Impact Factor
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2016; 6(5):666-678. doi:10.7150/thno.14068 This issue Cite
Research Paper
A Comparison Study of iTEP Nanoparticle-Based CTL Vaccine Carriers Revealed a Surprise Relationship between the Stability and Efficiency of the Carriers
Shuyun Dong1, Tiefeng Xu2, Peng Zhao1, Kristin N. Parent3, Mingnan Chen1 ![]()
1. Department of Pharmaceutics and Pharmaceutical Chemistry, University of Utah, Salt Lake City, Utah 84112, U.S.A.
2. Cancer Institute of Hainan Medical College, Affiliated Hospital of Hainan Medical College, Haikou City, Hainan Province 570102, China.
3. Dept. of Biochemistry and Molecular Biology, Michigan State University, East Lansing, MI 48824, U.S.A.
Received 2015-10-7; Accepted 2016-2-8; Published 2016-3-10
Abstract

Vaccine carriers have been shown to enhance cytotoxic T lymphocyte (CTL) epitope peptide vaccines by addressing intrinsic limitations of the vaccines. We have previously developed an immune-tolerant elastin-like polypeptide (iTEP)-based nanoparticle (NP) as an effective and unique CTL vaccine carrier. The NP is unique for its humoral immune tolerance, flexible structure, and ability to deliver CTL vaccines as polypeptide fusions.
Here, we aimed to improve the NP by increasing its stability since we found it was not stable. We thus generated a more stable iTEP NP (ST-NP) and used it to deliver a CTL peptide vaccine, SIINFEKL. However, we surprisingly found that the ST-NP had a lower efficiency than the previously developed, marginally stable iTEP NP (MS-NP) in terms of promoting vaccine presentation and vaccine-induced CTL responses. On the other hand, dendritic cells (DCs) showed preferential uptake of the ST-NP but not the MS-NP. To develop an iTEP vaccine carrier that outperforms both the MS-NP and the ST-NP, we devised an iTEP NP that has a changeable stability responsive to a cytosolic, reductive environment, termed reductive environment-dependent NP or RED-NP. The RED-NP showed an intermediate ability to promote vaccine presentation and T cell responses in vitro between the MS-NP and the ST-NP. However, the RED-NP induced the strongest CTL responses in vivo among all three NPs. In conclusion, iTEP NPs that have a dynamically changeable stability are most effective to deliver and enhance CTL peptide vaccines. The work also demonstrated the versatile nature of iTEP vaccine carriers.
Keywords: Cytotoxic T lymphocyte (CTL) vaccine, iTEP nanoparticle, changeable carrier stability, vaccine processing.
Introduction
Cytotoxic T lymphocyte (CTL) epitope peptide-based vaccines are broadly tested preclinically and clinically for the treatment of diseases ranging from cancer to viral infections [1-11]. These types of vaccines edge out other forms of CTL vaccines for their unparalleled specificity and low production cost [12-14]. However, the potency of peptide vaccines is hindered by their size, usually 8-9 residues. The small size has been linked to several limitations of the vaccines: short half-lives, inefficient uptake or processing by dendritic cells (DCs), and poor cross-presentation by DCs [14]. Recently, nanocarriers were utilized to increase the size of the vaccines and hence resolve the limitations [15-19]. Indeed, these nanocarriers increase the potency of the vaccines [16, 18, 20]. However, the prospect of clinically applying these carriers is plagued by the limitations of the carriers such as toxicity and non-biodegradability [17, 21]. In addition, some of these carriers have low reproducibility due to the complex components of the carriers [21, 22].
We have striven to design and generate a robust CTL peptide vaccine carrier that is devoid of the aforementioned limitations. Recombinant polypeptides such as elastin-like polypeptides are an appealing candidate material for the vaccine carrier. The polypeptides are safe and biodegradable [23]. The vaccine peptides can be fused with the polypeptide carriers through peptide bonds in the same way these vaccines are linked with their parent antigen proteins in nature. This natural linkage between the vaccine and carriers has at least two advantages. First, the vaccines can be released efficiently from the carriers by s-proteasomes and immunoproteasomes in antigen presenting cells (APCs) because APCs are programmed to process antigens and generate CTL epitopes using these proteasomes [24]. Second, the fusions consisting of the vaccines and the polypeptide carriers may be easily and consistently produced using expression systems such as E.coli. Compared to synthetic polymer-based carriers, polypeptide-based carriers are composed of clearly defined and monodispersed materials [25].
Previously, we developed immune-tolerant elastin-like polypeptides (iTEPs) and engineered an iTEP-based NP as CTL peptide vaccine carriers [26]. We found that the NP enhanced the presentation of the peptide vaccine by DCs and boosted the vaccine-induced response [26]. The iTEP NP shares a similar chemical nature and size as virus-like particles (VLPs), the most developed polypeptide-based CTL vaccine carriers [27-29]. However, there are at least two advantages of the iTEP NP over VLPs. First, the iTEP NP is non-immunogenic; more specifically, it is humorally tolerated. There are studies showing that humoral responses against CTL vaccine carriers compromise immune responses toward the CTL vaccines [30-32]. Second, the iTEP NP has a flexible, micelle-like structure, which allows it to accommodate large vaccine payloads or adjuvants. For example, multiple copies of the same or different types of vaccines may be loaded to the iTEP NP without compromising its structure. In contrast, the VLPs can only load peptides smaller than 50 residues [28].
Our further study showed that the aforementioned iTEP NP carrier enhanced the CTL vaccines but was only marginally stable. We wondered whether a more stable NP carrier would be able to boost the vaccine enhancement effect of the marginally stable carrier. We, thus, generated and used a more stable iTEP NP (ST-NP) carrier to deliver a CTL vaccine. However, we found that the vaccine delivered by the ST-NP was poorly presented by DCs and that the vaccine-induced responses were weaker when compared to the vaccines delivered by the aforementioned marginally stable NP (MS-NP). Meanwhile, we found that DCs preferred to uptake the ST-NP over the MS-NP. Further, we noticed that although the MS-NP outperformed the ST-NP both in vitro and in vivo, the superiority was less intense in vivo than in vitro. We reasoned that while the high stability of the ST-NP was beneficial in delivering its vaccine payloads to DCs in vivo and promoting the DC uptake of the vaccines, the high stability, however, hindered vaccine processing in DCs. The hindrance, most likely, happened at the step where the vaccine payloads were released from the ST-NP. To test this conjecture, we designed a new iTEP NP that remained stable until it reached a reductive environment, in our case, cytosol. We termed the new NP the reductive-environment dependent NP or RED-NP. We found that the RED-NP was indeed more effective than either the MS-NP or the ST-NP in the enhancement of the CTL vaccines. We thus concluded that the stability of iTEP NP carriers played an important role in the delivery and processing of CTL peptide vaccines, and more importantly, that a CTL vaccine carrier performed at its best if it had kinetically changeable stability. The study also demonstrated that iTEPs were versatile vaccine carriers and had great potential to improve CTL peptide vaccines.
Materials and Methods
Cell lines and mice
The DC2.4 DC line (H-2Kb) was kindly provided by Dr. Kenneth Rock (University of Massachusetts, USA). The DC2.4 cells were cultured in RPMI-1640 medium supplemented with 10% heat inactivated fetal calf serum, 2 mM glutamine, 1% non-essential amino acids, 1% Hepes, 50 μM β-Mercaptoethanol, 100 units/ml penicillin and 100 μg/mL streptomycin (Invitrogen, USA). The B3Z T-cell hybridoma specific for H-2Kb: OVA257-264 (SIIFEKL) was kindly provided by Dr. Nilabh Shastri (University of Califorlia, USA). The B3Z cells were cultured in RPMI-1640 medium supplemented with 10% heat inactivated fetal calf serum, 2 mM glutamine, 1mM pyruvate, 50 μM β-Mercaptoethanol, 100 units/mL penicillin and 100 μg/mL streptomycin (Invitrogen, USA). EA.hy926, bEnd.3 and 3T3 cells were obtained from ATCC and were maintained in in DMEM medium supplemented with 10% heat inactivated fetal calf serum. C57BL/6 female mice, 6-8 weeks of age, were obtained from Jackson Laboratories. This study followed an approved protocol by the Institutional Animal Care and Use Committee (IACUC) at the Utah University.
Other materials will be described in the following individual studies.
Construction of the expression plasmids of iTEP-vaccine fusions
The genes encoding iTEP vaccines were synthesized on a modified pET25b(+) vector using a previously described method [26]. Specifically, genes that encoded subunits of iTEPA (GVLPGVG)4 and iTEPB (GAGVPG)5 were generated by annealing the sense and antisense oligonucleotides of these genes together (Table S1). Then these annealed iTEP genes were inserted to the vector at its BseRI site. Lastly, iTEP genes were polymerized by the PRe-RDL method until a desired length of iTEP genes were achieved. iTEPA contained 28 repeats of GVLPGVG and was named iTEPA28. iTEPB had 70 repeats of GAGVPG and was named iTEPB70. The genes encoding the fusions of iTEPB70-iTEPA28, iTEPB70-iTEPA56, iTEPB70-iTEPA28-pOVA, iTEPB70-iTEPA56-pOVA, iTEPB70-iTEPA28-(G8C)4-pOVA and iTEPB70-iTEPA28-(G1C)4-pOVA were generated in a similar manner. The sequences of the oligonucleotides used for constructing these genes are listed in Table S1. It was noteworthy that the sequences for the pOVA fusion had two copies of the CTL epitope, SIINFEKL, and one natural flanking residue on the each side of SIINFEKL. After the resulting expression vectors were transformed into DH5α for their amplification, the lengths of the coding genes were confirmed by an Xba I and BamH I double digestion and followed agarose gel analysis (Figure S1). The coding genes were also verified by DNA sequencing (Genewiz, USA).
Production and purification of iTEP-vaccine fusions and iTEPs
The fusions and iTEPs were produced and purified as previously described [26]. The sonication was done with Sonic Dismembrator Model 500 (Fisher Scientific). The endotoxin was also removed as previously described [26]. The purity of the fusions and iTEPs was assessed by SDS-PAGE (Figure S2).
Preparation of reductive environment-responsive iTEP-vaccine fusions
The protein purification of iTEPB70-iTEPA28-(G1C)4-pOVA and iTEPB70- iTEPA28-(G8C)4-pOVA was carried out in PBS with 10mM TCEP-HCl, pH 7.0 to keep the reductive condition. The final purified protein was dissolved in water before lyophilization. The purified proteins were then treated with 0.3% H2O2 at 37°C for 15 min for oxidization to generate disulfide bond stabilized particles. The H2O2 was then removed by Amicon centrifugal filter devices (Millipore, USA).
Characterization of hydrodynamic diameters of iTEP-vaccine fusions
Hydrodynamic diameters of iTEP-vaccine fusions were determined by dynamic light scattering (DLS) using a Zetasizer Nano-ZS instrument (Malvern Instruments, Malvern, UK) as previously described [33]. The samples were resuspended at concentrations of 5, 25, 50, and 100 μM in PBS and sat at room temperature overnight before measurement. Some samples were kept in 5% CO2 at 37°C for 16 h before DLS measurement. Only hydradynamic diameter results of the sample of 5 μM concentration were reported because hydradynamic diameters were not affected by the used sample concentrations. Hydrodynamic diameters were primarily presented using the size-by-number distribution approach. Z-average values and sizes-by-intensity data, however, were also reported.
Negative-stain, transmission electron microscopy (TEM) of iTEP-vaccine fusions
TEM was done as previously described [26].
Measurement of critical micelle concentrations (CMCs) of iTEP-vaccine fusions by pyrene assay
The CMCs of iTEP-vaccine fusions (NPs) were measured through fluorescence spectra by using pyrene as a hydrophobic fluorescent probe as described previously with some adjustments [5, 34]. Briefly, the fusions were serially 2X time diluted in PBS starting from 250 µM in 96-well black clear-bottom plates. The final volume for each dilution was 150 µL. 3 µL of a stock solution of 30 µM pyrene in ethanol was added to each well and mixed well. Fluorescence of these samples were scanned using an Infinite M1000 PRO plate reader (Tecan Trading AG, Switzerland) with the setting: Ex: 334 nm with a slit of 10 nm, Em: ranging 360-400 nm with a slit of 2.5 nm. There were 4 peaks recorded for pyrene between 360-400 nm. The ratio of emission peak number 1 (I1; 370-373 nm) and emission peak number 3 (I3; 381-384 nm) of pyrene (I1/I3) was plotted as a function of iTEP concentration. The data points were fitted to a sigmoidal-dose response curve, and the CMC was defined as the inflection point of the curve.
Quantification of free sulfhydryl groups of RED-NP after reducing treatments
20 mg of RED-NP were dissolved in 2mL of H2O and treated with 1 mM of GSH for 16 h. The solution was then dialyzed (membrane cutoff MW: 3,500 Dalton) in deionized water at 4°C for 24 h. Then the sample was concentrated to 0.5 mL using Amicon centrifugal filter devices (Millipore, USA). Numbers of free sulfhydryl groups of RED-NP were determined by Ellman's method [35]. Briefly, 250 μL of sample and 50 μL of Ellman's reagent (Thermo Scientific, USA) were added to 2.5 mL of reaction buffer (0.1 M sodium phosphate, pH 8.0, 1mM EDTA). After 15 min of incubation at room temperature, the OD412 was measured. To generate a standard curve of free sulfhydryl groups, OD412 of serial dilutions of cysteine hydrochloride monohydrate was measured. The serial OD412 values were plotted against concentrations of cysteine hydrochloride monohydrate to generate a standard curve. Concentrations of free sulfhydryl groups in the RED-NP samples were determined from the standard curve and used to calculate numbers of free sulfhydryl groups per iTEP-vaccine fusion molecules.
Fluorescent labeling of iTEP-vaccine fusions
For the flow cytometry study, the fusions were labeled with the amine-reactive coumarin, 7-diethylaminocoumarin-3-carboxylic acid, through succinimidyl ester to form blue-fluorescent bioconjugates. For the microscropy-based cellular uptake study and the cellular degradation study, the fusions were labeled with Alexa Fluo 488 5-SDP ester (Alexa-488) to form green-fluorescent bioconjugates. The reactions were performed based on the manufacturer's instructions (Molecular probes by life technologies). Briefly, 10mg of iTEPs were reacted with 1mg of coumarin or 0.2 mg of Alexa-488 in 0.1 M sodium bicarbonate buffer, pH 8.3 at a final volume of 1mL. The reaction was carried out with continuous stirring in the dark and at room temperature for 1.5 h. The protein-dye conjugates were purified using Amicon Ultra-15 (10k) centrifugal filters (Millipore). The coumarin conjugate was measured for their absorbances of at 205 nm (A205) and 433 nm (A433, λmax for coumarin); the Alexa-488 conjugate was measured at 205 nm and 495 nm (A495, λmax for Alexa-488). The iTEP-vaccine fusion concentrations and the degree of labeling (DOL) were calculated based on these equations:



where ε is the extinction coefficient in cm-1 M-1. DOLcoumarins for the coumarin conjugate were between 8-10% and adjusted to 3% by adding the corresponding, unlabeled iTEP-vaccines fusions before usage. DOLAlexa-488s for Alexa-488 conjugate were between 0.02-0.03% and adjusted to 0.02% before usage.
Presentation of SIINFEKL epitope vaccine by DCs
The presentation was studied as previously described [26]. The concentration of iTEP-vaccine fusions (NPs) used in the study was 5 μM. The results of presentation were shown as mean fluorescence intensity (MFI) of 5000 treated, live cells in a given treatment, which were normalized to the MFI value of untreated DC2.4 cells.
Activation of B3Z hybridoma (CD8+ T) cells
This assay was done with a protocol described previously [26].
Cellular uptake of iTEP-vaccine fusions
DC2.4, EA.hy926, bEnd.3, 3T3 cells were plated at 1.5 × 105 /500 μL/well in 24-well plates. When the cells attached to the bottom of the plate (4 h or overnight), the media were replaced with 500 μL of fresh media with or without 5 μM of coumarine labeled iTEP-vaccine fusions. After being cultured for 4 h at 37°C in 5% CO2, the cells were collected and washed twice with PBS. The fluorescence of the cells was measured by flow cytometry (collecting 5 ×104 events per sample).
For the florescent microscopy, DC 2.4 cells were seeded onto a 12 mm glass coverslip-bottomed 24-well plate at 250,000 per well in complete RPMI-1640 medium. When the cells reached 80% confluence, they were washed with warm medium followed by incubation with 5 μM of iTEP-vaccine fusions at 37°C for 1 to 4 h. Then the cells were rinsed twice with warm medium, and incubated with 75nM of LysoTracker Red DND-99 (Invitrogen, USA). Afterwards, the cells were washed twice with PBS and fixed with 4% of paraformaldehyde. The cells were mounted on a glass slide and imaged using a Nikon ECLIPSE Ti (Nikon Instruments Inc., USA).
Cellular degradation (processing) of iTEP-vaccine fusions in DC 2.4 cells
The study was carried out similarly to an ELP biodegradation study [36]. Specifically, we incubated 40 μM of the iTEP fusions with DC 2.4 cells at a density of 250,000 cells per well in 48-well plates for 1 h. Then, the fusions were washed away. After the wash, the cells were harvested at different time points, and the cell lysates were resolved using SDS-PAGE. The iTEP-vaccine fusions on the gel were imaged using FluorChem FC2 (Alpha-Innotech, USA).
Characterization of cytotoxicity of iTEP-vaccine fusions in vitro
The cytotoxicity was evaluated by an MTS assay as previously described [33]. The concentration range of iTEP-vaccine fusions were from 0 to 100 μM. The cells were incubated with the fusions in culture medium for 24 h.
Animal immunization
C57BL/6 mice were immunized by the subcutaneous injection of 2 nmol iTEP NP vaccines and incomplete Freunds Adjuvans (IFA; Sigma, USA) at their left flanks. Immunization was repeated at their right flanks after 1 week. On day 10 after the second immunization, mice were sacrificed, and the spleens were harvested.
ELISPOT IFN-γ assay
This assay was done with a protocol described previously [26]. The spot images were automatically counted using ImageJ software.
Results
Design and generation of a highly stable iTEP NP
Previously, we generated an iTEP NP, iTEPB70-iTEPA28-pOVA (schematic in Figure 1A), that enhanced the potency of a CTL peptide vaccine, SIINFEKL, when compared with free vaccine peptide or the vaccines delivered as soluble ovalbumin protein [26]. However, we found that this NP was not stable. The NP structure dissolved after it was kept in 5% CO2 at 37°C for 16 h. At 0 h, the hydrodynamic diameter of the NP sample was 71.94±20.81 nm according to DLS measurement (the major peak by number distribution); after 16 h of incubation, the hydrodynamic diameter of the sample became 8.39±1.20 nm, which was similar to the diameters of soluble iTEPs (Figure 1B) [26]. We termed this NP the marginally stable NP (MS-NP).
A. A schematic showing that the iTEPB70-iTEPA28-pOVA and iTEPB70-iTEPA56-pOVA fusions self-assemble in MS-NP and ST-NP. B. Hydrodynamic diameters by number distribution of two NPs before and after 16 h incubations at 37°C. The measurements were obtained by DLS. Green line was for MS-NP and blue line was for ST-NP. Z-averages for MS-NP and ST-NP were 101.00 nm and 74.59 nm before the incubation. Z-averages for MS-NP and ST-NP were 422.90 nm and 70.42 nm after the incubation. C. A representative TEM micrograph of a negatively stained sample of iTEPB70-iTEPA56-pOVA (ST-NP) confirmed NP structure of the sample. Three particles were pointed out by arrows. D. I1/I3, the ratio between two peaks [peak1 (I370-373nm); peak3 (I381-384nm)] of pyrene fluorescence emissions was plotted as a function of log10 concentrations of iTEP-vaccine fusions. The error bars indicated the standard deviation of each data point (n=3). The inflection point of the sigmoid fit of one fusion was defined as the CMC of that fusion.
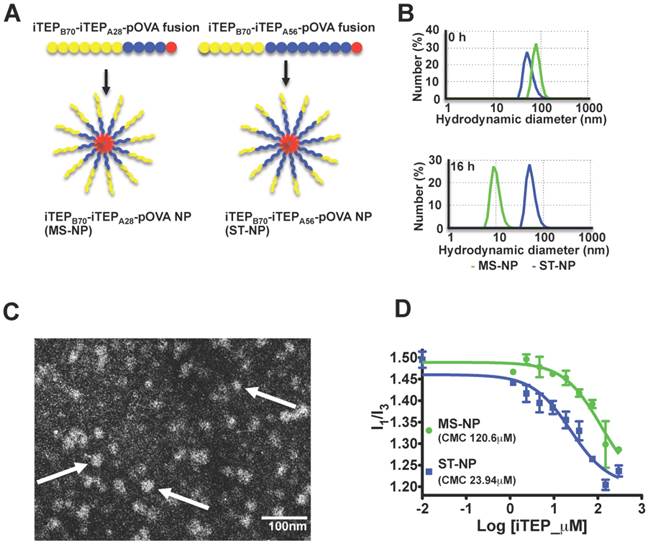
We wondered whether we could expand the benefit of the MS-NP if we used a more stable NP. To this end, we generated a new amphiphilic iTEP diblock copolymer fused with the SIINFEKL vaccine, iTEPB70-iTEPA56-pOVA (Figure 1A). This new fusion had a longer hydrophobic block than the fusion used for the MS-NP, iTEPA56 versus iTEPA28. According to DLS analysis, iTEPB70-iTEPA56-pOVA formed a NP with a hydrodynamic diameter of 56.01±13.54 nm (the major peak by number distribution) (Figure 1B). The size of this NP is comparable to the MS-NP. The TEM images of this new fusion also confirmed its particle structure (Figure 1C). The new fusion kept its particle structure after incubation in cell culture medium in 5% CO2 at 37°C for 16 h (diameter, 55.36±12.88 nm) (Figure 1B), indicating the particle of the new fusion is more stable than the MS-NP. We, thus, termed the NP of this new fusion the stable NP or ST-NP. It is the noteworthy that the difference between the MS-NP and the ST-NP was confirmed when sizes-by-intensity data from DLS measurement were used for analysis (Figure S3A).
To further compare the stability of the MS-NP and the ST-NP, their critical micelle concentrations (CMCs) were measured by a pyrene assay [34]. Results of the assay revealed that the CMC of the MS-NP was 120.6 μM and the CMC of the ST-NP was 23.94 μM (Figure 1D), The CMC results also suggested that the ST-NP was much more stable than the MS-NP.
The ST-NP fails to expand the benefit of the MS-NP to CTL vaccines
We compared in vitro and in vivo activity of the SIINFEKL vaccine that was delivered by the ST-NP and the MS-NP. First, the SIINFEKL epitope presentation by DCs was assessed after the cells were treated with the ST-NP or the MS-NP. The results of the assessment showed that the ST-NP led to a significantly weaker presentation than the MS-NP (Figure 2A). Second, we compared the activation of B3Z cells after the cells were incubated with the DCs that were pretreated with either the ST-NP or the MS-NP. B3Z cells are a genetically engineered CD8+ T cell hybridoma line restricted to the H-2Kb/SIINFEKL complex [37]. We found that the ST-NP was much less robust in activating B3Z cells than the MS-NP (Figure 2B). Last, we immunized C57BL/6 mice using either the ST-NP or the MS-NP and then examined the SIINFELKL-specific CTL response the NPs induced. The ST-NP did elicit the response. The response, however, was significantly weaker than the one elicited by the MS-NP (Figure 2C).
A. Presentation of pOVA by DC 2.4 cells after the cells were incubated with pOVA delivered in different NPs. The data are presented as MFI means ± SD of DC cells in each treatment; each treatment had three repeats (N=3). ★ p < 0.05 (t-test). The graph represents data collected from four independent experiments. B. The activation of B3Z cells by DC 2.4 cells which were pre-incubated with different NPs. The shown values are mean ODs ± SD of samples of each treatment (N=3). The OD values of these treatments had been normalized to the mean OD of the NP-free treatment. ★ p < 0.05 (t-test). The graph represents data collected from three independent experiments. C. Ex vivo analysis of active, SIINFEKL-restricted splenocytes cells from mice (N=5) immunized with MS-NP and ST-NP. The activation of the cells was evaluated by an IFNγ-based ELISPOT assay. Data are presented as Spot Forming Units (SFU)/million cells ± SD. ★ p < 0.05 (t-test). D. The comparison of uptake of ST-NP and MS-NP by DCs and various control cells. The uptake ratios between ST-NP and MS-NP by a given cell type are presented as MFI ratios of the cells after they were incubated with fluorescence-labeled ST-NPs and MS-NPs, respectively. The ratio data are presented as ratio mean ± SD (N=3). ★ p < 0.05 (t-test).
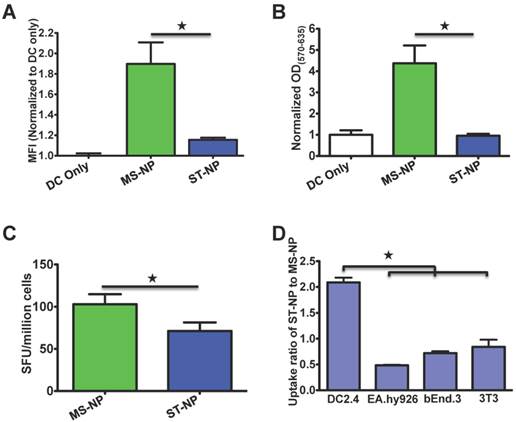
DCs preferentially internalize the ST-NP
Although the MS-NP is better than the ST-NP in promoting SIINFEKL presentation, B3Z activation, and in vivo CTL responses, we noticed that the superiority was much weaker for in the CTL responses than the presentation and the B3Z activation (Figure 2C versus Figures 2A or 2B). The discrepancy between the in vitro and in vivo results suggested that the ST-NP might have certain unrevealed advantages over the MS-NP in vivo. We hypothesized that one advantage of the ST-NP is that DCs preferentially uptake the ST-NP over the MS-NP. When we compared the uptake of the MS-NP and the ST-NP by DCs and other cells, we found that DCs internalized two times more of the ST-NP than the MS-NP (Figure 2D). In contrast, endothelial cells (EA.hy926 and bEnd.3) and fibroblasts (3T3) internalized either the same amount or less of the ST-NP than the MS-NP. These observations suggested that DCs but not other common cells in the body favored the uptake of particles. The ST-NP is more stable than the MS-NP, therefore, the ST-NP sample should have more iTEP-vaccine fusion molecules in NP structure than the MS-NP sample during above uptake study, resulting the more uptake of the ST-NP sample over the MS-NP sample. Practically, the NP structure exerts a passive DC-targeting effect.
Since the ST-NP was better than the MS-NP for DC uptake, the low SIINFEKL presentation and B3Z activation associated with the ST-NP must be due to inefficient processing of the NP in DCs. Because the stability is the major difference, if not the only difference, between the ST-NP and the MS-NP, it is plausible to link the impeded intracellular processing of the ST-NP with the high stability of the NP. It is possible that the high stability prevents the dissociation of the NP inside DCs. Without the dissociation, the linkage between pOVA and iTEP will not be exposed to s-proteasomes and immunoproteasomes in DCs since pOVA is in the hydrophobic core of the ST-NP. Thus, the SIINFEKL epitope cannot be effectively released from the ST-NP.
Generation of an iTEP NP carrier having a reductive environment-responsive stability
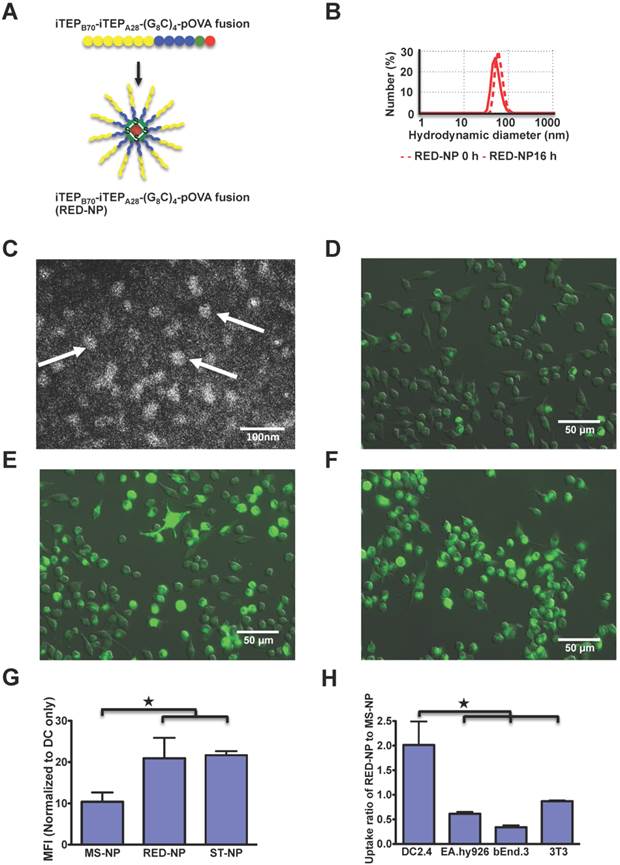
Based on comparisons between the ST-NP and the MS-NP, we reasoned that a more effective carrier of CTL peptide vaccines should be the one that is as stable as the ST-NP during its systematic circulation in the body but becomes as unstable as the MS-NP after its internalization by DCs. To generate such a carrier, we modeified the MS-NP by forming disulfide bonds inside the NP and using the bonds to stabilize the NP (Figure 3A). The new NP was expected to behave as a stable NP unless its disulfide bonds were reduced in a reductive environment such as cytosol [38, 39]. Specifically, we inserted four cysteines between the amphiphilic copolymer iTEPB70-iTEPA28 and pOVA and separated the adjacent cysteines by eight glycines. The resultant iTEP fusion, iTEPB70-iTEPA28-(G8C)4-pOVA, self-assembled in a NP according to a DLS measurement of the fusion (Figure 3B). The NP had a hydrodynamic diameter of 61.94±12.71 nm (the major peak by number distribution) and maintained its particle structure after an overnight incubation in cell culture medium in 5% CO2 at 37°C. The hydrodynamic diameter after the incubation was 53.05±12.38 nm (Figure 3B). The same conclusion was reached when sizes-by-intensity data were used for the analysis (Figure S3B). The TEM image of the iTEPB70-iTEPA28-(G8C)4-pOVA fusion also confirmed the particle structure of the fusion (Figure 3C).
A. A schematic showing that the iTEPB70-iTEPA28-(G8C)4-pOVA fusion self-assembles in RED-NP. B. Hydrodynamic diameters by number distribution of two NPs before and after 16 h incubations at 37°C. Z-averages for RED-NP before and after the incubation were 87.59 nm and 76.89 nm, respectively. C. A representative TEM micrograph of RED-NP. Three particles were pointed out by arrows. D-F. Fluorescence microscopy of MS-NP (D), ST-NP (E) and RED-NP (F) that were internalized by DC2.4 cells. Alexa-488 labeled NPs were incubated with DCs for 4 h before imaging. G. The uptake of MS-NPs, ST-NPs and RED-NPs by DC2.4 cells was shown as normalized MFI of the cells. The data were collected by flow cytometry. Data are presented as mean MFI ± SD (N=3). ★ p < 0.05 (t-test). H. The comparison of uptake of RED-NP to MS-NP by DCs and various control cells. The data were processed and presented as Figure 2D (N=3). ★ p < 0.05 (t-test).
We tested if disulfide bonds inside the new NP were reducible and if the NP structure was dissolvable in a reductive environment like cytosol (glutathione, GSH 1~10 mM) [38, 39]. We treated the new NP and the ST-NP using two concentrations of GSH, at 37°C overnight and then analyzed particle structure changes of both NPs by DLS. These concentrations mimic GSH concentrations in either cytoplasm (GSH 1-10 mM) or the extracellular (GSH 1-10 μM) environment [38, 40, 41]. Both NPs were stable at 10 μM of GSH. The new NP lost its particle structure after being treated with 1 mM of GSH (Table 1). Consistent with this result, free sulfhydryl groups increased, on average, from zero per iTEP molecule to two per iTEP molecule after the treatment of 1 mM of GSH. In contrast, the ST-NP, as a control, maintained its particle structure regardless of its incubation with any concentration of GSH. Similar results emerged when dithiothreitol (DTT) was used as the reductant except that the new iTEP-NP was not stable in 10 μM of DTT (Table 1). The drastic difference between the new NP and the ST-NP suggested that the new NP had stability responsive to the reductive environment. We thus named the new NP the reductive-environment dependable NP or RED-NP.
Comparison of ST-NP and RED-NP for their response to reductants
| ST-NP | RED-NP | ||
|---|---|---|---|
| Reductant concentration (μM) | Hydrodynamic diameter (nm) | ||
| 0 | 54.82±12.89 (NP) | 47.11±13.18 (NP) | |
| GSH | 10 | 52.23±13.59 (NP) | 37.98±6.72 (NP) |
| 1000 | 50±13.62 (NP) | 8.35±1.04 | |
| DTT | 10 | 52.56±13.67 (NP) | 8.75±2.03 |
| 1000 | 53.10±13.56 (NP) | 6.43±2.01 | |
ST-NP and RED-NP were treated with or without reductants for 16 h. Hydrodynamic diameters by number distribution of these two NPs were compared.
We also used SDS-PAGE to analyze responses of the RED-NP, the MS-NP, and the ST-NP to the treatment of 1 mM GSH. We found that the RED-NP can be reduced by 1 mM but not by 10 μM of GSH (Figure S4A). In contrast, the MS-NP and the ST-NP did not respond to the treatment (Figure S4B). This result reinforced the aforementioned conclusion.
Last, we investigated whether DCs would internalize the RED-NP as efficiently as the ST-NP, or as inefficiently as the MS-NP. We found the internalization of the RED-NP to DCs was as efficient as the ST-NP and much more robust than the MS-NP according to both fluorescence microscopy results (Figure 3D-F for 4h of uptake and Figure S5 for 1 h of uptake) and flow cytometry results (Figure 3G). In addition, DCs, but not endothelial and fibroblasts, preferentially internalized the RED-NP over the MS-NP (Figure 3H), which was similar to the ST-NP (Figure 2D). Therefore, the RED-NP has a passive DC-targeting capability, just as the ST-NP.
The RED-NP is a more effective vaccine carrier than both the ST-NP and the MS-NP
We first compared the effect of the RED-NP, the MS-NP, and the ST-NP on CTL epitope presentation by DCs. Although the SIINFEKL epitope delivered by the RED-NP was not presented as well as when it was delivered by the MS-NP, the presentation that resulted from the RED-NP was significantly better than that of the ST-NP (Figure 4A). The comparative results implied that the environment-dependent stability of the RED-NP facilitated processing and presentation of its vaccine payloads by DCs. On the other hand, there was a gap between the presentation results of the RED-NP and the MS-NP, indicating the reduction of the disulfide bonds inside the RED-NP may not be complete so that not all the RED-NP behaved like the MS-NP. Consistent with the presentation results, the RED-NP failed to activate B3Z cells as effectively as the MS-NP but, again, fared better than the ST-NP (Figure 4B). It is worth noting that all these DC presentation and B3Z activation results were SIINFEKL vaccine-specific since incubations of empty carrier NPs with DCs and B3Z cells did not lead to any antigen presentation or T cell activation (Figure S6).
A Presentation of pOVA by DC 2.4 cells after the cells were incubated with three NPs. The data are presented as MFI means ± SD of DC cells in each treatment; each treatment had three repeats (N=3). ★ p < 0.05 (t-test). The graph represents data collected from three independent experiments. B. The activation of B3Z cells by DC 2.4 cells after the DCs were pre-incubated with different NPs. The shown values are mean ODs±SD of samples of each treatment (N=3). ★ p < 0.05 (t-test). The graph represents data collected from three independent experiments. C. Ex vivo analysis of active, SIINFEKL-restricted splenocytes cells from mice (N=5) immunized with MS-NP and RED-NP. Data are presented as Spot Forming Units (SFU)/million cells ± SD. ★ p<0.05 (t-test).
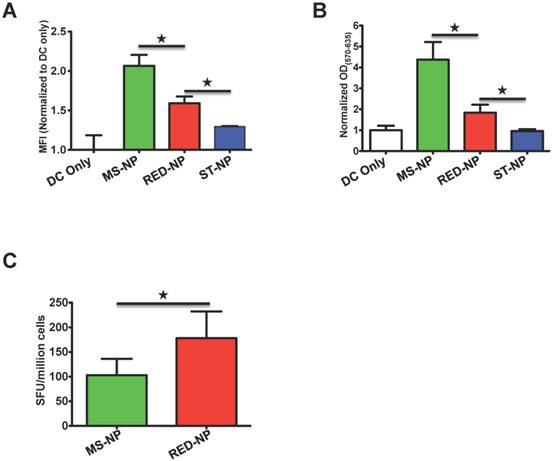
When we immunized mice with either the RED-NP or the MS-NP and compared their induced CTL responses, we found that the RED-NP outperformed the MS-NP and induced stronger CTL responses to the SIINFEKL epitope (Figure 4C). It was noteworthy that another reductive environment-dependent NP, RED-NP2 (assembled from the fusion, iTEPB70-iTEPA28-(G1C)4-pOVA) brought about similar results as RED-NP and was also superior than the MS-NP in vivo (Figure S7).
Table 2 summarizes the stability, uptake, and CTL responses of the three iTEP NPs. Among the NPs, RED-NP, the one that had a changeable stability between the extracellular non-reductive and the cytosolic reductive environments was the most effective in delivering CTL peptide vaccines. It was noted that all three NPs together with their vaccines were safe. They were not toxic to DC 2.4 cells or EA.hy926 cells even at 50 μM (Figure S8). They did not cause any adverse response during our lengthy vaccination study.
Summary of three iTEP NPs in this study.
| Fusion design | Stability | Uptake by DC 2.4 cells | CTL response (in vitro) | CTL response (in vivo) | |
|---|---|---|---|---|---|
| MS-NP | iTEPB70-iTEPA28-pOVA | + | + | +++ | ++ |
| ST-NP | iTEPB70-iTEPA56-pOVA | ++ | ++ | + | + |
| RED-NP | iTEPB70-iTEPA28-(G8C)4-pOVA | ++ (unstable under reductive environment) | ++ | ++ | +++ |
The RED-NP is trafficked to cytosol and degraded
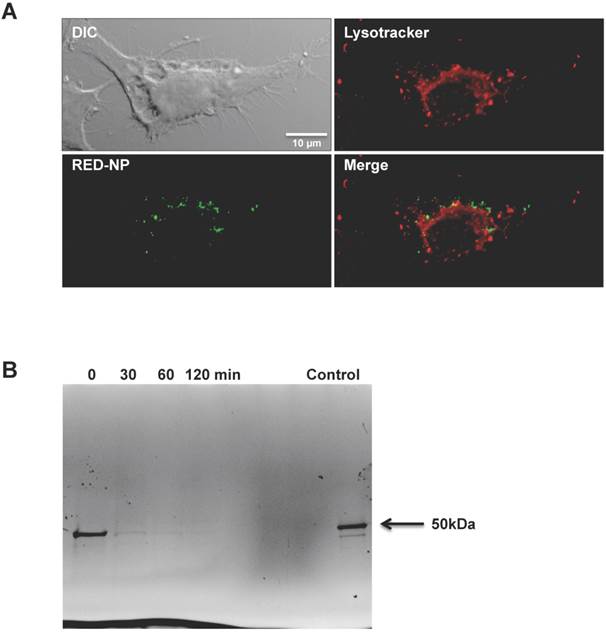
Since the RED-NP has stability responsive to a reductive environment and performed better than the ST-NP in vaccine delivery, it is very likely that the RED-NP reached the reductive cytosol during the intracellular trafficking and leveraged the reductive environment of cytosol. To investigate whether this conjecture is true, we studied intracellular partition of RED-NP after it was internalized by DCs. We found that, after 1 h incubation of RED-NP with DCs, a good fraction of the internalized NPs had reached the cytosol while the remaining NPs were in the phagolysosomes (Figure 5A). This observation suggests a fairly fast escape of the NP from the phagolysosomes to the cytosol of DCs. We also found that processing and degradation of RED-NP happened quickly inside DCs. We analyzed the integrity of intracellular RED-NP by SDS-PAGE after the NP was incubated with DCs for 1 h (Figure 5B). iTEP-vaccine fusions of RED-NP showed a ~50 kDa band on the SDS-PAGE gel immediately after the incubation. This band, however, mostly faded away within 30 min after the incubation. At 1 h after incubation, this band disappeared completely. The fast degradation is consistent with the fast trafficking from phagolysosomes to the cytosol as s-proteasomes and immunoproteasomes for peptide degradation are in cytosol [42-44]. Meanwhile, an efficient biodegradation of the epitope-containing proteins facilitates the MHC class I antigen presentation pathway [24, 42, 45].
A. Intracellular distribution of RED-NP in DC 2.4 cells revealed by fluorescent microscopy. The red dye stains low pH lysosome compartments in DCs. The green dye was used to label RED-NP. Yellow staining in the merged image indicated the co-localization of RED-NPs and lysosomes. B. SDS-PAGE picture of DC 2.4 cell lysate after the cells was incubated with Alexa-488 labeled RED-NP for 1h and washed away. Cell lysate was collected at different time points after the incubation. The ~50 kDa band was iTEP-vaccine fusions from the RED-NP.
Discussion
CTL epitope peptide vaccines have unparalleled specificity compared with other forms of CTL vaccines [12-14]. However, for this type of vaccines to induce CTL responses, there are several critical conditions to be met: the vaccines have to be efficiently delivered to DCs; once the vaccines are internalized by DCs, they have to be translocated from phagolysosomes to cytosol for antigen cross-presentation; and the vaccines need to be released from their carriers for antigen processing and presentation. Meanwhile, DCs and T helper cells need to be activated to create pro-inflammatory milieu to support CTL vaccines [14, 46-48]. In this study, we used iTEPs to fabricate three NPs as vaccine carriers. These NP carriers helped to passively target the vaccines to DCs. Our observations were consistent with the previous understanding of using nanocarriers to target DCs [46].
The major effort of this study was to generate iTEP NP carriers with different stability. Our main conclusion was that a desirable NP should have changeable stability that responds to different physiological environments. Vaccines should be packaged inside the NP carriers before they reach the cytosolic compartment of DCs and are ready for proteasomal degradation. There are at least two advantages of keeping the vaccines inside their carriers. First, the carriers are useful for passive DC-targeting of the vaccines. Second, the carriers protect the vaccines from degradation and clearance during the systematic circulation of the vaccines just like what carriers do for other peptide therapeutics [49]. When the vaccines reach cytosol, the carriers should dissociate to expose the peptide bond linkages between the vaccines and the carriers so that the vaccines can be released from the carriers by proteasomes and presented by DCs. Indeed, our conclusion regarding changeable stability applies to all CTL vaccine carriers that package vaccine payloads inside them, including VLPs. It is interesting that there has been no report about whether possessing changeable stability would improve the CTL vaccine delivery function of VLPs. The absence of this type of report might be because it is not as easy to modulate stability of VLPs as iTEP NPs.
There are successful examples of delivering CTL peptide vaccines on the surface of particle carriers and linking the peptides with the particles through disulfide bonds [50, 51]. The peptides are released from the carriers in cytosol, a reductive environment, while the carriers still maintain their particle structures after the release. This strategy of vaccine delivery and release, however, has two potential issues. First, peptide vaccines on the surface of the carriers are subject to degradation by peptidases during systematic circulation [14]. Second, although the particle carriers are expected to be degraded inside cells eventually, the process might not be efficient. The degradation of the carriers may be hindered by their particle structures just as revealed by our results of ST-NP. The inefficient degradation can lead to accumulation of the carriers in cells, which could have some adverse effects. In contrast, although our approach similarly utilizes disulfide bonds and the cytosolic reductive environment as aforementioned studies [50, 51], it has a temporal and spatial control over the integrity of both vaccines and their carriers. With the control, our approach less likely has the two issues mentioned above. First, we package CTL vaccines inside the RED-NP and keep the NP stable before the NP is transported to the cytosol in DCs. In this way, the vaccines were protected from undesired enzymatic degradation during systematic circulation. Second, once the RED-NP reaches to the cytosol, it disassembles since cytosolic reductive environment promotes the cleavage of disulfide bonds inside the NP. The disassembly of the NP into iTEP-vaccine fusion molecules is desired because the vaccines need to be released and processed, and the carriers need to be degraded in the cytosol. The iTEP fusions molecules are presumably degraded like other intracellular proteins, and consequently, iTEP NPs will less likely accumulate inside cells. In summary, we leverage the cytosolic reductive environment and disulfide bonds to address the different needs of CTL vaccines at their different processing stages.
It is acknowledged that the reductive-environment induced destabilization of the RED-NP may not be complete. The RED-NP did not result in same level of in vitro antigen presentation as the MS-NP (Figure 4A). The most possible reason of RED-NP's deficiency is that only a fraction of the RED-NP had their disulfide bonds reduced and transformed into the MS-NP. Our data also showed that there were unreduced disulfide bonds in RED-NP. Therefore, further optimization of disulfide bonds in the RED-NP is needed to optimize the sensitivity of the bonds to the cytosolic, reductive environment. Nevertheless, this deficiency of RED-NP was compensated in vivo by the selective uptake of the NP by DCs. In contrast, DCs do not show this selectivity to the MS-NP. Compared to the RED-NP, a larger fraction of the MS-NP might have been internalized by other cells instead of dendritic cells in body. Therefore, the RED-NP fared best in eliciting CTL responses in vivo among the three NPs studied.
In summary, we leveraged the recombinant nature of iTEPs and generated three NPs with very different stability. Using these NPs, we showed that a vaccine carrier with changeable stability was effective to facilitate two critical steps toward CTL vaccine-induced responses, targeting the vaccines to DCs and releasing the vaccines from their carriers. In addition, the RED-NP is a promising platform to further develop CTL vaccine carriers so they can address other critical barriers of CTL peptide vaccines. The RED-NP may improve therapeutic and prophylactic effects of CTL vaccines when it is used to deliver disease-relevant CTL vaccines.
Supplementary Material
Supplementary Tables and Figures.
Acknowledgements
The authors thank Drs. Kenneth Rock and Nilabh Shastri for providing cell lines and Mrs. James Marvin and Chris Leukel for flow cytometry work. The flow cytometry work was supported by the University of Utah Flow Cytometry Facility in addition to the National Cancer Institute through Award Number 5P30CA042014-24. The research work was supported by Ministry of Human Resources and Social Security of the People's Republic of China Fund: 2013-277 to T. X., and the University of Utah Start-up Fund, University of Utah Seed Grant, and NIH R00CA153929 to M.C.
Competing Interests
The Authors claim no competing interest.
References
1. Gjertsen MK, Buanes T, Rosseland AR, Bakka A, Gladhaug I, Soreide O. et al. Intradermal ras peptide vaccination with granulocyte-macrophage colony-stimulating factor as adjuvant: Clinical and immunological responses in patients with pancreatic adenocarcinoma. International journal of cancer Journal international du cancer. 2001;92:441-50
2. Hunger RE, Brand CU, Streit M, Eriksen JA, Gjertsen MK, Saeterdal I. et al. Successful induction of immune responses against mutant ras in melanoma patients using intradermal injection of peptides and GM-CSF as adjuvant. Experimental dermatology. 2001;10:161-7
3. Epstein JE, Tewari K, Lyke KE, Sim BK, Billingsley PF, Laurens MB. et al. Live attenuated malaria vaccine designed to protect through hepatic CD8(+) T cell immunity. Science. 2011;334:475-80
4. Wu YL, Park K, Soo RA, Sun Y, Tyroller K, Wages D. et al. INSPIRE: A phase III study of the BLP25 liposome vaccine (L-BLP25) in Asian patients with unresectable stage III non-small cell lung cancer. BMC cancer. 2011;11:430
5. Ohyanagi F, Horai T, Sekine I, Yamamoto N, Nakagawa K, Nishio M. et al. Safety of BLP25 liposome vaccine (L-BLP25) in Japanese patients with unresectable stage III NSCLC after primary chemoradiotherapy: preliminary results from a Phase I/II study. Japanese journal of clinical oncology. 2011;41:718-22
6. Butts C, Maksymiuk A, Goss G, Soulieres D, Marshall E, Cormier Y. et al. Updated survival analysis in patients with stage IIIB or IV non-small-cell lung cancer receiving BLP25 liposome vaccine (L-BLP25): phase IIB randomized, multicenter, open-label trial. Journal of cancer research and clinical oncology. 2011;137:1337-42
7. Yoshitake Y, Fukuma D, Yuno A, Hirayama M, Nakayama H, Tanaka T. et al. Phase II clinical trial of multiple peptide vaccination for advanced head and neck cancer patients revealed induction of immune responses and improved OS. Clinical cancer research: an official journal of the American Association for Cancer Research. 2015;21:312-21
8. Yamada A, Sasada T, Noguchi M, Itoh K. Next-generation peptide vaccines for advanced cancer. Cancer science. 2013;104:15-21
9. Asahara S, Takeda K, Yamao K, Maguchi H, Yamaue H. Phase I/II clinical trial using HLA-A24-restricted peptide vaccine derived from KIF20A for patients with advanced pancreatic cancer. Journal of translational medicine. 2013;11:291
10. Yoshimura K, Minami T, Nozawa M, Uemura H. Phase I clinical trial of human vascular endothelial growth factor receptor 1 peptide vaccines for patients with metastatic renal cell carcinoma. British journal of cancer. 2013;108:1260-6
11. Ichihashi T, Yoshida R, Sugimoto C, Takada A, Kajino K. Cross-protective peptide vaccine against influenza A viruses developed in HLA-A*2402 human immunity model. PloS one. 2011;6:e24626
12. Gridelli C, Rossi A, Maione P, Ferrara ML, Castaldo V, Sacco PC. Vaccines for the treatment of non-small cell lung cancer: a renewed anticancer strategy. The oncologist. 2009;14:909-20
13. Kanaly CW, Ding D, Heimberger AB, Sampson JH. Clinical applications of a peptide-based vaccine for glioblastoma. Neurosurgery clinics of North America. 2010;21:95-109
14. Purcell AW, McCluskey J, Rossjohn J. More than one reason to rethink the use of peptides in vaccine design. Nature reviews Drug discovery. 2007;6:404-14
15. Skwarczynski M, Toth I. Peptide-based subunit nanovaccines. Current drug delivery. 2011;8:282-9
16. Moon JJ, Huang B, Irvine DJ. Engineering nano- and microparticles to tune immunity. Advanced materials. 2012;24:3724-46
17. Krishnamachari Y, Geary SM, Lemke CD, Salem AK. Nanoparticle delivery systems in cancer vaccines. Pharmaceutical research. 2011;28:215-36
18. Joshi MD, Unger WJ, Storm G, van Kooyk Y, Mastrobattista E. Targeting tumor antigens to dendritic cells using particulate carriers. Journal of controlled release: official journal of the Controlled Release Society. 2012;161:25-37
19. Skwarczynski M, Toth I. Recent advances in peptide-based subunit nanovaccines. Nanomedicine. 2014;9:2657-69
20. Akagi T, Baba M, Akashi M. Biodegradable Nanoparticles as Vaccine Adjuvants and elivery systems: Regulation of Immune Responses by Nanoparticle-Based Vaccine. Adv polym Sci. 2012:31-64
21. Gregory AE TR, Williamson D. Vaccine delivery using nanoparticles. Frontiers in Cellular and Infection Microbiology. 2013:3
22. Zhao L, Seth A, Wibowo N, Zhao CX, Mitter N, Yu C. et al. Nanoparticle vaccines. Vaccine. 2014;32:327-37
23. MacEwan SR, Chilkoti A. Elastin-like polypeptides: biomedical applications of tunable biopolymers. Biopolymers. 2010;94:60-77
24. Kruger E, Kloetzel PM. Immunoproteasomes at the interface of innate and adaptive immune responses: two faces of one enzyme. Current opinion in immunology. 2012;24:77-83
25. Chilkoti A, Dreher MR, Meyer DE. Design of thermally responsive, recombinant polypeptide carriers for targeted drug delivery. Advanced drug delivery reviews. 2002;54:1093-111
26. Cho S, Dong S, Parent KN, Chen M. Immune-tolerant elastin-like polypeptides (iTEPs) and their application as CTL vaccine carriers. Journal of drug targeting. 2015:1-12
27. Grgacic EV, Anderson DA. Virus-like particles: passport to immune recognition. Methods. 2006;40:60-5
28. Plummer EM, Manchester M. Viral nanoparticles and virus-like particles: platforms for contemporary vaccine design. Wiley interdisciplinary reviews Nanomedicine and nanobiotechnology. 2011;3:174-96
29. Win SJ, Ward VK, Dunbar PR, Young SL, Baird MA. Cross-presentation of epitopes on virus-like particles via the MHC I receptor recycling pathway. Immunology and cell biology. 2011;89:681-8
30. Da Silva DM, Pastrana DV, Schiller JT, Kast WM. Effect of preexisting neutralizing antibodies on the anti-tumor immune response induced by chimeric human papillomavirus virus-like particle vaccines. Virology. 2001;290:350-60
31. Ruedl C, Schwarz K, Jegerlehner A, Storni T, Manolova V, Bachmann MF. Virus-like particles as carriers for T-cell epitopes: limited inhibition of T-cell priming by carrier-specific antibodies. Journal of virology. 2005;79:717-24
32. Liu XS, Xu Y, Hardy L, Khammanivong V, Zhao W, Fernando GJ. et al. IL-10 mediates suppression of the CD8 T cell IFN-gamma response to a novel viral epitope in a primed host. J Immunol. 2003;171:4765-72
33. Zhao P, Dong S, Bhattacharyya J, Chen M. iTEP nanoparticle-delivered salinomycin displays an enhanced toxicity to cancer stem cells in orthotopic breast tumors. Molecular pharmaceutics. 2014;11:2703-12
34. Dreher MR, Simnick AJ, Fischer K, Smith RJ, Patel A, Schmidt M. et al. Temperature triggered self-assembly of polypeptides into multivalent spherical micelles. Journal of the American Chemical Society. 2008;130:687-94
35. Ellman GL. Tissue sulfhydryl groups. Archives of biochemistry and biophysics. 1959;82:70-7
36. Shah M, Hsueh PY, Sun G, Chang HY, Janib SM, MacKay JA. Biodegradation of elastin-like polypeptide nanoparticles. Protein Sci. 2012;21:743-50
37. Karttunen J, Sanderson S, Shastri N. Detection of rare antigen-presenting cells by the lacZ T-cell activation assay suggests an expression cloning strategy for T-cell antigens. Proceedings of the National Academy of Sciences of the United States of America. 1992;89:6020-4
38. Smith CV, Jones DP, Guenthner TM, Lash LH, Lauterburg BH. Compartmentation of glutathione: implications for the study of toxicity and disease. Toxicology and applied pharmacology. 1996;140:1-12
39. Wu G, Fang YZ, Yang S, Lupton JR, Turner ND. Glutathione metabolism and its implications for health. The Journal of nutrition. 2004;134:489-92
40. Cantin AM, North SL, Hubbard RC, Crystal RG. Normal alveolar epithelial lining fluid contains high levels of glutathione. J Appl Physiol (1985). 1987;63:152-7
41. Jones DP, Carlson JL, Samiec PS, Sternberg P Jr, Mody VC Jr, Reed RL. et al. Glutathione measurement in human plasma. Evaluation of sample collection, storage and derivatization conditions for analysis of dansyl derivatives by HPLC. Clinica chimica acta; international journal of clinical chemistry. 1998;275:175-84
42. Yewdell JW, Bennink JR. Cut and trim: generating MHC class I peptide ligands. Current opinion in immunology. 2001;13:13-8
43. Kloetzel P-M, Ossendorp F. Proteasome and peptidase function in MHC-class-I-mediated antigen presentation. Current opinion in immunology. 2004;16:76-81
44. Brooks P, Fuertes G, Murray RZ, Bose S, Knecht E, Rechsteiner MC. et al. Subcellular localization of proteasomes and their regulatory complexes in mammalian cells. Biochemical Journal. 2000;346:155-61
45. Amigorena S, Savina A. Intracellular mechanisms of antigen cross presentation in dendritic cells. Current opinion in immunology. 2010;22:109-17
46. Kreutz M, Tacken PJ, Figdor CG. Targeting dendritic cells-why bother? Blood. 2013;121:2836-44
47. Banchereau J, Briere F, Caux C, Davoust J, Lebecque S, Liu YJ. et al. Immunobiology of dendritic cells. Annual review of immunology. 2000;18:767-811
48. Hu Y, Litwin T, Nagaraja AR, Kwong B, Katz J, Watson N. et al. Cytosolic delivery of membrane-impermeable molecules in dendritic cells using pH-responsive core-shell nanoparticles. Nano letters. 2007;7:3056-64
49. Fosgerau K, Hoffmann T. Peptide therapeutics: current status and future directions. Drug discovery today. 2015;20:122-8
50. Hirosue S, Kourtis IC, van der Vlies AJ, Hubbell JA, Swartz MA. Antigen delivery to dendritic cells by poly(propylene sulfide) nanoparticles with disulfide conjugated peptides: Cross-presentation and T cell activation. Vaccine. 2010;28:7897-906
51. Nembrini C, Stano A, Dane KY, Ballester M, van der Vlies AJ, Marsland BJ. et al. Nanoparticle conjugation of antigen enhances cytotoxic T-cell responses in pulmonary vaccination. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:E989-97
Author contact
![]() Corresponding author: Mingnan.chenedu.
Corresponding author: Mingnan.chenedu.