Impact Factor
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Subjects and Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
Conflicts of Interest
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2016; 6(5):638-649. doi:10.7150/thno.14596 This issue Cite
Research Paper
Spectrum and Classification of ATP7B Variants in a Large Cohort of Chinese Patients with Wilson's Disease Guides Genetic Diagnosis
Yi Dong1*, Wang Ni1*, Wan-Jin Chen2*, Bo Wan3*, Gui-Xian Zhao4, Zhu-Qing Shi5, Yue Zhang4, Ning Wang2, Long Yu3, Jian-Feng Xu5, Zhi-Ying Wu1 ![]()
1. Department of Neurology and Research Center of Neurology in Second Affiliated Hospital, and the Collaborative Innovation Center for Brain Science, Zhejiang University School of Medicine, Hangzhou, China;
2. Department of Neurology and Institute of Neurology, First Affiliated Hospital, Fujian Medical University, Fuzhou, China;
3. State Key Laboratory of Genetic Engineering, Institutes of Biomedical Sciences, School of Life Science, Fudan University, Shanghai, China;
4. Department of Neurology and Institute of Neurology, Huashan Hospital, Shanghai Medical College, Fudan University, Shanghai, China;
5. Center for Genomic Translation and Health Intervention, School of Public Health, Fudan University, Shanghai, China.
* These authors contributed equally to this work.
Received 2015-12-3; Accepted 2016-1-27; Published 2016-3-3
Abstract

Background: Wilson's disease (WD) is an autosomal recessive disorder of copper metabolism caused by ATP7B pathogenic mutations. The symptoms of WD can be effectively prevented if the affected individuals are identified and intervened early. However, clinical utility of this molecular analysis is challenging due to hundreds of variants with various clinical effects in the gene. Here, we aim to describe the spectrum of ATP7B variants and assess their clinical effects in the Han Chinese population.
Methods: The ATP7B gene was directly sequenced in 632 unrelated WD patients and 503 unrelated healthy individuals. The effects of identified variants were classified according to the American College of Medical Genetics and Genomics (ACMG) standards and guidelines. Different frequency of variants observed in both cases and controls were tested using Chi-square or Fisher's exact tests.
Results: We detected 161 non-synonymous variants in these 632 WD patients, 58 of which were novel. Among these variants, 78, 64, 8, 4, and 7 were classified as 'pathogenic variants', 'likely pathogenic variants', 'variants with uncertain significance', 'likely benign variants', and 'benign variants', respectively. Ninety percent (569/632) of these WD patients can be genetically diagnosed with two or more 'pathogenic' or 'likely pathogenic' variants. The 14 most common disease-causing variants were found at least once in 94% (537/569) of genetically diagnosed patients.
Conclusions: These data expand the spectrum of ATP7B variants and facilitate effective screening for ATP7B variants for early diagnosis of WD and development of individualized treatment regimens.
Keywords: Wilson's disease, ATP7B, variants classification.
Introduction
Wilson's disease (WD, OMIM #277900), or progressive hepatolenticular degeneration, is an autosomal recessive disease caused by pathogenic mutations within the ATP7B gene. 1-3ATP7B encodes a P-type ATPase that is involved in the transport of copper into the plasma protein ceruloplasmin (Cp) and in the excretion of copper from the liver. ATP7B protein malfunction leads to massive accumulation of copper in the liver, brain, and other tissues. The accumulation of copper in these areas can present a wide spectrum of symptoms, including liver cirrhosis; neuronal degeneration of the brain, particularly in the basal ganglia; Kayser-Fleischer (K-F) rings at the corneal limbus; and kidney damage.4 The incidence of WD is estimated to be 1 in 30,000 individuals, and the heterozygous carrier rate is about 1 in 90 among many ethnic groups.5 However, a recent study identified the genetic prevalence of WD as 1:7,026 in the United Kingdom by sequencing ATP7B in 1000 control subjects,6 much higher than the typically reported prevalence of WD of 1:30,000. In addition, WD is thought to be more frequent in Asian populations. The presumed prevalence of WD is approximately 1 in 3,000 in the Korean population7 and the expected frequency of WD is 1 in 5,400 in the Hong Kong Han Chinese population.8
WD is among a limited number of inherited diseases for which symptoms can be prevented if the affected individuals can be identified and intervened early. However, accurate and early clinical diagnoses are often difficult to make due to the wide array of phenotypic variations.9 While genetic analysis is feasible, its clinical utility has been limited due to more than 700 ATP7B reported variants with various clinical effects (https://portal.biobase-international.com/hgmd/pro/gene.php?gene=ATP7B, Human Gene Mutation Database Professional, access date: 20 October, 2015). Therefore, better understanding of the spectrum of ATP7B variants and the clinical effects of these variants are necessary for clinical application.
Here, we reported variants identified from a study that directly sequenced the ATP7B gene in 632 consecutively treated WD patients at three Chinese academic medical centers between 2004 and 2015, and 503 unrelated phenotypically normal individuals. The clinical effects of identified variants were classified according to the American College of Medical Genetics and Genomics (ACMG) standards and guidelines.
Subjects and Methods
Participants
Consecutive patients who sought for diagnosis and treatment of WD between January 15, 2004 and April 30, 2015 at the Departments of Neurology at three leading Chinese academic medical centers were recruited, including Second Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou; First Affiliated Hospital, Fujian Medical University, Fuzhou; Huashan Hospital, Fudan University, Shanghai. Each patient was examined by at least two senior neurologists, including medical history review and physical exams. The patients were clinically diagnosed with WD according to the diagnostic criteria described previously.4 Blood samples of 632 unrelated patients (346 males and 286 females, aged 9 to 56) who have average onset age of 18.5 ± 0.6 ranging from 3 to 48 years and 503 unrelated phenotypically normal controls with no known family history of WD were included in the study. Family history were detected in 16 out of 632 WD patients. All participants are of Han Chinese descent. The probands, or their legal guardians, provided informed consents. This study was approved by three institutional review boards.
Direct DNA Sequencing for Mutation Analysis of ATP7B
Genomic DNA was isolated from peripheral blood lymphocytes using a DNA isolation kit (Qiagen Inc, Valencia, CA). All 21 exons and their flanking regions as well as the 5'-untranslated region (UTR) and promoter regions of the ATP7B gene were sequenced. Exon 2, which is 1,234 base pair (bp) in length, was covered by three overlapping PCR fragments. The primer sequences, annealing temperatures, and sizes of the PCR products of exons 1, 2, 4, 16, and 18 were listed in Table S1. Information related to the remaining primers has been described previously.10 PCR amplification and direct sequencing were performed as previously reported.11 Obtained sequences were compared, and nucleotide changes were numbered according to their position in ATP7B mRNA (NM_000053). Novel variants were further confirmed by sequencing both the forward and reverse strands. The sequence variants were interpreted and classified according to ACMG Standards and Guidelines.12
Statistical Analysis
Different frequency of variants observed in both WD patients and controls was tested using Chi-square test or the Fisher's exact test (for variants with the expected number of subjects below 5). The Bonferroni correction was used to declare statistical significance.
Results
Variants identified in the ATP7B Gene
A total of 173 variants in the coding region and the adjacent splice sites of the ATP7B gene were identified among 632 unrelated WD patients and 503 controls, including 161 non-synonymous and 12 synonymous variants. Among the 161 non-synonymous variants, 150 were detected only in WD patients and 11 were detected in both WD patients and normal controls. To assess link of the latter group of variants with WD, we compared allele frequency for 10 of these 11 variants between WD patients and controls (one of the 11 variants, p.R778L, was excluded because it is a well-established pathogenic variant13). Three variants (p.I390V, p.T935M, and p.V1106I) were considerably more common in WD patients than controls and the differences reached a statistical significance level (p<0.005) after adjusting for 10 multiple tests (0.05/10) (Table 1). The estimated odds ratios (ORs) of these three variants greater than 10 (p.I390V, OR=11.26; p.T935M, OR=77.04; p.V1106I, OR=10.44). For the remaining 7 variants, the allele frequency was similar between cases and controls (p>0.005), with ORs close to 1.00. Homozygotes in controls were found for 5 of these 7 variants (Table 1).
The Allele Frequency of 10 Non-Synonymous Variants in 632 WD Patients and 503 Controls.
| Variants | Controls | WD Patients | Probabilities | OR(95%CI) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Wildtype | Heterozygote | Homozygote | Allele Frequency | Wildtype | Heterozygote | Homozygote | Allele Frequency | |||
| p.I390V | 502 | 1 | 0 | 0.001 | 619 | 12 | 1 | 0.011 | 0.003* | 11.256(1.478-85.743) |
| p.S406A | 146 | 223 | 134 | 0.488 | 153 | 271 | 208 | 0.544 | 0.044 | 1.249(1.058-1.474) |
| p.L456V | 150 | 240 | 113 | 0.463 | 157 | 304 | 171 | 0.511 | 0.085 | 1.211(1.026-1.430) |
| p.R832K | 339 | 59 | 105 | 0.267 | 441 | 47 | 144 | 0.265 | 0.045 | 0.988(0.819-1.192) |
| p.I929V | 488 | 15 | 0 | 0.015 | 624 | 8 | 0 | 0.006 | 0.042 | 0.421(0.178-0.997) |
| p.T935M | 502 | 1 | 0 | 0.001 | 543 | 88 | 1 | 0.071 | 1.40E-22* | 77.044(10.716-553.910) |
| p.R952K | 164 | 261 | 78 | 0.415 | 206 | 335 | 91 | 0.409 | 0.862 | 0.978(0.826-1.157) |
| p.V1106I | 502 | 1 | 0 | 0.001 | 619 | 13 | 0 | 0.010 | 0.0049* | 10.444(1.364-79.969) |
| p.V1140A | 195 | 240 | 68 | 0.374 | 258 | 286 | 88 | 0.366 | 0.706 | 0.965(0.813-1.146) |
| p.V1297I | 498 | 5 | 0 | 0.005 | 628 | 4 | 0 | 0.003 | 0.520 | 0.636(0.17-2.373) |
*Based on Fisher's Exact test due to small observed number of variants.
Among 12 synonymous variants, 5 were novel, including c.2145c>T (p.Y715Y), c.3243G>A (p.E1081E), c.3861C>G (p.G1287G), c.4014T>A (p.I1338I), c.4194c>T (p.S1398S). The remaining variants have been previously identified (http://asia.ensembl.org/Homo_sapiens/Transcript/Sequence_cDNA?db=core;g=ENSG00000123191;r=13:51932673-52011494;t=ENST00000242839, access date: 20, October, 2015), including c.747G>A (p.L249L, rs554554415), c.1620c>T (p.L540L,rs145798966), c.2292c>T (p.F764F, rs372979339), c.2310C>G (p.L770L, rs398123136), c.2973G>A (p.T991T, rs1801246), c.3009G>A (p.A1003A, rs1801247) and c.4251A>G (p.T1417T, rs546721020).
Classification of Variants
Among 161 non-synonymous variants, 7 were similar between cases and controls (Table 1), thus classified as benign variants according to the ACMG Standards and Guidelines.12 The remaining 154 non-synonymous variants were only observed in WD patients or significantly more frequent in WD patients (Table 2). They are distributed throughout the ATP7B gene exons 1 to 21 (Figure 1). Among these 154 non-synonymous variants, 18 (11.7%) are nonsense variants, 25 (16.2%) are small deletions or insertions, 11 (7.1%) are splice site variants, and 100 (64.9%) are missense variants. According to the ACMG Standards and Guidelines,12 18 nonsense variants can be classified as 'pathogenic variants'. Among 25 small deletions/insertions, three shift variants including c.2316_2317insCTCTTTGTG (p.V772insLFV), c.2790_2792del(p.I930del) and c.4005_4006insTTATAATGGGTTGCG (p.G1335insLXWVA) can only be classified as 'likely pathogenic variants' due to its in-frame characteristics, other 22 ones can be classified as 'pathogenic variants' as well. For the 11 splice site mutations, six (c.51+1g>a, c.1543+1g>t, c.1708-1g>c, c.1870-2a>g, c.2356-1g>c, and c.3557-2a>g) are predicted to result in exon skipping and lead to the production of a defective protein, therefore can also be classified as 'pathogenic variants'. For the other five splice site variants (c.1543+4a>g, c.1708-5t>g, c.1946+5g>a, c.2447+5g>t, and c.3903+5g>a), because additional functional analysis is required to assess their impact on RNA and protein, they can be classified as 'variants with uncertain significance' at this stage. For the 100 missense variants (Table 3), 32, 61, 4, and 3 can be classified as 'pathogenic variants', 'likely pathogenic variants'', 'likely benign variants'', and 'variants with uncertain significance', respectively, based on results of SIFT, PolyPhen-2, 1000 Genomes Project, Exome Aggregation Consortium and the ACMG Standards and Guidelines. In total, among 161 non-synonymous variants, 78 are classified as 'pathogenic variants', 64 as 'likely pathogenic variants', 8 as 'variants with uncertain significance', 4 as 'likely benign variants' and 7 as 'benign variants'. Therefore, 142 variants could be considered as potential disease-causing variants ('pathogenic variants' and 'likely pathogenic variants') at this stage.
One Hundred and Fifty-Four Non-Synonymous Variants were Only Observed in WD Patients or Significantly More Frequent in WD Patients.
| Mutation analysis | Domain | Frequency of MU (%) | No. of patients | |||
|---|---|---|---|---|---|---|
| Nucleotide mutation | Protein alteration | Exon | MU/MU | WT/MU | ||
| c.51+1g>a | Na | 1 | before Cu1 | 0.08 | 0 | 1 |
| c.268_271del | p.K90FfsX10 | 2 | Cu2 | 0.08 | 0 | 1 |
| c.314C>A | p.S105X | 2 | Cu2 | 0.08 | 0 | 1 |
| c.367delG | p. A123PfsX30 | 2 | Cu2 | 0.08 | 0 | 1 |
| c.525dupA | p.V176SfsX28 | 2 | Cu2 | 1.50 | 1 | 17 |
| c.588C>A | p.D196E | 2 | Cu2 | 0.40 | 0 | 5 |
| c.592A>G | p.R198G | 2 | Cu2 | 0.08 | 0 | 1 |
| c.695delC | p.P232QfsX30 | 2 | Cu3 | 0.08 | 0 | 1 |
| c.994G>T | p.E332X | 2 | Cu3/Cu4 | 0.55 | 0 | 7 |
| c.1168A>G | p.I390V | 2 | Cu3/Cu4 | 1.11 | 1 | 12 |
| c.1403_1416del | p.A468GfsX33 | 3 | Cu4/Cu5 | 0.08 | 0 | 1 |
| c.1426G>A | p.A476T | 3 | Cu4/Cu5 | 0.08 | 0 | 1 |
| c.1449_1456del | p.R483SfsX20 | 3 | Cu5 | 0.16 | 0 | 2 |
| c.1470C>A | p.C490X | 3 | Cu5 | 0.24 | 0 | 3 |
| c.1531C>T | p.Q511X | 3 | Cu5 | 2.37 | 2 | 26 |
| c.1543+1g>t | Na | 3 | Cu5 | 0.47 | 0 | 6 |
| c.1543+4a>g | Na | 3 | Cu5 | 0.08 | 0 | 1 |
| c.1544G>T | p.G515V | 4 | Cu5 | 0.08 | 0 | 1 |
| c.1545delT | p.G515GfsX9 | 4 | Cu5 | 0.08 | 0 | 1 |
| c.1552_1553delTC | p.S518RfsX15 | 4 | Cu5 | 0.08 | 0 | 1 |
| c.1639C>T | p.Q547X | 4 | Cu5 | 0.08 | 0 | 1 |
| c.1708-5t>g | Na | 5 | Cu6 | 1.03 | 1 | 11 |
| c.1708-1g>c | Na | 5 | Cu6 | 1.50 | 2 | 15 |
| c.1760C>T | p.T587M | 5 | Cu6 | 0.08 | 0 | 1 |
| c.1782T>A | p.Y594X | 5 | Cu6 | 0.08 | 0 | 1 |
| c.1803delC | p.S602AfsX46 | 5 | Cu6 | 0.08 | 0 | 1 |
| c.1817T>G | p.V606G | 5 | Cu6 | 0.16 | 0 | 2 |
| c.1820dupA | p.F608VfsX2 | 5 | Cu6 | 0.16 | 0 | 2 |
| c.1846C>T | p.R616Y | 5 | Cu6 | 0.08 | 0 | 1 |
| c.1870-2a>g | na | 6 | Cu6 | 0.08 | 0 | 1 |
| c.1925A>G | p.D642G | 6 | Cu6 | 0.08 | 0 | 1 |
| c.1946+5g>a | na | 6 | Cu6/TM1 | 0.08 | 0 | 1 |
| c.1950G>A | p.W650Term | 7 | Cu6/TM1 | 0.08 | 0 | 1 |
| c.2038C>T | p.Q680X | 7 | Cu6/TM1 | 0.08 | 0 | 1 |
| c.2043delC | p.S681SfsX15 | 7 | TM2 | 0.08 | 0 | 1 |
| c.2078C>G | p.S693C | 7 | TM2 | 0.16 | 0 | 2 |
| c.2128G>A | p.G710S | 8 | TM2/TM3 | 0.08 | 0 | 1 |
| c.2156A>G | p.Y719C | 8 | TM2/TM3 | 0.08 | 0 | 1 |
| c.2157C>A | p.Y719X | 8 | TM2/TM3 | 0.24 | 0 | 3 |
| c.2192T>A | p.V731E | 8 | TM3 | 0.08 | 1 | 0 |
| c.2195T>C | p.L732P | 8 | TM3 | 0.08 | 0 | 1 |
| c.2223T>A | p.Y741X | 8 | TM3 | 0.08 | 0 | 1 |
| c.2251G>T | p.A751S | 8 | TM3 | 0.08 | 0 | 1 |
| c.2261A>G | p.E754G | 8 | TM3/TM4 | 0.08 | 0 | 1 |
| c.2267C>G | p.A756G | 8 | TM3/TM4 | 0.08 | 0 | 1 |
| c.2294A>G | p.D765G | 8 | TM4 | 0.24 | 0 | 3 |
| c.2297C>T | p.T766M | 8 | TM4 | 0.16 | 0 | 2 |
| c.2304dupC | p.M769HfsX26 | 8 | TM4 | 0.87 | 0 | 11 |
| c.2305A>G | p.M769V | 8 | TM4 | 0.08 | 0 | 1 |
| c.2308C>T | p.L770F | 8 | TM4 | 0.08 | 0 | 1 |
| c.2316_2317ins CTCTTTGTG | p.V772insLFV | 8 | TM4 | 0.08 | 0 | 1 |
| c.2332C>T | p.R778W | 8 | TM4 | 0.16 | 0 | 2 |
| c.2333G>T | p.R778L | 8 | TM4 | 29.67 | 64 | 247 |
| c.2333G>A | p.R778Q | 8 | TM4 | 1.98 | 3 | 19 |
| c.2336G>A | p.W779X | 8 | TM4 | 0.08 | 0 | 1 |
| c.2341G>A | p.E781K | 8 | TM4 | 0.08 | 0 | 1 |
| c.2356-1g>c | na | 9 | TM4/Td | 0.08 | 0 | 1 |
| c.2383C>T | p.L795F | 9 | TM4/Td | 0.08 | 0 | 1 |
| c.2390C>T | p.S797F | 9 | TM4/Td | 0.08 | 0 | 1 |
| c.2447+5g>t | Na | 9 | TM4/Td | 0.08 | 0 | 1 |
| c.2455C>T | p.Q819X | 10 | TM4/Td | 0.08 | 0 | 1 |
| c.2525A>G | p.D842G | 10 | Td | 0.08 | 0 | 1 |
| c.2587C>T | p.P863S | 11 | Td | 0.08 | 0 | 1 |
| c.2605G>A | p.G869R | 11 | Td | 0.16 | 0 | 2 |
| c.2620G>C | p.A874P | 11 | Td/TM5 | 0.08 | 0 | 1 |
| c.2621C>T | p.A874V | 11 | Td/TM5 | 3.56 | 3 | 39 |
| c.2648_2649del | p.V883AfsX3 | 11 | Td/TM5 | 0.08 | 0 | 1 |
| c.2662A>C | p.T888P | 11 | Td/TM5 | 0.32 | 0 | 4 |
| c.2668G>A | p.V890M | 11 | Td/TM5 | 0.16 | 0 | 2 |
| c.2740C>T | p.Q914X | 12 | Td/TM5 | 0.08 | 0 | 1 |
| c.2755C>G | p.R919G | 12 | Td/TM5 | 1.98 | 0 | 25 |
| c.2790_2792del | p.I930del | 12 | TM5 | 0.47 | 0 | 6 |
| c.2794_2795insGT | p.S932CfsX4 | 12 | TM5 | 0.08 | 0 | 1 |
| c.2804C>T | p.T935M | 12 | TM5 | 7.12 | 1 | 88 |
| c.2810delT | p.V937GfsX5 | 12 | TM5 | 0.63 | 0 | 8 |
| c.2827G>A | p.G943S | 12 | TM5 | 0.55 | 1 | 5 |
| c.2828G>A | p.G943D | 12 | TM5 | 2.21 | 1 | 26 |
| c.2848G>T | p.V950F | 12 | TM5/TM6 | 0.08 | 0 | 1 |
| c.2853_2856del | p.Q951HfsX15 | 12 | TM5/TM6 | 0.08 | 0 | 1 |
| c.2905C>T | p.R969W | 13 | TM6 | 0.08 | 0 | 1 |
| c.2924C>A | p.S975Y | 13 | TM6 | 0.79 | 0 | 10 |
| c.2930C>T | p.T977M | 13 | TM6 | 0.08 | 0 | 1 |
| c.2957C>T | p.S986F | 13 | TM6 | 0.08 | 0 | 1 |
| c.2975C>T | p.P992L | 13 | TM6/Ph | 14.56 | 26 | 132 |
| c.3007G>A | p.A1003T | 13 | TM6/Ph | 0.16 | 0 | 2 |
| c.3008C>T | p.A1003V | 13 | TM6/Ph | 0.08 | 0 | 1 |
| c.3010C>T | p.Q1004X | 13 | TM6/Ph | 0.08 | 0 | 1 |
| c.3029A>C | p.K1010T | 13 | TM6/Ph | 0.16 | 0 | 2 |
| c.3044T>C | p.L1015P | 13 | TM6/Ph | 0.08 | 0 | 1 |
| c.3053C>T | p.A1018V | 13 | TM6/Ph | 0.24 | 0 | 3 |
| c.3083A>G | p.K1028R | 14 | Ph | 0.08 | 0 | 1 |
| c.3087delT | p.G1030AfsX91 | 14 | Ph | 0.08 | 0 | 1 |
| c.3089G>A | p.G1030D | 14 | Ph | 0.40 | 0 | 5 |
| c.3095T>C | p.I1032T | 14 | Ph | 0.08 | 0 | 1 |
| c.3098C>T | p.T1033I | 14 | Ph | 0.08 | 0 | 1 |
| c.3104G>T | p.G1035V | 14 | Ph | 0.08 | 0 | 1 |
| c.3122G>C | p.R1041P | 14 | ATP loop | 0.16 | 1 | 0 |
| c.3121C>T | p.R1041W | 14 | ATP loop | 0.08 | 0 | 1 |
| c.3140A>T | p.D1047V | 14 | ATP loop | 0.24 | 1 | 1 |
| c.3155C>T | p.P1052L | 14 | ATP loop | 0.16 | 0 | 2 |
| c.3209C>G | p.P1070R | 14 | ATP loop | 0.08 | 0 | 1 |
| c.3221C>T | p.A1074V | 14 | ATP loop | 0.08 | 0 | 1 |
| c.3263T>C | p.L1088S | 15 | ATP loop | 0.32 | 0 | 4 |
| c.3271T>C | p.C1091R | 15 | ATP loop | 0.08 | 0 | 1 |
| c.3274A>C | p.T1092P | 15 | ATP loop | 0.08 | 1 | 0 |
| c.3311G>A | p.C1104Y | 15 | ATP loop | 0.08 | 0 | 1 |
| c.3316G>A | p.V1106I | 15 | ATP loop | 1.03 | 0 | 13 |
| c.3368C>T | p.P1123L | 15 | ATP loop | 0.08 | 0 | 1 |
| c.3377_3378delAC | p.H1126PfsX3 | 15 | ATP loop | 0.08 | 0 | 1 |
| c.3426G>C | p.Q1142H | 16 | ATP loop | 1.58 | 1 | 18 |
| c.3443T>C | p.I1148T | 16 | ATP loop | 3.32 | 1 | 40 |
| c.3446G>A | p.G1149E | 16 | ATP loop | 0.08 | 0 | 1 |
| c.3451C>T | p.R1151C | 16 | ATP loop | 0.08 | 0 | 1 |
| c.3451C>G | p.R1151G | 16 | ATP loop | 0.08 | 0 | 1 |
| c.3452G>A | p.R1151H | 16 | ATP loop | 0.16 | 0 | 2 |
| c.3459G>T | p.W1153C | 16 | ATP loop | 0.08 | 0 | 1 |
| c.3517G>A | p.E1173K | 16 | ATP loop | 0.79 | 1 | 8 |
| c.3532A>G | p.T1178A | 16 | ATP loop | 0.16 | 0 | 2 |
| c.3557-2a>g | na | 17 | ATP loop | 0.08 | 0 | 1 |
| c.3563T>G | p.L1188R | 17 | ATP loop | 0.08 | 0 | 1 |
| c.3587A>G | p.D1196G | 17 | ATP loop | 0.08 | 0 | 1 |
| c.3605C>G | p.A1202G | 17 | ATP loop | 0.08 | 0 | 1 |
| c.3646G>A | p.V1216M | 17 | ATP bind | 1.98 | 2 | 21 |
| c.3653T>C | p.L1218P | 17 | ATP bind | 0.08 | 0 | 1 |
| c.3700delG | p.V1234LfsX96 | 18 | ATP bind | 0.32 | 0 | 4 |
| c.3715G>T | p.V1239F | 18 | ATP bind | 0.24 | 0 | 3 |
| c.3733C>G | p.P1245A | 18 | ATP bind | 0.08 | 0 | 1 |
| c.3741C>G | p.H1247Q | 18 | ATP bind | 0.08 | 0 | 1 |
| c.3766_3767dupCA | p.Q1256PfsX75 | 18 | ATP bind | 0.08 | 0 | 1 |
| c.3776G>T | p.G1259V | 18 | ATP hinge | 0.08 | 1 | 1 |
| c.3791T>C | p.M1264T | 18 | ATP hinge | 0.08 | 0 | 1 |
| c.3802G>A | p.G1268R | 18 | ATP hinge | 0.08 | 0 | 1 |
| c.3809A>G | p.N1270S | 18 | ATP hinge | 2.22 | 0 | 28 |
| c.3818C>T | p.P1273L | 18 | ATP hinge | 0.08 | 0 | 1 |
| c.3818C>A | p.P1273Q | 18 | ATP hinge | 0.16 | 0 | 2 |
| c.3836A>G | p.D1279G | 18 | ATP hinge | 0.24 | 0 | 3 |
| c.3848C>T | p.A1283V | 18 | ATP hinge | 0.08 | 0 | 1 |
| c.3859G>A | p.G1287S | 18 | ATP hinge | 0.08 | 0 | 1 |
| c.3877G>A | p.E1293K | 18 | ATP hinge | 0.08 | 0 | 1 |
| c.3884C>T | p.A1295V | 18 | ATPhinge/TM7 | 0.40 | 0 | 5 |
| c.3896T>G | p.L1299R | 18 | ATP hinge/TM7 | 0.08 | 0 | 1 |
| c.3901_3902insA | p.R1301KfsX3 | 18 | ATP hinge/TM7 | 0.08 | 0 | 1 |
| c.3903+5g>a | na | 18 | ATP hinge/TM7 | 0.08 | 0 | 1 |
| c.3955C>T | p.R1319X | 19 | ATP hinge/TM7 | 0.24 | 0 | 3 |
| c.3982G>A | p.A1328T | 19 | TM7 | 0.16 | 0 | 2 |
| c.4003G>C | p.G1335R | 19 | TM7 | 0.24 | 0 | 3 |
| c.4005_4006insTTATAATGGGTTGCG | p.G1335insLXWVA | 19 | TM7 | 0.16 | 0 | 2 |
| c.4057T>C | p.W1353R | 20 | TM8 | 0.16 | 0 | 2 |
| c.4059G>A | p.W1353X | 20 | TM8 | 0.08 | 0 | 1 |
| c.4064G>A | p.G1355D | 20 | TM8 | 0.08 | 0 | 1 |
| c.4112T>C | p.L1371P | 20 | TM8 | 0.40 | 0 | 5 |
| c.4114C>T | p.Q1372X | 20 | TM8 | 0.63 | 0 | 8 |
| c.4162delG | p.A1388RfsX5 | 21 | after TM8 | 0.08 | 0 | 1 |
| c.4272T>G | p.Y1424X | 21 | after TM8 | 0.08 | 0 | 1 |
MU: mutant; WT: wild type; Cu: copper binding domain; Td: transduction domain converting energy from ATP hydrolysis to cation transportation; Tm: transmembrane domain; Ch: ion channel; Ph: phosphorylation loop.
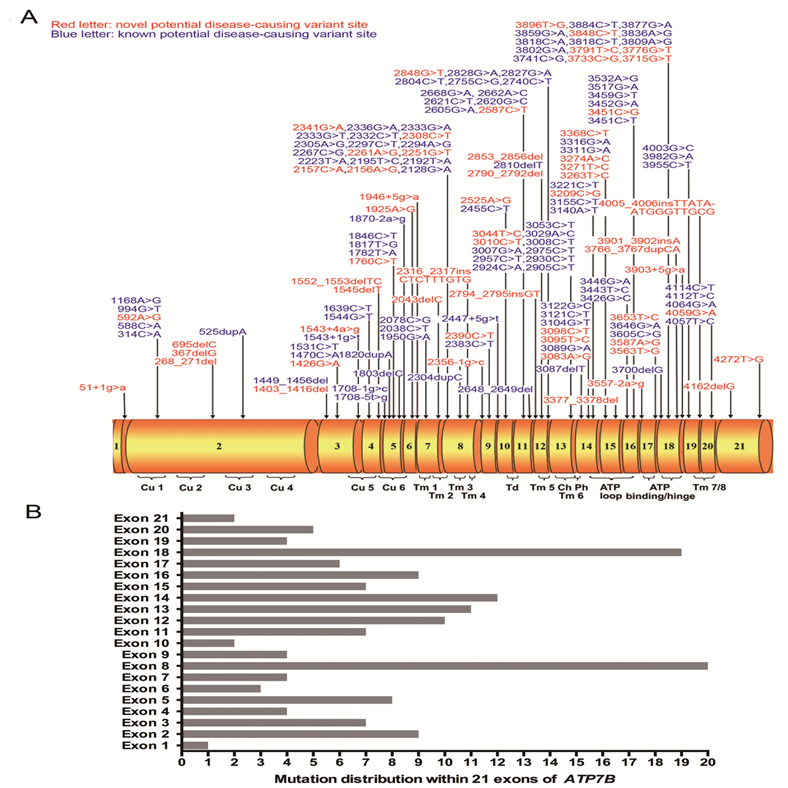
(A) Schematic representation of the ATP7B gene with 21 exons. All here identified variants in WD patients with Chinese Han descent are visualized in the corresponding ATP7B protein regions. Fifty-eight novel variants are indicated in red characters. Cu 1~6, six copper binding domains; Tm 1~8, eight transmembrane domains; Td, a transduction domain converting the ATP hydrolysis energy to the impetus of transporting copper cation; Ch, the transmembrane cation channel; Ph, the phosphorylation domain; ATP loop/binding/hinge, the ATP binding domain. (B) Distribution of ATP7B variants is shown within 21 exons of ATP7B gene.
Classified mutations within 100 missense mutations.
| Nucleotide mutation | Protein alteration | Exon | Sift | PolyPhen 2 | 1000g | ExAc | Classification | Evidence of pathogenicity | ||
|---|---|---|---|---|---|---|---|---|---|---|
| Score | Prediction | Score | Prediction | |||||||
| c.588C>A | p.D196E | 2 | 1 | Tolerated | 0.924 | Probably damaging | 0 | 5 | US | 4*PP |
| c.592A>G | p.R198G | 2 | 0.05 | Damaging | 0.994 | Probably damaging | 0 | 0 | LP | 1*PM,4*PP |
| c.1168A>G | p.I390V | 2 | 0.57 | Tolerated | 0.001 | Benign | 0 | 2 | LB | 1*BS,1*BP |
| c.1426G>A | p.A476T | 3 | 0.63 | Tolerated | 0.503 | Possibly damaging | 2 | 53 | US | 1*BS |
| c.1544G>T | p.G515V | 4 | 0 | Damaging | 1 | Probably damaging | 0 | 0 | P | 1*PS,2*PM,5*PP |
| c.1760C>T | p.T587M | 5 | 0.19 | Tolerated | 0.099 | Benign | 0 | 0 | LB | 1*BS,1*BP |
| c.1817T>G | p.V606G | 5 | 0 | Damaging | 1 | Probably damaging | 0 | 0 | LP | 1*PM,5*PP |
| c.1846C>T | p.R616Y | 5 | 0 | Damaging | 1 | Probably damaging | 0 | 4 | LP | 2*PM,5*PP |
| c.1925A>G | p.D642G | 6 | 0.01 | Damaging | 1 | Probably damaging | 0 | 0 | LP | 1*PM,4*PP |
| c.2078C>G | p.S693C | 7 | 0.01 | Damaging | 1 | Probably damaging | 0 | 0 | P | 1*PS,2*PM,5*PP |
| c.2128G>A | p.G710S | 8 | 0.02 | Damaging | 1 | Probably damaging | 0 | 0 | P | 1*PS,2*PM,5*PP |
| c.2156A>G | p.Y719C | 8 | 0 | Damaging | 0.991 | Probably damaging | 0 | 0 | LP | 2*PM,4*PP |
| c.2192T>A | p.V731E | 8 | 0 | Damaging | 1 | Probably damaging | 0 | 0 | P | 1*PS,2*PM,5*PP |
| c.2195T>C | p.L732P | 8 | 0 | Damaging | 1 | Probably damaging | 0 | 0 | P | 1*PS,2*PM,5*PP |
| c.2251G>T | p.A751S | 8 | 0.16 | Tolerated | 0.999 | Probably damaging | 0 | 0 | LP | 2*PM,3*PP |
| c.2261A>G | p.E754G | 8 | 0.34 | Tolerated | 0.999 | Probably damaging | 0 | 0 | LP | 2*PM,3*PP |
| c.2267C>G | p.A756G | 8 | 0.21 | Tolerated | 1 | Probably damaging | 0 | 0 | P | 1*PS,2*PM,5*PP |
| c.2294A>G | p.D765G | 8 | 0 | Damaging | 1 | Probably damaging | 0 | 0 | LP | 3*PM,5*PP |
| c.2297C>T | p.T766M | 8 | 0 | Damaging | 1 | Probably damaging | 0 | 1 | P | 1*PS,2*PM,5*PP |
| c.2305A>G c.2308C>T | p.M769V p.L770F | 8 8 | 00 | Damaging Damaging | 1 1 | Probably damaging Probably damaging | 00 | 8 0 | LP LP | 3*PM,5*PP 2*PM,4*PP |
| c.2332C>T | p.R778W | 8 | 0 | Damaging | 1 | Probably damaging | 0 | 5 | P | 2*PS,3*PM |
| c.2333G>A | p.R778Q | 8 | 0 | Damaging | 1 | Probably damaging | 0 | 5 | P | 2*PS,3*PM |
| c.2333G>T | p.R778L | 8 | 0 | Damaging | 1 | Probably damaging | 0 | 5 | P | 2*PS,3*PM |
| c.2341G>A | p.E781K | 8 | 0 | Damaging | 1 | Probably damaging | 0 | 0 | P | 1*PS,2*PM,4*PP |
| c.2383C>T | p.L795F | 9 | 0 | Damaging | 1 | Probably damaging | 0 | 4 | P | 1*PS,3*PM |
| c.2390C>T | p.S797F | 9 | 0 | Damaging | 1 | Probably damaging | 0 | 0 | LP | 1*PM,4*PP |
| c.2525A>G | p.D842G | 10 | 0 | Damaging | 1 | Probably damaging | 0 | 0 | LP | 1*PM,4*PP |
| c.2587C>T | p.P863S | 11 | 0 | Damaging | 1 | Probably damaging | 0 | 0 | LP | 1*PM,4*PP |
| c.2605G>A | p.G869R | 11 | 0 | Damaging | 1 | Probably damaging | 5 | 82 | LP | 2*PM,5*PP |
| c.2620G>C | p.A874P | 11 | 0.01 | Damaging | 1 | Probably damaging | 0 | 0 | LP | 2*PM,5*PP |
| c.2621C>T | p.A874V | 11 | 0.01 | Damaging | 1 | Probably damaging | 0 | 9 | LP | 2*PM,5*PP |
| c.2662A>C | p.T888P | 11 | 0 | Damaging | 1 | Probably damaging | 0 | 0 | LP | 2*PM,5*PP |
| c.2668G>A | p.V890M | 11 | 0.01 | Damaging | 1 | Probably damaging | 0 | 0 | LP | 2*PM,5*PP |
| c.2755C>G | p.R919G | 12 | 0.01 | Damaging | 0.988 | Probably damaging | 0 | 2 | P | 1*PS,2*PM,5*PP |
| c.2804C>T | p.T935M | 12 | 0 | Damaging | 1 | Probably damaging | 0 | 21 | P | 1*PS,1*PM,5*PP |
| c.2827G>A | p.G943S | 12 | 0.16 | Tolerated | 1 | Probably damaging | 0 | 2 | P | 1*PS,1*PM,4*PP |
| c.2828G>A | p.G943D | 12 | 0 | Damaging | 1 | Probably damaging | 0 | 2 | P | 1*PS,2*PM,5*PP |
| c.2848G>T | p.V950F | 12 | 0.01 | Damaging | 0.994 | Probably damaging | 0 | 0 | LP | 2*PM,4*PP |
| c.2905C>T | p.R969W | 13 | 0.02 | Damaging | 1 | Probably damaging | 0 | 5 | LP | 2*PM,5*PP |
| c.2924C>A | p.S975Y | 13 | 0.01 | Damaging | 1 | Probably damaging | 0 | 1 | LP | 2*PM,5*PP |
| c.2930C>T | p.T977M | 13 | 0 | Damaging | 1 | Probably damaging | 0 | 10 | P | 1*PS,2*PM,5*PP |
| c.2957C>T | p.S986F | 13 | 0 | Damaging | 1 | Probably damaging | 0 | 0 | LP | 2*PM,5*PP |
| c.2975C>T | p.P992L | 13 | 0 | Damaging | 1 | Probably damaging | 0 | 5 | P | 2*PS,3*PM |
| c.3007G>A | p.A1003T | 13 | 0 | Damaging | 1 | Probably damaging | 1 | 2 | LP | 2*PM,5*PP |
| c.3008C>T | p.A1003V | 13 | 0 | Damaging | 1 | Probably damaging | 0 | 7 | LP | 2*PM,5*PP |
| c.3029A>C | p.K1010T | 13 | 0 | Damaging | 1 | Probably damaging | 0 | 0 | P | 1*PS,2*PM,5*PP |
| c.3044T>C | p.L1015P | 13 | 0 | Damaging | 1 | Probably damaging | 0 | 0 | LP | 2*PM,4*PP |
| c.3053C>T | p.A1018V | 13 | 0.07 | Tolerated | 1 | Probably damaging | 0 | 3 | LP | 2*PM,5*PP |
| c.3083A>G | p.K1028R | 14 | 0.02 | Damaging | 0.998 | Probably damaging | 0 | 0 | LP | 2*PM,4*PP |
| c.3089G>A | p.G1030D | 14 | 0 | Damaging | 1 | Probably damaging | 0 | 0 | LP | 2*PM,5*PP |
| c.3095T>C | p.I1032T | 14 | 0 | Damaging | 0.998 | Probably damaging | 0 | 0 | LP | 2*PM,4*PP |
| c.3098C>T | p.T1033I | 14 | 0.01 | Damaging | 1 | Probably damaging | 0 | 0 | P | 1*PS,2*PM,4*PP |
| c.3104G>T | p.G1035V | 14 | 0 | Damaging | 1 | Probably damaging | 0 | 0 | LP | 2*PM,5*PP |
| c.3121C>T | p.R1041W | 14 | 0 | Damaging | 1 | Probably damaging | 0 | 5 | LP | 2*PM,5*PP |
| c.3122G>C | p.R1041P | 14 | 0.01 | Damaging | 1 | Probably damaging | 0 | 0 | LP | 2*PM,5*PP |
| c.3140A>T | p.D1047V | 14 | 0.13 | Tolerated | 0.997 | Probably damaging | 0 | 0 | LP | 2*PM,4*PP |
| c.3155C>T | p.P1052L | 14 | 0.06 | Tolerated | 0.998 | Probably damaging | 0 | 1 | LP | 2*PM,4*PP |
| c.3209C>G | p.P1070R | 14 | 0 | Damaging | 1 | Probably damaging | 0 | 0 | LP | 2*PM,4*PP |
| c.3221C>T | p.A1074V | 14 | 0 | Damaging | 1 | Probably damaging | 0 | 0 | LP | 2*PM,5*PP |
| c.3263T>C | p.L1088S | 15 | 0.2 | Tolerated | 1 | Probably damaging | 0 | 0 | LP | 1*PS,1*PM,3*PP |
| c.3271T>C | p.C1091R | 15 | 0 | Damaging | 0.96 | Probably damaging | 0 | 0 | LP | 1*PM,4*PP |
| c.3274A>C | p.T1092P | 15 | 0.11 | Tolerated | 0.832 | Possibly damaging | 0 | 0 | US | 1*PM,3*PP |
| c.3311G>A | p.C1104Y | 15 | 0 | Damaging | 1 | Probably damaging | 0 | 1 | P | 1*PS,2*PM,5*PP |
| c.3316G>A | p.V1106I | 15 | 0.15 | Tolerated | 0.984 | Probably damaging | 2 | 16 | P | 1*PS,2*PM,4*PP |
| c.3368C>T | p.P1123L | 15 | 0.31 | Tolerated | 0.025 | Benign | 0 | 25 | LB | 1*BS,1*BP |
| c.3426G>C | p.Q1142H | 16 | 0.16 | Tolerated | 0.007 | Benign | 0 | 3 | LB | 1*BS,1*BP |
| c.3443T>C | p.I1148T | 16 | 0 | Damaging | 0.999 | Probably damaging | 0 | 5 | LP | 1*PM,5*PP |
| c.3446G>A | p.G1149E | 16 | 0 | Damaging | 1 | Probably damaging | 0 | 0 | P | 1*PS,2*PM,5*PP |
| c.3451C>G | p.R1151G | 16 | 0 | Damaging | 1 | Probably damaging | 0 | 0 | P | 1*PS,1*PM,4*PP |
| c.3451C>T | p.R1151C | 16 | 0 | Damaging | 1 | Probably damaging | 0 | 5 | P | 1*PS,1*PM,4*PP |
| c.3452G>A | p.R1151H | 16 | 0.01 | Damaging | 1 | Probably damaging | 0 | 2 | LP | 3*PM,5*PP |
| c.3459G>T | p.W1153C | 16 | 0 | Damaging | 1 | Probably damaging | 0 | 0 | LP | 3*PM,5*PP |
| c.3517G>A | p.E1173K | 16 | 0 | Damaging | 1 | Probably damaging | 0 | 1 | LP | 3*PM,5*PP |
| c.3532A>G | p.T1178A | 16 | 0 | Damaging | 0.988 | Probably damaging | 0 | 0 | LP | 1*PM,5*PP |
| c.3563T>G | p.L1188R | 17 | 0 | Damaging | 0.998 | Probably damaging | 0 | 0 | LP | 1*PM,4*PP |
| c.3587A>G | p.D1196G | 17 | 0 | Damaging | 1 | Probably damaging | 0 | 0 | LP | 1*PM,4*PP |
| c.3605C>G | p.A1202G | 17 | 0 | Damaging | 1 | Probably damaging | 0 | 0 | LP | 1*PM,4*PP |
| c.3646G>A | p.V1216M | 17 | 0 | Damaging | 1 | Probably damaging | 0 | 5 | LP | 1*PM,4*PP |
| c.3653T>C | p.L1218P | 17 | 0 | Damaging | 0.999 | Probably damaging | 0 | 0 | LP | 1*PM,4*PP |
| c.3715G>T | p.V1239F | 18 | 0 | Damaging | 0.998 | Probably damaging | 0 | 2 | P | 1*PS,1*PM,4*PP |
| c.3733C>G | p.P1245A | 18 | 0 | Damaging | 1 | Probably damaging | 0 | 0 | LP | 1*PM,4*PP |
| c.3741C>G | p.H1247Q | 18 | 0.17 | Tolerated | 1 | Probably damaging | 0 | 1 | LP | 1*PM,4*PP |
| c.3776G>T | p.G1259V | 18 | 0 | Damaging | 0.997 | Probably damaging | 0 | 0 | LP | 1*PM,4*PP |
| c.3791T>C | p.M1264T | 18 | 0 | Damaging | 0.998 | Probably damaging | 0 | 0 | LP | 1*PM,4*PP |
| c.3802G>A | p.G1268R | 18 | 0 | Damaging | 1 | Probably damaging | 0 | 0 | LP | 2*PM,5*PP |
| c.3809A>G | p.N1270S | 18 | 0 | Damaging | 1 | Probably damaging | 0 | 18 | P | 1*PS,2*PM,5*PP |
| c.3818C>A | p.P1273Q | 18 | 0 | Damaging | 1 | Probably damaging | 0 | 0 | P | 1*PS,2*PM,5*PP |
| c.3818C>T | p.P1273L | 18 | 0 | Damaging | 1 | Probably damaging | 0 | 4 | P | 1*PS,2*PM,5*PP |
| c.3836A>G | p.D1279G | 18 | 0.01 | Damaging | 1 | Probably damaging | 0 | 2 | LP | 2*PM,5*PP |
| c.3848C>T | p.A1283V | 18 | 0 | Damaging | 1 | Probably damaging | 0 | 0 | LP | 2*PM,5*PP |
| c.3859G>A | p.G1287S | 18 | 0 | Damaging | 1 | Probably damaging | 0 | 3 | P | 1*PS,2*PM,5*PP |
| c.3877G>A | p.E1293K | 18 | 0 | Damaging | 1 | Probably damaging | 0 | 1 | LP | 2*PM,5*PP |
| c.3884C>T | p.A1295V | 18 | 0 | Damaging | 1 | Probably damaging | 0 | 0 | P | 1*PS,2*PM,4*PP |
| c.3896T>G | p.L1299R | 18 | 0 | Damaging | 0.999 | Probably damaging | 0 | 0 | P | 1*PS,2*PM,4*PP |
| c.3982G>A | p.A1328T | 19 | 0 | Damaging | 1 | Probably damaging | 0 | 0 | LP | 1*PM,5*PP |
| c.4003G>C | p.G1335R | 19 | 0 | Damaging | 1 | Probably damaging | 0 | 0 | LP | 1*PM,5*PP |
| c.4057T>C | p.W1353R | 20 | 0 | Damaging | 1 | Probably damaging | 0 | 0 | LP | 2*PM,5*PP |
| c.4064G>A | p.G1355D | 20 | 0 | Damaging | 1 | Probably damaging | 0 | 0 | LP | 2*PM,5*PP |
| c.4112T>C | p.L1371P | 20 | 0.01 | Damaging | 1 | Probably damaging | 0 | 0 | LP | 1*PM,5*PP |
PolyPhen 2/ SIFT: Software prediction programs used for sequence variant effect explanation; 1000g: 1000 Genomes Project; ExAc: Exome Aggregation Consortium.
Novel Variants
Among the 161 non-synonymous variants, 58 are novel and the chromatograms of these variants were illustrated in Figures 2A, 2B, 2C and 2D, grouped by the type of variation. None of which was found in the 503 normal individuals. Of these 58 novel variants, 16 are small deletions or insertions, 4 are nonsense variants, 6 are splice site variants, and 32 are missense variants. The other 103 variants have been recorded in the professional version of HGMD (Human Gene Mutation Database Professional, access date: 20 October, 2015).
Chromatograms of 58 novel ATP7B variants identified in the present study. The lower chromatogram in each frame represents the variant, while the upper one represents the normal sequence. The c.4162delG variant is shown in reverse sequence, while the other 57 variants are illustrated in forward sequence. 2A, 2B, 2C and 2D respectively illustrates 32 missense changes, 16 small deletions or insertions, 6 splicing site variants as well as 4 nonsense variants.
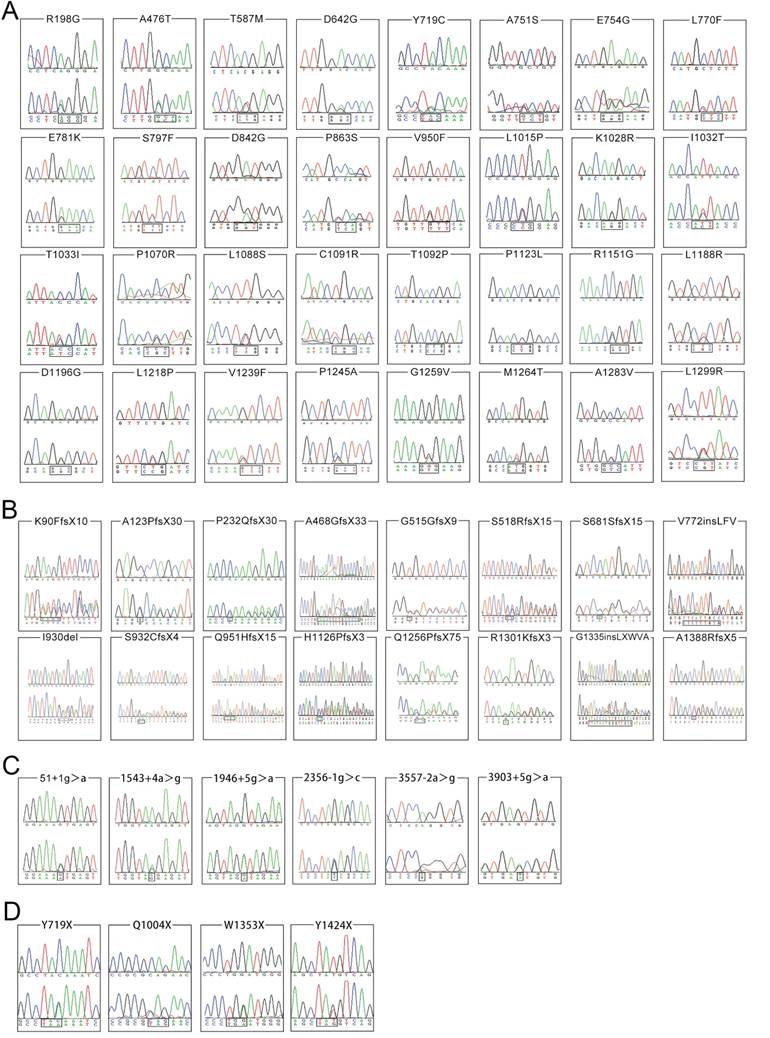
Among the 16 small deletions or insertions, 13 alter the reading frame of ATP7B and result in production of a truncated dysfunctional protein. They are c.268_271del (p.K90FfsX10), c.367delG (p.A123PfsX30), c.695delC (p.P232QfsX30), c.1403_1416del (p.A468GfsX33), c.1545delT (p.G515GfsX9), c.1552_1553delTC (p.S518RfsX15), c.2043delC (p.S681SfsX15), c.2794_2795insGT (p.S932CfsX4), c.2853_2856delGAGA (p.Q951HfsX15), c.3377_3378delAC (p.H1126PfsX3), c.3766_3767dupCA (p.Q1256PfsX75), c.3901_3902insA (p.R1301KfsX3) and c.4162delG (p.A1388RfsX5). The other 3 small deletions or insertions, c.2316_2317insCTCTTTGTG (p.V772insLFV),c.2790_2792delCAT (p.I930del) and c.4005_4006insTTATAATGGGTTGCG (p.G1335insLXWVA) do not change the reading frame of ATP7B, but cause prolonged (p.V772insLFV and p.G1335insLXWVA) or shortened (p.I930del) dysfunctional ATP7B protein. Similarly, the 4 nonsense variants could result in production of a shortened, dysfunctional protein.
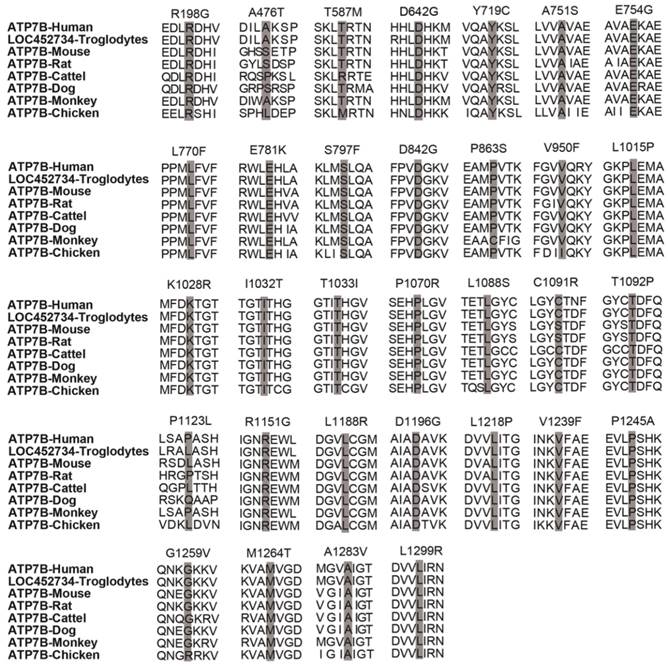
For six splice site variants, three (c.51+1g>a, c.2356-1g>c and c.3557-2a>g) are classified as 'pathogenic variants', the other three (c.1543+4a>g, c.1946+5g>a, 3903+5g>a) are 'variants with uncertain significance'. In addition, among the 32 novel missense variants, 2 (p.T587M, p.P1123L) are classified as 'likely benign variants', 2 (p.A476T, p.T1092P) as 'variants with uncertain significance', 5 (p.E781K, p.T1033I, p.R1151G, p.V1239F, p.L1299R) as 'pathogenic variants' and the other 23 as 'likely pathogenic variants' (Table 3). A homology search of the ATP7B protein in different species demonstrated that 32 missense variants occur within highly conserved regions of ATP7B protein (Figure 3). In total, among 58 novel variants, 25 are classified as 'pathogenic variants', 26 as 'likely pathogenic variants', 5 as 'variants with uncertain significance' and 2 as 'likely benign variants'.
Homology comparisons of ATP7B protein sequences. The highlighted zones respectively indicate 32 novel missense variant sites among 8 species.
Most Common Variants
Among the 632 WD patients, 569 patients (90%) were identified with homozygous or compound heterozygous potential disease-causing variants, 58 patients (9%) with one heterozygous variant and other 5 patients (1%) did not have any potential disease-causing variant. Therefore, 90% (569/632) of patients can be genetically diagnosed with WD. Among the 142 potential disease-causing variants including 78 'pathogenic variants' and 64 'likely pathogenic variants', 14 were relatively common in the WD patient cohort, each with an allelic frequency 1% or higher. These 14 most common disease-causing variants were found in 94% (537/569) of genetically diagnosed WD patients with two or more 'pathogenic' or 'likely pathogenic' variants. The allelic frequencies and numbers of patients with each of these variant are presented in Table 4. Notably, the three most prevalent variants, p.R778L, p.P992L and p.T935M, were detected at least once in 78% (445/569) of genetically diagnosed WD patients. The allelic frequencies of p.R778L, p.P992L and p.T935M are 0.319, 0.155 and 0.077, respectively; all of which are 'pathogenic variants'.
Common disease-causing variants within ATP7B among 569 WD patients.
| Mutation | Domain affected | Number of patients | Allelic Frequencies | Classification | ||
|---|---|---|---|---|---|---|
| WW | WM | MM | ||||
| p.R778L | TM4 | 270 | 235 | 64 | 0.319 | Pathogenic |
| p.P992L | TM6/Ph | 419 | 124 | 26 | 0.155 | Pathogenic |
| p.T935M | TM5 | 482 | 86 | 1 | 0.077 | Pathogenic |
| p.A874V | Td/TM5 | 529 | 37 | 3 | 0.038 | Likely pathogenic |
| p.I1148T | ATP loop | 530 | 38 | 1 | 0.035 | Likely pathogenic |
| p.Q511X | Cu5 | 541 | 26 | 2 | 0.026 | Pathogenic |
| p.G943D | TM5 | 543 | 25 | 1 | 0.024 | Pathogenic |
| p.N1270S | ATP hinge | 544 | 25 | 0 | 0.022 | Pathogenic |
| p.R778Q | TM4 | 548 | 18 | 3 | 0.021 | Pathogenic |
| p.R919G | Td/TM5 | 545 | 24 | 0 | 0.021 | Pathogenic |
| p.V1216M | ATP bind | 547 | 20 | 2 | 0.021 | Likely pathogenic |
| p.V176SfsX28 | Cu2 | 551 | 17 | 1 | 0.017 | Pathogenic |
| c.1708-1g>c | Cu6 | 553 | 14 | 2 | 0.016 | Pathogenic |
| p.V1106I | ATP loop | 556 | 13 | 0 | 0.011 | Pathogenic |
WW: neither of chromosome carries mutation; WM: one chromosome carries mutation;
MM: both chromosomes carry mutations.
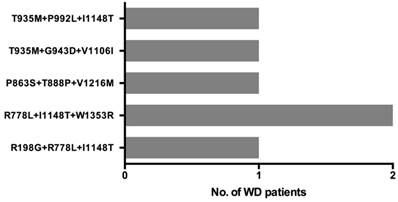
Patients with More than Two Variants
Six patients carried three disease-causing variants and their 3-variant genotypes are described in Figure 4. As two patients had the same genotype, there are 5 unique 3-variant genotypes. Four of these 3-variant genotype include at least one 'likely pathogenic' variant, while one genotype (T935M+G943D+V1106I) consists of three 'pathogenic' variants.
Three distinct ATP7B disease-causing variants co-occur in 6 WD patients.
Discussion
In the present study, the prevalence of ATP7B variants was systematically investigated in the largest Chinese WD cohort to date, with 632 patients and 503 normal controls. One hundred and sixty-one non-synonymous variants within the ATP7B gene were found in WD patients, including 58 novel variants. This study catalogs ATP7B variants in Han Chinese WD patients and expands the spectrum of ATP7B variants.
Another major contribution of the study is the classification of ATP7B variants. Based on the type of alterations, their predicted impact, and frequency between WD cases and controls, these 161 non-synonymous variants are classified as 'pathogenic variants' (N=78), 'likely pathogenic variants' (N=64), 'variants with uncertain significance' (N=8), 'likely benign variants' (N=4), and 'benign variants' (N=7). The observation that 90% (569/632) of these WD patients had two or more 'pathogenic' or 'likely pathogenic' variants demonstrates the clinical utility of the catalog and classification of ATP7B variants.
The 14 most common disease-causing variants were found at least once in 94% (537/569) of genetically diagnosed patients. Notably, the three most prevalent pathogenic variants, p.R778L, p.P992L and p.T935M, were detected in 78% (445/569) of the patients. These results demonstrate the feasibility of developing a rapid and cost-effective genetic test such as multiplex allele-specific PCR to screen for WD.
We found 6 unrelated WD patients (1.1%) carrying three disease-causing variants, and which was reported previously in Caucasian populations.6 This observation is important because it suggests that two disease-causing variants may reside in a chromosome. The implication is that if an individual carries two disease-causing variants but his/her clinical features do not support the diagnosis of WD, it is necessary to test these variants in parents to identify the specific location of them. If the two variants reside in a chromosome, the individual is a heterozygous carrier and cannot be genetically diagnosed. If two variants reside in different chromosomes, then a genetic diagnosis of WD can be made.
According to ACMG Standards and Guidelines,12 we classified 4 variants as 'likely benign' variants, one of which (p.Q1142H) was previously considered as a pathogenic variant.14 Tsai et al. found that Q1142 was located at the ATP pocket of ATP7B protein, and the amino acid replacement of the site would directly disrupt ATP7B function. However, the amino acid change in p.Q1142H was predicted to be tolerated by SIFT (score: 0.16) and benign by PolyPhen-2 (score: 0) software programs, this variant should be considered as 'likely benign'. This finding is important because the variant is relatively frequent and found in 19 WD patients (18 heterozygous and one homozygous). Caution should be made in interpreting the carrier of the Q1142 variant to avoid misdiagnosis and unnecessary treatment.
Sixty-three clinically diagnosed WD patients cannot be genetically diagnosed with WD because they were detected to carry only one disease-causing variant or none. Several factors may induce the observation. First, a subset of these 63 patients may have been misdiagnosed. For example, these patients may have been included in the patient cohort on the basis of false positive K-F rings. Second, large hemizygous deletions may occur in a subset of these patients and these alterations are difficult to be detected using the Sanger sequencing method. In fact, a hemizygous large deletion spanning the exon 20 and adjacent introns has previously been reported.15 Other methods such as multiplex ligation-dependent probe amplification (MLPA) may be used to detect large deletions. Third, other genetic alterations outside the ATP7B coding region and adjacent splice sites as well as other cellular factors associated with WD may contribute to the clinical development of WD in these patients.16
In conclusion, the current study considerably expands the spectrum of ATP7B variants and provides classification of their clinical effects. These results improve the genetic diagnosis of suspected WD patients and facilitate genetic screening for WD among asymptomatic children in the general Chinese population.
Abbreviations
Cp: ceruloplasmin; K-F: Kayser-Fleischer; ACMG: American College of Medical Genetics and Genomics; HGMD: Human Gene Mutation Database; MLPA: multiplex ligation-dependent probe amplification.
Supplementary Material
Supplementary Table S1.
Acknowledgements
This work was supported by a grant from the National Natural Science Foundation of China to Zhi-Ying Wu (81125009). We gratefully acknowledge all participants for their help and willingness to participate this study.
Conflicts of Interest
All authors reported no biomedical financial interests or potential conflicts of interest.
References
1. Bull PC, Thomas GR, Rommens JM, Forbes JR, Cox DW. The Wilson disease gene is a putative copper transporting P-type ATPase similar to the Menkes gene. Nat Genet. 1993;5:327-337
2. Tanzi RE, Petrukhin K, Chernov I, Pellequer JL, Wasco W, Ross B. et al. The Wilson disease gene is a copper transporting ATPase with homology to the Menkes disease gene. Nat Genet. 1993;5:344-350
3. Yamaguchi Y, Heiny ME, Gitlin JD. Isolation and characterization of a human liver cDNA as a candidate gene for Wilson disease. Biochem Biophys Res Commun. 1993;197:271-277
4. Scheinberg IH, Sternlieb I. Wilson's disease. In: (ed.) Smith LH Jr. Major Problems in Internal Medicine. Vol 23. Philadelphia, Pa: WB Saunders Co. 1984
5. Ala A, Walker AP, Ashkan K, Dooley JS, Schilsky ML. Wilson's disease. Lancet. 2007;369:397-408
6. Coffey AJ, Durkie M, Hague S, Mclay K, Emmerson J, Lo C. et al. A genetic study of Wilson's disease in the United Kingdom. Brain. 2013;136:1476-1487
7. Park HD, Ki CS, Lee SY, Kim JW. Carrier frequency of the R778L, A874V, and N1270S mutations in the ATP7B gene in a Korean population. Clin Genet. 2009;75:405-407
8. Mak CM, Lam CW, Tam S, Lai CL, Chan LY, Fan ST. et al. Mutational analysis of 65 Wilson disease patients in Hong Kong Chinese: Identification of 17 novel mutations and its genetic heterogeneity. J Hum Genet. 2008;53:55-63
9. Saito T. An assessment of efficiency in potential screening in Wilson disease. J Epidemiol Community Health. 1981;35:274-280
10. Thomas GR, Forbes JR, Roberts EA, Walshe JM, Cox DW. The Wilson disease gene: spectrum of mutations and their consequences. Nat Genet. 1995;9:210-217
11. Wu ZY, Zhao GX, Chen WJ, Wang N, Wan B, Lin MT. et al. Mutation analysis of 218 Chinese patients with Wilson disease revealed no correlation between the canine copper toxicosis gene MURR1 and Wilson disease. J Mol Med. 2006;84:438-442
12. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al.Standards, guidelines for the interpretation of sequence variants. a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405-424
13. Wu ZY, Wang N, Lin MT, Fang L, Murong SX, Yu L. Mutation analysis and the correlation between genotype and phenotype of Arg778Leu mutation in Chinese patients with Wilson disease. Arch Neurol. 2001;58:971-976
14. Tsai CH, Tsai FJ, Wu JY, Chang JG, Lee CC, Lin SP. et al. Mutation analysis of Wilson disease in Taiwan and description of six new mutations. Hum Mutat. 1998;12:370-376
15. Moller LB, Ott P, Lund C, Horn N. Homozygosity for a gross partial gene deletion of the C-terminal end of ATP7B in a Wilson patient with hepatic and no neurological manifestations. Am J Med Genet A. 2005;138:340-343
16. Mufti AR, Burstein E, Csomos RA. XIAP Is a copper binding protein deregulated in Wilson's disease and other copper toxicosis disorders. Mol Cell. 2006;21:775-785
Author contact
![]() Corresponding author: Prof. Zhi-Ying Wu, Department of Neurology and Research Center of Neurology in Second Affiliated Hospital, and the Collaborative Innovation Center for Brain Science, Zhejiang University School of Medicine, 88 Jiefang Rd, Hangzhou, 310009, China. E-mail: zhiyingwuedu.cn.
Corresponding author: Prof. Zhi-Ying Wu, Department of Neurology and Research Center of Neurology in Second Affiliated Hospital, and the Collaborative Innovation Center for Brain Science, Zhejiang University School of Medicine, 88 Jiefang Rd, Hangzhou, 310009, China. E-mail: zhiyingwuedu.cn.