Impact Factor
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and methods
Results and discussion
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2016; 6(6):795-807. doi:10.7150/thno.13725 This issue Cite
Research Paper
Histone Deacetylase Inhibitors Delivery using Nanoparticles with Intrinsic Passive Tumor Targeting Properties for Tumor Therapy
Fatima el Bahhaj1#, Iza Denis2#, Loic Pichavant3#, Régis Delatouche1, Floraine Collette3, Camille Linot2, Daniel Pouliquen2, Marc Grégoire2, Valérie Héroguez3*, Christophe Blanquart2,4*, Philippe Bertrand1,4* ![]()
1. Institut de Chimie des Milieux et Matériaux de Poitiers, University of Poitiers, CNRS UMR 7285, 4 rue Michel Brunet TSA 51106, B27, Poitiers. F-86073 Poitiers cedex 9, France
2. Centre de Recherche contre le Cancer Nantes et Angers, University of Nantes, CNRS UMR 6299, Inserm, U892, 8 quai Moncousu, F-44000 Nantes cedex 1, France
3. Laboratoire de Chimie des Polymères Organiques, University of Bordeaux, CNRS UMR 5629, 16 Avenue Pey-Berland, F-33607 Pessac, France
4. Réseau Epigénétique du Cancéropôle Grand Ouest.
# These authors have equally contributed to this work.
* These authors have equally contributed to this work.
Received 2015-9-2; Accepted 2015-12-31; Published 2016-3-25
Abstract

Fast clearance, metabolism and systemic toxicity are major limits for the clinical use of anti-cancer drugs. Histone deacetylase inhibitors (HDACi) present these defects despite displaying promising anti-tumor properties on tumor cells in vitro and in in vivo model of cancers. Specific delivery of anti-cancer drugs into the tumor should improve their clinical benefit by limiting systemic toxicity and by increasing the anti-tumor effect. In this work, we describe a simple and flexible polymeric nanoparticle platform highly targeting the tumor in vivo and triggering impressive tumor weight reduction when functionalized with HDACi. Our nanoparticles were produced by Ring-Opening Metathesis Polymerization of azido-polyethylene oxide-norbornene macromonomers and functionalized using click chemistry. Using an orthotopic model of peritoneal invasive cancer, a highly selective accumulation of the particles in the tumor was obtained. A combination of epigenetic drugs involving a pH-responsive histone deacetylase inhibitor (HDACi) polymer conjugated to these particles gave 80% reduction of tumor weight without toxicity whereas the free HDACi has no effect. Our work demonstrates that the use of a nanovector with theranostic properties leads to an optimized delivery of potent HDACi in tumor and then, to an improvement of their anti-tumor properties in vivo.
Keywords: polymeric nanoparticle, epigenetic, HDAC, cancer, theranostics, peritoneal, mesothelioma.
Introduction
Post-translational modifications (PTM) of histone proteins and DNA methylation play major roles in the epigenetic regulation of gene expression.(1),(2) The enzymes regulating epigenetic modifications are classified as writers, erasers and readers, respectively adding, removing and “decoding” PTM. Overexpression or abnormal activities of these regulators have been linked to human disease like cancers (3) for many years. Drugs targeting dysfunctional epigenetic regulators represent a widespread innovative therapeutic strategy against cancers.(4) The overexpression of histone deacetylases (HDAC) in many cancer types induces histones hypoacetylation, and in turn chromatin compaction resulting in tumor suppressor genes (TSG) repression. (5),(6),(7) HDAC inhibitors (HDACi), able to stimulate TSG expression, are evaluated in many clinical trials, with Vorinostat (8), Istodax (9) and Belinostat (10) being currently approved for clinical use for the treatment of cutaneous lymphomas. Recently Panobinostat has also been approved for multiple myelomas.(11) Except Istodax, Vorinostat, Belinostat and Panobinostat are all hydroxamic acid-based HDACi. However, anti-cancer applications of HDACi are still limited mainly because of dose limiting toxicity,(12) acquired resistance (13) fast metabolism and variable clinical results.(14) Clinical data have shown that sustained delivery and low drug dosing (15) of epigenetic target inhibitors can reduce off-target effects such as systemic toxicity. Besides HDAC, the deregulated activities of DNA methyl transferases (DNMT) also participates in tumorigenesis and the combination of epigenetic inhibitors has emerged as a highly valuable option in many cases.(16),(17),(18)
With epigenetic targeting drugs, side effects or systemic toxicity can be reduced with highly active compounds used alone or in combination, at low dosage, in association with the tumor targeting mediated by drug delivery systems (DDS).(19) In contrast to the reported DDS applications in epigenetics,(20) focus should also be set on low drug loading and sustained release to comply with clinical results. The selection of the DDS is also an important issue. Micellar DDS have a heterogeneous composition and the undesired “burst release” effect,(21) whereas polymeric DDS can be stable during the administration schedule, and designed for chemically stimulated drug release circumventing resistance.(22) Passive tumor targeting due to the enhanced permeability and retention (EPR) effect (23) requires a long circulation time, which can be obtained by DDS pegylation to introduce 'stealth' properties.
The Ring-Opening Metathesis Polymerization (ROMP) of functional norbornene (NB) produces well-defined nanoparticles (NP). Post-functionalization (24),(25) is used to gain stealth properties, tumor cell targeting, and drug release avoiding the liposomal burst release effect. All these functionalities increase the chemical complexity of DDS. In this study, we implemented a more simple and flexible approach with the azido-polyethylene oxide (PEO)-NB macromonomer 6 (Fig. 1).(26) The stealth properties for passive targeting are natively present thanks to the PEO chain. The terminal azide group can be used to build a library of functional macromonomers by click chemistry with alkyne-bearing functions. The NB group is used for (co)polymerization in dispersed media to give a (multi)functional core-shell spherical NP. The plasticity and stealth property of this DDS made it convenient for high tumor targeting and internalization by cancer cells without additional modifications. Using a clickable pH-responsive prodrug,(27) NP can be prepared for drug loading with high stability during blood circulation and release of the drug after NP endocytosis by cells.(28) Selecting an aggressive cancer model for testing this novel DDS increases the challenge and positive results could open the way for the treatment of other cancers. For that purpose, we selected the asbestos exposure-related cancer, malignant pleural mesothelioma (MPM), which has a poor prognostic and a limited clinical response to therapy.(29) This cancer is located mainly in the pleura and in the peritoneum. Epigenetic errors (30) observed in mesothelioma contribute to ineffective chemotherapy treatment and characterize this cancer as a candidate for epigenetic therapeutic strategies. Despite promising effects in MPM models, epigenetic-driven therapies in the clinic have to date been unsuccessful.(31)
Synthesis of compound 2, its prodrugs 4 and nanoparticle 8. Reaction conditions: i) KHCO3 3eq., CH3I 6 eq. rt, 1 week. ii) four steps, reference (30). iii) EDC, NH2OH (freshly prepared from NH2OH.HCl, KOH, MeOH). iv) a) AcCl 5eq., toluene, reflux 3h. b) 2, NEt3/CH3CN 1/1 v/v, 45 min. room temperature. v) PMDETA, DMF, CuBr 1.7eq., inert atmosphere, 4 days, room temperature. vi) CH2Cl2/EtOH 35/65 v/v, Grubbs I catalyst 1% relative to NB, NEt3, 24h, room temperature.
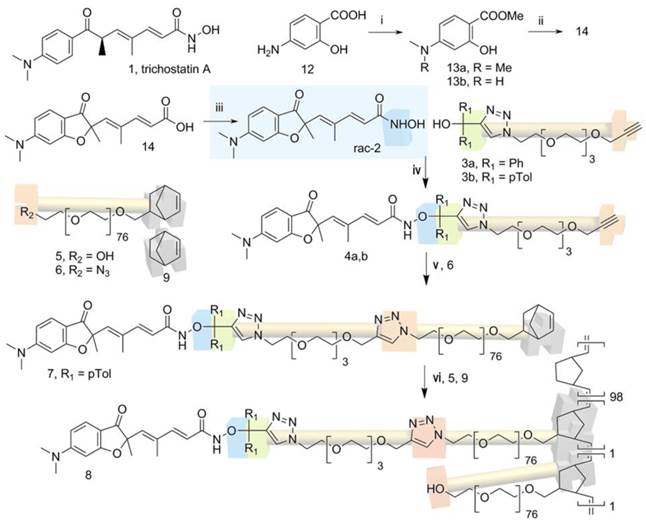
pH-mediated delivery of HDAC inhibitor SAHA with dendrimers was demonstrated in vitro.(32) In this communication we showed for the first time that an epigenetic strategy combining pH-responsive ROMP-nanoparticles functionalized with a nanomolar active hydroxamic acid HDACi and free decitabine, a hypomethylating agent, can induce an 80% mesothelioma tumor weight reduction without toxicity while the combination of the free HDACi with decitabine had no effect. These results could also be extended to the increasing number of approved HDAC inhibiting molecules belonging to the hydroxamic acid family.
Materials and methods
Reagents
All commercial chemicals and solvents were reagent grade and were used without further purification unless otherwise noted. Reactions were carried out with the use of standard techniques for the exclusion of moisture. Ethanol (96%, purissium grade pur, Xilab), dichloromethane (96%, purissium grade pur, Xilab) and dimethyl formamide (99.8%, Panreac) were degassed before use. Tetrahydrofuran (J.T. Baker) diethyl ether anhydrous (J.T. Baker), N,N,N',N'',N''-pentamethyldiethylenetriamine (PMDETA, 99%, Aldrich), Na2SO4 (99%, Alfa Aesar), norbornene (NB, 99% GC, Aldrich), Grubbs first generation catalyst Cl2(PCy3)2Ru=CHPh (Aldrich, stored in a glovebox under Argon atmosphere), dodecane (99%, Aldrich), triethylamine (99%, Acros Organics), ethyl vinyl ether (99% stab. with ca. 0.1% N,N-diethylalanine, Alfa Aesar), Trizma®base (99.9%, Aldrich) were used without further purification. CuBr (98%, Aldrich) was purified in acetic acid and stored under inert atmosphere (glovebox), α-norbornenyl-ω-azide-poly(ethylene oxide) macromonomer 6 bare nanoparticles 10 (33) and Rhodamine B labeled nanoparticles 11 (26) were prepared as previously described. ACN: acetonitrile, DCM: dichloromethane, DMF: dimethyl formamide, EA: ethyl acetate, TEA: trimethylamine, THF: tetrahydrofuran, PE: petroleum ether. PMDETA= pentamethyldiethylenetetramine.
Methods
Reactions were monitored by thin layer chromatography when applicable with 0.25 mm silica gel plates (60F-254. E. Merck) and revealed with phosphomolybdic acid 5% weight in ethanol. 1H NMR spectra were recorded in 5 mm diameter tubes with a Bruker spectrometer (1H 400 MHz. 13C 100 MHz), in CDCl3. acetone D6 or DMSO D6 (for compound 2) at 25 °C. The chemical shift scale expressed in ppm was calibrated on the basis of the deuterated solvent or tetramethylsilane as reference. For compounds 4 peak assignments was made with DEPT135, COSY and HSQC experiments. High resolution mass spectra were recorded at Centre Régional des Mesures Physiques de l'Ouest for compounds 4 on a Waters Micro-TOF-Q and Q-2. HPLC for compounds 2, 3 and 4 analyses were performed on Hitachi equipped with an auto-sampler, a diode array detection DAD L-2455 and with a reversed phase ACE® C18 (5 μm. 4.6 x 250 mm). Analyses were recorded at 230 nm. Size exclusion chromatography (SEC) equipment consists of a JASCO HPLC pump type 880-PU, TOSOHAAS TSK gel columns, a Varian refractive index detector, and a JASCO 875 UV-vis absorption detector, with THF as the mobile phase and the calibration curve was performed using polystyrene standards. The conversion of macromonomers was determined by SEC with dodecane as internal standard (SEC retention times: tSECmacromonomers = 18.75 min; tSECdodecane = 31.70 min). The conversion of norbornene 9 was determined by gas chromatography with dodecane as internal standard (GC retention times: tGCNb = 1.77 min; tGCdodecane = 8.55 min). DLS measurements were performed using a MALVERN zetasizer Nano ZS equipped with He-Ne laser (4 mW and 633 nm). Before measurements, latexes were diluted about 800 times to minimize multiple scatterings caused by high concentration. The scattering angle used was 173°. TEM pictures were performed with a Hitachi H7650 microscope operating at an accelerating voltage of 120 kV. For the particle size and morphology observation, samples diluted about 100 times were deposited on a 200 mesh carbon film-coated copper grids surface (3 x 5 μl).
Synthesis of compound 2 and its prodrugs 4
4-N.N-dimethylamino-2-hydroxy-benzoic acid methyl ester (13a) and 4-N-methylamino-2-hydroxy-benzoic acid methyl ester (13b). To a solution of amino-hydroxybenzoic acid 12 (30.62 g, 0.2 mol) in DMF (200 mL) were added CH3I (62 mL, 1 mol. 5eq.) and KHCO3 (60 g, 0.6 mol, 3eq.). After one week stirring at room temperature the solution was diluted with water (500 mL) and extracted with DCM (3x200 mL). The combined organic layers were dried (MgSO4) and solvent removed under vacuum. The crude product was purified by chromatography (silica gel, EA:PE gradient 0:100, 100 mL; 1.25:98.75, 500 mL; 2.5:97:5, 500 mL and finished with 5:95) to afford compound 13a as a white solid (27.3 g, 70%) and compound 13b as a light ivory solid (3.3 g, 9%). For compound 13a (identical to reference 30): TLC EA/PE 5/95 Rf = 0.5, 1H NMR (400 MHz; CDCl3) δ ppm: 10.93 (1H, s), 7.63 (1H, d. J = 9.0 Hz), 6.20 (1H, dd. J = 9.0. 2.4 Hz), 6.12 (1H, d. J = 2.4 Hz), 3.87 (3H, s), 3.0 (6H, s).
5-(6-Dimethylamino-2-methyl-3-oxo-2.3-dihydro-benzofuran-2-yl)-4-methyl-penta-2.4-dienoic acid hydroxamide (2). To a 0°C cooled solution of the acid 14 (34) (6.14 g, 20.4 mmol) in anhydrous THF, ethyl chloroformiate (0.253 L, 30.6 mmol) and TEA (5.01 mL, 30.6 mmol) were added, and the mixture was stirred for 30 min. The solid was filtered and the filtrate was added to a freshly prepared solution of hydroxylamine in methanol. To prepare the solution of hydroxylamine, at 0°C, a solution of hydroxylamine hydrochloride (3.95 g, 61.3 mmol) in methanol (60 mL) was added to a stirred solution of potassium hydroxide (3.44 g, 61.3 mmol) in methanol (60 mL). The mixture was stirred for 15 min, the precipitate was then removed by filtration to have the solution the hydroxylamine in methanol. The mixture was stirred at room temperature for 30 min and then was evaporated. The resulting crude was purified by combi-flash (DCM/MeOH: 95/5) and the residue was recrystallized from a mixture of DCM/PE (2/8). The solution was filtered to have a white pink solid of 2 (2.31 g, 36%). The inhibitor 2 prepared with this new synthesis and its intermediates gave analytical data consistent with previously reported longer synthesis (Supplementary information, NMR data for compound 2 and HPLC). TLC MeOH/DCM 5/95 Rf = 0.05, 1H NMR (400 MHz. MeOH-d4) δ ppm: 1.45 (3H, s), 1.9 (3H, s), 3.1 (6H, s), 5.8 (1H, s), 5.80 (1H, d, J = 15.6 Hz), 6.16 (1H, d, J = 2.1 Hz), 6.45 (1H, dd, J = 2.1, 8.9 Hz), 7.05 (1H, d, J = 15.6 Hz), 7.30 (1H, d, J = 8.9 Hz). 13C NMR (100 MHz. MeOH-d4) δ ppm: 14.0, 25.0, 41.0, 90.8, 93.5, 108.5, 110.3, 119.1, 126.9, 136.9, 138.6, 146.1, 160.4, 166.6, 175.2, 200.7.
5-(6-Dimethylamino-2-methyl-3-oxo-2.3-dihydro-benzofuran-2-yl)- [[1-[2-[2-[2-(2-prop-2-ynoxyethoxy) ethoxy] ethoxy] ethyl] triazol-4-yl]-bis(phenyl)methoxy]-4-methyl-penta-2.4-diene-amide (4a). To a solution of 3a (67 mg, 0.144 mmol) in dry toluene (1 mL) under nitrogen atmosphere was added AcCl (54 μL, 720 μmol, 5 eq.). After 2h reflux, the solution was evaporated to give the crude intermediate chloride as brown oil. To a solution of 2 (52 mg, 172 μmol, 1.2 eq.) in a mixture of dry NEt3/ACN (2 mL, 1:1) was added the crude chloride dissolved in a minimum of dry ACN at ambient temperature. The resulting solution was stirred 45 min at room temperature, the solvent was then evaporated under vacuum and purified by combi-flash (MeOH/ DCM: 0 to 3% MeOH) to afford the compound 4a as a colorless oil (38.5 mg, 35%). TLC MeOH/DCM 5/95 Rf = 0.34; 1H NMR (400 MHz, Acetone-d6) δ ppm: 9.83 (1H, s, NH), 7.78 (1H, s, Ha), 7.47 (4H, d, J = 8.1 Hz, Hb), 7.39 (2H, d, J = 8.3 Hz, Hc), 7.31 (6H, m, Hd+p), 6.63 (1H, dd, J = 12.0, 3.9 Hz, Hg), 6.22 (1H, s, He), 5.95 (1H, d, J = 16.0 Hz, Hh), 5.88 (1H, s, Hi), 4.54 (2H, t, J = 4.0 Hz, Hj), 4.15 (2H, s, Hk), 3.84 (2H, t, J = 4.1 Hz, Hl), 3.47 (12H, m, Hm), 3.11 (6H, s, Hn), 2.94 (1H, s, Ho), 1.81 (3H, s, Hq), 1.52 (3H, s, Hr); 13C NMR (400 MHz, Acetone-d6) δ ppm: 197.5, 173.7, 158.8, 136.4, 136.5, 129.6-128.2 (10C), 128.1, 127.8, 127.5, 126.6, 126.0, 108.9, 108.1, 93.1, 89.5, 80.9, 75.8, 74.5, 71.1-69.7 (7C), 58.5, 50.7, 40.4 (2C), 24.4, 13.6; HRMS ESI: found 814.3776; calculated 814.3786 for [M+Na]+ C43H49N5O8Na.
5-(6-Dimethylamino-2-methyl-3-oxo-2.3-dihydro-benzofuran-2-yl)- [[1-[2-[2-[2-(2-prop-2-ynoxyethoxy) ethoxy] ethoxy] ethyl] triazol-4-yl]-bis(p-toluyl)methoxy]-4-methyl-penta-2.4-diene-amide (4b). To a solution of 3b (0.3 g, 0.63 mmol) in dry toluene under nitrogen atmosphere was added AcCl (0.24 mL, 3.14 mmol). After 2h reflux, the solution was evaporated to give the intermediate crude chloride as brown oil. To a solution of 2 (0.188 g, 0.63 mmol) in a mixture of dry NEt3/ACN (1:1) (6 mL) was added the crude chloride dissolved in a minimum of dry ACN at ambient temperature. The resulting solution was stirred 45 min at room temperature, the solvent was then evaporated under vacuum and purified by combi-flash (MeOH/ DCM: 0 to 3% MeOH) to afford the compound 4b as a colorless oil (80 mg, 18%). TLC MeOH/DCM 5/95 Rf = 0.34; 1H NMR (400 MHz. Acetone-d6) δ ppm: 9.80 (1H, s, NH), 7.74 (1H, s, Ha), 7.36 (1H, d, J = 12.1 Hz, Hc), 7.30 (4H, d, J = 8.3 Hz, Hd), 7.12 (4H, d, J = 8.1 Hz, He), 6.54 (1H, dd, J = 12.1, 4.2 Hz, Hf), 6.23 (1H, s, Hg), 5.95 (1H, d, J = 16.0 Hz, Hh), 5.88 (1H, s, Hi), 4.53 (2H, t, J = 4.0 Hz, Hj), 4.15 (2H, d, J = 0.8 Hz, Hk), 3.84 (2H, t, J = 8.0 Hz, Hl), 3.52 (12H, m, Hm), 3.13 (6H, s, Hn), 2.93 (1H, t, J = 0.8 Hz, Ho), 2.31 (6H, s, Hp), 1.82 (3H, s, Hq), 1.52 (3H, s, Hr); 13C NMR (400 MHz, Acetone-d6) δ ppm: 197.8, 173.7, 158.9 (2C), 128.9-126.03 (10C), 108.9, 108.2, 93.1, 89.6, 75.8, 71.1-69.8 (7C), 58.6, 50.1, 40.5 (2C), 24.5, 20.9 (2C), 13.6; HRMS ESI: found 786.3495; calculated 786.3473 for [M+Na]+.C45H53N5O8Na.
Hydrolysis rates of compounds 4
A solution of 1 mg compound 4b (or 4a) dissolved in 0.2 mL acetonitrile was mixed with aqueous 0.8 mL buffered solutions with pH 3.0, 4.3, 5.0 and 7.3. The samples were injected at various times to monitor the formation of compounds 3b (or 3a) and 2. The binary eluting system was composed of acetonitrile and water with 0.1% TFA 85:15 v:v and the flow rate was gradually modified over the time of analysis (Time:Flow compositions: (A) 0-5min: 0.25 to 0.5 mL/min (B) 5-10 min:0.5 to 1 mL/min (C) 10-15 min:1mL/min. Table 1 shows the half-life of compounds 4 at the selected pHs (for detailed kinetic see Table S1).
Half-lives of prodrugs 4 at selected pHs.
| compounds | pH 3.0 | pH 4.3 | pH 5.0 | pH 7.3 |
|---|---|---|---|---|
| 4a | n.d. | stable | stable | stable |
| 4b | 3h | 13h | 21h | 3.2 days |
Synthesis of the prodrug 4b functionalized macromonomer 7
The prodrug-functionalized macromonomer 7 was obtained by a Huisgen 1.3-cycloaddition between 6 and 4b. 251 mg of 4b (Mn=3600 g.mol-1, n=7 10-5 mol, 1 eq.), 98 mg of 7 (M=792 g.mol-1, 1.2 10-4 mol, 1.7 eq.) and 35µL of PMDETA (d=0.83, M=173 g.mol-1, n=1.8 10-4 mol, 1.7 eq.) were dissolved in 4.3 mL of DMF. Then the mixture was degassed according to the freeze-pump-thaw procedure. 24 mg of CuBr (M=143 g.mol-1, n=1.8 10-4 mol, 1.7 eq.) were then added under inert atmosphere (glovebox). The mixture was stirred during 4 days under argon at room temperature. Then, 45 mL of dichloromethane was added to the reaction mixture and the solution was washed ten times with 25 mL of water and dried with Na2SO4. The solution was filtrated, the solvent was evaporated and the macromonomer was dissolved in 30 mL of THF and precipitated in 200 mL of diethyl ether, filtrated, dried under vacuum and finally lyophilized in dioxane. The macromonomer 7 was stored under argon before use. rdt: 56%. functionalization: F=75%. 1H NMR (400 MHz. CDCl3) δ ppm for peaks that were clearly attributed : 7.65 (1H, s, Ha), 7.4 (1H, d, J = 12.0 Hz, Hc), 7.05 (1H, d, J = 12.0 Hz, Hd), 7.0 (1H, m, He 6.35 (1H, d, J = 4.0 Hz, Hf), 6.0 (4H, m, Hg.h.u), 5.80 (1H, s, Hi), 4.6-4.2 (m, 4H, Hjj',kk',ll'), 3.0 (s, 6H ,Hn), 2.25 (s, 6H, Hp), 1.5 (3H, s, Hr), 0.4 (0.6H, m, Ht); Mn;RMN=4410 g/mol. SEC: Mn;SEC= 3850 g/mol; PDI=1.12.
Synthesis of prodrug 4b-functionalized nanoparticles 8
NPs 8 were obtained by Ring-Opening Metathesis coPolymerization of norbornene 9 with macromonomers 5 and 7 in dispersion. Solvents were degassed according to the freeze-pump-thaw procedure. The reaction was carried out at room temperature under inert atmosphere (glovebox). In a typical experiment, 4 mg (M=823 g/mol, n=4.8 10-5 mol) of Grubbs first generation complex were dissolved in 2.2 mL of dichloromethane/ethanol mixture (50/50% v/v). Both norbornene 9 (192 mg, M=94 g/mol, n=2.0 10-3 mol) and macromonomers 5 and 7 (m5=63 mg, Mn;5=3150 g/mol, m7= 88 mg, Mn;7=4410 g/mol, n5=n7=2.0 10-5mol) were first dissolved in 3.5 mL of CH2Cl2/EtOH solution (35/65% v/v) and added to the catalyst solution. 0.1 mL of NEt3 were added to maintain the pH solution higher than 7. The mixture was stirred during 24 h. At the end of polymerization Ruthenium end-capped chains were deactivated by addition of 0.2 mL of ethyl vinyl ether.
Composition of the nanoparticles
The composition of the latex was determined according to consumed starting monomer and macromonomers. NB 9 exhibits a total conversion while global macromonomer conversion (π) was 90%. It is assumed than both macromonomers 5 and 7 are consumed with the same rate (we approximate than the ω-functionalization does not act on their reactivity). By this way, we can determine the amount of compound 2 n2 per gram of polymer with the following equation:
n2/gpolym = (F7 x n7 x π)/(π x (m5+m7)+m9) = 40 μmol/g
with F7 the ω-functionalization yield of macromonomer 7 (F7=0.75), n7 the amount of macromonomer 7 introduced for the NPs synthesis (µmol), π the global conversion ratio of the macromonomers 5 and 7 (π=0.9), mi the weight of the compound i introduced for the synthesis of the NPs (g).
The polymer concentration can be estimated with the equation:
[polymer] = (π x (m5+m7)+m9) / V = 0.05 g.mL-1.
with V the volume of the latex solution (5.7 mL).
Taking into account the amount of 2 per gram of polymer and the polymer concentration the concentration of 2 can be evaluated with the equation:
[Cpd 2] = [polymer] x n2/gpolym = 2 μmol/mL
The amount of linked prodrug of compound 2 molecule per NP (n2/NP) can then be calculated by multiplying the amount of 2 per gram of polymer with the weight of one NP using the following equation:
n2/NP = n2/gpolym x VNP x ρNP x NA =350 000 molecules per NP
with VNP the volume of one NP (VNP=πDNP3/6). ρNP the density of the NPs approximated to equal to 1 g/mL. NA the Avogadro constant.
The NPs were transferred in an aqueous solution of Trizma®base (10 mM) by successive evaporation and an ultrafiltration steps to give a final latex concentration of 20 mg/mL and a final NODH concentration of 0.8 µmol/mL (0.8mM).
BRET measurements
All BRET measurements were performed at room temperature using the Mithras LB 940 microplate analyzer (Berthold Technologies). Cells were pre-incubated for 15 min in PBS in the presence of 2.5 µM coelenterazine (Interchim), following which light-emission acquisition at 485 and 530 nm was carried out. Plates were measured five times. The BRET signal was expressed in milliBRET units (mBu). The BRET unit has been defined previously as the ratio 530/485 nm obtained when the two partners are present, corrected by the ratio 530/485 nm obtained under the same experimental conditions, when only the partner fused to Renilla luciferase is present in the assay.
Cell culture
The pleural mesothelial cell line, MeT-5A, was obtained from American Type Culture Collection (ATCC). The mesothelioma cell lines Meso13, Meso34 and Meso56 were established from the pleural fluids of mesothelioma patients (42). All cell lines were maintained in RPMI medium (Invitrogen) supplemented with 2 mM L-glutamine. 100 IU/ml penicillin, 0.1 mg/mL Streptomycine and 10% heat inactivated fetal calf serum (FCS) (Eurobio).
Transfections studies
MeT-5A cells were seeded at a density of 1.5x105 cells per 35 mm dish. Transient transfections were performed 1 day later using Attractene (Qiagen), according to the manufacturer's protocol. For BRET experiments, MeT-5A cells were transfected with 0.6 µg Rluc-Brd cDNA and 1 µg YFP-fused histone H3 cDNA (40). One day after transfection, cells were transferred into 96-well microplates (microplate 96 well, white, Berthold Technologies) at a density of 3x104 cells per well. The following day, BRET measurements were performed as described below.
Determination of cell viability
Cell growth was monitored using Uptiblue reagent (Interchim). Reduction of this compound by the cell results in the formation of a fluorescent compound quantified by measuring fluorescence at 595 nm after excitation at 532 nm using a Typhoon apparatus (GE Healthcare). Cells were seeded in 96-well plates at a density of 5x103 cells/well in culture medium. Twenty-four hours later, compounds solutions or nanoparticles were added for 72 h. Uptiblue (5%. v/v) was then added to the culture medium for 2 h at 37 °C. Fluorescence was quantified by measuring emission at 595 nm after excitation at 532 nm using a Typhoon apparatus (GE Healthcare).
Animal experiments
These experiments were carried out in compliance with the guidelines of the European Union for the care and use of animals in research protocols. The experiments were approved by ethical committee for animal experiment (CEEA.PdL 2013.6).
Bio-distribution experiments - 3x106 AK7 murine mesothelioma cells (AK7) per mouse were administered intra-peritoneally to five C57Bl/6 mice (Charles River) (day 0). Mice were given one i.v. injection of NPs 11 (60 µg/g) for 1h, 6h, 24h, 72h or 96h. Then, animals were necropsied and tumor, blood, liver, ovary, brain, spleen and kidneys were collected and analyzed for fluorescence emission. Fluorescence was observed at 630 nm using Photon Imager (Biospace Lab) after excitation at 580 nm and pictures were analyzed using PhotoVision+ software (Biospace Lab).
Anti-tumor activity of NPs 8 - 3x106 AK7 murine mesothelioma cells (AK7) per mouse were administered intra-peritoneally to four groups of five C57Bl/6 mice (Charles River) (day 0). Group 1 did not receive any further treatment. Groups 2, 3 and 4 received two injections of decitabine 4 mg/kg at days 7 and 9. Then, groups 2, 3 and 4 were respectively given four successive i.v. injections of bare-NPs 10 (80 µg/g), compound 2 (0.25 µg/g) or NPs 8 (16 µg/g. 0.25 µg/g cpd 2 equivalent) at days 12, 14, 16 and 19. All animals were necropsied on day 21. Liver, kidneys and spleen were weighted. Blood analyses, using a MS9-5 analyzer (Melet Schloesing laboratories), were performed and tumors were collected to perform histochemical analyses.
Histology
Tissues were fixed in 4% CH2O in PBS, embedded in paraffin, cut into 5-µm sections. Histology was performed on tissues slices (paraffin-embedded) by Cellular and Tissue Imaging Core Facility of Nantes University (MicroPICell) using hematoxylin, eosin and safran staining (HES). Pictures were obtained using a NanoZoomer 2.0HT (Hamamatsu).
Results and discussion
We previously reported that NP loaded with 1% mol Vorinostat triggered histone re-acetylation without tumor weight reduction with no associated toxicity.(35) To achieve tumor weight reduction without toxicity while maintaining a low percentage inhibitor loading, a more potent compound could be used. The highly active (R)-trichostatin A 1 (36) (TSA, Fig. 1) is not used for clinical purposes. The (S)-isomer is 70-fold less potent.(37) The racemic TSA analogue (34) 2 exhibited an activity equivalent to TSA, a result that could be explained by molecular docking in HDAC1 and HDAC6, showing that for the two separate enantiomers the aromatic group has no preferred orientation in the active site. Compound 2 has limited toxicity, stimulates TSG expression in non-small lung cancer cells (34) and reduces resistance to cisplatin treatment in MPM.(38) Its shorten synthesis and potential TSA-like metabolism to inactive compounds made it a good candidate for delivery with results valuable for other anti-cancer compounds.
Synthesis of compound 2
Analogue 2 was synthesized in 6 steps by direct trimethylation (39) of the acid 12 (Fig. 1) to the ester 13a easily separated from monomethylamino ester 13b. The conversion of the ester 13a to the acid 14 as already reported allowed accessing compound 2 by direct hydroxamic acid preparation in up to 4 g synthesis.
Synthesis and characterization of nanoparticles 8
The clickable pH-responsive prodrugs 4 needed for our NP synthesis were prepared by reacting compound 2 with alcohols 3 (27) (Fig. 1). The two prodrugs 4a,b gave analytical data consistent with their structure and purity (Supplementary information, NMR spectra and HPLC for 4a,b). The half-lives of the two prodrugs 4a,b were determined at selected pHs with HPLC (Fig. 2A, Table 1, and in supplementary data Table S1 and Fig. S1). Prodrug 4a was stable at all pHs and prodrug 4b showed satisfying stability for blood stream (stable more than 3 days at pH >= 7) and for the release at lysosomal equivalent pH (13 and 21h, pH 4.3 and 5 respectively). The prodrug 4b was selected for further development and reacted with azide 6 under copper catalysis to afford the macromonomer 7 (Fig. 1). 1H NMR analysis revealed a synthetic yield of 56% for a functional yield of 75% (Fig. 2C). The missing 25% were the residual macromonomer 5. Functional core-shell NPs 8 were formed in dispersed media by mixing norbornene 9 with macromonomers 5 and 7 in a 100:1:1 molar ratio (9 n=2.0 mmol, 5 and 7 n=20 μmol). The composition of the copolymer blocks 9:5:7 was 110:1.25:0.75, calculated from 75% functional yield of macromonomer 7 (+25% macromonomer 5) and 90% incorporation of both macromonomers 5 and 7 compared to 100% incorporation of NB (see Composition of the particle in Materials and Methods section). The concentration of releasable compound 2 from NPs 8 was finally 2.25 mM (13.5 μmol in 6 mL total reaction volume). The NPs 8 were transferred in an aqueous solution (Trizma®base 10 mM) by successive evaporation and an ultrafiltration (26) steps to give 20 mg/mL of NPs 8 corresponding to 0.8 μmol/mL (0.8 mM) compound 2. In these conditions the diameter of the spherical nanoparticles was 305 nm in the organic solvent and 380 nm in water (Fig. 2B), as demonstrated by DLS measurements. This result was also confirmed by TEM imaging (Fig. S2) clearly showing homogeneous sizes within the samples, with an average diameter also around 380 nm. The increase in size is a common result due to water molecules insertion in the nanoparticles PEO shell. The absence of surface charges limits potential opsonization.
Characterization of the prodrug 4b and synthetic nanoparticles 8. (A) Formation of compound 2 from hydrolysis of prodrug 4b at selected pHs. The sub-panel indicates the stability at longer time for pH 7.3. (B) Size distribution of NP 8 in the two solvent systems used. (C) NMR characterization of intermediate compounds and nanoparticles 8.
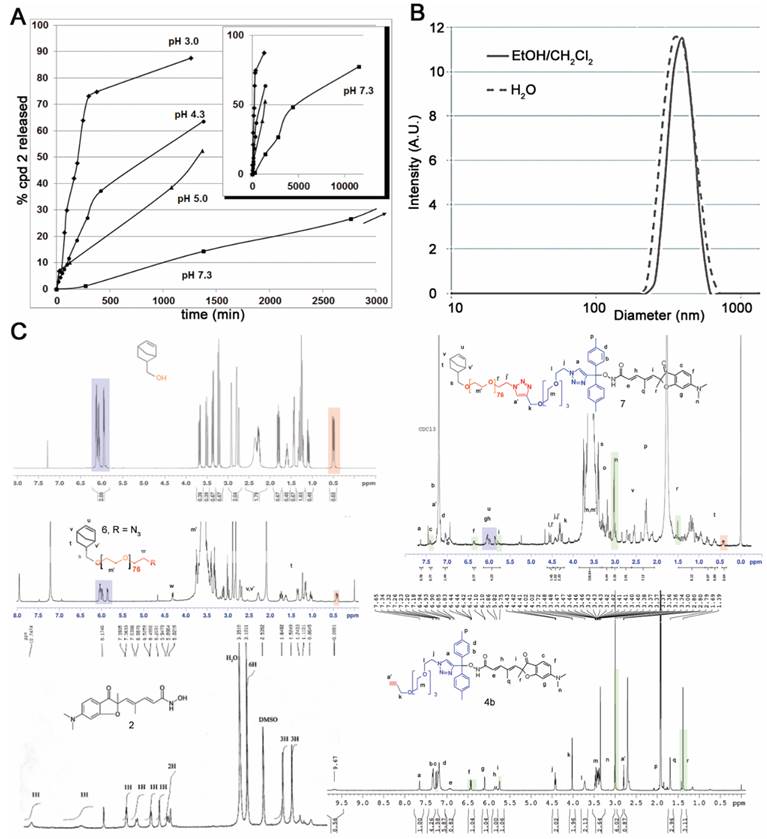
Key hydrogen signals found in starting compounds and products are highlighted with colors (Fig. 2C). The 1H NMR of norbornene methanol alone showed that the signal at 0.4 ppm represents 0.64 H when the ethylenic protons at 6.0 ppm are calibrated to integrate 2H. This signal at 0.4 ppm is common in all macromonomer spectra and the 0.64 calibration was used as a reference in the 1H NMR spectra calibration of functional macromonomer 7, leading to a confirmation of the 75% functionalization found by GC methods whereas the PEO signal integration was close to 318, corresponding approximatively to 79 units (PEO76 + PEO3 in prodrug part), other methylene signals being more specific.
HDAC inhibition and cell viability
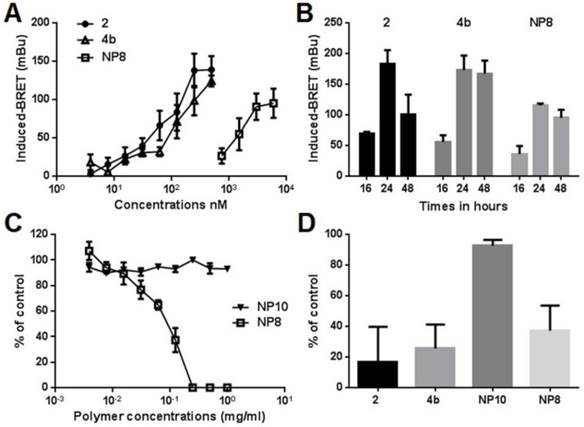
The ability of compounds 2, 4b and NPs 8 to enter cancer cells and inhibit cellular HDAC was then determined with a bioluminescence resonance energy transfer (BRET) assay in living cells (40). HDAC inhibition is correlated to higher histone acetylation level resulting in higher BRET signals. BRET signals induced by compounds 2, 4b and 8 were measured 16h post treatment in MeT-5A cells (Fig. 3A). A dose dependent increased BRET signals and thus HDAC inhibition was observed for all compounds, demonstrating they have entered cells. The IC50 concentrations were 500 nM for compounds 2 or 4b and 0.125 μg/mL for NPs 8 (equivalent to 6.0 μM compound 2). The lowest activity of NPs 8, despite its highest equivalent concentration of inhibitor 2, showed that there is no free inhibitor in the particle solution used to treat the cells. Such situation should have led to significant BRET signal at higher NPs dilution. NPs internalization in acidic vesicles is thus the only way for the slow release of compound 2 as previously demonstrated,(27) also explaining the lower activity. The kinetics of histone reacetylation after the treatment with compounds 2, 4b and 8 at their IC50 concentrations was then determined (Fig. 3B). Compounds showed a maximum BRET signal at 24h. NPs 8 gave 60-75% response compared to compounds 2 and 4b. Activity of compounds 4b and 8 was maintained for more than 48h while molecule 2 showed 50% activity decrease 48h after treatment. This result highlighted a protecting role of the delivery strategy, lowering metabolic activity and prolonging the reacetylation effect. Associated with a selective tumor delivery, this strategy should give sustained inhibitor release in vivo. Indeed, maintaining the reacetylation of histone as long as possible is an important issue in epigenetic treatment as renormalization is supposed to occur after hours. The toxicity of compounds 2, 4b, prodrug-bearing NPs 8 and hydroxyl NPs 10 (bare NP) (26) was evaluated on three mesothelioma cell lines established in our laboratory from patients pleural effusions (Fig. 3C,D). The unfunctional NPs 10 (showed in Fig. 4) induced no toxicity for all tested polymer concentrations whereas NPs 8 led to a rapid cell viability decrease even at 0.05 mg/ml of polymer. The compound 2 and its prodrug induced toxic effects at low nanomolar concentrations as expected owing to the already reported activities of compound 2. The BRET and viability data demonstrated that compound 2 grafted on the nanoparticle can be delivered in MPM cells in sufficient amount according to the histone acetylation and toxicity induction and that toxicity is not due to the nanoparticle itself but to HDACi release. The toxicity of NPs 8 compared to NPs 10 provided a wide safe window for in vivo experiments. The lower activity of NPs 8 compared with compound 2 and 4b is probably related to the delivery rate of the active compound into the cells. Indeed, compound 2 and 4b can diffuse rapidly into the cells and then can reach high intracellular concentrations. NPs 8 is progressively internalized, thus intracellular concentration of active compounds is the result of the balance between delivery and metabolism reducing it. This hypothesis is supported by BRET results. Indeed, maximal BRET induced by NPs 8 is lower than maximal BRET induced by compound 2 and 4b.
Pharmacological characterization of compound 2, prodrug 4b and nanoparticles 8. (A and B) MeT-5A cells were transfected with phRluc-C1 BrD and pEYFP-C1 histone H3. (A) Cells were treated for 16 h with increasing doses of the different molecules. For NPs 8 the concentration corresponds to the amount of releasable compound 2. (B) Cells were treated with 500nM of compound 2 or 4b or 0.125 mg/mL NPs 8 (6 µM compound 2) during 16h, 24h, or 48h. Results were expressed as the induced-BRET signal. (C) Viability of MM cells was evaluated following 72h of treatments with increasing amounts of NPs 8 and bare-NPs 10 for 72h. (D) Cells were treated with 125nM of compound 2 or 4b, 0.125 mg/mL NPs 10 or 0.125 mg/mL NPs 8 (6 µM compound 2) during 72h. Results are the means ± S.E.M of three independent experiments.
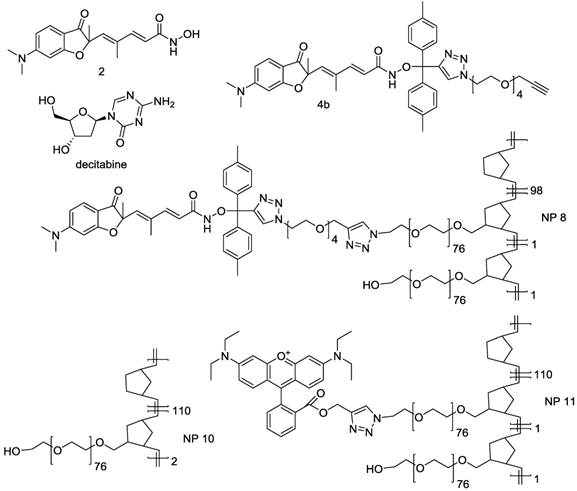
Summary of compounds used in this work. Free compounds were decitabine and HDAC inhibitor 2, prodrug of inhibitor 2 was compound 4b and nanoparticles were NP8 bearing HDAC inhibitor 2 prodrug 4b, NP10 with no functionalities and NP11 bearing a fluorescent dye as reporter for biodistribution studies.
Biodistribution of nanoparticles
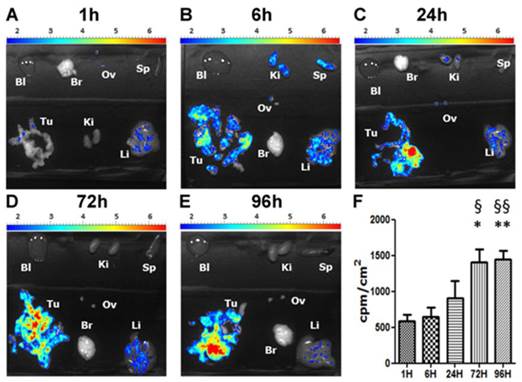
We then determined the biodistribution of our DDS in an orthotopic mouse model of peritoneal mesothelioma (PM) which presents the advantage of being more relevant due to a better reconstitution of the tumor-host interactions compared to subcutaneous xenograft models of cancers (41). For these experiments a fluorescent version of our NPs, NP 11, was used. Five groups of five C57Bl/6 mice received intraperitoneal injections of AK7 murine mesothelioma cells (protocol Fig. S3). After 7 days, a solution of the rhodamine-bearing NP 11 (26) (60 μg NP 11/g of mice, Fig. 4) was injected in the tail vein of mice. Mice were euthanized 1h, 6h, 24h, 72h and 96h after injection and the organs collected for biodistribution studies. Fluorescence imaging of isolated organs (Fig. 5A-E) and quantification (Fig. 5F) showed a strong and specific accumulation of NP 11 in tumors increasing over the first 24h after injection, reaching a plateau at 72h, and then remaining stable until 96h. The quantification of NP 11 accumulation in liver, spleen, kidneys, brain, ovaries and blood is provided in figure S4. A transient fluorescence was detected in liver 6h after NP 11 injection corresponding probably to the presence of NP 11 in blood. Then, NP 11 are cleared from the bloodstream, fluorescence in liver decreases and NP 11 continue to accumulate in tumor. These results demonstrated that the high tumor selectivity of these NP injected i.v. observed previously in xenografts is maintained in this more relevant diffuse tumor model, allowing tumor identification by fluorescence imaging.
Biodistribution of NPs 11. C57Bl/6 mice bearing orthotopic AK7 tumors were injected with 60 μg of NPs 11 per g of mice in the tail vein. (A-E) Pictures of time dependent fluorescence observed on dissected tumor (Tu), kidneys (Ki), liver (Li), ovary (Ov), brain (Br), spleen (Sp) and in blood (Bl). (F) Quantification of NPs 11 accumulation in tumors over time. Results are expressed as the fluorescence intensity corrected by the organ surface. Values are means ± S.E.M. of results obtained on 5 mice. * p<0.05; ** p<0.01.
Anti-cancer effect of nanoparticle 8 and toxicity
In the laboratory, we previously demonstrated that decitabine in combination with valproic acid induced a stronger decrease of the tumor weight compared to drugs used alone in our orthotopic model of mesothelioma.(42) In addition, the observed synergy of decitabine and TSA in myeloid leukemia (43) and the clinical data obtained in a phase II trial using hypomethylating agent in combination with HDACi prompted us to use decitabine in combination with compound 2 and its pH-responsive NP version. Compounds 2, NP 8 and NP 10 were evaluated in combination with decitabine to treat PM tumors (Fig. 6 and Fig. S3 for mice administration schedule). As for biodistribution studies, AK7 murine mesothelioma cells were administered by intra-peritoneal injection to four groups of five C57Bl/6 mice at day 0 (Fig. S3). Group 1 did not receive any treatment and groups 2 to 4 received two intraperitoneal injection of 4µg/g of decitabine at day 7 and 9, followed by four successive intravenous injections of hydroxyl NP 10 for group 2 (80 µg/g), the free analogue 2 (0.25 µg/g) for group 3 and NP 8 for group 4 (0.25 µg/g 2, 16 µg/g polymer). All mice survived and were euthanized at day 21 to collect tumors. Tumors weight were not modified following injections in mice treated with bare-NP 10 (Fig. 6A) showing the absence of effect of this polymer in vivo. We confirmed that decitabine in these conditions has no anti-tumor effect by itself in our model. Interestingly, whereas iv injections of analogue 2 showed no anti-tumor effect, iv injections of NP 8 (0.25 µg/g equivalent of analogue 2) decreased strongly (80%) and significantly the tumors weights (Fig. 6A).
In vivo anti-tumor activity of nanoparticles 8. C57Bl6 mice bearing orthotopic AK7 tumors were treated (except control group 1) with intraperitoneal injection of decitabine (4μg/g) at days 7 and 9, followed by intravenous injection of bare-NPs 10 (hydroxyl NPs 10, group 2) (80 µg/g), compound 2 (0.25 µg/g, group 3), or NPs 8 (0.25 µg/g compound 2, 16 µg/g polymer, group 4). (A) Graphic represents the tumor weight measured at the end of the experiment. Results are means ± S.E.M. of results obtained on 5 mice. * p<0.05; ** p<0.01. (B to D) Representative HES stained histology slices of pancreas from the control group (B), group treated with analogue 2 (C) or group treated with NPs 8 (D). Arrows indicate areas of pancreatic invasion by tumor cells.
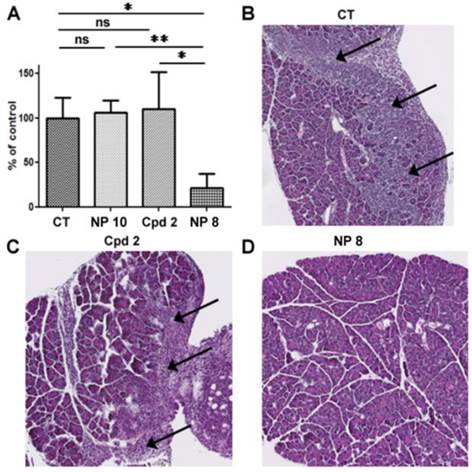
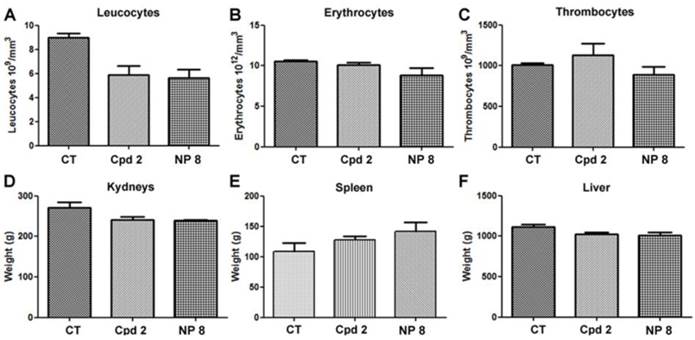
Moreover, in untreated mice and in free analogue 2 injected mice, we observed large and numerous pancreatic tumor invasion areas (Fig. 6B and C). In mice treated with NP 8 (Fig. 6D), the tumor weight reduction was associated with the absence of tumor cells in pancreas for a majority of mice (4/5). These anti-tumor effects of NP 8 were associated with an increase of histone H3 acetylation in residual tumors demonstrating the in vivo activity of the HDACi (Fig. S5). All the tested conditions did not induce apparent toxicity in animals, as determined by liver, kidneys and spleen weight and by blood formula (Fig. 7). This is also correlated with the absence of modification in acetylated histone H3 level (Fig.S6) in liver and kidneys. Thus, once loaded onto our pH-responsive DDS, in vivo study clearly showed an improved activity of compound 2, suggesting an effective and specific delivery in the tumor site by the EPR effect followed by acidic mediated release. This vectorization strategy gave an optimal antitumoral effect without apparent toxicity despite a very low drug loading (1%).
Toxic and haematological effects of compound 2 and NPs 8. The day of mice euthanasia, blood samples were collected and analyzed to determine (A) leucocytes (B) erythrocytes and (C) thrombocytes. (D) kidneys, (E) spleen, (F) liver were collected and weighted. Graphics represent the means ± S.E.M of results obtained on 5 mice.
These results support our proposal that tumors can be identified with this type of NP made fluorescent, although PET or MRI techniques can be envisioned with the corresponding functional entities. Our ROMP-based conjugate 8 circumvented the lack of efficacy of the highly potent nanomolar inhibitor 2 in an orthotopic model of PM and represent a viable alternative to current strategies. This ROMP-DDS is an innovative, flexible, simplified, safe and potentially cost-effective technology that could be used in targeted non-epigenetic or epigenetic personalized medicine both for therapy or identification of tumors. With the increasing number of molecules targeting epigenetic regulators, this work paves the way for efficient delivery of epidrugs, as well as other chemotherapeutics.
Supplementary Material
Supplementary tables and figures.
Acknowledgements
Authors thank Agence Nationale de la Recherche (ANR) for RD and FC grants (ANR-08-PCVI-030), Région Poitou-Charentes for FEB grant, the Ligue National Contre la Cancer for ID grant, the Ligue Contre le Cancer: committees of Morbihan, Sarthe, Vendée, Loire-Atlantique and Charente-Maritime, Centre National de la Recherche Scientifique (CNRS), ARSMESO44, and Nantes University Hospital. The authors thank MicroPICell core facilities for histology.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Arrowsmith CH, Bountra C, Fish PV, Lee K, Schapira M. Epigenetic protein families: a new frontier for drug discovery. Nat Rev Drug Discov. 2012;11:384-400
2. Dawson MA, Kouzarides T. Cancer Epigenetics: From Mechanism to Therapy. Cell. 2012;150:12-27
3. Suva ML, Riggi N, Bernstein BE. Epigenetic reprogramming in cancer. Science. 2013;339:1567-70
4. DeWoskin VA, Million RP. The epigenetics pipeline. Nat Rev Drug Discov. 2013;12:661-2
5. Feng D, Wu J, Tian Y, Zhou H, Zhou Y, Hu W. et al. Targeting of Histone Deacetylases to Reactivate Tumour Suppressor Genes and Its Therapeutic Potential in a Human Cervical Cancer Xenograft Model. PLoS ONE. 2013;8:e80657
6. Wagner T, Brand P, Heinzel T, Krämer OH. Histone deacetylase 2 controls p53 and is a critical factor in tumorigenesis. Biochim Biophys Acta BBA - Rev Cancer. 2014;1846:524-38
7. Ma Y, Jin H, Wang X, Shin VY, Chen D, Chen Y. et al. Histone deacetylase 3 inhibits new tumor suppressor gene DTWD1 in gastric cancer. Am J Cancer Res. 2015;5:663-73
8. Duvic M, Vu J. Vorinostat: a new oral histone deacetylase inhibitor approved for cutaneous T-cell lymphoma. Expert Opin Investig Drugs. 2007;16:1111-20
9. Grant C, Rahman F, Piekarz R, Peer C, Frye R, Robey RW. et al. Romidepsin: a new therapy for cutaneous T-cell lymphoma and a potential therapy for solid tumors. Expert Rev Anticancer Ther. 2010;10:997-1008
10. Poole R. Belinostat: First Global Approval. Drugs. 2014;74:1543-54
11. Fenichel MP. FDA Approves New Agent for Multiple Myeloma. J Natl Cancer Inst. 2015;107:djv165
12. How J, Minden MD, Brian L, Chen EX, Brandwein J, Schuh AC. et al. A phase I trial of two sequence-specific schedules of decitabine and vorinostat in patients with acute myeloid leukemia. Leuk Amp Lymphoma. 2015;56:2793-802
13. Robey RW, Chakraborty AR, Basseville A, Luchenko V, Bahr J, Zhan Z. et al. Histone Deacetylase Inhibitors: Emerging Mechanisms of Resistance. Mol Pharm. 2011;8:2021-31
14. Mithraprabhu S, Khong T, Spencer A. Overcoming inherent resistance to histone deacetylase inhibitors in multiple myeloma cells by targeting pathways integral to the actin cytoskeleton. Cell Death Dis. 2014;5:e1134
15. Azad N, Zahnow CA, Rudin CM, Baylin SB. The future of epigenetic therapy in solid tumours[mdash]lessons from the past. Nat Rev Clin Oncol. 2013;10:256-66
16. Momparler R, Cote S, Momparler L, Idaghdour Y. Epigenetic therapy of acute myeloid leukemia using 5-aza-2'-deoxycytidine (decitabine) in combination with inhibitors of histone methylation and deacetylation. Clin Epigenetics. 2014;6:19
17. Kalac M, Scotto L, Marchi E, Amengual J, Seshan VE, Bhagat G. et al. HDAC inhibitors and decitabine are highly synergistic and associated with unique gene-expression and epigenetic profiles in models of DLBCL. Blood. 2011;118:5506-16
18. Ahrens TD, Timme S, Hoeppner J, Ostendorp J, Hembach S, Follo M. et al. Selective inhibition of esophageal cancer cells by combination of HDAC inhibitors and Azacytidine. Epigenetics. 2015;10:431-45
19. Sun T, Zhang YS, Pang B, Hyun DC, Yang M, Xia Y. Engineered Nanoparticles for Drug Delivery in Cancer Therapy. Angew Chem Int Ed. 2014;53:12320-64
20. Bahhaj F el, Dekker FJ, Martinet N, Bertrand P. Delivery of epidrugs. Drug Discov Today. 2014;19:1337-52
21. Wang Y, Byrne JD, Napier ME, DeSimone JM. Engineering nanomedicines using stimuli-responsive biomaterials. Adv Drug Deliv Rev. 2012;64:1021-30
22. Yin Q, Shen J, Zhang Z, Yu H, Li Y. Reversal of multidrug resistance by stimuli-responsive drug delivery systems for therapy of tumor. Adv Drug Deliv Rev. 2013;65:1699-715
23. Maeda H. Toward a full understanding of the EPR effect in primary and metastatic tumors as well as issues related to its heterogeneity. Adv Drug Deliv Rev. 2015;91:3-6
24. Miki K, Oride K, Inoue S, Kuramochi Y, Nayak RR, Matsuoka H. et al. Ring-opening metathesis polymerization-based synthesis of polymeric nanoparticles for enhanced tumor imaging in vivo: Synergistic effect of folate-receptor targeting and PEGylation. Biomaterials. 2010;31:934-42
25. Johnson JA, Lu YY, Burts AO, Lim Y-H, Finn MG, Koberstein JT. et al. Core-Clickable PEG-Branch-Azide Bivalent-Bottle-Brush Polymers by ROMP: Grafting-Through and Clicking-To. J Am Chem Soc. 2011;133:559-66
26. Gueugnon F, Denis I, Pouliquen D, Collette F, Delatouche R, Héroguez V. et al. Nanoparticles Produced by Ring-Opening Metathesis Polymerization Using Norbornenyl-poly(ethylene oxide) as a Ligand-Free Generic Platform for Highly Selective In Vivo Tumor Targeting. Biomacromolecules. 2013;14:2396-402
27. Delatouche R, Denis I, Grinda M, Bahhaj FE, Baucher E, Collette F. et al. Design of pH responsive clickable prodrugs applied to histone deacetylase inhibitors: A new strategy for anticancer therapy. Eur J Pharm Biopharm. 2013;85(Part B):862-72
28. Scita G, Di Fiore PP. The endocytic matrix. Nature. 2010;463:464-73
29. Arrieta O, Medina LA, Estrada-Lobato E, Hernandez-Pedro N, Villanueva-Rodriguez G, Martinez-Barrera L. et al. First-line chemotherapy with liposomal doxorubicin plus cisplatin for patients with advanced malignant pleural mesothelioma: phase II trial. Br J Cancer. 2012;106:1027-32
30. Vandermeers F, Sriramareddy SN, Costa C, Hubaux R, Cosse J-P, Willems L. The role of epigenetics in malignant pleural mesothelioma. Lung Cancer. 2013;81:311-8
31. Krug LM, Kindler HL, Calvert H, Manegold C, Tsao AS, Fennell D. et al. Vorinostat in patients with advanced malignant pleural mesothelioma who have progressed on previous chemotherapy (VANTAGE-014): a phase 3, double-blind, randomised, placebo-controlled trial. Lancet Oncol. 2015;16:447-56
32. Zong H, Shah D, Selwa K, Tsuchida RE, Rattan R, Mohan J. et al. Design and Evaluation of Tumor-Specific Dendrimer Epigenetic Therapeutics. ChemistryOpen. 2015;4:335-41
33. Collette F, Delatouche R, Blanquart C, Gueugnon F, Grégoire M, Bertrand P. et al. Easy and effective method to produce functionalized particles for cellular uptake. J Polym Sci Part Polym Chem. 2013;51:176-89
34. Charrier C, Clarhaut J, Gesson J-P, Estiu G, Wiest O, Roche J. et al. Synthesis and Modeling of New Benzofuranone Histone Deacetylase Inhibitors that Stimulate Tumor Suppressor Gene Expression. J Med Chem. 2009;52:3112-5
35. Denis I, el Bahhaj F, Collette F, Delatouche R, Gueugnon F, Pouliquen D. et al. Vorinostat-Polymer Conjugate Nanoparticles for Acid-Responsive Delivery and Passive Tumor Targeting. Biomacromolecules. 2014;15:4534-43
36. Olaharski AJ, Ji Z, Woo J-Y, Lim S, Hubbard AE, Zhang L. et al. The Histone Deacetylase Inhibitor Trichostatin A Has Genotoxic Effects in Human Lymphoblasts In Vitro. Toxicol Sci. 2006;93:341-7
37. Yoshida M, Hoshikawa Y, Koseki K, Mori K, Beppu T. Structural Specificity For Biological Activity Of Trichostatin A, A Specific Inhibitor Of Mammalian Cell Cycle With Potent Differentiation-Inducing Activity In Friend Leukemia Cells. J Antibiot (Tokyo). 1990;43:1101-6
38. Gueugnon F, Cartron P-F, Charrier C, Bertrand P, Fonteneau J-F, Gregoire M. et al. New histone deacetylase inhibitors improve cisplatin antitumor properties against thoracic cancer cells. Oncotarget. 2014;5:4504-15
39. Wang P, Wang Y, Hu H, Spencer C, Liang X, Pan L. Sequential Removal of Photolabile Protecting Groups for Carbonyls with Controlled Wavelength. J Org Chem. 2008;73:6152-7
40. Blanquart C, François M, Charrier C, Bertrand P, Grégoire M. Pharmacological characterization of histone deacetylase inhibitor and tumor cell-growth inhibition properties of new benzofuranone compounds. Curr Cancer Drug Targets. 2011;11:919-28
41. Kung AL. Practices and Pitfalls of Mouse Cancer Models in Drug Discovery. Adv Cancer Res. 2007;96:191-212
42. Leclercq S, Gueugnon F, Boutin B, Guillot F, Blanquart C, Rogel A. et al. A 5-aza-2′-deoxycytidine/valproate combination induces cytotoxic T-cell response against mesothelioma. Eur Respir J. 2011;38:1105-16
43. Shaker S, Bernstein M, Momparler LF, Momparler RL. Preclinical evaluation of antineoplastic activity of inhibitors of {DNA} methylation (5-aza-2′-deoxycytidine) and histone deacetylation (trichostatin A, depsipeptide) in combination against myeloid leukemic cells. Leuk Res. 2003;27:437-44
Author contact
![]() Corresponding author: Dr. Philippe Bertrand, Institut de Chimie des Milieux et Matériaux de Poitiers, University of Poitiers, CNRS UMR 7285, 4 rue Michel Brunet TSA 51106, B27, Poitiers. F-86073 Poitiers cedex 9, France. Phone number: +33 (0)5 49 45 41 92. philippe.bertrandfr.
Corresponding author: Dr. Philippe Bertrand, Institut de Chimie des Milieux et Matériaux de Poitiers, University of Poitiers, CNRS UMR 7285, 4 rue Michel Brunet TSA 51106, B27, Poitiers. F-86073 Poitiers cedex 9, France. Phone number: +33 (0)5 49 45 41 92. philippe.bertrandfr.