Theranostics
13.3
Impact Factor
- Current Issue
- Advance Articles
- Volume 16; 2026
- Volume 15; 2025
- Volume 14; 2024
- Volume 13; 2023
- Volume 12; 2022
- Archive
- Cover Images
- Cover Suggestion
- Special Issues
Top
Introduction
Materials and Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References
Introduction
Materials and Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2016; 6(3):357-368. doi:10.7150/thno.13621 This issue Cite
Research Paper
Image-aided Suicide Gene Therapy Utilizing Multifunctional hTERT-targeting Adenovirus for Clinical Translation in Hepatocellular Carcinoma
Yun-Hee Kim 1,2, Kyung Tae Kim 1, Sang-Jin Lee 1,2, Seung-Hee Hong 3, Ju Young Moon 1, Eun Kyung Yoon1,4, Sukyoung Kim1, Eun Ok Kim1, Se Hun Kang 1, Seok Ki Kim 1, Sun Il Choi1,4, Sung Ho Goh 1, Daehong Kim1, Seong-Wook Lee 5, Mi Ha Ju6, Jin Sook Jeong 6 ![]() , In-Hoo Kim 1,2
, In-Hoo Kim 1,2 ![]()
1. Research Institute and hospital, National Cancer Center, Goyang 410-769, Republic of Korea
2. Graduate School of Cancer Science & Policy, National Cancer Center, Goyang 410-769, Republic of Korea
3. Division of Food Science and Culinary Arts, Food and Nutrition Major, Shinhan University, Uijeonbu 480-857, Republic of Korea
4. Department of Life Science, Ewha Womans University, Seoul 120-750, Republic of Korea
5. Department of Molecular Biology, Institute of Nanosensor and Biotechnology, Dankook University, Youngin 448-701, Republic of Korea
6. Department of Pathology, Dong-A University College of Medicine, Busan 602-714, Republic of Korea
Received 2015-8-21; Accepted 2015-11-21; Published 2016-1-6
Citation:
Kim YH, Kim KT, Lee SJ, Hong SH, Moon JY, Yoon EK, Kim S, Kim EO, Kang SH, Kim SK, Choi SI, Goh SH, Kim D, Lee SW, Ju MH, Jeong JS, Kim IH. Image-aided Suicide Gene Therapy Utilizing Multifunctional hTERT-targeting Adenovirus for Clinical Translation in Hepatocellular Carcinoma. Theranostics 2016; 6(3):357-368. doi:10.7150/thno.13621. https://www.thno.org/v06p0357.htm
Other stylesAbstract

Trans-splicing ribozyme enables to sense and reprogram target RNA into therapeutic transgene and thereby becomes a good sensing device for detection of cancer cells, judging from transgene expression. Previously we proposed PEPCK-Rz-HSVtk (PRT), hTERT targeting trans-splicing ribozyme (Rz) driven by liver-specific promoter phosphoenolpyruvate carboxykinase (PEPCK) with downstream suicide gene, herpes simplex virus thymidine kinase (HSVtk) for hepatocellular carcinoma (HCC) gene therapy. Here, we describe success of a re-engineered adenoviral vector harboring PRT in obtaining greater antitumor activity with less off-target effect for clinical application as a theranostics. We introduced liver-selective apolipoprotein E (ApoE) enhancer to the distal region of PRT unit to augment activity and liver selectivity of PEPCK promoter, and achieved better transduction into liver cancer cells by replacement of serotype 35 fiber knob on additional E4orf1-4 deletion of E1&E3-deleted serotype 5 back bone. We demonstrated that our refined adenovirus harboring PEPCK/ApoE-Rz-HSVtk (Ad-PRT-E) achieved great anti-tumor efficacy and improved ability to specifically target HCC without damaging normal hepatocytes. We also showed noninvasive imaging modalities were successfully employed to monitor both how well a therapeutic gene (HSVtk) was expressed inside tumor and how effectively a gene therapy took an action in terms of tumor growth. Collectively, this study suggests that the advanced therapeutic adenoviruses Ad-PRT-E and its image-aided evaluation system may lead to the powerful strategy for successful clinical translation and the development of clinical protocols for HCC therapy.
Keywords: trans-splicing ribozyme, in vivo imaging, cancer gene therapy, hepatocellular carcinoma
Introduction
Hepatocellular carcinoma (HCC) represents a major histological subtype (70-85 %) of liver cancer and globally ranks as the fifth most common cancer in men, seventh in women, and the third most common cause of death among all cancers worldwide [1]. Despite major progress in the understanding of the disease and therapeutic options in the past 2 decades, HCC responds poorly to conventional therapies and frequently relapses shortly after surgical and nonsurgical treatment [2,3]. Moreover, the presence of multiple tumors in the liver, i.e., multifocal hepatocellular carcinoma, is a strong risk factor for patient mortality and has no alternative treatment due to its potential for invasion and intrahepatic metastasis [4]. Therefore, there remains an urgent need for new approaches to treat this dreaded disease.
Transcriptionally targeted cancer gene therapy using cancer- or tissue-specific promoters provides one such approach. This approach exploits the fact that cancer cells tend to activate or overexpress many genes that are important for uncontrolled proliferation and cell survival [5]. However, the successful clinical translation of experimental gene therapies has been hampered by their poor transcriptional efficiency and specificity [6,7]. To overcome these drawbacks of the poor transcriptional control of therapeutic genes, our group previously developed adenoviral vectors harboring the Tetrahymena Group I intron-based trans-splicing ribozymes (Rz) specifically targeting and effectively converting human telomerase reverse transcriptase (hTERT) RNA into herpes simplex virus thymidine kinase (HSVtk) in hTERT-abundant cancer cells, showing the therapeutic efficacy on the basis of HSVtk/GCV system [8,9]. This Rz achieved enhanced tumor selectivity with nearly equal efficacy relative to the standard cytomegalovirus (CMV) promoter regardless of the tumor origin [9,10]. Furthermore, the ribozyme-based hTERT-targeting strategy is an excellent candidate for investigating HCC therapeutic options because telomerase activity, which is negligible in normal liver tissues, becomes detectable in 89.5% of HCC cells [11]. However, this approach has limitation in clinical setting because telomerase is expressed in regenerative liver lesions, hematopoietic stem cells and germ cells in addition to HCC[12,13]. This lack of specificity led to the design of a liver-specific Rz promoted by phosphoenolpyruvate carboxykinase (PEPCK) promoter to control HSVtk. This approach, when tested in a mouse model for intraperitoneal carcinomatosis using HCC cells demonstrated high anti-tumor activity without affecting normal liver tissues [14].
Thus, the RNA replacement approach for targeting liver cancer cells in combination with a liver-selective promoter appears to have a high potential for clinical application in HCC gene therapy. However, further evaluation in human HCC mimicking model and refinement of vector system for successful clinical setting is required. In addition, it became clear that inclusion of real-time monitoring of in vivo gene delivery by in vivo imaging is needed as part of preclinical protocols leading to fully fledged clinical trials.
We describe here re-engineering of the previously developed adenovirus gene therapy vector containing PEPCK promoter-driven ribozyme with: 1) the addition of an apolipoprotein E (ApoE) enhancer to the distal region of the PEPCK promoter for enhanced transcriptional activity without specificity loss; 2) creation of a serotype 5-based shaft with a serotype 35 knob to circumvent poor infectivity due to an inefficient coxsackievirus and adenovirus receptor (CAR)-dependent endocytosis pathway; and 3) deletion of E1, E3, and E4 regions of the adenovirus backbone to accommodate larger exogenous therapeutic genes. Then, we conducted evaluation whether the new therapeutic vector administered intravenously could inhibit tumor growth without off-target effect in a mouse model of HCC with concurrent monitoring, employing MRI, PET/CT, and other multimodal imaging tools throughout the treatment period, of the levels of gene transfer and therapeutic effects.
Materials and Methods
Cell studies
Human cancer cells and the human dermal fibroblast (HDF) used in this study were purchased from American Type Culture Collection. Hep3B cells and the HDFs were maintained in MEM medium supplemented with 10% FBS (Invitrogen, Carlsbad, CA) and 1% penicillin/streptomycin (Invitrogen). All other cell lines were maintained in RPMI1640 medium containing 10% FBS and 1% penicillin/streptomycin. Cell survival was determined using the crystal violet assay.
Plasmid and adenoviral vector construction
Adenovirus Ad5/35PEPCK-Rz-HSVtk controlled by hTERT-specific trans-splicing ribozyme under PEPCK promoter was constructed as previously described with multiple structural modifications [14]. The hTERT pre-mRNA-targeting Rib21AS trans-splicing ribozyme targeting was designed to have the extended internal guided sequence that included an extension of the P1 helix, an additional 6-nt-long P10 helix and a 325-nt-long antisense sequence. The Rib21AS trans-splicing ribozyme was cloned into pCDNA3.1.PEPCK.HSVtk vector to make pCDNA3.1.PEPCK.Rz.HSVtk. The resultant pCDNA3.1.PEPCK.Rz.HSVtk vector was ligated with SmaI/NotI fragment containing ApoE enhancer DNA fragment from (pPEPCK-LCR (kind gift from Dr. Oka in Baylor College of Medicine). Next, Ad-PRT-E (Ad5/35PEPCK.Rz.HSVtk.Enh) was generated according to the protocol established by OD260 (Boise, ID). In brief, the SpeI/EcoRV fragment of pPEPCK.Rz.HSVtk.Enh was cloned into the XbaI/EcoRV sites of pZAP1.1 (OD260), becoming pZAP.PEPCK.Rz.HSVtk.Enh. The shuttle vector pZAP.PEPCK.Rz.HSVtk.Enh was digested with PflMI and ligated with RightZAP1.4. RightZap1.4 was made 1) by cloning into RightZap1.2 the Ad5/35 fiber from NdeI/AflII fragment containing serotype 5 shaft /serotype 35 fiber knob of Ad5EGFP/F35 (a gift from Dr. Andre Lieber, University of Washington, Seattle, WA) and 2) deleting E4orf(open-reading-frame)1-4 adjacent to the right-end of RightZap1.2 vector. Adenovirus Ad-PRT-E was generated in HEK293 cells following procedures instructed from OD260 (Boise, ID).
Cell survival assay
The cell survival was determined by crystal violet assay. After transfection of plasmids or infection of adenovirus, cells were treated with gancyclovir (GCV) for 5 days and then stained with 1% crystal violet, and the survival cells were determined by OD405.
PCR analysis
The production of TSMs by Ad-PRT was investigated using nested RT-PCR. The first PCR was performed using a 5' primer specific for the junction area of the trans-splicing reaction (5'-GGG GAA TTC AGC GCT GCG TCC TGC T-3') and a 3' primer for HSVtk (5'-GTT ATC TGG GCG CTT GTC AA-3'). The second PCR was performed using primers for an internal sequence of HSVtk (forward 5'-CGT CCT GCT AAA GTT GGC CGC-3' and reverse 5'-GCA GTT GCG TGG TGG TGG TT-3') [15]. A detailed description of the PCR assays is provided in the “Supplementary Experimental Procedures” section. The endogenous hTERT concentration in Hep3B cells was quantified by quantitative PCR (qPCR) using SYBR Tag (TaKaRa BIO INC, Japan) and ABI PRISM 7900HT (Applied biosystems, Foster City, CA). The sequence of primers used for the hTERT amplification in Ad-CRT-infected cells were 5'-GGGAAGAGTGCTGGACAA-3' and 5'-GGATGAAGCGGAGTCTGGA-3' and performed under conditions as 94 °C for 30 s, 55 °C for 40 s and 72 °C for 1 min.
FISH/ Immunofluorescence method
Hep3B cells were cultured with 30 MOI of Mock, Ad-CT, or Ad-CRT adenoviruses on a coverslip for 4 days. Cells on the coverslip were incubated with specific primary antibody, mouse monoclonal anti-TRF2 (IMGENEX, San Diego, CA, USA) and rabbit polyclonal anti-53BP1, anti-γ-H2AX (Bethyl Laboratories, Inc., Montgomery, AL, USA) after permeabilization and blocking, washed, and secondary antibody added. For the telomere ends FISH, cells were fixed again, dehydrated, and incubated with denatured telomere probe (Cy3-oo-(TTAGGG)3 PNA, Panagene, Daejeon, Korea) in 70% formamide in 2× SSC, 0.25% nucleotide blocking reagent, 5% MgCl2 at 90°C, washing, and 4'-6-diamidino-2-phenylindole (DAPI) staining.
Generation of the intrahepatic multifocal HCC mouse model and animal studies
A multifocal mouse liver cancer model, which mimics human HCC, was established by the splenic subcapsular inoculation of 3 x 106 Hep3B cells into Balb/c-nude mice (Orient Bio Inc., Korea) as previously described [16]. This study was reviewed and approved by the Institutional Animal Care and Use Committee (IACUC) of National Cancer Center Research Institute. NCCRI is an Association for Assessment and Accreditation of Laboratory Animal Care International (AAALAC International) accredited facility and abide by the Institute of Laboratory Animal Resources (ILAR) guide. (accredited unit—National Cancer Center Research Institute: unit number-001392). Nearly all of the mice demonstrated multiple tiny tumor nodules in their livers, particularly along the margin, at 12 or 14 days after Hep3B cell injection, which were detected by PET-CT scan (fig. S3). Treatment in HCC animal model were performed in accordance with an experimental proposal (No. NCC-10-036D and NCC-12-163). Briefly, 14 days after the splenic subcapsular inoculation of the Hep3B cells, the mice were administered with 5 x108 pfu of Ad-PRT-E, Ad-PT-E or a control (Mock) in 100 μl of PBS via tail vein. After one day, the mice were given intra-peritoneal injections of 50 mg/kg ganciclovir (GCV) twice a day for 14 days. Histological studies and in vivo imaging experiments were performed at each indicated time point. TUNEL assays were performed using the in situ Cell Detection Kit (Roche, Germany) according to the manufacturer's protocol. Paraffin-embedded liver samples were treated with primary antibodies (anti-AKT; 1:200, anti-VEGF-C; 1:50, anti-CD34; 1:200, AB Biotech), and stained with 3,3'-diaminobenzidine substrate system. To measure the anti-HCC efficacy, the animals were euthanized 14 days after the first GCV treatment, whole liver lobes were removed, measured, and serially sectioned and stained with hematoxylin and eosin. The tumor fractions were calculated using the Aperio Imagescope v10.2.2.2319 software, and the tumor weights were estimated by multiplying the liver weights by the tumor fractions.
Monitoring of transgene expression and tumor growth by noninvasive in vivo imaging
For image scanning, animals were fasted for at least 6 h and anesthetized with 2% isoflurane in 100% oxygen. Tumor growth or HSVtk expression in the animals were monitored by PET imaging using 14.8 MBq of [18F]fluorodeoxyglucose ([18F]FDG) or 9-(4-[18F]fluoro-3-hydroxymethylbutyl) guanine ([18F]FHBG) administered through tail vein during anesthesia, respectively. PET-CT fusion imaging was performed in a three-dimensional acquisition mode (eXplore VistaCT, GE, Fairfield, CT) under x-ray conditions for CT (i.e., 300 μA and 40 kV for 6 min; resolution = 200 µm; acquired projection number =360). The images were normalized to standardized uptake values (SUV) using the following formula: SUV = decay corrected mean tissue activity concentration (in Bq/ml)/ (injected dose (in Bq) x body weight (in g)).
All MRI images were acquired using a 7T Biospec spectrometer (Bruker, Germany). A T2-weighted fast spin echo pulse sequence was recorded with the following settings: repetition time (TR) = 2,500 ms; echo time (TE) = 30 ms; 256*256 matrix; field of view (FOV) = 3*3 cm; slice thickness = 0.7 mm; number of average = 2; RARE factor = 4. For the PET/MR fusion experiment, MRI imaging was performed following the PET-CT scan using a common bed. PET-MR image fusion processing was performed using OsiriX imaging software.
Statistical analysis
All data are presented as the mean and standard error of the mean (SEM). A nonparametric statistical t test was used to compare between two groups. A value of P < 0.05 was considered to be statistically significant. Kaplan-Meier analysis was used to determine the survival rate.
Results
HCC-specific cytotoxicity of the newly engineered adenoviral vector Ad-PRT-E
We constructed chimeric Ad5/35 adenoviral vector encoding suicide HSVtk gene driven by hTERT-targeting trans-splicing ribozyme under the control of liver-specific PEPCK promoter with an ApoE enhancer placed in the distal region of the HSVtk expression cassette (Ad5/35-PRT-E, Ad-PRT-E, Fig. 1A; top). Moreover, we generated adenovirus by deleting Rz from Ad-PRT-E as a control for the analysis of target RNA specificity of the Rz (Ad-PT-E, Fig. 1A; middle). We also used empty serotype 5 adenoviruses as a negative control (Mock, Fig. 1A; bottom). We produced chimeric Ad5/35 vector by replacing the serotype 5 fiber knob with that of the serotype 35 to increase the transfection efficacy of adenoviral vector (fig. S1), and deleting E4 region to accommodate a larger exogenous HSVtk expression cassette than conventional Ad5 vector does (Fig. 1A). Prior to insertion of therapeutic cassette into Ad5/35, we checked whether ApoE enhancer increases transcriptional activity of HSVtk, using pPT-E plasmid containing PEPCK-HSVtk-Enhancer construct against Enhancer-deleted pPT (Fig. 1B). The pPT-E showed a 30-fold higher HSVtk expression than unenhanced pPT did in PEPCK-positive Hep3B hepatoma cells (Fig. 1C). However, the transcriptional activities of them were exceedingly week in PEPCK-negative SK-OV3 ovarian cancer cells. These results indicate that ApoE enhancer successfully increased transcriptional activity of PEPCK promoter in a liver-specific manner.
Following the vector construction, we examined whether the Ad-PRT-E mediated HSVtk could induce HCC-specific sensitivity to prodrug ganciclovir (GCV). Cells were treated with varying multiplicities of infection (MOI) of Ad-PRT-E, Ad-PT-E and Mock plus 100 μM of GCV, then subjected to crystal violet assays to determine the percentage of cells that survived at 5 days after the treatment. As expected, both Ad-PRT-E and Ad-PT-E caused significant dose-dependent cell death in Hep3B cells (Fig. 1D) but not in SK-OV3 (Fig. 1E), indicating that the ApoE-enhanced PECK promoter activity was specific to liver cells. Interestingly, we observed that Ad -PT-E is more effective than Ad-PRT-E with regard to the cytotoxicity in the Hep3B cells (Fig. 1D), since HSVtk expression of Ad -PRT-E is attenuated when it is mediated by the trans-spicing ribozyme [17]. To confirm that trans-splicing reaction by Rz is working, next we determined the production of trans-spliced molecules (TSMs) by RT-PCR. The Ad-PRT-E generated TSM adenoviral dose-dependently in hTERT+ Hep3B cells, and we also have proved that the trans-splicing reaction of Ad5/35-PRT-E is under the control of liver-specific promoter/enhancer complex, showing undetectable TSM in Ad-PRT-E transfected hTERT-positive non-liver SK-OV3 cells (Fig. 1F). Rz activity that inhibits hTERT transcription and replaces with thymidine kinase was controlled by PEPCK promoter, which agreed with the result of the reduced telomerase activity and hTERT expression only in Hep-3B cells from TRAP assay (fig. S2). We then determined and compared the cytotoxicity after treatment with 10 MOI of Ad-PRT-E plus GCV in a variety of cells including PEPCK+/hTERT+ (HepG2, SNU398, and SNU739), PEPCK-/hTERT+ (HCT116 and SBC-5) and PEPCK-/hTERT- (HDF; normal human fibroblast) cells. Noticeably, the Ad-PRT-E was cytotoxic solely to hTERT+ liver cells (HepG2, SNU398, and SNU739), whereas not in hTERT+ non-liver (lung carcinoma SBC-5 and colon carcinoma HCT116), nor hTERT- non-liver (HDF) cells (Fig. 1G). These results strongly suggest that Ad-PRT-E exerted HCC-specific anti-tumor activity via dual targeting mechanism, i.e., liver-specific by PEPCK promoter/ApoE enhancer complex and tumor-selective by Rz.
Fig 1
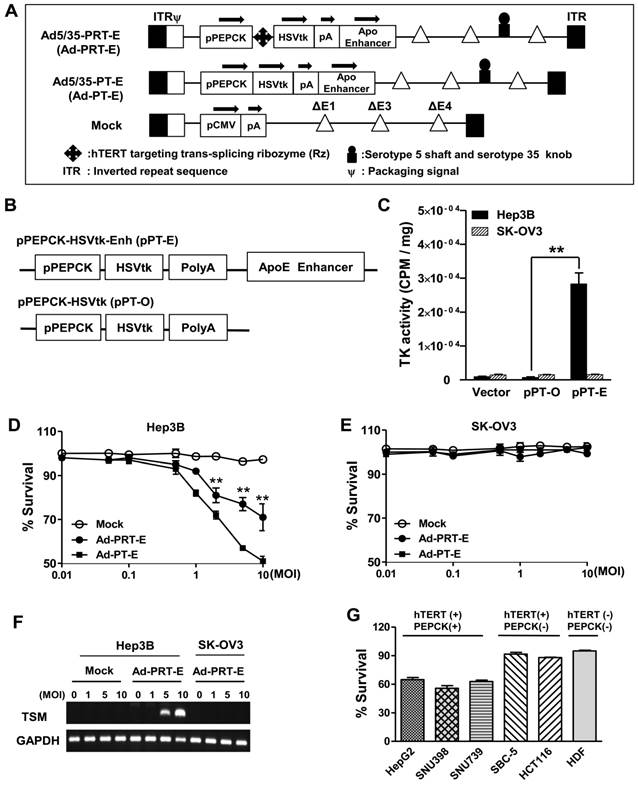
Construction of the engineered Ad-PRT-E and its HCC-specific cytotoxicity (A) Schematic diagram of the refined adenoviral vector with the Ad5/35 backbone and E4 deletion containing the PEPCK promoter and enhancer. Abbreviations: pPEPCK, promoter PEPCK; pA, poly A; Enh, ApoE enhancer. (B) Schematic diagram of HSVtk expressing a construct containing the PEPCK promoter and enhancer. (C) PEPCK-driven HSVtk activity in the absence or presence of enhancer in hepatoma cell line Hep3B and ovarian cancer cell line SK-OV3. To examine the activity of HSVtk using GCV uptake assay, 48 hrs after transfection with each plasmid in both cell lines, the cells were incubated at 37°C for 2 hours with [8-3H] PCV (1.0 μCi/ml), and the radioactivity was measured using cell pellets. Values are means as cpm/ cell protein (mg) ± SEM for the triplicate assays. (** p < 0.01). (D-E) Liver-specific cell death induced by the PEPCK promoter of Ad-PRT-E. Hep3B cells (D) and SK-OV3 cells (E) were infected with Mock, Ad-PT-E and Ad-PRT-E at various MOIs. After 5 days of daily GCV treatment, cell survival was determined using crystal violet assays. The error bars indicate the mean value ± SEM for the triplicate assays. (** p < 0.01) (F) RT-PCR of the trans-spliced product in the Ad-PRT-E-infected cells. TSM is represented by the amplification of the spliced RNA; human GAPDH RNA was amplified as an internal control. (G) The efficacy and HCC-specificity of Ad-PRT-E in various cell lines in the liver cancer, non-liver cancer and normal epithelial cells (HDF). Cells were infected with Mock and Ad-PRT-E at 10 MOI, and cell survival was determined using crystal violet assays 5 days after infection and GCV treatment. Error bars indicate the mean value relative to Ad-Mock ± SEM for the triplicate assays.
HCC-specific targeting ability of Ad5/35-PRT-E
To evaluate the tumor-specific targeting ability of Ad-PRT-E, we monitored the distribution of [18F]FHBG related to the HSVtk gene expression two days after injecting 5 x 108 pfu of each adenovirus (n=2/group) into non tumor-bearing mice via the tail vein. As expected, the [18F]FHBG uptake signal was present in the broad area of the liver, when injected with Ad-PT-E and Ad-CT. The SUV maximal uptake (1.2 ±0.3) of the Ad-PT-E-mediated HSVtk activity was the approximate half of that observed in the Ad-CT-injected mice (2.5 ± 0.4) (Fig. 2A-B). In contrast, the Ad-PRT-E-injected group exhibited no [18F]FHBG uptake signal in their livers despite the strong inborn tropism of the systemically injected adenoviral vectors to the liver (Fig. 2A-B). Ad-PRT-E appears to be inactive in the non-tumor cells because Rz curbed the PEPCK-driven transcription read-through in a tumor-specific manner. These results suggest that Ad-PRT-E is equipped with a highly efficient HCC-targeting device by utilizing dual controller, Rz and a tissue-specific promoter PEPCK.
Next, we examined whether HSVtk expression delivered by Ad-PRT-E was localized and expressed in tumors in a HCC-specific manner. We established a multifocal HCC mouse model and monitored tumor formation using in vivo imaging modalities (fig. S3), which included useful systems for assessing both the targeting efficiency and treatment efficacy of Ad-PRT-E. To confirm HCC-specific targeting of Ad-PRT-E, we examined the coincidence of the [18F]FHBG signal from PET, which reflected the thymidine kinase expression, and MRI signals at each tumor locus. As shown in Fig. 2C-E, [18F]FHBG was distributed in the tumor sites (white arrows) shown in the T2-weighted MRI. In addition, RT-PCR analysis with liver tissues separated into normal and tumor regions showed the expression of thymidine kinase, implying that Rz specifically targeted tumor sites and selectively converted hTERT RNA to HSVtk in the hTERT-abundant cancer region (Fig. 2F).
Contribution of Rz-mediated hTERT depletion to improve therapeutic efficacy of Ad-PRT-E
To find the additional mechanism that improve therapeutic efficacy of Ad-PRT-E based on the HSVtk/GCV system, we examined possible changes by Rz-mediated hTERT depletion after delivery of Ad-CRT for 96 hrs. The concentration of hTERT was decreased approximately 60 % by Ad-CRT treatment (Fig. 3A-B). We first analyzed telomeric ending with Cy3-fluorescence tagged-(TTAGGG)3 PNA and co-stained with telomeric repeat-binding factor 2 (TRF2), a shelterin family that bind to telomere double strand DNA. Double-staining telomere with TRF2 remained normal after infection with Mock or Ad-CT (Fig. 3C). While, after infection with Ad-CRT, there was faint telomere staining and overlapping TRF2 staining was weaker (Fig. 3C). Furthermore, many TRF2 staining spot did not co-localize with telomeric end from FISH-immunofluorescence staining. Because failure of telomere integrity induces foci formation those DNA damage signaling proteins, we look for 53BP1 and γ-H2AX foci after Ad-CRT infection. We found that 53BP1 and γ -H2AX foci were clearly present in Ad-CRT infected cells (Fig. 3D-E). In addition, MDC1 foci as well as less phosphorylation of ATR which is necessary for maintaining telomere integrity were detected in Ad-CRT-infected cells (fig. S4A-B). These findings suggest that hTERT depletion by Rz contribute to disruption of telomere integrity and DNA damaging.
Fig 2
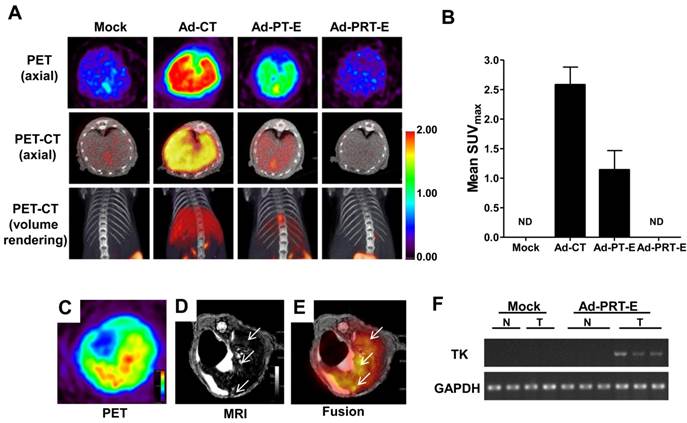
The HCC-specific targeting ability of Ad-PRT-E. (A) microPET/CT imaging of adenoviral vector-mediated thymidine kinase gene expression in the normal liver. A total of 5 x 108 pfu of each adenovirus was injected via the tail vein, and microPET/CT imaging was performed 2 days after injection without GCV treatment. The color scale is expressed based on the SUVmax value for the [18F] FHBG uptake. (B) The graph demonstrates the average [18F] FHBG retention in the liver (n=2/group). (C-E) microPET-MR fusion images of the HCC-specific targeting capacity of Ad-PRT-E. Three weeks after the splenic inoculation of the HEP-3B cells, 5 x 108 pfu of Ad-PRT-E was systemically injected. A representative Ad-PRT-E mouse was scanned using axial [18F] FHBG microPET imaging of the HSVtk expression (C) and T2-weighted MR imaging for the tumor locus (white arrow) (D). Colocalized spots were determined based on PET-MR fusion imaging (E). (F) Selective expression of HSVtk in the Ad-PRT-E-treated tumors from the RT-PCR analysis. RNAs were isolated from normal and tumor regions of liver tissues of mice treated with Mock (n=2) and Ad-PRT-E (n=3).
Fig 3
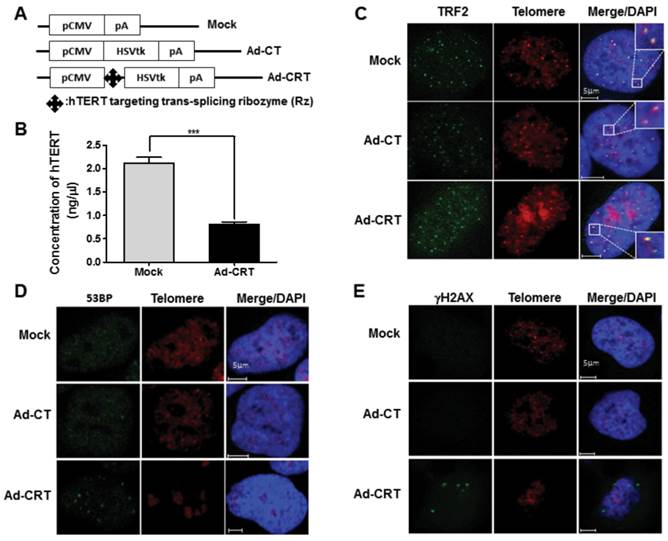
Effect of Rz-mediated hTERT depletion on telomere integrity. (A) Schematic diagram of HSVtk expressing construct containing CMV promoter, Ad-CRT, Ad-CT, and Mock vector. (B) Decrease of hTERT concentration in Ad-CRT-infected Hep-3B cells, determined with real-time PCR. A detailed description of PCR condition is provided in the “Materials and Methods”. Error bars indicate the mean values ± SEM for triplicate assays. (***p < 0.001) (C) Telomere G end staining was lighter or unclear and TRF2 co-localization was weaker in Ad-CRT-infected cells. TRF2 was detected using FITC labelled secondary antibody and the telomere G ends were detected with Cy3-tagged (TTAGGG)3 PNA using telomere FISH with immunofluorescence after Mock, Ad-CT, and Ad-CRT infection. DNA damage protein foci related to telomere dysfunction induced by Ad-CRT infection. The 53BP1 (D) and γ-H2AX (E) foci were detected in Ad-CRT-infected cells. Staining for 53BP1 and γ-H2AX protein foci was green fluorescence and telomere G end PNA was red.
Next, to examine hTERT depletion by Rz influences tumor cell invasion, Boyden chamber-matrigel assays were performed in adenovirus-treated cells without GCV treatment. A strong inhibition of invasive property of HCC cells was observed in the Ad-PRT-E treatment group different from no treatment or Mock and Ad-PT-E infection (Fig. 4A-B). In addition, as shown in Fig. 4C, the HCC cells in the Mock-infected group were strongly stained with AKT and VEGF rather than in Ad-PRT-E group. Especially, VEGF expression was dramatically reduced in the Ad-PRT-E group; thus, vessel formations in the tumor region appeared to be nearly completely blocked in the Ad-PRT-E group compared to the Mock group, as featured by CD34 immunostaining. These results suggest that Rz-mediated hTERT depletion contributes to the in vivo anti-tumor activity via suppression of metastasis- or angiogenesis-related pathway.
Safety evaluation of Ad-PRT-E in vivo
For the safety evaluation, we intravenously injected 5 x 108 pfu of adenoviruses into tumor-bearing mice followed by GCV treatment for 14 days (n=10/group). All of the mice in the Mock and Ad-PRT-E groups survived, whereas those injected with Ad-PT-E barely survived 10 days (Fig. 5A). During virus injection, the levels of liver-specific enzymes ALT and AST did not change in the Ad-PRT-E and Mock groups, although they were markedly higher in the PT-E group, particularly at day 6 (Fig. 5B). Overall, these results were indicative of the improved safety of Ad-PRT-E compared with Ad-PT-E. A histological examination confirmed that normal liver cells in the Ad-PRT-E and Mock groups were pathologically unharmed. The TUNEL assay showed that the appearance of apoptotic figures (i.e., strong brown spots) was most prominent in the tumor nodules of the Ad-PRT-E-treated group (Fig. 5C-D). In contrast, the livers from mice in the Ad-PT-E-treated group showed diffuse ballooning degeneration, spotty necrosis and apoptosis of the hepatocytes that were more extensive on day 6 (Fig. 5C-D). Apoptotic figures were frequently observed in both the neoplastic and non-neoplastic hepatocytes in the Ad-PT-E group. The apoptotic frequency in the tumor nodules in the Ad-PT-E group was less than that in the Ad-PRT-E group. These results support the hypothesis that the introduction of a liver-specific promoter PEPCK would confer additional safety for Rz-mediated tumor targeting, and Ad-PRT-E would specifically target HCC without injuring non-neoplastic hepatocytes.
Fig 4
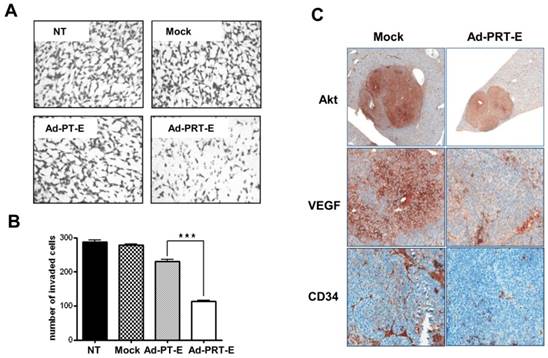
The inhibition of invasive and angiogenic potentials of HCC cells by Ad-PRT-E. (A-B) Cells were counted that invaded the matrigel in chamber (8.0 μm pore size) after infection with 10 MOI of each adenovirus. Experimental details are described in “Supplementary Experimental Procedure”. NT, no treatment. Representative invaded cells were imaged in A. The graph in B is expressed as the mean value ± SEM for each group (*** p < 0.001). The number of invaded cells was quantified in at least five regions in triplicate in independent experiments, and the statistical significance was assessed using a t-test. (C) Immunohistochemical evaluation of the decreased AKT, VEGF and CD34 expression in the multifocal mouse HCC xenografts treated with 5 x 108 pfu of Ad-PRT-E for 14 days (Magnitude x200). A detailed description of the immunostaining procedure is provided in the “Materials and Methods”.
Fig 5
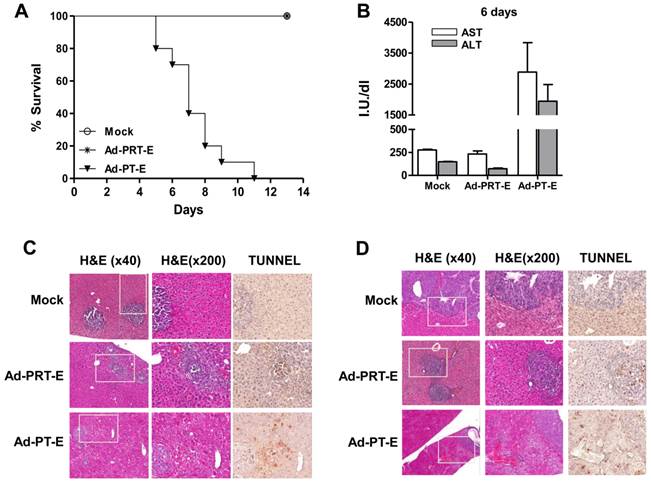
Tumor-specific targeting and safety of the systemic administration of Ad-PRT-E plus GCV in a multifocal HCC mouse model. (A) Survival plots of groups treated with Ad-Mock, Ad-PT-E and Ad-PRT-E assessed according the Kaplan-Meier method. For each group, tumor-bearing Balb/c nude mice were administered 5 x 108 pfu of adenoviruses by injection into the tail veins followed by GCV treatment for 14 days (n=10/group). (B) The liver function of the mice in fig. 5D, which was measured as serum ALT and AST. Liver enzymes were measured by UV spectrometry at 6 days after GCV treatment. Error bars indicate the mean value ± SEM. (C-D) Representative histological observations using hematoxylin and eosin staining and evaluations of apoptosis using TUNEL assays in the tumor-bearing liver tissues 3 days (C) and 6 days (D) after adenoviruses treatment. Mice were injected with 5 x 108 pfu of the indicated adenoviruses via the tail veins and treated with GCV (n=6/group). The dark-brown-colored nuclear or cellular staining in the TUNEL assay is indicative of apoptosis.
Marked anti-HCC effect by systemic delivery of Ad-PRT-E
The anti-tumor effect of Ad-PRT-E in the mouse model bearing multifocal intrahepatic liver tumors was monitored in parallel using in vivo imaging (n=10) and autopsy (n=30) for 14 days after the intravenous administration of adenoviruses. First, we imaged all animals using micro-PET/CT at day 14 and randomly divided mice into two groups with similar tumor-initiating stages based on the splenic uptake of FDG for two different treatments (Fig. 6A). The SUVmax of the Ad-PRT-E-treated group (n=5) was 2.3512 ± 0.194 compared to 2.6916 ±0.561 for the Mock-treated group (n=5) (fig. S5-6). Fourteen days after adenovirus plus GCV treatment, the Mock-treated animals showed that [18F]FDG PET signals were two to four times stronger in their livers than prior to treatment. As expected, mice treated with Ad-PRT-E exhibited significantly weaker signals compared with Mock treatment (2.281 ± 0.475 vs. 3.850 ± 0.564 in the liver, p=0.00531) (Fig. 6B). These findings suggest that the HSVtk expressed from Ad-PRT-E triggered Hep3B hepatoma cell death in the liver and spleen. PET images constructed from a representative mouse from the Mock and Ad-PRT-E groups are shown in fig. S5-6. After monitoring, animals in both groups were sacrificed, and their livers were examined to confirm histopathological concordance with the in vivo imaging results. The Mock group demonstrated remarkable abdominal distension. The livers of the control group animals were completely invaded by HCC, whereas the livers from the Ad-PRT-E group animals demonstrated only occasional, dispersed, tiny, whitish-pink tumor nodules as shown in the PET imaging results (Fig. 6C). In addition, we observed multifocal eosinophilic homogenous infarcts in the livers in the Mock group animals, which were attributable to extensive tumor growth and tumor emboli. Random grouping based on autopsy experiments demonstrated that Ad-PRT-E had a strong anti-HCC effect. Thirty nude mice injected with Hep3B cells were randomly grouped into Ad-PRT-E (n=20) and Mock (n=10) groups. The average weight of the liver tumors and the weight of the total liver multiplied by the histologically estimated tumor fraction was 4.60 ± 1.11 for the control group and 0.49 ± 0.62 for the Ad-PRT-E group; this difference was highly significant (p= 0.00254) (Fig. 6D).
Fig 6
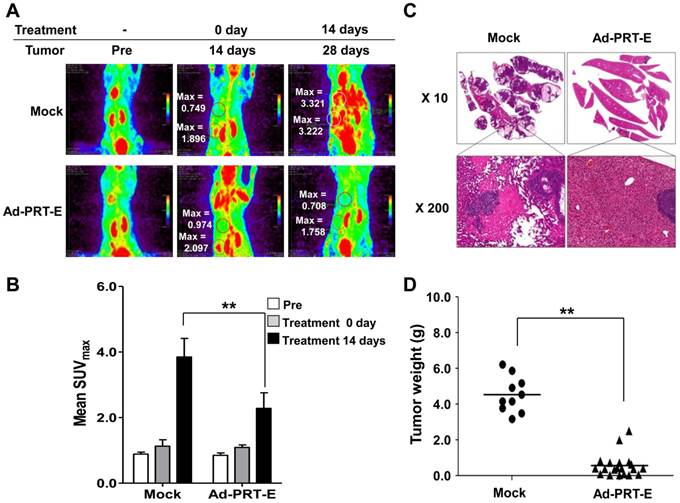
Anti-HCC efficacy of Ad-PRT-E in vivo. (A) Monitoring of the therapeutic efficacy of Ad-PRT-E using 18[F]-FDG PET/CT imaging. Representative 18[F]-FDG PET images of each treatment group are shown with SUVmax in the liver (upper region). The “Pre” images demonstrate the mice status just prior to tumor cell inoculation. The starting point of the adenovirus treatment was day 14 after the tumor cell inoculation, and the two treatment groups were sorted by similar SUVmax values in the spleen and liver. (B) The bar graph shows the values of tumor SUVmax in liver of the Mock (n=5) and Ad-PRT-E (n=5) groups in (A). The error bars indicate the mean value ± SEM (** p < 0.01). (C) Representative microscopic observations of the liver slices of each group stained with hematoxylin and eosin. (D) On day 14 after tumor cell inoculation, the mice were randomly sorted for the systemic administration of the Ad-PRT-E (n=20) or control/Mock (n=10) treatment. The HCC tumor weights of the Mock and Ad-PRT-E groups were determined based on the product of the liver weights and the histologically assessed tumor fractions. (** p < 0.01)
Taken together, these findings demonstrate that Ad-PRT-E has strong anti-tumor activity against HCC. Furthermore, the close correspondence between results of the repeated analyses by autopsy and noninvasive in vivo monitoring proves the effectiveness of in vivo monitoring systems in developing preclinical protocols.
Discussion
Here, we shed new light on advanced HCC anticancer treatment by depleting hTERT transcripts with trans-splicing ribozyme. Upon stimulation using novel therapeutic effects, we modified PEPCK promoter with a goal of increasing the likelihood of obtaining better results in future clinical trials. The restriction of toxic gene expression to tumor cells through use of tissue-specific promoters has been well recognized. The choice of promoters is governed by the transcriptional activity and tissue specificity of a promoter. The PEPCK promoter is a well-defined liver-specific promoter, and because its activity is attenuated when it is mediated by the trans-splicing ribozyme, the ApoE transcriptional enhancer was used to compensate for the low PEPCK activity [18]. As a result, the transcriptional activity of PEPCK with an ApoE enhancer was greater than that of a promoter lacking this element as shown in Fig. 1.
In this study, the adenoviral vector backbone was refined to overcome several drawbacks. Adenovirus serotype 5 is a vector of choice in clinical trials worldwide. Ad5 has been shown to infect host cells through the Coxsakie and adenovirus receptor (CAR) [19]. However, the performance of Ad5 has been disappointing in clinical trials due to a lack of CAR expression on malignant cells [20]. A number of studies have modified the infective viral capsid fiber knob to overcome poor infectivity in the CAR-dependent pathway [21]. Thus, we re-engineered a previous construct to possess the target cell determinant fiber knob, i.e., that of serotype 35. The new Ad5/35 construct infects cells via CD46, which is a membrane cofactor protein that is highly up-regulated in tumor cells [22]. As shown in fig. S1, the transduction efficiency of the modified-Ad5/35 was dramatically higher than that of Ad5. This improved level of expression enabled us to systemically administer lower Ad5/35 doses without sacrificing its anti-tumor efficacy, thereby avoiding the toxicity encountered when using Ad5. Moreover, to accommodate such a large exogenous gene (PEPCK-Rz-HSVtk-Enhancer), we performed an additional genetic modification by deleting E4 in the chimeric Ad5/35 adenovirus (fig. S1). An E1 or E1/E3-deleted replication-deficient adenovirus has been the most extensively used vehicle for the delivery of therapeutic genes. However, E1 deletion could not completely block the expression of early and late viral gene products or eliminate unwanted toxic effects on normal cells [23]. As observed in the strategy used in this study, E4 deletion can reduce the toxicity to normal cells, delay the host immune response and achieve a longer duration of transgene expression in cancer cells.
PET/MR monitoring demonstrated that Ad-PRT-E-mediated HSVtk expression only occurred in the tumor nodules of liver and not in non-tumor liver tissue (Fig. 2C-E). Enhanced targeting efficiency commonly guarantees better safety. A viral dose of 5x 108 pfu, which achieved effective tumor growth suppression and was a tolerable titer in the Ad-PRT-E-treated mice (compared with Ad-PT-E), resulted in death due to liver toxicity in the normal cells, which was later confirmed by histological examination (Fig. 5). Compared to Ad-PT-E, Ad-PRT-E was safer due to its high tumor selectivity. In addition, the Ad-PRT-E in our refined vector system was highly efficacious in its anti-tumor activity and also demonstrated a tumor-specific targeting capacity that was greater than expected based on its cytotoxic activity in vitro, suggesting additional anti-tumor activity in vivo, as well as cell killing effect by HSVtk/GCV system as a major functional mechanism of Rz. Three potential reasons elicited by Rz-mediated hTERT depletion may be proposed, i.e., 1) disruption of telomere integrity 2) blocking invasion and angiogenesis, and 3) recruit of host anti-tumoral immunity [24]. A sudden decrease in telomerase activity or senescence induces a change in the telomere ends, shelterin structure disruption, and activates the DNA damage checkpoint response [25]. We found the function of Rz-mediated hTERT depletion itself on telomere dysfunction. Ad-CRT-infected cells showed damage to shelterin structure integrity because (AATGGG)3 PNA labelling telomere FISH was faint and TRF2 was not co-localized with telomere-end labelling (Fig. 3C). The DNA damage response foci, γ-H2AX- and 53BP1-positive cells were detected in Ad-CRT-infected cells (Fig. 3D-E) and Ad-CRT infection decreased ATR phosphorylation for telomere protection from chromosome fusion (fig. S4A) [26]. Based on these Ad-CRT-based findings, we would suggest that hTERT depletion by Rz might affect to telomere integrity and cell damage as an additive plausible mechanism of Ad-PRT-E-induced anti-tumor activity in vivo. Second concept that the depletion of hTERT by Ad-PRT-E blocks the invasiveness of the tumor cells and angiogenesis, is based on report demonstrating that RNA interference targeting hTERT is involved in the regulation of tumor growth and cell motility [27,28]. In addition, ribozyme-induced reduction in telomerase activity has been reported to decrease glycolytic pathway genes in human melanoma cells and these genes possibly regulated by PI3K/Akt pathway [29,30]. Here, we found new evidence that major determinants of tumor cell growth and angiogenesis, i.e., PI3K/AKT/mTOR and VEGF signaling [31,32], were suppressed and significantly inhibited in vitro cell invasion by Rz (Ad-CRT without GCV) treatment alone, corresponding to suppression of AKT and VEGF in Ad-PRT-E treatment (fig. S7A and Fig. 4C). Moreover, Ad-PRT-E appeared to have a strong inhibitory effect on invasion compared with Ad-CRT (Fig. 4A and fig. S7B). These data demonstrate that Ad-PRT-E may be the correct choice when powerful systemic therapies for anti-metastatic and anti-proliferative effects in HCC are urgently needed, although specific information on the mechanisms underlying these phenomena remains unknown
Another possible mechanism is that the immune response of the immune cells recruited into the Ad-PRT-E-induced hTERT-depleted environments appear to play a critical role in tumor regression, because the interior of the tumor that was treated with only Ad-PRT-E included patchy areas that featured infiltrating immune cells, and because most of the cell death occurred in only those areas (Fig. 5C-D). The entry of immune cells into a tumor is not induced by a general cellular immune response against adenoviruses because tumors treated with Ad-PT-E and Mock did not demonstrate characteristic immune cell infiltration, which was unlike that associated with Ad-PRT-E. Although Ad-PRT-E produced lower HSVtk transcript levels due to its weaker transcriptional activity in vitro, Ad-PRT-E is an effective anti-tumor reagent that is comparable with Ad-PT-E considering in vivo efficacy.
Invasive procedures, such as the repeated collection of biopsy specimens, have been employed to estimate treatment efficacy. Such invasive procedures are incapable of answering several important questions concerning the specific delivery of a vector to a target, which involves a sufficient transgene expression level for a therapeutic effect and the proper timing to initiate suicide gene therapy. This study demonstrates the usefulness of the real-time parallel monitoring of transgene expression and tumor status, which is important in clinical situation. PET imaging using [18F]FHBG facilitated the estimation of the delivery and expression of adenoviral-mediated HSVtk to target sites (Fig. 2), and would be critical information to determine optimal treatment condition dependent on the level of therapeutic transgene expression [33]. The information obtained via noninvasive imaging was reliable because it provided comparable information to that provided by an autopsy (Fig. 6). This study provides protocols that are potentially useful in other preclinical and clinical studies of hepatocellular carcinoma as well as other cancers. Furthermore, the overall results suggest that Ad-PRT-E is a possible approach to be a viable alternative to conventional treatments inducing resistance, which lead to a realizable therapeutic strategy like as a direct delivery using catheter into intra-hepatic artery.
In conclusion, we described an ideal adenoviral therapeutic construct with enhanced PEPCK promoter-driven Rz and useful system for evaluating therapeutic efficacy using noninvasive imaging modalities. Without further refining or modifying it, the therapeutic module introduced here would be applied for patients who suffers from HCC insults due to the limited therapeutic effect of current treatment modalities. In addition, these image-aided analyses could be equally translated to clinic, enabling us to evaluate how well individual HCC patient respond to this gene therapy in the future clinical trial. We believe that this study provides strategies for designing advanced therapeutic adenoviruses to attack multiple hepatocellular carcinoma at growth and metastasis sites and for obtaining extensive information through noninvasive monitoring of adenovirus-mediated hepatocellular carcinoma treatment.
Abbreviations
hTERT: human telomerase reverse transcriptase; HSVtk: herpes simplex virus thymidine kinase gene; PEPCK: phosphoenolpyruvate carboxylase; HCC: hepatocellular carcinoma; Rz: hTERT-targeting trans-splicing ribozyme; ApoE: apolipoprotein E; GCV: ganciclovir.
Supplementary Material
Supplementary Methods, Figs. S1-S7.
Acknowledgements
We thank Kyoung-Sook Cho for providing assistance with the animal experiments, the Molecular Imaging Core in the Korea National Cancer Center for assistance with the in vivo imaging, and Dr. J. Sri Ram for editorial assistance. Funding: This work was supported by research grants from the National Cancer Center Grants (NCC1010220 and NCC1410040), the National R&D program for Cancer Control, Ministry for Health and Welfare (grant No. 0720520) and the National Research Foundation of Korea (NRF) grant funded by the Korea Government (MISP) (No. 2010-0028684).
Author Contributions
Y.H.K. designed and performed the overall experimental work, and wrote the manuscript. S.J.L established adenoviral system for Rz; K.T.K. designed and performed cellular experiments; S.H.H., K.T.K., J.Y.M., E.K.Y., S.K., S.I.C. and S.H.G. conducted most of the experiments and analyzed the data; J.S.J., E.O.K. and M.H.J. provided expertise tissue histology & immunohistochemistry examination with mouse model; S.H.K., S.K.K. and D.K. contributed to in vivo imaging study and interpretation of data; S.W.L. provided the technical support for vector construction; J.S.J. and I.H.K. designed the research concept, and provided critical review and edits to the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69-90
2. Faloppi L, Scartozzi M, Maccaroni E. et al. Evolving strategies for the treatment of hepatocellular carcinoma: from clinical-guided to molecularly-tailored therapeutic options. Cancer Treat Rev. 2011;37:169-77
3. Rahbari NN, Mehrabi A, Mollberg NM. et al. Hepatocellular carcinoma: current management and perspectives for the future. Ann Surg. 2011;253:453-69
4. Lou L, Ye W, Chen Y. et al. Ardipusilloside inhibits survival, invasion and metastasis of human hepatocellular carcinoma cells. Phytomedicine. 2012;19:603-8
5. Lo HW, Day CP, Hung MC. Cancer-specific gene therapy. Adv Genet. 2005;54:235-55
6. Nettelbeck DM, Jerome V, Muller R. A strategy for enhancing the transcriptional activity of weak cell type-specific promoters. Gene Ther. 1998;5:1656-64
7. Wu L, Johnson M, Sato M. Transcriptionally targeted gene therapy to detect and treat cancer. Trends Mol Med. 2003;9:421-9
8. Sullenger BA, Cech TR. Ribozyme-mediated repair of defective mRNA by targeted, trans-splicing. Nature. 1994;371:619-22
9. Kwon BS, Jung HS, Song MS. et al. Specific regression of human cancer cells by ribozyme-mediated targeted replacement of tumor-specific transcript. Mol Ther. 2005;12:824-34
10. Hong SH, Jeong JS, Lee YJ. et al. In vivo reprogramming of hTERT by trans-splicing ribozyme to target tumor cells. Mol Ther. 2008;16:74-80
11. Nagao K, Tomimatsu M, Endo H, Hisatomi H, Hikiji K. Telomerase reverse transcriptase mRNA expression and telomerase activity in hepatocellular carcinoma. J Gastroenterol. 1999;34:83-7
12. Kotoula V, Hytiroglou P, Pyrpasopoulou A, Saxena R, Thung SN, Papadimitriou CS. Expression of human telomerase reverse transcriptase in regenerative and precancerous lesions of cirrhotic livers. Liver. 2002;22:57-69
13. Forsyth NR, Wright WE, Shay JW. Telomerase and differentiation in multicellular organisms: turn it off, turn it on, and turn it off again. Differentiation. 2002;69:188-97
14. Song MS, Jeong JS, Ban G. et al. Validation of tissue-specific promoter-driven tumor-targeting trans-splicing ribozyme system as a multifunctional cancer gene therapy device in vivo. Cancer Gene Ther. 2009;16:113-25
15. Lee SJ, Lee SW, Jeong JS, Kim IH. In vivo reprogramming of human telomerase reverse transcriptase (hTERT) by trans-splicing ribozyme to target tumor cells. Methods Mol Biol. 2010;629:307-21
16. Giavazzi R, Jessup JM, Campbell DE, Walker SM, Fidler IJ. Experimental nude mouse model of human colorectal cancer liver metastases. J Natl Cancer Inst. 1986;77:1303-8
17. Kwon BS, Jeong JS, Won YS. et al. Intracellular efficacy of tumor-targeting group I intron-based trans-splicing ribozyme. J Gene Med. 2011;13:89-100
18. van der Poel HG, McCadden J, Verhaegh GW. et al. A novel method for the determination of basal gene expression of tissue-specific promoters: an analysis of prostate-specific promoters. Cancer Gene Ther. 2001;8:927-35
19. Ghosh SS, Gopinath P, Ramesh A. Adenoviral vectors: a promising tool for gene therapy. Appl Biochem Biotechnol. 2006;133:9-29
20. Li D, Duan L, Freimuth P, O'Malley BW Jr. Variability of adenovirus receptor density influences gene transfer efficiency and therapeutic response in head and neck cancer. Clin Cancer Res. 1999;5:4175-81
21. Koski A, Kangasniemi L, Escutenaire S. et al. Treatment of cancer patients with a serotype 5/3 chimeric oncolytic adenovirus expressing GMCSF. Mol Ther. 2010;18:1874-84
22. Sova P, Ren XW, Ni S. et al. A tumor-targeted and conditionally replicating oncolytic adenovirus vector expressing TRAIL for treatment of liver metastases. Mol Ther. 2004;9:496-509
23. Ji L, Bouvet M, Price RE, Roth JA, Fang B. Reduced toxicity, attenuated immunogenicity and efficient mediation of human p53 gene expression in vivo by an adenovirus vector with deleted E1-E3 and inactivated E4 by GAL4-TATA promoter replacement. Gene Ther. 1999;6:393-402
24. Edukulla R, Woller N, Mundt B. et al. Antitumoral immune response by recruitment and expansion of dendritic cells in tumors infected with telomerase-dependent oncolytic viruses. Cancer Res. 2009;69:1448-58
25. d'Adda di Fagagna F, Reaper PM, Clay-Farrace L. et al. A DNA damage checkpoint response in telomere-initiated senescence. Nature. 2003;426:194-8
26. McNees CJ, Tejera AM, Martinez P. et al. ATR suppresses telomere fragility and recombination but is dispensable for elongation of short telomeres by telomerase. J Cell Biol. 2010;188:639-52
27. George J, Banik NL, Ray SK. Knockdown of hTERT and concurrent treatment with interferon-gamma inhibited proliferation and invasion of human glioblastoma cell lines. Int J Biochem Cell Biol. 2010;42:1164-73
28. Shen Y, Zhang YW, Zhang ZX, Miao ZH, Ding J. hTERT-targeted RNA interference inhibits tumorigenicity and motility of HCT116 cells. Cancer Biol Ther. 2008;7:228-36
29. Bagheri S, Nosrati M, Li S. et al. Genes and pathways downstream of telomerase in melanoma metastasis. Proc Natl Acad Sci U S A. 2006;103:11306-11
30. DeBerardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB. The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 2008;7:11-20
31. Whittaker S, Marais R, Zhu AX. The role of signaling pathways in the development and treatment of hepatocellular carcinoma. Oncogene. 2010;29:4989-5005
32. Semela D, Piguet AC, Kolev M. et al. Vascular remodeling and antitumoral effects of mTOR inhibition in a rat model of hepatocellular carcinoma. J Hepatol. 2007;46:840-8
33. Penuelas I, Mazzolini G, Boan JF. et al. Positron emission tomography imaging of adenoviral-mediated transgene expression in liver cancer patients. Gastroenterology. 2005;128:1787-95
Author contact
![]() Corresponding authors: In-Hoo Kim (I.H.Kim), Tel.: +82-31-920-2720; fax: +82-31-920-1511; e-mail: ikimre.kr; Jin Sook Jeong (J.S.Jeong), Tel.: +82-51-240-5352; fax: +82-51-243-7396; E-mail:jsjung1ac.kr.
Corresponding authors: In-Hoo Kim (I.H.Kim), Tel.: +82-31-920-2720; fax: +82-31-920-1511; e-mail: ikimre.kr; Jin Sook Jeong (J.S.Jeong), Tel.: +82-51-240-5352; fax: +82-51-243-7396; E-mail:jsjung1ac.kr.
Citation styles
APA
Kim, Y.H., Kim, K.T., Lee, S.J., Hong, S.H., Moon, J.Y., Yoon, E.K., Kim, S., Kim, E.O., Kang, S.H., Kim, S.K., Choi, S.I., Goh, S.H., Kim, D., Lee, S.W., Ju, M.H., Jeong, J.S., Kim, I.H. (2016). Image-aided Suicide Gene Therapy Utilizing Multifunctional hTERT-targeting Adenovirus for Clinical Translation in Hepatocellular Carcinoma. Theranostics, 6(3), 357-368. https://doi.org/10.7150/thno.13621.
ACS
Kim, Y.H.; Kim, K.T.; Lee, S.J.; Hong, S.H.; Moon, J.Y.; Yoon, E.K.; Kim, S.; Kim, E.O.; Kang, S.H.; Kim, S.K.; Choi, S.I.; Goh, S.H.; Kim, D.; Lee, S.W.; Ju, M.H.; Jeong, J.S.; Kim, I.H. Image-aided Suicide Gene Therapy Utilizing Multifunctional hTERT-targeting Adenovirus for Clinical Translation in Hepatocellular Carcinoma. Theranostics 2016, 6 (3), 357-368. DOI: 10.7150/thno.13621.
NLM
Kim YH, Kim KT, Lee SJ, Hong SH, Moon JY, Yoon EK, Kim S, Kim EO, Kang SH, Kim SK, Choi SI, Goh SH, Kim D, Lee SW, Ju MH, Jeong JS, Kim IH. Image-aided Suicide Gene Therapy Utilizing Multifunctional hTERT-targeting Adenovirus for Clinical Translation in Hepatocellular Carcinoma. Theranostics 2016; 6(3):357-368. doi:10.7150/thno.13621. https://www.thno.org/v06p0357.htm
CSE
Kim YH, Kim KT, Lee SJ, Hong SH, Moon JY, Yoon EK, Kim S, Kim EO, Kang SH, Kim SK, Choi SI, Goh SH, Kim D, Lee SW, Ju MH, Jeong JS, Kim IH. 2016. Image-aided Suicide Gene Therapy Utilizing Multifunctional hTERT-targeting Adenovirus for Clinical Translation in Hepatocellular Carcinoma. Theranostics. 6(3):357-368.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) License. See http://ivyspring.com/terms for full terms and conditions.