Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2016; 6(4):533-544. doi:10.7150/thno.14315 This issue Cite
Research Paper
Mifepristone Suppresses Basal Triple-Negative Breast Cancer Stem Cells by Down-regulating KLF5 Expression
Rong Liu1 ![]() , Peiguo Shi1, Zhi Nie2, Huichun Liang1, Zhongmei Zhou1, Wenlin Chen3, Haijun Chen4, Chao Dong5, Runxiang Yang5, Suling Liu6, Ceshi Chen1
, Peiguo Shi1, Zhi Nie2, Huichun Liang1, Zhongmei Zhou1, Wenlin Chen3, Haijun Chen4, Chao Dong5, Runxiang Yang5, Suling Liu6, Ceshi Chen1 ![]()
1. Kunming Institute of Zoology, Chinese Academy of Sciences, Key Laboratory of Animal Models and Human Disease Mechanisms of Chinese Academy of Sciences & Yunnan Province, Kunming, 650223, China;
2. Department of Neurology, First Affiliated Hospital of Kunming Medical University, Kunming, 650032, China;
3. Department of Breast surgery, the 3rd affiliated Hospital of Kunming Medical University, Kunming, Yunnan, 650118, China;
4. College of Chemistry and Chemical Engineering, Fuzhou University, Fuzhou, 350108, China;
5. Department of the second medical oncology, the 3rd affiliated Hospital of Kunming Medical University, Kunming, Yunnan, 650118, China;
6. Institute for Cancer Research, CAS Key Laboratory of Innate Immunity and Chronic Disease, School of Life Sciences and Medical Center, University of Science & Technology of China, Hefei, Anhui, 230027, China.
Received 2015-11-4; Accepted 2016-1-9; Published 2016-2-13
Abstract

Triple-negative breast cancer (TNBC) is currently the most malignant subtype of breast cancers without effective targeted therapies. Mifepristone (MIF), a drug regularly used for abortion, has been reported to have anti-tumor activity in multiple hormone-dependent cancers, including luminal type breast cancers. In this study, we showed that MIF suppressed tumor growth of the TNBC cell lines and patient-derived xenografts in NOD-SCID mice. Furthermore, MIF reduced the TNBC cancer stem cell (CSC) population through down-regulating KLF5 expression, a stem cell transcription factor over-expressed in basal type TNBC and promoting cell proliferation, survival and stemness. Interestingly, MIF suppresses the expression of KLF5 through inducing the expression of miR-153. Consistently, miR-153 decreases CSC and miR-153 inhibitor rescued MIF-induced down-regulation of the KLF5 protein level and CSC ratio. Taken together, our findings suggest that MIF inhibits basal TNBC via the miR-153/KLF5 axis and MIF may be used for the treatment of TNBC.
Keywords: Triple-negative Breast Cancer, Mifepristone, Cancer Stem Cell, KLF5, miR-153.
Introduction
Breast cancer is a highly heterogeneous disease clinically classified into several subtypes: the luminal subtype (estrogen receptor ERα+ and/or PR (progesterone receptor)+), the HER2 subtype (human epidermal growth factor 2+), and triple-negative subtype (ER-/PR-/HER2-) [1, 2]. TNBC is associated with higher rates of cell proliferation, metastasis and higher tumor grade. It also has a poorer prognosis than other breast cancer subtypes, as demonstrated by diminished progression-free survival and overall survival rates [3, 4]. Recent studies suggest that TNBC is also heterogeneous and is classified into different subtypes, including basal-like TNBC [5, 6]. Compared to the luminal and HER2 subtypes, TNBC does not have effective targeted therapies and adjuvant chemotherapy is the only choice. Even so, more than 70% of women with metastatic TNBC die within 5 years [3]. It is urgently required to develop effective treatments for this aggressive type of breast cancer.
Increasing evidence indicated that cancer stem cells (CSCs) play important roles in breast cancer metastasis, recurrence, and drug resistance. Traditional cancer therapies, although are effective initially and kill the majority of tumor cells, fail ultimately due to not targeting and eliminating the minority of CSCs, which lead to drug-resistance and relapse of cancers [7]. Gupta et al. reported that salinomycin, which specifically targets breast CSCs, effectively inhibits breast cancer growth [7]. Therefore, it's important to develop more therapies to target CSCs to achieve cures for breast cancer, especially for TNBC, which displays a less differentiated “stem/progenitor” cell phenotype [8].
Human Krüppel-like factor 5 (KLF5) has been implicated in promoting breast cell proliferation, survival and tumor growth [9, 10]. Additionally, KLF5 promotes the self-renewal of embryonic stem cell (ESC) and to maintain ESC in an undifferentiated state [11, 12]. Interestingly, the gene expression signature of basal-like breast cancer cells is similar to that of ESC and KLF5 is highly expressed in basal-type breast tumors [13]. Consistently, we found that KLF5 is specifically highly expressed in basal TNBC cell lines [14]. Positive KLF5 expression is an unfavorable prognostic marker correlated with shorter survival for breast cancer patients [15, 16]. We recently demonstrated that depletion of KLF5 significantly suppresses basal TNBC cell proliferation, survival and tumor growth in vivo [17, 18].
Mifepristone (MIF), a synthetic progesterone receptor (PR) antagonist, has been safely used as an abortifacient in the first month of pregnancy, and in smaller doses as an emergency contraceptive for decades [19]. In recent years, MIF has been found to have anti-tumor activity against different types of cancers, including endometrial cancer [20], ovarian cancer [21], prostate cancer [22], and luminal type breast cancers [23]. It appears that MIF suppresses cell growth at relatively high dosage (IC50: 9-30 μM) regardless of the PR status [24]. Although MIF has been reported to inhibit the CDK2 activity [24] and to regulate the NF-κB signaling pathway [25], the mechanisms by which MIF suppresses tumor cell growth have not been fully elucidated yet.
In this study, we firstly demonstrated that MIF has anti-tumor activity in a TNBC xenograft model and a patient-derived xenograft model in vivo. Following that, we found that MIF has anti-proliferation and pro-apoptosis activity. Interestingly, MIF suppresses CSC through inducing expression of miR-153 to suppress the protein expression of KLF5. Our findings may provide new therapeutic strategies for the treatment of basal-like TNBCs.
Materials and Methods
Cell culture and transfection
The immortalized breast epithelial cell line MCF10A was maintained in DMEM/Ham's F-12 50/50 medium supplemented with 5 % horse serum, 0.5 µg/ml hydrocortisone, 10 µg/ml insulin, 20 ng/ml epidermal growth factor, 0.1 µg/ml cholera enterotoxin, and 2 mM L-glutamine. Basal type TNBC cell lines HCC1937 were cultured in PRI-1640 containing 5% fetal bovine serum (FBS), 1.5 g/L sodium bicarbonate, and 1 mM sodium pyruvate. These cells were maintained in a humidified atmosphere with 5 % CO2 at 37 °C.
All microRNA mimics and inhibitors (Ribobio, Guangzhou, China) were transfected into different cell lines using Lipofectamine 2000 following the manufacturer's manual (Invitrogen, Carlsbad, CA, USA).
Drugs and antibodies
MIF was purchased from Sigma-Aldrich (St. Louis, MO). The anti-KLF5 antibody has been described in our previous study [26]. The anti-FGF-BP antibody was purchased from R&D Systems (Minneapolis, MN), anti-Ki-67 was from Thermo Fisher Scientific (Fremont, CA, USA). The anti-Mcl-1, anti-PARP, anti-cleaved-caspase3 antibodies and all secondary antibodies were from Cell Signaling (Danvers, MA, USA). The anti-β-actin antibody was purchased from Sigma-Aldrich (St. Louis, MO, USA).
Cell proliferation and apoptosis assays
Cell proliferation of HCC1937 and MCF10A cells was measured using Click-iT EdU Alexa Fluor 647 Imaging Kit (Invitrogen), following the manufacturer's instructions. The apoptotic biomarkers, cleaved PARP and cleaved caspase-3, were detected by Western blotting (WB).
Flow Cytometry analysis of CSCs
Cancer cells were trypsinized into single cells, washed once using Hanks Balanced Salt Solution (HBSS) with 2% FBS (HF solution), and stained freshly with antibodies against CD24 (BD biosciences, CA, USA, Cat#555428) and CD44 (BD biosciences, CA, USA, Cat#555478, #559942) or applied for Aldefluor assay (Stemcell technologies, Vancouver, BC, Canada) according to the manufacturer's manuals. Briefly, for CD24/CD44 staining, the cells were incubated with anti-CD24 and anti-CD44 antibodies on ice for 25 minutes; after centrifuging at 500g for 5 minutes, cells were collected, washed once using HF solution, and applied for flow cytometry analysis. For the Aldefluor assay, the cells were centrifuged and resuspended in the Aldefluor assay buffer containing a fluorescent substrate for aldehyde dehydrogenase (ALDH), BODIPY-aminoacetaldehyde, and incubated at 37 °C for 45 minutes. A specific inhibitor of ALDH, diethylaminobenzaldehyde, was used to control the background fluorescence.
Patient-derived xenograft (PDX) model
Xenotransplants from two independent human breast cancers (MC1, UM1) were maintained in fat-pads of NOD/SCID mice, without culture in vitro [27]. Both tumors were ER-PR-HER2- invasive ductal carcinomas.
Xenograft tissue was minced and dissociated in F12/DMEM supplemented with 5% FBS, 300 U/ml collagenase and 100 U/ml hyaluronidase (Sigma) at 37°C for 2h. After centrifuging at 400 g for 5 minutes, the pellet was resuspended in RBC lysis buffer (eBioscience, San Diego, CA) and incubated at 37°C for 5 min to remove red blood cells. Following centrifugation at 400 g, the pellet was reuspended in F12/DMEM supplemented with 10% FBS and was filtered through a 40-mm nylon mesh (BD Biosciences).
After counting, around 5x105 cells were plated in 12-well plates followed by treating with mifepristone at indicated concentrations for 24 h. The cells were collected and stained with anti-CD44 antibody labeled with FITC (BD Biosciences), anti-CD24 antidoby labeled with PE-Cy7 (BD Biosciences), and lineage markers anti-CD2, CD3, CD16, CD18, CD19, CD31, CD45, CD140b (all antibodies were labeled with PE, BD Biosciences). Fresh cells were stained with 1 μg/ml PI (Sigma) to exclude dead cells.
Mammosphere culture
Single cancer cells were plated in ultra-low attachment plated (Corning Incorporated, Tewksbury, MA, USA) and cultured with mammosphere culture medium (Stemcell technologies, Vancouver, BC, Canada) according to manufacturer's guidelines. The number of spheres was counted 8-14 days after plating.
Stable knockdown of KLF5
The KLF5 and control luciferase (Luc) shRNAs were expressed using the pSIH1-H1-puro lentiviral vector. Target sequences for KLF5 and Luc are listed in Table S1. HCC1937 cells were infected with lentiviruses and selected using 1 μg/ml puromycin to generate stable populations.
Stable over-expression of KLF5
The KLF5 cDNA was amplified and cloned into FUCGW-GFP lentiviral vector, which allowed separate expression of KLF5 and GFP proteins, using forward primer, 5'-ACTCTAGAGGATCTGAATTC ACC ATG GCT ACA AGG GTG CTG-3' and reverse primer 5'-CGGACGCACTCAATGAATTC G TCAGTTCTGGTGCCTCTTC-3'. Lentiviruses were prepared according to published protocols [28]. HCC1937 cells were infected with lentiviruses and GFP-positive cells were checked using flow cytometry assays 72 hours after infection.
Electroporation
Electroporation was performed using the Neon transfection system (Life Technologies). 1×106 HCC1937 cells were prepared follows the manufacturer's manuals and were mixed with 5-10 μg pBabe-KLF5 plasmids or pBabe vector control for electroporation. After electroporation, the cells were plated in 12-well plate at a density of 2×105/well immediately. Thirty-six hours later, the cells were treated with MIF for 24 hours for either WB or FACS analysis.
microRNA reverse-transcription and qPCR
HCC1937 and MCF10A cells were treated with MIF at indicated dosage for 24 hours. Total mRNAs were isolated using TRIzol® reagent (Invitrogen). Reverse transcription was performed using the QuantiMir RT Kit Small RNA Quantitation System (SBI, CA) and miRNA levels were quantified using RT Real-TimeTM SYBR Green/Rox PCR master mix (SAbiosciences, CA) on the ABI-7300 system. Primers for U6, miR-21, miR-152 and miR-153 are provided in Table S2.
Dual luciferase assays
The 1,652 bp KLF5 3'UTR was amplified using normal human cDNA as templates. The PCR products were cloned into the pMIR-REPORT™ miRNA Expression Reporter Vector (a gift from Dr. Jiliang Zhou at Georgia Regents University, Augusta, GA, USA). The inserts were confirmed by DNA sequencing. HEK293FT cells were seeded in 24-well plates at 1×105 cells per well. On next day, the cells were transfected with the KLF5 3'UTR luciferase reporter constructs (0.3 μg per well) and an internal control pRL-CMV (0.1 μg per well) in triplicate together with miR-153 mimics or negative controls. At 48h after transfection, luciferase activities were measured by using the dual luciferase reporter assay system (Promega).
Cell viability assay
The cell viability was measured by sulforhodamine B (SRB) assays [29]. Briefly, HCC1937 and MCF10A cells were plated in 24-well plate at a density of 5×104/well, the day after cell plating, the cells were transfected with either miR-153 mimics or negative controls. The cells were fixed using 10% trichloroacetic acid (TCA) two days after transfection, and stained with 0.4% SRB. After dissolving SRB from the cells using 10 mM unbuffered Tris-base, optical results were read by an automated spectrophotometric plate reader at a single wavelength of 530 nm.
Tumorigenesis in SCID mice
This study was approved by the institutional ethics committees of Kunming Institute of Zoology, Chinese Academy of Sciences.
Eighteen 6 week old female NOD SCID mice were purchased from Harlan Laboratories (Indianapolis, USA). HCC1937 cells of 5×106 were implanted into mammary fat pads of the mice. Tumor size was measured using Vernier calipers once tumors became palpable. Tumor volumes were calculated using the following equation: tumor volume (cm3) = л(length×width2)/6. When the tumor size reached 50mm3, mice were randomly and equally distributed into three groups, which were treated with the placebo, 30mg or 60mg MIF pellet (60 days release; Innovative Research of America, Sarasota, FI, USA). Tumor size was monitored twice/week for 7 weeks after drug pallet was implanted. All mice were sacrificed at the end of the experiment and tumors were harvested for analysis.
MC1 [27] and UM1 PDX tissues were collected and dissociated as described above. 1×106 cells were implanted into mammary fat pads of the mice. Twelve mice carried MC1 xenografts were randomly and equally distributed into two groups when the tumor size reached about 50mm3, and were given 1 mg MIF (dissolved in tea oil) or vehicle control per day by i.p. All mice were sacrificed at the end of the experiment and tumors were harvested for analysis.
MIF or tea oil control treated UM1 xenografts were collected and dissociated. UM1 cell suspension was injected to the fat pad of nude mice at designed numbers. 5 weeks later, when the tumors reached approximately 1.2 cm in the largest diameter, all mice were sacrificed and tumors were collected for analysis.
Statistical analysis
The luciferase reporter assay and the cell viability assay were conducted in triplicate. When appropriate, the data were pooled to generate means ± standard deviation and were analyzed by t-test. P-values of less than 0.05 were considered to be significant.
Results
MIF suppresses HCC1937 and MC1 PDX growth in mice
Whether MIF inhibits basal TNBC cell growth in vivo has not been tested before. We found that MIF significantly suppressed the basal TNBC cell line HCC1937 xenograft growth in immunodeficient NOD-SCID mice in a dosage-dependent manner (Fig. 1A). Seven weeks after MIF treatment, the average tumor weight from MIF treated groups, especially the high dose group (60 mg/60 d release), was significantly lighter than those from the placebo group (Fig. 1B). It is worth pointing out that the MIF doses used in this study did not decrease the body weight of the mice (Fig. 1C), indicating there are no significant toxicity. Immunohistochemistry (IHC) staining for Ki67 and cleaved-caspase-3 in these xenografts showed that MIF significantly decreased cell proliferation (Fig. S1A) and slightly increased apoptosis (Fig. S1B).
MIF suppresses triple-negative breast cell growth and induces apoptosis in vivo and in vitro. A. MIF suppressed HCC1937 tumor growth in NOD SCID mice in a dosage dependent manner. HCC1937 cells were injected into the fat pat of female NOD SCID mice. When the average tumor size reached about 50 mm3 after inoculation, the mice were randomly and equally distributed into three groups (n=6/group): placebo, 30 mg MIF pellet (60 days release), and 60 mg MIF pellet (60 days release). Tumor size were measured weekly for 7 weeks. B. MIF significantly decreased tumor weights compared to the placebo group (p<0.001, t-test) in a dosage dependent manner. Tumors were collected 7 weeks after MIF implantation. C. MIF did not affect the body weight of mice. The mice were weighted at the end of the experiment. D. MIF suppressed MC1 PDX growth in vivo. MC1 cells were injected into the fat pat of female NOD SCID mice. Tumor size was measured twice per week once tumors became palpable. Twelve mice carried MC1 xenografts were randomly distributed into two groups equally when the tumor size reached around 50 mm3 and were given 1 mg MIF (dissolved in tea oil) or vehicle control per day by i.p. for 25 days. E. MIF significantly decreased tumor weights compared to the vehicle control (p<0.05, t-test). Tumors were collected 25 days after MIF treatment. F. MIF did not affect the body weight of mice. The mice were weighted at the end of the experiment. G. MIF significantly inhibited DNA synthesis in both HCC1937 and MCF10A by using the Click-iTTM EdU Alexa Fluor® 647 Imaging Kit. The quantitative results are shown in the right graphs. *P<0.05; **P<0.01, t-test. H. MIF down-regulated the KLF5 protein levels and induced the cleavage of PARP and caspase 3 in a dosage-dependent manner in both HCC1937 and MCF10A cells. Protein levels were detected by WB. β-actin was used as the loading control.
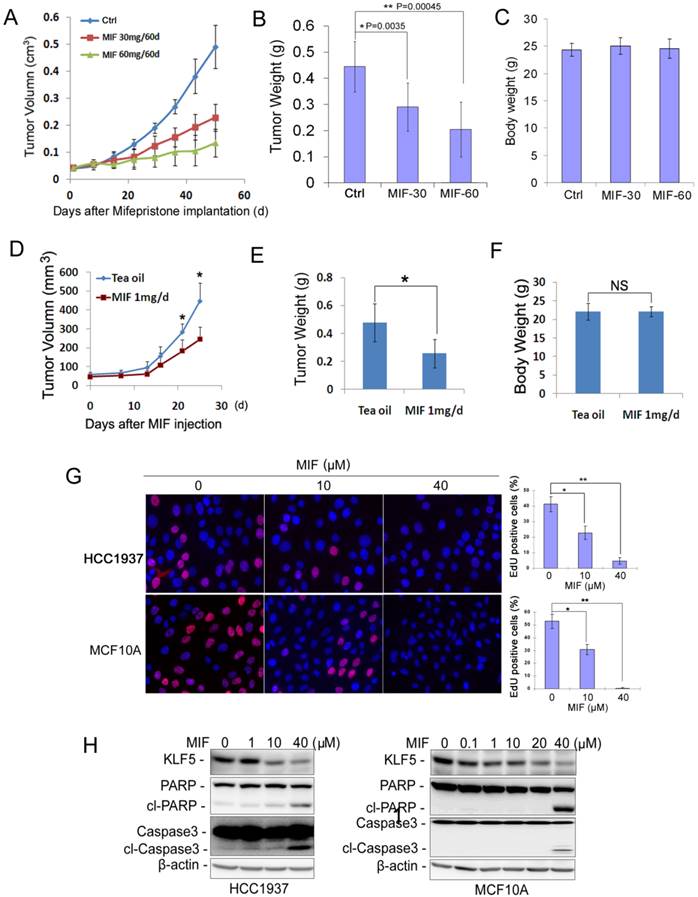
To further confirm this result in clinical relevant models, we employed a triple-negative PDX model to test whether MIF could also inhibit tumor growth. As shown in Fig. 1D-F, MIF (1 mg/day) significantly inhibited MC1 xenografts growth in vivo without reducing the body weight of the mice compared to the vehicle control.
MIF suppresses basal breast epithelial cell proliferation and induces apoptosis in vitro
We further investigated whether MIF suppresses cell proliferation and/or induces apoptosis in vitro. We examined DNA synthesis in HCC1937 and MCF10A, an immortalized basal triple negative breast epithelial cell line. As shown in Fig. 1G, MIF treatment dramatically reduced the EdU-positive cells in both cell lines in a dosage-dependent manner. We also examined apoptosis by detecting the cleavage of PARP and Caspase-3 and found that MIF significantly induced the cleavage of these two proteins in both cell lines at a high concentration (20-40 μM) (Fig. 1H).
MIF suppresses the CSC population in HCC1937 and PDX
To test whether MIF decreases breast cancer stem cells, which are CD24low/CD44+ or ALDH+ cells [27, 30], we analyzed the HCC1937 CSC populations after MIF treatment. As shown in Figure 2, MIF significantly reduced the proportion of CSC at a dose-dependent manner by either CD24/CD44 markers (Fig. 2A) or Aldefluor assays (Fig. S3A). These results were further confirmed by the mammosphere formation assay (Fig. 2B). Both the sphere size and number were significantly decreased by the MIF treatment. We also confirmed this result in another triple-negative cancer cell line SUM149PT (Fig. S2A-B). To determine if MIF inhibits CSC self-renewal, we treated HCC1937 cells with MIF or vehicle for 24 hours and performed two rounds of mammosphere culture in the absence of MIF. As shown in Fig. 2B, MIF suppressed the CSC self-renewal.
MIF decreases CSC of HCC1937 and PDX cells. A. The percentage of CD24low/CD44+ cells was assessed using the flow cytometry analysis. HCC1937 cells were treated with MIF for 24 h at indicated concentration. **, P<0.01, t-test. B. MIF suppresses sphere formation and CSC self-renewal in HCC1937 cells. HCC1937 cells were treated with 10 or 20μM MIF or vehicle control for 24 h. The cells were stained with typan blue and counted to eliminate dead cells before seeding in ultra-low attachment dishes for sphere culture. 14 days later, mammospheres were collected and dissociated for passaging and the 2nd round of sphere culture. Data are shown as averages ± SD. **, P<0.01, t-test. C&D. The percentage of lin-CD24low/CD44+ cells was assessed using flow cytometry. MC1 and UM1 PDX derived cells were treated with MIF for 24 h at indicated dosage or applied for sphere culture together with MIF or vehicle control. *, P<0.05; **, P<0.01, t-test. E. MIF suppressed UM1 PDX growth in vivo. UM1 cells were injected into the fat pat of female NOD-SCID mice. Mice carried UM1 xenografts were randomly distributed into two groups equally when the tumor size reached around 50 mm3 and were given 1 mg MIF (dissolved in tea oil) or vehicle control per day by i.p. *, P<0.05, t-test. F. UM1 xenografts collected from F were dissociated, and the percentage of lin-CD24low/CD44+ cells was analyzed. G. Dissociated UM1 cells from MIF or control group were submitted to sphere culture in ultra-low attachment dishes for 2 weeks. *, P<0.05, t-test. H. Dissociated UM1 xenograft cells were injected to the fat pad of nude mice at indicated numbers, 5 weeks later, xenografts were collected for incidence analysis.
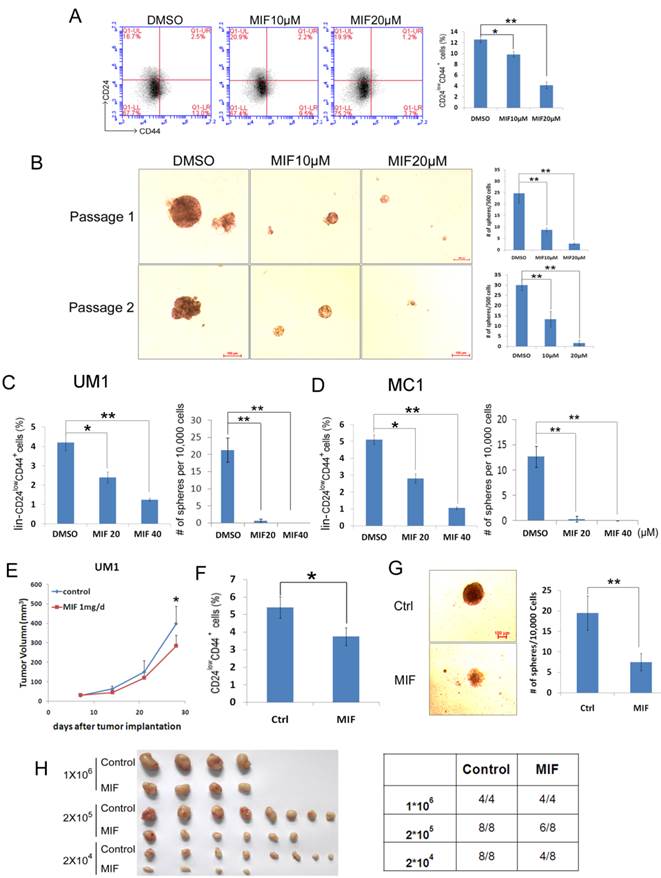
Furthermore, we collected cells from triple-negative PDX models (MC1 and UM1) [27], and treated them with MIF at indicated concentration for 24 hours in vitro. As shown in Fig. 2C-D and S3B&C, MIF also suppressed the lin-CD24lowCD44+ and the lin-ALDH+ CSC populations in a dosage-dependent manner in both MC1 and UM1 PDX models.
To further confirm that MIF targets the CSC population in vivo, we collected UM1 xenografts from MIF or vehicle control treated mice (Fig. 2E), and dissociated xenografts into single cells for CD24/CD44 analysis (Fig. 2F), mammosphere culture (Fig. 2G) and limited dilution assays (Fig. 2H). Indeed, MIF treatment significantly suppressed the CSC population in vivo.
MIF suppresses CSCs partially through KLF5
Previously, we have demonstrated that the stem cell transcription factor KLF5 is specifically expressed in basal TNBC cell lines, but not in other subtypes of breast cancer and MIF blocked the progesterone-induced KLF5 transcription in the PR+ T47D breast cancer cell line [14]. Interestingly, MIF also significantly decreased the protein levels of KLF5 in triple negative HCC1937, SUM149PT, PDX cells (MC1 and UM1) and MCF10A in time and dosage-dependent manners (Fig. 3A, S2C and S3D-E). Furthermore, MIF suppressed KLF5 expression in HCC1937 xenograft tumors in vivo (Fig. S3H).
MIF suppresses KLF5 expression in time and dosage-dependent manners in basal breast epithelial cells and KLF5 depletion decreases CSC. A. The KLF5 protein level was suppressed by MIF in time- and dosage-dependent manners in MCF10A and HCC1937. The cells were treated with MIF for 24 h at indicated concentration or at 40 μM for indicated time. β-actin was used as the loading control. B. KLF5 is highly expressed in CSC population. The CSC-enriched CD24low/CD44+ population and the non-CSC CD24+/CD44- population were sorted for WB analysis. GAPDH was used as loading control. C. KLF5 protein level was significantly silenced by three different shRNAs in HCC1937. D. Stable knockdown of KLF5 by different shRNAs decreased the percentage of CD24low/CD44+ CSC in HCC1937. *, P<0.05, t-test. E. Stable depletion of KLF5 in HCC1937 by different shRNAs significantly suppressed mammosphere formation compared to the Lucsh control. The quantitative results are shown in the right graph. *, P<0.05, t-test.
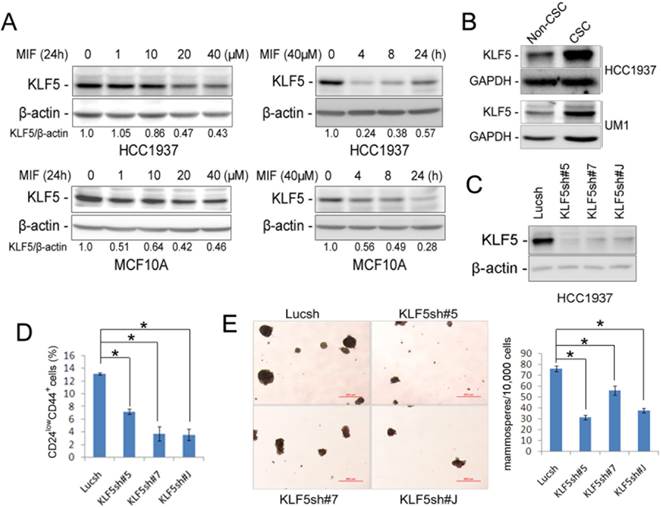
Since MIF suppressed the KLF5 expression and the CSC population in TNBC, we wondered whether KLF5 promotes CSC. We sorted the CSC-enriched CD24low/CD44+ population and the non-CSC CD24+/CD44- population and examined the KLF5 protein levels. As shown in figure 3B, KLF5 protein levels are higher in CSC than non-CSC populations in both HCC1937 and the UM1 PDX model. Since KLF5 is predominately expressed in CSC, we wondered whether depletion of KLF5 will decrease CSC. When KLF5 was knocked down in HCC1937 using three different shRNAs (Fig. 3C), the CD24low/CD44+ CSC population and the mammosphere formation ability were significantly reduced (Fig. 3D-E).
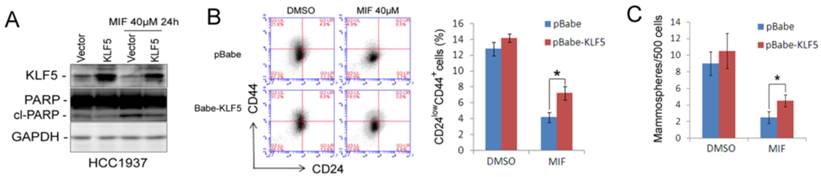
Subsequently, we over-expressed KLF5 by electroporating HCC1937 to investigate whether KLF5 could rescue the MIF-induced CSC loss. As shown in Fig. 4, KLF5 over-expression partially but significantly rescued MIF-induced apoptosis (Fig. 4A) and CSC reduction (Fig. 4B-C). These findings indicated that MIF induces TNBC apoptosis and inhibits CSC at least partially through suppressing the KLF5 expression.
Ectopic over-expression of KLF5 partially rescues MIF-induced apoptosis and CSC reduction in HCC1937. A. KLF5 over-expression decreases MIF-induced PARP cleavage in HCC1937. HCC1937 cell were transiently electroporated with pBabe-KLF5 or pBabe vector control, followed by treating with 40 μM MIF for 24 h. The apoptosis marker cl-PARP was detected by WB. B. Over-expression of KLF5 significantly rescued the MIF-induced CD24low/CD44+ CSC reduction in HCC1937. The quantitative results are shown in the right graph. *, P<0.05, t-test. C. Ectopic expression of KLF5 in HCC1937 partially rescued the MIF-induced mammosphere formation reduction. Mammospheres were counted 12 days after seeding. *, P<0.05, t-test.
MIF suppresses KLF5 by inducing the expression of miR-153
MIF significantly suppresses the KLF5 protein expression in basal type breast epithelial cells. To explore the mechanisms, we first tested KLF5 mRNA levels. To our surprise, the mRNA level of KLF5 was not decreased by MIF (Fig. S4A). We then switched to miRNAs, which can suppress the protein translation without reducing the mRNA level. The KLF5 3'-UTR contains binding sites for several miRNAs, including miR-21, -143, -145, -152, and -153, according to the TargetScan prediction software (http://www.targetscan.org/). Among these miRNAs, the expression levels of miR-21, -152 and -153 were significantly induced by MIF in both HCC1937 and MCF10A cell lines (Fig. 5A and Fig. S4B). We then transfected HCC1937 and MCF10A cells with mimics of these miRNAs and found that only miR-153 mimics suppressed the KLF5 expression (Fig. 5B). Furthermore, MIF also induced miR-153 expression and miR-153 mimics suppressed KLF5 expression in UM1 PDX derived cells (Fig. S3F-G). To further test whether miR-153 inhibits the KLF5 expression through putative binding site at KLF5 3'UTR, we performed the dual luciferase reporter assay. As expected, miR-153 significantly suppressed the luciferase activity when the reporter gene linked with KLF5 3'-UTR, but not the miR-153 binding site mutated one (Fig. 5C).
MIF suppresses the expression of KLF5 through inducing the miR-153 expression. A. MIF increased the expression of miR-21, miR-152 and miR-153 in both HCC1937 and MCF10A cells. U6 was used as the internal control. B. miR-153 mimics (50 nM) decreased the KLF5 protein level in HCC1937 and MCF10A cells. The cells were transfected with miRNA mimics for two days. KLF5 protein levels were detected by WB. C. miR-153 suppressed the KLF5 3'-UTR luciferase reporter activity. HEK293FT cells were transfected with miR-153 mimics (50 nM) and pMIR-KLF5 3'-UTR or pMIR-KLF5 3'-UTRm reporter together with pCMV-renilla control. Two days after transfection, the cells were collected for the dual-luciferase reporter assay. **, P<0.001, t-test. D. miR-153 inhibitor rescued the MIF-induced KLF5 protein decrease in MCF10A cells. The cells were transfected with either miR-153 inhibitor or negative control miRNA inhibitor. One day after transfection, the cells were treated with 20 μM MIF for 24 h. E. The miR-153 inhibitor rescued the MIF-induced KLF5 protein decrease in HCC1937 cells.
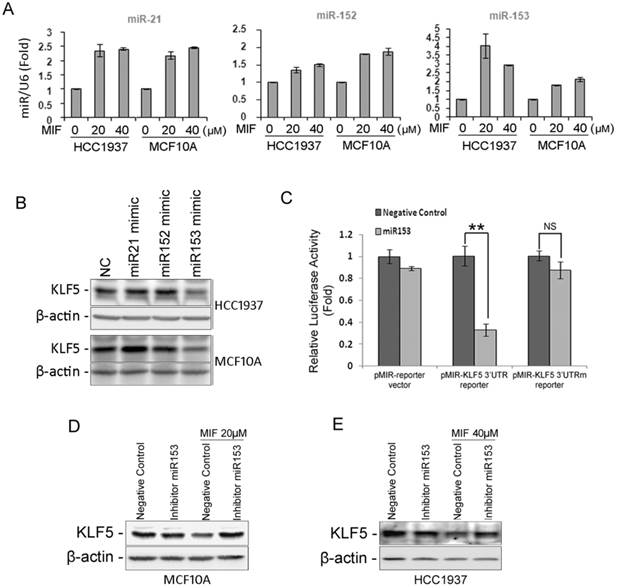
Since MIF induces the miR-153 expression and miR153 inhibits the KLF5 expression, we further validated whether MIF inhibits the KLF5 expression predominately through inducing the miR-153. MCF10A and HCC1937 cells were transfected with miR-153 inhibitors followed by treating with MIF. Indeed, miR-153 inhibitors almost completely rescued MIF-induced KLF5 decrease (Fig. 5D-E).
miR-153 suppresses cell survival, proliferation and CSC
miR-153 has been reported to inhibit cell proliferation and to promote apoptosis via suppressing the expression of Mcl-1 and other downstream target genes in glioblastoma [31] and leukemia [32]. However, the role of miR-153 in breast cancer has not been fully assessed. As expected, miR-153 suppressed the expression of both KLF5 and Mcl-1 and induced cleavage of PARP in both HCC1937 and MCF10A cells (Fig. 6A). Additionally, miR-153 significantly suppressed cell proliferation, as detected by the EdU incorporation assay (Fig. 6B), and dramatically reduced cell viability, as detected by the SRB assay (Fig. 6C), in both HCC1937 and MCF10A. Importantly, miR-153 significantly decreased the CSC population of HCC1937 by both the CD24/CD44 analysis (Fig. 6D), the Aldefluor assay (Fig. 6E), and the mammosphere formation assay (Fig. S5).
miR-153 suppresses basal breast epithelial cell proliferation, survival and CSC. A. miR-153 suppressed the expression of KLF5 and Mcl-1 and induced the cleavage of PARP in HCC1937 and MCF10A cells. The cells were transfected with 50 nM negative control (NC) or miR-153 mimics for two days. B. miR-153 inhibits DNA synthesis using the EdU assay in HCC1937 and MCF10A cells. The quantitative results are shown in the right graphs. C. The cell viability was significantly reduced by miR-153 in HCC1937 and MCF10A cells. The cells were plated in 24-well plate at a density of 5×104 in triplicates. One day after plating, the cells were transfected with 50 nM NC or miR-153 mimics. Two days after microRNA transfection, the cells were fixed for SRB assays. **, P<0.01, t-test. D&E. miR-153 suppressed the percentage of CD24low/CD44+ (D) and ALDH+ (E) cells in HCC1937 in a dosage dependent manner. The cells were transfected with 50 nM NC or miR-153 mimics for 2 days. *, P<0.05; **, P<0.001, t-test. F. Ectopic over-expression of KLF5 decreases MIF-induced PARP cleavage in HCC1937. HCC1937 cell were infected with FUCGW-KLF5 or FUCGW vector control, followed by treating with 20 μM MIF for 24 h. The apoptosis marker cl-PARP was detected by WB. β-actin was used as loading control. G. FUCGW-KLF5 or vector control lentivirus-infected HCC1937 cells were plated in 48-well plate at 3×104/well and transfected with miR-153 mimic or negative control for 2 days. Cell viability was analyzed using SRB assays. H. FUCGW-KLF5 or vector control lentivirus-infected HCC1937 cells were transfected with miR-153 mimic or negative control for 2 days before being subjected for analyzing the percentage of CD24low/CD44+ populations. I-K. miR-153 inhibitor or negative control transfected cells were treated with MIF or vehicle control for 24 h. The cells were collected for WB (I), CD24low/CD44+ populations analysis (J) or mammosphere culture for 2 weeks (K). *, P<0.05, t-test.
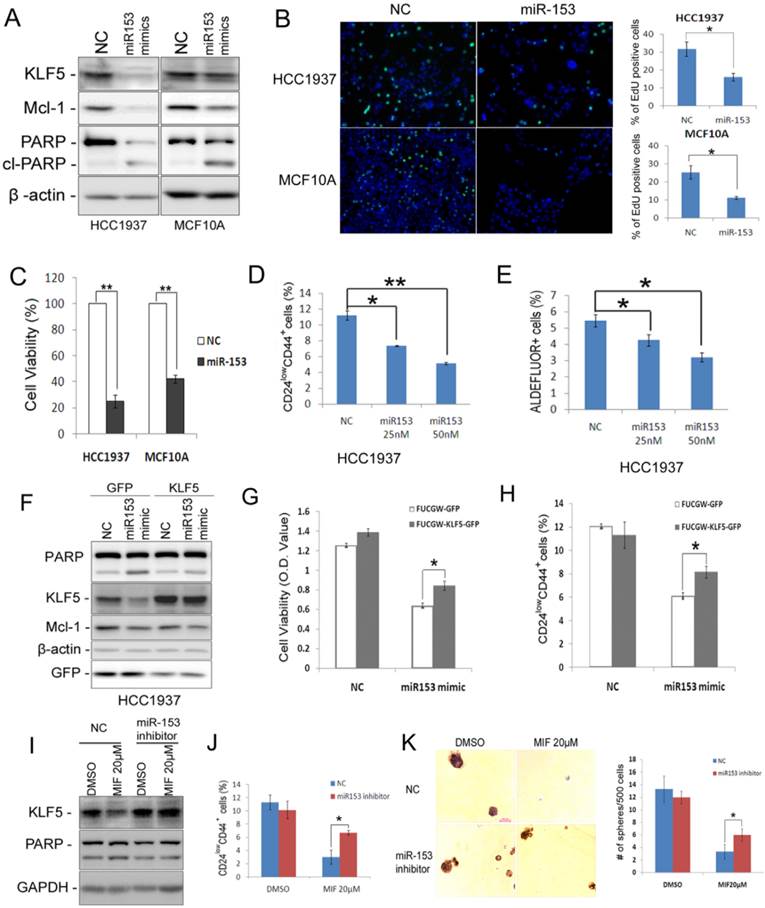
To investigate whether miR-153 function through KLF5, we transfected miR-153 mimics into KLF5 overexpression cells. Indeed, ectopic overexpression of KLF5 significantly rescued miR-153-induced apoptosis and CD24low/CD44+ population (Fig. 6F-H). Furthermore, miR-153 inhibitor significantly rescued MIF-induced apoptosis and the reduction of CD24low/CD44+ population and mammosphere formation (Fig. 6I-K).
Discussion
Among all subtypes of breast cancers, TNBC has the poorest prognosis but does not have effective targeted therapies. In our study, for the first time, we demonstrated that MIF, a synthetic PR antagonist that has been safely used as an abortifacient and an emergency contraceptive for decades [19], has an anti-tumor activity in basal TNBC cells through inhibiting KLF5 expression. First, MIF significantly suppresses the HCC1937 xenograft and MC1 PDX growth in NOD-SCID mice (Fig. 1). Secondly, MIF inhibits cell proliferation, promotes apoptosis, and suppresses CSC in vitro and in vivo (Fig. 1&2). Moreover, MIF functions partially through suppressing the expression of KLF5, a key transcription factor for maintaining basal TNBC CSC (Fig. 3&4). MIF suppresses the expression of KLF5 through inducing miR-153 and miR-153 inhibited cell proliferation, survival and CSC in TNBC cells (Fig. 5-6). Thus, our findings provide preclinical evidence that the MIF-miR-153-KLF5 axis may be used for basal TNBC treatment.
Our previous studies suggested that KLF5 is highly expressed in basal TNBC cell lines and promotes breast cancer cell proliferation, survival and tumorigenesis [9, 10, 14, 17]. High levels of KLF5 mRNA [15] or protein [33] are correlated with worse clinical outcome of breast cancer patients. Importantly, depletion of KLF5 significantly inhibits HCC1937 xenograft growth in vivo [17]. Yagi et al delivered KLF5 siRNA into prostate cancer-bearing mice and significant suppressed tumor growth [34]. Bialkowska et al. identified two small molecules suppressing the KLF5 expression and significantly inhibited colorectal cancer cell proliferation [35]. Interestingly, depletion of KLF5 causes mouse embryonic stem cell differentiation and suppresses intestine stem cell proliferation and survival [36, 37]. Consistently, depletion of KLF5 significantly decreases CSC in HCC1937 (Fig. 3). Although the mechanism by which KLF5 promotes CSC in basal TNBC is unclear at present, KLF5 is a potential therapeutic target for basal TNBC and other cancers.
MIF suppresses KLF5 expression, cell proliferation, survival and CSC in basal TNBC cells, at least partially, through inducing the expression of miR-153 because miR-153 is sufficient and necessary for MIF's functions. For the first time, we demonstrated that miR-153 suppresses CSC in basal TNBC and targeting KLF5 and Mcl1. Consistently, miR-153 suppresses glioblastoma stemness [38], sensitizes leukemia cell to As2O3-induced apoptosis [32], and inhibits the epithelial-mesenchymal transition (EMT) and metastasis [39]. Besides KLF5, miR-153 has previously been shown to target several important oncogenes, such as Mcl1, Bcl2, IRS-2, SNAI, and ZEB2 [39, 40]. These findings suggest that miR-153 is a tumor suppressor and might hold promise for cancer therapy.
Although miR-153 induction is essential for MIF to suppress the KLF5 expression and CSC in basal TNBC cells, how miR-153 is regulated by MIF is unknown at present. The expression of miR-153 is known to be co-regulated with islet-associated protein (IA) 2 and IA-2β by glucose in pancreas and brain [41]. It has been reported that miR-153 gene promoters methylated and DNA demethylating agent 5-aza-2'-deoxycytidine increases the miR153 expression [40, 42]. In addition, nicotine stimulates miR-153 expression through nicotinic acetylcholine receptor [43].
Whether these mechanisms are involved in the induction of miR-153 by MIF requires investigation in the future. Since HCC1937 is a PR-negative cell line and MIF can antagonize glucocorticoid receptor (GR) [44] and androgen receptor (AR) [45], we tested whether MIF functions through GR or AR to suppress KLF5 expression. As a result, silencing neither GR nor AR affects the KLF5 expression down-regulation by MIF (Fig. S6 and data not shown). These findings suggest that MIF may regulate miR-153 and KLF5 through a non-canonical pathway. Previous studies reported that MIF activated NF-κB signaling at concentrations from 20-100 μM [25]. And according to unpublished data, it is also possible that MIF activates ER stress pathway [46]. Whether MIF induces the expression of miR-153 through NF-κB or ER stress needs to be investigated in the future.
As shown in Figures 4& 6, over-expression of either KLF5 or miR-153 inhibitor could only partially rescue the inhibitory effects of MIF on breast CSCs. Compared to KLF5 over-expression (Fig 4B), the miR-153 inhibitor (Fig 6J) rescued the CSC loss better (35% vs 25%). It's possible that other targets of miR-153 are involved. In addition, the miR-153 inhibitor still could not completely rescue the MIF-induced CSC loss. It means that MIF also reduces CSC in a miR-153 independent mechanism, and other molecular pathways might also be involved. We checked the expression of several other CSC-related molecules, including ERK [47], AKT [48], Slug, Sox9 [49], and TAZ [50], in TNBC cell lines HCC1937 and SUM149PT. As shown in Figure S7, MIF suppresses the expression levels of pERK, Slug, Sox9 and TAZ in a dosage-dependent manner. These findings suggest that these molecular pathways might also be involved in mediating the CSC-suppressing effects of MIF in breast CSCs.
As a well tolerated abortifacient drug, MIF has been reported to prevent BRCA1-mediated mammary tumorigenesis via suppressing PR accumulation in mice [51]. MIF in combination with paclitaxel significantly increased cytotoxicity in GR positive breast cancer cells than paclitaxal alone via inhibiting the GR signaling [52]. Besides the canonical hormone-dependent effects, MIF also inhibits cell growth through targeting NF-κB [25] and CDK2 [24] regardless of the PR status. MIF should be further tested for basal TNBC patients in clinic. MIF significantly suppressed HCC1937 xenograft, MC1 and UM1 PDX growth (Fig. 1A-F & Fig. 2E). The doses we used did not cause any body weight loss (Fig. 1C&F). The peak MIF plasma concentration is about 10 μM 1-2 hours and remains in the micromolar range for the next 24-48 hours after a single dose of 200 mg MIF administration in health female volunteers [53]. This concentration is sufficient to significantly suppress proliferation, survival, KLF5 expression and CSC in HCC1937, UM1 and clinical TNBC cells (Fig. 1-2 and Fig. S8). Nevertheless, the human primary mammary epithelial cell line 48RS [54] is much less sensitive to MIF (Fig. S8A). A Phase II study of MIF (200 mg daily) in 28 postmenopausal patients with PR-positive breast cancer suggested that only 10.7% of patients responded to MIF alone [55]. The toxic effects were generally mild to moderate [55]. It is warranted to perform the study in basal TNBC patients with MIF (200 mg daily) alone or in combination with traditional chemotherapeutic drugs in the future.
In conclusion, MIF inhibits basal TNBC cell growth in vitro and in NOD SCID mice. MIF induces the expression of miR-153 to suppress the expression of KLF5, which promotes basal TNBC cell proliferation, survival and CSC maintenance. These findings suggest that MIF and miR-153 could be used for basal TNBC treatment.
Supplementary Material
Supplementary tables and figures.
Acknowledgements
This study was supported by Strategic Priority Research Program of the Chinese Academy of Sciences, Stem Cell and Regenerative Medicine Research (XDA01040406), National Nature Science Foundation of China (81322038, 81272930, 81325016, 81120108019, U1132605 and U1502222), Yunnan Applied Basic Research Key Projects (2015FA027), West Light Foundation of Chinese Academy of Sciences (to R.L.), Youth Innovation Promotion Association, Chinese Academy of Sciences (to R.L.), and Changsha Science and Technology Program Key Projects (K1406209-31).
Author contributions
RL designed and performed experiments and wrote the draft manuscript. PS and HL helped perform the animal experiments, and they received help from SL for PDX mouse model establishment, and HC for drug delivery. ZZ assisted in the experiments. ZN, WC, CD and RY collected clinical samples. All PDX models are provided by SL. CC designed experiments and revised the paper.
Competing Interests
Authors have no conflicts of interest.
References
1. Desmedt C, Ruiz-Garcia E, Andre F. Gene expression predictors in breast cancer: current status, limitations and perspectives. Eur J Cancer. 2008;44:2714-20
2. Goldhirsch A, Wood WC, Coates AS. et al. Strategies for subtypes-dealing with the diversity of breast cancer: highlights of the St. Gallen International Expert Consensus on the Primary Therapy of Early Breast Cancer 2011. Annals of oncology: official journal of the European Society for Medical Oncology / ESMO. 2011;22:1736-47
3. Dent R, Trudeau M, Pritchard KI. et al. Triple-negative breast cancer: clinical features and patterns of recurrence. Clinical cancer research: an official journal of the American Association for Cancer Research. 2007;13:4429-34
4. Rakha EA, El-Sayed ME, Green AR. et al. Prognostic markers in triple-negative breast cancer. Cancer. 2007;109:25-32
5. Lehmann BD, Bauer JA, Chen X. et al. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. The Journal of clinical investigation. 2011;121:2750-67
6. Metzger-Filho O, Tutt A, de Azambuja E. et al. Dissecting the heterogeneity of triple-negative breast cancer. J Clin Oncol. 2012;30:1879-87
7. Gupta PB, Onder TT, Jiang G. et al. Identification of selective inhibitors of cancer stem cells by high-throughput screening. Cell. 2009;138:645-59
8. Valent P, Bonnet D, De Maria R. et al. Cancer stem cell definitions and terminology: the devil is in the details. Nature reviews Cancer. 2012;12:767-75
9. Liu R, Zheng HQ, Zhou Z. et al. KLF5 promotes breast cell survival partially through fibroblast growth factor-binding protein 1-pERK-mediated dual specificity MKP-1 protein phosphorylation and stabilization. J Biol Chem. 2009;284:16791-8
10. Zheng HQ, Zhou Z, Huang J. et al. Kruppel-like factor 5 promotes breast cell proliferation partially through upregulating the transcription of fibroblast growth factor binding protein 1. Oncogene. 2009;28:3702-13
11. Jiang J, Chan YS, Loh YH. et al. A core Klf circuitry regulates self-renewal of embryonic stem cells. Nature cell biology. 2008;10:353-60
12. Parisi S, Passaro F, Aloia L. et al. Klf5 is involved in self-renewal of mouse embryonic stem cells. J Cell Sci. 2008;121:2629-34
13. Ben-Porath I, Thomson MW, Carey VJ. et al. An embryonic stem cell-like gene expression signature in poorly differentiated aggressive human tumors. Nature genetics. 2008;40:499-507
14. Liu R, Zhou Z, Zhao D. et al. The induction of KLF5 transcription factor by progesterone contributes to progesterone-induced breast cancer cell proliferation and dedifferentiation. Mol Endocrinol. 2011;25:1137-44
15. Tong D, Czerwenka K, Heinze G. et al. Expression of KLF5 is a prognostic factor for disease-free survival and overall survival in patients with breast cancer. Clinical cancer research: an official journal of the American Association for Cancer Research. 2006;12:2442-8
16. Takagi K, Miki Y, Onodera Y. et al. Kruppel-like factor 5 in human breast carcinoma: a potent prognostic factor induced by androgens. Endocrine-related cancer. 2012;19:741-50
17. Zhao D, Zhi X, Zhou Z. et al. TAZ antagonizes the WWP1-mediated KLF5 degradation and promotes breast cell proliferation and tumorigenesis. Carcinogenesis. 2012;33:59-67
18. Zhi X, Zhao D, Zhou Z. et al. YAP promotes breast cell proliferation and survival partially through stabilizing the KLF5 transcription factor. The American journal of pathology. 2012
19. Spitz IM. Mifepristone: where do we come from and where are we going? Clinical development over a quarter of a century. Contraception. 2010;82:442-52
20. Schneider CC, Gibb RK, Taylor DD. et al. Inhibition of endometrial cancer cell lines by mifepristone (RU 486). J Soc Gynecol Investig. 1998;5:334-8
21. Goyeneche AA, Caron RW, Telleria CM. Mifepristone inhibits ovarian cancer cell growth in vitro and in vivo. Clinical cancer research: an official journal of the American Association for Cancer Research. 2007;13:3370-9
22. Song LN, Coghlan M, Gelmann EP. Antiandrogen effects of mifepristone on coactivator and corepressor interactions with the androgen receptor. Mol Endocrinol. 2004;18:70-85
23. Fjelldal R, Moe BT, Orbo A. et al. MCF-7 cell apoptosis and cell cycle arrest: non-genomic effects of progesterone and mifepristone (RU-486). Anticancer research. 2010;30:4835-40
24. Tieszen CR, Goyeneche AA, Brandhagen BN. et al. Antiprogestin mifepristone inhibits the growth of cancer cells of reproductive and non-reproductive origin regardless of progesterone receptor expression. BMC cancer. 2011;11:207
25. Han S, Sidell N. RU486-induced growth inhibition of human endometrial cells involves the nuclear factor-kappa B signaling pathway. The Journal of clinical endocrinology and metabolism. 2003;88:713-9
26. Rossouw JE, Anderson GL, Prentice RL. et al. Risks and benefits of estrogen plus progestin in healthy postmenopausal women: principal results From the Women's Health Initiative randomized controlled trial. Jama. 2002;288:321-33
27. Ginestier C, Hur MH, Charafe-Jauffret E. et al. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell. 2007;1:555-67
28. Bu W, Xin L, Toneff M. et al. Lentivirus vectors for stably introducing genes into mammary epithelial cells in vivo. Journal of mammary gland biology and neoplasia. 2009;14:401-4
29. Chen C, Zhou Z, Ross JS. et al. The amplified WWP1 gene is a potential molecular target in breast cancer. Int J Cancer. 2007;121:2834-41
30. Al-Hajj M, Wicha MS, Benito-Hernandez A. et al. Prospective identification of tumorigenic breast cancer cells. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:3983-8
31. Xu J, Liao X, Wong C. Downregulations of B-cell lymphoma 2 and myeloid cell leukemia sequence 1 by microRNA 153 induce apoptosis in a glioblastoma cell line DBTRG-05MG. International journal of cancer Journal international du cancer. 2010;126:1029-35
32. Liu L, Chen R, Huang S. et al. miR-153 sensitized the K562 cells to As2O3-induced apoptosis. Med Oncol. 2012;29:243-7
33. Takagi K, Miki Y, Onodera Y. et al. Kruppel-like factor 5 in human breast carcinoma: a potent prognostic factor induced by androgens. Endocrine-related cancer. 2012
34. Yagi N, Manabe I, Tottori T. et al. A nanoparticle system specifically designed to deliver short interfering RNA inhibits tumor growth in vivo. Cancer research. 2009;69:6531-8
35. Bialkowska AB, Crisp M, Bannister T. et al. Identification of small-molecule inhibitors of the colorectal cancer oncogene Kruppel-like factor 5 expression by ultrahigh-throughput screening. Molecular cancer therapeutics. 2011;10:2043-51
36. Nandan MO, Yang VW. The role of Kruppel-like factors in the reprogramming of somatic cells to induced pluripotent stem cells. Histol Histopathol. 2009;24:1343-55
37. Nakaya T, Ogawa S, Manabe I. et al. KLF5 regulates the integrity and oncogenicity of intestinal stem cells. Cancer research. 2014;74:2882-91
38. Zhao S, Deng Y, Liu Y. et al. MicroRNA-153 is tumor suppressive in glioblastoma stem cells. Molecular biology reports. 2013;40:2789-98
39. Xu Q, Sun Q, Zhang J. et al. Downregulation of miR-153 contributes to epithelial-mesenchymal transition and tumor metastasis in human epithelial cancer. Carcinogenesis. 2013;34:539-49
40. Xu J, Liao X, Lu N. et al. Chromatin-modifying drugs induce miRNA-153 expression to suppress Irs-2 in glioblastoma cell lines. Int J Cancer. 2011;129:2527-31
41. Mandemakers W, Abuhatzira L, Xu H. et al. Co-regulation of intragenic microRNA miR-153 and its host gene Ia-2 beta: identification of miR-153 target genes with functions related to IA-2beta in pancreas and brain. Diabetologia. 2013;56:1547-56
42. Bao B, Rodriguez-Melendez R, Zempleni J. Cytosine methylation in miR-153 gene promoters increases the expression of holocarboxylase synthetase, thereby increasing the abundance of histone H4 biotinylation marks in HEK-293 human kidney cells. J Nutr Biochem. 2012;23:635-9
43. Balaraman S, Winzer-Serhan UH, Miranda RC. Opposing Actions of Ethanol and Nicotine on MicroRNAs are Mediated by Nicotinic Acetylcholine Receptors in Fetal Cerebral Cortical-Derived Neural Progenitor Cells. Alcohol Clin Exp Res. 2012
44. Agarwai MK. The antiglucocorticoid action of mifepristone. Pharmacology & therapeutics. 1996;70:183-213
45. Lin MF, Kawachi MH, Stallcup MR. et al. Growth inhibition of androgen-insensitive human prostate carcinoma cells by a 19-norsteroid derivative agent, mifepristone. The Prostate. 1995;26:194-204
46. Maria B. Hapon CDG-L, Alicia A. Goyeneche, Eduardo A. Callegari, Kathleen M. Eyster, and Carlos M. Telleria. Induction of the unfolded protein response in ovarian cancer cells exposed to cytostatic concentrations of antiprogestin/antiglucocorticoid mifepristone. [abstract]. Clinical cancer research: an official journal of the American Association for Cancer Research. 2013;19:Abstract_nr_A23
47. Chang CJ, Yang JY, Xia W. et al. EZH2 promotes expansion of breast tumor initiating cells through activation of RAF1-beta-catenin signaling. Cancer cell. 2011;19:86-100
48. Dubrovska A, Kim S, Salamone RJ. et al. The role of PTEN/Akt/PI3K signaling in the maintenance and viability of prostate cancer stem-like cell populations. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:268-73
49. Guo W, Keckesova Z, Donaher JL. et al. Slug and Sox9 cooperatively determine the mammary stem cell state. Cell. 2012;148:1015-28
50. Cordenonsi M, Zanconato F, Azzolin L. et al. The Hippo transducer TAZ confers cancer stem cell-related traits on breast cancer cells. Cell. 2011;147:759-72
51. Poole AJ, Li Y, Kim Y. et al. Prevention of Brca1-mediated mammary tumorigenesis in mice by a progesterone antagonist. Science. 2006;314:1467-70
52. Skor MN, Wonder EL, Kocherginsky M. et al. Glucocorticoid receptor antagonism as a novel therapy for triple-negative breast cancer. Clinical cancer research: an official journal of the American Association for Cancer Research. 2013;19:6163-72
53. Sitruk-Ware R, Spitz IM. Pharmacological properties of mifepristone: toxicology and safety in animal and human studies. Contraception. 2003;68:409-20
54. Garbe JC, Bhattacharya S, Merchant B. et al. Molecular distinctions between stasis and telomere attrition senescence barriers shown by long-term culture of normal human mammary epithelial cells. Cancer research. 2009;69:7557-68
55. Perrault D, Eisenhauer EA, Pritchard KI. et al. Phase II study of the progesterone antagonist mifepristone in patients with untreated metastatic breast carcinoma: a National Cancer Institute of Canada Clinical Trials Group study. J Clin Oncol. 1996;14:2709-12
Author contact
![]() Corresponding authors: Ceshi Chen, Tel: 86-871-65181944, E-mail: chenckiz.ac.cn Or Rong Liu, Tel: 86-871-65181945, E-mail: liurongkiz.ac.cn.
Corresponding authors: Ceshi Chen, Tel: 86-871-65181944, E-mail: chenckiz.ac.cn Or Rong Liu, Tel: 86-871-65181945, E-mail: liurongkiz.ac.cn.