Impact Factor
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2016; 6(5):650-665. doi:10.7150/thno.14479 This issue Cite
Research Paper
Multimodal Somatostatin Receptor Theranostics Using [64Cu]Cu-/[177Lu]Lu-DOTA-(Tyr3)octreotate and AN-238 in a Mouse Pheochromocytoma Model
Martin Ullrich1, Ralf Bergmann1, Mirko Peitzsch2, Erik F. Zenker1, Marc Cartellieri3, Michael Bachmann1,3, Monika Ehrhart-Bornstein4, Norman L. Block5, Andrew V. Schally5, Graeme Eisenhofer2,4, Stefan R. Bornstein4, Jens Pietzsch1,6, Christian G. Ziegler7,8 ![]()
1. Dept. of Radiopharmaceutical and Chemical Biology, Institute of Radiopharmaceutical Cancer Research, Helmholtz-Zentrum Dresden-Rossendorf, Dresden, Germany;
2. Institute for Clinical Chemistry and Laboratory Medicine, Technische Universität Dresden, Dresden, Germany;
3. University Cancer Center, Tumorimmunology, Technische Universität Dresden, Dresden, Germany;
4. Dept. of Medicine III, University Hospital Carl Gustav Carus, Dresden, Germany.
5. VA Medical Center Miami FL and Dept. of Pathology and Medicine, Div. of Endocrinology and Hematology-Oncology and Sylvester Comprehensive Cancer Center, University of Miami Miller School of Medicine, Miami FL, USA.
6. Dept. of Chemistry and Food Chemistry, Technische Universität Dresden, Dresden, Germany;
7. Paul Langerhans Institute Dresden of the Helmholtz Center Munich at the University Hospital and faculty of Medicine, Technische Universität Dresden, Dresden, Germany;
8. German Center for Diabetes Research e.V., Neuherberg, Germany.
Received 2015-11-19; Accepted 2016-2-10; Published 2016-3-10
Abstract

Pheochromocytomas and extra-adrenal paragangliomas (PHEO/PGLs) are rare catecholamine-producing chromaffin cell tumors. For metastatic disease, no effective therapy is available. Overexpression of somatostatin type 2 receptors (SSTR2) in PHEO/PGLs promotes interest in applying therapies using somatostatin analogs linked to radionuclides and/or cytotoxic compounds, such as [177Lu]Lu-DOTA-(Tyr3)octreotate (DOTATATE) and AN-238. Systematic evaluation of such therapies for the treatment of PHEO/PGLs requires sophisticated animal models. In this study, the mouse pheochromocytoma (MPC)-mCherry allograft model showed high tumor densities of murine SSTR2 (mSSTR2) and high tumor uptake of [64Cu]Cu-DOTATATE. Using tumor sections, we assessed mSSTR2-specific binding of DOTATATE, AN-238, and somatostatin-14. Therapeutic studies showed substantial reduction of tumor growth and tumor-related renal monoamine excretion in tumor-bearing mice after treatment with [177Lu]Lu-DOTATATE compared to AN-238 and doxorubicin. Analyses did not show agonist-dependent receptor downregulation after single mSSTR2-targeting therapies. This study demonstrates that the MPC-mCherry model is a uniquely powerful tool for the preclinical evaluation of SSTR2-targeting theranostic applications in vivo. Our findings highlight the therapeutic potential of somatostatin analogs, especially of [177Lu]Lu-DOTATATE, for the treatment of metastatic PHEO/PGLs. Repeated treatment cycles, fractionated combinations of SSTR2-targeting radionuclide and cytotoxic therapies, and other adjuvant compounds addressing additional mechanisms may further enhance therapeutic outcome.
Keywords: neuroendocrine tumors, catecholamines, metanephrines, DOTATATE, PET, SPECT, optical in vivo imaging, doxorubicin.
Introduction
Pheochromocytomas and extra-adrenal paragangliomas (PHEO/PGLs) are rare catecholamine-producing tumors arising from sympathetic adrenal and extra-adrenal chromaffin cells [1]. Diagnosis of malignant pheochromocytoma continues to rely on the identification of metastases at sites where chromaffin cells are normally absent (e.g., liver, bones and lungs). At that stage, therapeutic options are limited [2, 3].
To date, systemic chemotherapy combining cyclophosphamide, vincristine, and dacarbazine as well as radiotherapy with meta-[131I]iodobenzylguanidine represent the two primary modes of treatment for metastatic PHEO/PGLs [4]. Other therapeutic approaches include combinations of etoposide and cisplatin, cisplatin and 5-fluorouracil, cytosine arabinoside, anthracyclines such as doxorubicin, and more recently, temozolomide and thalidomide [5, 6], as well as radiofrequency ablation [7]. However, complete remissions are rare and these therapies can only be regarded as palliative [8]. Thus, there is a need for identifying new targets for the treatment of metastatic PHEO/PGLs along with the development of appropriate therapeutic agents and treatment strategies.
Similar to other neuroendocrine tumors, PHEOs/PGLs abundantly express somatostatin receptors, promoting strong interest in applying peptide receptor-targeting radionuclide or cytotoxic therapies using radionuclide- or toxin-labeled somatostatin analogs [9, 10]. Some support for the radionuclide approach is provided by clinical reports on [177Lu]Lu- and [90Y]Y-labeled somatostatin analogs leading to longer progression-free survival, mainly in gastroenteropancreatic neuroendocrine tumors [11], but also in metastatic neuroendocrine tumors, including PHEO/PGLs [12-15]. Preclinical reports on targeted cytotoxic therapy using AN-238, a somatostatin analog conjugated to 2-pyrrolinodoxorubicin, have shown inhibition of somatostatin receptor-positive tumor cells, primary tumors, and their metastases in various models of neuroendocrine tumors and prostate cancer [16-20]. However, due to the limited number of cases, there is still no systematic evaluation of somatostatin receptor-targeting therapies focusing on the treatment of metastatic PHEO/PGLs. Therefore, preclinical investigations using sophisticated animal models provide a valuable alternative.
The mouse pheochromocytoma (MPC) cell line has been developed from neurofibromatosis type 1 knockout mice [21]. The recently established MPC-mCherry tumor allograft model of pheochromocytoma is based on the subcutaneous engraftment of a genetically modified MPC cell line expressing mCherry [22]. This model exhibits functional similarities to human PHEO/PGLs and allows for monitoring tumor burden by fluorescence imaging and measurements of tumor-related renal monoamine excretion [22].
This study had two objectives: 1. characterization of the MPC-mCherry tumor model for preclinical evaluation of somatostatin type 2 receptor (SSTR2)-targeting theranostic applications; and 2. use of the model for preclinical evaluation of receptor-targeted therapies using [177Lu]Lu-DOTA-(Tyr3)octreotate (DOTATATE) and AN-238 compared to systemic therapy using doxorubicin for the treatment of PHEO/PGLs. We employed a comprehensive approach involving several strategies: (i) investigation of the SSTR2 status in murine tumor cell cultures and tumors; (ii) evaluation of cellular uptake, tumor binding and functional in vivo imaging using the radiotracer [64Cu]Cu-DOTATATE; (iii) assessment of the therapeutic outcome on reduction of tumor growth, reduction of renal monoamine excretion, and tumor morphology; and (iv) assessment of potential adverse effects of the various therapies. Using this comprehensive approach, our goal was to identify potential opportunities for the further development and refinement of SSTR2-targeted and combined therapeutic strategies for the treatment of metastatic PHEO/PGLs and other, so far untreatable, neuroendocrine tumors.
Materials and methods
Cell culture
MPC cells consecutively expressing mCherry (MPC-mCherry cells deriving from lentivirally gene-modified MPC cells; clone 4/30PRR [21], passage 32) were cultured and prepared for in vivo application as described previously [22].
Animal experiments
All animal experiments were carried out at the Helmholtz-Zentrum Dresden-Rossendorf according to the guidelines of German Regulations for Animal Welfare and have been approved by the Landesdirektion Dresden. Transplantation of MPC-mCherry cells (passage 13) into NMRI nu/nu mice (Medical Faculty Carl-Gustav Carus Experimental Center, TU Dresden, Dresden, Germany), anesthesia, determination of tumor volume and body weight, intervals of optical imaging and urine collection were carried out as described previously [22]. A terminal white blood cell count was performed after 30 days post cell injection (pci). Animals were sacrificed after 30 days pci or after 38 days pci (extended follow-up) using CO2 inhalation and cervical dislocation, tumors and organs were excised. For histologic investigations, tumors were fixed for 72 hours at 4 °C using 4% (w/v) paraformaldehyde in PBS containing 2% (w/v) sucrose. For autoradiography, tumors were frozen in 2-methylbutane at -20 °C. For Western blot analyses, tumors were frozen in liquid nitrogen and stored at -66 °C.
Immunoblotting
Cells and tissues (n = 6) were added to ice-cold RIPA-buffer (Sigma-Aldrich Chemie, Munich, Germany) containing 1 µg/mL leupeptin, 1 mmol/L phenylmethylsulfonyl fluoride, 5 mmol/L NaF, 1 mmol/L Na3VO4 and 1 mmol/L dithiothreitol and disrupted in a TissueLyzer (Qiagen, Hilden, Germany). Proteins (40 µg) were separated on 10% (w/v) sodium dodecyl sulfate polyacrylamide gels and transferred to polyvinylidene difluoride membranes (Whatman, Dassel, Germany). Membranes were blocked with PBS containing 0.5% (v/v) Tween 20, 5% (w/v) fat-free milk and 2% (w/v) bovine serum albumin. Protein bands were detected using the primary antibodies anti-SSTR2 [UMB1, ab134152] (Abcam, Cambridge, UK), anti-chromogranin A [C-20, sc1488] (Santa Cruz Biotechnology, Heidelberg, Germany), anti-ß-actin [A5316] (Sigma-Aldrich). The peroxidase-conjugated secondary antibodies anti-mouse IgG [A9044] and anti-rabbit IgG [A0545] were purchased from Sigma-Aldrich. Specific binding was detected using SuperSignal™ West Pico/Dura Substrate (Life Technologies, Carlsbad, CA, USA). Densitometric analysis was performed using the TL100 image analysis software (TotalLab, Newcastle upon Tyne, UK) and normalized to ß-actin.
Immunocyto-/histochemistry
Paraformaldehyde-fixed tumor cell monolayers and tissue sections were permeabilized with PBS containing 1% (v/v) Triton-X 100 (AplliChem, Darmstadt, Germany). Murine somatostatin type 2 receptor (mSSTR2) was detected with the primary anti-SSTR2 [UMB1 ab134152] antibody (Abcam) and the secondary Alexa Fluor donkey anti-rabbit FITC 488 [A21206] antibody (Thermo Fisher Scientific, Waltham, MA, USA). Nuclei were stained with Hoechst 33258. Fluorescence images were captured using the FluoView™ FW1000 confocal laser scanning microscope (Olympus Europe, Hamburg, Germany).
Paraformaldehyde-fixed tumor samples were dehydrated in a graded series of propane-2-ol, embedded in paraffin and cut in 3-micron sections. Antigen retrieval was performed in boiling 10 mmol/L citrate buffer, pH 6.0. Endogenous peroxidase was blocked with PBS containing 0.3% (v/v) H2O2. mSSTR2 was detected with the primary anti-SSTR2 [UMB1, ab134152] antibody (Abcam), the secondary biotinylated goat anti-rabbit antibody [A0545] (Sigma-Aldrich). For negative control, sections were co-incubated with primary antibody and the human SSTR2 fragment [ab171899] (Abcam). Sections were stained with 3,3'-diaminobenzidine, counterstained with eosin, and imaged using the AXIO Imager A1 microscope (Carl Zeiss, Oberkochen, Germany).
Labeling of DOTATATE
[64Cu]Cu was produced at the Helmholtz-Zentrum Dresden-Rossendorf on the Cyclone 18/9 cyclotron (IBA, Louvain-la-Neuve, Belgium) by 64Ni(p,n)64Cu nuclear reaction as reported previously [23, 24]. [177Lu]Lu was produced by indirect 176Yb(n,γ)177Yb → 177Lu nuclear reaction (ITG, Garching, Germany). Anhydrous [natLu]LuCl3 was purchased from Sigma-Aldrich.
For [64Cu]Cu labeling, an aqueous solution of [64Cu]CuCl2 (1 GBq, 120 µL, 0.01 mol/L HCl, 0.3 mol/L NH4OAc, pH 5.0) was added to 14 nmol of DOTATATE (ABX, Radeberg, Germany) and tempered at 80 °C for 45 minutes, resulting in a labeling yield and a radiochemical purity (decay-corrected) > 97% as determined by analytical high performance liquid chromatography (HPLC). For [177Lu]Lu labeling, an aqueous solution of [177Lu]LuCl3 (0.9 GBq, 420 µL, 0.01 mol/L HCl, 0.1 mol/L NH4OAc, pH 5.2) was added to 14 nmol of DOTATATE and tempered at 90 °C for 20 minutes, resulting in a labeling yield and a radiochemical purity (decay-corrected) of > 97% as determined by HPLC. Radiolysis of peptides [25] was not observed until subsequent experimental application. For the synthesis of [natLu]Lu-DOTATATE, an aqueous solution of [natLu]LuCl3 (348 nmol, 1 mL, 0.01 mol/L HCl, 0.1 mol/L NH4OAc, pH 5.2) was added to 348 nmol of DOTATATE and tempered at 90 °C for 20 minutes, resulting in a labeling yield and a radiochemical purity (decay-corrected) of > 98% as determined by mass spectrometry.
Analytical HPLC was performed on a Series 1200 device (Agilent Technologies, Santa Clara, CA, USA) equipped with a Ramona ß/γ-ray detector (Raytest, Straubenhardt, Germany). Eluent A: 0.1% (v/v) trifluoroacetic acid in H2O; eluent B: 0.1% (v/v) trifluoroacetic acid in acetonitrile; HPLC system: Zorbax SB-C18, 300 Å, 4 µm, 250 × 9.4 mm (Agilent); gradient elution using 95% eluent A for 5 minutes, 95% eluent A to 95% eluent B in 10 minutes, 95% eluent B for 5 minutes and 95% eluent B to 95% eluent A in 5 minutes, 3 mL/minute, 50 °C, recovery of activity (decay-corrected) was > 95 %. Mass spectrometry was performed with the Xevo TQ-S tandem quadrupole mass spectrometry system (Waters, Milford, MA, USA ) using ultrapure acetonitrile and methanol (Sigma-Aldrich).
Binding studies and autoradiography
Tumor samples (n = 2) were serially cut in 10-micron cryosections on a CM1850 cryostat (Leica Biosystems, Wetzlar, Germany), mounted on polytetrafluorethylene-coated diagnostic slides (Thermo Fisher) and stored at -80 °C. Sections were pre-incubated at room temperature for 10 minutes with 50 mol/L Tris-HCl, pH 7.4 containing 2 mmol/L CaCl2 and 5 mmol/L KCl and washed with salt-free 50 mmol/L Tris-HCl, pH 7.4. Sections were incubated with [64Cu]Cu-DOTATATE (As = 10 GBq/µmol) for 120 minutes in 50 mmol/L Tris-HCl, pH 7.4 containing 1% (w/v) bovine serum albumin and cOmplete™ ethylenediaminetetraacetic acid-free protease inhibitor (Roche, Basel, Switzerland). Sections were washed twice with ice-cold Tris-HCl, pH 7.4 containing 0.25% (w/v) bovine serum albumin, rinsed in H2O, and air flow-dried. Sections and serially diluted radioligand standards were exposed to photostimulatable phosphor imaging plates (Fujifilm Europe, Düsseldorf, Germany) for 12-16 hours. Photostimulated luminescence was detected using the BAS-5000 system (Fujifilm). Autoradiograms were quantified using the AIDA Image Analyzer software (Raytest).
Saturation binding was performed with increasing concentrations of radioligand (0.625-40 nmol/L). Non-specific binding was determined by adding 5 µmol/L non-labeled DOTATATE. Competition binding was performed with increasing concentrations (0.1 nmol/L-10 µmol/L) of various somatostatin analogs and a radioligand concentration of 1.9 nmol/L. Binding data were evaluated using Prism 5.02 (GraphPad Software, San Diego CA, USA). Saturation binding data were fitted with the equation for one site-specific binding. Maximum number of binding sites (Bmax) and dissociation constant (Kd) were determined in Scatchard plots. Competition binding data were fitted with the equation for one site-specific binding-IC50 for calculation of half maximal inhibitory concentrations (IC50).
Cellular uptake of somatostatin analogs
A number of 105 MPC-mCherry cells were pre-cultured for 2 days. Attached cells (n = 4 wells) were incubated with 0.4 GBq/L (40 nmol/L) of [64Cu]Cu-DOTATATE (As = 10 GBq/µmol) in 0.5 mL serum-supplemented medium for 1 hour at 37 °C and 4 °C, respectively. Blocking experiments were performed with somatostatin-14, DOTATATE, and AN-238 at 1 µmol/L. Incubated cells were washed with ice-cold PBS containing 0.9 mol/L CaCl2 and 0.5 mol/L MgCl2. Washed cells were lysed in 0.1 mol/L NaOH containing 0.1% SDS. Decay-corrected 64Cu activity of lysates was quantified using the gamma counter Wizard (PerkinElmer, Waltham, MA, USA). Protein was quantified using the Quant-iT protein assay (Life Technologies). Cellular uptake was expressed as % dose per mg of protein.
Preparation of therapeutic peptides and cytotoxic agents
AN-238 (Ӕterna Zentaris, Frankfurt, Germany) and doxorubicin hydrochloride (Sigma-Aldrich) were dissolved in 20 µL of 0.01 mol/L acetic acid. [177Lu]Lu-DOTATATE (As = 65 GBq/µmol) was dissolved in 80 µL of 0.01 mol/L chloric acid containing 0.01 mol/L ammonium-acetate. Each compound was diluted with Infesol®15 (Serumwerk Bernburg, Bernburg, Germany); 0.3 mL/30 g body weight were injected into the tail vein, corresponding to 0.2 µmol/kg of AN-238, 6.9 µmol/kg of doxorubicin and 80 MBq/animal (1.2 nmol/animal) of [177Lu]Lu-DOTATATE. Applied doses were based on therapeutic studies of Szepeshazi et al. [17, 18] and Schmitt et al. [26], as well as on pilot studies in our laboratory.
Animal treatment protocol
Mice bearing continually growing tumors of 5 ± 3 mm in diameter were grouped according to a similar average tumor volume per treatment group. Control and therapy groups were selected at random. Three consecutive treatment experiments (E1, E2, E3) were performed according to the identical protocol. Mice received vehicle only (control; nE1 = 4; nE2 = 5; nE3 = 5), doxorubicin (nE1 = 2; nE2 = 2; nE3 = 3), [177Lu]Lu-DOTATATE (nE1 = 2; nE2 = 2; nE3 = 3), AN-238 (nE1 = 2; nE2 = 2; nE3 = 3), [177Lu]Lu-DOTATATE + doxorubicin (nE1 = 2; nE2 = 2; nE3 = 3) simultaneously after 15 days pci, or [177Lu]Lu-DOTATATE + AN-238 (nE1 = 1; nE2 = 2; nE3 = 2) consecutively after 15 and 18 days pci to avoid competitive effects. Collective data of all 3 independent experiments were analyzed statistically. Reduction of tumor growth (TGred) was calculated between 15 days pci (start) and 30 days pci (end) with TGred = 100% - 100% × (Vtumor (therapy, end) - Vtumor (therapy, start)) / (Vtumor (control, end) - Vtumor (control, start)). Extended follow-up was done in randomly selected animals (n = 3).
Optical in vivo imaging
Optical in vivo imaging of mice was performed using the In-Vivo Xtreme system (Bruker, Billerica, MA, USA) equipped with a 400 watts xenon illuminator and a back-illuminated 4 megapixel charge-coupled device detector. For detection of mCherry, fluorescence imaging (FLI) was performed for 5 s at λEx/Em = 600/700 nm. Reference images were captured at λEx/Em = 480/535 nm. Photostimulated luminescence imaging (PLI) was performed for 2 minutes at 4 × 4 binning. X-ray imaging, was performed for 1.2 s. Images were analyzed using the MI 7.2 software (Bruker). For demarcation of tumors, regions of interest were determined in mCherry/reference-divided fluorescence images by thresholding (signal/background = 2). For demarcation of muscles, regions of interest were determined manually in X-ray images at the position of the masseters. Accumulation of radioligands was determined in PLI images as the maximum uptake value (max UVPLI = [photons/s/mm2] / [MBq injected activity/g body weight]).
Functional in vivo imaging
Independent of the therapy experiments, in vivo positron emission tomography (PET) was performed in an additional group of untreated mice (n = 3) using the microPET® P4 scanner (Siemens Preclinical Solutions, Knoxville, TN, USA). When tumor diameters reached 8 ± 2 mm, a transmission scan was carried out using a rotating 57Co point source and 12-15 MBq (0.3-0.4 nmol) of [64Cu]Cu-DOTATATE (As = 40 GBq/µmol) in 0.5 mL of E153 (Serumwerk Bernburg) were administered as a tail vein infusion (0.5 mL/min) using a syringe pump (Harvard Apparatus, Holliston, MA, USA). Acquisition was started simultaneously with radiotracer infusion. Emission data were collected continuously for 120 minutes. Three-dimensional list mode data were sorted into sinograms with 38 frames (15 × 10 s, 5 × 30 s, 5 × 60 s, 4 × 300 s, 9 × 600 s). Acquisition data were corrected for decay, scatter, and attenuation. Frames were reconstructed as published elsewhere [27]. PET images are presented as maximum intensity projections between 13 and 115 minutes.
In vivo single-photon emission computed tomography (SPECT) and X-ray computed tomography (CT) were performed in [177Lu]Lu-DOTATATE-treated mice from the therapeutic investigation using the NanoSPECT system (BioScan, Washington, DC, USA). Static SPECT/CT images are presented as maximum intensity projections 24 hours after treatment.
Image volume data were converted to Siemens ECAT7 format and further processed with the software Rover (ABX). In PET images, three-dimensional regions of interest were defined by signal-to-background thresholding and time-activity curves were generated. Demarcation of tumors was done at a signal-to-background threshold of 39% in parametric images after Patlak analysis. Standardized uptake values (SUVPET = [MBq activity/mL tissue] / [MBq injected activity/g body weight], mL/g) were calculated for each region of interest averaging the frames 26-38 (13 - 115 minutes) and presented as maximum SUV (max SUV).
Radiotracer distribution studies
Ex vivo radiotracer distribution studies in mice (n = 3) were performed using the same animals, and radiotracer injection protocol as for the PET studies. After PET acquisition, blood samples were collected by heart puncture and animals were sacrificed 150 minutes after radiotracer injection. Organs and tissues were excised and weighed, and radioactivity was determined using the gamma counter Wizard (PerkinElmer). Accumulated radioactivity was calculated as the standardized uptake value (SUVRD = [MBq activity/g tissue] / [MBq injected activity/g body weight]).
Determination of urinary free monoamines
Monoamine excretion was monitored in all animals described in the animal treatment protocol. Urinary concentrations of free monoamines were determined simultaneously by liquid chromatography tandem mass spectrometry, as described elsewhere [28]. Reduction of renal monoamine excretion (MAred) was calculated between 14 (start) and 28 (end) days pci with MAred = 100% - 100% × (cMA (therapy end) - cMA (therapy start)) / (cMA (control end) - cMA (control start)).
Statistics
Statistical analysis was performed using Prism version 5.02 (GraphPad). Data are presented as mean ± standard error of the mean (SEM) and n represents the number of data sets investigated. Tumor volume and 177Lu activity accumulation data were fitted with the equations for exponential growth or for exponential 1-phase decay; doubling times (tD) or half-lives (t½) were calculated, respectively. Significance of differences was tested using Šídák's post hoc multiple comparison test. Differences were considered significant at p-values < 0.05. Significance of relationships between reduction of tumor growth and renal monoamine excretion (n = 35) was tested using Spearman's linear correlation test and displayed as Spearman's correlation coefficient (rs). Performance of diagnostic tests was evaluated using receiver operating characteristic (ROC) curve analysis (comparison of control group vs. therapy groups) and displayed as area under curve (AUC) with 95% confidence interval. Endpoint parameters were considered to significantly discriminate animals in the therapy groups from controls at p-values < 0.05.
Results
mSSTR2 status of murine pheochromocytoma cells and tumors
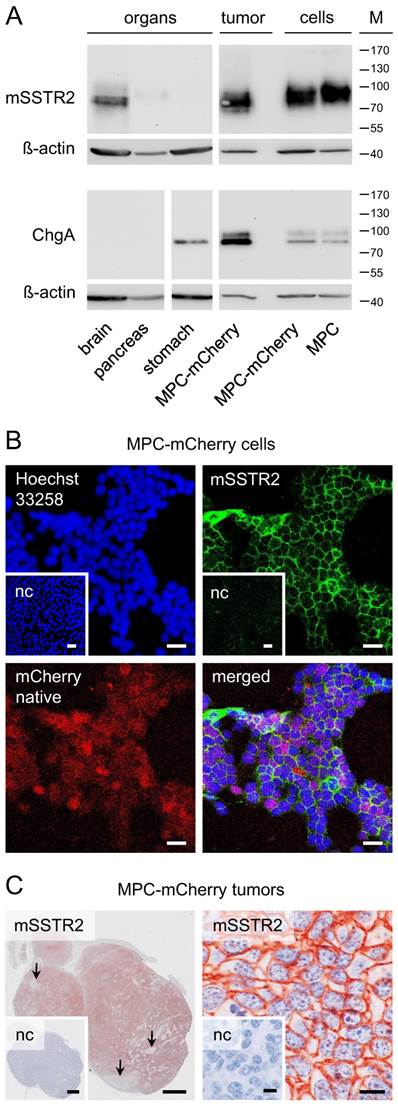
Western blot analysis showed mSSTR2 between 70 and 85 kDa in murine brain, pancreas, and tumors (Figure 1 A). The antibody also recognized mSSTR2 from the cell cultures between 85 and 100 kDa. Furthermore, we detected the chromaffin cell marker chromogranin A predominantly between 80 and 85 kDa in stomach, tumors, and cell cultures. In tumors and cell cultures, the antibody recognized an additional chromogranin A band between 95 and 100 kDa. Notably, chromogranin A levels were higher in tumors compared to cell cultures.
In cell cultures, immunohistochemistry showed a homogeneous membranous mSSTR2 distribution among the red-fluorescent tumor cell monolayers (Figure 1 B). In tumors, mSSTR2 was widely distributed across the tissue sections, though showing inhomogeneous membranous levels and partial absence in necrotic areas infiltrated with other cell types (Figure 1 C).
Density of mSSTR2 and binding of somatostatin analogs to MPC-mCherry tumor sections
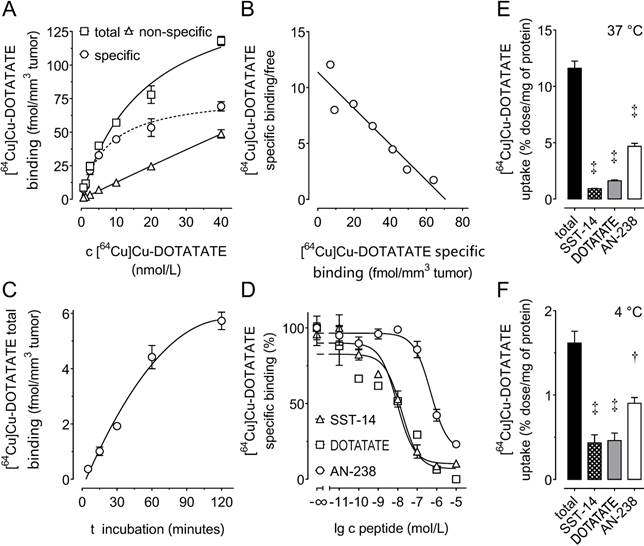
Saturation and Scatchard analysis (Figure 2 A, B) revealed specific [64Cu]Cu-DOTATATE binding to mSSTR2 on MPC-mCherry tumor sections showing a Bmax of 71 fmol/mm3 tissue (8.9 × 104 mSSTR2 sites/tumor cell) and a Kd of 6.2 nmol/L. Receptor-ligand interaction reached equilibrium after 120 minutes (Figure 2 C). Competition analyses revealed an IC50 of 9.3 nmol/L for somatostatin-14, a 1.7-fold higher IC50 of 15.9 nmol/L for DOTATATE, and a 52.9-fold higher IC50 of 492 nmol/L for AN-238 (Figure 2 D). The affinity order of somatostatin analogs was also reflected by the efficiency order of blocking [64Cu]Cu-DOTATATE uptake in MPC-mCherry cell cultures, showing inhibition of 92.4% for somatostatin-14, 86.2% for DOTATATE, and 59.1% for AN-238 at 37 °C (Figure 2 E) as well as 68.8% for somatostatin-14, 68.8% for DOTATATE, and 44.3% for AN-238 at 4 °C (Figure 2 F).
mSSTR2 status of murine pheochromocytoma cells and tumors. (A) Western blot analyses of murine organs, tumors and cell cultures; (B) immunocytochemistry in MPC-mCherry cell cultures in vitro; bars: 30 µm; (C) immunohistochemistry on tissue sections of MPC-mCherry tumors; (left) overview of a tumor 4 weeks after cell injection, arrows indicate the absence of mSSTR2 in necrotic regions; bars: 1 mm; (right) membranous distribution of mSSTR2 in tumors; bars: 10 µm; (M) molecular weight of proteins in kDa; (nc) negative control in presence of blocking peptide (human SSTR2 fragment).
(A-D) Binding of somatostatin analogs to mSSTR2 on MPC-mCherry tumor sections. (A) mSSTR2 saturation binding analysis at increasing concentrations of the radioligand [64Cu]Cu-DOTATATE; non-specific binding was determined in presence of 5 µmol/L non-labeled DOTATATE; (B) Scatchard analysis of radioligand binding; (C) time-dependence of radioligand binding; (D) competition of radioligand with increasing concentrations of various SSTR2 agonists; (E-F) cellular uptake of [64Cu]Cu-DOTATATE in MPC-mCherry cell cultures at various temperatures; inhibition of radioligand uptake by co-incubation with various somatostatin agonists at 1 µmol/L; data are presented as means ± SEM; significance of differences was tested as compared to control; † p < 0.01; ‡ p < 0.001.
Uptake of somatostatin analogs in MPC-mCherry tumors in vivo
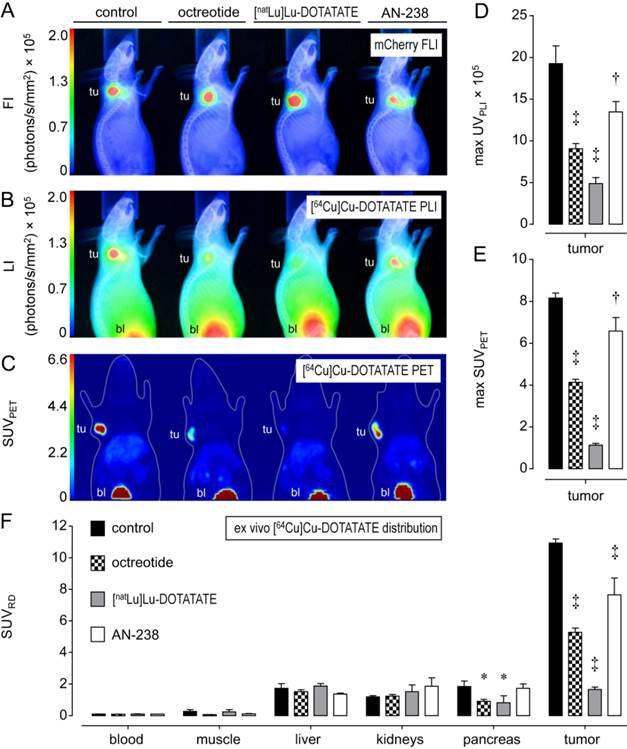
Functional [64Cu]Cu-DOTATATE in vivo imaging and ex vivo distribution studies in excised tumors and organs allowed for investigating the tumor targeting efficiency of octreotide, [natLu]Lu-DOTATATE, and AN-238 by co-injection in MPC-mCherry tumor-bearing animals. FLI demarcated the red-fluorescent primary tumors in mice (Figure 3 A). PLI, PET, and ex vivo radiotracer distribution studies revealed strong radiotracer accumulation in tumors of control animals but only weak accumulation in blood and muscles, as well as in visceral organs such as liver, kidneys, and pancreas (Figure 3 B-D, Table S3).
Most importantly, by co-injection of somatostatin analogs, all imaging and distribution studies similarly showed significantly reduced radiotracer uptake in tumors (Figure 3 D-F) in the order of [natLu]Lu-DOTATATE > octreotide > AN-238. Ex vivo radiotracer distribution revealed octreotide and [natLu]Lu-DOTATATE to significantly inhibit radiotracer uptake also in pancreas due to physiologic mSSTR expression. For all somatostatin analogs investigated, the different methods showed similar results regarding their relative inhibition of radiotracer uptake in tumors (Table 1).
Relative inhibition (%) of [64Cu]Cu-DOTATATE accumulation in MPC-mCherry tumors after co-injection of octreotide, [natLu]Lu-DOTATATE and AN-238 at 0.2 µmol/kg as determined by different methods; (RD) radiotracer distribution; data are presented as means ± SEM; significance of differences was tested as compared to control; * p<0.05; † p < 0.01; ‡ p < 0.001.
| control | octreotide | [natLu]Lu-DOTATATE | AN-238 | |
|---|---|---|---|---|
| PLI | 0 ± 11.1 | 52.9 ± 3.2† | 74.7 ± 3.8‡ | 30.0 ± 6.3* |
| PET | 0 ± 3.0 | 49.3 ± 1.8‡ | 86.2 ± 1.1‡ | 19.3 ± 7.8* |
| ex vivo RD | 0 ± 2.4 | 51.8 ± 2.5‡ | 84.9 ± 1.4‡ | 30.0 ± 9.7‡ |
Progression of MPC-mCherry tumors in animals undergoing doxorubicin, AN-238, and [177Lu]Lu-DOTATATE treatment
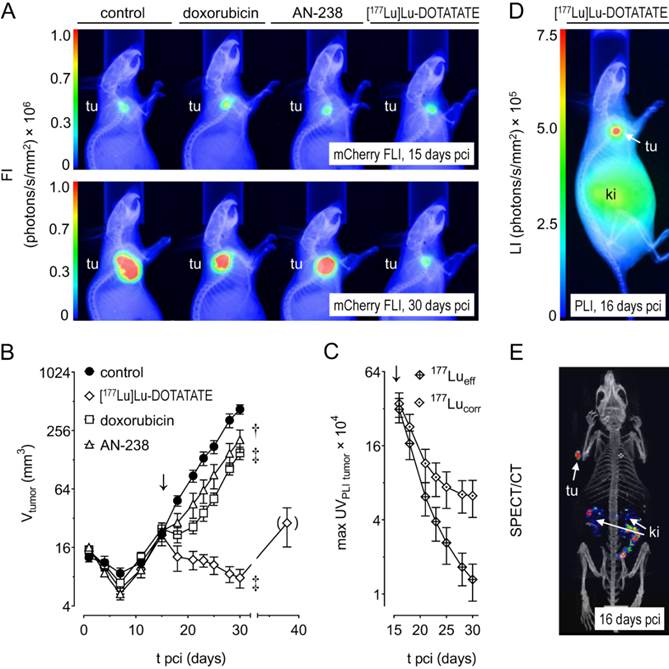
Monitoring MPC-mCherry tumor volume in mice allowed for evaluating therapeutic effects on tumor progression. Size and fluorescence intensity of tumors (Figure 4 A) appeared similar at therapy start (15 days pci) and reduced after treatment (30 days pci) with doxorubicin, AN-238, and [177Lu]Lu-DOTATATE compared to the control.
Tumor volume increased exponentially in control animals (tD = 4.0 days) between 7 and 30 days pci (Figure 4 B). After doxorubicin treatment, tumors initially declined, but regained exponential growth after 21 days pci (tD = 3.4 days). After [177Lu]Lu-DOTATATE treatment, tumors declined continuously until 30 days pci (t½.= 2.8 days). However, extended monitoring of three randomly selected animals revealed that, after 30 days pci, [177Lu]Lu-DOTATATE-treated tumors finally regained growth. After AN-238 treatment, tumor growth initially decelerated, though regaining exponential growth between 21 and 30 days pci (tD = 4.1 days). Notably, doubling time of tumor volume was significantly lower in animals treated with AN-238 compared to doxorubicin.
Distribution of the [64Cu]Cu-DOTATATE in mice bearing MPC-mCherry tumors; radiotracer accumulation was blocked by co-injection of octreotide, [natLu]Lu-DOTATATE and AN-238 at 0.2 µmol/kg; (A) FLI of tumors; (B) PLI of radiotracer distribution; (C) PET maximum intensity projections of radiotracer distribution; (D) radiotracer accumulation in tumors as determined in PLI studies; (E) radiotracer accumulation in tumors as determined in PET studies; (F) radiotracer accumulation in organs and tumors as determined in ex vivo radiotracer distribution studies; (tu) tumor; (bl) bladder; data are presented as means ± SEM; significance of differences was tested as compared to control; * p < 0.05; † p < 0.01; ‡ p < 0.001.
In relation to the control group, single treatment with doxorubicin, AN-238, or [177Lu]Lu-DOTATATE produced significant reduction of tumor growth in vivo (Table 2). Additionally, combinations of [177Lu]Lu-DOTATATE with doxorubicin (simultaneously) or AN-238 (consecutively) exerted similar effects on tumor growth compared to single [177Lu]Lu-DOTATATE treatment (Table 2, Figure S8 A-B, Table S4). As determined by ROC curve analysis, reduction of tumor growth significantly discriminated animals in therapy groups from the controls (Figure S9 F-H, Table S5).
Accumulation of 177Lu activity in MPC-mCherry tumors after [177Lu]Lu-DOTATATE treatment
After [177Lu]Lu-DOTATATE treatment of MPC-mCherry tumor-bearing animals, in vivo PLI and SPECT imaging (Figure 4 C-E) revealed strong accumulation of 177Lu activity in tumors and, to a lesser extent, in kidneys one day after therapy start (≙ 16 days pci). 177Lu activity in tumors decreased exponentially over time (Figure 4 C). Effective and decay-corrected 177Lu activity showed a half-live of 2 days and 2.3 days, respectively. Notably, decay-corrected 177Lu activity in tumors as well as tumor volume decreased with similar half-lives (Table S4). Additionally, combinations of [177Lu]Lu-DOTATATE with doxorubicin or AN-238 exerted similar effects on 177Lu activity in tumors compared to single [177Lu]Lu-DOTATATE treatment (Figure S8 D, Table S4).
Renal monoamine excretion in MPC-mCherry tumor-bearing animals undergoing doxorubicin, AN-238, and [177Lu]Lu-DOTATATE treatment
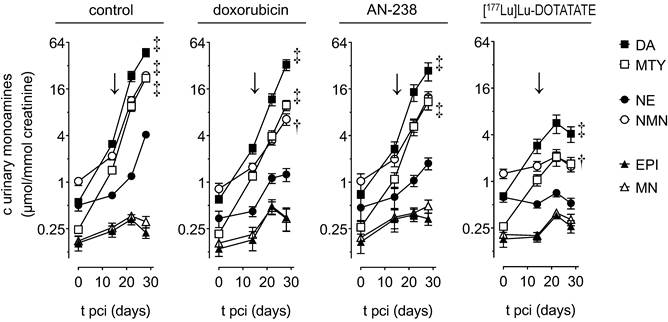
In order to evaluate therapeutic effects on tumor-related catecholamine release, we monitored renal monoamine excretion in MPC-mCherry tumor-bearing mice (Figure 5). Before treatment (0-14 days pci), animals initially showed slightly increasing excretion of dopamine, 3-methoxytyramine, norepinephrine, and normetanephrine, though a permanently low excretion of epinephrine and metanephrine.
After treatment (15-28 days pci), control, doxorubicin-, and AN-238-treated animals still showed considerably further increasing excretion of dopamine, 3-methoxytyramine, norepinephrine, and normetanephrine until 28 days pci. In contrast, animals undergoing [177Lu]Lu-DOTATATE treatment merely showed slightly increasing dopamine and 3-methoxytyramine excretion, though almost constant norepinephrine and normetanephrine until 22 days pci. Later on, overall monoamine excretion even decreased. Notably, epinephrine and metanephrine continuously retained low excretion levels in all treatment groups.
In relation to the control group, all animals undergoing therapies showed substantially reduced renal excretion of dopamine, 3-methoxytyramine, norepinephrine, and normetanephrine (Table 2). Notably, [177Lu]Lu-DOTATATE was 2-fold more effective in reducing the overall renal monoamine excretion compared to doxorubicin and AN-238. Reduction of tumor growth showed a significant positive correlation (p < 0.001) with the reduction of renal dopamine (rs = 0.864), 3-methoxytyramine (rs = 0.930), norepinephrine (rs = 0.938), normetanephrine (rs = 0.935), as well as the overall monoamine excretion (rs = 0.868).
Additionally, combination of [177Lu]Lu-DOTATATE with doxorubicin or AN-238 exerted similar effects on renal monoamine excretion compared to single [177Lu]Lu-DOTATATE treatment (Table 2, Figure S8 C). As determined by ROC curve analysis, reduction of norepinephrine and normetanephrine showed superior efficacy in discriminating animals in therapy groups from the controls (Figure S9 F-H, Table S5).
Therapeutic endpoint parameters after doxorubicin (6.9 µmol/kg), AN-238 (0.2 µmol/kg), and [177Lu]Lu-DOTATATE (80 MBq/animal) treatment of MPC-mCherry tumor-bearing mice; (red) reduction compared to control; (TG) tumor growth; (DA) dopamine; (MTY) 3-methoxytyramine; (NE) norepinephrine; (NMN) normetanephrine; (∑MA) overall monoamines DA + MTY + NE + NMN; data are presented as means ± SEM; significance of differences was tested as compared to control; * p < 0.05, † p < 0.01, ‡ p < 0.001.
| control | doxorubicin | AN-238 | [177Lu]Lu-DOTATATE | |||
|---|---|---|---|---|---|---|
| - | + doxorubicin | + AN-238 | ||||
| Reduction of tumor growth [15-30 days pci] (%) | ||||||
| TGred | 0 ± 11.3 | 68.8 ± 4.7‡ | 54.7 ± 12.0† | 103 ± 0.8‡ | 104 ± 0.7‡ | 104 ± 1.0‡ |
| Reduction of renal monoamine excretion [14-28 days pci] (%) | ||||||
| DAred | 0 ± 12.3 | 31.5 ± 11.3 | 43.9 ± 15.3* | 97.2 ± 0.9‡ | 96.4 ± 1.7‡ | 100 ± 1.1‡ |
| MTYred | 0 ± 9.9 | 57.7 ± 6.7‡ | 52.3 ± 10.8‡ | 96.9 ± 1.1‡ | 98.2 ± 1.2‡ | 101 ± 1.3‡ |
| NEred | 0 ± 9.2 | 74.8 ± 7.5‡ | 67.7 ± 8.9‡ | 99.6 ± 1.9‡ | 93.1 ± 5.3‡ | 84.1 ± 3.9‡ |
| NMNred | 0 ± 8.3 | 77.3 ± 5.1‡ | 51.8 ± 9.9‡ | 100 ± 0.7‡ | 100 ± 1.4‡ | 98.6 ± 0.7‡ |
| ∑MAred | 0 ± 10.2 | 50.2 ± 8.1† | 48.7 ± 12.3† | 97.9 ± 0.6‡ | 97.6 ± 1.0‡ | 97.9 ± 1.1‡ |
Progression of MPC-mCherry tumors in mice undergoing doxorubicin (6.9 µmol/kg), AN-238 (0.2 µmol/kg), and [177Lu]Lu-DOTATATE (80 MBq/animal) treatment; (A) mCherry FLI/X-ray overlays of tumor-bearing mice after 15 and 30 days pci; (B) monitoring of tumor volume; (C) monitoring of 177Lu activity in tumors after [177Lu]Lu-DOTATATE treatment as determined by PLI; (177Lueff/corr) effective and decay-corrected 177Lu activity; (D) [177Lu]Lu-DOTATATE PLI after 16 days pci; (E) [177Lu]Lu-DOTATATE SPECT after 16 days pci; (↓) treatment start; data are presented as means ± SEM; significance of differences was tested as compared to control after 30 days pci; * p < 0.05; † p < 0.01; ‡ p < 0.001.
Renal monoamine excretion in MPC-mCherry tumor-bearing mice undergoing doxorubicin (6.9 µmol/kg), AN-238 (0.2 µmol/kg), and [177Lu]Lu-DOTATATE (80 MBq/animal) treatment; (↓) treatment start; (DA) dopamine; (NE) norepinephrine; (MTY) 3-methoxytyramine; (NMN) normetanephrine; (EPI) epinephrine; (MN) metanephrine; data are presented as means ± SEM; significance of differences was tested as compared to renal monoamine excretion before cell injection (0 days pci); † p < 0.01; ‡ p < 0.001.
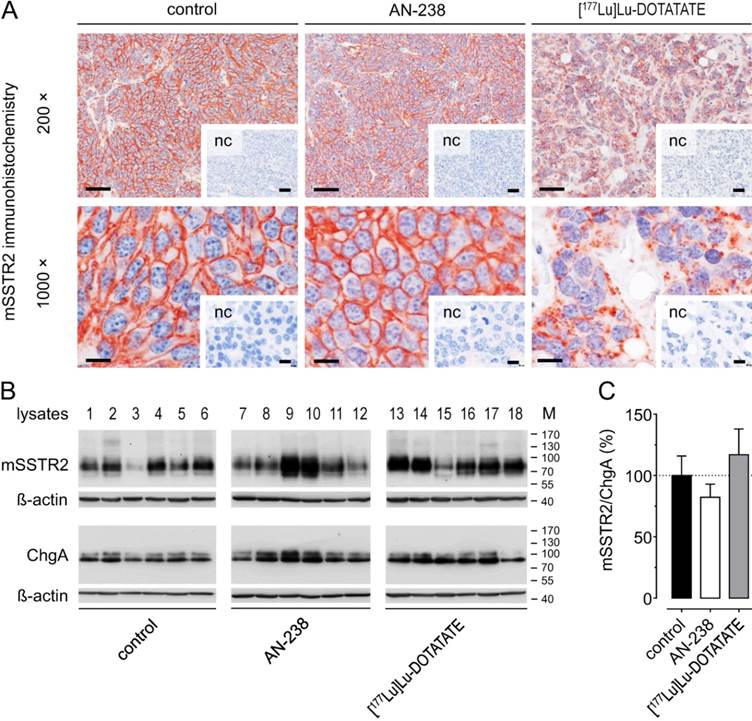
Morphology and mSSTR2 levels of tumors after AN-238 and [177Lu]Lu-DOTATATE treatment
Morphology and subcellular mSSTR2 distribution were examined in MPC-mCherry tumors after receptor-targeting therapies (30 days pci). Immunohistochemistry revealed strong membranous mSSTR2 staining and a densely united tumor cell structure in control tumors (Figure 6 A). The mSSTR2 pattern and morphology of AN-238-treated tumors appeared similar to control. In contrast, [177Lu]Lu-DOTATATE-treated tumors showed condensed mSSTR2 spots irrespective of any membranous localization as well as a dispersed tumor cell structure with irregularly-shaped nuclei and large intercellular spaces. Notably, Western blot analysis revealed no significant differences in the relative mSSTR2 tumor levels per chromogranin A-positive tumor cells after AN-238 and [177Lu]Lu-DOTATATE treatment compared to control (Figure 6 B-C).
Adverse effects of doxorubicin, AN-238, and [177Lu]Lu-DOTATATE on body weight and white blood cells
Most importantly, we did not observe any severe changes and no significant differences in body weight between treatment groups (Figure 7 A). However, body weight of control animals initially tended to remain constant until it slightly decreased after 25 days pci. In therapy groups, body weight tended to decrease immediately after both doxorubicin and AN-238, but not after [177Lu]Lu-DOTATATE treatment.
mSSTR2 status of MPC-mCherry tumors after AN-238 (0.2 µmol/kg) and [177Lu]Lu-DOTATATE (80 MBq/animal) treatment; (A) mSSTR2 immunohistochemistry on MPC-mCherry tumor sections; bars (200x) 50 µm, bars (1000x): 10 µm; (B-C) Densitometric evaluation of relative mSSTR2 levels per chromogranin A-positive tumor cells; (nc) negative control; (M) molecular weight of proteins in kDa; data are presented as means ± SEM; significance of differences was tested as compared to control.
Adverse effects of doxorubicin (6.9 µmol/kg), AN-238 (0.2 µmol/kg), and [177Lu]Lu-DOTATATE (80 MBq/animal) treatment in mice; (A) monitoring of body weight, (B) white blood cell count after 30 days pci; (↓) treatment start; data are presented as means ± SEM; significance of differences was tested as compared to control; * p < 0.05.
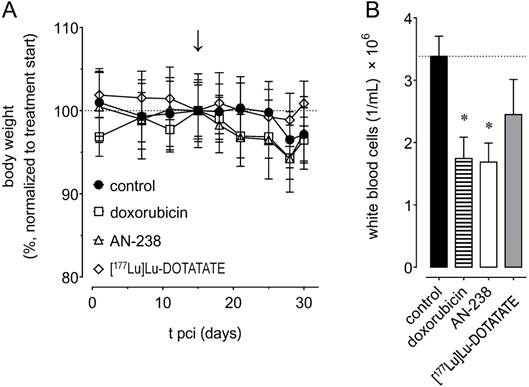
Control animals showed an average white blood cell number of 3.4 × 106/mL after 30 days pci (Figure 7 B). We found white blood cells to be significantly reduced to 1.7 × 106/mL after both doxorubicin and AN-238 treatment, but only non-significantly lowered to 2.4 × 106/mL after [177Lu]Lu-DOTATATE treatment. Additionally, combinations of [177Lu]Lu-DOTATATE with doxorubicin or AN-238 tended to increase adverse effects (Figure S8 E-F).
Discussion
This study shows that the MPC-mCherry allograft model of pheochromocytoma is a uniquely powerful tool for investigating SSTR2-targeting theranostic applications. The majority of MPC-mCherry tumors in mice showed high mSSTR2 levels, similar to SSTR2 levels in human pheochromocytoma [29]. Most importantly, [177Lu]Lu-DOTATATE treatment was by far the most efficient therapeutic strategy in the MPC-mCherry model compared to AN-238 and doxorubicin. Moreover, diagnostic testing validated the reduction of renal monoamine excretion, especially of norepinephrine and normetanephrine, as highly reliable endpoint parameters for evaluating therapeutic outcome in MPC-mCherry tumor-bearing mice.
We assessed the density and affinity of mSSTR2 in MPC-mCherry tumors using the well-established radiotracer [64Cu]Cu-DOTATATE. Among other somatostatin analogs, radiolabeled DOTATATE has proven effective for targeting human SSTR2 in tumor cell lines and tumors [30-32]. Moreover, small animal imaging and autoradiographic applications benefit from [64Cu]Cu-labeled radiotracers in terms of long radionuclide half-life and low maximal ß+ emission energy [33], allowing for higher imaging resolution and longer experimental durations.
Binding studies using [64Cu]Cu-DOTATATE on MPC-Cherry tumor sections showed high binding affinity in the low-nanomolar range and a high density of mSSTR2, similar to the density of binding sites found in various human SSTR-overexpressing tumor cell lines [34]. However, higher receptor densities have been described in some human SSTR2-transfected cell lines commonly used for preclinical investigations [35]. For efficient PET imaging, a Bmax/Kd ratio of 10 or greater is generally preferable [36]. The MPC-mCherry model meets this requirement in terms of tumor imaging using [64Cu]Cu-DOTATATE (Bmax/Kd = 11.3). In line with the in vitro binding results, PET studies in mice revealed high [64Cu]Cu-DOTATATE uptake in tumors confirming that MPC-mCherry cells maintain their mSSTR2-positive phenotype after subcutaneous engraftment in mice.
However, a certain variation of mSSTR2-levels between tumors of different animals, as well as between clonal tumor cell sub-populations within individual lesions reflect the heterogeneity of MPC-mCherry tumors, as has been described for human PHEO/PGLs as a consequence of genetic lesions and epigenetic changes [37], as well as specific copy number alterations [38]. Genetic heterogeneity of MPC-mCherry tumors remains to be investigated. In Western blot analyses, varying molecular mass of mSSTR2 between tumor cell cultures and tumors may be due to differential N-linked glycosylation, as has been described for most human SSTRs [39]. Normalization on the MPC-mCherry cell content of tumors was done by assessing chromogranin A as it forms the principal component of the soluble core of dense-core secretory granules in neuroendocrine cells [40]. Higher chromogranin A levels in tumors compared to cell cultures may indicate for alterations of the secretory granules.
Given the high mSSTR2 density, the MPC-mCherry model met the requirements for being employed in the preclinical evaluation of [177Lu]Lu-DOTATATE and AN-238 therapies for the treatment of pheochromocytoma. The peptide component DOTATATE showed high mSSTR2 binding affinity to MPC-mCherry tumor sections in the low-nanomolar range similar to the naturally-occurring ligand somatostatin-14 whereas AN-238 showed a considerably lower affinity, though, still in the nanomolar range. Since it is known that the chosen radionuclide changes the binding affinity of peptide analogs [30], the IC50 of DOTATATE in these tumors does not entirely reflect the binding affinity of [177Lu]Lu/[natLu]Lu-DOTATATE complexes employed in the in vivo experiments. As a characteristic feature of binding studies on tissue sections, affinities appear to be generally lower compared to other studies using genetically modified human SSTR2-overexpressing cell lines or pituitary membrane preparations [29, 41-43].
In mice, PLI, PET and ex vivo radiotracer distribution studies using [64Cu]Cu-DOTATATE similarly showed that, after co-injection, [natLu]Lu-DOTATATE exhibit superior tumor targeting efficiency to octreotide and AN-238. Apparently, these differences reflect the order of mSSTR2 binding affinities of the compounds as well as their plasma half-lives [44-46]. In the in vivo targeting experiments, we preferentially used octreotide as a control, due to its higher serum stability but similar SSTR2 binding affinity compared to somatostatin-14 [47]. In the context of methodology, our studies confirmed small-animal PLI as a reliable in vivo radionuclide imaging strategy for evaluating radiotracer uptake in subcutaneous tumors, featuring advantages such as lower cost, higher throughput and lower imaging time. However, quantitative evaluation and parametric imaging of radiotracers in vivo remain unique features of PET/SPECT.
In terms of evaluating therapeutic efficacy, reduction of both tumor growth and tumor-related renal monoamine excretion were considered to be the important endpoint parameters. The observed pattern of renal monoamine excretion in control animals confirmed previous findings [22]. In response to therapies, reduction of renal dopamine, 3-methoxytyramine, norepinephrine, and normetanephrine excretion correlated significantly with the reduction of tumor growth. Moreover, reduction of renal norepinephrine and normetanephrine excretion as well as the reduction of tumor growth showed similar performance in discriminating animals of the therapy groups from the controls. These results demonstrate that the reduction of tumor growth and of certain urinary monoamines are highly reliable endpoint parameters for the preclinical evaluation of therapeutic strategies in MPC-mCherry bearing animals.
In the therapy experiments, we evaluated therapeutic effects of [177Lu]Lu-DOTATATE and AN-238 in comparison with doxorubicin. Since AN-238 contains a 2-pyrrolinodoxorubicin-moiety, cytotoxic mechanisms share similarities to doxorubicin after entering the tumor cells [48, 49]. Most importantly, all therapeutic strategies evaluated here exerted significant temporary or sustaining therapeutic effects although, no therapy was able to achieve permanent cure. The superior therapeutic efficacy of [177Lu]Lu-DOTATATE reflects its ability for high-affinity mSSTR2 binding as well as for exerting 177Lu-related bystander and crossfire effects assumed to allow for overcoming tumor-specific escape strategies based on, e.g., multi-drug resistance, hypoxia, and irregular vascularization [50].
In tumors, we also observed similar half-lives of 177Lu activity and tumor volume demonstrating that trapped [177Lu]Lu-DOTATATE is actively removed over time, most likely due the elimination of apoptotic, necrotic, and senescent tumor cells. Supporting this, only [177Lu]Lu-DOTATATE therapy severely affected the histologic integrity of tumors. After radiation therapy in T cell deficient NMRI-nu/nu mice, immunologic dying cell clearance is attributed to amateur phagocytes such as fibroblasts, endothelial cells, mesothelial cells, neutrophils, and natural killer cells [51].
Both AN-238 and doxorubicin are assumed to exhibit cytotoxic effects exclusively on single tumor cells after cellular uptake [48, 49]. In this study, doxorubicin was applied at a 34-fold higher dose in vivo compared to AN-238. However, both compounds showed similar therapeutic effects. These results reflect the benefit of the mSSTR2-targeting properties of AN-238 as well as the effectiveness of the 2-pyrrolinodoxorubicin moiety, which itself is known to be more potent than doxorubicin [52].
Due to the fact that 2-pyrrolinodoxorubicin is no longer available for research, it was not possible to investigate its sole therapeutic effect here. However, superior anti-tumor efficacy of AN-238 to 2-pyrrolinodoxorubicin on tumor cells with elevated SSTR2 levels has been demonstrated in many studies [16-20]. AN-238 was applied in nude mice at its maximum tolerated dose [53], whereas doxorubicin was applied at one third of its maximum tolerated dose [54]. Hence, there is no room for further increasing the AN-238 dose in the MPC-mCherry model.
In the MPC-mCherry model, all single therapies were tolerated well. Reduced white blood cells result from the cytotoxic effects of doxorubicin on rapidly dividing multipotent hematopoietic progenitor cells as well as from the capability of AN-238 and [177Lu]Lu-DOTATATE for targeting mSSTR2 on lymphocytes and inflammatory cells [39]. Importantly, agonist-induced downregulation of mSSTR2 was not present in tumors after [177Lu]Lu-DOTATATE and AN-238 treatment. These results suggest that repeated treatment cycles and combinations of SSTR2-targeting radionuclide and cytotoxic therapies may be effective. However, our pilot studies on combining [177Lu]Lu-DOTATATE with doxorubicin (simultaneously) or AN-238 (in quick succession) revealed similar therapeutic effects compared to single [177Lu]Lu-DOTATATE, thereby tending to increase adverse effects. These results indicate that especially fractionated therapy combinations should be preferred to prevent cumulative adverse effects.
Although the MPC-cell models resemble at least in part biochemical features and characteristics of sporadic and familial human PHEO/PGLs [22, 55] the underlying neurofibromatosis type 1 gene mutation and downstream pathways do not accurately reflect the typical mutations and pathways responsible for human metastatic disease [56, 57]. Subcutaneous tumor models may also not be a predictor of organ metastatic lesion responses to treatment. Considering that the sensitivity to cytotoxic drugs frequently depends on the proliferative activity, therapeutic efficacy of doxorubicin and AN-238 may turn out higher in the MPC-mCherry model due to the more rapidly growing murine tumors compared to human PHEO/PGLs [58, 59].
Nevertheless, the model does recapitulate the clinical situation in which [68Ga]Ga-DOTATATE has been shown to be a superior imaging agent compared to others for metastatic PHEO/PGLs due to mutations of the succinate dehydrogenase subunit 2 (SDHB) gene [10, 60]. This is also in agreement with other findings of high SSTR2 expression in PHEO/PGLs due to SDHB mutations [61]. These clinical findings support the possibility that utility of [177Lu]Lu-DOTATATE reported here in the MPC-mCherry model may indeed translate to the clinic, and particularly to metastatic PHEO/PGLs due to SDHB mutations, which carry a high metastatic risk responsible for 40-50% of all cases of malignant disease [62, 63].
Taken together, the present preclinical and accumulating clinical findings provide a basis for trials directed to radionuclide therapies targeting SSTR2 in metastatic PHEO/PGL. At the same time given the likelihood of therapeutic escape, reflected here by a regain in tumor growth at 15 days after treatment, it seems that further preclinical studies will also be important for refinement of SSTR2-directed radiotherapies. Especially, fractionated combinations of [177Lu]Lu-labeled somatostatin analogs with compounds addressing additional therapeutic mechanisms, such as radiosensitizers, angiogenesis inhibitors, mitogen-activated protein kinase inhibitors or systemic chemotherapeutics may substantially enhance the therapeutic outcome and the duration of tumor remission.
Abbreviations
As: specific activity; c: concentration; DOTA: 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid; FI: fluorescence intensity; GBq: gigabecquerel; kDa: kilodalton; LI: luminescence intensity; MBq: megabecquerel; PBS: phosphate-buffered saline; pci: post cell injection; w/v: weights per volume; v/v: volume per volume.
Supplementary Material
Supplementary Tables and Figures.
Acknowledgements
The authors greatly acknowledge the excellent technical assistance of Andrea Suhr, Regina Herrlich, Sonja Lehnert, Peggy Wecke, and Sebastian Meister. The authors further thank the staff of the Production of Radiopharmaceuticals Department at the Institute of Radiopharmaceutical Cancer Research, Helmholtz-Zentrum Dresden-Rossendorf for the production of [64Cu]Cu. MPC 4/30PRR cells were kindly provided by Prof. Arthur Tischler, Dr. James Powers and Prof. Karel Pacak. This work was supported by the Deutsche Forschungsgemeinschaft (Grants ZI-1362/2-1/2-2 (C.G.Z. & G.E.) and BE-2607/1-1/1-2 (R.B. & J.P)). C.G.Z. was further supported by the German Ministry for Education and Research (BMBF). S.R.B., G.E. and C.G.Z. further acknowledge the "transcampus initiative" between Kings College London and Technische Universität Dresden for combining forces for the treatment of rare diseases.
Author contributions
C.G.Z., R.B., G.E. and J.P. jointly conceived and supervised the study. A.V.S. gave advice on performing cytotoxic therapies in mice and supported the supply of reagents. G.E. provided analytical tools. S.R.B., M.E.B., N.L.B. and M.B. coordinated international collaborations and gave conceptual and editorial advice. M.C. installed the mCherry reporter gene in MPC cells. E.F.Z. acquired Western blot and immuncyto- /histochemistry data. R.B. conducted the PET/SPECT experiments. M.P. performed the liquid chromatography tandem mass spectrometry analyses. M.U. designed and performed experiments, analyzed data and wrote the manuscript. All authors discussed the results and implications and commented on the manuscript at all stages.
Conflict of Interest
The authors have declared that no conflict of interest exists.
References
1. Harding JL, Yeh MW, Robinson BG, Delbridge LW, Sidhu SB. Potential pitfalls in the diagnosis of phaeochromocytoma. Med J Aust. 2005;182:637-40
2. Lenders JW, Eisenhofer G, Mannelli M, Pacak K. Phaeochromocytoma. Lancet. 2005;366:665-75
3. Zinnamosca L, Petramala L, Cotesta D, Marinelli C, Schina M, Cianci R. et al. Neurofibromatosis type 1 (NF1) and pheochromocytoma: prevalence, clinical and cardiovascular aspects. Arch Dermatol Res. 2011;303:317-25
4. Chrisoulidou A, Kaltsas G, Ilias I, Grossman AB. The diagnosis and management of malignant phaeochromocytoma and paraganglioma. Endocr Relat Cancer. 2007;14:569-85
5. Eisenhofer G, Siegert G, Kotzerke J, Bornstein SR, Pacak K. Current progress and future challenges in the biochemical diagnosis and treatment of pheochromocytomas and paragangliomas. Horm Metab Res. 2008;40:329-37
6. Ayala-Ramirez M, Feng L, Habra MA, Rich T, Dickson PV, Perrier N. et al. Clinical benefits of systemic chemotherapy for patients with metastatic pheochromocytomas or sympathetic extra-adrenal paragangliomas: insights from the largest single-institutional experience. Cancer. 2012;118:2804-12
7. Pacak K, Fojo T, Goldstein DS, Eisenhofer G, Walther MM, Linehan WM. et al. Radiofrequency ablation: a novel approach for treatment of metastatic pheochromocytoma. J Natl Cancer Inst. 2001;93:648-9
8. Adjalle R, Plouin PF, Pacak K, Lehnert H. Treatment of malignant pheochromocytoma. Horm Metab Res. 2009;41:687-96
9. Ziegler CG, Brown JW, Schally AV, Erler A, Gebauer L, Treszl A. et al. Expression of neuropeptide hormone receptors in human adrenal tumors and cell lines: antiproliferative effects of peptide analogues. Proc Natl Acad Sci U S A. 2009;106:15879-84
10. Janssen I, Blanchet EM, Adams K, Chen CC, Millo C, Herscovitch P. et al. Superiority of [68Ga]-DOTATATE PET/CT to other functional imaging modalities in the localization of SDHB-associated metastatic pheochromocytoma and paraganglioma. Clin Cancer Res. 2015
11. Kwekkeboom DJ, de Herder WW, Kam BL, van Eijck CH, van Essen M, Kooij PP. et al. Treatment with the radiolabeled somatostatin analog [177Lu-DOTA0,Tyr3]octreotate: toxicity, efficacy, and survival. J Clin Oncol. 2008;26:2124-30
12. Imhof A, Brunner P, Marincek N, Briel M, Schindler C, Rasch H. et al. Response, survival, and long-term toxicity after therapy with the radiolabeled somatostatin analogue [90Y-DOTA]-TOC in metastasized neuroendocrine cancers. J Clin Oncol. 2011;29:2416-23
13. Forrer F, Riedweg I, Maecke HR, Mueller-Brand J. Radiolabeled DOTATOC in patients with advanced paraganglioma and pheochromocytoma. Q J Nucl Med Mol Imaging. 2008;52:334-40
14. Zovato S, Kumanova A, Dematte S, Sansovini M, Bodei L, Di Sarra D. et al. Peptide receptor radionuclide therapy (PRRT) with 177Lu-DOTATATE in individuals with neck or mediastinal paraganglioma (PGL). Horm Metab Res. 2012;44:411-4
15. Puranik AD, Kulkarni HR, Singh A, Baum RP. Peptide receptor radionuclide therapy with (90)Y/ (177)Lu-labelled peptides for inoperable head and neck paragangliomas (glomus tumours). Eur J Nucl Med Mol Imaging. 2015;42:1223-30
16. Kiaris H, Schally AV, Nagy A, Szepeshazi K, Hebert F, Halmos G. A targeted cytotoxic somatostatin (SST) analogue, AN-238, inhibits the growth of H-69 small-cell lung carcinoma (SCLC) and H-157 non-SCLC in nude mice. Eur J Cancer. 2001;37:620-8
17. Szepeshazi K, Schally AV, Halmos G, Armatis P, Hebert F, Sun B. et al. Targeted cytotoxic somatostatin analogue AN-238 inhibits somatostatin receptor-positive experimental colon cancers independently of their p53 status. Cancer Res. 2002;62:781-8
18. Szepeshazi K, Schally AV, Halmos G, Sun B, Hebert F, Csernus B. et al. Targeting of cytotoxic somatostatin analog AN-238 to somatostatin receptor subtypes 5 and/or 3 in experimental pancreatic cancers. Clin Cancer Res. 2001;7:2854-61
19. Plonowski A, Schally AV, Nagy A, Sun B, Szepeshazi K. Inhibition of PC-3 human androgen-independent prostate cancer and its metastases by cytotoxic somatostatin analogue AN-238. Cancer Res. 1999;59:1947-53
20. Kidd M, Schally AV, Pfragner R, Malfertheiner MV, Modlin IM. Inhibition of proliferation of small intestinal and bronchopulmonary neuroendocrine cell lines by using peptide analogs targeting receptors. Cancer. 2008;112:1404-14
21. Powers JF, Evinger MJ, Tsokas P, Bedri S, Alroy J, Shahsavari M. et al. Pheochromocytoma cell lines from heterozygous neurofibromatosis knockout mice. Cell Tissue Res. 2000;302:309-20
22. Ullrich M, Bergmann R, Peitzsch M, Cartellieri M, Qin N, Ehrhart-Bornstein M. et al. In vivo fluorescence imaging and urinary monoamines as surrogate biomarkers of disease progression in a mouse model of pheochromocytoma. Endocrinology. 2014;155:4149-56
23. Szelecsényi F, Blessing G, Qaim SM. Excitation functions of proton induced nuclear reactions on enriched 61Ni and 64Ni: Possibility of production of no-carrier-added 61Cu and 64Cu at a small cyclotron. Applied Radiation and Isotopes. 1993;44:575-80
24. McCarthy DW, Shefer RE, Klinkowstein RE, Bass LA, Margeneau WH, Cutler CS. et al. Efficient production of high specific activity 64Cu using a biomedical cyclotron. Nucl Med Biol. 1997;24:35-43
25. de Blois E, Chan HS, Konijnenberg M, de Zanger R, Breeman WA. Effectiveness of quenchers to reduce radiolysis of (111)In- or (177)Lu-labelled methionine-containing regulatory peptides. Maintaining radiochemical purity as measured by HPLC. Curr Top Med Chem. 2012;12:2677-85
26. Schmitt A, Bernhardt P, Nilsson O, Ahlman H, Kolby L, Maecke HR. et al. Radiation therapy of small cell lung cancer with 177Lu-DOTA-Tyr3-octreotate in an animal model. J Nucl Med. 2004;45:1542-8
27. Kniess T, Laube M, Bergmann R, Sehn F, Graf F, Steinbach J. et al. Radiosynthesis of a 18F-labeled 2,3-diarylsubstituted indole via McMurry coupling for functional characterization of cyclooxygenase-2 (COX-2) in vitro and in vivo. Bioorg Med Chem. 2012;20:3410-21
28. Peitzsch M, Pelzel D, Glockner S, Prejbisz A, Fassnacht M, Beuschlein F. et al. Simultaneous liquid chromatography tandem mass spectrometric determination of urinary free metanephrines and catecholamines, with comparisons of free and deconjugated metabolites. Clin Chim Acta. 2013;418:50-8
29. Reubi JC, Waser B, Schaer JC, Laissue JA. Somatostatin receptor sst1-sst5 expression in normal and neoplastic human tissues using receptor autoradiography with subtype-selective ligands. Eur J Nucl Med. 2001;28:836-46
30. Reubi JC, Schar JC, Waser B, Wenger S, Heppeler A, Schmitt JS. et al. Affinity profiles for human somatostatin receptor subtypes SST1-SST5 of somatostatin radiotracers selected for scintigraphic and radiotherapeutic use. Eur J Nucl Med. 2000;27:273-82
31. Breeman WA, de Blois E, Sze Chan H, Konijnenberg M, Kwekkeboom DJ, Krenning EP. 68Ga-labeled DOTA-peptides and 68Ga-labeled radiopharmaceuticals for positron emission tomography: current status of research, clinical applications, and future perspectives. Semin Nucl Med. 2011;41:314-21
32. Haug AR, Auernhammer CJ, Wangler B, Schmidt GP, Uebleis C, Goke B. et al. 68Ga-DOTATATE PET/CT for the early prediction of response to somatostatin receptor-mediated radionuclide therapy in patients with well-differentiated neuroendocrine tumors. J Nucl Med. 2010;51:1349-56
33. Bergmann R, Ruffani A, Graham B, Spiccia L, Steinbach J, Pietzsch J. et al. Synthesis and radiopharmacological evaluation of 64Cu-labeled bombesin analogs featuring a bis(2-pyridylmethyl)-1,4,7-triazacyclononane chelator. Eur J Med Chem. 2013;70:434-46
34. Smith-Jones PM, Bischof C, Leimer M, Gludovacz D, Angelberger P, Pangerl T. et al. DOTA-lanreotide: a novel somatostatin analog for tumor diagnosis and therapy. Endocrinology. 1999;140:5136-48
35. Zhang H, Moroz MA, Serganova I, Ku T, Huang R, Vider J. et al. Imaging expression of the human somatostatin receptor subtype-2 reporter gene with 68Ga-DOTATOC. J Nucl Med. 2011;52:123-31
36. Patel S, Gibson R. In vivo site-directed radiotracers: a mini-review. Nucl Med Biol. 2008;35:805-15
37. Dahia PL. Pheochromocytoma and paraganglioma pathogenesis: learning from genetic heterogeneity. Nat Rev Cancer. 2014;14:108-19
38. Crona J, Backman S, Maharjan R, Mayrhofer M, Stalberg P, Isakson A. et al. Spatio-temporal heterogeneity characterizes the genetic landscape of pheochromocytoma and defines early events in tumourigenesis. Clin Cancer Res. 2015
39. Patel YC. Somatostatin and its receptor family. Front Neuroendocrinol. 1999;20:157-98
40. Giovinazzo F, Schimmack S, Svejda B, Alaimo D, Pfragner R, Modlin I. et al. Chromogranin A and its fragments as regulators of small intestinal neuroendocrine neoplasm proliferation. PLoS One. 2013;8:e81111
41. Nagy A, Schally AV, Halmos G, Armatis P, Cai RZ, Csernus V. et al. Synthesis and biological evaluation of cytotoxic analogs of somatostatin containing doxorubicin or its intensely potent derivative, 2-pyrrolinodoxorubicin. Proc Natl Acad Sci U S A. 1998;95:1794-9
42. Schottelius M, Simecek J, Hoffmann F, Willibald M, Schwaiger M, Wester HJ. Twins in spirit - episode I: comparative preclinical evaluation of [68Ga]DOTATATE and [68Ga]HA-DOTATATE. EJNMMI Res. 2015;5:22
43. Reubi JC. Peptide receptors as molecular targets for cancer diagnosis and therapy. Endocr Rev. 2003;24:389-427
44. Baum RP, Wahl RL. Third Theranostics World Congress on Gallium-68 and PRRT: Abstracts. J Nucl Med. 2015;56(Suppl 2):2A-30
45. Maecke HR, Reubi JC. Somatostatin receptors as targets for nuclear medicine imaging and radionuclide treatment. J Nucl Med. 2011;52:841-4
46. Lasfer M, Vadrot N, Schally AV, Nagy A, Halmos G, Pessayre D. et al. Potent induction of apoptosis in human hepatoma cell lines by targeted cytotoxic somatostatin analogue AN-238. J Hepatol. 2005;42:230-7
47. Kumar U, Grant M. Somatostatin and somatostatin receptors. Results Probl Cell Differ. 2010;50:137-84
48. Schally AV, Szepeshazi K, Nagy A, Comaru-Schally AM, Halmos G. New approaches to therapy of cancers of the stomach, colon and pancreas based on peptide analogs. Cell Mol Life Sci. 2004;61:1042-68
49. Emons G, Sindermann H, Engel J, Schally AV, Grundker C. Luteinizing hormone-releasing hormone receptor-targeted chemotherapy using AN-152. Neuroendocrinology. 2009;90:15-8
50. Gulenchyn KY, Yao X, Asa SL, Singh S, Law C. Radionuclide therapy in neuroendocrine tumours: a systematic review. Clin Oncol (R Coll Radiol). 2012;24:294-308
51. Lauber K, Ernst A, Orth M, Herrmann M, Belka C. Dying cell clearance and its impact on the outcome of tumor radiotherapy. Front Oncol. 2012;2:116
52. Nagy A, Armatis P, Schally AV. High yield conversion of doxorubicin to 2-pyrrolinodoxorubicin, an analog 500-1000 times more potent: structure-activity relationship of daunosamine-modified derivatives of doxorubicin. Proc Natl Acad Sci U S A. 1996;93:2464-9
53. Nagy A, Plonowski A, Schally AV. Stability of cytotoxic luteinizing hormone-releasing hormone conjugate (AN-152) containing doxorubicin 14-O-hemiglutarate in mouse and human serum in vitro: implications for the design of preclinical studies. Proc Natl Acad Sci U S A. 2000;97:829-34
54. Miyazaki M, Nagy A, Schally AV, Lamharzi N, Halmos G, Szepeshazi K. et al. Growth inhibition of human ovarian cancers by cytotoxic analogues of luteinizing hormone-releasing hormone. J Natl Cancer Inst. 1997;89:1803-9
55. Korpershoek E, Pacak K, Martiniova L. Murine models and cell lines for the investigation of pheochromocytoma: applications for future therapies? Endocr Pathol. 2012;23:43-54
56. Brems H, Beert E, de Ravel T, Legius E. Mechanisms in the pathogenesis of malignant tumours in neurofibromatosis type 1. Lancet Oncol. 2009;10:508-15
57. Cascon A, Robledo M. MAX and MYC: a heritable breakup. Cancer Res. 2012;72:3119-24
58. Walther MM, Reiter R, Keiser HR, Choyke PL, Venzon D, Hurley K. et al. Clinical and genetic characterization of pheochromocytoma in von Hippel-Lindau families: Comparison with sporadic pheochromocytoma gives insight into natural history of pheochromocytoma. Journal of Urology. 1999;162:659-64
59. Amar L, Baudin E, Burnichon N, Peyrard S, Silvera S, Bertherat J. et al. Succinate dehydrogenase B gene mutations predict survival in patients with malignant pheochromocytomas or paragangliomas. J Clin Endocrinol Metab. 2007;92:3822-8
60. Tan TH, Hussein Z, Saad FF, Shuaib IL. Diagnostic Performance of 68Ga-DOTATATE PET/CT, 18F-FDG PET/CT and 131I-MIBG Scintigraphy in Mapping Metastatic Pheochromocytoma and Paraganglioma. Nucl Med Mol Imaging. 2015;49:143-51
61. Elston MS, Meyer-Rochow GY, Conaglen HM, Clarkson A, Clifton-Bligh RJ, Conaglen JV. et al. Increased SSTR2A and SSTR3 expression in succinate dehydrogenase-deficient pheochromocytomas and paragangliomas. Hum Pathol. 2015;46:390-6
62. Brouwers FM, Eisenhofer G, Tao JJ, Kant JA, Adams KT, Linehan WM. et al. High frequency of SDHB germline mutations in patients with malignant catecholamine-producing paragangliomas: implications for genetic testing. J Clin Endocrinol Metab. 2006;91:4505-9
63. Sue M, Martucci V, Frey F, Lenders JM, Timmers HJ, Peczkowska M. et al. Lack of utility of SDHB mutation testing in adrenergic metastatic phaeochromocytoma. Eur J Endocrinol. 2015;172:89-95
Author contact
![]() Corresponding author: Dr. Christian G. Ziegler, PhD. Molecular Endocrinology, Medical Clinic III, University Hospital Carl Gustav Carus, Fiedlerstr. 42, 01307 Dresden, Germany. Phone: +49-351-4586615, Fax: +49-351-4586336 E-mail: Christian.Zieglerde.
Corresponding author: Dr. Christian G. Ziegler, PhD. Molecular Endocrinology, Medical Clinic III, University Hospital Carl Gustav Carus, Fiedlerstr. 42, 01307 Dresden, Germany. Phone: +49-351-4586615, Fax: +49-351-4586336 E-mail: Christian.Zieglerde.