Impact Factor
 Global reach, higher impact
Global reach, higher impact
Theranostics 2016; 6(5):610-626. doi:10.7150/thno.13757 This issue Cite
Research Paper
Nano-zymography Using Laser-Scanning Confocal Microscopy Unmasks Proteolytic Activity of Cell-Derived Microparticles
Aurélien Briens1*, Maxime Gauberti1*, Jérôme Parcq1, Joan Montaner2, Denis Vivien1, Sara Martinez de Lizarrondo1 ![]()
1. Institut National de la Santé et de la Recherche Médicale (INSERM), INSERM UMR-S U919, Serine Proteases and Pathophysiology of the Neurovascular Unit, GIP Cyceron, Université Caen-Normandie, Caen, France.
2. Neurovascular Research Laboratory, Vall d'Hebron Research Institute, Barcelona, Spain.
*A.B. and M.G participated equally to this work.
Received 2015-9-4; Accepted 2016-1-27; Published 2016-3-2
Abstract

Cell-derived microparticles (MPs) are nano-sized vesicles released by activated cells in the extracellular milieu. They act as vectors of biological activity by carrying membrane-anchored and cytoplasmic constituents of the parental cells. Although detection and characterization of cell-derived MPs may be of high diagnostic and prognostic values in a number of human diseases, reliable measurement of their size, number and biological activity still remains challenging using currently available methods. In the present study, we developed a protocol to directly image and functionally characterize MPs using high-resolution laser-scanning confocal microscopy. Once trapped on annexin-V coated micro-wells, we developed several assays using fluorescent reporters to measure their size, detect membrane antigens and evaluate proteolytic activity (nano-zymography). In particular, we demonstrated the applicability and specificity of this method to detect antigens and proteolytic activities of tissue-type plasminogen activator (tPA), urokinase and plasmin at the surface of engineered MPs from transfected cell-lines. Furthermore, we were able to identify a subset of tPA-bearing fibrinolytic MPs using plasma samples from a cohort of ischemic stroke patients who received thrombolytic therapy and in an experimental model of thrombin-induced ischemic stroke in mice. Overall, this method is promising for functional characterization of cell-derived MPs.
Keywords: Extracellular vesicles, Phosphatidylserine, Optical Imaging, fibrinolysis, MPIO, exosomes, inflammation
Introduction
Cell-derived microparticles (MPs) are nano-sized vesicles released by virtually all eukaryotic cells after stimulation or apoptosis[1, 2]. They originate from the budding of the plasma membrane, leading to the formation of 100 nm to 1 µm vesicles with a double layered membrane of phospholipids that are released in body fluids (blood, cerebrospinal fluid, urine, tears and saliva)[1]. Since secreted MPs harbour proteins from their parental cell, they can be used as biomarkers of cellular activation, which are of high diagnostic and prognostic values in a number of human diseases[3, 4]. For instance, red-blood cells MPs predict severe clinical outcomes after dengue virus infection[5], high plasmatic levels of leukocyte MPs are associated with unstable plaque in asymptomatic patients with high-grade carotid stenosis[6] and endothelial MPs are elevated in chronic obstructive pulmonary disease[7]. MPs are distinct from exosomes, another subtype of extracellular vesicles, with sizes ranging from 40 to 100 nm.
Besides their interest as biomarkers, MPs are also active players in biological processes such as blood coagulation, cell-cell communications or immunity[8-10]. MPs are especially essential for spreading the activity of surface proteins which require membrane binding to exert their biological functions. In particular, cells expressing proteases or protease receptors on their surface are able to generate active and circulating proteolytic MPs[10-13]. For instance, fibrinolytic MPs corresponding to urokinase (uPA) bearing MPs from leukocytes and tissue-type plasminogen activator (tPA) bearing MPs from endothelial cells have been described in patients presenting thrombotic thrombocytopenic purpura (TTP) [14]. Moreover, MPs bearing the endothelial protein C receptor (EPCR) have been reported in sepsis patients and are capable to acquire proteolytic activity upon activated protein C (APC) treatment[15, 16]. Proteolytic MPs play also an important role in cancer dissemination[17]. Notably, fractionation of malignant ascites revealed that extracellular matrix-degrading proteinases including matrix-metalloproteinases (MMPs) and uPA are localized preferentially in membrane vesicles.[18]
Because of the suspected widespread implication of MPs in human pathophysiology, new methods allowing MPs characterization are awaited. Indeed, the detection of cell-derived MPs is made difficult by their small size, high concentration, low refractive index and heterogeneity[19]. Most studies used flow-cytometry to detect MPs after antibody and/or annexin-V labelling[20]. Although this method allows high-throughput detection of MPs in fluids, its ability to measure MP size, number and antigens remains limited and prone to false positives/negatives[21]. Other methods are available, such as electron microscopy, resistive pulse sensing or nanoparticle tracking analysis but are of limited availability, do not allow complete phenotypic characterization of MPs and often require extensive sample preparation. Moreover, there is no available method to measure the proteolytic activity of individual MPs.
With recent advances in optical imaging, fluorescence microscopy appears promising for MPs characterization, but its sensitivity to detect nano-sized vesicles is hampered by the rapid Brownian motion of MPs in fluids. The aim of the present study was to develop a method allowing labelling, immobilization, antigen detection and measurement of proteolytic activity of cell-derived MPs by high-resolution laser-scanning confocal microscopy (LSCM). We illustrated the potential uses of this method by studying fibrinolytic MPs from several cultured cell-lines and plasma from mice and ischemic stroke patients.
Methods
Chemicals
Plasmin fluorescent substrate came from SensoLyte AFC Plasmin Activity Assay Kit *Fluorimetric* (Anaspec) and tPA Spectrofluor 444FL substrate was from American Diagnostica (ADF Biomedical, Neuville-sur-oise, France). Hygromycin B, bovine thrombin, carboxyfluorescein succinidimyl ester (CFSE), EDTA and lipopolysaccharide (LPS 0111:B4) were from Sigma-Aldrich (L'Isle d'Abeau, France). Bovine fibrinogen used for fibrin-agar zymography came from Bovogen Labs (Australia). Plasminogen used for plasmin substrate activity studies was from Enzyme Research Laboratories (ERL, UK). Sc-uPA, purified from human cell lines, was from Biopool AB (UK). The uPA standards for fibrin-agar zymographies were obtained using ACTOSOLV® (Eumedica). Lipofectamine 2000, fetal bovine sera were from Invitrogen (Cergy Pontoise). tPA (Actilyse) was from Boehringer-Ingelheim (Paris, France). Human PAI-1 (N-terminal cysteine, active fraction) came from Gentaur. Sheep antiserum raised against human tPA was prepared at the national institute for agronomic research (INRA, Clermont-Theix, France). Tumor Necrosis Factor-α was purchased from PeproTech (Rocky Hill, NJ). Recombinant annexin-V came from Abcam (France). Aprotinin (Trasylol) was a gift from Bayer HealthCare AG.
Animals
Animals were maintained under specific pathogen-free conditions at the Centre Universitaire de Ressources Biologiques (CURB, Basse-Normandie, France) and all had free access to food and tap water before and after the procedures.
Acute systemic inflammation
Systemic inflammation was performed by intra-peritoneal (i.p.) injection of LPS from E. coli (2 mg/kg) in 8-week-old male tPA wt mice (Janvier, France). Control mice received an equivalent volume of saline. Blood sampling was performed 6 hours after LPS administration by intracardiac puncture under deep isoflurane anaesthesia. MPs purification was then performed as described below.
Thromboembolic stroke model
We performed in situ thromboembolic occlusion in anesthetized mice (2% isoflurane in a mixture of O2/N2O 33%/67% with a rectal temperature maintained at 37°C) by intra-arterial injection of thrombin (1 UI/µL, 1 µL, Kordia, Leyden) as previously described[22, 23].
Magnetic resonance imaging (MRI)
Experiments in mice were carried out on a Pharmascan 7 T/12 cm system using surface coils (Bruker, Germany). T2-weighted images were acquired using a multi-slice-multi-echo sequence: echo-time (TE)/ repetition-time (TR) 51 ms/2500 ms with 70 × 70 × 500 µm3 spatial resolution. Magnetic resonance angiographies (MRA) were performed using a 2D-TOF sequence (TE/TR 10/50 ms).[24] Diffusion weighted imaging (DWI) was performed using a standard spin-echo sequence modified with Stejskal-Tanner diffusion gradient scheme (TR/TE 2500/30ms).[25]
Collection and human sample processing
Patients admitted to the emergency department of the Vall d'Hebron University Hospital (Barcelona, Spain) with an acute ischemic stroke within the first 4.5 h after symptoms onset were recruited. Stroke diagnosis was performed based on a standardized protocol of clinical and neuroradiological assessments[26]. Before administration of any treatment, peripheral blood samples were drawn from each patient in EDTA collection tubes (Before). All patients received intravenous recombinant tissue-type plasminogen activator (tPA) in a standard 0.9 mg/Kg dose (10% bolus, 90% continuous infusion during 1 h). Plasma samples were recollected at the end of tPA infusion (1h), and at 2, 12 and 24h after thrombolysis. All plasmas were immediately separated by serial centrifugation at 1,500 g for 15 min at +4°C and at 14,000g for 2min to remove any residual platelet. Aliquots were immediately stored at -80°C until use.
Study approval
Animal experiments were approved by the local ethical committee (C-31.555-07, US006 CREFRE (CEEA-122)) and were performed in accordance with the French (Decree 87/848) and the European Communities Council (Directive 86/609) guidelines. For human samples, the local ethical committee (Vall d'Hebron Hospital, Barcelona, Spain) approved the study and written consent was obtained from all patients or relatives in accordance with the Helsinki declaration.
Cell culture
The human monocytic cell line THP-1 (ATCC) was grown in RPMI-1640 medium supplemented with 10% MP-free fetal bovine serum, 4mM glutamine, 0.5mM sodium pyruvate, 0.5% nonessential amino acids, and 1% antibiotics (penicillin, streptomycin). The human microvascular endothelial cell line (HMEC-1) was cultured in DMEM medium (Invitrogen) supplemented with 10% MP-free fetal bovine serum, 2mM glutamine and 1% antibiotics (penicillin, streptomycin). Human Embryonic Kidney (HEK)-293 cells were grown in RPMI-1640 medium supplemented with 10% fetal bovine serum, 2mM glutamine, 0.5mM sodium pyruvate, 0.5% nonessential amino acids, and 1% antibiotics (penicillin, streptomycin). Cells were maintained at 37°C in a humidified atmosphere of 5% CO2.
Construction of human wild-type tPA in the pcDNA5/FRT vector
The human tPA cDNA sequence was amplified by PCR with the following primers: 5′-CCGGGATCCTCTTACCAAGTGATCTGC-3' and 5'-GGCAAGCTTTCACGGTCGCATGTTGTCACG-3′. PCR products were inserted into a pcDNA5/FRT vector (Invitrogen). Final constructs were automatically sequenced.
Bioreactor production of HEK-293 cell cultures and stable transfection with human tPA encoding plasmid
Stable HEK-293 cells were co-transfected with the tPA-pcDNA5/FRT and pFRT/lacZeo vectors (HEK-FlpIn; Invitrogen) by Lipofectamine 2000. Positive clones were isolated by hygromycin B selection. To produce high amounts and stable transfected HEK-293 cells were grown in a laboratory-scale bioreactor (CELLine AD 1000; Dominique Dutscher SAS, Brumath, France).
In vitro MPs generation
Culture media from stable tPA-HEK-293 and baseline HEK-293 cells (not subjected to transfection, which were used as controls) were collected twice a week. Culture supernatants were then centrifuged at 300g for 5 min and at 12,000g for 5 min to remove detached cells and debris. Supernatants were collected from monocytic THP-1 and endothelial HMEC-1 cell line cultures after 48h stimulation with 100 ng/ml Tumor Necrosis Factor-α (PeproTech, Rocky Hill, NJ), as previously described[12, 14].
Microparticles isolation
Frozen samples from patients, mice and from cell cultures were thawed and processed. They were then centrifuged at 20,000 g for 90 min at 4°C[12]. Pelleted MPs were washed twice using the same protocol of centrifugation and resuspended in NaCl/Hepes buffer (150 mM NaCl and 10 mM HEPES, pH=7.4).
MPs immobilization, immunolabeling and proteolytic activity assessment
In order to immobilize the MPs, micro-wells (µ-Slides, Ibidi) were first coated overnight with 50 µg/ml recombinant annexin-V (Abcam). Purified MPs, diluted in NaCl/Hepes buffer, were incubated at 37°C during 30 minutes in the presence of carboxyfluorescein succinidimyl ester (CFSE, 0.2 mM, Sigma). This protocol allows labelling virtually all cell-derived MPs (data not shown). Then, MPs diluted in NaCl/Hepes buffer (with or without CaCl2, 10mM) were then seeded in coated micro-wells and were immobilized in the dark at room temperature in the absence of stirring. When appropriate, experiments were also performed and in the presence or in the absence of the calcium chelator EDTA (20mM). Immunofluorescence was performed using a polyclonal sheep antiserum raised against human tPA (1: 5000) prepared at the National institute for agronomic research (INRA, Clermont-Theix, France). MPs proteolytic activity was assessed by adding a plasmin fluorescent substrate (SensoLyte AFC Plasmin Activity Assay Kit *Fluorimetric*, 10 µM, Anaspec) in the presence or in the absence of 2 µM exogenous human plasminogen (Enzyme Research Laboratories). Reaction specificity was confirmed by using aprotinin (plasmin inhibitor, 20 IU/ml) and, when stated, with the addition of active fraction of human PAI-1. When stated, tPA-MPs proteolytic activity was also confirmed using a specific tPA-fluorogenic substrate, (Spectrofluor (Spectrofluor tPA: CH3SO2-D-Phe-Gly-Arg-AMC.AcOH in Tris/NaCl buffer, pH 8.0), 100 µM, American Diagnostica).
Fibrin-Agar Zymography
The presence/abscence of active tPA on HEK-derived MPs (coming from tPA-transfected and control HEK cells) was detected by direct fibrin autography following sodium dodecylsulphate polyacrylamide gel electrophoresis (SDS-PAGE) performed as previously described[14]. Purified tPA and control MPs (tPA and ctl-MPs, respectively) were subjected to SDS electrophoresis (8% polyacrylamide gels, under non-reducing conditions). SDS was then exchanged with 2.5% Triton X-100. After washing off excess Triton X-100 with distilled water, the gel was carefully overlaid on a 1% agarose gel containing 1 mg/mL bovine fibrinogen, 100 nM plasminogen and 0.2 NIH U/mL of bovine thrombin. Zymograms were allowed to develop at 37°C for 12h and photographed at regular intervals using dark-ground illumination. Active proteins in tPA-MP sample were identified by reference to the migration of known markers (tPA, 0,25nM).
Western blot
Recombinant tPA and MPs samples (tPA-transfected and control, ctrl-MPs coming from HEK supernatants) were resolved for the western blot on 4-12 % Nupage gels (Invitrogen) gels in reduced conditions and transferred onto iBlot, a polyvinylidene difluoride (PVDF) membrane by iBLOT transfer stacks (Invitrogen). Blots were blocked with 5% BSA (bovine serum albumin, Sigma-Aldrich, L'Isle d'Abeau, France) in Tris-buffered saline containing 0.05% Tween-20 and then incubated overnight with the same buffer at 1% BSA with anti-tPA antibody (1/5000), followed by incubation with a peroxidase-conjugated goat anti-sheep secondary antibody (1/50000, sigma) and developed by a Amersham ECL Western blotting detection reagents and analysis system (GE Healthcare, France) using ImageQuant™ LAS 4000 camera (GE healthcare, France).
tPA and plasminogen conjugation to MPIO
Microparticles of iron oxide (MPIO; diameter 1.08 μm) with p-toluenesulphonyl reactive surface groups (Invitrogen) were covalently conjugated to dialysed (against a 0.3M bicarbonate buffer) recombinant tPA (Actilyse) [27-29]. The tosyl reactive surface allows covalent binding of MPIO to proteins by primary amine (NH2) or sulphydryl (SH) groups[30]. Briefly, dialysed tPA (in order to eliminate amine-containing excipients), purified plasminogen from human plasma (purified using sepharose-lysine columns, as previously described[31]) or BSA were covalently conjugated to MPIOs in borate buffer (pH 9.5), by incubation at 37 °C for 24 hours. 40 μg of proteins were used for the coating of 1 mg of reactive MPIOs. MPIOs were then washed in phosphate buffered saline (PBS) containing 0.5% bovine serum albumin (BSA) and incubated for 24 hours at 37°C, to block the remaining active groups. MPIOs were rinsed in PBS (0.1% BSA). To disperse MPIO aggregates, a sonication procedure at low intensity was performed one time, immediately after antibody labelling, for 60 seconds. Thereafter, MPIO were stored at 4°C in a PBS buffer under constant agitation to prevent settling and aggregate formation. Immunofluorescence (for tPA and plasminogen) and MPIOs proteolytic activities were performed following the same protocol than the one used for cell-derived MPs.
Laser scanning confocal microscopy
Laser-scanning confocal microscopy (LSCM) was performed using an inverted Leica SP5 confocal microscope (Leica Microsystems SAS) equipped with an Argon Gas laser and a X40 NA=1.4 oil immersion objective. For CFSE detection, excitation was set at 488 nm and emission filters between 500 and 540 nm. For Cy3 detection, excitation was set at 561 nm and emission filters between 575 and 625 nm. For plasmin substrate detection, excitation was set at 405 nm and emission filters between 475 and 525 nm. For tPA substrate detection, excitation was set at 405 nm and emission filters between 425 and 475 nm. Field of view was set at 1 µm x 1 µm with a 1024x1024 planar matrix (pixel size= 97.6 nm x 97.6 nm). Autofluorescence of MPIOs was detected using the CFSE filter settings and increased laser power.
Image analysis
An automated segmentation method was developed with the ImageJ software (v1.45r, NIH). CFSE staining was first used to identify MPs and estimate their number and size using automatic Otsu-thresholding and the integrated “particle analysis” setting. Fluorescence intensity of tPA immunostaining and plasmin substrate was then measured in CFSE positive particles using the “ROI manager” of ImageJ. In particular, the Feret's diameters of the MPs were computed.
Statistics
Results are presented as the mean ± SD. Statistical analyses were performed using Kruskal-Wallis (for multiple comparisons) followed by Mann-Whitney's U-test. The nonparametric Wilcoxon signed-rank test was used to examine differences in MPs density and MPs content in tPA in stroke patients over time. When comparing two groups, a p-value < 0.05 was considered to be significant.
Results
Isolation and detection of MPs using Laser-Scanning Confocal Microscopy (LSCM)
Among extracellular vesicles, MPs are commonly characterized by their size (100 nm to 1 µm) and the presence of phosphatidylserine (PS) on their surface [8, 32]. We took advantage of the high-affinity binding of PS to annexin-V in the presence of calcium to immobilize MPs on annexin-V coated surface and therefore, prevent Brownian motion (Additional File 2: Movie S1). First, MPs from THP-1 cells were purified by sequential ultra-centrifugation as previously reported (Additional File 1: Figure S1) [33, 34]. To allow their visualisation using fluorescent microscopy, we used a fixable-cell-permeant, fluorescein-based tracer (carboxyfluorescein succinidimyl ester, CFSE). After cleavage by intravesicular esterases, CFSE bind to proteins via amine labelling, ensuring long-term and homogeneous dye retention inside MPs with minimal background fluorescence. Then, CFSE-labelled MPs were immobilized on annexin-V coated micro-wells and detected on high-resolution LSCM images using an automated analysis procedure in ImageJ (Figure 1).
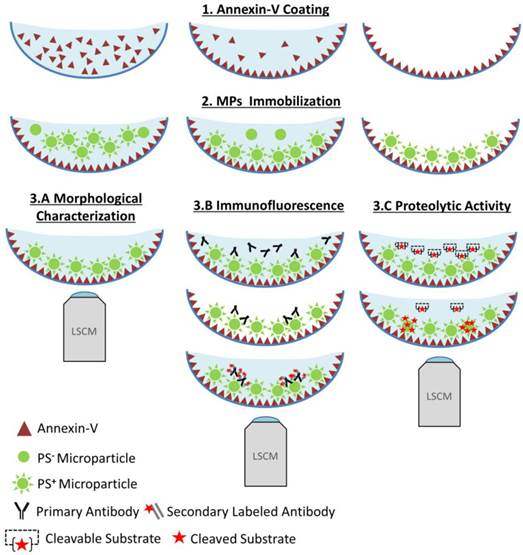
Functional characterization of microparticles (MPs) by Laser Scanner Confocal Microscopy (LSCM). Schematic representation of the LSCM method allowing individual MPs immobilization, characterization (including labeling and functional studies). Step 1-annexin-V coating of the micro-wells. Step 2- PS+-MPs (CFSE+) immobilization on the coated surface in the presence of Ca2+ thanks to the high affinity binding of PS present to annexin-V. Step 3- Immobilized MPs characterization: 3A. Morphological MPs characterization, including number and size distribution. 3B. Immunofluorescence analyses, using specific primary (against proteins present on the MP surface) and secondary-labeled antibodies. 3C. Proteolytic activity studies (nano-zymography), after addition of specific fluorogenic substrates. PS=Phosphatidylserine. LSCM=laser scanning Confocal Microscopy.
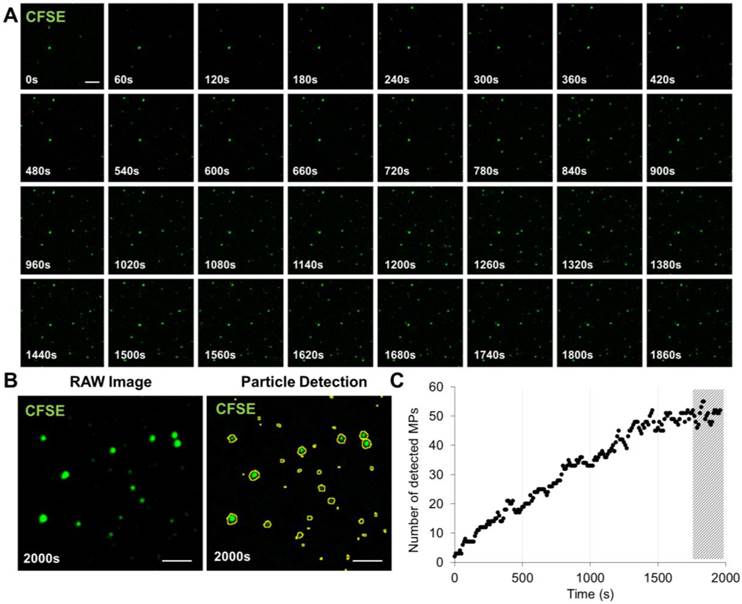
Time-lapse imaging revealed that >95% of MPs bind to the annexin-V surface in the first 25 minutes of incubation (Figure 2 and Additional File 3: Movie S2). All LSCM acquisitions were thus performed after 30 minutes incubation in the micro-wells. Importantly, the number of detected MPs dramatically dropped (~10-fold) when binding was performed in the absence of calcium (Figure 3A and B), confirming that the binding to the micro-wells surface was dependent on annexin-V interaction with PS. Chelation of calcium using EDTA did not further reduce unspecific binding (Additional File 1: Figure S2), suggesting that the low binding-rate in the absence of added calcium is annexin-V independent. Importantly, there was a linear relationship between the number of surface-immobilized MPs and their concentration that reached saturation only at very high concentrations, supporting the ability of the present method to provide semi-quantitative results (Figure 3A and B).
Time-Lapse of MPs immobilization on annexin-V surface. (A) Time-lapse (from 0-2000s) was performed in order to study the time needed to efficiently immobilize to the annexin-V coated surface the maximum of PS+-MPs present in our sample. Images were obtained by high-resolution LSCM every 60s. (B) Example of raw image at 2000s before and after MPs detection using ImageJ software. (C) Automated quantification of the number of detected MPs in the field of view showing that, after ~30 min (1800s), the amount of immobilized MPs is stable (representative of n=3 independent experiments). The hashed area represents the imaging window starting after 1800s. (Bars, 5 µm).
Laser Scanner Confocal Microscopy of immobilized MPs from THP-1 cells allows measurement of MP size and concentration. (A) Dose-dependent detection of THP-1-derived MPs (CFSE+, green) in the presence or in the absence of Ca2+. (n=3) (B) CFSE+ MPs quantification according to their concentration and the presence (black) or absence (grey) of Ca2+. (n=3) (C) CFSE+ MPs distribution depending on their size, including the apparent median diameter (0.43 µm). (n=4) (D) Loss of CFSE staining by THP-1 MPs after destruction by a detergent (Triton X-100, 1%) as compared to control-untreated MPs. (n=3) (Bars, 10µm). * p<0.05 vs Control. Statistical analyses were performed using Mann-Whitney's U-test.
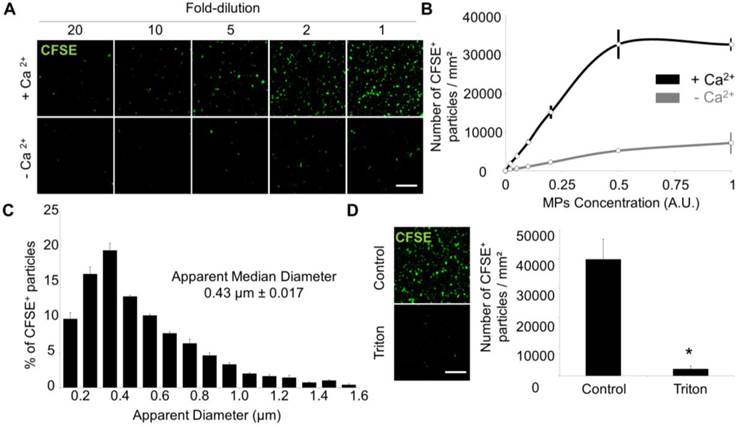
Using an automated segmentation method in ImageJ, we measured the apparent size of THP-1 derived MPs immobilized on the annexin-V coated micro-wells. 95% of the apparent MP diameters ranged from 100-200 nm to 1.2 µm with a median size of 452 nm, which is similar to previously reported MP sizes[35] and confirms the ability of the present method to detect both small and large MPs (Figure 3C). The few detected MPs with apparent diameter >1 µm may correspond to aggregates of smaller MPs. After addition of a detergent (Triton X-100) to the micro-wells, the number of detected MPs dramatically dropped (Figure 3D), confirming the lipid nature of the MPs. Altogether, these data demonstrated that annexin-V coated micro-wells allow immobilization of PS+ MPs and subsequent morphological characterization using LSCM.
It is interesting to note that some fluorescently labelled vesicles interacted with the annexin-V coated surface but were not able to bind (Additional File 1: Figure S3), suggesting that they did not express PS (or in insufficiently high amount). We performed a time-lapse using fast LSCM imaging, in order to detect these transient events and therefore, to be able to calculate the number of binding versus non-binding vesicles (representing the number of PShigh versus PSlow vesicles) using murine plasma samples. In these conditions, about two-third of extracellular vesicles were not able to bind annexin-V (contact without binding), supporting recent studies claiming that most extracellular vesicles in the plasma do not expose PS on their surfaces[36]. In the following experiments, only the vesicles bound to the annexin-V coated micro-wells corresponding to PS+ MPs were analysed.
Detection of MPs antigens using immunolabelling and LSCM
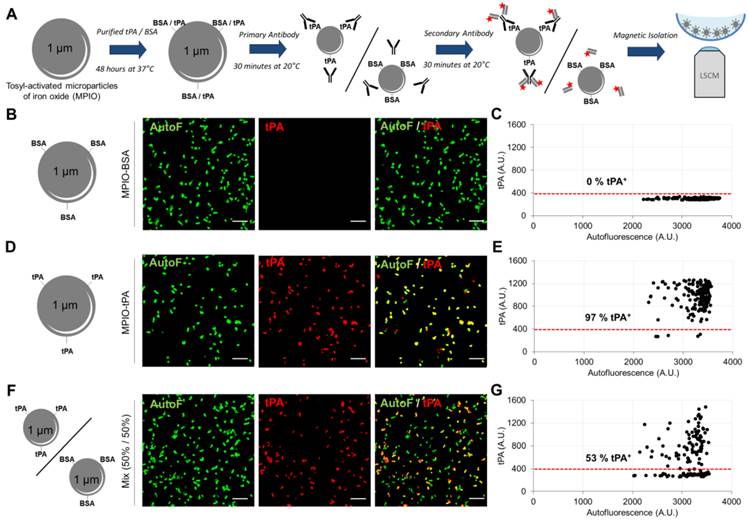
In addition to morphological characterization of immobilized MPs, LSCM imaging offers an opportunity for specific detection of MPs antigens by fluorescent immunolabeling. However, suitable primary antibodies selection and optimisation of binding conditions remain challenging, since there is no well-characterized MPs population to be used as positive control for most proteins. We therefore developed an assay based on the binding of a known recombinant protein on the surface of synthetic 1-µm-sized particles (micro-sized particles of iron oxide, MPIO) that can be magnetically immobilized in the micro-wells and detected with LSCM (Figure 4A)[37]. We performed this method to validate the feasibility of detecting human recombinant tPA on the surface of MPs. Bovine serum albumin (BSA, used as a control for tPA with approximately the same molecular mass of ~70 KDa) and tPA-bearing MPIOs (MPIO-tPA) were synthetized and labelled using a primary anti-tPA antibody, followed by a secondary Cy3-labelled Fab'2. Thereafter, MPIO-BSA and MPIO-tPA were imaged using high-resolution LSCM and the generated images were analysed using an automated macro in ImageJ. As shown on Figure 4, no significant binding of anti-tPA antibody was detected on MPIO-BSA (Figure 4B and C), whereas binding was detectable on MPIO-tPA (Figure 4D and E), allowing the differentiation of MPIO-tPA and MPIO-BSA in the same sample using LSCM and automated analysis (Figure 4F and G). These results supported the use of this anti-tPA antibody for the detection of human tPA on the surface of cell-derived MPs.
Immunolabeling of tPA coupled to Micro-sized particles of iron oxide (tPA-MPIO) as a model for tPA-MPs. (A) Schematic representation of the tPA-MPIO production, immunolabeling and imaging procedures. MPIOs (known diameter of 1.08 μm) with p-toluenesulphonyl reactive surface groups were covalently conjugated to dialysed recombinant tPA (MPIO-tPA) or control (Bovine Serum Albumin, MPIO-BSA) and then labelled with anti-tPA antibodies and fluorescently labelled secondary antibodies. (B) Schematic representation of control microparticles (MPIO-BSA, visible in green by autofluorescence, AutoF) and representative images of tPA immunofluorescence with primary and secondary antibodies (tPA, red). (C) Quantification of tPA immunoreactivity (tPA+ events). Red horizontal bar indicates cutoff value for tPA positivity (mean + 2*standard deviation of control condition). No fluorescence was detected on the control MPIO (0% tPA+). (D) Schematic representation of a tPA microparticles (tPA-MPIO) and representative images of tPA immunofluorescence with primary and secondary antibodies. (E) Quantification of tPA immunoreactivity (tPA+ events). 97% of MPIO fluorescence values were above the cutoff in this MPIO mixture, confirming antibody sensitivity. (F) Schematic representation of a 50%/50% mix of tPA/BSA microparticles (MPIO-BSA and tPA-MPIO) and representative images of tPA immunofluorescence with primary and secondary antibodies. (G) Quantification of tPA immunoreactivity (tPA+ events). 53% of MPIO fluorescence values were above the cutoff in this MPIO mixture, confirming the ability of this LSCM method to distinguish tPA-MPIO from BSA-MPIO in the same sample. All the images and quantifications are representative of n=3 independent experiments. BSA= Bovine Serum Albumine.
Thereafter, we wanted to perform anti-tPA immunofluorescence labelling on cell-derived MPs presenting human tPA on their surface (tPA-MPs). In order to obtain these engineered tPA-MPs, we transfected human epithelial kidney cells (HEK-293) with a human tPA encoding plasmid containing the entire tPA cDNA sequence (Figure 5A-D). We used the HEK Flp-In® system for generating HEK-293 cells with constitutive and stable expression of human tPA. Moreover, to produce high yields of these MPs, stable transfected HEK-293 cells were grown in a laboratory-scale bioreactor and MPs were purified from the supernatants (Figure 5E). We confirmed that these MPs harboured active tPA by immunoblot and zymography (Figure 5F and G). Therefore, this protocol allowed us to produce large amounts of proteolytically active tPA-MPs for further studies.
Generation, detection and characterization of engineered cell-derived fibrinolytic MPs by LSCM. (A) Primers sequence (Forward and Reverse) that allowed whole human tPA cDNA sequence amplification. (B) Structure of human cDNA tPA sequence, including all tPA domains. (C) tPA plasmid. (D) Plasmid allowing stable integration of the human tPA in the cell genome. (E) Schematic representation of the method that allows tPA-MPs production from HEK293 cell cultures through stable transfection with plasmids containing tPA sequence with subsequent purification of released MPs by sequential ultracentrifugations. (F) Representative tPA immunoblot after migration of the proteins of MPs purified from tPA-expressing (tPA-MPs) or control non-transfected HEK-293 (Ctl-MPs). (G) Same as in (F) but representing a fibrin-agar zymography, demonstrating that the tPA in tPA-MPs is proteolytically active. (H) Representative images of tPA-MPs immunofluorescence with primary and secondary antibodies (1°+2° AB, red). (I) Quantification of tPA immunoreactivity in MPs incubated without (black) or with (grey) tPA primary antibody. Red vertical bar indicates baseline value (mean + 2*standard deviation of control condition) (n=3). tPA domains= F: finger, EGF: Epithelial Growth Factor, K: Kringle, SP: Serine Protease.
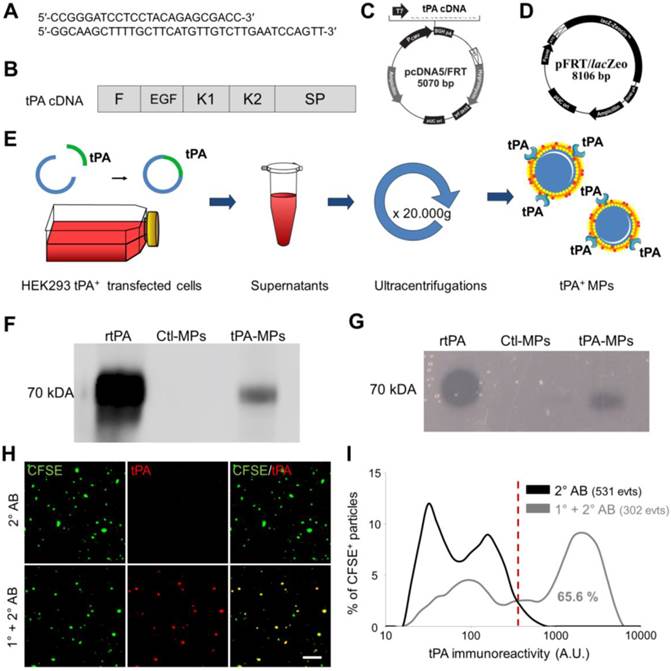
We immobilized these tPA-MPs on annexin-V coated micro-wells and performed high resolution LSCM with or without anti-tPA primary antibodies (no detergent was added to preserve MP membrane integrity and prevent antibody to reach intravesicular antigens). As shown on Figure 5H, we were able to detect tPA at the surface of these MPs using anti-tPA antibodies and fluorescently labelled secondary antibodies (1° and 2° AB). Using this method, 65.6% of the purified MPs from tPA-transfected HEK-293 cells appeared tPA+ (Figure 5I).
Nano-zymography by LSCM allows the detection of specific surface proteolytic activities of individual MPs
The recent descriptions of the presence of proteases at the surface of MPs (such as tPA, uPA, ADAM17, APC…) suggest that MPs can disseminate the proteolytic activity of the parent cells. Therefore, in addition to the detection of protease antigen, measurement of the proteolytic activity of cell-derived MPs may be of diagnostic value. Here, we aimed to reveal the proteolytic activity of our engineered tPA-MPs (Figure 6). To reveal their proteolytic activities, we incubated tPA-MPs with plasminogen (Plg) and a plasmin (Pln)-specific fluorogenic substrate and performed LSCM[38]. Whereas in the absence of exogenous plasminogen or in the presence of an inhibitor of plasmin activity (aprotinin) no substrate cleavage occurred, a significant plasmin-related proteolytic activity was detected on the surface of tPA-bearing MPs (Figure 6A and B). Using a direct substrate of tPA, we were also able to reveal the presence of active tPA at the surface of the engineered MPs (Figure 6C and D). This activity was completely inhibited by the addition of plasminogen activator inhibitor-1 (PAI-1), suggesting that plasminogen activators at the surface of MPs are susceptible to inhibition by circulating inhibitors.
Nano-zymography of proteolytic activities (tPA and plasmin) at the surface of different sources of fibrinolytic-MPs (tPA and uPA-MPs). (A) Schematic representation of tPA-bearing MPs and representative LSCM images of HEK-derived tPA-MPs (CFSE, green) in the presence or in the absence of exogenous plasminogen (Plg, 2µM). In red is represented the fluorescence from cleaved plasmin substrate (Pln substrate, red). Loss of the fluorescence from the plasmin substrate in the presence of aprotinin (plasmin inhibitor, 20 IU/ml) confirmed reaction specificity. (B) Quantification of cleaved plasmin substrate fluorescence by tPA-MPs alone (blue), in the presence of plg (red) and with plg+aprotinin (green). (C) Schematic representation of tPA-bearing MPs and representative images of cleaved tPA fluorogenic substrate (tPA substrate, Spectrofluor tPA, 0.1mM, red) on tPA-MPs. Loss of staining in the absence of substrate or in the presence of the tPA inhibitor PAI-1 (Human PAI-1 (N-terminal cysteine, active fraction, Gentaur), 3.5µM) confirmed reaction specificity. (C) Quantification of the fluorescence from cleaved tPA substrate by tPA-MPs in the presence (red) or in the absence (blue) of tPA substrate or in the presence of the tPA inhibitor PAI-1 (green). (E) Schematic representation of control and uPA-bearing MPs (uPA-MPs) and representative images of cleaved plasmin fluorescent substrate (Pln substrate, red) in both subtypes of MPs (incubated in the presence of plasminogen). Loss of staining in control MPs or in the presence of the uPA inhibitor PAI-1 (Human PAI-1 (N-terminal cysteine, active fraction, Gentaur), 3.5µM) confirmed reaction specificity. (F) Quantification of the fluorescence from cleaved plasmin substrate by control MPs (blue) or uPA-MPs in the absence (red) or in the presence of an uPA inhibitor, PAI-1 (green). All results are representative of n=3 independent experiments. Plg=plasminogen, Pln=plasmin. Bars, 10µm.
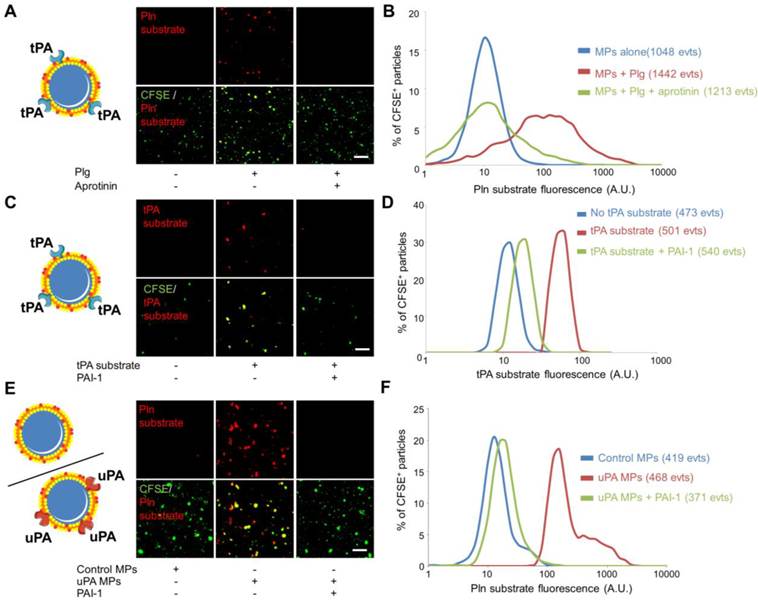
Since uPA has been described at the surface of circulating MPs, we also generated urokinase (uPA)-bearing MPs. To this aim, we purified MPs from HMEC-1 cells that are known to present uPA-receptor on their surface (Additional File 1: Figure S4) [12]. After a short incubation with sc-uPA, the MPs were washed to remove unbound uPA, as previously described[12]. The proteolytic activity of uPA-bearing MPs was detected using nano-zymography after addition of a plasmin specific fluorogenic substrate in the presence of plasminogen. Again, this proteolytic activity was inhibited by the addition of PAI-1, confirming the susceptibility of plasminogen activator-bearing MPs to inhibition by circulating inhibitors. These in vitro data demonstrated the feasibility of visualizing proteolytic activities of individual MPs from different sources using nano-zymography by LSCM.
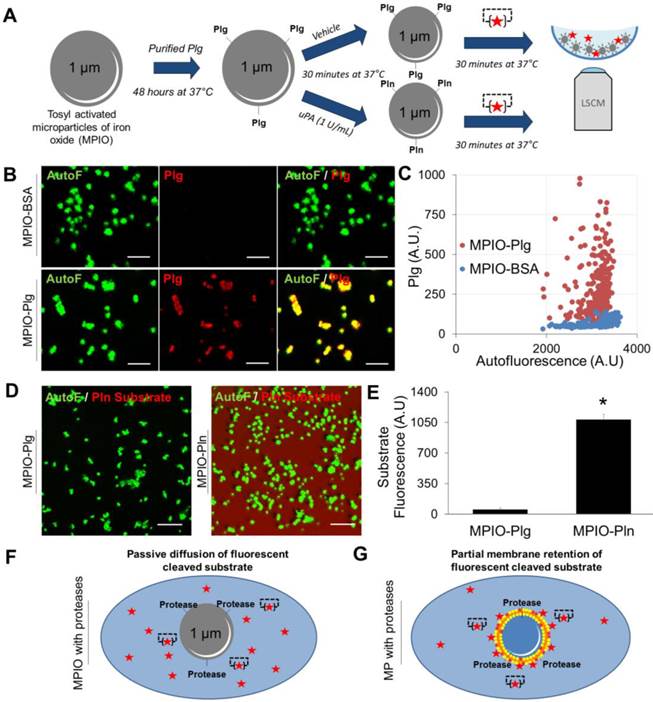
It should however be acknowledged that sufficient membrane retention of the cleaved fluorogenic substrate appears mandatory for efficient nano-zymography. Indeed, the proteolytic activity of synthetic MPIO harbouring active plasmin cannot be detected in situ using LSCM (Figure 7A). When MPIO-Plasmin (MPIO-Pln, generated by activation of MPIO coated with plasminogen by uPA, Figure 7A-C) were incubated with the fluorogenic plasmin substrate, the fluorescence appeared diffuse and not localized on the MPIO surface (Figure 7D and E). This is probably related to the free diffusion of the fluorescent dye that is not as efficiently retained by the MPIOs than by naturally-occurring cell-derived MPs (Figure 7F and G).
Detection of the proteolytic activity of artificial MPIOs harbouring plasmin on their surface. (A) Schematic representation of the MPIO-Plg (Plasminogen) and MPIO-Pln (Plasmin) production and imaging procedures. MPIOs (known diameter of 1.08 μm) with p-toluenesulphonyl reactive surface groups were covalently conjugated to purified human Plg (MPIO-Plg) and eventually activated in the presence of uPA into MPIO-Pln. (B) Representative images from immunolabeling studies demonstrating the presence of Plg at the surface of MPIO-Plg. (Scale Bar= 5 µm) (C) Corresponding quantification of the fluorescence intensity after Plg immunolabeling in MPIO-Plg and control MPIO-BSA. Results are representative of n=3 independent experiments. (D) Representative LSCM imaging of MPIO-Plg and MPIO-Pln after 30 minutes of incubation in the presence of a fluorogenic plasmin substrate. Although significant cleavage of the substrate occurred (diffuse red fluorescence), it was not possible to reliably detect plasmin activity at the surface of artificial MPIO-Pln. (Scale Bar= 10 µm) (E) Corresponding quantification of the substrate fluorescence in the supernatant after 30 minutes of incubation (n=3 per group). (F) and (G) Schematic representations of the LSCM imaging results between proteolytic MPIO (artificial) and proteolytic MPs (cell-derived). Retention of the plasmin substrate along biological membranes immediately after cleavage may explain the ability of nano-zymography by LSCM to detect plasmin activity in situ.
LSCM of immobilized MPs allows the detection of circulating tPA-bearing MPs in ischemic stroke mice
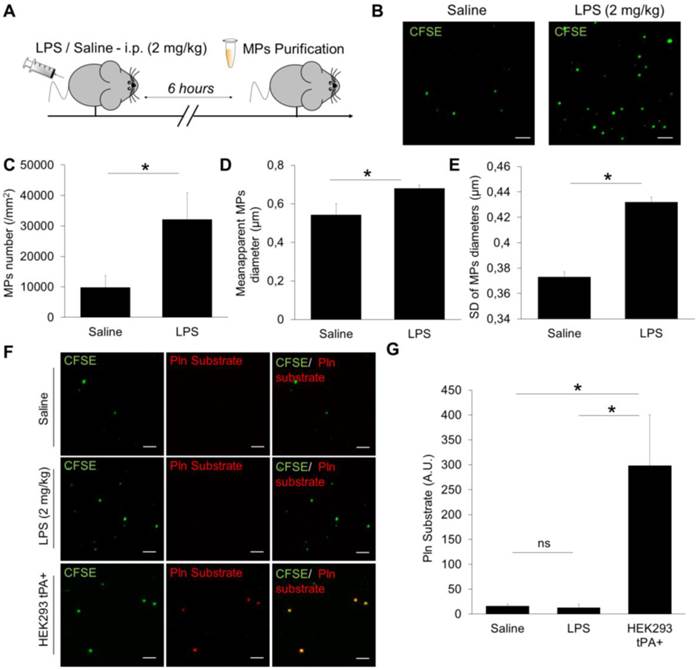
In order to investigate the feasibility of detecting endogenous proteolytic MPs using nano-zymography, we obtained plasma samples from two pathological conditions where fibrinolytic MPs could be generated. First, in mice with LPS-induced systemic inflammation (an experimental model for sepsis), and second in mice after thromboembolic stroke with or without thrombolytic treatment. MPs were purified by ultracentrifugation, fluorescently labelled using CFSE, immobilized on annexin-V coated micro-wells and detected by high resolution LSCM.
In mice (Figure 8A), the number of detected MPs increased by ~300% after LPS treatment as compared to control saline-treated mice (Figure 8B and C). Interestingly, these LPS-induced MPs were also larger (Figure 8D) and their sizes were more heterogeneous than MPs from control mice (Figure 8E). These results demonstrated that the developed method of MPs imaging is able to detect MPs from murine plasma and characterize their morphology. However, we failed to detect any plasmin-dependent proteolytic activity of the purified MPs in either control or LPS-treated mice, as compared to a positive control (HEK293 tPA-MPs) (Figure 8F and G).
Detection and characterization of plasma MPs in an experimental model of systemic inflammation in mice. (A) Schematic representation of the experimental design of systemic inflammation (LPS administration at 2 mg/kg i.p), known to trigger plasmatic MPs generation. MPs were purified from plasma samples obtained 6 hours after saline/LPS administration. (B) Representative images of MPs (CFSE, green) from saline (control) and LPS-treated mice. (C) Quantification of plasmatic MPs number from controls (saline) and LPS-treated mice (n=4 per group). (D) Quantification of apparent MPs diameter in both groups, showing that MPs coming from LPS-treated mice are slightly larger than those from control mice. (E) Mean SD of MPs diameter, showing that MPs in LPS-treated mice are more heterogeneous in size than in control mice. (F) In order to study the possible generation of fibrinolytic MPs after systemic inflammation, we performed analyses of proteolytic activity (plasmin) from purified MPs in both control and LPS-treated mice (nano-zymography). Representative images of cleaved plasmin fluorescent substrate (Pln substrate, red) in MPs (CFSE, green) from control and LPS-treated mice in the presence of exogenous plasminogen (2 µM). No signal of cleaved substrate was detected in both conditions, as compared to the positive control (HEK-derived tPA-MPs). (G) Corresponding quantification (n=5 per group). Data are means ± SD. (Bars, 10µm). Statistical analyses were performed Kruskal-Wallis (for multiple comparisons) followed by Mann-Whitney's U-test.
Then, we induced stroke in mice by in situ intra-arterial injection of thrombin, which leads to the formation of an intracranial thrombus (Figure 9A) leading to a superficial ischemic stroke in the middle cerebral artery territory, as it occurs frequently in humans. Twenty-minutes after ischemic onset, the mice received an infusion of either saline or recombinant tPA. We performed MRI 1 hour after stroke onset, to ensure the success of the procedure, and start blood sampling (Figure 9B). As shown in Figure 9C, stroke mice presented an occlusion of the right middle cerebral artery, leading to an acute ischemic stroke (visible on diffusion weighted images but not on T2-weighted imaging, corresponding to the actual clinical definition of acute ischemic stroke). We purified the circulating MPs from the blood (30 minutes after the end of thrombolysis) and measured their concentrations and their sizes in three different experimental groups: control mice, stroke mice and stroke mice that received thrombolytic therapy. As shown on Figure 9D-F, stroke mice presented higher MP levels in their blood than control mice. When stroke mice received tPA, this concentration was even higher. To investigate whether the generated MPs displayed tPA on their surface, we performed immunolabeling coupled to LSCM to measure the concentration of tPA-MPs in the different experimental groups. We found that, tPA administration in ischemic stroke mice leads to the formation of tPA-MPs (Figure 9G-H), that can be readily identified on LSCM images.
Detection and characterization of plasma MPs in an experimental model of ischemic stroke in mice. (A) Schematic representation of the experimental stroke model induced by intra-arterial injection of thrombin. (B) Timeline of the experiments. MPs were purified from plasma samples obtained 30 minutes after the end of saline or tPA infusion. (C) Representative T2-weighted (T2W), Diffusion-weighted (DWI) and magnetic resonance angiography (MRA) images obtained in control mice and 1 hour after ischemic onset in a saline treated mouse. The yellow arrowhead represents the ischemic core, only visible on DWI and not in T2W images, which is a characteristic of an acute ischemic lesion. On the MRA, the left middle cerebral artery is circled in green and the right in red. In the stroke mouse, we cannot see anymore the right middle cerebal artery which is occluded by a thrombin-triggered thrombus. (D) Representative LSCM images of MPs (CFSE, green) from control, stroke and stroke+tPA mice. (E) Quantification of plasmatic MPs number from from control, stroke and stroke+tPA mice (n=5 per group). (F) Quantification of the apparent MPs diameter in the three groups, showing the lack of significant differences (n=5 per group). (G) Representative images of anti-tPA immunolabelled MPs from a tPA-treated stroke mouse showing numerous tPA positive (arrowhead) and tPA negative (arrow) MPs in this mouse. (H) Corresponding quantification (n=5 per group). Data are means ± SD. Statistical analyses were performed Kruskal-Wallis (for multiple comparisons) followed by Mann-Whitney's U-test.
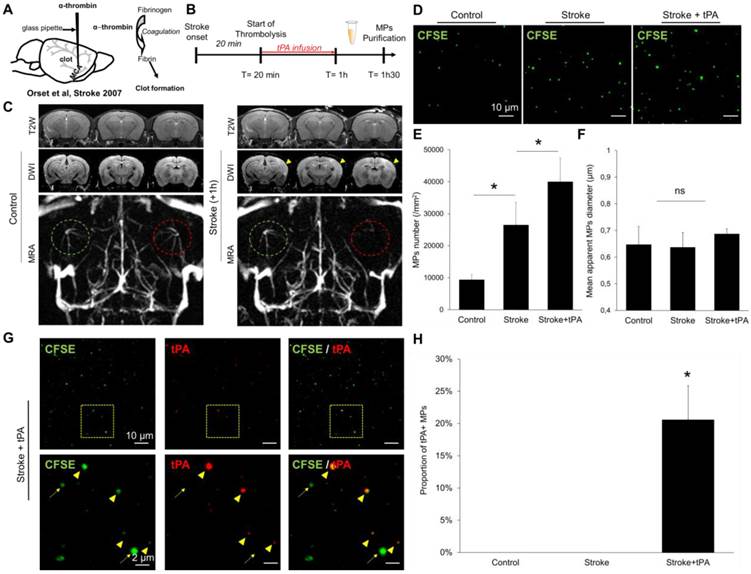
Altogether, these additional experiments demonstrate that, in a murine model of acute ischemic stroke, tPA administration leads to the formation of tPA-MPs. Interestingly, these tPA-MPs were still present in the blood 30 minutes after the end of the thrombolytic treatment, whereas the plasmatic half-life of tPA is <5 minutes in mice (we previously demonstrated that there is no detectable plasmatic tPA activity 30 minutes after tPA administration in the same mouse strain [39]). This suggests that the tPA binding to the surface of MPs allows to prolong its plasmatic half-life.
LSCM of immobilized MPs allows the detection of circulating fibrinolytic MPs in ischemic stroke patient
Given our results in the ischemic stroke model, we wanted to study by LSCM the circulating MPs from ischemic stroke patients who received thrombolytic treatment (intravenous injection of recombinant human tPA). Therefore, MPs were purified at different time-points post-stroke onset and analysed using LSCM. In this ischemic stroke patient cohort (Figure 10A), CFSE-labelled MPs imaging first revealed that the plasmatic concentration of MPs peaks at 1-2 hours after the start of thrombolytic treatment (Figure 10B and C). Immunofluorescence of tPA antigen on immobilized MPs revealed that a few number of tPA-MPs were present in the plasma of ischemic stroke patients before thrombolytic treatment (Figures 10B and C). Moreover, we observed an increase in the number of tPA-MPs after thrombolytic treatment which represented up to 60% of the total number of MPs at 2 hours (Figure 10D). Interestingly, these MPs were still present 1 hour after the end of tPA infusion (+2 h time point) when there is virtually no more exogenous tPA in the plasma (the half-life of tPA in the plasma in humans is <5 min). Nano-zymography revealed that these MPs are able to generate detectable amount of plasmin in situ (Figure 10E and F). This result was confirmed in a fibrin-agar zymography assay showing that the tPA contents of MPs from stroke patients is able to induce fibrin degradation (Additional File 1: Figure S5).
Detection and characterization of fibrinolytic MPs in ischemic stroke patients by nano-zymography. (A) Time-course of plasma samples collection in ischemic stroke patients that received thrombolytic therapy. MPs were purified from samples obtained at 0h-before, 1h, 2h, 12h and 24h after tPA infusion. (B) Representative images of MPs (CFSE, green) from stroke patients immunostained for tPA (red). (n=6) (C) Quantification of tPA immunoreactivity in MPs from patient 218 before (black) and 2h post-thrombolysis (grey). Red vertical bar indicates baseline value (mean + 2DS of control condition). (D) Quantification of total MPs (grey) and tPA-positive MPs (black) in stroke patients over time. Data are means ± SD. (n=6). (E) Representative images of cleaved plasmin fluorescent substrate (Pln substrate, red) in MPs (CFSE, green) from patient 218 in the presence of exogenous plasminogen (2 µM) before and 2h post-thrombolysis. (F) Quantification of cleaved substrate fluorescence in MPs from patient 218 before (black) and 2h post-thrombolysis (grey). (Bars, 10µm). The nonparametric Wilcoxon signed-rank test was used to examine differences in MPs concentration and MPs content in tPA in stroke patients over time.
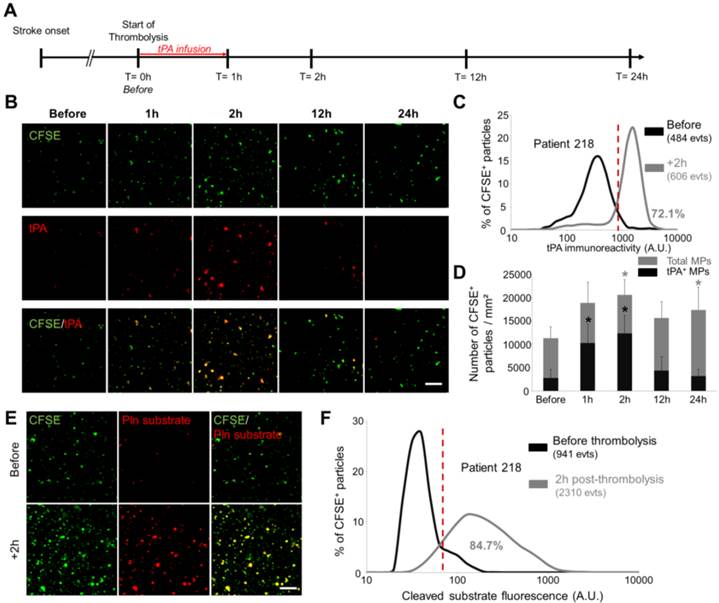
Altogether, these results in mice and humans illustrate the potential of LSCM imaging of immobilized MPs for experimental and clinical studies aiming at detecting and characterizing cell-derived MPs, especially regarding their fibrinolytic potential.
Discussion
In the present study, we developed an original method allowing imaging of cell-derived MPs at high resolution after immobilization on annexin-V coated micro-wells using LSCM. After trapping of MPs on the coated surface, we demonstrated that further characterizations such as antigen detection by fluorescent immunolabeling and measurement of proteolytic activity using fluorescent reporters are possible (Figure 11). Using this method, we were able, for the first time to our knowledge, to identify fibrinolytic MPs in ischemic stroke patients, therefore unveiling tPA-MPs as potential fibrinolytic players in acute ischemic stroke.
Schematic representation of the main methods that can be used after MPs binding to the micro-wells.
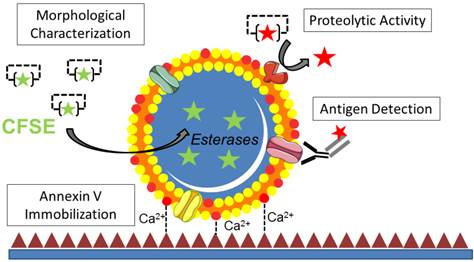
Among the available methods to detect MPs, high-resolution LSCM on immobilized MPs present several advantages. First, it allows direct visualisation and rapid assessment of the morphology and number of the immobilized MPs. Second, sample processing is fast and straightforward to perform and imaging requires only small sample volumes (MPs from 20 µL of plasma were used in Figures 7 and 8). This last characteristic could be of particular interest when analysing small volume samples, such as cerebrospinal fluid. Moreover, the present method can benefit from the recent advances in optical imaging including nanoscopy (such as stimulated emission depletion microscopy, STED)[40], brighter imaging probes (such as quantum dots) and combination of both[41]. These improvements could theoretically allow detecting even smaller vesicles (resolution limit ~50 nm) with lower antigen density (single molecule imaging on MPs could be achieved using quantum dots).
The ability to detect proteolytic activity of individual MPs is also unique to the present method. Since we demonstrated that proteases present at the surface of MPs can be inhibited by circulating inhibitors, the ability to measure activity rather than concentration provide unique information about their potential biological effects. Moreover, multiplexed fluorescent LSCM could allow, on a single MP, to simultaneously characterize proteolytic activity and antigen exposure. This may prove especially useful to study membrane anchored proteases such as ADAM10, ADAM17 or membrane-type MMPs that are present on extracellular vesicles[42-44]. In fact, the described method can be adapted to virtually all proteases for which specific fluorogenic substrates can be developed[45], provided there is sufficient retention of the cleaved substrate by MPs. Among the methods that could be used to increase the sensitivity of the nano-zymography assay[46], we believe that optimisation of the fluorogenic substrate and longer incubation times will be the most efficient. In particular, the use of amplification procedures, such as polymer-based fluorochrome[47], the use of aptamer exonuclease protection and exonuclease III (Exo III)-assisted recycling amplification-responsive cascade ZnPPIX/G-quadruplex supramolecular fluorescent labels[48] or carbon nanotube-enhanced polarization of fluorescent peptides[49], may be of great value to detect MP-associated proteases.
Among the limits of this method, two appears particularly important: first, the size of the detected particles is limited by the resolution of confocal microscopy (~200 nm). The use of super-resolution microscopy (such as STED or STORM) could solve this issue[40]. Second, only PS positive extracellular vesicles can be isolated using annexin-V coating[36]. One possibility is the use of duramycin as an immobilizing agent, which is able to bind phosphatidylethanolamine (PE), another phospholipid which is externalized on MPs membrane [50, 51]. Interestingly, there is a mismatch in the populations that present PS and PE on their surface, therefore, to further increase the percentage of immobilized MPs, a double coating using both annexin V and duramycin could be used. On the other hand, the ability to select the subpopulation of MPs that will be immobilized could be interesting for diagnostic purposes: for instance, coating with anti-T cell antibodies (such as anti-CD3) could be used as a mean to investigate membrane markers specifically on these cells, without requiring co-labelling with multiple secondary antibodies. Comparison of the morphological results obtained with the LSCM after CFSE staining to the results obtained by more classical methods (such as MP-optimized flow-cytometry) remains also to be performed.
The results obtained in the LPS model show that after systemic inflammation there is an increased number of immobilized MPs on the Annexin-V coated surface. Therefore, there are two main possible explanations: either the actual concentration of MPs in the blood samples does not change, but there is a higher annexin-V exposure on their surfaces. Or, the actual number of MPs increases. As previously reported, LPS induces the release of PS+ MPs from leucocytes[52] and endothelial cells[53], two important sources of circulating MPs in vivo. Moreover, LPS treated mice have increased thrombin activation, leading to platelet activation, thereby inducing the release of platelet-derived PS+ MPs[54]. Altogether, these results suggest that after LPS administration, both the number of MPs and the fraction that presents PS on their surface are increased.
We mainly focused our study on fibrinolytic MPs that we and other recently described in vitro and in humans[10, 12, 14]. In the present study, we demonstrated in particular that the number of fibrinolytic MPs significantly increases after tPA-administration in ischemic stroke patients. This adds to the numerous roles of tPA that have been described in acute ischemic stroke.[55] A similar phenomenon has been described for APC since recombinant APC treatment in patients with sepsis significantly increases circulating APC-MPs[15, 16, 56]. Regarding tPA-MPs, the most likely explanation is that recombinant tPA administration induces MPs release that are themselves able to bind tPA, thereby leading to the formation of new tPA-MPs. Interestingly, this mechanism seems to prolong tPA plasmatic half-life, since significant amounts of tPA were still detectable at the surface of MPs one hour after the end of thrombolysis despite a plasmatic half-life for tPA in humans of 5 minutes. The fibrinolytic activity of these tPA-MPs may be clinically relevant since exogenous tPA administration saturates inhibition systems (such as PAI-1, Protease Nexin-1, alpha2-macroglobulin) thus potentially enhancing the tPA activity on the surface of MPs[57]. Although we demonstrated that the proteolytic activity tPA- and uPA-MPs can be inhibited by PAI-1, it remains to be investigated whether the binding of plasminogen activators to a MP surface provides a relative protection to serpins compared to their soluble counterparts. Further studies will be needed to understand how tPA administration generates MPs, what is the receptor mediating the binding of tPA to MPs and what are the roles of these fibrinolytic MPs[58-60]. Moreover, precise quantitative measurement of tPA activity on the surface of MPs could be of diagnostic value. We are currently planning to investigate whether the number and activity of tPA-MPs before, during and after thrombolysis are associated with stroke outcome (including the risk of haemorrhagic transformation).
The presence of circulating tPA+ MPs in stroke patients before thrombolysis should be further confirmed, using confocal microscopy by comparing the tPA content of MPs from stroke patients to MPs from control patients, free of stroke. If confirmed, it would suggest that these patients present an underlying chronic pathological process leading to the formation of fibrinolytic MPs. Because we previously demonstrated that the majority of tPA+ MPs in the plasma comes from endothelial cells (and uPA+ MPs from leucocytes), the main hypothesis is that chronically activated endothelial cells generate MPs that bear tPA on their surface. However, this remains to be definitively proven by MPs subgroup analysis and further case-control studies. Moreover, later time-points after stroke onset could be of interest, especially to evaluate the contribution of post-stroke neuroinflammation to the generation of circulating MPs.
Overall, the present method allows to functionally characterize cell-derived MPs, especially in terms of antigen exposure and proteolytic activity (nano-zymography). Since a growing body of evidence suggests that proteolytic MPs play a role in numerous biological processes, the new information provided by nano-zymography over classical methods of MPs characterization may be of high diagnostic and prognostic value.
Supplementary Material
Additional File 1Figures S1-S5.
Movie S1.
Movie S2.
Acknowledgements
This work was supported by the Institut National de la Santé Et de la Recherche Médicale, the Conseil Régional de Basse-Normandie, the Eranet-ProteA Program (2011-2014) and the L'Oréal-UNESCO For Women in Science French Fellowship Program.
Conflict of Interest Statement
The authors have declared that no conflict of interest exists.
References
1. Yanez-Mo M, Siljander PR, Andreu Z, Zavec AB, Borras FE, Buzas EI. et al. Biological properties of extracellular vesicles and their physiological functions. Journal of extracellular vesicles. 2015;4:27066
2. Rothmeier AS, Marchese P, Petrich BG, Furlan-Freguia C, Ginsberg MH, Ruggeri ZM. et al. Caspase-1-mediated pathway promotes generation of thromboinflammatory microparticles. The Journal of clinical investigation. 2015;125:1471-84
3. Mause SF, Weber C. Microparticles: protagonists of a novel communication network for intercellular information exchange. Circulation research. 2010;107:1047-57
4. Owens AP 3rd, Mackman N. Microparticles in hemostasis and thrombosis. Circulation research. 2011;108:1284-97
5. Punyadee N, Mairiang D, Thiemmeca S, Komoltri C, Pan-Ngum W, Chomanee N. et al. Microparticles provide a novel biomarker to predict severe clinical outcomes of dengue virus infection. Journal of virology. 2015;89:1587-607
6. Sarlon-Bartoli G, Bennis Y, Lacroix R, Piercecchi-Marti MD, Bartoli MA, Arnaud L. et al. Plasmatic level of leukocyte-derived microparticles is associated with unstable plaque in asymptomatic patients with high-grade carotid stenosis. Journal of the American College of Cardiology. 2013;62:1436-41
7. Thomashow MA, Shimbo D, Parikh MA, Hoffman EA, Vogel-Claussen J, Hueper K. et al. Endothelial microparticles in mild chronic obstructive pulmonary disease and emphysema. The Multi-Ethnic Study of Atherosclerosis Chronic Obstructive Pulmonary Disease study. American journal of respiratory and critical care medicine. 2013;188:60-8
8. Loyer X, Vion AC, Tedgui A, Boulanger CM. Microvesicles as cell-cell messengers in cardiovascular diseases. Circulation research. 2014;114:345-53
9. Tian Y, Salsbery B, Wang M, Yuan H, Yang J, Zhao Z. et al. Brain-derived microparticles induce systemic coagulation in a murine model of traumatic brain injury. Blood. 2015;125:2151-9
10. Lacroix R, Dignat-George F. Microparticles: new protagonists in pericellular and intravascular proteolysis. Seminars in thrombosis and hemostasis. 2013;39:33-9
11. Canault M, Leroyer AS, Peiretti F, Leseche G, Tedgui A, Bonardo B. et al. Microparticles of human atherosclerotic plaques enhance the shedding of the tumor necrosis factor-alpha converting enzyme/ADAM17 substrates, tumor necrosis factor and tumor necrosis factor receptor-1. The American journal of pathology. 2007;171:1713-23
12. Dejouvencel T, Doeuvre L, Lacroix R, Plawinski L, Dignat-George F, Lijnen HR. et al. Fibrinolytic cross-talk: a new mechanism for plasmin formation. Blood. 2010;115:2048-56
13. Martinez de Lizarrondo S, Roncal C, Calvayrac O, Rodriguez C, Varo N, Purroy A. et al. Synergistic effect of thrombin and CD40 ligand on endothelial matrix metalloproteinase-10 expression and microparticle generation in vitro and in vivo. Arteriosclerosis, thrombosis, and vascular biology. 2012;32:1477-87
14. Lacroix R, Plawinski L, Robert S, Doeuvre L, Sabatier F, Martinez de Lizarrondo S. et al. Leukocyte- and endothelial-derived microparticles: a circulating source for fibrinolysis. Haematologica. 2012;97:1864-72
15. Perez-Casal M, Downey C, Fukudome K, Marx G, Toh CH. Activated protein C induces the release of microparticle-associated endothelial protein C receptor. Blood. 2005;105:1515-22
16. Perez-Casal M, Downey C, Cutillas-Moreno B, Zuzel M, Fukudome K, Toh CH. Microparticle-associated endothelial protein C receptor and the induction of cytoprotective and anti-inflammatory effects. Haematologica. 2009;94:387-94
17. Dolo V, D'Ascenzo S, Violini S, Pompucci L, Festuccia C, Ginestra A. et al. Matrix-degrading proteinases are shed in membrane vesicles by ovarian cancer cells in vivo and in vitro. Clinical & experimental metastasis. 1999;17:131-40
18. Graves LE, Ariztia EV, Navari JR, Matzel HJ, Stack MS, Fishman DA. Proinvasive properties of ovarian cancer ascites-derived membrane vesicles. Cancer research. 2004;64:7045-9
19. van der Pol E, Coumans F, Varga Z, Krumrey M, Nieuwland R. Innovation in detection of microparticles and exosomes. Journal of thrombosis and haemostasis: JTH. 2013;11(Suppl 1):36-45
20. Arraud N, Gounou C, Linares R, Brisson AR. A simple flow cytometry method improves the detection of phosphatidylserine-exposing extracellular vesicles. Journal of thrombosis and haemostasis: JTH. 2015;13:237-47
21. van der Pol E, Coumans FA, Grootemaat AE, Gardiner C, Sargent IL, Harrison P. et al. Particle size distribution of exosomes and microvesicles determined by transmission electron microscopy, flow cytometry, nanoparticle tracking analysis, and resistive pulse sensing. Journal of thrombosis and haemostasis: JTH. 2014;12:1182-92
22. Gauberti M, Martinez de Lizarrondo S, Orset C, Vivien D. Lack of secondary microthrombosis after thrombin-induced stroke in mice and non-human primates. Journal of thrombosis and haemostasis: JTH. 2014;12:409-14
23. Gakuba C, Gauberti M, Mazighi M, Defer G, Hanouz JL, Vivien D. Preclinical evidence toward the use of ketamine for recombinant tissue-type plasminogen activator-mediated thrombolysis under anesthesia or sedation. Stroke; a journal of cerebral circulation. 2011;42:2947-9
24. Le Behot A, Gauberti M, Martinez De Lizarrondo S, Montagne A, Lemarchand E, Repesse Y. et al. GpIbalpha-VWF blockade restores vessel patency by dissolving platelet aggregates formed under very high shear rate in mice. Blood. 2014;123:3354-63
25. Montagne A, Gauberti M, Macrez R, Jullienne A, Briens A, Raynaud JS. et al. Ultra-sensitive molecular MRI of cerebrovascular cell activation enables early detection of chronic central nervous system disorders. NeuroImage. 2012;63:760-70
26. Mendioroz M, Fernandez-Cadenas I, Rosell A, Delgado P, Domingues-Montanari S, Ribo M. et al. Osteopontin predicts long-term functional outcome among ischemic stroke patients. Journal of neurology. 2011;258:486-93
27. Gauberti M, Montagne A, Marcos-Contreras OA, Le Behot A, Maubert E, Vivien D. Ultra-sensitive molecular MRI of vascular cell adhesion molecule-1 reveals a dynamic inflammatory penumbra after strokes. Stroke; a journal of cerebral circulation. 2013;44:1988-96
28. Melemenidis S, Jefferson A, Ruparelia N, Akhtar AM, Xie J, Allen D. et al. Molecular magnetic resonance imaging of angiogenesis in vivo using polyvalent cyclic RGD-iron oxide microparticle conjugates. Theranostics. 2015;5:515-29
29. Jefferson A, Ruparelia N, Choudhury RP. Exogenous microparticles of iron oxide bind to activated endothelial cells but, unlike monocytes, do not trigger an endothelial response. Theranostics. 2013;3:428-36
30. Gauberti M, Montagne A, Quenault A, Vivien D. Molecular magnetic resonance imaging of brain-immune interactions. Frontiers in cellular neuroscience. 2014;8:389
31. Deutsch DG, Mertz ET. Plasminogen: purification from human plasma by affinity chromatography. Science (New York, NY). 1970;170:1095-6
32. Nieuwland R, Berckmans RJ, Rotteveel-Eijkman RC, Maquelin KN, Roozendaal KJ, Jansen PG. et al. Cell-derived microparticles generated in patients during cardiopulmonary bypass are highly procoagulant. Circulation. 1997;96:3534-41
33. Lacroix R, Sabatier F, Mialhe A, Basire A, Pannell R, Borghi H. et al. Activation of plasminogen into plasmin at the surface of endothelial microparticles: a mechanism that modulates angiogenic properties of endothelial progenitor cells in vitro. Blood. 2007;110:2432-9
34. Wang JG, Williams JC, Davis BK, Jacobson K, Doerschuk CM, Ting JP. et al. Monocytic microparticles activate endothelial cells in an IL-1beta-dependent manner. Blood. 2011;118:2366-74
35. Bernimoulin M, Waters EK, Foy M, Steele BM, Sullivan M, Falet H. et al. Differential stimulation of monocytic cells results in distinct populations of microparticles. Journal of thrombosis and haemostasis: JTH. 2009;7:1019-28
36. Connor DE, Exner T, Ma DD, Joseph JE. The majority of circulating platelet-derived microparticles fail to bind annexin V, lack phospholipid-dependent procoagulant activity and demonstrate greater expression of glycoprotein Ib. Thrombosis and haemostasis. 2010;103:1044-52
37. Belliere J, Martinez de Lizarrondo S, Choudhury RP, Quenault A, Le Béhot A, Delage C. et al. Unmasking Silent Endothelial Activation in the Cardiovascular System Using Molecular Magnetic Resonance Imaging. Theranostics. 2015;5:1187-202
38. Lojda Z. The use of substrates with 7-amino-3-trifluoromethylcoumarine (AFC) leaving group in the localization of protease activities in situ. Acta histochemica. 1996;98:215-28
39. Lemarchand E, Gauberti M, Martinez de Lizarrondo S, Villain H, Repesse Y, Montagne A. et al. Impact of alcohol consumption on the outcome of ischemic stroke and thrombolysis: role of the hepatic clearance of tissue-type plasminogen activator. Stroke; a journal of cerebral circulation. 2015;46:1641-50
40. Hell SW, Wichmann J. Breaking the diffraction resolution limit by stimulated emission: stimulated-emission-depletion fluorescence microscopy. Optics letters. 1994;19:780-2
41. Hanne J, Falk HJ, Gorlitz F, Hoyer P, Engelhardt J, Sahl SJ. et al. STED nanoscopy with fluorescent quantum dots. Nature communications. 2015;6:7127
42. Bertram A, Lovric S, Engel A, Beese M, Wyss K, Hertel B. et al. Circulating ADAM17 Level Reflects Disease Activity in Proteinase-3 ANCA-Associated Vasculitis. Journal of the American Society of Nephrology: JASN. 2015
43. Lee JH, Wittki S, Brau T, Dreyer FS, Kratzel K, Dindorf J. et al. HIV Nef, paxillin, and Pak1/2 regulate activation and secretion of TACE/ADAM10 proteases. Molecular cell. 2013;49:668-79
44. Taraboletti G, D'Ascenzo S, Borsotti P, Giavazzi R, Pavan A, Dolo V. Shedding of the matrix metalloproteinases MMP-2, MMP-9, and MT1-MMP as membrane vesicle-associated components by endothelial cells. The American journal of pathology. 2002;160:673-80
45. Vandooren J, Geurts N, Martens E, Van den Steen PE, Opdenakker G. Zymography methods for visualizing hydrolytic enzymes. Nature methods. 2013;10:211-20
46. Lee S, Kim K. Protease activity: meeting its theranostic potential. Theranostics. 2012;2:125-6
47. Wosnick JH, Mello CM, Swager TM. Synthesis and application of poly(phenylene ethynylene)s for bioconjugation: a conjugated polymer-based fluorogenic probe for proteases. Journal of the American Chemical Society. 2005;127:3400-5
48. Lv Y, Xue Q, Gu X, Zhang S, Liu J. A label-free fluorescence assay for thrombin based on aptamer exonuclease protection and exonuclease III-assisted recycling amplification-responsive cascade zinc(II)-protoporphyrin IX/G-quadruplex supramolecular fluorescent labels. The Analyst. 2014;139:2583-8
49. Huang Y, Hu K, Zhao S, Li M, Chen ZF, Lv Q. et al. Carbon nanotube-enhanced polarization of fluorescent peptides: a novel amplification strategy for homogeneous detection of proteases. Chemistry, an Asian journal. 2014;9:87-92
50. Wehman AM, Poggioli C, Schweinsberg P, Grant BD, Nance J. The P4-ATPase TAT-5 inhibits the budding of extracellular vesicles in C. elegans embryos. Current biology: CB. 2011;21:1951-9
51. Larson MC, Woodliff JE, Hillery CA, Kearl TJ, Zhao M. Phosphatidylethanolamine is externalized at the surface of microparticles. Biochimica et biophysica acta. 2012;1821:1501-7
52. Satta N, Toti F, Feugeas O, Bohbot A, Dachary-Prigent J, Eschwege V. et al. Monocyte vesiculation is a possible mechanism for dissemination of membrane-associated procoagulant activities and adhesion molecules after stimulation by lipopolysaccharide. Journal of immunology (Baltimore, Md: 1950). 1994;153:3245-55
53. Simak J, Holada K, Vostal JG. Release of annexin V-binding membrane microparticles from cultured human umbilical vein endothelial cells after treatment with camptothecin. BMC cell biology. 2002;3:11
54. Zhang Y, Meng H, Ma R, He Z, Wu X, Cao M. et al. Circulating Microparticles, Blood Cells and Endothelium Induce Procoagulant Activity in Sepsis Through Phosphatidylserine Exposure. Shock (Augusta, Ga). 2015
55. Vivien D, Gauberti M, Montagne A, Defer G, Touze E. Impact of tissue plasminogen activator on the neurovascular unit: from clinical data to experimental evidence. Journal of cerebral blood flow and metabolism: official journal of the International Society of Cerebral Blood Flow and Metabolism. 2011;31:2119-34
56. Perez-Casal M, Thompson V, Downey C, Welters I, Wyncoll D, Thachil J. et al. The clinical and functional relevance of microparticles induced by activated protein C treatment in sepsis. Critical care (London, England). 2011;15:R195
57. Declerck PJ, Gils A. Three decades of research on plasminogen activator inhibitor-1: a multifaceted serpin. Seminars in thrombosis and hemostasis. 2013;39:356-64
58. Herz J, Strickland DK. LRP: a multifunctional scavenger and signaling receptor. The Journal of clinical investigation. 2001;108:779-84
59. Doeuvre L, Plawinski L, Goux D, Vivien D, Angles-Cano E. Plasmin on adherent cells: from microvesiculation to apoptosis. The Biochemical journal. 2010;432:365-73
60. Zhu H, Fan X, Yu Z, Liu J, Murata Y, Lu J. et al. Annexin A2 combined with low-dose tPA improves thrombolytic therapy in a rat model of focal embolic stroke. Journal of cerebral blood flow and metabolism: official journal of the International Society of Cerebral Blood Flow and Metabolism. 2010;30:1137-46
Author contact
![]() Corresponding author: Sara Martinez de Lizarrondo, Inserm UMR-S U919, GIP Cyceron, Bd H. Becquerel, BP5229, 14074 Caen, France; e-mail: smartinezfr. phone/fax: 33231470160/+33231470222.
Corresponding author: Sara Martinez de Lizarrondo, Inserm UMR-S U919, GIP Cyceron, Bd H. Becquerel, BP5229, 14074 Caen, France; e-mail: smartinezfr. phone/fax: 33231470160/+33231470222.