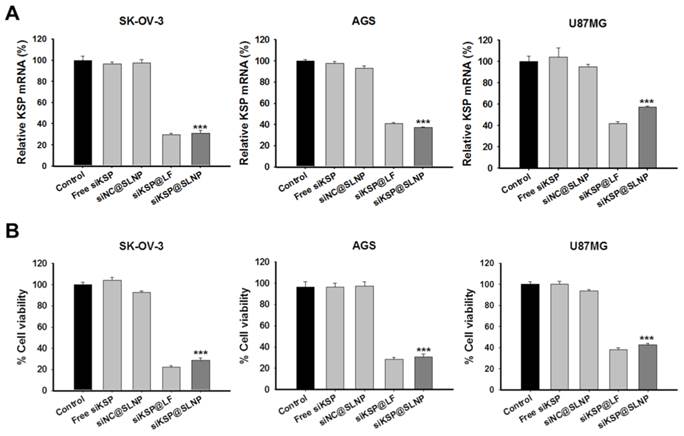
In vitro effects of siKSP@SLNPs in various cancer cell lines
To examine the feasibility of siKSP@SLNPs as a delivery platform, its gene silencing efficacy was assessed in human ovarian cancer (SK-OV-3), gastric cancer (AGS), and glioma (U87MG) cell lines. These cells were treated for 4 h with free siKSP, siNC@SLNPs, siKSP@SLNPs, and siKSP@LF at a siRNA concentration of 100 nM. qRT-PCR analyses after 48 h indicated that siKSP@SLNPs were able to effectively knock down KSP mRNA expression in all three cancer cell lines, showing efficacies of ~70% for SK-OV-3, ~63% for AGS, and ~43% for U87MG cells (Figure 4A). The overall efficacy of these SLNPs was similar to that of lipofectamine 2000. As expected from the mRNA knockdown data, siKSP@SLNPs resulted in the death of substantial numbers of all three cancer cells (Figure 4B). This cytotoxic effect was not due to non-specific siRNA knockdown or toxicity of the vehicle because SLNPs carrying negative control siRNA (siNC@SLNPs) did not induce cancer cell death.
In vitro efficacy and gene silencing of siKSP@SLNPs in human SK-OV-3, AGS, and U87MG cancer cell lines. Cells were treated with siKSP(100 nM)@SLNPs for 4 h and assayed after 48 h. (A) KSP mRNA levels analyzed by qRT-PCR 48 h after transfection. KSP mRNA expression was notably knocked down in siKSP@SLNPs-treated cells. Results shown are mean ± s.e. (n=6). ***p < 0.001. (B) Cell viability assays, showing that siKSP@SLNPs were significantly more cytotoxic than controls. Results shown are means ± s.e. (n=9). ***p < 0.001.
Intracellular uptake and biological function of siKSP@SLNPs
Uptake of siRNA@SLNPs by PC-3 cells was examined using TAMRA-labeled siKSP for tracking by fluorescence. At 4 h after transfection, an appreciably intense TAMRA signal was observed in these cells, mostly in the cytosol (Figure 5A), suggesting that siKSP@SLNPs had been successfully internalized. After 24 h, many of these cells became circularized, with abnormally fragmented nuclei, indicating massive cell death owing to the knockdown of the target KSP (Figure 5A). This finding was consistent with previous reports,[31] showing that knockdown of KSP mRNA leads to a monoastral spindle arrangement, culminating in cell death. Although the mechanism responsible for the endosomal escape of siKSP@SLNPs remains to be elucidated, biologically active siKSP is released into the cytosol, silencing KSP mRNA. In contrast, cells treated with free naked siKSP did not show any fluorescence or morphologic changes and did not die. Immunostaining with antibody to α-tubulin confirmed the change in morphology and monoastral phenotype of cells treated with siKSP@SLNPs, as shown by cytoskeletal alterations; by contrast, control cells showed a bipolar spindle phenotype or cytoplasmic localization of α-tubulin (Figure 5B). Similar findings were observed in human U87MG glioma cells (Supplementary Information: Figure S6). This result indicates that SLNPs-mediated delivery of siKSP to the cytosol silenced the target KSP, resulting in cancer cell death.
In vitro uptake by PC-3 cells of TAMRA-labeled siKSP@SLNPs and induction of a monoastral spindle arrangement. (A) Cells were treated with TAMRA-siKSP(100 nM)@SLNPs for 4 h and monitored for 24 h by confocal microscopy. After 24 h, the cells showed a circularized morphology characteristic of dead cells, as well as abnormally fragmented nuclei, indicating that siRNA had been released from the endosome or lysosome within 24 h. (B) Immunostaining of cells with antibody to α-tubulin 48 h after transfection with KSP siRNA@SLNPs, showing a monoastral spindle arrangement.
In vivo biodistribution and antitumor effect of siKSP@SLNPs
The therapeutic efficacy of siKSP@SLNPs was investigated in mice bearing human prostate tumors. To assess whether siKSP@SLNPs could extravasate and localize within the tumor using the enhanced permeability and retention (EPR) effect, free Cy5.5-siKSP and Cy5.5-siKSP@SLNPs were intravenously injected into nude mice bearing prostate tumors (n = 3 per group), followed by live animal fluorescence imaging 24 h later. Although little fluorescence intensity was observed in the tumor area in mice injected with free Cy5.5-siKSP, strong fluorescence was evident in the tumors of mice injected with Cy5.5-siKSP@SLNPs (Figure 6A). To further determine the biodistribution of Cy5.5-siKSP@SLNPs, several major organs and tumor tissue were dissected and assessed by optical fluorescence imaging. The tumors of all three mice injected with Cy5.5-siKSP@SLNPs exhibited high fluorescence intensity, with appreciable but lower intensity observed in their livers (Figure 6B). In contrast, the signal from mice injected with free Cy5.5-siKSP was present mostly in the kidneys, with little at the tumor site. These findings indicate that SLNPs may be able to preferentially accumulate in the tumor site due to an EPR effect associated with their small size (< ~50 nm), neutral surface charge, and PEGylated corona.
Biodistribution and in vivo antitumor activity of siKSP@SLNPs. (A) In vivo fluorescence imaging of PC-3 tumor-bearing nude mice after a single intravenous injection of 30 µg of Cy5.5-labeled free siRNA or an equivalent amount of Cy5.5-siRNA@SLNPs. (B) Ex vivo fluorescence imaging of the tumor and major vital organs (liver, heart, kidney, spleen, and lung) harvested from euthanized mice 24 h after injection. (C) Antitumor activity of low- and high-dose siKSP@SLNPs (0.5 and 1 mg/kg, respectively) in PC-3 tumor-bearing nude mice (n=5 mice/ group). *p < 0.05, **p < 0.01. (D) TUNEL assays to detect apoptotic cells in tumor sections. Tumor tissues were collected 24 h after final administration. (E) KSP mRNA levels in tumor tissues were evaluated by qRT-PCR and normalized relative to GAPDH mRNA levels in the same samples (*p < 0.05, ***p < 0.001). (F) KSP protein expression in tumor tissues was analyzed by western blotting.
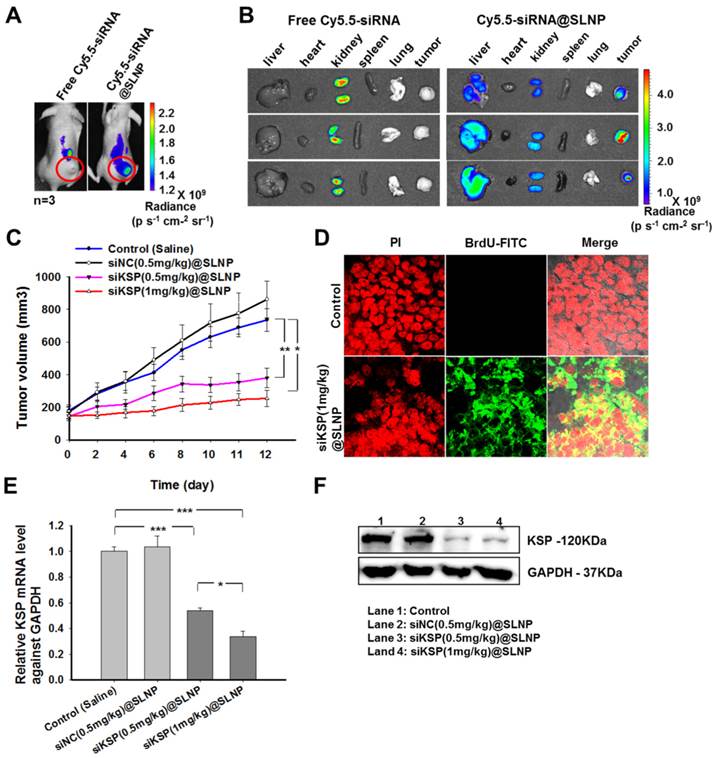
The antitumor effects of siKSP@SLNPs were subsequently analyzed in the same tumor model. When the prostate tumors in nude mice reached approximately 150-170 mm3 in size, the mice were randomly divided into four groups of five mice each and injected intravenously into the tail vein with (1) saline, (2) siNC(0.5 mg/kg)@SLNPs (negative control siRNA), (3) siKSP(0.5 mg/kg)@SLNPs (low dose siKSP), and (4) siKSP(1 mg/kg)@SLNPs (high dose siKSP) every other day for a total of six injections. Tumor volumes and body weights were monitored throughout the treatment. Tumor growth inhibition was greater in mice injected with siKSP@SLNPs than in mice injected with saline or siNC@SLNPs (Figure 6C). High dose siKSP@SLNPs showed the highest efficacy, inhibiting tumor growth ~81% relative to the saline control, although low dose siKSP@SLNPs also showed substantial efficacy, inhibiting tumor growth by ~58%. The therapeutic efficacy of siKSP@SLNPs was also shown by weighing the tumors (Supplementary Information: Figure S7A). In addition, body weights in all four groups did not change significantly during the treatment period (Supplementary Information: Figure S7B). TUNEL (terminal deoxynucleotidyl transferase dUTP nick-end labeling) assays also showed a high level of tumor cell apoptosis in mice treated with siKSP@SLNPs (Figure 6D).
To investigate whether the regression of tumor growth was caused by the specific silencing effect of siKSP, tumor expression of KSP mRNA and protein was assessed by qRT-PCR and western blotting, respectively. The levels of KSP mRNA in the tumors of mice treated with low and high-dose siKSP@SLNPs were 53% and 33%, respectively, compared with untreated mice, and much lower than tumor expression in mice treated with saline and siNC (Figure 6E). Similarly, tumor expression of KSP protein was significantly lower when mice were treated with siKSP@SLNPs (Figure 6F).
Hemolysis study was performed to verify the potential toxicity of siRNA@SLNPs after intravenous injection in vivo. SLNPs at a dose equivalent to the single injection used for the anticancer therapy experiment did not induce any hemolysis (Supplementary Information: Figure S8). Although DC-Chol/DOPE-based lipid nanoparticles also did not exhibit hemolytic effect at the same dose as SLNPs, its high toxicity on cells as confirmed in Figure 3 may limit the potential use in vivo. Taken together, these results indicate that MA-Chol/DOPE-based SLNPs are suitable nanocarriers for systemic delivery of siRNA in the treatment of cancers, showing preferential accumulation and effective silencing of a target gene within tumors.
Conclusions
Efforts to develop a new nanocarrier for RNAi-based cancer therapy led to our development of nontoxic SLNPs, which resulted in the systemic delivery of siRNA and significantly inhibited tumor growth in vivo. A new cationic cholesterol derivative, MA-Chol, was able to form complexes with siRNA with high efficiency, resulting in the formation of SLNPs with a neutral surface charge. Furthermore, as MA-Chol, unlike DOTAP and DC-Chol, was designed to generate two nontoxic endogenous compounds, arginine and cholesterol, upon degradation in vivo, the resulting SLNPs showed little cytotoxicity, both in vitro and in vivo, even at relatively high concentrations. The siRNA-loaded SLNPs (siKSP@SLNPs) showed a high level of target gene silencing in several cancer cell lines. Furthermore, systemically injected siKSP@SLNPs showed preferential accumulation at the tumor site and dose-dependently inhibited tumor growth in a prostate cancer model. Taken together, these findings indicate that these cationic cholesterol derivative-based SLNPs possess many of the attributes required of a delivery platform for RNAi-based cancer therapy, including small size, lack of toxicity, neutral surface charge, and highly efficient complex formation. This SLNP platform may therefore be applicable to RNAi-based treatment of cancers and other hard-to-cure diseases.
Supplementary Material
Figures S1- S8, Table S1.
Acknowledgements
This work was supported by Global Research Laboratory (no. 2014044002) through the National Research Foundation of Korea (NRF), Intelligent Synthetic Biology Center of the Global Frontier Project (no. 2013M3A6A8073557), and KAIST Future Systems Healthcare Project funded by the Ministry of Science, ICT (Information and Communication Technologies) and Future Planning.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Whitehead KA, Langer R, Anderson DG. Knocking down barriers: advances in siRNA delivery. Nat Rev Drug Discov. 2009;8:129-38
2. Pecot CV, Calin GA, Coleman RL, Lopez-Berestein G, Sood AK. RNA interference in the clinic: challenges and future directions. Nature reviews Cancer. 2011;11:59-67
3. Dykxhoorn DM, Lieberman J. Knocking down disease with siRNAs. Cell. 2006;126:231-5
4. Kurreck J. RNA Interference: From Basic Research to Therapeutic Applications. Angew Chem Int Edit. 2009;48:1378-98
5. Draz MS, Fang BA, Zhang PF, Hu Z, Gu SD, Weng KC. et al. Nanoparticle-Mediated Systemic Delivery of siRNA for Treatment of Cancers and Viral Infections. Theranostics. 2014;4:872-92
6. Ozcan G, Ozpolat B, Coleman RL, Sood AK, Lopez-Berestein G. Preclinical and clinical development of siRNA-based therapeutics. Advanced drug delivery reviews. 2015
7. Zhu X, Xu YJ, Solis LM, Tao W, Wang LZ, Behrens C. et al. Long-circulating siRNA nanoparticles for validating Prohibitin1-targeted non-small cell lung cancer treatment. P Natl Acad Sci USA. 2015;112:7779-84
8. Simoes S, Filipe A, Faneca H, Mano M, Penacho N, Duzgunes N. et al. Cationic liposomes for gene delivery. Expert opinion on drug delivery. 2005;2:237-54
9. Han SE, Kang H, Shim GY, Suh MS, Kim SJ, Kim JS. et al. Novel cationic cholesterol derivative-based liposomes for serum-enhanced delivery of siRNA. International journal of pharmaceutics. 2008;353:260-9
10. Lu JJ, Langer R, Chen JZ. A Novel Mechanism Is Involved in Cationic Lipid-Mediated Functional siRNA Delivery. Molecular pharmaceutics. 2009;6:763-71
11. Hong CA, Nam YS. Functional Nanostructures for Effective Delivery of Small Interfering RNA Therapeutics. Theranostics. 2014;4:1211-32
12. Morrissey DV, Lockridge JA, Shaw L, Blanchard K, Jensen K, Breen W. et al. Potent and persistent in vivo anti-HBV activity of chemically modified siRNAs. Nature biotechnology. 2005;23:1002-7
13. Zimmermann TS, Lee ACH, Akinc A, Bramlage B, Bumcrot D, Fedoruk MN. et al. RNAi-mediated gene silencing in non-human primates. Nature. 2006;441:111-4
14. Li Y, Cheng Q, Jiang Q, Huang Y, Liu H, Zhao Y. et al. Enhanced endosomal/lysosomal escape by distearoyl phosphoethanolamine-polycarboxybetaine lipid for systemic delivery of siRNA. Journal of controlled release: official journal of the Controlled Release Society. 2014;176:104-14
15. Wolff JA, Rozema DB. Breaking the bonds: Non-viral vectors become chemically dynamic. Mol Ther. 2008;16:8-15
16. Schroeder A, Levins CG, Cortez C, Langer R, Anderson DG. Lipid-based nanotherapeutics for siRNA delivery. Journal of internal medicine. 2010;267:9-21
17. Sun TM, Zhang YS, Pang B, Hyun DC, Yang MX, Xia YN. Engineered Nanoparticles for Drug Delivery in Cancer Therapy. Angew Chem Int Edit. 2014;53:12320-64
18. Cui MJ, Au JLS, Wientjes MG, O'Donnell MA, Loughlin KR, Lu Z. Intravenous siRNA Silencing of Survivin Enhances Activity of Mitomycin C in Human Bladder RT4 Xenografts. J Urology. 2015;194:230-7
19. Wang J, Lu Z, Wang J, Cui M, Yeung BZ, Cole DJ. et al. Paclitaxel tumor priming promotes delivery and transfection of intravenous lipid-siRNA in pancreatic tumors. Journal of controlled release: official journal of the Controlled Release Society. 2015;216:103-10
20. Stylianopoulos T, Poh MZ, Insin N, Bawendi MG, Fukumura D, Munn LL. et al. Diffusion of Particles in the Extracellular Matrix: The Effect of Repulsive Electrostatic Interactions. Biophys J. 2010;99:1342-9
21. Cabral H, Matsumoto Y, Mizuno K, Chen Q, Murakami M, Kimura M. et al. Accumulation of sub-100 nm polymeric micelles in poorly permeable tumours depends on size. Nat Nanotechnol. 2011;6:815-23
22. Lin Q, Chen J, Zhang Z, Zheng G. Lipid-based nanoparticles in the systemic delivery of siRNA. Nanomedicine (Lond). 2014;9:105-20
23. Ozpolat B, Sood AK, Lopez-Berestein G. Liposomal siRNA nanocarriers for cancer therapy. Advanced drug delivery reviews. 2014;66:110-6
24. Dokka S, Toledo D, Shi XG, Castranova V, Rojanasakul Y. Oxygen radical-mediated pulmonary toxicity induced by some cationic liposomes. Pharmaceutical research. 2000;17:521-5
25. Lv HT, Zhang SB, Wang B, Cui SH, Yan J. Toxicity of cationic lipids and cationic polymers in gene delivery. Journal of Controlled Release. 2006;114:100-9
26. Spagnou S, Miller AD, Keller M. Lipidic carriers of siRNA: Differences in the formulation, cellular uptake, and delivery with plasmid DNA. Biochemistry. 2004;43:13348-56
27. Gjetting T, Arildsen NS, Christensen CL, Poulsen TT, Roth JA, Handlos VN. et al. In vitro and in vivo effects of polyethylene glycol (PEG)-modified lipid in DOTAP/cholesterol-mediated gene transfection. International journal of nanomedicine. 2010;5:371-83
28. Ozpolat B, Sood AK, Lopez-Berestein G. Nanomedicine based approaches for the delivery of siRNA in cancer. Journal of internal medicine. 2010;267:44-53
29. Tao W, South VJ, Zhang Y, Davide JP, Farrell L, Kohl NE. et al. Induction of apoptosis by an inhibitor of the mitotic kinesin KSP requires both activation of the spindle assembly checkpoint and mitotic slippage. Cancer cell. 2005;8:49-59
30. Judge AD, Robbins M, Tavakoli I, Levi J, Hu L, Fronda A. et al. Confirming the RNAi-mediated mechanism of action of siRNA-based cancer therapeutics in mice. The Journal of clinical investigation. 2009;119:661-73
31. Gujrati V, Kim S, Kim SH, Min JJ, Choy HE, Kim SC. et al. Bioengineered bacterial outer membrane vesicles as cell-specific drug-delivery vehicles for cancer therapy. Acs Nano. 2014;8:1525-37
32. Saw PE, Kim S, Lee I-h, Park J, Yu M, Lee J. et al. Aptide-conjugated liposome targeting tumor-associated fibronectin for glioma therapy. Journal of Materials Chemistry B. 2013;1:4723
33. Letsch M, Schally AV, Busto R, Bajo AM, Varga JL. Growth hormone-releasing hormone (GHRH) antagonists inhibit the proliferation of androgen-dependent and -independent prostate cancers. P Natl Acad Sci USA. 2003;100:1250-5
34. Bennett MJ, Nantz MH, Balasubramaniam RP, Gruenert DC, Malone RW. Cholesterol Enhances Cationic Liposome-Mediated DNA Transfection of Human Respiratory Epithelial-Cells. Bioscience Rep. 1995;15:47-53
35. Wasungu L, Hoekstra D. Cationic lipids, lipoplexes and intracellular delivery of genes. Journal of Controlled Release. 2006;116:255-64
36. Nie Y, Ji L, Ding H, Xie L, Li L, He B. et al. Cholesterol derivatives based charged liposomes for doxorubicin delivery: preparation, in vitro and in vivo characterization. Theranostics. 2012;2:1092-103
37. Dar GH, Gopal V, Rao NM. Systemic Delivery of Stable siRNA-Encapsulating Lipid Vesicles: Optimization, Biodistribution, and Tumor Suppression. Molecular pharmaceutics. 2015;12:610-20
38. Choi JS, Lee EJ, Jang HS, Park JS. New cationic liposomes for gene transfer into mammalian cells with high efficiency and low toxicity. Bioconjugate Chem. 2001;12:108-13
39. Choi WJ, Kim JK, Choi SH, Park JS, Ahn WS, Kim CK. Low toxicity of cationic lipid-based emulsion for gene transfer. Biomaterials. 2004;25:5893-903
40. Zhang Y, Li HM, Sun J, Gao J, Liu W, Li BH. et al. DC-Chol/DOPE cationic liposomes: A comparative study of the influence factors on plasmid pDNA and siRNA gene delivery. International journal of pharmaceutics. 2010;390:198-207
41. Tseng YC, Mozumdar S, Huang L. Lipid-based systemic delivery of siRNA. Advanced drug delivery reviews. 2009;61:721-31
42. Sakai N, Matile S. Anion-mediated transfer of polyarginine across liquid and bilayer membranes. Journal of the American Chemical Society. 2003;125:14348-56
43. Du ZX, Munye MM, Tagalakis AD, Manunta MDI, Hart SL. The Role of the Helper Lipid on the DNA Transfection Efficiency of Lipopolyplex Formulations. Scientific reports. 2014:4
44. Saw PE, Ko YT, Jon S. Efficient Liposomal Nanocarrier-mediated Oligodeoxynucleotide Delivery Involving Dual Use of a Cell-Penetrating Peptide as a Packaging and Intracellular Delivery Agent. Macromol Rapid Comm. 2010;31:1155-62
45. Hak S, Helgesen E, Hektoen HH, Huuse EM, Jarzyna PA, Mulder WJ. et al. The effect of nanoparticle polyethylene glycol surface density on ligand-directed tumor targeting studied in vivo by dual modality imaging. Acs Nano. 2012;6:5648-58
46. Saw PE, Park J, Lee E, Ahn S, Lee J, Kim H. et al. Effect of PEG pairing on the efficiency of cancer-targeting liposomes. Theranostics. 2015;5:746-54
47. Jang JY, Choi Y, Jeon YK, Aung KCY, Kim CW. Over-expression of adenine nucleotide translocase 1 (ANT1) induces apoptosis and tumor regression in vivo. Bmc Cancer. 2008:8
Author contact
![]() Corresponding author: syjonac.kr
Corresponding author: syjonac.kr