Impact Factor
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and methods
Results and discussions
Conclusions
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2016; 6(4):470-484. doi:10.7150/thno.14184 This issue Cite
Research Paper
Polydopamine-Based Surface Modification of Novel Nanoparticle-Aptamer Bioconjugates for In Vivo Breast Cancer Targeting and Enhanced Therapeutic Effects
Wei Tao1,2,3*, Xiaowei Zeng1,2*, Jun Wu4, Xi Zhu3, Xinghua Yu1, Xudong Zhang5, Jinxie Zhang1,2, Gan Liu1,2, Lin Mei1,2 ![]()
1. The Shenzhen Key Lab of Gene and Antibody Therapy, and Division of Life and Health Sciences, Graduate School at Shenzhen, Tsinghua University, Shenzhen 518055, P.R. China;
2. School of Life Sciences, Tsinghua University, Beijing 100084, P.R. China.
3. Brigham and Women's Hospital, Harvard Medical School, Boston, Massachusetts 02115, United States.
4. Department of Biomedical Engineering, School of Engineering, Sun Yat-sen University, Guangzhou 510006, P.R. China.
5. Department of Molecular Microbiology and Immunology, University of Southern California, Los Angeles, CA 90033, USA.
* The two authors contributed equally to this work.
Received 2015-10-20; Accepted 2015-12-30; Published 2016-2-11
Abstract

In this study, we reported a simple polydopamine (pD)-based surface modification method to prepare novel nanoparticle-aptamer bioconjugates (Apt-pD-DTX/NPs) for in vivo tumor targeting and enhanced therapeutic effects of breast cancer. With simple preparation procedures, the new functionalized Apt-pD-DTX/NPs could maximumly increase the local effective drug concentration on tumor sites, achieving enhanced treatment effectiveness and minimizing side effects. The dopamine polymerization and aptamer conjugation barely changed the characters of NPs. Both in vitro cell experiments (i.e. endocytosis of fluorescent NPs, in vitro cellular targeting and cytotoxicity assays) and in vivo animal studies (i.e. in vivo imaging, biodistribution and antitumor effects of NPs) demonstrated that the Apt-pD-DTX/NPs could achieve significantly high targeting efficiency and enhanced therapeutic effects compared with clinical Taxotere® and NPs without functional modification. Above all, the Apt-pD-DTX/NPs showed great potential as a promising nanoformulation for in vivo breast cancer therapy and the construction of pD-modified NP-aptamer bioconjugates could be of great value in medical use.
Keywords: Nanomedicine, Dopamine, Aptamer, Cancer targeting, Enhanced therapeutic effects.
Introduction
Nanotechnology, especially NPs applied in drug delivery system, has shown significant prospects for fundamentally changing the landscape of biotechnology and pharmaceutical industries [1, 2]. A feasible method to improve the therapeutic efficacy and reduce the side effects of cytotoxic anticancer drugs is to increase the local effective pharmaceutical concentration in the tumor site, which can be achieved by targeted drug delivery NPs [3-5]. By incorporating various active targeting ligands (i.e. peptides, antibodies, nucleic acids and small molecules), NPs can ferry drugs to specific cells, tissues or organs [6]. Aptamers are single-stranded RNA or DNA oligonucleotides with specific 3D structures, which can specifically bind a wide variety of biological targets with high affinity [7-10]. With significant properties (such as readily availability, lack of immunogenicity, smaller size, lower molecular weights, higher ratio of target accumulation, easier production without the use of animal, and higher in vivo stability) [11, 12], aptamers can offer distinct advantages over antibodies and be emerged as good targeting moieties for imaging and therapeutic applications [13-15]. AS1411 aptamer is one of the DNA aptamers can specifically bind to nucleolin with high affinity, while nucleolin is a protein overexpressed in the plasma membrane of cancer cells (e.g. cervical cancer, breast cancer, liver cancer, glioma and so on) [16, 17]. The AS1411 has been successfully exploited as an efficient targeting ligand for tracking breast cancer cells such as MCF-7 and MDA-MB-231 [18-20]. Furthermore, as reported by Keefe et al [21], AS1411 can also inhibit the viabilities of various cancer cells, such as breast cancer cells (MCF-7) prostate cancer cells (DU145), and cervical cancer cells (Hela).
To conjugate aptamers to NPs, there are primarily two schemes: surface modification of NPs or using pre-functionalized polymers to produce NPs [22]. However, surface modification is sometimes cumbersome for NPs lack of reactive functional groups, which need to be active with coupling agents or reactive linkers followed by exhaustive purification processes to get rid of excess reactants and catalysts [23]. For NPs prepared by pre-functionalized polymers, the synthesis of polymer-aptamer conjugates may be inefficient, lengthy with high cost, at a risk of altering the chemical properties and compromising the ability of the polymer to encapsulate drugs [24]. Here we adopted a novel, simple and versatile modification technique based on dopamine polymerization in this research. Dopamine catechol can be oxidized to quinone which reacts with other catechols or quinones to form polydopamine in weak alkaline conditions (approximately pH 8 - pH 8.5), eventually leading to a water-insoluble polymer film on the surface of NPs [25]. After that, ligands possessing nucleophilic functional groups (e.g. amine and thiol) can be incorporated into the surface layer via Michael addition or Schiff base reactions [26, 27].
Star-shaped copolymers have many advantages (e.g. drug loading content, encapsulation efficiency and so on) for application of drug-delivery NPs [28]. In our previous research [29], a star-shaped CA-PLGA-b-TPGS copolymer was successfully synthesized and characterized, and it was demonstrated that docetaxel (DTX)-loaded CA-PLGA-b-TPGS NPs have many superiorities such as increased bioavailability of anticancer drugs (solubility, permeability and stability), improved drug loading content and encapsulation efficiency, accelerated drug release, good in vitro and in vivo antitumor efficiency. Herein, we continued our work on CA-PLGA-b-TPGS NPs for breast cancer therapy and inferred that aptamers could be conjugated to pD-modified CA-PLGA-b-TPGS NPs. The functionalized NPs were characterized and the anti-tumor effects were evaluated both in vitro and in vivo. Additionally, the pharmacokinetics and biodistribution analysis of the modified NPs were also investigated.
Materials and methods
Materials
D-α-tocopheryl polyethylene glycol 1000 succinate (TPGS), coumarin-6 (C6), IR-780, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), 4',6-diamidino-2-phenylindole (DAPI) and 2-(3,4-dihydroxyphenyl) ethylamine (dopamine) hydrochloride were purchased from Sigma-Aldrich (St. Louis, MO, USA). Docetaxel (purity: 99.9%) was bought from Shanghai Jinhe Bio-Technology Co., Ltd. (Shanghai, China). Star-shaped copolymer CA-PLGA-b-TPGS was synthesized as described previously [29]. All other agents used were of the highest commercial grade available. The human breast carcinoma cell line MCF-7, MDA-MB-231, human prostate adenocarcinoma cell line LNCaP and human embryonic kidney cell line HEK 293 were from American Type Culture Collection (ATCC, Rockville, MD).
Fabrication of DTX-loaded CA-PLGA-b-TPGS NPs
A modified nano-precipitation method with an acetone/water system was used to prepare the DTX-loaded CA-PLGA-b-TPGS NPs (DTX/NPs) [30, 31]. Briefly, 100 mg CA-PLGA-b-TPGS copolymer and 10 mg DTX powder were dissolved in 8 ml of acetone. The mixed solution was dropwise added into 100 ml of 0.03% (w/v) TPGS aqueous solution under stirring. After stirring overnight (600 rpm) at room temperature to remove acetone, the DTX/NPs were centrifuged at 20,000 rpm for 20 min and washed three times in 20 ml of deionized water to remove unencapsulated drug and TPGS emulsifier. At last, the precipitated DTX/NPs were weighed by electronic scale and two-thirds of the final weight of DTX/NPs were dispersed in 0.1 mg/ml dopamine hydrochloride/Tris buffer solution (pH 8.5, dopamine hydrochloride dissolved in a 10 mM Tris buffer) for pD coating, while the left DTX/NPs were dispersed in deionized water and lyophilized 2 days for further use.
Prime-Coating with pD
The pD-coated DTX/NPs (pD-DTX/NPs) were synthesized by incubating DTX/NPs in 0.1 mg/ml dopamine hydrochloride/Tris buffer solution under stirring at room temperature. After magnetic stirring for 3 h, the mixed solution turned dark slightly. Then the pD-DTX/NPs were centrifuged at 20,000 rpm for 20 min to get rid of Tris buffer solution and unreacted dopamine hydrochloride. Then the deposition was collected and resuspended by deionized water. Another centrifugation was conducted at 5,000 rpm for 20 min to separate the particulate pD with pD-DTX/NPs. This washing procedure was repeated for 3 times to guarantee the purity of pD-DTX/NPs. Finally, the precipitated pD-DTX/NPs were weighed by electronic scale and one half of the final weight of pD-DTX/NPs were dispersed in Tris buffer (pH 8.0) for aptamer-conjugation, while the left pD-DTX/NPs were dispersed in deionized water and lyophilized 2 days for further use.
Conjugation of aptamers to pD-coated NPs
AS1411 aptamer (5'-GGT GGT GGT GGT TGT GGT GGT GGT GGT TTT TTT TTT-thiol-3'), whose sequence contains ten extra T bases at the 3-terminus, was applied in this study. A random DNA control with sequence of 5'-GAG AAC CTG AGT CAG TAT TGC GGA GAT TTT TTT TTT-thiol-3' was used as a control aptamer. The aptamers were connected to the surface of pD-DTX/NPs via the Michael addition reaction [32]. In brief, the pD-DTX/NPs were resuspended in Tris buffer (pH 8.0) containing aptamers. The final concentration of pD-DTX/NPs and aptamers was 1 and 0.5 mg/ml, respectively. After magnetic stirring for 2 h at room temperature, the resulting NPs (designate as Apt-pD-DTX/NPs) were centrifuged, washed three times with deionized water and lyophilized for 2 days.
All the fluorescent NPs (C6- or IR-780-loaded NPs) were fabricated in a similar manner at each of the three steps described above. All the lyophilized NPs were redispersed in PBS before use.
Particle Size and Zeta Potential
The size and zeta potential of DTX/NPs, pD-DTX/NPs and Apt-pD-DTX/NPs were performed by Malvern Mastersizer 2000 (Zetasizer Nano ZS90, Malvern Instruments Ltd., UK). The NPs were resuspended in deionized water before experiments. The data were gained with the average of three times.
Surface Morphology
A transmission electron microscopy (TEM, Tecnai G2 F30, FEI Company, Hillsboro, Oregon, USA) was used to observe the surface morphology of NPs. To prepare samples for TEM, the optimized NPs resuspension solution was dropped onto a copper grid coated with a carbon membrane. The grid was allowed to dry before observation.
Fourier transform infrared spectroscopy analysis
The Fourier transform infrared spectroscopy analysis (FT-IR) spectra of drug-free NPs, drug-free pD/NPs and drug-free Apt-pD/NPs was recorded by FT-IR spectrophotometer (Thermo Nicolet, Madison, Wisconsin) using KBr.
X-ray photoelectron spectroscopy
A Kratos Axis Ultra DLD spectrometer with monochromatic Al Kα radiation (hν = 1486.58 eV) was used to obtain X-ray photoelectron spectroscopy (XPS). Survey and high-resolution spectra were collected at a fixed analyzer pass energy of 160 and 20 eV, respectively. Binding energy values were referenced to the Fermi edge, and charge correction was performed setting the C1s peak at 284.80 eV.
Drug loading content and encapsulation efficiency
Drug loading content (LC) and encapsulation efficiency (EE) of the modified NPs was determined by HPLC (LC 1200, Agilent Technologies, Santa Clara, CA) using previously published methods [33]. In short, 5 mg NPs were dissolved in 1 ml of dichloromethane (DCM) under vigorous vortexing. This solution was transferred to 5 ml of mobile phase consisting of acetonitrile and deionized water (50:50, v/v). To obtain a clear solution for HPLC, a nitrogen stream was introduced to evaporate DCM for about 15 min. A reverse-phase C-18 column (150 × 4.6 mm, 5μm, C18, Agilent Technologies, CA, USA) was used at 35 °C. The flow rate of mobile phase was set at 1.0 ml/min. The column effluent was detected at 227 nm using a UV/VIS detector. The calibration curve, with a correlation coefficient of R2 = 0.9999, was linear. The concentration of DTX from the dissolved NPs could be calculated through this curve and corresponding peak area from HPLC. Measurements were carried out three times for each batch. The drug LC and EE of modified NPs were calculated by the following equations respectively.


In vitro drug release profiles
To investigate the in vitro DTX release profiles, 5 mg of the lyophilized NPs were accurately weighed and dispersed in 1 ml of phosphate buffer solution (PBS, pH 7.4, containing 0.1% w/v Tween 80) to form a suspension solution. Tween 80 was used to increase the solubility of DTX in the buffer and to avoid the binding of DTX to the tube wall. The suspension solution was then transferred into a dialysis membrane bag (MWCO=3,500, Shanghai Sangon, China) that immersed in 15 ml of PBS release medium in a centrifuge tube. Afterwards, the tube was transferred into an orbital water bath and shaken at 200 rpm at 37°C. At designated time intervals, the release buffer outside the dialysis bag was replaced with fresh buffer and subjected to HPLC analysis.
Endocytosis of fluorescent NPs
MCF-7 cells or LNCaP cells were cultured in a chambered cover glass system in Dulbecco's Modified Eagle's Medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 100 U/ml penicillin, and 100 mg/ml streptomycin in 5% CO2 at 37°C. The culture was stayed in 95% air humidified atmosphere. The cells were incubated with 250 μg/ml C6-loaded NPs for 2 h, washed with cold PBS three times and fixed by cold methanol for 20 min. After that, the nuclei were counterstained with DAPI for 10 min. PBS was used to wash the stained cells three times thus removing free DAPI. In order to visualize the cells, the chambers were mounted onto the confocal laser scanning microscope (CLSM, Olympus Fluoview FV-1000, Tokyo, Japan) with the following channels: a blue channel excited at 340 nm and a green channel excited at 485 nm. For quantitative analysis, MCF-7 cells (initial density: 1×104 cells/well) were plated in 96-well black plates and incubated overnight. Then the MCF-7 cells were equilibrated with Hank's buffered salt solution (HBSS) for 1 h at 37°C, and C6-loaded NPs were added at concentrations of 100, 250 and 500 μg/ml, respectively. After incubation for 2 h, the medium was removed and the cells were washed three times with cold PBS. At last, 50 μl 0.5% Triton X-100 in 0.2 N sodium hydroxide was put into each sample well to lyse the cells. The same cell experiments were conducted on MDA-MB-231 cells with the same method.
For flow cytometric (FCM) study, MCF-7 cells (initial density:1×105 cells/well) were seeded in 6-well culture plates, and treated with 500 μg/ml C6/NPs, pD-C6/NPs, and Apt-pD-C6/NPs for 1 h at 37 °C, respectively. After removing the medium, the cells were washed with PBS twice, digested by trypsin, and finally harvested by centrifugation. The fluorescence intensity of C6 was detected by a flow cytometer (BD Biosciences, San Jose, CA, USA) at the excitation wavelength of 488 nm and the emission wavelength of 530 nm.
Cell viability evaluation
MCF-7 cells or MDA-MB-231 cells were seeded at a density of 5,000 cells/well in 96-well plates and incubated overnight. Afterwards, the cells were incubated with drug-free NPs, drug-free pD/NPs, drug-free Apt-pD/NPs, commercial Taxotere®, DTX/NPs, pD-DTX/NPs, Apt-pD-DTX/NPs or Control-Apt-pD-DTX/NPs suspension at 0.25, 2.5, 12.5 and 25 μg/ml equivalent DTX concentrations for 24 h and 48 h, respectively. At a determined time, the formulations were changed with MTT-contained DMEM (5 mg/ml) and an additional 4 h-incubation was conducted for the cells. Then MTT was removed and DMSO was dropped in to dissolve the formazan crystals at 37 °C (dark, lasted for 2 h). A microplate reader (Bio-Rad Model 680, UK) was used to measure the absorbance at 570 nm. Untreated cells represented for 100% viability and cells incubated with MTT-free medium were taken as blank to calibrate the spectrophotometer to zero absorbance. The drug concentration, at which the growth of 50% cells was inhibited (defined as IC50), was detected by curve fitting of the cell viability data in comparison with that of the control groups.
Pharmacokinetic studies
Male Sprague-Dawley (SD) rats of 200 ± 10 g (4-5 weeks old) were used for the in vivo pharmacokinetic studies. All the protocols for animal experiments were approved by the Administrative Committee on Animal Research in the Tsinghua University and Harvard Medical School. The animals were acclimatized at temperature of 25 °C under natural light/dark conditions for 7 days before experiments. After that, the SD rats were randomly divided into four groups (n = 5) and intravenously injected via tail with Taxotere®, DTX/NPs, pD-DTX/NPs or Apt-pD-DTX/NPs (DTX dose of 10 mg/kg in PBS) respectively. Blood samples of all groups were collected from the rim of eyes at 5 min, 15 min, 30 min, 1 h, 2 h, 4 h, 8 h, 12 h and 24 h after intravenous administration. Plasma samples were harvested by centrifugation at 4,000 rpm for 10 min and stored at -20 °C for HPLC analysis. DTX in the plasma samples was extracted into 1 ml of diethyl ether and was allowed to evaporate in separate tubes. 100 ml of HPLC mobile phase A (Vacetonitrile : Vmethanol : Vwater = 45: 5: 50) was added to the dried tubes and centrifuged at 12,000 rpm for 15 min. 90 ml of the supernatant was transferred to HPLC vial inserts and 50 ml was injected into the column. By increasing the proportion of mobile phase A from 0 % to 100% in 50 min, an elution gradient was applied. The system was then brought to initial condition of 100% mobile phase B (Vacetonitrile : Vmethanol : Vwater = 40: 5: 50) and equilibrated for 4 min by holding at 100% mobile phase B. The flow rate was kept at 1 ml/min and the total run time was 55 min. Through the standard curve obtained for known concentrations of DTX in plasma, the DTX concentrations in plasma of all groups were determined similarly. All the concentration data were dose-normalized and plotted as plasma DTX concentration-time curves.
Xenograft tumor model
The Institute of Laboratory Animal Sciences, Chinese Academy of Medical Science provided the female severe combined immunodeficient (SCID) mice aged 4-5 weeks old. The Administrative Committee on Animal Research in Tsinghua University approved the protocols for animal assays. About 100 μl of MCF-7 cells or MDA-MB-231 cells in PBS were implanted subcutaneously into the backs of mice (15-20 g) at a dosage of 2×106 cells per mouse. After that, we observed the tumor growth in each mouse at frequent intervals. A vernier caliper was used to survey the tumor size and tumor volume (V) was calculated as 4π/3×(length/2)×(width/2)2. 95% of the mice injected developed a tumor with an average volume of about 80 mm3 after ten days.
In vivo imaging and biodistribution analysis
After the tumor size reaching about 80 mm3, the mice were randomly divided into three groups (n = 3 mouse per group), i.e. free dye group (1 mg/kg IR-780), non-targeted pD-IR-780/NPs and targeted Apt-pD-IR-780/NPs (1 mg/kg equivalent IR-780 for NPs). The excitation wavelength for IR-780 dye is 704 nm, and the emission spectrum is 740-950 nm. With free IR-780 as control, the pD-IR-780/NPs and Apt-pD-IR-780/NPs were intravenously injected to the mice via tails. The in vivo images were recorded by the Maestro™ Automated In-Vivo Imaging system (CRi Maestro™, USA) at 0.5, 6, and 24 h post-injection respectively. Mice were humanely killed at 24 h post-injection, and then we collected the heart, liver, spleen, lung, kidney and tumor from each mouse without delay. The fluorescence intensity in all organs was further analyzed by the Maestro™ Automated In-Vivo Imaging system.
In vivo antitumor effects
Treatments were started after the tumor size reached approximately 80 mm3. Two experienced researchers randomly divided the mice into four groups (n = 5), which were intravenously injected with saline as a control, pD-DTX/NPs, Apt-pD-DTX/NPs, and Taxotere® respectively at a DTX doses of 10 mg/kg in 100 μl PBS on Day 0, 4, 8, 12 and 16. The body weights and tumor size of each mouse were recorded every other day. The mice were humanely killed after 20 days of treatment. The antitumor efficacy was investigated via tumor growth and terminal tumor weight in this study.
The antitumor efficacy of the DTX-loaded NPs was also investigated on female Tientsin Albino 2 (TA2) mice, a kind of spontaneous breast cancer-bearing mouse, by measuring their survival time after treatments [9, 34, 35]. The female TA2 mice were purchased from the Experimental Animal Center of Tianjin Medical University. After choosing 20 mice with similar body status (i.e. age, weight and tumor size), the mice were randomly divided into four groups (n = 5), and injected with Taxotere®, pD-DTX/NPs, Apt-pD-DTX/NPs (DTX dose of 10 mg/kg) and saline via tail vein at Day 0, 4, 8, 12 and 16. The survival data were presented as Kaplane-Meier plots and analyzed with a log-rank test.
Statistical methodology
At least three repetitions were carried out in all experiments unless otherwise stated. The results are expressed as mean ± SD, and the statistical significance of all the results was determined by the Student's t-test. p< 0.05 was considered to be statistically significant.
Results and discussions
Fabrication of Apt-pD-DTX/NPs
The fabrication of DTX/NPs and the surface modification of polymeric NPs through dopamine polymerization is schematized in Figure 1. As a mild and facile pathway for drug loading of polymeric NPs, the modified nano-precipitation method was used to prepare the DTX/NPs [36, 37]. A promising method to form thin pD films onto the surfaces of NPs with control over film thickness is based on the oxidative self-polymerization of dopamine. Originally inspired by the adhesive properties displayed in mussels [38], this approach is recently demonstrated to generate a range of pD-coated substrates, carbon nanotubes and particles [38-40]. It is also reported by Park et al that the dopamine polymerization method is a simple and versatile surface modification pathway, applicable to a variety of NP drug carriers irrespective of their chemical reactivity and the types of ligands (e.g. small molecules, peptides, and polymers) [32].
(A) Synthesis of aptamer-conjugated polydopamine film through Oxidative Polymerization and Michael Addition Reaction. (B) Schematic illustration of the preparation procedure of the targeted Apt-pD-DTX/NPs.
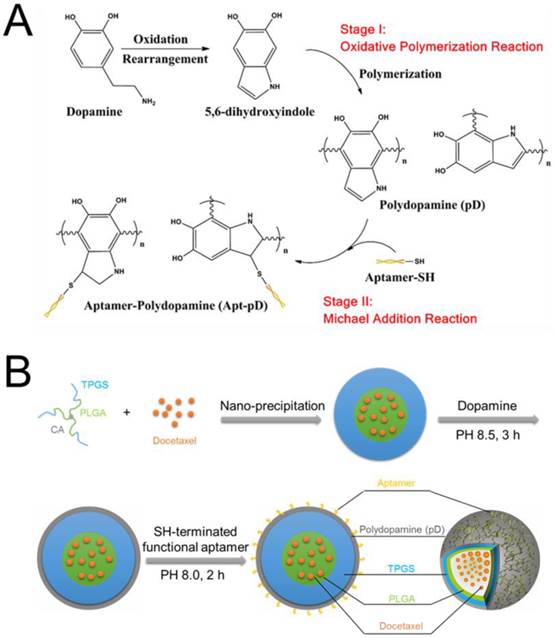
As shown in Figure 1, the final production of functionalized Apt-pD-DTX/NPs followed by two further processes: prime-coating of pD with an oxidative polymerization reaction (Stage I) and conjugation of aptamer to pD-coated NPs with a Michael addition reaction (Stage II) in a weak alkaline solution. The suspensions turned dark when dopamine hydrochloride was added, indicating dopamine was successfully polymerized. To provide cancer-targeting capability to pD-coated NPs, a 26-mer SH-terminated DNA aptamer (AS1411), which contains a thiol group at the 3'- end to allow conjugation to the pD-coated NPs, was used to be highly specific to interact with breast cancer cells [41].
Characteristics of Apt-pD-DTX/NPs
The size and surface properties of NPs have a vital position in cellular uptake, drug release, in vivo pharmacokinetics and biodistribution [42, 43]. As presented in Table 1, the average particle size of DTX/NPs was 99.3 ± 2.9 nm (PDI 0.117), while that of pD-DTX/NPs was 108.5 ± 5.7 nm (PDI 0.123) and that of Apt-pD-DTX/NPs was 112.1 ± 5.3 nm (PDI 0.119). The increased size (approximately 10 nm) of pD-DTX/NPs or Apt-pD-DTX/NPs may be contributed to the thin pD films that form around the DTX/NPs, which could also be an evidence of the successful coating of pD through the oxidative polymerization reaction. The mean hydrodynamic size of DTX-loaded NPs is about 100~110 nm in diameter, which is an appropriate size range for easily accumulating in tumor vasculature under the influence of the enhanced permeability and retention (EPR) effect [44]. These new functionalized NPs could be dissolved in organic solvent (e.g. acetone and dichloromethane).
Characterization of DTX/NPs, pD-DTX/NPs and Apt-pD-DTX/NPs.
| Samples (n=3) | Size (nm) | PDI | ZP (mV) | LC (%) | EE (%) |
|---|---|---|---|---|---|
| DTX/NPs pD-DTX/NPs Apt-pD-DTX/NPs | 99.3 ± 2.9 108.5 ± 5.7 112.1 ± 5.3 | 0.117 0.123 0.119 | -12.7 ± 3.2 -13.8 ± 4.1 -14.3 ± 3.9 | 10.13 ± 0.39 10.01 ± 0.43 9.98 ± 0.52 | 96.97 ± 1.78 95.89 ± 2.24 95.63 ± 2.32 |
PDI = polydispersity index, ZP = zeta potential, LC = loading content, EE = encapsulation efficiency, n=3.
The data in Table 1 showed that the absolute value of zeta potential of Apt-pD-DTX/NPs (-14.3 ± 3.9 mV) was slightly increased or similar compared with DTX/NPs (-12.7 ± 3.2 mV) and pD-DTX/NPs (-13.8 ± 4.1 mV). As a result of the slightly negative charge of NPs, needless clearance by the reticuloendothelial system (RES) such as liver could be reduced, followed by perfecting the blood compatibility and the accumulation of the NPs in tumor sites [45]. The size and zeta potential of fluorescent NPs (C6- and IR-780-loaded NPs) were similar as the DTX-loaded NPs (Supplementary Information, Table S1). It also can be seen from Table 1 that the LC and EE of Apt-pD-DTX/NPs (9.98 ± 0.52 %, 95.63 ± 2.32 %) were nearly the same as those of DTX/NPs (10.13 ± 0.39 %, 96.97 ± 1.78 %) and pD-DTX/NPs (10.01 ± 0.43 %, 95.89 ± 2.24 %). Also, the redispersed NPs kept a good uniformity with the NPs before lyophilization (Supplementary Information, Table S2).
In order to access the morphology of NPs, TEM were used in this study. Figure 1 (A)-(F) show the TEM images of DTX/NPs, pD-DTX/NPs and Apt-pD-DTX/NPs. As can be observed, DTX/NPs were spherical with a single surface, while thin films or membranes could be obviously visualized both on the surface of pD-DTX/NPs and Apt-pD-DTX/NPs, suggesting that pD films were successfully deposited on the surface of NPs via an oxidative polymerization reaction. The average particle size is around 90-100 nm in TEM images, reaching a smaller size than that obtained from DLS experiments. This difference may be induced by a tendency to shrink and collapse while the NPs are in the dry state [46]. As shown in Figure 2 (B) - (F) and Figure S1 (Supplementary Information), the increasing thickness of pD film (caused by the extended reaction time of pD modification) can be obviously observed by the naked eye. The growth of the pD film was monitored in Figure S2 (Supplementary Information). Furthermore, the average size of the NPs hardly changed during the 14 days of investigation (redispersion in PBS), indicating the good redispersion stability of the DTX/NPs, pD-DTX/NPs and Apt-pD-DTX/NPs (Figure 2 (G)).
Characterization of all kinds of NPs. TEM images of (A) DTX/NPs, (B) pD-DTX/NPs and (C) Apt-pD-DTX/NPs. (D) - (F) Local images in the box are shown in the low panel. (G) Stability of DTX/NPs, pD-DTX/NPs and Apt-pD-DTX/NPs in phosphate-buffered saline (PBS), respectively, by monitoring particle size over a span of two weeks (n = 3). (H) FTIR spectra of NPs, pD/NPs and Apt-pD/NPs.
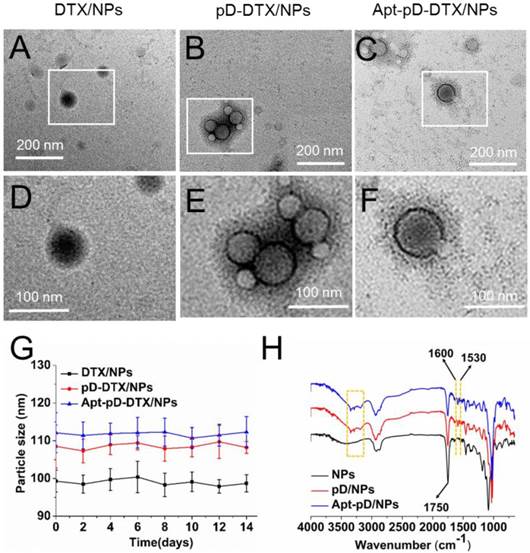
FT-IR spectroscopy was employed to verify the successful modification of pD film and the surface chemical group composition of NPs. As displayed in Figure 2 (H), several new absorption signals appeared after the surface modification. The absorption band at 1,750 cm-1 is attributed to the carbonyl band of TPGS and PLGA [47]. The overlapping of the CH stretching band of PLGA at 2,945 cm-1 was observed. But for the curves of pD/NPs and Apt-pD/NPs, the broad absorbance between 3390 and 3150 cm-1 corresponded to the stretching vibrations of N-H/O-H [48]. The absorption peaks at 1600 and 1530 cm-1 were caused by the overlap of the C=C resonance vibrations in the aromatic ring and the N-H bending vibrations [49]. These characteristic absorption bands were all from pD layers, indicating the successful incorporation of the pD layer on the surface of NPs.
To further demonstrate the successful modification of pD and conjugation of aptamers, XPS was studied and displayed in Figure 3. Both pD/NPs and Apt-pD/NPs showed two nitrogen peaks (N1s) at ∼399.6 eV (non-protonated amine groups) and ~401.4 eV (protonated amine groups) in XPS spectra, but bare NPs did not, verifying the presence of pD films [50, 51]. Moreover, the nitrogen peaks of Apt-pD/NPs were more intense than that of pD/NPs, indicating the conjugation of aptamers on the surface of pD/NPs. As for the carbon peaks (C1s), the increase intensity (pD/NPs or Apt-pD/NPs v.s. bare NPs) at ~286.2 eV and ~284.8 eV are contributed by the C-O groups and C-C groups from pD films on NP surface. In addition, the intensity of peaks at ~286.2 eV and ~284.8 eV for Apt-pD/NPs are both larger than those for pD/NPs, indicating the aptamers were conjugated to pD/NPs via the Michael addition reaction [52]. All taken together, the FT-IR spectroscopy and XPS studies indicated the successful incorporation of pD film and conjugation of aptamers on the NPs.
XPS spectra of NPs, pD/NPs and Apt-pD/NPs. (A) - (C) narrow scan for N1s peaks. (D) - (F) narrow scan for C1s peaks.
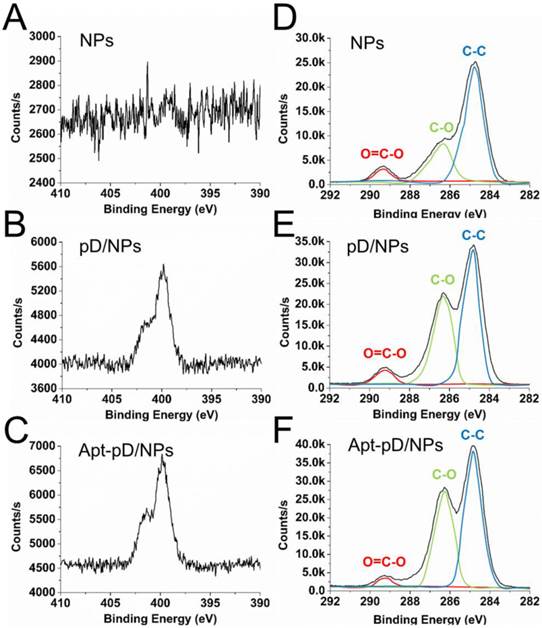
For quantitative analysis, the Wt (%) of Apt on NP surface could be calculated from XPS elemental analysis (Supplementary Information). As displayed in Table S3, the ratio of C and O atoms for drug-free NPs is 73.62 % and 26.38 %, while that of C, O and N atoms for pD/NPs is 69.36 %, 28.29 % and 2.36 %, respectively. The nitrogen could be ascribed to the successful modification of pD film. As for Apt-pD/NPs, the ratio of nitrogen (2.92 %) was slightly increased compared with that of pD/NPs, also demonstrating the successful conjugation of Apt. In the meanwhile, the Wt (%) of Apt on NP surface could also be calculated through the changes of elemental ratio, i.e. 1.65 %, which is a significant effective amount for active targeting assays. For biological application, the affinity of aptamers on NP surface and biocompatibility study of pD films were further evaluated in Figure S3 and Figure S4 (Supplementary Information).
In vitro drug release kinetics
The DTX release from NPs is an intricate course involving physicochemical properties, structure of particles and DTX diffusion after degradation [53, 54]. Figure 4 shows the in vitro DTX release profiles of DTX/NPs, pD-DTX/NPs and Apt-pD-DTX/NPs in the release medium with different pH (pH = 7.4, 6.5, and 5.0; PBS containing 0.1% w/v Tween 80) at 37 °C. The pH value of blood stream (i.e. long term circulation) is approximately 7.4, that of tumor extracellular microenvironment is approximately 6.5, and that of late endosomes/lysosomes is about 5.0 [55, 56]. Different pH values represent for different stages of NPs during in vivo delivery and cellular uptake. As displayed in the figure, the pH changes do not seem to have much influence on the in vitro DTX release profiles of this NP system. However, it is interesting that the DTX release profiles of pD-coated NPs (i.e. pD-DTX/NPs and Apt-pD-DTX/NPs) are getting close to those of uncoated NPs (i.e. DTX/NPs) with the dropping of the pH. This may be attributed to the fall-off of pD film in the acid environment.
In vitro drug release profile of DTX/NPs, pD-DTX/NPs and Apt-pD-DTX/NPs in media with different pH value: (A) pH 7.4; (B) pH 6.5; (C) pH 5.0. (D) Photos of DTX/NPs (left), pD-DTX/NPs (middle) and Apt-pD-DTX/NPs (right) solutions.
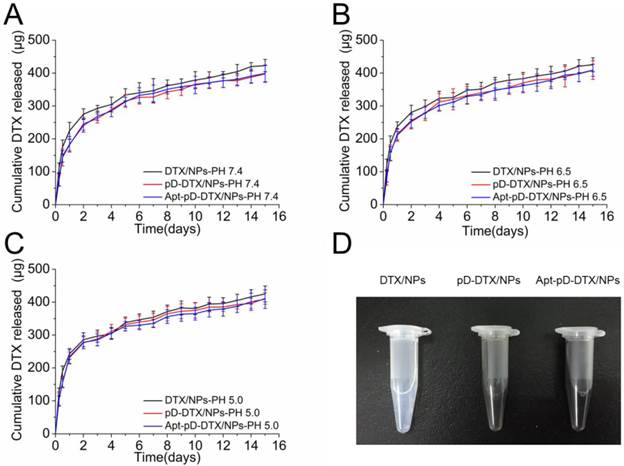
All the NPs exhibited a typically biphasic release pattern: a burst release of DTX at the initial stage and a subsequent sustained release of DTX last for more than 10 days. Take pH 5.0 for example, 285.77 ± 21.27, 276.43 ± 18.02, and 278.08 ± 19.46 μg of the encapsulated drug was released from DTX/NPs, pD-DTX/NPs and Apt-pD-DTX/NPs in the first 2 days, respectively. While 424.91 ± 24.31, 411.06 ± 17.52, and 408.98 ± 28.44 μg of drugs were released from DTX/NPs, pD-DTX/NPs and Apt-pD-DTX/NPs after two weeks, respectively. All types of the NPs had a similar DTX release profile, indicating that the surface modification reaction did not have much influence on the drug release properties of the DTX/NPs (since the pD film on NP surface is thin enough), thus making the Apt-pD-DTX/NPs a more possible and promising drug delivery vector for cancer therapy since exciting results of DTX/NPs have been achieved in our previous study [29].
Cell uptake of fluorescent NPs and in vitro cellular targeting
Due to the reason that the cellular internalization and sustained retention play important roles on therapeutic effects of the drug-loaded NPs, we then investigate the cellular uptake profile of the NPs [57, 58]. DTX was replaced by a fluorescent probe C6 to assess the extents of internalization of the prepared NPs into MCF-7 cells and observed by CLSM [33]. The CLSM images of the cells after 2 h-incubation with the suspension of 250 μg/ml C6-loaded NPs in DMEM were illustrated in Figure 5 (A). As shown in the figure, the fluorescence intensity (i.e. cellular uptake efficiency) was nearly equivalent in the cells incubated with C6/NPs, pD-C6/NPs and Control-Apt-pD-C6/NPs. However, the fluorescence intensity increased significantly in MCF-7 cells after 2 h-incubated with Apt-pD-C6/NPs, indicating the existence of AS1411 aptamers on the surface of NPs may be the essential reason for high-efficiency in vitro cellular targeting. Similar results were also obtained in MDA-MB-231 cells (Supplementary Information, Figure S5). To demonstrate the significant role of AS1411 aptamers in the cellular uptake of Apt-pD-C6/NPs, we performed a receptor competition assay by selecting AS1411 aptamers as the competitive reagent. Consisted with our hypothesis, the fluorescence was significantly quenched when Apt-pD-C6/NPs and AS1411 aptamers were added to wells at the same time. Furthermore, to investigate the tumor targeting specificity of Apt-pD-C6/NPs, LNCaP cells (AS1411 non-specific cancer cells) were used as control. As a result, the fluorescence observed in LNCaP cells was also significantly decreased compared with that of MCF-7 cells incubated with Apt-pD-C6/NPs.
Endocytosis of C6/NPs, pD-C6/NPs and Apt-pD-C6/NPs. (A) CLSM images of MCF-7 and LNCaP cells after 2 h-incubation. (B) Cellular uptake efficiency of C6/NPs, pD-C6/NPs and Apt-pD-C6/NPs in MCF-7 cells after 2 h-incubation. (C) FCM histograms for C6/NPs, pD-C6/NPs and Apt-pD-C6/NPs in MCF-7 cells after 1 h-incubation. Cytotoxicity of NPs detected by MTT assays. Viability of MCF-7 cells cultured with DTX-loaded NPs in comparison with that of Taxotere® at the same DTX dose and drug-free NPs with the same amount of NPs for (D) 24 h and (E) 48 h (t-test, *p< 0.05, **p< 0.01, ***p< 0.001).
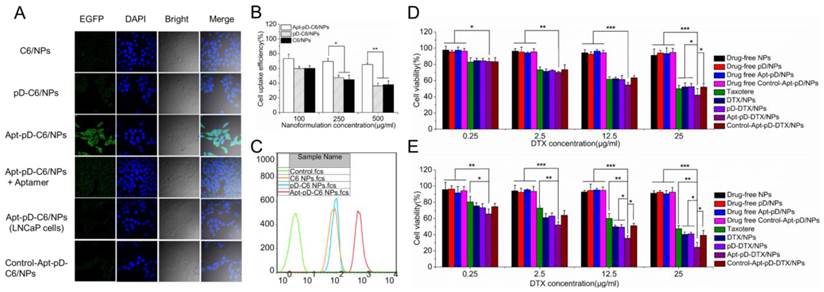
For quantitatively investigation, MCF-7 cells were incubated with 50, 250 and 500 μg/ml C6/NPs, pD-C6/NPs, and Apt-pD-C6/NPs for 2 h, respectively. As displayed in Figure 5 (B), the cellular uptake efficiency of all fluorescent NPs decreased with the increasing concentration of NPs from 100 to 500 μg/ml. For C6/NPs, the cellular uptake efficiency was nearly the same as that of pD-C6/NPs at all NPs concentration. However, it was only 81.99%, 64.28%, and 58.01% of the Apt-pD-C6/NPs group at the concentration of 100, 250, and 500 μg/ml, respectively. Therefore, we consider that the high uptake efficiency may be attributed to the targeting ligand (i.e. AS1411) on the surface of the NPs. The endocytosis of C6/NPs, pD-C6/NPs and Apt-pD-C6/NPs was further confirmed by FCM assay (Figure 5 (C)). The fluorescence intensity of cells treated with Apt-pD-C6/NPs exceeds those of cells treated with C6/NPs and pD-C6/NPs, reinforcing that the endocytosis of NPs can be improved by AS1411 aptamers through the specific interactions with breast tumor cells.
Effects of NPs on cell viability
In order to evaluate the cytotoxicity of DTX/NPs, pD-DTX/NPs and Apt-pD-DTX/NPs, MTT assay was performed on MCF-7 cells. Taxotere®, a kind of DTX formulation which is clinically available, was selected as the contrast. The cytotoxicity of drug-free NPs was also investigated to exclude any non-specific effect. MCF-7 cells were treated with Drug-free or DTX-loaded NPs suspensions and Taxotere® at 0.25, 2.5, 12.5 and 25 μg/ml equivalent DTX concentrations for 24 h and 48 h respectively. We can conclude from Figure 5 (D) and (E): (a) None of the drug-free NPs exhibited obvious cytotoxity to MCF-7 cells at various concentrations from 0.25 to 25 μg/ml, declaring both the copolymer and the modification of NPs (pD coating and conjugation of aptamers) are biocompatible and nontoxic to cells and tissues; (b) the cellular viability decreased with the increase of incubation time for both Taxotere® and nanoformulations, exhibiting a dose-dependent and time-dependent effect, especially for Apt-pD-DTX/NPs; (c) DTX/NPs and pD-DTX/NPs showed similar cytotoxicity at virous concentrations from 0.25 to 25 μg/ml, further indicating that pD film on the surface of the NPs did not influence the cell viability, being biocompatible and non-toxicity; (d) Apt-pD-DTX/NPs exhibited better in vitro antitumor efficacy than Taxotere®, DTX/NPs, pD-DTX/NPs and Control-Apt-pD-DTX/NPs on MCF-7 cells. For example, the MCF-7 cellular viability (48 h, 12.5 μg/ml) was 59.93 % for Taxotere®, 49.62 % for DTX/NPs, 49.26 % for pD-DTX/NPs, 50.91 % for Control-Apt-pD-DTX/NPs, and 35.61 % for Apt-pD-DTX/NPs. Furthermore, the viability of MCF-7 cells (48 h, 25 μg/ml) after incubation with Apt-pD-DTX/NPs was 22.80 %, 15.82 %, 16.57 %, or 14.75 % less than that of Taxotere®, DTX/NPs, pD-DTX/NPs, or Control-Apt-pD-DTX/NPs, respectively. These data also demonstrated the Apt-pD-DTX/NPs (with the specific targeting effect of AS1411 aptamers) have better performance on in vitro antitumor efficiency than clinical available Taxotere® and other non-target NPs. Therefore, it is possible that Apt-pD-DTX/NPs could be used as a promising drug delivery system with extremely high efficiency. Similar conclusions could be achieved on MDA-MB-231 cells (Supplementary Information, Figure S6).
The in vitro therapeutic effects of a pharmaceutical formulation could be quantitatively studied by IC50 value, which is defined as the drug inhibitory concentration causing 50 % tumor cell mortality in a designated period. Table 2 lists the IC50 values of MCF-7 cells after 24 and 48 h-incubation with clinical available Taxotere®, DTX/NPs, pD-DTX/NPs and Apt-pD-DTX/NPs (calculated from Figure 5 (D) and (E)). As revealed in Table 2, the IC50 values of DTX/NPs and pD-DTX/NPs for MCF-7 cells were 20.13 μg/ml and 20.64 μg/ml after 24 h-incubation respectively, and 6.28 μg/ml and 6.88 μg/ml after 48 h-incubation respectively, which was also consist with the results got from cell uptake experiments and indicating no obvious effect would be brought after pD coating. In the meantime, the IC50 value of Apt-pD-DTX/NPs for MCF-7 cells was 12.72 μg/ml, only a little smaller than Taxotere® (18.39 μg/ml) after 24 h-incubation. However, that of Apt-pD-DTX/NPs was 8.1-fold smaller than that of Taxotere® (1.69 μg/ml v.s. 13.69 μg/ml) after 48 h-incubation. The advantages in tumor inhibition of Apt-pD-DTX/NPs > DTX/NPs (or pD-DTX/NPs) > clinical Taxotere® could be concluded, demonstrating significant in vitro therapeutic effects and great possibilities for in vivo treatment of targeted Apt-pD-DTX/NPs.
IC50 values of Taxotere®, DTX/NPs, pD-DTX/NPs and Apt-pD-DTX/NPs on MCF-7 cells after 24 h- and 48 h-incubation.
| Incubation time (h) | IC50 (μg/ml) | |||
|---|---|---|---|---|
| Taxotere® | DTX/NPs | pD-DTX/NPs | Apt-pD-DTX/NPs | |
| 24 | 18.39 ± 1.41 | 20.13 ± 1.48 | 20.64 ± 1.67 | 12.72 ± 0.59 |
| 48 | 13.69 ± 0.98 | 6.28 ± 0.47 | 6.88 ± 0.51 | 1.69 ± 0.09 |
Pharmacokinetic studies
Figure 6 displays the in vivo pharmacokinetics, i.e. the plasma drug concentration versus time curve after i.v. injection of Taxotere®, DTX/NPs, pD-DTX/NPs and Apt-pD-DTX/NPs to SD rats at the same DTX concentration of 10 mg/kg (n = 5). The key pharmacokinetics parameters were analyzed according to Figure 6 and the results are listed in Table 3, including Cmax (the maximum DTX concentration encountered after injection), AUC0-inf (the total area under the curve, representing the in vivo therapeutic effects), Mean Residence Time (MRT, the average time of the DTX remaining in the body after i.v. injection) and T1/2 (the time at which the DTX concentration reached 50 % of its peak value in the plasma) [59]. As shown in Figure 6 and Table 3, the DTX of Taxotere® formulation was rapidly cleared through systemic circulation (clearance half-life: 0.193 h). However, the DTX encapsulation in NPs achieved much longer circulation time, i.e. the clearance half-life of DTX/NPs, pD-DTX/NPs and Apt-pD-DTX/NPs was 2.432 h, 2.139 h, and 2.246 h, respectively. The higher plasma concentration and longer circulation time could be caused by the enhanced stability and the stealth effects of NP surface [60]. Both the clearance of pD-DTX/NPs and that of Apt-pD-DTX/NPs were slightly faster than that of DTX/NPs, in the meantime, the AUC0-inf of pD-DTX/NPs and that of Apt-pD-DTX/NPs were also slightly smaller than that of DTX/NPs. The slightly decreased of T1/2 and AUC0-inf may be caused by the pD-coating on the surface of NPs, which may partly compromise the stealth effect of PEG. However, no statistical significance was detected and the parameters were quite close, indicating that the pD-coating and aptamer-decorating led to little influence on the in vivo pharmacokinetics of NPs. Taken all together, the results showed the NPs possessed significant advantages on long-term circulation performance compared with clinical Taxotere®, ensuring the possibility of excellent in vivo targeting efficacy and antitumor effect of Apt-pD-DTX/NPs.
DTX concentration-time profile following intravenous administration of Taxotere®, DTX/NPs, pD-DTX/NPs and Apt-pD-DTX/NPs in SD rats at the DTX dose of 10 mg/kg (n = 5).
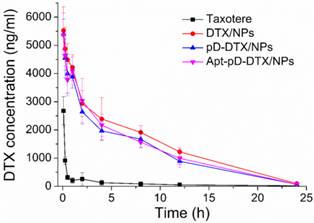
Pharmacokinetics in SD rats after i.v. administration of Taxotere®, DTX/NPs, pD-DTX/NPs and Apt-pD-DTX/NPs at the same 10 mg/kg drug dose.
| Parameters | Taxotere® | DTX/NPs | pD-DTX/NPs | Apt-pD-DTX/NPs |
|---|---|---|---|---|
| Cmax (ng/ml) | 1028.935 | 5950.842 | 5366.556 | 5875.360 |
| T1/2 (h) | 0.193 | 2.432 | 2.139 | 2.246 |
| AUC0-inf (ng/ml*h) | 1028.935 | 52059.121 | 46045.746 | 48568.713 |
| MRT (h) | 0.216 | 22.118 | 19.032 | 19.726 |
In vivo imaging and biodistribution of NPs
A whole animal near-infrared imaging system was employed to study the in vivo distribution and tumor targeting of Apt-pD-DTX/NPs. IR-780 is a lipophilic cation heptamethine dye which has a stable chemical property. With strong fluorescence intensity, IR-780 could be used for near-infrared imaging and observation [61]. The in vivo fluorescence intensity distribution of free IR-780, pD-IR-780/NPs (non-targeted) and Apt-pD-IR-780/NPs (targeted) was displayed in Figure 7 (A) and (B). At 0.5 h post-injection, it could be observed that the fluorescent NPs were whole body distributed in all groups, but a large number of the fluorescence intensity was located at the joint of the tail and the body. At 6 h post-injection, the fluorescence intensity at the joint of the tail and the body began to wane, while the signals all over the body tend to increase, indicating the IR-780 or NPs were transported in the body through long-term circulation of the nude mice. In the same time, the fluorescence signals were beginning to be accumulated in tumors of all groups. But the signal fluorescence signals of Apt-pD-IR-780/NPs in tumor tissues were stronger than those of free IR-780 and pD-IR-780/NPs. After 24 h post-injection, the signal intensity in all groups was observed much stronger in tumor tissues. Nevertheless, the tumor signal intensity of Apt-pD-IR-780/NPs was significantly higher than that of IR-780 and pD-IR-780/NPs, indicating Apt-pD-IR-780/NPs had the best in vivo targeting effects.
In vivo imaging and biodistribution analysis of SCID mice bearing MCF-7 cells xenograft after tail vein injected of free IR-780, pD-IR-780/NPs and Apt-pD-IR-780/NPs. (A) Time-lapse NIR fluorescence images of nude mice. The tumors were circled with dotted line. (B) NIR fluorescence intensity of tumors was quantified at indicated time points. (C) NIR fluorescence images of major organs and tumors after injection at 24 h. (D) Semiquantitative biodistribution of free IR-780, pD-IR-780/NPs and Apt-pD-IR-780/NPs in nude mice determined by the averaged fluorescence intensity of organs and tumors.
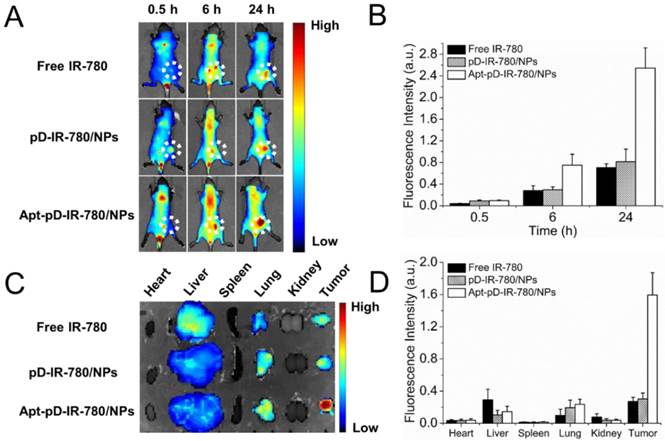
Figure 7 (C) and (D) show the biodistribution of IR-780, pD-IR-780/NPs and Apt-pD-IR-780/NPs at 24 h post-injection in the MCF-7 xenograft, which further demonstrating that Apt-pD-IR-780/NPs exhibit the highest tumor accumulation compared with IR-780 and pD-IR-780/NPs. It also could be observed that some fluorescence intensity was detected in the liver, especially for the free IR-780 group. This situation may be explained by the fact that kupffer cells in liver play an important role on uptaking and degrading extra-phagocytosis, i.e. IR-780 dye is finally metabolized in the liver [62]. The larger size particle of pD-IR-780/NPs and Apt-pD-IR-780/NPs may lead to the mechanical retention of NPs in the lung capillaries.
In vivo antitumor effects of NPs
Based on the positive results from above experiments, we continued in vivo antitumor evaluation to verify the immense value of Apt-pD-DTX/NPs for breast cancer chemotherapy. As shown in Figure 8 (B), both clinical available Taxotere® and DTX-loaded NPs (i.e. pD-DTX/NPs and Apt-pD-DTX/NPs) inhibited tumor growth compared with saline (control group). However, the therapeutic effect of DTX-loaded NPs was significantly better than clinical Taxotere®. In the meantime, that of targeted Apt-pD-DTX/NPs was also superior to that of non-targeted pD-DTX/NPs, which was consistent with our previous cell uptake and cytotoxicity results. Notably, treatment with DTX-loaded NPs did not reduce body weight of the mice, while Taxotere® treatment can cause serious side effects such as weight loss and physical deterioration (Figure 8 (D)). At the end of the 20-day treatment, all mice were sacrificed the tumors were separated from bodies. Figure 8 (A) and (B) clearly show the morphology and average weight of the tumors in all groups, directly confirming the excellent in vivo tumor-suppressing effects of targeted Apt-pD-DTX/NPs. The MDA-MB-231 xenograft model also provided evidence for this conclusion (Supplementary Information, Figure S7).
Anti-tumor efficacy of Taxotere®, pD-DTX/NPs, and Apt-pD-DTX/NPs on the SCID nude mice bearing MCF-7 xenograft. (A) Images of tumors in each group taken out from the sacrificed mice at the end point of research. Lane 1, Saline; Lane 2, Taxotere®; Lane 3, pD-DTX/NPs; Lane 4, Apt-pD-DTX/NPs. (B) Tumor growth curve after intravenously injected with Saline, Taxotere®, pD-DTX/NPs, and Apt-pD-DTX/NPs (t-test, *p<0.05, **p<0.01, ***p<0.001). (C) Tumor weight of each group taken out from the sacrificed mice at the end of the study (t-test, *p<0.05, ***p<0.001). (D) Weight change curve of the SCID nude mice bearing MCF-7 cells xenograft after intravenously injected with Saline, Taxotere®, pD-DTX/NPs, and Apt-pD-DTX/NPs.
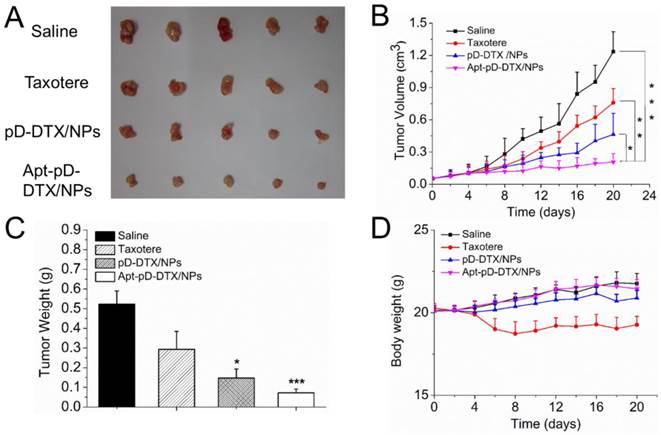
Breast cancer-bearing model in TA2 mice was also applied as another animal model for in vivo antitumor efficacy evaluation by providing survival data. The dosing schedule utilize for the TA2 breast cancer-bearing model was adopted from that used in the xenograft MCF-7 model. As can be seen from Figure S8 (Supplementary Information), the increased survival time of targeted Apt-pD-DTX/NPs compared with non-targeted pD-DTX/NPs, Taxotere® and saline was 10, 16, and 21 days respectively, which was significantly meaningful in biological value, and signally prolonged the lifetime of the mice (Apt-pD-DTX/NPs v.s. pD-DTX/NPs, 22.22 % of lifetime extension; Apt-pD-DTX/NPs v.s. Taxotere®, 41.03 % of lifetime extension; Apt-pD-DTX/NPs v.s. saline, 61.76 % of lifetime extension). All above, Apt-pD-DTX/NPs worked significantly better than clinical used Taxotere® and non-targeted pD-DTX/NPs, suggesting that Apt-pD-DTX/NPs could be used as a promising and high-efficiency drug delivery system for breast cancer therapy. Moreover, this in vivo antitumor effect of Apt-pD-DTX/NPs reported here was superior to that of our previous studies using DTX/NPs [29], DTX-loaded M-PLGA-TPGS NPs [63], DTX-loaded CA-(PCL-ran-PLA)-b-PEG1k NPs [31] or DTX-loaded H40-PLA-b-TPGS [30] to mediate DTX treatment in tumor xenograft model.
Conclusions
In this work, novel pD-modified NP-aptamer bioconjugates (Apt-pD-DTX/NPs) were synthesized successfully for the first time, and applied as a targeted drug carrier for breast cancer therapy. By using the facile dopamine polymerization method, CA-PLGA-b-TPGS NPs were simply functionalized by aptamers. The pD film on the surface of NPs could be clearly observed through TEM and the growth rate of the film was further studied. The endocytosis of fluorescent NPs and in vitro cellular targeting assay demonstrated that the Apt-pD-C6/NPs managed to target MCF-7 and MDA-MB-231 cells specifically with high efficacy compared to C6/NPs and pD-C6/NPs. The in vitro cytotoxicity assay showed the Apt-pD-DTX/NPs significantly inhibit cell proliferation in comparison with DTX/NPs and pD-DTX/NPs. The pharmacokinetic studies proved that the Apt-pD-DTX/NPs have excellent long-term circulation effects compared with clinical used Taxotere®. All data obtained from the in vivo imaging and biodistribution of NPs were consist with the in vitro experiments, declaring that Apt-pD-DTX/NPs could be a potentially qualified drug delivery system targeting breast cancer. The Apt-pD-DTX/NPs were demonstrated to have enhanced therapeutic effects through in vivo antitumor assays, further reducing the side effects of anti-tumor drugs and improving survival quality during treatment. Moreover, construction of pD-modified NP-aptamer bioconjugates is a simple modification method for NPs and can be applied in various NPs.
Supplementary Material
Supplementary Tables and Figures.
Acknowledgements
This work was supported by the Tsinghua Scholarship for Overseas Graduate Studies (No. 2013159), National Natural Science Foundation of China (No. 31270019), Guangdong Natural Science Funds for Distinguished Young Scholar (No. 2014A030306036), Natural Science Foundation of Guangdong Province (No. 2015A030313848), China Postdoctoral Science Foundation (No. 2015M580109), and Science, Technology & Innovation Commission of Shenzhen Municipality (Nos. JCYJ20150430163009479, JCYJ20150529164918738, CYZZ 20130320110255352).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Shi J, Xiao ZY, Votruba AR, Vilos C, Farokhzad OC. Differentially Charged Hollow Core/Shell Lipid-Polymer-Lipid Hybrid Nanoparticles for Small Interfering RNA Delivery. Angew Chem Int Ed Engl. 2011;50:7027-31
2. Shi J, Votruba AR, Farokhzad OC, Langer R. Nanotechnology in drug delivery and tissue engineering: from discovery to applications. Nano Lett. 2010;10:3223-30
3. Porta F, Lamers GEM, Morrhayim J, Chatzopoulou A, Schaaf M, den Dulk H. et al. Folic Acid-Modified Mesoporous Silica Nanoparticles for Cellular and Nuclear Targeted Drug Delivery. Adv Healthc Mater. 2013;2:281-6
4. Yu C, Hu Y, Duan J, Yuan W, Wang C, Xu H. et al. Novel Aptamer-Nanoparticle Bioconjugates Enhances Delivery of Anticancer Drug to MUC1-Positive Cancer Cells In Vitro. PloS one. 2011;6:e24077
5. Xiao Z, Levy-Nissenbaum E, Alexis F, Lupták A, Teply BA, Chan JM. et al. Engineering of Targeted Nanoparticles for Cancer Therapy Using Internalizing Aptamers Isolated by Cell-Uptake Selection. ACS nano. 2012;6:696-704
6. Locatelli E, Broggi F, Ponti J, Marmorato P, Franchini F, Lena S. et al. Lipophilic Silver Nanoparticles and Their Polymeric Entrapment into Targeted-PEG-Based Micelles for the Treatment of Glioblastoma. Adv Healthc Mater. 2012;1:342-7
7. Farokhzad OC, Jon S, Khademhosseini A, Tran TN, Lavan DA, Langer R. Nanoparticle-aptamer bioconjugates: a new approach for targeting prostate cancer cells. Cancer Res. 2004;64:7668-72
8. Dhar S, Gu FX, Langer R, Farokhzad OC, Lippard SJ. Targeted delivery of cisplatin to prostate cancer cells by aptamer functionalized Pt(IV) prodrug-PLGA-PEG nanoparticles. Proc Natl Acad Sci U S A. 2008;105:17356-61
9. Guo J, Gao X, Su L, Xia H, Gu G, Pang Z. et al. Aptamer-functionalized PEG-PLGA nanoparticles for enhanced anti-glioma drug delivery. Biomaterials. 2011;32:8010-20
10. Aravind A, Jeyamohan P, Nair R, Veeranarayanan S, Nagaoka Y, Yoshida Y. et al. AS1411 aptamer tagged PLGA-lecithin-PEG nanoparticles for tumor cell targeting and drug delivery. Biotechnol Bioeng. 2012;109:2920-31
11. Jiang F, Kumar RA, Jones RA, Patel DJ. Structural basis of RNA folding and recognition in an AMP-RNA aptamer complex. Nature. 1996;382:183-6
12. Sun H, Zhu X, Lu PY, Rosato RR, Tan W, Zu Y. Oligonucleotide Aptamers: New Tools for Targeted Cancer Therapy. Mol Ther Nucleic Acids. 2014;3:e182
13. Kim MG, Shon Y, Lee J, Byun Y, Choi BS, Kim YB. et al. Double stranded aptamer-anchored reduced graphene oxide as target-specific nano detector. Biomaterials. 2014;35:2999-3004
14. Liu Q, Jin C, Wang Y, Fang X, Zhang X, Chen Z. et al. Aptamer-conjugated nanomaterials for specific cancer cell recognition and targeted cancer therapy. NPG Asia Mater. 2014;6:e95
15. Reinemann C, Strehlitz B. Aptamer-modified nanoparticles and their use in cancer diagnostics and treatment. Swiss Med Wkly. 2014;144:w13908
16. Soundararajan S, Chen W, Spicer EK, Courtenay-Luck N, Fernandes DJ. The nucleolin targeting aptamer AS1411 destabilizes Bcl-2 messenger RNA in human breast cancer cells. Cancer Res. 2008;68:2358-65
17. Kim JK, Choi K-J, Lee M, Jo M-h, Kim S. Molecular imaging of a cancer-targeting theragnostics probe using a nucleolin aptamer- and microRNA-221 molecular beacon-conjugated nanoparticle. Biomaterials. 2012;33:207-17
18. Li LL, Yin Q, Cheng JJ, Lu Y. Polyvalent Mesoporous Silica Nanoparticle-Aptamer Bioconjugates Target Breast Cancer Cells. Adv Healthc Mater. 2012;1:567-72
19. Zhou W, Zhou Y, Wu J, Liu Z, Zhao H, Liu J. et al. Aptamer-nanoparticle bioconjugates enhance intracellular delivery of vinorelbine to breast cancer cells. J Drug Target. 2013;22:57-66
20. Xing H, Tang L, Yang X, Hwang K, Wang W, Yin Q. et al. Selective Delivery of an Anticancer Drug with Aptamer-Functionalized Liposomes to Breast Cancer Cells in Vitro and in Vivo. J Mater Chem B. 2013;1:5288-97
21. Keefe AD, Pai S, Ellington A. Aptamers as therapeutics. Nat Rev Drug Discov. 2010;9:537-50
22. Liu AX, Hong ZK, Zhuang XL, Chen XS, Cui Y, Liu Y. et al. Surface modification of bioactive glass nanoparticles and the mechanical and biological properties of poly(L-lactide) composites. Acta Biomater. 2008;4:1005-15
23. Takahashi M, Yoshino T, Matsunaga T. Surface modification of magnetic nanoparticles using asparagines-serine polypeptide designed to control interactions with cell surfaces. Biomaterials. 2010;31:4952-7
24. Ho HA, Leclerc M. Optical sensors based on hybrid aptamer/conjugated polymer complexes. J Am Chem Soc. 2004;126:1384-7
25. Jiang JH, Zhu LP, Zhu LJ, Zhu BK, Xu YY. Surface Characteristics of a Self-Polymerized Dopamine Coating Deposited on Hydrophobic Polymer Films. Langmuir. 2011;27:14180-7
26. Lee H, Dellatore SM, Miller WM, Messersmith PB. Mussel-inspired surface chemistry for multifunctional coatings. Science. 2007;318:426-30
27. Lee H, Rho J, Messersmith PB. Facile Conjugation of Biomolecules onto Surfaces via Mussel Adhesive Protein Inspired Coatings. Adv Mater. 2009;21:431-4
28. Mei L, Jiang Y, Feng S-S. Star-shaped block polymers as a molecular biomaterial for nanomedicine development. Nanomedicine (Lond). 2013;9:9-12
29. Zeng XW, Tao W, Mei L, Huang LG, Tan CY, Feng SS. Cholic acid-functionalized nanoparticles of star-shaped PLGA-vitamin E TPGS copolymer for docetaxel delivery to cervical cancer. Biomaterials. 2013;34:6058-67
30. Zeng X, Tao W, Wang Z, Zhang X, Zhu H, Wu Y. et al. Docetaxel-Loaded Nanoparticles of Dendritic Amphiphilic Block Copolymer H40-PLA-b-TPGS for Cancer Treatment. Part Part Syst Char. 2015;32:112-22
31. Tao W, Zeng X, Zhang J, Zhu H, Chang D, Zhang X. et al. Synthesis of cholic acid-core poly([varepsilon]-caprolactone-ran-lactide)-b-poly(ethylene glycol) 1000 random copolymer as a chemotherapeutic nanocarrier for liver cancer treatment. Biomater Sci-UK. 2014;2:1262-74
32. Park J, Brust TF, Lee HJ, Lee SC, Watts VJ, Yeo Y. Polydopamine-based simple and versatile surface modification of polymeric nano drug carriers. ACS nano. 2014;8:3347-56
33. Tao W, Zeng X, Liu T, Wang Z, Xiong Q, Ouyang C. et al. Docetaxel-loaded nanoparticles based on star-shaped mannitol-core PLGA-TPGS diblock copolymer for breast cancer therapy. Acta Biomater. 2013;9:8910-20
34. Gu Y, Zhang S, Wu Q, Xu S, Cui Y, Yang Z. et al. Differential expression of decorin, EGFR and cyclin D1 during mammary gland carcinogenesis in TA2 mice with spontaneous breast cancer. J Exp Clin Cancer Res. 2010;29:6
35. Zhang DF, Sun BC, Zhao XL, Ma YM, Ji R, Gu Q. et al. Twist1 expression induced by sunitinib accelerates tumor cell vasculogenic mimicry by increasing the population of CD133(+) cells in triple-negative breast cancer. Mol Cancer. 2014:13
36. Zhang YQ, Tang L, Sun LL, Bao JB, Song CX, Huang LQ. et al. A novel paclitaxel-loaded poly(epsilon-caprolactone)/Poloxamer 188 blend nanoparticle overcoming multidrug resistance for cancer treatment. Acta Biomater. 2010;6:2045-52
37. Tao W, Zhang J, Zeng X, Liu D, Liu G, Zhu X. et al. Blended Nanoparticle System Based on Miscible Structurally Similar Polymers: A Safe, Simple, Targeted, and Surprisingly High Efficiency Vehicle for Cancer Therapy. Adv Healthc Mater. 2015
38. Liu Y, Ai K, Lu L. Polydopamine and its derivative materials: synthesis and promising applications in energy, environmental, and biomedical fields. Chem Rev. 2014;114:5057-115
39. Postma A, Yan Y, Wang Y, Zelikin AN, Tjipto E, Caruso F. Self-Polymerization of Dopamine as a Versatile and Robust Technique to Prepare Polymer Capsules. Chemistry of Materials. 2009;21:3042-4
40. Fei B, Qian B, Yang Z, Wang R, Liu WC, Mak CL. et al. Coating carbon nanotubes by spontaneous oxidative polymerization of dopamine. Carbon. 2008;46:1795-7
41. Cao Z, Tong R, Mishra A, Xu W, Wong GCL, Cheng J. et al. Reversible Cell-Specific Drug Delivery with Aptamer-Functionalized Liposomes. Angew Chem Int Ed Engl. 2009;48:6494-8
42. Perrault SD, Walkey C, Jennings T, Fischer HC, Chan WCW. Mediating Tumor Targeting Efficiency of Nanoparticles Through Design. Nano Lett. 2009;9:1909-15
43. Zhu X, Xu Y, Solis LM, Tao W, Wang L, Behrens C. et al. Long-circulating siRNA nanoparticles for validating Prohibitin1-targeted non-small cell lung cancer treatment. Proc Natl Acad Sci U S A. 2015;112:7779-84
44. Yan F, Zhang C, Zheng Y, Mei L, Tang LN, Song CX. et al. The effect of poloxamer 188 on nanoparticle morphology, size, cancer cell uptake, and cytotoxicity. Nanomedicine. 2010;6:170-8
45. Xiao K, Li YP, Luo JT, Lee JS, Xiao WW, Gonik AM. et al. The effect of surface charge on in vivo biodistribution of PEG-oligocholic acid based micellar nanoparticles. Biomaterials. 2011;32:3435-46
46. Zhang CN, Wang W, Liu T, Wu YK, Guo H, Wang P. et al. Doxorubicin-loaded glycyrrhetinic acid-modified alginate nanoparticles for liver tumor chemotherapy. Biomaterials. 2012;33:2187-96
47. Bernabeu E, Helguera G, Legaspi MJ, Gonzalez L, Hocht C, Taira C. et al. Paclitaxel-loaded PCL-TPGS nanoparticles: In vitro and in vivo performance compared with Abraxane (R). Colloid Surface B. 2014;113:43-50
48. Zhang M, Zhang XH, He XW, Chen LX, Zhang YK. A self-assembled polydopamine film on the surface of magnetic nanoparticles for specific capture of protein. Nanoscale. 2012;4:3141-7
49. Iqbal Z, Lai EPC, Avis TJ. Antimicrobial effect of polydopamine coating on Escherichia coli. J Mater Chem. 2012;22:21608-12
50. Eby DM, Artyushkova K, Paravastu AK, Johnson GR. Probing the molecular structure of antimicrobial peptide-mediated silica condensation using X-ray photoelectron spectroscopy. J Mater Chem. 2012;22:9875-83
51. Metwalli E, Haines D, Becker O, Conzone S, Pantano CG. Surface characterizations of mono-, di-, and tri-aminosilane treated glass substrates. J Colloid Interface Sci. 2006;298:825-31
52. Liu Q, Wang NY, Caro J, Huang AS. Bio-Inspired Polydopamine: A Versatile and Powerful Platform for Covalent Synthesis of Molecular Sieve Membranes. J Am Chem Soc. 2013;135:17679-82
53. Guduru R, Khizroev S. Magnetic Field-Controlled Release of Paclitaxel Drug from Functionalized Magnetoelectric Nanoparticles. Part Part Syst Char. 2014;31:605-11
54. Fedoryshin LL, Tavares AJ, Petryayeva E, Doughan S, Krull UJ. Near-Infrared-Triggered Anticancer Drug Release from Upconverting Nanoparticles. ACS Appl Mater Interfaces. 2014;6:13600-6
55. Zhou KJ, Wang YG, Huang XN, Luby-Phelps K, Sumer BD, Gao JM. Tunable, Ultrasensitive pH-Responsive Nanoparticles Targeting Specific Endocytic Organelles in Living Cells. Angew Chem Int Ed Engl. 2011;50:6109-14
56. Angelos S, Khashab NM, Yang YW, Trabolsi A, Khatib HA, Stoddart JF. et al. pH Clock-Operated Mechanized Nanoparticles. J Am Chem Soc. 2009;131:12912 -+
57. Wu J, Zhao L, Xu X, Bertrand N, Choi W II, Yameen B. et al. Hydrophobic Cysteine Poly(disulfide)-based Redox-Hypersensitive Nanoparticle Platform for Cancer Theranostics. Angew Chem Int Ed Engl. 2015;127:9350-5
58. Shi J, Xu Y, Xu X, Zhu X, Pridgen E, Wu J. et al. Hybrid lipid-polymer nanoparticles for sustained siRNA delivery and gene silencing. Nanomedicine. 2014;10:e897-e900
59. Feng SS, Mei L, Anitha P, Gan CW, Zhou W. Poly(lactide)-vitamin E derivative/montmorillonite nanoparticle formulations for the oral delivery of Docetaxel. Biomaterials. 2009;30:3297-306
60. Yu DH, Lu Q, Xie J, Fang C, Chen HZ. Peptide-conjugated biodegradable nanoparticles as a carrier to target paclitaxel to tumor neovasculature. Biomaterials. 2010;31:2278-92
61. Yue CX, Liu P, Zheng MB, Zhao PF, Wang YQ, Ma YF. et al. IR-780 dye loaded tumor targeting theranostic nanoparticles for NIR imaging and photothermal therapy. Biomaterials. 2013;34:6853-61
62. Zhang EL, Luo SL, Tan X, Shi CM. Mechanistic study of IR-780 dye as a potential tumor targeting and drug delivery agent. Biomaterials. 2014;35:771-8
63. Tao W, Zeng XW, Liu T, Wang ZY, Xiong QQ, Ouyang CP. et al. Docetaxel-loaded nanoparticles based on star-shaped mannitol-core PLGA-TPGS diblock copolymer for breast cancer therapy. Acta Biomater. 2013;9:8910-20
Author contact
![]() Corresponding author: Lin Mei: Tel/Fax: +86 75526036736, E-mail: mei.lintsinghua.edu.cn.
Corresponding author: Lin Mei: Tel/Fax: +86 75526036736, E-mail: mei.lintsinghua.edu.cn.