Impact Factor
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
1. Introduction
2. DNA MTase activity assays
3. Challenges and prospects
4. Conclusion
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2016; 6(3):369-391. doi:10.7150/thno.13438 This issue Cite
Review
DNA Methyltransferase Activity Assays: Advances and Challenges
Wan Jun Poh, Cayden Pang Pee Wee, Zhiqiang Gao ![]()
Department of Chemistry, National University of Singapore, Singapore 117543
Received 2015-8-3; Accepted 2015-10-12; Published 2016-1-6
Abstract

DNA methyltransferases (MTases), a family of enzymes that catalyse the methylation of DNA, have a profound effect on gene regulation. A large body of evidence has indicated that DNA MTase is potentially a predictive biomarker closely associated with genetic disorders and genetic diseases like cancer. Given the attention bestowed onto DNA MTases in molecular biology and medicine, highly sensitive detection of DNA MTase activity is essential in determining gene regulation, epigenetic modification, clinical diagnosis and therapeutics. Conventional techniques such as isotope labelling are effective, but they often require laborious sample preparation, isotope labelling, sophisticated equipment and large amounts of DNA, rendering them unsuitable for uses at point-of-care. Simple, portable, highly sensitive and low-cost assays are urgently needed for DNA MTase activity screening. In most recent technological advances, many alternative DNA MTase activity assays such as fluorescent, electrochemical, colorimetric and chemiluminescent assays have been proposed. In addition, many of them are coupled with nanomaterials and/or enzymes to significantly enhance their sensitivity. Herein we review the progress in the development of DNA MTase activity assays with an emphasis on assay mechanism and performance with some discussion on challenges and perspectives. It is hoped that this article will provide a broad coverage of DNA MTase activity assays and their latest developments and open new perspectives toward the development of DNA MTase activity assays with much improved performance for uses in molecular biology and clinical practice.
Keywords: DNA methyltransferase, DNA methylation, fluorometry, colourimetry, electrochemistry, chemiluminescence, electrochemiluminescence.
1. Introduction
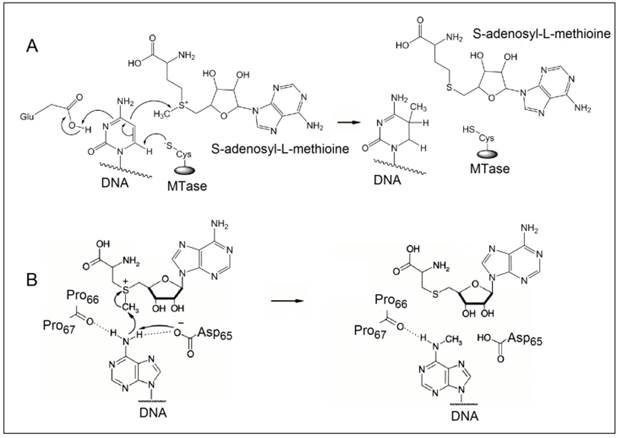
The methylation of DNA, referring to the covalent modification of nucleotides in DNA with methyl groups, is one of the most common epigenetic events taking place in mammal genome [1,2]. Two of the four nucleotides, i.e. cytosine and adenine, can be methylated, but adenine methylation is only found in prokaryotes [3]. Consequently, methyltransferases (MTases) are categorised into three groups on the basis of the nucleotides and positions they methylate, namely, those methylate N6 of adenine (m6A EC 2.1.1.72), those methylate N4 of cytosine (m4C EC 2.1.1.113) and those methylate C5 of cytosine (m5C EC 2.1.1.37). All the known DNA methyltransferases engage S-adenosyl methionine as the methyl donor in their methylation processes. It is known that the most common DNA methylation in occurs not only at the 5-carbon position of the pyrimidine ring of cytosine within CpG sites, but also at the 6-nitrogen position of the purine ring of adenine in the symmetric tetranucleotides 5'-G-A-T-C-3' [4,5]. Three different DNA MTases, namely DNMT1, DNMT3A and DNMT3B, have been identified in mammals [6]. Among them, DNMT1 is the most abundant DNA and considered as the key methyltransferase in mammals. DNA methylation mainly occurs at the 5-carbon position of the pyrimidine ring of cytosine at the CpG sites in mammal genome [7]. The methyl moiety is grafted onto cytosine at the CpG sites by transferring a methyl group from S-adenosyl-L-methionine to cytosine catalysed by DNA MTases [8,9]. Figure 1A details the reaction mechanism of cytosine methylation in DNA catalysed by DNA MTase. As illustrated in Figure 1A, the methylation cytosine employs a conjugate addition reaction between a nucleophilic thiolate (deprotonated thiol) of the cysteine residue of MTase and 6-carbon of the pyrimidine ring of cytosine, forming a covalent intermediate between the thiolate and the 6-carbon to activate the 5-carbon for methyl addition. The thiolate in the cysteine residue acts a strong nucleophile, attacking the 6-carbon atom of the pyrimidine ring of cytosine to form a covalent bond between the thiolate atom and the 6-carbon atom. A glutamate residue in the vicinity of the reaction site stabilises the negative charge on cytosine. Nucleophilic attack then occurs on the methyl group of S-adenosyl-L-methionine, which is converted to S-adenosyl-L-homocysteine. Finally, β-elimination takes place across the 5-carbon and 6-carbon bond, dislodging MTase form the methylated cytosine. On the other hand, the methylation of exocyclic amino nitrogen atoms of DNA takes rather different pathways although the same methyl donor is involved in the process. Taking adenine as an example, instead of employing a conjugate addition reaction to activate the 5-carbon for methyl addition, methylation at the exocyclic amino position proceeds with the deprotonation of the nitrogen being methylated followed by a direct methyl transfer from S-adenosyl-L-methionine to the exocyclic amino group (Figure 1B) [10-13]. The carbonyl of the peptide chain between two prolines interacts with the target amino group via hydrogen bonding and the deprotonation of the target amino group is facilitated by the aspartate(65) residue of MTase. Methyl transfer then takes place by the nucleophilic attack of the deprotonated amino group to the methylsulfonium group of S-adenosyl-L-methionine.
The reaction mechanism of the methylation of (A) cytosine and (B) adenine. (Reproduced with permission from reference [10].)
DNA methylation is one of the central regulating processes in the epigenetics of living organisms. Increasing evidence has suggested that methylated DNA is an important regulator in many biological processes like X-chromosome inactivation, genomic imprinting and gene expression [14-16]. For instance, genes with high levels of methylation in their promoter regions are transcriptionally silent [17]. Aberrant DNA MTase activity, which often leads to aberrant DNA methylation patterns, is closely associated with many types of genetic disorders and a large number of human malignancies [18,19]. Both hypomethylation (under-methylation) and hypermethylation (over-methylation) have been identified in various types of cancer [20,21], such as breast cancer [22], ovarian cancer [23], lung cancer [24], colon cancer [25], urological cancer [26] and leukemia [27]. Hypermethylation of CpG islands present in the respective promoter regions silence tumour suppressor genes [28]. Consequently, the absence of tumour suppressing proteins strongly encourages the onset of tumour formation. On the other hand, due to the abnormal loss of methylation status at the repetitive regions of genes that are not part of the CpG islands [28,29], hypomethylation affects the genomic stability and correlates with the progression and severity of the disease [30]. The epigenetic effect of DNA methylation on oncogenesis is therefore believed by either the activation of protooncogenes due to hypomethylation or the inactivation of tumour suppressor genes due to hypermethylation. Recent studies have also indicated that DNA MTase is potentially a predictive biomarker and therapeutic target in the diagnosis and treatment of various types of cancer [31-34] since aberrant DNA MTase activity usually occurs far before other signs of malignancy [35].
As the first step toward improving human health, the activity of DNA MTase at cellular level has to be screened in order to gain an insight into the regulation of methylation and determine therapeutic strategy [36-39]. Conventional techniques used in screening the activity of DNA MTase include high-performance liquid chromatography (HPLC) [40], methylation-specific polymerase chain reaction (PCR) [41], gel electrophoresis [42], high performance capillary electrophoresis (HPCE) [43], as well as using isotopically labelled S-adenosyl-L-methionine for methylation [44,45]. Even though these techniques still prove to be useful and employed in laboratory practice, they are not without limitations. For example, most of the above-mentioned techniques not only employ bulky and expensive equipment, but also require complicated sample preparation and an undesirable amount of time for data analysis. With these limitations, there have been continuous efforts to develop new DNA MTase activity screening assays hoping to overcome some of the limitations. In recent years, such new assays utilising other signal transduction strategies such as fluorometry, chemiluminescence, electrochemistry and colourimetry have been developed. This article reviews the progress in the research and development of DNA MTase activity assays and their applications. It starts off with a brief summary of conventional techniques and then moves onto introducing newly developed assays with great details and with typical examples illustrating their working principles and possible advantages they could bring to DNA MTase activity screening along with some discussion on challenges and perspectives. We hope that this article will provide a comprehensive overview of the current status of the field and open new perspectives toward the development of novel concepts and strategies for DNA MTase activity assays with much improved performance and reliability.
2. DNA MTase activity assays
2.1 Radio DNA MTase assays
As one of the earliest platforms and the standard assay for screening DNA MTase activity, the isotope-labelling was widely used in the detection of the activity of DNA MTase and methylated DNA in early days [46,47]. During methylation catalysed by DNA MTase, isotope-labelled methyl moiety is grafted on DNA and thus granting the methylated DNA radioactivity. This radioactivity can be conveniently detected either by a scintillation counter or by means of an autoradiograph. Unfortunately, the involvement of radioactive agents is the biggest drawback which has limited this assay in centralised laboratories. The search for radiation-free DNA MTase activity assays led to the development of alternatives like methylation-specific PCR [48], HPCE [43] and HPLC [41,48], while methylation-specific PCR is considered as the “gold standard'. In methylation-specific PCR, a substrate DNA is first treated by sodium bisulphite during which all unmethylated, but not methylated, cytosines are converted to uracils. Subsequently, PCR is carried out with primers specific for methylated versus unmethylated DNA. Owing to the exceptional power of PCR amplification and the high accuracy of sequencing, methylation-specific PCR is sensitive to 0.1% methylated alleles of a given CpG island locus and can be performed on minute amounts of DNA samples extracted from various specimens [48]. On the other hand, the excellent separation efficiency of HPCE and HPLC enables the detection of individual nucleotides including methylated nucleotides after the substrate DNA is enzymatically hydrolysed. Although non-radioactive, the above-mentioned DNA MTase activity assays require bulky and often expensive equipment, tedious and time-consuming sample preparation and data analysis, and cumbersome detection schemes that are undesirable in clinical practice. Therefore, there is a great demand in the research and development of DNA MTase activity assays that possess the attractive features of conventional DNA MTase activity assays and yet overcome their drawbacks. Over last decade, several detection strategies like fluorometry, colourimetry, electrochemistry, chemiluminescence and electrochemiluminescence (ECL) have been engaged in the construction of DNA MTase activity assays. In the following sections, different types of DNA MTase activity assays will be discussed with typical examples elaborating on their working principle and performance.
2.2 Colourimetric DNA MTase activity assays
Colourimetric DNA MTase activity assays rely on the visual observation of a colour change or the spectrometric measurement of an absorption spectrum associated with DNA MTase. They have the advantages of low cost, simplicity, portability and in some cases instrument-free. Although DNA can be quantify by UV-vis spectrometry, the low sensitivity and an insignificant difference between methylated and unmethylated DNA in UV-vis absorption characteristics basically negate UV-vis spectrometry in the direct detection of DNA MTase activity and necessitate the engagement of more sensitive chromogens and/or amplification strategies. In the following section, both approaches will be discussed in detail with representative examples to illustrate their working principles. There have not been reported cases of colorimetric assays apart from the usage of gold nanoparticles or amplification strategies according to the best of our knowledge. For colorimetric methods to execute sensitively, there must be an absorption in the UV-visible range with a high extinction coefficient, which limits the choice of chromophores.
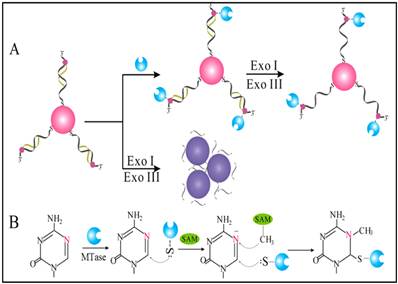
Owing to their ultrahigh extinction coefficient (108-109 M-1 cm-1) of the surface plasmon resonance absorption with a strong dependence on inter-particle distance, gold nanoparticles (AuNPs) have attracted much attention in the development of colourimetric assays for DNA MTase activity screening. Since the distance-dependent plasmonic absorption exhibits a distinct change in UV-vis absorption or solution colour upon altering the distance among the AuNPs, strategies that can associate DNA MTase activity with the distance among the AuNPs have been proposed in DNA MTase activity assays [49]. One of such strategies is to coat the AuNPs with DNA MTase substrate and build a mechanism associated with DNA methylation (Figure 2) [50]. As seen in Figure 2, the AuNPs coated with doubles-stranded DNA (ds-DNA) that contains a DNA MTase recognition sequence and a terminal thiol at 5′-end in one of the two strands. The high negative charge density on the surface of the AuNPs ensures the AuNPs stably and homogeneously dispersed in solution. When the coated AuNPs are incubated with exonucleases I and III, the DNA layer on the AuNP surface is enzymatically digested to mononucleotides and hence significantly diminish the negative charges on the AuNPs, thereby resulting in the aggregation of the AuNPs accompanying with a concomitant red-to-purple colour change of the solution. On the other hand, in the presence of DNA MTase, the DNA MTase is covalently tagged at the 6-carbon position of the 5-aza-C base ring in the ds-DNA because the absence of a proton at the 5-nitrogen position prevented β elimination (Figure 1B). The methylated DNA strongly resists exonuclease digestion, and thus the AuNPs remain homogeneously dispersed in solution. It was observed that the absorbance of the AuNP aggregates at 526 nm shows a linear correlation with the activity of the DNA MTase between 2 and 32 U/mL, achieving a detection limit of 0.5 U/mL. The ability of MTase to methylate specific DNA sequence has not yet been tested in real biological samples. In another report, AuNPs decorated with single-stranded DNA (ss-DNA) complementary to the two single-stranded ends of the substrate DNA were employed in the construction of a colourimetric DNA MTase assay [51]. Because of the strong electrostatic repulsion among the AuNPs arising from the negative charges on the surface of the AuNPs, the DNA-coated AuNPs are homogeneously dispersed in solution in the absence of the substrate DNA, thus showing a wine-red colour. On the other hand, in the presence of the substrate DNA, hybridisation between the DNA-coated AuNPs and the substrate DNA induces the aggregation of the AuNPs, giving the solution a blue colour. The DNA-coated AuNPs again are homogeneously dispersed in solution when both the substrate DNA and an endonuclease are added to the AuNP solution because of the instantaneous cleavage of the hybridised substrate DNA by the endonuclease. However, if the hybridisation is followed by a period of incubation with DNA MTase and then the addition of the endonuclease, the AuNPs firmly remain aggregated since the methylation of the hybridised substrate DNA inhibits its cleavage by the endonuclease. During methylation, a covalent complex forms between the sulfhydryl group of cysteine in the Pro-Cys motif of MTase and the 6-carbon position of 5-aza-dC base ring followed by the transfer of a methyl group from S-adenosyl-L-methionine to the 5-nitrogen position, where the absence of a proton prevents β elimination and thus causes the trapping of DNA MTase.
(A) Schematic illustration of the AuNP-based colourimetric DNA MTase activity assay and (B) Trapping mechanism of MTase. (Reproduced with permission from reference [50].)
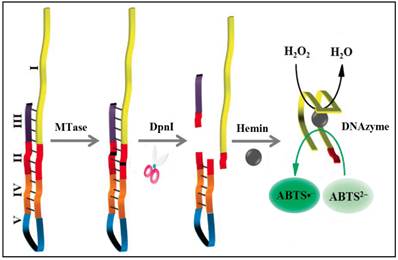
To significantly enhance the sensitivity, signal amplification mechanism by a DNAzyme (G-quadruplex-hemin complex) is incorporated in colourimetric DNA MTase assays [52-54]. This DNAzyme acts like peroxidase capable of catalysing the oxidation 2,2'-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid) (ABTS2-) in the presence of hydrogen peroxide to produce a colour change. For example, based on the fact that the methylated DNA can be cleaved by certain endonucleases like DpnI, a methylation-responsive DNAzyme strategy for the detection of DNA MTase activity was proposed by Li and co-workers (Figure 3) [53]. As illustrated in Figure 3, the key in the design of the hairpin probe is the integration of both methylation-responsive sequence (segment III) and DNAzyme sequence (segment I) into one probe together with two spacers (segment II and IV) and a loop (segment V) with the pre-requite that no G-quadruplex is formed before methylation and DpnI cleavage. Therefore, in the hairpin probe the DNAzyme sequence (region I) is partially caged in the stem of the hairpin probe by spacer III, thus preventing the formation of G-quadruplex. When both MTase and DpnI are introduced, successive enzymatic actions on the hairpin probe (MTase catalytic methylation and DpnI catalysed hairpin cleavage) release the complete DNAzyme sequence to solution. The freed DNAzyme sequence spontaneously folds into G-quadruplex and acquires its peroxidase-like activity upon binding to hemin. Finally, catalysed oxidation of ABTS2- to its coloured product ABTS•- is realised in the presence of H2O2. The same authors went one step further to incorporate target-cycling amplification into the assay to enhance its sensitivity. In the presence of DpnI, methylation-induced scission of the substrate DNA initiates the target-cycling amplification and consequently the production of the DNAzyme. A detection limit of 0.25 U/mL was obtained. Biological sample matrix was not incorporated into this highly sensitive DNAzyme-based assay to monitor MTase activity.
Schematic illustration of the DNAzyme-amplified MTase activity assay. (Reproduced with permission from reference [53].)
Colourimetry has also attracted considerable attention of biotechnical industry in the development of MTase activity/inhibition kits [55-59]. To significantly enhance assay sensitivity and throughput, enzymatic amplifications such as horse reddish peroxidase (HRP) in a microplate format are usually adopted in those kits. The specificity of the assays is safeguarded by the use of antibodies. For example, In the EpiQuik™ DNA methyltransferase activity/inhibition assay kit, cytosine-rich DNA substrates are first immobilized on strip wells of a microplate [55]. After methylation, 5-methylcytosine-specific antibody is introduced to specifically bind to the methylated DNA substrate. After which, an enzyme-linked immunosorbent assay-like reaction is performed to colourimetrically quantify the methylated DNA and hence the activity of MTase.
2.3 Fluorescent DNA MTase activity assays
Fluorescent DNA MTase activity assays involve the absorption of light which excites fluorophores to promote electrons from ground state to excited states. The electrons rapidly relax to the lowest energy levels of the excited states and then emit light of longer wavelength(s) when the electrons eventually return to the ground state. Comparing to other DNA MTase activity assays, the advantages of fluorescent assays are their straightforwardness in the detection procedures and high sensitivity [60], but the complicated optics of fluorometer often limits their use in centralised laboratory. Moreover, neither DNA nor methylated DNA is intrinsically fluorescent. It is necessary to build a fluorescence generation mechanism in fluorescent MTase assays so that the methylation events can be detected by a fluorometer. Fluorescent DNA MTase activity assays can be broadly classified into direct and amplified assays. Generally, the performance of the amplified DNA MTase activity assays is better than their unamplified counterparts. On the other hand, the downsides of the amplified DNA MTase activity assays are their high cost and sometime time-consuming multistep procedures.
2.3.1 Direct fluorescent DNA MTase activity assays
Fluorometry is one of the most frequently used forms of detection in DNA MTase activity assays. Fluorophores and quenchers are usually attached either to ss-DNA or a ds-DNA strands which manifest as hairpin probes or simply as double helix probes. These fluorescent probes have been extensively used in assays for drug development [61], in the detection of nucleic acids, proteins and enzymes [62,63]. In most fluorescent MTase activity assays, fluorescence signal generation is accomplished by fluorescence resonance energy transfer (FRET). In most cases, the fluorophores receive energy from light and when the quenchers are in close proximity, the quenchers absorb the energy and emit as heat, thus preventing the fluorophores from fluorescing [64].
A number of different types of direct fluorescent DNA MTase activity assays have been creatively developed by various research groups around the world. It can be broadly sub-categorized into two groups, with and without incorporating nanomaterials, which were discussed below. Though these innovative assays have relatively low detection limits, there still remains a driving force to construct simple, easy to operate and highly sensitive MTase activity assays.
A) Assays without the usage of nanomaterials
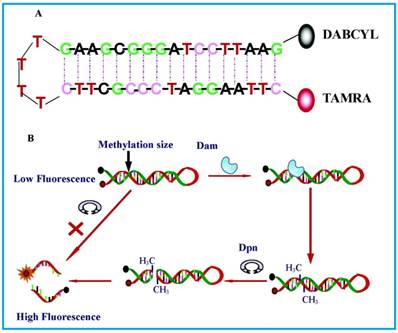
The most basic form of such assays were shown by Li and co-workers [65] in 2007 and Wood et al.[66] in 2010 in which only a fluorophore and a quencher are attached to the substrate DNA. Both groups designed DNA hairpin probes with fluorophores and quenchers respectively attached at their ends (Figure 4). The hairpin probes emit no fluorescence since the fluorophores are completely quenched by the quenchers through FRET. In order to ensure minimum false negatives, there are only a few bases at the loop section of the hairpin probes. Otherwise, a complementary sequence can hybridise with the loop, it might cause the emission of fluorescence and thus, no means of detecting DNA MTase activity. Once the hairpin probes are catalytically methylated by DNA MTase, a restriction endonuclease DpnI is activated to cleave the probes at the methylated sites, producing ss-DNA strands and very short ds-DNA strands (<10 base pairs) with the fluorophores and quenchers attached. However, due to the instability of the short ds-DNA strands (Their melting temperature is lower than the reaction temperature), they readily dissociate for the fluorescence to be restored. One major requirement for this kind of assays to be carried out successfully is the proper design of the DNA probes. The probes must have optimal number of base pairs in the double-stranded segment for them to dissociate to ss-DNA strands during enzymatic actions. Consequently, this assay enables real-time monitoring with a detection limit of 0.8 U/mL[65] and the study of the methylation kinetics with measurable rates of DNA methylation down to 0.34 ± 0.06 fmol/s [66]. It is also worth mentioning that the assay in Li's group took 30 min to complete, in contrast with 3 hours of using gel electrophoresis. However when this assay was investigated as a potential screening for inhibitory drugs, restriction endonuclease Dpn I was particularly sensitive to the antitumour drugs concentration at 100 μM, resulting in the inability of the enzyme to cleave the methylated sequence. This drawback may limit the versatility of such assays in clinical settings. The testing of MTase activity has not been done in real biological samples, which is crucial to assess the assay applicability.
Schematic representation of the DNA MTase activity assay based on fluorophore (TAMRA) and quencher (DABCYL)-tagged DNA hairpin probes. (Reproduced with permission from reference [65].)
Also without the assistance of nanomaterials, Chen and co-workers implemented an emission-based approach using induced aggregation of polyphenyl systems [67]. They proposed a DNA MTase activity assay using a positively charged tetraphenylethene derivative as an aggregation-induced emission (AIE) fluorogen [67]. Electrostatic interactions occur between AIE molecules and negatively charged DNA strands which emit strong fluorescence. In this assay, a short strand of DNA duplex containing a methylation sequence is tagged with quencher molecules at its two ends. This form of aggregation exhibits efficient super-quenching until DNA MTase and the restriction endonuclease DpnI perform their functions. Thus, the cleavage of the methylated DNA strands results in four separate single strands, two of which contain the quencher. Fluorescence is observed due to the induced aggregation of the tetraphenylethene derivative and the other two single strands without the quencher molecules which serves as a means to detect DNA MTase activity. Although this assay does not require additional enzymes to amplify the signal, its performance is impressive with a detection limit of 0.25 U/mL while maintaining a simple and economical approach. In the screening application of enzyme MTase inhibitors, a low drug concentration of 1 μM was employed, thus eliminating the possibility of enzyme Dpn I inhibition. This also confines the assay application in clinical settings, which may be a disadvantage. Activity of MTase enzyme was also evaluated in 1% calf serum at different concentrations and were shown that it was able to function relatively well in complex sample mixtures.
Only in the work of Wang et al. [68], one can find an example of label-free assay without any fluorescent labels. There are particularly several crucial issues of such merging and combinations. For example, the inclusion of nanoparticles in the strategies necessitated cumbersome nanomaterial preparation and derivatisation procedures which will be discussed in the following section. Some required complex designs of labelled DNA probes which are readily impaired by external nonspecific events. These potential concerns are avoided by adopting perylene bisimide derivatives into this assay.
Perylene bisimide derivatives are currently of considerable interest in optical applications for their remarkable properties of high fluorescence quantum yield and resistance against photooxidation [69,70]. Furthermore, their ability to form self-assembled aggregates harnesses their potential in the construction of supramolecular functionalities [71]. More interestingly, perylene excimers exhibit strong emission during their aggregation. At monomeric level, however, they emit weak fluorescence at a distinctly different wavelength. As such, switching between monomer molecules and aggregates (monomer-excimer transition) is an effective means of fluorescence signal generation. Perylene, being negatively charged, aggregates at the introduction of a cationic polymer via electrostatic interaction. The addition of ss-DNA strands of adequate length initiates competition for the cationic polymer with perylene since it is highly anionic in nature. Wang and co-workers in 2014 thus proposed a DNA MTase activity assay to use excimer emission as a platform of detecting enzymatic action [68]. In addition to perylene molecules and the cationic polymer, DNA duplexes without hydroxyl groups at their 3' termini, a terminal deoxyribonucleotidyl transferase (TdT) and deoxyribonucleoside triphosphates (dNTPs) are deployed in a MTase activity assay. TdT acts as a DNA polymerase without specificity to any oligonucleotides and incorporates mononucleotides into a growing primer at 3'-OH end [71]. In the absence of DNA MTase, there is excimer emission due to the 3'-OH-removed substrate DNA which is an inactivated primer for TdT. The cationic polymer thus favourably binds to perylene aggregates. At the enzymatic action of DNA MTase and restriction endonuclease DpnI, the substrate DNA formerly unable to be lengthened by TdT, now is cleaved to give two fragments with 3'-OH groups. Elongation of these two strands occurs with the assistance of TdT at 3'-OH ends, which eventually leads to cationic polymer binding, restoring the fluorescence of perylene monomers. Thus, the excimer-monomer transition mechanism is established and consequently the fluorescence signal associated with DNA methylation is used to monitor the methylation event with the lowest detectable activity of 0.2 U/mL. To figure out MTase activity in real biological sample, it was tested in 5% calf serum and was concluded the complex sample matrix does not affect its activity significantly.
B) Assays with the usage of nanomaterials
In the increasing popularity of nanomaterials, some groups incorporated nanoparticles such as silver nanoclusters [61], carbon nanotubes [73] and gold nanorods [74] into the assays based on the strong affinity each type of nanomaterials has with either a particular base present in the substrate DNA, with ss-DNA or ds-DNA. Consequently, the review below seeks to give an overview yet detailed explanation of these novel assays.
A dedicate design of DNA probes for the detection of DNA MTase activity has always been vital. A good manifestation is found in the work of Liu and co-workers published in 2015 [61]. They reported the construction of the DNA probes by designing hairpin-shaped probes with 5'-C-rich and G-rich-3' and incorporating silver nanoclusters (AgNCs) onto them, a fluorescence turn-off DNA MTase activity assay was developed. It is a simple and cost-effective way with a detection limit of 1 U/mL without any amplification. In recent advancements, metal nanoparticles have been a great interest and the AgNCs in particular, have been used in the detection of miRNA [75,76], protein kinase [77], etc. Based on this interest, Liu and co-workers have synthesised a hairpin probe with 5'-C-rich as Ag+ is capable of binding itself to the cytosine residues [61]. The complementary 3'-G-rich end acts as a fluorescence enhancer when in close proximity to the AgNCs. As a result, the hairpin DNA-AgNCs exhibits a fluorescence emission peak when it is intact. As DNA MTase executes its role in methylating the adenine of 5'-G-A-T-C-3' sequence, the endonuclease DpnI then cleaves specifically at the methylated site. A ss-DNA and a highly unstable ds-DNA containing the AgNCs with poly-C and poly-G tails are formed because of the enzymatic action. The dissociation of the ds-DNA occurs and fluorescence emission is thus quenched by the increased distance between the 3'-G-rich sequence and the AgNCs. Thus, the change in fluorescence intensity enables the detection of DNA MTase activity. This assay was tested out as a method for MTase inhibitor screening against 5-fluorouracil drug molecule and a mediocre IC50 value (half maximal inhibitory concentration) of 75 μM was obtained. Since the interest of MTase activity is found in real biological fluids, the implication of matrix effect on the MTase activity is of valid concern. However, the authors have not assessed this aspect of MTase activity.
Besides integrating silver in the form of nanoparticles, carbon nanotubes with their intricate nanostructure and unique optoelectronic properties, have gained much attention in recent years. Their interaction with biological materials has thus been investigated and found that multi-walled carbon nanotubes (MWCNTs) have a strong interaction with ss-DNA but not to ds-DNA [78]. This type of interaction is due to π-π stacking since both the MWCNTs and nucleobases are made up of aromatic rings. In 2014, Huang et al. harnessed these aspects of the MWCNTs' properties to DNA MTase activity assay [73]. To match the requirement of the MWCNTs, a carefully-designed DNA has to be synthesised such as it comprises both ds-DNA and ss-DNA segments. The former is vital for the catalytic action of methylation-sensitive endonucleases while the latter is used to 'wrap around' the MWCNTs. As for the mode of detection, fluorescence polarization (FP) as a signal generator is integrated into this assay. FP quantifies the fluorescence intensity of rotational motion of the fluorophore tagged on the DNA. When free in solution, the fluorescent molecule rotates at a rapid rate which leads to a low FP value. Only at a slow motion when it binds with another material or molecule does the FP value goes up. As such, a fluorescein amidite (FAM) molecule is tagged to the double-stranded end while its single-stranded end is bound to a MWCNT. DNA MTase functions itself to methylate at the specific recognition sequence and due to the inability of the restriction endonuclease DpnII to cleave the methylated site, the DNA-MWCNT conjugate continues to rotate at a slow rate with a high FP value. By the same mechanism, in the absence of the DNA MTase, DpnII will then cleave at the unmethylated site, leaving a small fragment bound to the FAM which drastically decreases the FP value. On this account, this design serves as an effective detection strategy for DNA MTase activity with a very low detection limit of 1.0 x 10-4 U/mL. The authors also verified the application of this assay with another DNA MTase, EcoRI MTase. After replacing the duplex DNA substrate specifically for EcoRI MTase, the MWCNT-based amplified assay was also able to achieve the same detection limit of 1.0 x 10-4 U/mL. This proof gives rise to the generality of this assay with different DNA MTase, obtaining an edge over the other fluorescent assays. On the downside, the method of using FP is greatly dependent on its conjugation chemistry. The size and amount of MWCNTs has to be optimised in the paper to effectuate significant and sensitive FP value. To evaluate this in real biological samples, MTase at three different concentrations were spiked into buffer solution and in diluted human serum (both containing the required reagents) and comparable responses were obtained.
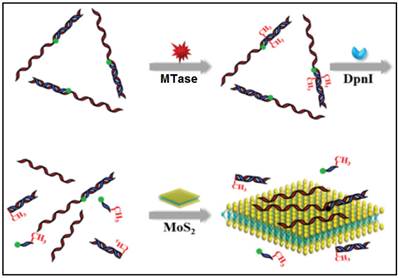
In other cases, graphene oxide nanosheets, a type of carbon nanomaterials, also found applications in DNA MTase activity assays arising from their remarkable structural and electronic properties [79]. Likewise, inorganic graphene analogues such as two-dimensional MoS2 nanosheets have also been applied in the construction of DNA MTase activity assays. For example, Deng and colleagues proposed a signal-on fluorescent DNA MTase activity assay employing a MoS2 nanosheet-mediated fluorescence quenching strategy (Figure 5) [80]. A fluorophore-tagged substrate DNA comprising a double-stranded segment containing the recognition sequence of DNA MTase and a single-stranded segment for interacting with the MoS2 nanosheets was configured in the assay. The fluorescence of the fluorophore is completely quenched by the MoS2 nanosheets before methylation due to close proximity of the fluorophore and the MoS2 nanosheets. When the substrate DNA is methylated, the fluorophore together with the single-stranded segment is cleaved by the methylation-sensitive restriction endonuclease DpnI, thereby effectively recovering the fluorescence. Based on this strategy, DNA MTase activity in the range from 0.2 to 20 U/mL can be accurately quantified with high selectivity. This assay can also be applied to screen potential DNA MTase inhibitors. The authors tried with 5-fluorouracil, an existing recognized inhibitor and reported a relatively low IC50 of 17.7 μM. In addition, the combination of methylation-resistant endonucleases and fluorescent intercalating dyes was tapped on to construct signal-on fluorescent DNA MTase activity assays [81.82]. With the usage of such novel MoS2 nanosheets, it poses some difficulty in obtaining low background signal and additional step of centrifugation is required to decrease the signal. This assay has not yet been utilised in real biological fluids, thus the implication of matrix effect on the MTase activity is still unknown.
Schematic representation of the signal-on fluorescence DNA MTase activity assay. (Reproduced with permission from reference [80].)
In a recent report, FRET was employed between gold nanorods (AuNRs) and fluorescein FAM-tagged substrate ds-DNA by Wang and colleagues [74]. Due to high affinity or aggregation formation between the AuNRs and the substrate ds-DNA, the substrate ds-DNA thus forms a hybrid with the FAM-tagged DNA which will have electrostatic interaction with the AuNRs. This causes the fluorescence of the FAM-tagged DNA to be quenched by the AuNRs, showing a much diminished fluorescence intensity. The restoration of the fluorescence of FAM occurs when the substrate DNA is methylated and digested by the endonuclease, resulting in weak electrostatic attraction between the AuNRs and the digested DNA fragments. This change in fluorescence intensity indicates the presence of the activity of DNA MTase down to the activity level of 0.25 U/mL. In their investigation, the authors altered the DNA substrate sequence in order to prove the selectivity of Dam MTase. This could become a major concern as it was unlike in other papers, where those researchers tested their assays with a few different MTases with the objective of ensuring the high selectivity of specific MTase with the same DNA substrate. In addition, in using this assay as a screening test for potential drug to inhibit MTase, there is no reported IC50 value for 5-fluorouracil, which is crucial for assessing drug effectiveness. This assay has not yet been operated in real biological fluids, thus the resultant activity of enzyme MTase is still unknown.
2.3.2 Amplified fluorescent DNA MTase activity assays
The inclusion of enzyme such as exonuclease III could be harnessed for signal amplification through target recycling. Signal amplification progressed further by tapping on the advances made in DNA research like isothermal DNA amplification techniques. Consequently, rolling circle amplification (RCA) [83] and processes involving nicking enzymes [84] have evolved into DNA MTase activity assays for improvement purposes. On the other hand, the need for label-free DNA MTase activity assays has grown tremendously in recent years. In the work by Xue and colleagues [85], though there are no fluorescent tags attached to DNA, the assay utilises a few other exogenous materials while amplifying response by isothermal exponential amplification reaction (EXPAR). With that comes a few downsides in this approach. For example, the use of several exogenous reagents like DNA ligase, polymerase, exonuclease I and III adds on to the degree of complexity and non-specificity in RCA [86].
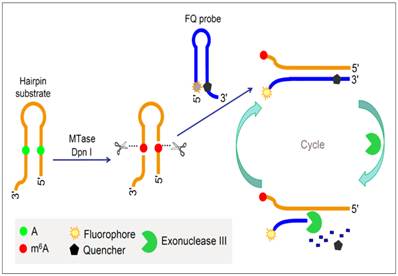
To improve the performance of fluorescent DNA MTase assays, various amplification schemes were proposed. For example, Xing et al. reported an approach of detecting DNA methylation by employing substrate DNA hairpin probes with methylation sequences in their stems and fluorophore (FAM)/quencher (BHQ) dually labelled hairpin reporting probes (FQ probes), and by utilising Exonuclease III (Exo III). The signal can be amplified by Exo III-assisted target recycling (Figure 6) [87]. The FQ probes are designed in such a way that the fluorescence of FAM is completely quenched by the quencher BHQ in their hairpin configuration. As a consequence of target recycling, the detection sensitivity increases and it is worth mentioning that a detection limit of 0.01 U/mL was obtained. As seen in Figure 6, upon successive methylation and cleavage of the stems of the substrate hairpin probes by DNA MTase and DpnI, ss-DNA strands are released. The released ss-DNA strands hybridise to the 3′ protruding termini of the FQ probes, opening up the FQ probes. Exo III then binds to the blunt 3′ termini and degrade the FQ probes in mononucleotides, releasing FAM to solution. Since Exo III only recognises and digests from the 3′ termini of ds-DNA strands with blunt or receding ends, the ss-DNA strands produced from the substrate hairpin probes remain intact during Exo III treatment because of lack of Exo III recognition sites (The ss-DNA strands is not fully hybridised to the PQ probes). Consequently, the ss-DNA strands avail themselves for the next round of hybridisation and Exo III digestion, thus establishing a target recycling mechanism. The fluorescence from FAM is therefore greatly amplified after a sufficient period of incubation with Exo III, thus allowing the sensitive detection of DNA MTase activity. Instead of the fluorophore-quencher pair, Ma and colleagues replaced the quencher with graphene oxide while keeping the exonuclease-mediated target recycling in their DNA MTase assay [88]. Leveraging on the efficient digestion ability of T7 exonuclease and the super fluorescence quenching efficiency of graphene oxide, this assay produced a high signal-to-background ratio and satisfactory sensitivity without the requirement of a specific recognition sequence for enzymatic cyclic amplification, thus greatly simplifying the design of the assay with much reduced cost. This method has not yet been brought forth in biological sample mixture.
Schematic illustration of the DNA MTase activity assay based on Exo III-mediated target recycling. (Reproduced with permission from reference [87].)
Since 2010, isothermal amplification techniques have been applied to DNA MTase activity assays, emerging much effort in the research field thereafter [83-85]. Their main attraction is that they could increase the detection sensitivity to an extremely high level in an exponential manner while retaining the flexibility to vary the type of detection methods. Consequently, the incorporation of colorimetry, fluorometry, electrochemical techniques or chemiluminescence is thus made possible. One of the most common isothermal amplification techniques is RCA whose function is to generate long DNA strands of repeated sequences with DNA polymerase as the catalytic enzyme in the synthesis. Bst DNA polymerase was selected due to its high fidelity in specificity, simplicity in protocol design and good precision in isothermal amplification process [89]. For instance, Liu and co-workers in 2014 developed a procedure which combines RCA with molecular beacons, or commonly known as fluorescence-quenched hairpin probes [83]. This procedure produced a detection limit of 0.18 U/mL and exhibited ease in operation and simplicity without having to include the usage of many enzymes. Nonetheless, it requires the specific methylation at the adenine in the 5'-G-A-T-C-3' sequence catalysed by DNA MTase and the specific cleavage at the methylated adenine catalysed by DpnI. The production of a shorter hairpin probes and ds-DNA strands then occurs, followed by the dissociation of the unstable ds-DNA strands. The long ss-DNA strand plays the role of a signal primer to hybridise to a circular DNA template and consequently triggers the replication of a much longer strand by Bst DNA polymerase (large fragment) [90] together with dNTPs. Each strand produced comprises of many repeated units which complements the sequence present in the molecular beacons. These fluorescence-quenched probes, upon short duplex formation emit fluorescence. Accordingly, the enzymatic actions of methylation and cleavage of the hairpin probes lead to an amplified signal during RCA and hence, much enhanced fluorescence intensity from the molecular beacons. This technique has not yet been tried out in real biological sample mixture.
In the realm of isothermal amplification technology, DNA nicking enzymes have been viably utilised in strand displacement amplification (SDA), nicking enzyme amplification reaction (NEAR) and EXPAR for their ability of great signal amplification. Being found within the category of restriction enzyme endonucleases, they recognise ds-DNA and specifically cleaves only one strand of the duplex [91,92]. Due to their high specificity and efficiency, they are frequently found operating in assays involving the detection of nucleic acids, protein and small biological molecules [93-96]. Thereby, Zhao and co-workers in 2013 developed a DNA MTase activity assay based on recurring cycles of the action of a nicking enzyme Nt.BbvCl [84]. In their assay, molecular beacons and Nt.BbvCl are critical participants besides the intricately designed hairpin-shaped DNA probes with 5' overhangs, DNA MTase and methylation-sensitive restriction endonuclease DpnI. Selective methylation and cleavage at the methylated site which is situated in the stems of the hairpin probes produce shorter hairpins and destabilised DNA duplexes at the reaction temperature of 37°C. The presence of the 5' overhangs pre-determines the length of the DNA duplexes. Initially, no fluorescence was observed due to the quenching ability of the close locations fluorophore and quencher as part of the design of the hairpin probes. The release of the longer ss-DNA permits the duplex formation to a signalling molecular beacon which consequently activates a turn-on fluorescence readout. Alongside with this is the mechanical action of the nicking enzyme Nt.BbvCl whose only active site is at the fluorescence-active molecular beacon. Thus, the long ss-DNA strands go through a series of recycling by triggering a series of hybridisation to the hairpin probes and nicking. With the effect of amplifying the signal as a direct consequence of DNA methylation, this assay produced in a detection limit of 0.06 U/mL within a period of 60 min of action. It is also worth mentioning that the proposed scheme has prospective in detecting MTase activity in complex biological fluids.
In a more recent work, Xue et al. coupled EXPAR to a supramolecular fluorescent zinc(II)-protoporphyrin IX (ZnPPIX)/G-quadruplex complex to significantly increase the signal arising from the methylation of adenine [85]. This approach has thus registered an extremely low detection limit of 8.6 x 10-5 U/mL. In addition to the usual hairpin DNA probes, MTase and S-adenosyl methionine, this assay uses an amplification template (denoted as “XaXaY” sequence), dNTPs, two kinds of enzymes (DNA polymerase and nicking enzyme) as well as ZnPPIX. The first step is the methylation and digestion of the palindromic sequence 5'-G-A-T-C-3' in the stem portion of the hairpin DNA probes, generating shorter hairpins and highly unstable DNA duplexes. The DNA duplexes readily dissociate into two single strands, one of which functions as a signal primer to the 'X' portion of the amplification template, which starts off the second step. The second step involves the activation of DNA polymerase to perform the replication on the template until it reaches the “a” portion (nicking site) when nicking enzyme Nt.BstNBI is activated. It cleaves at the nicking site on the newly formed strand, leaving the amplification template intact. However, only a short oligonucleotide sequence “T” forms and it has inadequate stability to remain as a duplex. This transient duplex dissociate instantaneously after its formation which then initiates the cycle of elongation and sequence “T” release at the two “X” portions. The formation of transient duplex at “Y” portion results in the production of reporter oligonucleotide R which serves as a scaffold for producing the G-quadruplex. In the third step, the G-quadruplex acts as the host to ZnPPIX and in so doing, emits a strong fluorescent signal upon interacting with ZnPPIX. It exhibits great potential in applying this system in real biological matrix since the authors had tried using five different concentrations of Dam MTase and achieved good quantification.
2.4 Chemiluminescent/bioluminescent DNA MTase activity assays
Chemiluminescent/bioluminescent DNA MTase activity assays engage chemical/biochemical reactions that produce one or more reaction intermediates in their electronically excited states, which eventually return to their ground states through the emission of light. Contrary to fluorescent and colorimetric DNA MTase assays where a light source and sophisticated optics are required, the main advantages of chemiluminescent DNA MTase assays are their simplicity of instrumentation and high sensitivity due to the absence of background light [97]. The main drawback of chemiluminescent platforms is that there is very limited choice [97]. Fortunately, for the development of chemiluminescent DNA MTase activity assays, a well-known chemiluminescent platform - luminol-peroxidase-H2O2 system can be conveniently adopted. Therefore, a commonly employed approach is the oxidation of luminol to 3-aminophthalate using hydrogen peroxide as the oxidant and horseradish peroxidase (HRP) as the catalyst [98,99]. Chemiluminescence is thus emitted and measured to quantify the level of DNA MTase activity with relatively good detection sensitivity. Isothermal signal amplification offers exponential signalling of the enzymatic action of DNA MTase and is therefore often operating together with chemiluminescent detection of DNA MTase activity [100]. Likewise, a well-known bioluminescent platform - luciferase-luciferin-adenosine triphosphate system can be conveniently adopted in the development of DNA MTase activity assays although there are only a few bioluminescent platforms available [101. In addition, the three methods discussed below did not test using real biological matrix, thus MTase activity cannot be established.
Being a peroxidase mimic, a G-quadruplex-hemin-based DNAzyme exhibits excellent peroxidase-like activity in catalysing the oxidation of peroxidase substrate in the presence of H2O2. Zeng and co-workers combined the effect of RCA to generate primers useful for DNAzyme production with the purpose of mimicking HRP to catalyse the chemiluminescence-generating oxidation of luminol [102]. Several enzymes were utilised in their specific and crucial roles in this assay, which includes DNA MTase, a restriction endonuclease DpnI, a DNA polymerase and a nicking enzyme Nb.BsmI. This assay encompasses three main processes. In the first and of high importance step, DNA MTase and DpnI dutifully perform their functions in methylation at adenine residues of the stem part of a hairpin DNA probe and in cleaving off the methylated adenine site. This gives rise to a shorter hairpin probe with a 5' blunt terminus and a ds-DNA of reasonable stability. The second step necessities the course of primer-generating RCA to be at 60°C. At this temperature, the shorter hairpin probe disassociates to two ss-DNA strands, which complementarily binds to the circular template and initiates the RCA process by adding mononucleotides with the assistance of DNA polymerase. With many repeated units formed on the elongated product, Nb.BsmI selectively cleaves at such and in so doing, produces many new primers as well as short DNA strands capable of forming G-quadruplexes to bind to hemin. This further amplifies the RCA process in an exponential manner and in the process, produces numerous DNAzyme molecules for the detection of DNA MTase activity. This leads to the third step whereby the multitude of DNAzyme catalyses the oxidation of luminol, yielding a substantial chemiluminescence signal. In the absence of DNA MTase, no chemiluminescence is detected since the unmethylated hairpin stays in its original state, preventing from hybridizing to the circular primer. The exponential power of the amplification produced an extremely low detection limit of 1.29 x 10-4 U/mL.
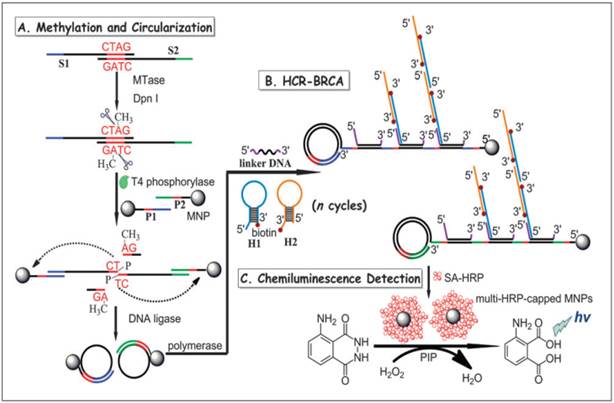
In another report, a combination of hybridisation chain reaction (HCR), branched rolling circle amplification (BRCA) and the coupling of magnetic nanoparticles with the aim of catalysing the chemiluminescence-generated reaction was reported by Bi and co-workers in 2013 (Figure 7) [103]. As shown in Figure 7, consecutive enzymatic actions of DNA MTase and restriction endonuclease DpnI efficiently resulted in four fragments after the hybridisation of DNA strands S1 and S2. At the 5' ends of two newly-formed shorter fragments, T4 phosphorylase then plays its part to add a padlock-like probe so as to prepare for RCA after T4 DNA ligase joins the complementary ends of the longer DNA fragment to produce a circular template. At the action of DNA polymerase, RCA commences to produce repeating sequences along the elongated strand which is designed to complement linker DNA to yield branched-like structures of overhanging ends of each linker DNA. HCR events can then be propagated between alternating hairpin probes H1 and H2. Since biotin molecules are tagged at 3' termini of H1 and H2 probes, they recognise streptavidin-HRP (SA-HRP) conjugates and bind on to them. At this point, multitudes of HRP-labelled RCA products are the outcome of the HCR-BRCA process of a single DNA MTase methylation and DpnI cleavage. The final stage of this assay requires the “fishing-out” of the HRP conjugates by the magnetic nanoparticles of which catalyse luminol oxidation enhanced by p-iodophenol, which manifests a powerful amplification of chemiluminescence readout.
Schematic illustration of the HCR-BRCA-based DNA MTase activity assay for by the production of multi-HRP-capped magnetic nanoparticles to catalyse the generation of chemiluminescence of the HRP- luminol-H2O2 system. (Reproduced with permission from reference [103].)
Similarly, by adopting the luciferase-luciferin-adenosine triphosphate bioluminescent platform coupled with a methylation-resistant cleavage and in vitro protein expression, a highly sensitive bioluminescent assay was developed for the detection of DNA MTase activity [104]. Using luciferase reporter DNA as substrate DNA for the DNA MTase and MboI as the methylation-resistant endonuclease, DNA MTase activity is quantified by measuring the bioluminescence of the expressed luciferase since methylated luciferase reporter DNA that resists Mbol cleavage could be expressed in cells to produce luciferase. The assay produced a wide dynamic range between 0.2-100 U/mL with a detection limit of 0.08 U/mL. Being isothermal in nature, the use of the methylation-resistant cleavage and protein expression approach offers the possibility of in vivo DNA MTase activity imaging and DNA MTase inhibitor screening.
2.5 Electrochemical DNA MTase activity assays
Electrochemical DNA MTase assays involve the measurements of electrical quantities, such as current, voltage, charge and resistance, to reflect the activity of DNA MTase. They are advantageous over many other types of DNA MTase activity assays because of their low cost, high sensitivity, the ability to perform on-site monitoring and great amenability to miniaturisation and integration with microfabrication technology. The development of electrochemical techniques for bioanalysis has always been helmed as one of the popular research areas in modern analytical chemistry [105,106]. Both direct and amplified electrochemical DNA MTase activity assays have been proposed. The following section details the development of electrochemical DNA MTase activity assays. Generally, two approaches, namely DNA methylation-initiated cleavage and the use of methylated DNA binding protein coupled with electrochemical reporters or electrochemical luminescence generators, are employed in the construction of electrochemical DNA MTase activity assays. Similar to fluorescent assays, to further enhance sensitivity, various enzymatic amplification strategies are incorporated in the electrochemical DNA MTase activity assays. However, comparing to fluorescent DNA MTase activity assays, the amplification strategies are rather limited because of the heterogeneous nature of electrochemical detection.
2.5.1 Direct electrochemical DNA MTase activity assays
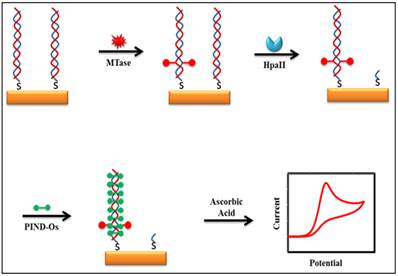
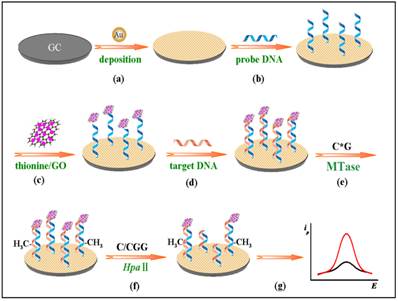
Some of the recently developed electrochemical platforms for screening and monitoring the activity of DNA MTase include electrochemical assays based on restriction endonucleases together with [Ru(NH3)6]3+[107], ferrocene and its derivatives [108,109], coomassie brilliant blue G250 [110], an electroactive and catalytic intercalator [111], methylene blue [112-116], carbon nanotubes [117], graphene [118] and graphene oxide [119], methylation sensitive cleavage utilising terminal transferase-mediated extension [120] and the use of methyl binding domain protein (MBD) protein [121-123] and antibody [124]. For instance, a simple and highly sensitive electrochemical DNA MTase activity assay was proposed by Deng and colleagues (Figure 8) [111]. After a monolayer of a substrate ds-DNA containing the endonuclease recognition sequence of 5′-CCGG-3′ is immobilised on a gold electrode, successive incubations of the substrate DNA-coated electrode with DNA MTase and endonuclease HapII result in the methylation of the substrate DNA and subsequent cleavage of unmethylated DNA off the electrode [125]. Since the methylated substrate DNA resists HapII digestion, only the methylated DNA remains on the electrode surface after the incubations. A final incubation of the treated electrode in a solution containing a threading intercalator-(N,N′-bis(3-propylimidazole)-1,4,5,8-naphthalene diimide functionalised with two electrocatalytic redox Os(bpy)2Cl+ moieties introduces the intercalator to the methylated DNA through threading intercalation. After a thorough rinsing, the electrode is tested in ascorbic acid solution where the intercalator on the methylated DNA catalyses the oxidation of ascorbic acid, producing a measurable current signal, thus enabling the assessment of the methylation event and specifically the activity of DNA MTase. Much better sensitivity was obtained by working with the electrocatalytic oxidation of ascorbic acid instead of the intercalator itself although the amount of the intercalator on the electrode surface also directly correlates to the activity of DNA MTase. A linear relationship between the activity of DNA MTase and the catalytic oxidation current of ascorbic acid was obtained between 0.08 and 120 U/mL with a current sensitivity of 0.046 μA mL/U. The concept behind this technique is based on the action of the restriction enzyme with specificity toward methylation, resulting in the electrochemical signal being detected by the conversion of the methylation state of the target DNA. This highly sensitive method has not yet been experimented with real biological sample matrix for more appropriate assessment.
Schematic representation of the electroactive and catalytic intercalator-based DNA MTase activity assay. (Reproduced with permission from reference [111].)
Moreover, a multiplexed signal-on electrochemical assay using DNA-modified electrodes has also been proposed by Muren and Barton [121]. As for real-world sample analysis, human DNA MTase1 (DNMT1) from the crude lysate of colorectal tumour biopsies was used as a target in the assay to measure the DNA MTase activity by the same group [126]. Hemimethylated substrate ds-DNA strands tagged with methylene blue molecules are immobilised onto a low-density electrode array, exposing the hemimethylated 5'-CG-3' methylation site where DNMT1 targets. The methylene blue labels are used as an electrochemical mediator to shuttle electrons from solution species to the electrode across the ds-DNA layer. The occurrence of a fully methylation by DNMT1 at the 5'-CG-3' site results in the inability of the restriction enzyme to successfully cleave the DNA. The methylene blue labels on the methylated ds-DNA strands are thus fully functional in mediating DNA charge transfer for electrochemical detection. As a result, a measureable current is recorded and its magnitude directly correlates to the activity of DNMT1. On the other hand, when the ds-DNA strands remains hemimethylated in the absence of DNMT1, there is clearance for specific binding by the restriction enzyme onto the ds-DNA for cleavage. The cleavage of the hemimethylated ds-DNA strands causes a significant reduction in the quantity of the ds-DNA strands along with their labels on the electrode surface, thereby producing a much-diminished current.
It is undeniable that researchers are now equipped with better understanding in cell biology. Coupling with extensive studies in molecular biology, researchers are now tapping on the vast potential of immunology in population screening and disease diagnosis. The high specificity arises from the sequence rearrangement of the variable chains in the different types of immunoglobulins upon adherence to foreign or abnormal biological receptors and extracellular matrix components [127]. Such an ability of the immunoglobulins enables researchers to take the advantage of the high specificity and completely compatibility with DNA MTase to produce immunoglobulins (antibodies) for DNA MTase [128,129]. One such technique in this context is the utilisation of electrochemical immunoassay to the molecular recognition of the epitopes of MBD protein. MBD protein is chosen as a good candidate in the detection of methylation because four out of the five polypeptides belonging to the MBD protein family have high affinity and specificity toward CpG motifs that have been symmetrically methylated [130-132]. Conventional methods in the detection of methylation involve chemical treatment and labelling of 5-methylcytosine. Those processes are in principle undesirable because sample treatment often leads to skewed data due partly to the loss and chemical changes of the analytes [133,134]. The high target specificity of MBD protein toward methylated CpG sequences offers an excellent opportunity for the construction of much simplified MBD protein-based assays for the detection of methylated NDA and DNA MTase activity by eliminating most of the sample processing steps.
Recently, Hiraoka et al. demonstrated the use of MBD protein in isolation and precipitation of methylated DNA and quantify the DNA methylation levels by introducing a substrate DNA to luciferase-fused zinc finger assay [135]. The advantage of the MBD-protein's specificity to methylation in conjunction with the elimination of sample treatment with bisulphate and PCR amplification, makes MBD protein a useful tool for probing DNA methylation and DNA MTase [135-138]. For instance, Xu et al. utilised a recombinant MBD1 protein acting as the DNA methylation recognition unit, coupling with a signal unit using a protein staining agent coomassie brilliant blue G250, to develop an electrochemical assay specifically targeting the detection of DNA MTase activity [121]. Another example can be found in the work of Yin et al [110]. After a substrate ds-DNA on an electrode is methylated at the symmetrical sequence of 5'-CCGG-3', a MBD protein MeCP2 is introduced which selectively binds to the methylated 5'-CCGG-3'. This mode of operation is similar to the previous assay. The methylated ds-DNA site is bound by the specific recognition unit of MeCP2 protein. Further, MeCP2 carries a His tag at its C-terminal end for selective binding by the anti-His tag antibody from HRP labelled and immunoglobulin G functionalised AuNPs (HRP-IgG-AuNPs). The HRP-IgG-AuNPs act as electrochemical signal amplifiers due to the loading of a large number of the IgG-HRP conjugates. Thus, HRP on the electrode efficiently catalyses the oxidation of hydroquinone to benzoquinone in the presence of H2O2. Current generated by the electrochemical reduction of benzoquinone is therefore utilised to analyse the activity of DNA MTase. Owing to the high specificity of MeCP2, excellent selectivity down a single base mismatch was observed. Upon further verification, it is believed that this assay could be applied for screening DNA MTase inhibitors and for developing new anticancer drugs.
2.5.2 Amplified electrochemical DNA MTase activity assays
With the rise of nanotechnology in biomedicine, bioassays are now utilising the beneficial traits of nanoparticles in their construction since the production of highly controllable nanoparticles can be accomplished although some of the nanoparticle-biomolecule conjugate preparation protocols are cumbersome and time-consuming. It is a desirable trait that the specifications and functional groups on the nanoparticles can also be controlled to increase their biocompatibility, specificity and functionalities [139-141]. AuNPs, for example, have been developed for a variety of applications ranging from biomedicine to catalysis due to their inertness, biocompatibility, stability and customisable functionalities [142-144]. As such, they have been used as nanocarriers, acting as probes by attaching many signal tags such as oligonucleotide [145], enzyme [146] and antibody [147]. When such AuNPs bind to a substrate or tag, they result in an amplification of signal due to agglomeration of numerous signal tags onto the AuNPs. This greatly increases the sensitivity of the assay. In addition to the nanoparticles, enzymes are also deployed to enhance the assay with the objective to increase detectability by generating a large number of electroactive species solution [148,149]. For example, alkaline phosphatase (ALP), one of the common enzymes that performs hydrolysis on various phosphate esters under alkaline conditions, is used in the construction of electrochemical assays [149]. APL has so far been used in applications such as electrochemical immunosensors for antibody detection to improve the linear range and sensitivity of the assay as reported by Yang and Gao [150]. Hence, by introducing these two entities into electrochemical immunoassay, a highly sensitive assay for DNA MTase activity is expected. Toward to this goal, the recombinant MBD1 protein, ALP labelled IgG tagged AuNPs (ALP-IgG-AuNPs) and anti-his-tag antibody tagged AuNPs (His-tag-AuNPs) were integrated into an electrochemical assay for the detection of DNA MTase activity [121]. A dual-amplification scheme is realised by the electrochemical oxidation of 1-naphthol produced from the hydrolysis process of enzyme ALP on its substrate. The mechanism of action involves the target recognition of methylated CpG sequences on ds-DNA by His-tagged recombinant MBD1 protein, followed by specific binding by His-tag-AuNPs. These His-tag-AuNPs then trigger the aggregation of the ALP-IgG-AuNPs through another specific binding with the antibody that is attached to the His-tag-AuNPs. The high loading capability of the antibody on the His-tag-AuNPs has resulted in a huge number of the ALP-IgG-AuNPs binding and tagging the methylated sequence of the substrate DNA. Hence, a much amplified electrochemical signal is produced due to the large production of 1-naphthol when ALP catalyses the hydrolysis of its substrate [121]. Another feature of the assay is that another enzyme, HpaII endonuclease, is used to increase the specificity of the electrochemical signal generation process. This is done through the digestion of un-methylated CpG DNA sequences by cleaving 5'-CCGG-3'. Meanwhile, this digestion is blocked by methylation, hence resulting in the inability in the formation of the ALP-IgG-AuNPs conjugates to interference or contribute to the detection level of actual amount of methylated CpG sequences [121]. In another study, a similar approach to the detection of DNA MTase activity was reported by Jing and co-workers [115]. In their assay, AuNPs are also used to enhance the electrochemical signals through amplification. The mechanism of action involve the accumulation of methylene blue onto the AuNPs conjugated with ds-DNA sequences that have been methylated by DNA MTase and digested by MboI restriction endonuclease. The intercalation of methylene blue onto the ds-DNA creates electron-transfer paths through methylene blue molecules through which a current signal is detected. Un-methylated ds-DNA strands are digested by MboI at the 5'-G-A-T-C-3' palindrome sequence site [151].
Other nanomaterials such as graphene oxide (GO) have all been tested as signal generators in electrochemical MTase activity assays. An example can be found in the work of Li and co-workers (Figure 9) [119]. In their assay, GO is utilised to amplify the signal and HpaII is engaged to improve the selectivity of the assay. The principle of the assay is similar to the electrochemical signal amplification by the electrocatalytic intercalator as discussed earlier [111]. In this assay, a significantly amount of the signal generator thionine is loaded onto GO. Consequently, as low as 0.05 U/mL MTase activity was detected. In addition, the excellent discrimination ability of HpaII between methylated and unmethylated substrate DNA strands produced single-base mismatch selectivity. No reported assessment was done in the presence of real biological matrix in this newly developed method.
Schematic illustration of the procedures of the gene-specific DNA methylation detection and DNA MTase activity assay based on the electrochemical signal amplification of GO and restriction endonuclease. (Reproduced with permission from reference [119].)
2.5.3 Photoelectrochemical DNA MTase activity assays
Besides engaging conventional electrochemical techniques for the detection of DNA MTase activity, Shen et al. reported the utility of photoelectrochemical detection in a DNA MTase activity assay [152]. The photoelectrochemical detection was realised by signal amplification of exciton energy transfer (EET) on the basis of interaction between AuNPs and cadmium selenate quantum dots (CdSe QDs). Because of the substantial overlapping of the plasmon energy band of the AuNPs with the exciton energy band of the CdSe QDs, a noticeable decrease in photocurrent of the CdSe QDs is expected. Utilising this phenomenon, a photoelectrochemical assay was proposed by depositing a thin film of titanium dioxide onto an indium tin oxide electrode together with the CdSe QDs so as to harvest the electron transfer from EET. Functional probes are constructed by attaching probe DNA onto the AuNPs which are then immobilised onto the electrode [152]. With this configuration, there is a competition between the photocurrent signal and EET effect due to the interaction of the AuNPs and CdSe QDs. As such, the presence of a strong EET effect when the AuNPs are in close proximity to the CdSe QDs effectively counters the photocurrent from external light source, resulting in a low photocurrent generation. This only occurs in the presence of ds-DNA methylation by DNA MTase after the lysates or analytes are introduced to the electrode for hybridisation with the probe DNA on the AuNPs. This is because methylation prevents binding of the restriction enzyme HpaII endonuclease onto the hybridised DNA for cleavage. Hence, strong EET is preserved [153]. On the other hand, unmethylated ds-DNA on the electrode is cleaved off by HpaII, resulting in the detachment of the AuNPs from the CdSe QDs, and consequently the reduction of EET. This, in turn, allows a high proportion of photocurrent to be received by the electrode and a high current signal registered [152]. This newly developed photoelectrochemical method has not been assessed using real biological fluids for quantification of MTase activity.
2.6 ECL DNA MTase activity assays
ECL, also known as electrogenerated chemiluminescence, is a marriage of electrochemistry and luminescence. Like chemiluminescence, to achieve ECL, one or more intermediates in electronically excited state must be produced in a highly exergonic reaction and upon relaxation to the ground state light is emitted. Instead of chemical reactions found in chemiluminescence, ECL generates the intermediates(s) via an electrochemical reaction usually electrooxidation. Since the discovery of ECL in the 1960s through the use of aromatic hydrocarbons [154,155], great advances have been made especially after the discovery of the tris-2,2'-bipyridylruthenium(III) ECL system in the 1970s [156]. Through introducing chemically active moieties to tris-2,2'-bipyridyl ruthenium(III), this ECL-generating species has been used extensively as an ECL label in the design of various bioanalytical platforms for highly sensitive and selective detections of a wide range of bioanalytes ranging from small biomolecules to proteins and nucleic acids [157,158]. Apart from sharing similar attractive features with chemiluminescence like simplicity of instrumentation and high sensitivity due to the absence of background light [159], ECL offers precise control of reaction kinetics by controlling the applied potential, compatibility with solution-phase and thin-film formats and excellent temporal and spatial control [157-160].
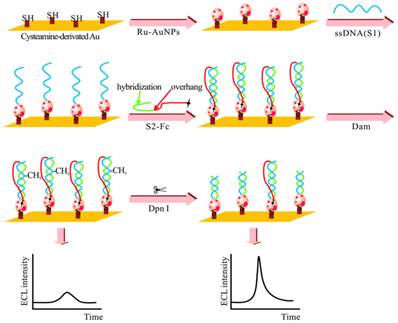
The adoption of ECL in DNA MTase activity assays is expected to deliver excellent performance in terms of simplicity, sensitivity and portability. Indeed, detection limits of ECL DNA MTase activity assays, ranging from 10-2 to 10-6 U/mL [161-167], which are at least one order of magnitude lower than most fluorescent, colourimetric and electrochemical assays. For example, a highly sensitive ECL DNA MTase activity assay was developed by labelling a substrate DNA with tris-2,2'-bipyridylruthenium(III) and the adoption of the popular DNA MTase/HpaII pair (Figure 10) [161]. First, ss-DNA strands together with tris-2,2'-bipyridylruthenium(III) labelled AuNPs are immobilised onto a gold electrode which subsequently form the substrate DNA through hybridisation with their complementary strands labelled with ferrocene moieties. Then, successive incubations in DNA MTase and Dpn I lead to the removal of all methylated substrate DNA together with their ferrocene labels from the electrode, leaving only the unmethylated ones with their ferrocene labels on the electrode. The amount of ferrocene labels found on the electrode and hence the degree of ECL suppression are directly associated with the activity of DNA MTase. Under optimal conditions, a detection limit of 0.03 U/mL was achieved. Real biological fluids were not included in the experiments to further assess MTase activity. Other ECL labels such as tris(1,10-phenanthroline) ruthenium [163-165] and luminol [166] were also utilised in the construction of ECL DNA MTase activity assays. Moreover, nanomaterials like graphene oxide [164,165] and silver nanoparticle/graphene oxide composites [166] were conjugated to the ECL labels in an attempt to further enhance the sensitivity of the ECL assays. Unfortunately, the enhancement in sensitivity was not as significant as expected.
Schematic representation of the ECL DNA MTase activity assay. (Reproduced with permission from reference [161].)
2.7 Other MTase assays
Leveraged on the nanopore technology developed to direct sequence DNA, several attempts were made in the development of nanopore-based devices to detect and quantify methylated DNA and correspondingly MTase activity [168-170]. For example, Laszloa and co-workers reported the use of engineered porin A to detect methylated DNA through recording the ion current through the nonopore [168]. Distinct differences in the magnitude of the ion current were observed between methylated and unmethylated DNA strands. A careful comparison of the ion currents and their frequencies reveals the precise location and abundance of methylated CpG sites. More recently, surface-enhanced Raman spectroscopy in conjunction with HCR was proposed for the detection of MTase activity [171]. Briefly, MTase substrate DNA tethered to magnetic beads was selectively methylated in the presence MTase. After cleaving all unmethylated substrate DNA strands on the magnetic beads by HpaII endonuclease, the methylated DNA on the magnetic beads triggered the HCR reaction and subsequent accumulation of silver nanoparticles through biotin-streptavidin interaction, thus leading to significantly amplified SERS response. A linear range from 0.1 to 10.0 U/ml with a correlation coefficient of 0.97 and a detection limit of 0.067 U/ml were obtained.
Comparison of the various DNA MTase activity assays
| MTase assay | Mechanism | Signalling Process | Advantages | Disadvantages | Sensitivity | Ref |
|---|---|---|---|---|---|---|
| Radio DNA MTase assays | Isotope-labelling detection | Isotope-labelled methyl moiety | Convenient detection | Harmful radioactive agents | NA | 46 |
| Polymerase Chain Reaction (PCR) | Methylation-specific PCR | Conversion of unmethylated cytosines to uracils | High accuracy of sequencing | Expensive equipment; tedious sample preparation; cumbersome detection scheme | NA | 48 |
| Colourimetric DNA MTase Activity assays using AuNPs | Visual observation of a colour change or the spectrometric measurement of an absorption spectrum associated with DNA MTase | Change in UV-vis absorption of distance-dependent plasmic absorption among AuNPs | Specificity of enzymes; signal amplification | Complex with the use of multiple enzymes; maintain physiological conditions | 0.5 U/mL | 50 |
| Catalysis of the oxidation of 2,2'-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid by DNAzyme | Specificity of enzymes; high sensitivity | Requires proper design of probe | 0.25 U/mL | 53 | ||
| Direct fluorescent DNA MTAse activity assays | Involvement of absorption of light which excites fluorophores to promote electrons from ground state to excited states | Fluorescence signal generated by fluorescence resonance energy transfer (FRET) | Real-time monitoring; direct detection | Requires proper design of probe | 0.8 U/mL | 65 |
| Emission spectrum of aggregation of perylene excimers | High fluorescence quantum yield; resistance against photooxidation; self-assembled aggregation | Requires proper design of probe | 0.2 U/mL | 72 | ||
| Fluorescence turn-off DNA MTase activity assay | Simple; cost-effective | Requires proper design of probe | 1 U/mL | 61 | ||
| Fluorescence polarization as signal generator | Highly sensitive | Complex with the use of multiple enzymes | 1.0x10-4 U/mL | 73 | ||
| MoS2 nano sheet-mediated fluorescence quenching strategy | A signal-on fluorescent DNA MTase activity assay; high sensitivity | Complex with the use of multiple enzymes | 0.14 U/mL | 80 | ||
| FRET employed between AuNRs and fluorescein FAM-tagged substrate ds-DNA | High sensitivity | Requires proper design of probe | 0.25 U/mL | 74 | ||
| Aggregation-induced emission (AIE) of fluorophores | High performance; simple; economical | Requires proper design of probe | 0.25 U/mL | 67 | ||
| Amplified fluorescent DNA MTase activity assays | Amplification of fluorescence to enhance detection | FRET dually labelled hairpin reporting probes (FQ probes) | Amplification of signal | Requires proper design of probe | 0.01 U/mL | 87 |
| Rolling circle amplification of DNA with molecular beacons | simple; does not require many enzymes; amplification of signal | Requires proper design of molecular beacons | 0.18 U/mL | 83 | ||
| Nicking enzyme with molecular beacons | Amplification of signal; fast | Requires proper design of probe | 0.06 U/mL | 84 | ||
| Supramolecular fluorescent zinc(II)-protoporphyrin IX/G-quadruplex complex | Extremely low detection limit | Use of multiple enzymes and complexes | 8.6x10-5 U/mL | 85 | ||
| Chemilumininescet/ bioluminescent DNA MTase activity assays | Engage chemical/biochemical reactions that produce one or more reaction intermediates in their electronically excited states, which eventually return to their ground states through the emission of light. | Oxidation of luminol to 3-aminophthalate | High sensitivity | Use of multiple enzymes and complexes | 1.29x10-4 U/mL | 102 |
| Bioluminescence expressed by luciferase reporter DNA | Isothermal; high sensitivity | Requires proper design of reporter DNA | 0.08 U/mL | 104 | ||
| MTase Electrochemical DNA MTase activity assays | The measurement of electrical quantities to reflect the activity of DNA | Electrocatalytic oxidation current of ascorbic acid | Low Cost; high sensitivity | Tedious treatment and preparation of electrodes | 0.046 uA mL/U | 111 |
| Amplified electrochemical DNA MTase activity assays | Amplification of electrical quantities to enhance detection | Signal amplification using graphene oxide | High sensitivity; signal amplification; improved selectivity | Need to functionalise graphene oxide | 0.05 U/mL | 119 |
| Photoelectrochemical DNA MTase activity assays | Signal amplification of exciton energy transfer | Measurement of photocurrent due to overlapping of plasmon energy band of AuNPs with exciton energy band of CdSe QDs | Signal amplification | Tedious treatment and preparation of electrodes; requires proper design of probe | 0.0042 U/mL | 152 |
| Electrochemiluminescence DNA MTase activity assays | Light emission in a highly exergonic reaction upon relaxation to the ground state | ECL suppression by ferrocene labels on substrate DNA | High sensitivity | Require functionalised AuNPs and substrate DNA | 0.03 U/mL | 161 |
3. Challenges and prospects
Epigenetic modifications play in a central role in creating diversity and maintaining specific cellular phenotypes and activities. They are also crucial in the stability of the DNA and chromosome structures. DNA methylation is one of the most important epigenetic modifications. It is of our best interest to understand and quantify DNA MTase activity as well as DNA methylation patterns in various cells to a deep understanding of the fundamentals and correlate them with human health. In addition, aberrant DNA MTase activity has been known to link to various disorders and diseases such as cancer. As such, a plethora of techniques and approaches have been developed to screen the activity of DNA MTase. Unlike conventional DNA MTase activity assays such as radiolabelling methylation-specific PCR and HPLC which can directly differentiate methylated and unmethylated DNA, these newly developed assays have to be coupled with other biological processes like enzymatic reactions and immunoreactions, thus bringing additional complexity and uncertainty into the assays. On the other hand, ingenious combinations of detection techniques and approaches also give rise to unique DNA MTase activity assays. Among them, fluorometry and electrochemical methods are the most studied detection techniques in DNA MTase activity assays. These two groups of detection techniques work on the principles of (i) direct detection of DNA MTase activity and (ii) detection of DNA MTase activity following an amplification process. Nonetheless, no one assay is superior to another, and the user should carefully analyse the suitability of the assays to the specific task before employing them. Despite impressive progress in the research and development of DNA MTase activity assays, none of them has clearly represented a fundamental breakthrough, becoming a much better alternative to conventional DNA MTase assays. Future research efforts should, therefore, be dedicated to the development of DNA MTase activity assays with much improved sensitivity, selectivity and robustness without engaging any complicated sample preparation and detection protocols. It is believed that the lack of available assays to measure DNA MTase activity in a highly reproducible, quantitative and sensitive manner continues to be the driving force in DNA MTase activity assay research.
Sensitivity is one of the most important criteria for a successful implementation of DNA MTase activity assays in molecular diagnostics. Biological specimens for analysis are usually limited by their quantities such that millilitres of blood can only yield a few nanograms of DNA. More importantly, the lack of highly sensitive DNA MTase activity assays has long plagued scientists from bypassing tedious and multistep sample preparation/sample enrichment procedures. In the last decade, extensive efforts have been directed to address this problem. Some of the reported assays are capable of detecting extremely low DNA MTase activity. Unfortunately, the high sensitivity is only achieved with highly purified sample or even pure synthetic samples. Significant discounts may have to be taken when working with clinical samples, or even worse, the clinical samples may completely disable some of the assays. So each assay must be thoroughly evaluated in complex biological matrices. DNA MTase activity assays intended for clinical use should tolerate samples of different matrices and origins ranging from cultured cells, tissues, to body fluids. One basic challenge associated with clinical applications is the complexity and variable composition of sample matrix. For example, clinical samples may have an inhibitory effect on the assay or have a significantly high background such as light absorption, light scattering or auto-florescence. Furthermore, sample collection protocols have to be carefully selected and examined as some may not yield reproducible results and other may introduce contaminants which either inhibit DNA MTase activity or are detrimental to the assay. To reduce human involvement, further work should also be directed to the development of automatic microfluidic devices to process clinical samples and possibly integrate with amplification and detection.
The reliability of DNA MTase assays is largely dependent on the precise evaluation of DNA MTase activity. However, a widely accepted reference assay that can quantitatively compare and assess DNA MTase activity regardless sample origin and matrix has not yet been developed. To achieve an acceptable precision and to minimise assay variability, a candidate reference material, such as methylated DNA could be simultaneously detected with the DNA MTase activity to assess whether the level of methylation is a direct consequence of the observed DNA MTase activity.
With increasing evidence showing that DNA MTase activity is closely associated with human health, DNA MTase activity assays will eventually advance into clinical use. In fact, there is the expectation that DNA MTase activity assays could serve as a prognostic or screening tool. However, accurate and quantitative measurements, standards and normalisation are needed to better evaluate the clinical relevance of DNA MTase. For instance, there is a need for a reliable and fully characterised threshold of signal-to-function ratio as the standard to validate test results across different platforms. Further fundamental research and large cohort screening with appropriate cases and controls are also needed. Although advances in the DNA MTase activity assays so far are very impressive and some of the proposed assays can quantify DNA MTase activity precisely in vitro, the direct visualisation of DNA MTase activity in vivo and in real-time will enable quantitative measurements at the single cell level. Conventional DNA MTase assays like radiolabelling and HPLC can only measure the activity of DNA MTase in cell lysates or purified samples at discrete time points, thus are not applicable in neither real-time nor in vivo applications. Toward this goal, fluorescence and bioluminescence imaging are two possible approaches for in vivo DNA MTase imaging since colourimetry and electrochemical techniques are not well-suited for in vivo applications. For example, transiently expressed fluorescent protein-fused DNA MTase coupled with a DNA methylation mechanism-based inhibitor (5-azacytidine) was developed for direct visualisation of DNA MTase activity in cultured cells. During methylation, 5-azacytidine is incorporated into the newly synthesised DNA at nuclear replication sites together with the fluorescent protein-fused DNA MTase at these sites, thus enabling the comparison of mutations and inhibitors that affect regulation and the catalytic activity of DNA MTase in single living cells in their native microenvironment [172]. More research should be dedicated toward DNA MTase imaging with much simplified protocols and affordable agents and probes. Only when the results obtained in multiple clinical centres are thoroughly validated, then as a biomarker, DNA MTase for early cancer diagnosis can be realised. Finally, DNA MTase will be best utilised as a biomarker for primary screening tool to diagnose and monitor disease status and to triage high-risk populations if clinical validation of DNA MTase activity assay eventually justifies the use of DNA MTase as a biomarker for diagnosis. However, to significantly enhance the reliability and the accuracy of early cancer diagnosis, instead of a single biomarker, like DNA MTase, a panel of biomarkers made up of both transcriptional and post-transcriptional biomarkers is essential. In addition to DNA MTase, microRNAs [173] and circulating tumour cells [174] in blood are closely associated to cancer incidence, as indicated in numerous studies, showing their potential as biomarkers in diagnosis and prognosis of cancer. We anticipate that a panel of biomarkers can be constructed by combining these three groups of biomarkers with other proteinic biomarkers such as carcinoembryonic antigen, alpha-fetoprotein and prostate-specific antigen. Advancing on the identification of diagnostic, prognostic and/or predictive biomarkers has enormous importance for the development of the next generation of diagnostic tools for cancer and the therapeutic options for cancer patients.
4. Conclusion
In this article, the advances and challenges of DNA MTase activity assay research are thoroughly reviewed. Over the past decade, significant progress has been made in DNA MTase activity assays. Several approaches have shown promise in the development of clinically viable assays for use in cancer diagnosis, prognosis and therapy. Nonetheless, this field is still in its infancy particularly with regard to its use as a biomarker in molecular diagnostics. Currently, researchers are working tirelessly to correlate DNA methylation with various malignant diseases. However, the low contents of DNA MTase in biological specimens imply that a sensitive, selective and cost effective DNA MTase assay for molecular biology research and biomedical applications at point-of-care is highly desirable. While conventional assays such as methylation-specific PCR and radiolabelling remain as the standard assays for the detection of DNA MTase activity, they are not without limitations. Therefore, emerging DNA MTase activity assays reviewed in this article offer many attractive features and could play a leading role in future DNA MTase research and applications. For example, fluorescent and bioluminescent assays may allow direct visualisation of DNA MTase activity in living cells. Colourimetric and electrochemical assays not only achieve comparable or improved sensitivity and specificity with respect to the standard radiolabelling assay, but also are considered to be more advantageous because of their simplicity and high portability. Such assays may eventually open the door to routine DNA MTase activity assays and molecular diagnostics with new perspectives toward the management and diagnostics of diseases. The outlook of DNA MTase activity assays certainly remains positive and breakthroughs and clinical applications of DNA MTase activity assays are realisable in future.
Acknowledgements
This work is supported by Ministry of Education (Grant No R-143-000-588-112).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Das PM, Singal R. DNA methylation and cancer. J. Clin. Oncol. 2004;22:4632-4642
2. Jurkowska RZ, Jurkowski TP, Jeltsch A. Structure and function of mammalian DNA methyltransferases. ChemBioChem. 2011;12:206-222
3. Suzuki MM, Bird A. DNA methylation landscapes: provocative insights from epigenomics. Nat. Rev. Genet. 2008;9:465-476
4. Lee GE, Kim JH, Taylor M, Muller MT. DNA methyltransferase 1-associated protein (dmap1) is a co-repressor that stimulates DNA methylation globally and locally at sites of double strand break repair. J. Biol. Chem. 2010;285:37630-37640
5. Liu SN, Tu YQ, Li W, Wu P, Zhang H, Cai CX. Assay Methods of DNA Methylation and Their Applications in Cancer Diagnosis and Therapy. Chinese J. Anal. Chem. 2011;39:1451-1458
6. HUGO Gene Nomenclature Committee. http://www.genenames.org/.
7. Smith ZD, Meissner A. DNA methylation: roles in mammalian development. Nat. Rev. Genet. 2013;14:204-220
8. Bestor TH. The DNA methyltransferases of mammals. Hum. Mol. Genet. 2000;9:2395-2402
9. Fauman EB, Blumenthal RM, Cheng X. S-Adenosylmethionine-dependent Methyltransferases: Structures and Functions. World Scientific Publishing, Singapore. 1999
10. Scavetta RD, Thomas CB, Walsh MA, Szegedi S, Joachimiak A, Gumport RI, Churchill MEA. Structure of Rsrl methyltransferase, a member of the N6-adenine β class of DNA methyltransferases. Nucleic Acids Res. 2000;28:3950-3961
11. Sheikhnejad G, Brank A, Christman JK, Goddard A, Alvarez E, Ford Jr H, Marquez VE, Marasco CJ, Sufrin JR, O'Gara M, Cheng X. Mechanism of inhibition of DNA (cytosine C5)-methyltransferases by oligodeoxyribonucleotides containing 5, 6-dihydro-5-azacytosine. J. Mol. Biol. 1999;285:2021-2034
12. Gong W, O'Gara M, Blumenthal RM, Cheng X. Structure of PvuII DNA-(cytosine N4) methyltransferase, an example of domain permutation and protein fold assignment. Nucleic Acids Res. 1997;25:2702-2715
13. Pogolotti AL Jr, Ono A, Subramaniam R, Santi DV. On the mechanism of DNA-adenine methylase. J. Biol. Chem. 1988;263:7461-7464
14. Csankovszki G, Nagy A, Jaenisch R. Synergism of xist RNA, DNA methylation, and histone hypoacetylation in maintaining x chromosome inactivation. J. Cell Biol. 2001;153:773-784
15. Li E, Beard C, Jaenisch R. Role for DNA methylation in genomic imprinting. Nature. 1993;366:362-365
16. Razin A, Cedar H. DNA methylation and gene expression. Microbiol. Rev. 1991;55:451-458
17. Huang WY, Hsu SD, Huang HY, Sun YM, Chou CH, Weng SL, Huang HD. MethHC: a database of DNA methylation and gene expression in human cancer. Nucleic Acids Res. 2015;43:D856-861
18. Lyko F, Brown R. DNA methyltransferase inhibitors and the development of epigenetic cancer therapies. J. Natl. Cancer Inst. 2005;97:1498-1506
19. Robertson KD. DNA methylation, methyltransferases, and cancer. Oncogene. 2001;20:3139-3155
20. Baylin SB, Herman JG, Graff JR, Vertino PM, Issa JP. Alterations in DNA methylation: a fundamental aspect of neoplasia. Adv. Cancer Res. 1997;72:141-182
21. Ehrlich M. DNA methylation in cancer: too much, but also too little. Oncogene. 2002;21:5400-5413
22. Szyf M. DNA methylation signatures for breast cancer classification and prognosis. Genome Med. 2012;4:26
23. BonDurant AE, Huang Z, Whitaker RS, Simel LR, Berchuck A, Murphy SK. Quantification detection of RASSF1A DNA promotor methylation in tumors and serum of patients with serious epithelial ovarian cancer. Gynecol. Oncol. 2011;123:581-587
24. Son JW, Jeong KJ, Jean WS, Park SY, Jheon S, Cho HM, Park CG, Lee HY, Kang J. Genome-wide combination profiling of DNA copy number and methylation for deciphering biomarkers in non-small cell lung cancer patients. Cancer Lett. 2011;311:29-37
25. Hong L, Ahuja N. DNA methylation biomarkers of stool and blood for early detection of colon cancer. Genet. Test. Mol. Biomarkers. 2013;17:401-406
26. Andrés G, Ashour N, Sánchez-Chapado M, Ropero S, Angulo JC. The study of DNA methylation in urological cancer: present and future. Actas Urol Esp. 2013;37:368-375
27. Schoofs T, Berdel WE, Müller-Tidow C. Origins of aberrant DNA methylation in acute myeloid leukemia. Leukemia. 2014;28:1-14
28. Jones PA, Baylin SB. The fundamental role of epigenetic events in cancer. Nat. Rev. Genet. 2002;3:415-428
29. Wilson AS, Power BE, Molloy PL. DNA hypomethylation and human diseases. Biochim. Biophys. Acta Rev. Cancer. 2007;1775:138-162
30. Widschwendter M, Jiang G, Woods C, Müller HM, Fiegl H, Goebel G, Marth C, Müller-Holzner E, Zeimet AG, Laird PW, Ehrlich M. DNA hypomethylation and ovarian cancer biology. Cancer Res. 2004;64:4472-4480
31. Issa JPJ, Vertino PM, Wu J, Sazawal S, Celano P, Nelkin BD, Hamilton SR, Baylin SB. Increased cytosine DNA-methyltransferase activity during colon cancer progression. J. Natl. Cancer Inst. 1993;85:1235-1240
32. Mutze K, Langer R, Schumacher F, Becker K, Ott K, Novotny A, Hapfelmeier A, Höfler H, Keller G. DNA methyltransferase 1 as a predictive biomarker and potential therapeutic target for chemotherapy in gastric cancer. Eur. J. Cancer. 2011;47:1817-1825
33. Belinsky SA, Nikula KJ, Baylin SB, Issa JPJ. Increased cytosine DNA-methyltransferase activity is target-cell-specific and an early event in lung cancer. Proc. Natl. Acad. Sci. U. S. A. 1996;93:4045-4050
34. Kobayashi Y, Absher DM, Gulzar ZG, Young SR, McKenney JK, Peehl DM, Brooks JD, Myers RM, Sherlock G. DNA methylation profiling reveals novel biomarkers and important roles for DNA methyltransferases in prostate cancer. Genome Res. 2011;21:1017-1027
35. Baylin SB, Herman JG. DNA hypermethylation in tumorigenesis: epigenetics join genetics. Trends Genet. 2000;16:168-174
36. Frigola J, Song J, Stirzaker C, Hinshelwood RA, Peinado MA, Clark SJ. Epigenetic remodeling in colorectal cancer results in coordinate gene suppression across an entire chromosome band. Nat. Genet. 2006;38:540-549
37. Choy JS, Wei S, Lee JY, Tan S, Chu S, Lee TH. DNA methylation increases nucleosome compaction and rigidity. J. Am. Chem. Soc. 2010;132:1782-1783
38. Scholten S. Genomic imprinting in plant embryos. Epigenetics. 2010;5:455-459
39. Rodenhiser D, Mann M. Epigenetics and human disease: translating basic biology into clinical applications. CMAJ. 2006;174:341-348
40. Boye E, Marinus MG, Løebner-Olesen A. Quantification of dam methyltransferase in Escherichia coli. J. Bacteriol. 1992;174:1682-1685
41. Eads CA, Danenberg KD, Kawakami K, Saltz LB, Danenberg PV, Laird PW. CpG island hypermethylation in human colorectal tumors is not associated with DNA methyltransferase overexpression. Cancer Res. 1999;59:2302-2306
42. Bergerat A, Guschlbauer W, Fazakerley GV. Allosteric and catalytic binding of s-adenosylmethionine to escherichia coli DNA adenine methyltransferase monitored by 3H NMR. Proc. Natl. Acad. Sci. U. S. A. 1991;88:6394-6397
43. Fraga MF, Rodríguez R, Cañal MJ. Rapid quantification of DNA methylation by high performance capillary electrophoresis. Electrophoresis. 2000;21:2990-2994
44. Yokochi T, Robertson KD. DMB (dnmt-magnetic beads) assay: measuring DNA methyltransferase activity in vitro. Methods Mol. Biol. 2004;287:285-296
45. Adams RLP, Rinaldi A, Seivwright C. Microassay for DNA methyltransferase. J. Biochem. Bioph. Methods. 1991;22:19-22
46. Jurkowska RZ, Ceccaldl A, Zhang Y, Arimondo PB, Jeltsch A. DNA methyltransferase assays. Methods Mol. Biol. 2011;791:157-177
47. Pradhan S, Bacolla A, Wells RD, Roberts RJ. Recombinant human DNA (cytosine-5) methyltransferase. J. Biol. Chem. 1999;274:33002-33010
48. Herman JG, Graff JR, Myöehänen S, Nelkin BD, Baylin SB. Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands. Proc. Natl. Acad. Sci. U. S. A. 1996;93:9821-9826
49. Friso S, Choi SW, Dolnikowski GG, Selhub J. A method to assess genomic DNA methylation using high-performance liquid chromatography/electrospray ionization mass spectrometry. Anal. Chem. 2002;74:4526-4531
50. Wu Z, Wu ZK, Tang H, Tang LJ, Jiang JH. Activity-based DNA-gold nanoparticle probe as colorimetric biosensor for DNA methyltransferase/glycosylase assay. Anal. Chem. 2013;85:4376-4383
51. Song G, Chen C, Ren J, Qu X. A simple, universal colorimetric assay for endonuclease/methyltransferase activity and inhibition based on an enzyme-responsive nanoparticle system. ACS Nano. 2009;3:1183-1189
52. Zhao Y, Chen F, Lin M, Fan C. A methylation-blocked cascade amplification strategy for label-free colorimetric detection of DNA methyltransferase activity. Biosens. Bioelectron. 2014;54:565-570
53. Li W, Liu Z, Lin H, Nie Z, Chen J, Xu X, Yao S. Label-free colorimetric assay for methyltransferase activity based on a novel methylation-responsive DNAzyme strategy. Anal. Chem. 2010;82:1935-1941
54. Zhu C, Wen Y, Peng H, Long Y, He Y, Huang Q, Li D, Fan C. A methylation-stimulated DNA machine: an autonomous isothermal route to methyltransferase activity and inhibition analysis. Anal. Bioanal. Chem. 2011;399:3459-3464
56. https://www.usbio.net/item/S0070-84.
57. http://www.abnova.com/products/products_detail.asp?catalog_id=KA1604.
58. http://www.abcam.com/histone-h3-k4-methyltransferase-activity-quantification-assay-kit-ab113452.html.
59. https://www.caymanchem.com/app/template/Product.vm/catalog/700140.
60. Chen F, Zhao Y. Methylation-blocked enzymatic recycling amplification for highly sensitive fluorescence sensing of DNA methyltransferase activity. Analyst. 2013;138:284-289
61. Liu W, Lai H, Huang R, Zhao C, Wang Y, Weng X, Zhou X. DNA methyltransferase activity detection based on fluorescent silver nanocluster hairpin-shaped DNA probe with 5'-C-rich/G-rich-3' tails. Biosens. Bioelectron. 2015;68:736-740
62. Wang K, Tang Z, Yang CJ, Kim Y, Fang X, Li W, Wu Y, Medley CD, Cao Z, Li J, Colon P, Lin H, Tan W. Molecular engineering of DNA: molecular beacons. Angew. Chem. Int. Ed. 2009;48:856-870
63. Han XX, Kitahama Y, Tanaka Y, Guo J, Xu WQ, Zhao B, Ozaki Y. Simplified protocol for detection of protein-ligand interactions via surface-enhanced resonance raman scattering and surface-enhanced fluorescence. Anal. Chem. 2008;80:6567-6572
64. Tyagi S, Kramer FR. Molecular beacons: probes that fluoresce upon hybridization. Nat. Biotechnol. 1996;14:303-308
65. Li J, Yan H, Wang K, Tan W, Zhou X. Hairpin fluorescence DNA probe for real-time monitoring of DNA methylation. Anal. Chem. 2007;79:1050-1056
66. Wood RJ, McKelvie JC, Maynard-Smith MD, Roach PL. A real-time assay for CpG-specific cytosine-C5 methyltransferase activity. Nucleic Acids Res. 2010;38:e107
67. Chen J, Wang Y, Li W, Zhou H, Li Y, Yu C. Nucleic acid-induced tetraphenylethene probe noncovalent self-assembly and the superquenching of aggregation-induced emission. Anal. Chem. 2014;86:9866-9872
68. Wang Y, Chen J, Chen Y, Li W, Yu C. Polymer-induced perylene probe excimer formation and selective sensing of DNA methyltransferase activity through the monomer-excimer transition. Anal. Chem. 2014;86:4371-4378
69. Chandrasekharan N, Kelly LA. A dual fluorescence temperature sensor based on perylene/exciplex interconversion. J. Am. Chem. Soc. 2001;123:9898-9899
70. Peneva K, Mihov G, Nolde F, Rocha S, Hotta JI, Braeckmans K, Hofkens J, Uji-i H, Herrmann A, Müllen K. Water-soluble monofunctional perylene and terrylene dyes: powerful labels for single-enzyme tracking. Angew. Chem. Int. Ed. 2008;47:3372-3375
71. Chen Z, Stepanenko V, Dehm V, Prins P, Siebbeles LDA, Seibt J, Marquetand P, Engel V, Wüerthner F. Photoluminescence and conductivity of self-assembled π-π stacks of perylene bisimide dyes. Chem. Eur. J. 2007;13:436-449
72. Chang LMS, Bollum FJ. Multiple roles of divalent cation in the terminal deoxynucleotidyltransferase reaction. J. Biol. Chem. 1990;265:17436-17440
73. Huang Y, Shi M, Zhao L, Zhao S, Hu K, Chen ZF, Chen J, Liang H. Carbon nanotube signal amplification for ultrasensitive fluorescence polarization detection of DNA methyltransferase activity and inhibition. Biosens. Bioelectron. 2014;54:285-291
74. Wang GL, Luo HQ, Li NB. Gold nanorods-based FRET assay for ultrasensitive detection of DNA methylation and DNA methyltransferase activity. Analyst. 2014;139:4572-4577
75. Hong B, Kang KA. Biocompatiable, nanogold-particle fluorescence enhancer for fluorophore mediated, optical immunosensor. Biosens. Bioelectron. 2006;21:1333-1338
76. Qiu X, Wang P, Cao Z. Hybridization chain reaction modulated DNA-hosted silver nanoclusters for fluorescent identification of single nucleotide polymorphisms in the let-7 miRNA family. Biosens. Bioelectron. 2014;60:351-357
77. Shen C, Xia X, Hu S, Yang M, Wang J. Silver nanoclusters-based fluorescence assay of protein kinase activity and inhibition. Anal. Chem. 2015;87:693-698
78. Tang X, Bansaruntip S, Nakayama N, Yenilmez E, Chang YL, Wang Q. Carbon nanotube DNA sensor and sensing mechanism. Nano Lett. 2006;6:1632-1636
79. Ji L, Cai Z, Qian Y, Wu P, Zhang H, Cai C. Highly sensitive methyltransferase activity assay and inhibitor screening based on fluorescence quenching of graphene oxide integrated with the site-specific cleavage of restriction endonuclease. Chem. Commun. 2014;50:10691-10694
80. Deng H, Yang X, Gao Z. MoS2 nanosheets as an effective fluorescence quencher for DNA methyltransferase activity detection. Analyst. 2015;140:3210-3215
81. Gao C, Li H, Liu Y, Wei W, Zhang Y, Liu S. Label-free fluorescence detection of DNA methylation and methyltransferase activity based on restriction endonuclease HpaII and exonuclease III. Analyst. 2014;139:6387-6392
82. Quach QH, Chung BH. A signal-on fluorescent assay for DNA methyltransferase activity using a methylation-resistant endonuclease. Analyst. 2014;139:2674-2677
83. Liu P, Yang XH, Wang Q, Huang J, Liu JB, Zhu Y, He LL, Wang KM. Sensitive detection of DNA methyltransferase activity based on rolling circle amplification technology. Chin. Chem. Lett. 2014;25:1047-1051
84. Zhao Y, Chen F, Wu Y, Dong Y, Fan C. Highly sensitive fluorescence assay of DNA methyltransferase activity via methylation-sensitive cleavage coupled with nicking enzyme-assisted signal amplification. Biosens. Bioelectron. 2013;42:56-61
85. Xue Q, Lv Y, Xu S, Zhang Y, Wang L, Li R, Yue Q, Li H, Gu X, Zhang S, Liu J. Highly sensitive fluorescence assay of DNA methyltransferase activity by methylation-sensitive cleavage-based primer generation exponential isothermal amplification-induced G-quadruplex formation. Biosens. Bioelectron. 2015;66:547-553
86. Cao A, Zhang CY. Sensitive and label-free DNA methylation detection by ligation-mediated hyperbranched rolling circle amplification. Anal. Chem. 2012;84:6199-6205
87. Xing XW, Tang F, Wu J, Chu JM, Feng YQ, Zhou X, Yuan BF. Sensitive detection of DNA methyltransferase activity based on exonuclease-mediated target recycling. Anal. Chem. 2014;86:11269-11274
88. Ma Y, Chen L, Zhang L, Liao S, Zhao J. A sensitive strategy for the fluorescence detection of DNA methyltransferase activity based on the graphene oxide platform and T7 exonuclease-assisted cyclic signal amplification. Analyst. 2015;140:4076-4082
89. Li N, Jablonowski C, Jin H, Zhong W. Stand-alone rolling circle amplification combined with capillary electrophoresis for specific detection of small RNA. Anal. Chem. 2009;81:4906-4913
90. Hafner GJ, Yang LC, Wolter LC, Stafford MR, Giffard PM. Isothermal amplification and multimerization of DNA by Bst DNA polymerase. BioTechniques. 2001;30:852-867
91. Chan SH, Stoddard BL, Xu SY. Natural and engineered nicking endonucleases-from cleavage mechanism to engineering of strand-specificity. Nucleic Acids Res. 2011;39:1-18
92. Gerasimova YV, Kolpashchikov DM. Enzyme-assisted target recycling (EATR) for nucleic acid detection. Chem. Soc. Rev. 2014;43:6405-6438
93. Gerasimova YV, Peck S, Kolpashchikov DM. Enzyme-assisted binary probe for sensitive detection of RNA and DNA. Chem. Commun. 2010;46:8761-8763
94. Zhang H, Li F, Dever B, Li XF, Le XC. DNA-mediated homogeneous binding assays for nucleic acids and proteins. Chem. Rev. 2013;113:2812-2841
95. Huang Y, Chen J, Zhao S, Shi M, Chen ZF, Liang H. Label-free colorimetric aptasensor based on nicking enzyme assisted signal amplification and DNAzyme amplification for highly sensitive detection of protein. Anal. Chem. 2013;85:4423-4430
96. Li J, Fu HE, Wu LJ, Zheng AX, Chen GN, Yang HH. General colorimetric detection of proteins and small molecules based on cyclic enzymatic signal amplification and hairpin aptamer probe. Anal. Chem. 2012;84:5309-5315
97. Garcia-Campana AM, Baeyens WRG. Chemiluminescence in Analytical Chemistry. Marcel Dekker, New York. 2001
98. Yang X, Guo Y, Wang A. Luminol/antibody labeled gold nanoparticles for chemiluminescence immunoassay of carcinoembryonic antigen. Anal. Chim. Acta. 2010;666:91-96
99. Creamer JI, Buck AM. The assaying of haemoglobin using luminol chemiluminescence and its application to the dating of human skeletal remains. Luminescence. 2009;24:311-316
100. Chen C, Li B. Chemiluminescence resonance energy transfer biosensing platform for site-specific determination of DNA methylation and assay of DNA methyltransferase activity using exonuclease III-assisted target recycling amplification. Biosens. Bioelectron. 2014;54:48-54
101. Hastings JW. Biological diversity, chemical mechanisms, and the evolutionary origins of bioluminescent systems. J. Mol. Evol. 1983;19:309-321
102. Zeng YP, Hu J, Long Y, Zhang CY. Sensitive detection of DNA methyltransferase using hairpin probe-based primer generation rolling circle amplification-induced chemiluminescence. Anal. Chem. 2013;85:6143-6150
103. Bi S, Zhao T, Luo B, Zhu JJ. Hybridization chain reaction-based branched rolling circle amplification for chemiluminescence detection of DNA methylation. Chem. Commun. 2013;49:6906-6908
104. Jiang C, Yan CY, Huang C, Jiang JH, Yu RQ. A bioluminescence assay for DNA methyltransferase activity based on methylation-resistant cleavage. Anal. Biochem. 2012;423:224-228
105. Teo AKL, Lim CL, Gao Z. The development of electrochemical assays for microRNAs. Electrochim. Acta. 2014;126:19-30
106. Ronkainen NJ, Halsall HB, Heineman WR. Electrochemical biosensors. Chem. Soc. Rev. 2010;39:1747-1763
107. Wang GL, Zhou LY, Luo HQ, Li NB. Electrochemical strategy for sensing DNA methylation and DNA methyltransferase activity. Anal. Chim. Acta. 2013;768:76-81
108. Yin H, Xu Z, Wang M, Zhang X, Ai S. An electrochemical biosensor for assay of DNA methyltransferase activity and screening of inhibitor. Electrochim. Acta. 2013;89:530-536
109. Liu S, Wu P, Li W, Zhang H, Cai C. An electrochemical approach for detection of DNA methylation and assay of the methyltransferase activity. Chem. Commun. 2011;47:2844-2846
110. Yin H, Zhou Y, Xu Z, Chen L, Zhang D, Ai S. An electrochemical assay for DNA methylation, methyltransferase activity and inhibitor screening based on methyl binding domain protein. Biosens. Bioelectron. 2013;41:492-497
111. Deng H, Yang X, Yeo SPX, Gao Z. Highly sensitive electrochemical methyltransferase activity assay. Anal. Chem. 2014;86:2117-2123
112. Xu Z, Wang M, Zhou T, Yin H, Ai S. Electrochemical biosensing method for the detection of DNA methylation and assay of the methyltransferase activity. Sens. Actuators, B. 2013;178:412-417
113. Liu P, Pang J, Yin H, Ai S. G-quadruplex functionalized nano mesoporous silica for assay of the DNA methyltransferase activity. Anal. Chim. Acta. 2015;879:34-40
114. Wang X, Liu X, Hou T, Li W, Li F. Highly sensitive homogeneous electrochemical assay for methyltransferase activity based on methylation-responsive exonuclease III-assisted signal amplification. Sens. Actuators, B. 2015;208:575-580
115. Jing X, Cao X, Wang L, Lan T, Li Y, Xie G. DNA-AuNPs based signal amplification for highly sensitive detection of DNA methylation, methyltransferase activity and inhibitor screening. Biosens. Bioelectron. 2014;58:40-47
116. Li W, Liu X, Hou T, Li H, Li F. Ultrasensitive homogeneous electrochemical strategy for DNA methyltransferase activity assay based on autonomous exonuclease III-assisted isothermal cycling signal amplification. Biosens. Bioelectron. 2015;70:304-309
117. Wang Y, He X, Wang K, Su J, Chen Z, Yan G, Du Y. A label-free electrochemical assay for methyltransferase activity detection based on the controllable assembly of single wall carbon nanotubes. Biosens. Bioelectron. 2013;41:238-243
118. Zhou J, Zhang X, Xiong E, Yu P, Chen J. A label-free electrochemical strategy for highly sensitive methyltransferase activity assays. Chem. Commun. 2015;51:5081-5084
119. Li W, Wu P, Zhang H, Cai C. Signal amplification of graphene oxide combining with restriction endonuclease for site-specific determination of DNA methylation and assay of methyltransferase activity. Anal. Chem. 2012;84:7583-7590
120. Wu H, Liu S, Jiang J, Shen G, Yu R. A sensitive electrochemical biosensor for detection of DNA methyltransferase activity by combining DNA methylation-sensitive cleavage and terminal transferase-mediated extension. Chem. Commun. 2012;48:6280-6282
121. Xu Z, Yin H, Huo L, Zhou Y, Ai S. Electrochemical immunosensor for DNA methyltransferase activity assay based on methyl CpG-binding protein and dual gold nanoparticle conjugate-based signal amplification. Sens. Actuators, B. 2014;192:143-149
122. Muren NB, Barton JK. Electrochemical assay for the signal-on detection of human DNA methyltransferase activity. J. Am. Chem. Soc. 2013;135:16632-16640
123. Yin H, Zhou Y, Xu Z, Wang M, Ai S. Ultrasensitive electrochemical immunoassay for DNA methyltransferase activity and inhibitor screening based on methyl binding domain protein of MeCP2 and enzymatic signal amplification. Biosens. Bioelectron. 2013;49:39-45
124. Wang M, Xu Z, Chen L, Yin H, Ai S. Electrochemical immunosensing platform for DNA methyltransferase activity analysis and inhibitor screening. Anal. Chem. 2012;84:9072-9078
125. Quint A, Cedar H. In vitro methylation of DNA with Hpa II methylase. Nucleic Acids Res. 1981;9:633-646
126. Furst AL, Muren NB, Hill MG, Barton JK. Label-free electrochemical detection of human methyltransferase from tumors. Proc. Natl. Acad. Sci. U. S. A. 2014;111:14985-14989
127. Hamawy MM, Mergenhagen SE, Siraganian RP. Cell adherence to fibronectin and the aggregation of the high affinity immunoglobulin E receptor synergistically regulate tyrosine phosphorylation of 105-115-kDa proteins. J. Biol. Chem. 1993;268:5227-5233
128. Du D, Zou Z, Shin Y, Wang J, Wu H, Engelhard MH, Liu J, Aksay IA, Lin Y. Sensitive immunosensor for cancer biomarker based on dual signal amplification strategy of graphene sheets and multienzyme functionalized carbon nanospheres. Anal. Chem. 2010;82:2989-2995
129. Wu J, Fu Z, Yan F, Ju H. Biomedical and clinical applications of immunoassays and immunosensors for tumor markers. Trends Anal. Chem. 2007;26:679-688
130. Hendrich B, Bird A. Identification and characterization of a family of mammalian methyl-CpG binding proteins. Mol. Cell. Biol. 1998;18:6538-6547
131. Cross SH, Meehan RR, Nan X, Bird A. A component of the transcriptional repressor MeCP1 shares a motif with DNA methyltransferase and HRX proteins. Nat. Genet. 1997;16:256-259
132. Baubec T, Ivánek R, Lienert F, Schübeler D. Methylation-dependent and -independent genomic targeting principles of the MBD protein family. Cell. 2013;153:480-492
133. Tanaka K, Tainaka K, Kamei T, Okamoto A. Direct labeling of 5-methylcytosine and its applications. J. Am. Chem. Soc. 2007;129:5612-5620
134. Wan Y, Wang Y, Luo JF, Lu ZH. Bisulfite modification of immobilized DNAs for methylation detection. Biosens. Bioelectron. 2007;22:2415-2421
135. Hiraoka D, Yoshida W, Abe K, Wakeda H, Hata K, Ikebukuro K. Development of a method to measure DNA methylation levels by using methyl CpG-binding protein and luciferase-fused zinc finger protein. Anal. Chem. 2012;84:8259-8264
136. Yu Y, Blair S, Gillespie D, Jensen R, Myszka D, Badran AH, Ghosh I, Chagovetz A. Direct DNA methylation profiling using methyl binding domain proteins. Anal. Chem. 2010;82:5012-5019
137. Buschdorf JP, Sträetling WH. A WW domain binding region in methyl-CpG-binding protein MeCP2: impact on rett syndrome. J. Mol. Med. 2004;82:135-143
138. Rauch T, Li H, Wu X, Pfeifer GP. MIRA-assisted microarray analysis, a new technology for the determination of DNA methylation patterns, identifies frequent methylation of homeodomain-containing genes in lung cancer cells. Cancer Res. 2006;66:7939-7947
139. Zhou Y, Tian XL, Li YS, Pan FG, Zhang YY, Zhang JH, Yang L, Wang XR, Ren HL, Lu SY, Li ZH, Chen QJ, Liu ZS, Liu JQ. An enhanced ELISA based on modified colloidal gold nanoparticles for the detection of Pb(II). Biosens. Bioelectron. 2011;26:3700-3704
140. Lin D, Wu J, Wang M, Yan F, Ju H. Triple signal amplification of graphene film, polybead carried gold nanoparticles as tracing tag and silver deposition for ultrasensitive electrochemical immunosensing. Anal. Chem. 2012;84:3662-3668
141. Lai G, Yan F, Wu J, Leng C, Ju H. Ultrasensitive multiplexed immunoassay with electrochemical stripping analysis of silver nanoparticles catalytically deposited by gold nanoparticles and enzymatic reaction. Anal. Chem. 2011;83:2726-2732
142. Dykman L, Khlebtsov N. Gold nanoparticles in biomedical applications: recent advances and perspectives. Chem. Soc. Rev. 2012;41:2256-2282
143. Stratakis M, Garcia H. Catalysis by supported gold nanoparticles: beyond aerobic oxidative processes. Chem. Rev. 2012;112:4469-4506
144. Tansil NC, Gao Z. Nanoparticles in biomolecular detection. Nano Today. 2006;1:28-37
145. Hu K, Lan D, Li X, Zhang S. Electrochemical DNA biosensor based on nanoporous gold electrode and multifunctional encoded DNA-Au bio bar codes. Anal. Chem. 2008;80:9124-9130
146. Zhou Y, Tian XL, Li YS, Zhang YY, Yang L, Zhang JH, Wang XR, Lu SY, Ren HL, Liu ZS. A versatile and highly sensitive probe for Hg(II), Pb(II) and Cd(II) detection individually and totally in water samples. Biosens. Bioelectron. 2011;30:310-314
147. Yin H, Xu Z, Zhou Y, Wang M, Ai S. An ultrasensitive electrochemical immunosensor platform with double signal amplification for indole-3-acetic acid determinations in plant seeds. Analyst. 2013;138:1851-1857
148. Xu Z, Wang M, Yin H, Ai S, Wang L, Pang J. A sensitive electrochemical method for DNA methyltransferase assay and inhibitor screening based on DNA methylation-sensitive cleavage. Electrochim. Acta. 2013;112:596-602
149. Akanda MR, Tamilavan V, Park S, Jo K, Hyun MH, Yang H. Hydroquinone diphosphate as a phosphatase substrate in enzymatic amplification combined with electrochemical-chemical-chemical redox cycling for the detection of E. coli O157:H7. Anal. Chem. 2013;85:1631-1636
150. Yang X, Gao Z. Enzyme-catalysed deposition of ultrathin silver shells on gold nanorods: a universal and highly efficient signal amplification strategy for translating immunoassay into a litmus-type test. Chem. Commun. 2015;51:6928-6931
151. Horton JR, Liebert K, Bekes M, Jeltsch A, Cheng X. Structure and substrate recognition of the escherichia coli DNA adenine methyltransferase. J. Mol. Biol. 2006;358:559-570
152. Shen Q, Han L, Fan G, Abdel-Halim ES, Jiang L, Zhu JJ. Highly sensitive photoelectrochemical assay for DNA methyltransferase activity and inhibitor screening by exciton energy transfer coupled with enzyme cleavage biosensing strategy. Biosens. Bioelectron. 2015;64:449-455
153. Duan AP, Ning LM, Li C, Hou YF, Yang NN, Sun LZ, Li GX. A novel strategy to inhibit the reproduction and translation of hepatitis C virus. Sci. China Life Sci. 2013;56:293-297
154. Visco RE, Chandross EA. Electroluminescence in solutions of aromatic hydrocarbons. J. Am. Chem. Soc. 1964;86:5350-5351
155. Santhanam KSV, Bard AJ. Chemiluminescence of electrogenerated 9, 10-diphenylanthracene anion radical. J. Am. Chem. Soc. 1965;87:139-140
156. Tokel-Takvoryan NE, Hemingway RE, Bard AJ. Electrogenerated chemiluminescence. XIII. electrochemical and electrogenerated chemiluminescence studies of ruthenium chelates. J. Am. Chem. Soc. 1973;95:6582-6589
157. Forster RJ, Bertoncello P, Keyes TE. Electrogenerated chemiluminescence. Annu. Rev. Anal. Chem. 2009;2:359-385
158. Liu Z, Qi W, Xu G. Recent advances in electrochemiluminescence. Chem. Soc. Rev. 2015;44:3117-3142
159. Richter MM. Electrochemiluminescence (ECL). Chem. Rev. 2004;104:3003-3036
160. Muzyka K. Current trends in the development of the electrochemiluminescent immunosensors. Biosens. Bioelectron. 2014;54:393-407
161. Luo X. et al. The determination of DNA methyltransferase activity by quenching of tris(2,2'-bipyridine)ruthenium electrogenerated chemiluminescence with ferrocene. Chem. Commun. 2015;51:9487-9490
162. Li Y, Huang C, Zheng J, Qi H. Electrogenerated chemiluminescence biosensing method for the detection of DNA methylation and assay of the methyltransferase activity. Biosens. Bioelectron. 2012;38:407-410
163. Li Y, Luo X, Yan Z, Zheng J, Qi H. A label-free supersandwich electrogenerated chemiluminescence method for the detection of DNA methylation and assay of the methyltransferase activity. Chem. Commun. 2013;49:3869-3871
164. Yan Z, Li Y, Zheng J, Zhou M. Electrogenerated chemiluminescence biosensing method for methyltransferase activity using tris(1, 10-phenanthroline) ruthenium-assembled graphene oxide. J. Electroanal. Chem. 2014;731:133-138
165. Li Y, Yan Z, Zheng J, Qi H. Label-free and amplified electrogenerated chemiluminescence biosensing method for the determination of DNA methyltransferase activity using signal reagent-assembled graphene oxide. Electrochim. Acta. 2014;137:454-461
166. Zhao HF, Liang RP, Wang JW, Qiu JD. One-pot synthesis of GO/AgNPs/luminol composites with electrochemiluminescence activity for sensitive detection of DNA methyltransferase activity. Biosens. Bioelectron. 2015;63:458-464
167. Zhang H, Li MX, Fan MX, Gu JX, Wu P, Cai CX. Electrochemiluminescence signal amplification combined with a conformation-switched hairpin DNA probe for determining the methylation level and position in the Hsp53 tumor suppressor gene. Chem. Commun. 2014;50:2932-2934
168. Laszlo AH, Derrington IM, Brinkerhoff H, Langford KW, Nova IC, Samson JM, Bartlett JJ, Pavlenok M, Gundlach JH. Detection and mapping of 5-methylcytosine and 5-hydroxymethylcytosine with nanopore MspA. Proc. Natl. Acad. Sci. U. S. A. 2013;110:18904-18909
169. Shim J, Humphreys GI, Venkatesan BM, Munz JM, Zou X, Sathe C, Schulten K, Kosari F, Nardulli AM, Vasmatzis G, Bashir R. Detection and quantification of methylation in DNA using solid-state nanopores. Sci. Rep. 2013;3:1389
170. Kang I, Wang Y, Reagan C, Fu Y, Wang MX, Gu LQ. Designing DNA interstrand lock for locus-specific methylation detection in a nanopore. Sci. Rep. 2013;3:2381
171. Hu PP, Liu H, Zhen SJ, Li CM, Huang CZ. Nanosilver-based surface-enhanced Raman spectroscopic determination of DNA methyltransferase activity through real-time hybridization chain reaction. Biosens. Bioelectron. 2015;73:228-233
172. Schermelleh L, Spada F, Easwaran HP, Zolghadr L, Margot JB, Cardoso MC, Leonhardt H. Trapped in action: direct visualization of DNA methyltransferase activity in living cells. Nat. Methods. 2005;2:751-756
173. Kosaka N, Iguchi H, Ochiya T. Circulating microRNA in body fluid: a new potential biomarker for cancer diagnosis and prognosis. Cancer Sci. 2010;101:2087-2092
174. Attard G, de Bono JS. Utilizing circulating tumor cells:challenges and pitfalls. Curr. Opin. Genet. Dev. 2011;21:50-58
Author contact
![]() Corresponding author: Tel: 6516-3887, Fax: 6779-1691, e-mail: chmgaozedu.sg
Corresponding author: Tel: 6516-3887, Fax: 6779-1691, e-mail: chmgaozedu.sg