Impact Factor
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Results
Discussion
Materials and methods
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2016; 6(6):862-874. doi:10.7150/thno.12890 This issue Cite
Research Paper
Imaging and Selective Elimination of Glioblastoma Stem Cells with Theranostic Near-Infrared-Labeled CD133-Specific Antibodies
Hua Jing1,3, Claudia Weidensteiner2, Wilfried Reichardt2, Simone Gaedicke1, Xuekai Zhu1,3, Anca-Ligia Grosu1,3, Hisataka Kobayashi4, Gabriele Niedermann1,3 ![]()
1. Dept. for Radiation Oncology, University Hospital Freiburg, D-79106 Freiburg, Germany
2. Radiology Medical Physics, University Hospital Freiburg, D-79106 Freiburg, Germany
3. German Cancer Consortium (DKTK), Freiburg, and German Cancer Research Centre (DKFZ), D-69121 Heidelberg, Germany
4. Molecular Imaging Program, Center for Cancer Research, National Cancer Institute, National Institutes of Health, Bethesda, MD 20892, USA
Received 2015-6-5; Accepted 2016-2-12; Published 2016-4-12
Abstract

Near-infrared photoimmunotherapy (NIR-PIT), which employs monoclonal antibody (mAb)-phototoxic phthalocyanine dye IR700 conjugates, permits the specific, image-guided and spatiotemporally controlled elimination of tumor cells. Here, we report the highly efficient NIR-PIT of human tumor xenografts initiated from patient-derived cancer stem cells (CSCs). Using glioblastoma stem cells (GBM-SCs) expressing the prototypic CSC marker AC133/CD133, we also demonstrate here for the first time that NIR-PIT is highly effective against brain tumors. The intravenously injected theranostic AC133 mAb conjugate enabled the non-invasive detection of orthotopic gliomas by NIR fluorescence imaging, and reached AC133+ GBM-SCs at the invasive tumor front. AC133-targeted NIR-PIT induced the rapid cell death of AC133+ GBM-SCs and thereby strong shrinkage of both subcutaneous and invasively growing brain tumors. A single round of NIR-PIT extended the overall survival of mice with established orthotopic gliomas by more than a factor of two, even though the harmless NIR light was applied through the intact skull. Humanised versions of this theranostic agent may facilitate intraoperative imaging and histopathological evaluation of tumor borders and enable the highly specific and efficient eradication of CSCs.
Keywords: near-infrared photoimmunotherapy, cancer stem cells, CD133, glioblastoma
Introduction
Near-infrared photoimmunotherapy (NIR-PIT) combines the application of a target-specific antibody conjugated with a water-soluble NIR photosensitizer such as the silica-phthalocyanine dye IR700 with the local application of NIR light [1, 2]. This novel technology has several desirable features: (i) It allows the targeting of antigens that are not absolutely tumor specific, which is important because only few tumor-specific cell surface antigens are currently known; (ii) cell elimination is strictly dependent on target antigen expression, leaving neighboring antigen-negative cells unharmed [1, 3]; (iii) the modification with a water-soluble NIR dye does not compromise the in vivo specificity of the antibody conjugate [1]; (iv) target cell elimination, which occurs via structural damage to the cell membrane, is very rapid [1]; (v) the same antibody conjugates can be used for imaging and therapy, i.e. theranostically [1, 4]; and (vi) because of the relatively high tissue penetration of NIR light [5], target cells can be detected and, at higher light doses, eliminated within 1-2 cm of tissue [1].
So far, the high efficacy of NIR-PIT has been demonstrated in mice by using conventional tumor cell lines in models with subcutaneous (s.c.) flank tumors and by employing models of peritoneal or pleural carcinomatosis [1, 6, 7]. Based on these promising preclinical studies, a phase 1 trial of NIR-PIT in patients with inoperable head and neck cancer targeting the epidermal growth factor receptor was recently approved by the US Food and Drug Administration (https://clinicaltrials.gov/ct2/show/NCT02422979). However, NIR-PIT has not yet been evaluated in preclinical models based on tumor stem cells, commonly called cancer stem cells (CSCs) [8-10], and has also not yet been evaluated for the treatment of brain tumors, which would be a perfect target of NIR-PIT because complete resection of highly malignant primary brain tumors, which usually grow invasively, is impossible in most cases [11, 12]. After surgery, NIR-PIT could selectively ablate the left-over tumor cells that invade the normal brain tissue.
Glioblastoma multiforme (GBM) is the most common and most malignant primary brain tumor in adults. Despite extensive surgery and intensive radio- and chemotherapy, the median survival of GBM patients is currently only 14.6 months [13]. One major reason for the incurability is the highly invasive growth pattern [14], which makes complete surgical resection of GBMs impossible. Another reason is the dependence on CSCs. There is indeed accumulating evidence that GBM is driven by CSCs [8, 9, 15, 16].
CSCs are undifferentiated tumor cells with high self-renewal capacity, limited differentiation capability and high motility; they often preferentially locate to the invasive front of highly malignant tumors [17-19] and are therefore thought to be crucial for tumor initiation, long-term propagation, invasion, and metastasis [18, 20]. We and others have indeed shown that only undifferentiated CSC-like but not differentiated GBM cells are tumorigenic upon xenotransplantation into the brains of immunodeficient mice [19-21]. In addition, CSCs are often resistant to conventional genotoxic therapies [22, 23]. Recently, it has been shown that CSC-like cells are also more resistant than differentiated tumor cells to conventional non-targeted photodynamic therapy (PDT) which relies on the production of oxidative stress; resistance was due to the induction of autophagy [24]. Because of the crucial role of CSCs in the pathogenesis and treatment resistance of GBM and other aggressive tumor entities, it is desirable to develop methods for the specific and effective elimination of CSCs.
Several surface markers expressed by CSCs that could be targeted by mAbs or other ligands have been described so far. AC133, a stem cell-specific glycosylation-dependent epitope of CD133 [25, 26], is a CSC marker for many tumor entities including highly malignant brain tumors such as GBM [15, 16, 27, 28]. We and others found that AC133+ CSCs preferentially locate to the invasive front of orthotopic patient-derived tumor xenografts in immunocompromised mouse models [18, 19]. Similar observations have been made for CD44, another well-known CSC surface marker [17, 29].
An important factor that could potentially limit the efficacy of mAb-based PIT of brain tumors is that systemically administered mAbs might not be capable of penetrating into brain tumors because of the blood-brain and blood-tumor barriers [30]. However, we and others have recently published evidence that systemically administered full-length mAbs can quite efficiently penetrate brain tumors and enable their non-invasive detection [19, 31, 32].
The main aims of the present study were to investigate whether NIR-PIT could be used (i) for the elimination of patient-derived CSCs and (ii) for the treatment of invasively growing, established brain tumors.
Results
Validation of the binding specificity of the AC133-IR700 conjugate in vitro and in vivo
The AC133 mAb, which recognizes the AC133 epitope of CD133 on human CSCs [21, 26], was conjugated with IR700. The AC133-IR700 preparation showed strong association between the AC133 mAb and IR700, as determined by polyacrylamide gel electrophoresis (Fig. S1A). No aggregates were detected by size exclusion chromatography (Fig. S1B). IR700 conjugation did not impair binding of the AC133 mAb to the AC133 epitope, as revealed by conventional flow-cytometric titration of AC133-IR700 against the native, unlabeled AC133 mAb, by using mixtures of AC133+ and AC133- HCT116 colon carcinoma cells (Fig. 1A). Strong specific binding was found when U251 glioma cells overexpressing CD133 (CD133-OE U251 cells) were analyzed by NIR flow cytometry in comparison to CD133-negative wild-type (WT) U251 cells. In agreement with the lower AC133 expression level on NCH421k GBM stem cells (GBM-SCs) [21], the NIR flow cytometry signal caused by binding of AC133-IR700 to these patient-derived GBM-SCs was lower, but significant (Fig. 1B).
Validation of the immunoreactivity of the AC133-IR700 conjugate. (A) Side-by-side flow-cytometric analysis of the binding specificity of the AC133-IR700 conjugate compared to the unmodified AC133 mAb. Mixtures of HCT116 WT cells (AC133 positive) and p53-deficient HCT116 cells (AC133 negative) were incubated with serial dilutions of unmodified or modified antibody and analyzed by flow cytometry using an anti-mouse PE-conjugated F(ab')2 fragment. (B) Flow-cytometric analysis demonstrating binding of AC133-IR700 to CD133-OE U251 glioma cells and NCH421k GBM-SCs. (C) Immunofluorescence staining demonstrating specific binding of the AC133-IR700 conjugate (red) to CD133-OE U251 cells but not to WT U251 cells. The cells were incubated with AC133-IR700 alone (left) or - to block its binding - together with the competitive unlabeled AC133 mAb (right) for 6 h. To visualize the CSC marker CD133, the cells were stained with AC141-PE (green). Nuclei were stained with DAPI (blue). Scale bar, 100 μm. (D) NIR-FMT imaging of mice bearing WT U251 (AC133-) and CD133-OE U251 (AC133+) tumors 24 h after i.v. injection of the AC133-IR700 conjugate.
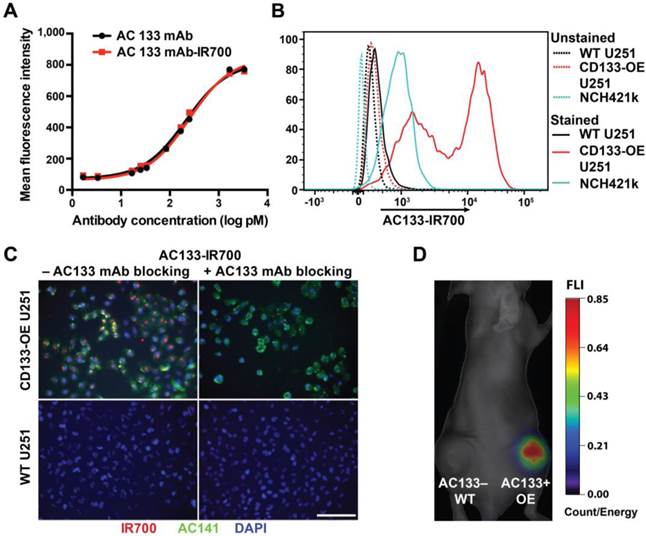
The binding specificity of the AC133-IR700 conjugate was further demonstrated by immunofluorescence. IR700 fluorescence was detected on CD133-OE U251 cells incubated with AC133-IR700, but not on WT U251 cells. Moreover, the IR700 signal co-localized with the fluorescence signal from a phycoerythrin (PE)-labeled mAb specific for another well-known stem cell-specific epitope of CD133 (AC141). No fluorescence signal was detected in the presence of the competitive unlabeled AC133 mAb (Fig. 1C).
To evaluate the in vivo binding specificity of the AC133-IR700 conjugate, we injected this mAb conjugate intravenously (i.v.), via the tail vein, into nude mice bearing WT U251 (AC133-) and CD133-OE U251 (AC133+) tumors in the left and right flank, respectively. Uptake of the mAb conjugate was non-invasively assessed by NIR-fluorescence molecular tomography (FMT) [33]. As shown in Fig. 1D, 24 h after AC133-IR700 injection, the AC133+ U251 tumor showed a considerably higher fluorescence signal than the AC133- tumor, indicating that the AC133-IR700 conjugate bound specifically to the AC133-expressing tumor cells in vivo.
AC133-IR700-mediated PIT efficiently kills CSCs in vitro
We first examined the specificity and extent of cell death, using the isogenic pair of AC133-negative and -positive U251 glioma cells (WT vs. CD133-OE). As shown in Fig. 2A, after incubation with the AC133-IR700 conjugate for 6 h, irradiation with NIR light (12 J/cm2) induced cell death in approximately 80% of the CD133-OE U251 glioma cells within a few minutes. In contrast, no significant cell death of CD133-OE U251 cells was observed upon incubation with an isotype control antibody conjugated with IR700 (Isotype-IR700). Microscopic changes in the CD133-OE U251 cells after PIT are shown in Fig. 2B. NIR light induced cell shrinkage and membrane blebbing, indicating necrotic cell death. WT U251 cells, which are AC133 negative, did not undergo cell death after NIR light exposure (Fig. 2C and Fig. S2).
Target-specific cell death induced by AC133-IR700-mediated PIT in vitro. (A) NIR light irradiation-induced death of CD133-OE U251 cells pre-incubated for 6 h with AC133-IR700, as determined by Trypan blue exclusion. No cell death was detected after incubation with IR700-conjugated isotype control mAb or in control cultures without antibody. Data are means ± SE (n ≥ 5, ****p < 0.0001 for cultures treated with AC133-IR700 compared to the control cultures using an unpaired t-test). (B) Microscopic analysis of CD133-OE U251 cells treated by AC133-IR700-mediated PIT in comparison to cultures treated with an IR700-labeled isotype control antibody or without antibody. Note the cell shrinkage and membrane blebbing in the AC133-IR700-containing cultures. Scale bar, 50 μm. (C) AC133-IR700-mediated PIT did not have phototoxic effects on WT U251 cells (n ≥ 5). (D) NIR light irradiation efficiently induced cell death of NCH421k GBM-SCs as determined by Trypan blue exclusion (n ≥ 5, ****p < 0.0001 for cultures treated with AC133-IR700 compared to the control cultures using an unpaired t-test). (E) PIT abolished sphere formation in cultures of NCH421k GBM-SCs treated as described in Materials and Methods. Scale bar, 200 μm. Except the cultures termed “untreated”, all cultures were exposed to NIR light as described in Materials and Methods.
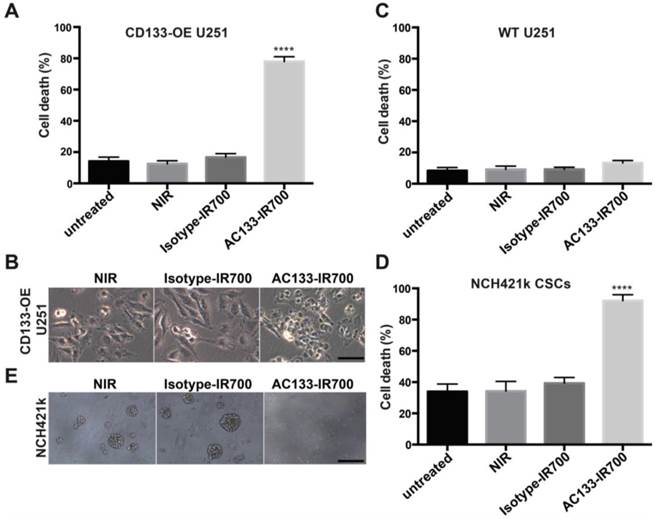
The well-characterized NCH421k GBM-SC line has been derived from a primary GBM (WHO grade IV) under stem cell culture conditions [21] and displays approximately 10-15-fold lower levels of AC133 than CD133-OE U251 glioma cells (see Fig. 1B and [19, 34]). AC133 mAb-based PIT effectively killed NCH421k GBM-SCs, despite the significantly lower AC133 expression level. NIR light induced cell death in more than 90% of the NCH421k GBM-SCs incubated with the AC133-IR700 conjugate. However, because of the low AC133 expression, efficient killing of the CSCs required an overnight pre-incubation with AC133-IR700. NIR light alone or together with the IR700-conjugated isotype control mAb was not cytotoxic (Fig. 2D); the relatively high level of basal cell death is typical of CSCs grown under sphere culture conditions [35]. In addition to the direct cell death assay, sphere formation, which is regarded as a stem cell surrogate assay, was addressed. Three days after NIR light exposure, spheres formed in significant numbers in cultures exposed to NIR light, alone or together with the IR700-conjugated isotype control mAb. In contrast, almost no spheres were found when NCH421k cells were treated with AC133 mAb-based PIT (Fig. 2E).
NIR-PIT delays CSC-driven tumor initiation in an s.c. mouse tumor model
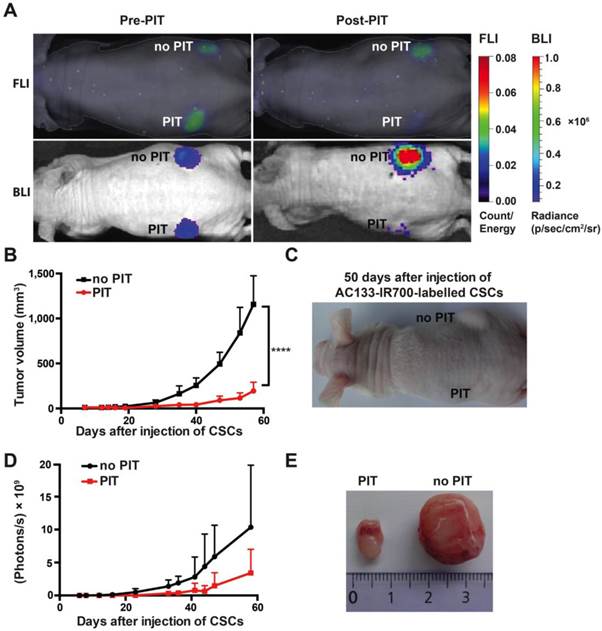
To assess the effect of NIR-PIT on the tumor-initiating capacity of CSCs, we conducted an experiment in an s.c. tumor model where tumor growth can be easily monitored by caliper measurement. To assess their tumor-initiating capacity, we pre-incubated NCH421k CSCs with the AC133-IR700 conjugate overnight and thereafter injected them s.c. into the left and right flanks of a T cell-deficient nude mouse; the left (but not the right) flank was then immediately irradiated with NIR light. As shown in Fig. 3A, both FMT (recording the AC133-IR700 signal) and bioluminescence imaging (BLI; recording the luminescence signal of the tumor cells) showed a significant signal reduction in the irradiated compared to the non-irradiated flank. Tumor growth was strongly delayed when the inoculated CSCs had been treated with AC133 mAb-based PIT compared to non-irradiated cells, as assessed by tumor volume measurements (Fig. 3B, C). The BLI signal strongly correlated with the tumor volume (Fig. 3D). Eight weeks after injection of the NCH421k CSCs, the control tumors in the non-irradiated flank were about six times larger in volume compared to the tumors in the irradiated flank (Fig. 3E).
AC133-IR700-mediated PIT delays tumor initiation by NCH421k GBM-SCs in an s.c. mouse model. (A) NIR-FMT and BLI of mice bearing s.c. inoculated NCH421k GBM-CSs before and after PIT. NCH421k cells were pre-incubated with the AC133-IR700 conjugate overnight before cell implantation. NIR light (100 J/cm2) was applied to the implantation site in the left, but not in the right, flank immediately after implantation. Representative images are shown. (B) Inhibition of tumor initiation assessed by tumor volume measurements (n = 4; ****p < 0.0001; two-way repeated measures ANOVA). (C) Representative image of a mouse bearing two NCH421k tumors 50 days after inoculation. (D) Tumor growth assessed by BLI correlates with the volumetric measurements (n = 4). (E) Excised tumors 60 days after cell implantation.
NIR-PIT of established s.c. tumors
An important factor for the in vivo efficacy of mAb conjugates is their stability in serum. Therefore, before using the AC133-IR700 conjugate in vivo, we evaluated its serum stability by incubation with mouse serum for 24, 48, or 72 h at 37 °C. As shown in Fig. S3A, the in vitro PIT efficacies of serum-pretreated and untreated conjugates were identical, indicating no loss of activity due to antibody degradation.
Thereafter, we assessed the effect of AC133 mAb-based NIR-PIT on established s.c. human xenograft tumors, first using CD133-OE U251 glioma cells, which were injected into both the left and the right flank of nude mice (Fig. S3B). Approximately 17 days after s.c. implantation, when the tumor volume was around 50 mm3, 100 μg of AC133-IR700 conjugate was injected i.v., and 24 h later the specific fluorescence signal could be detected at the tumor sites by NIR-FMT. After NIR light exposure, NIR-FMT and BLI revealed a significant reduction in signal intensity for the irradiated tumor compared to the control tumor, which was protected from the NIR light. CD133-OE U251 tumors treated with NIR-PIT showed significant growth inhibition, as demonstrated by tumor volume measurements (Fig. S3C-E). No growth inhibition was observed after NIR light irradiation in mice injected with IR700-conjugated isotype control antibody (IgG1-IR700) or in non-irradiated mice injected with AC133-IR700, indicating that the anti-tumor effects of AC133-IR700-mediated PIT were due to specific targeting of the AC133-IR700 conjugate (Fig. S3C).
Next, we evaluated AC133 mAb-based NIR-PIT in a nude mouse model with two established NCH421k CSC-derived tumors in the left and right flank, respectively. Despite the significantly lower AC133 expression of NCH421k cells compared to CD133-OE U251 cells, a tumor-associated fluorescence signal was detectable 24 h after i.v. injection of the AC133-IR700 conjugate, as shown in Fig. 4A. After exposure to a single dose of NIR light, NIR-FMT and BLI showed a significant decrease in signal intensity compared to the non-irradiated control tumor (Fig. 4A).
Strong therapeutic effect of a single round of AC133-IR700-mediated PIT on well-established s.c. xenograft tumors initiated by NCH421k GBM-SCs. (A) When the tumors had reached a size of 100 mm3 (approx. 40 days after implantation), 100 μg of AC133-IR700 was injected i.v. and FMT and BLI were performed 24 h later. Thereafter, the tumor in the left flank, but not the one in the right flank, was irradiated with NIR light at a dose of 100 J/cm2. Representative images are shown. (B) Reduced expression of the neural stem and progenitor cell markers Sox2, Nestin, and Musashi, and increased expression of the glial differentiation marker GFAP after AC133-IR700-mediated PIT, as assessed by Western blotting of tumor cell lysates. (C) Confocal microscopic images of tumor sections showing loss of cells expressing the stem and progenitor cell marker Nestin after NIR-PIT. The sections were stained with DAPI to visualize nuclei and with Alexa Fluor® 488-labeled Nestin mAb. Scale bar, 25 μm. In B and C, representative results are shown (n = 3). (D) Substantial tumor regression and strong growth delay after AC133-IR700-mediated PIT. BLI correlated with the volumetric measurements obtained with caliper readings (n = 5; ****p < 0.0001, **p < 0.01; two-way repeated measures ANOVA). (E) Representative photograph of a mouse 4.5 weeks after PIT. (F) T2-weighted axial MR images of a mouse bearing established s.c. NCH421k tumors before and 2 weeks after PIT. The left panels show the tumors on the left and right flanks before NIR light irradiation; the right panels show the non-irradiated (upper) and irradiated (lower) tumors 15 days after i.v. injection of AC133-IR700. (G) Tumors were harvested approx. 5 weeks after PIT. (H) Differences in final tumor weight between irradiated and non-irradiated tumors (n = 4).
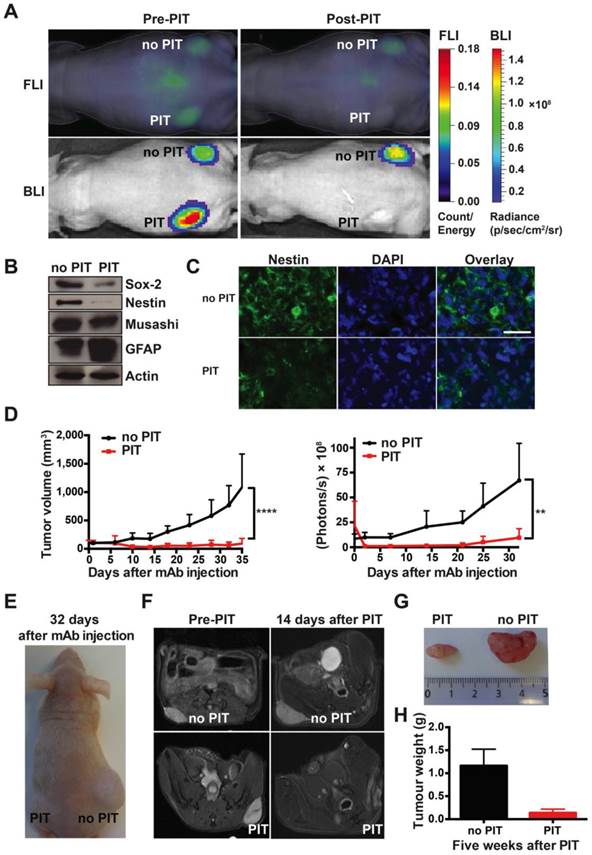
Western blot analyses showed that, 3 days after NIR-PIT, the expression of the neural stem and progenitor cell markers Sox2, Nestin, and Musashi were decreased, while the expression of the glial differentiation marker GFAP had increased (Fig. 4B), compatible with the specific killing of undifferentiated CSCs. Nestin immunofluorescence staining of tumor sections also indicated that only few CSCs were left after NIR-PIT (Fig. 4C). In line with a strong reduction of CSC numbers, tumor growth was strongly inhibited after AC133-IR700-mediated PIT, as shown by tumor volume measurements (Fig. 4D, E). BLI data for control and treated tumors strongly correlated with the tumor volume (Fig. 4D). In addition, magnetic resonance imaging (MRI) confirmed that tumors exposed to NIR light dramatically shrank and virtually completely disappeared 2 weeks after PIT, while the control tumors kept growing (Fig. 4F). The strong therapeutic effect of PIT was also confirmed by measuring the tumor weights 5 weeks after the single PIT. At that time, a dramatic difference in tumor weight was found between PIT-treated (0.137 ± 0.04 g) and control tumors (1.162 ± 0.18 g) (Fig. 4G, H). These experiments revealed a dramatic effect of a single PIT treatment on established s.c. tumors that had been initiated by AC133+ patient-derived GBM-SCs.
NIR-PIT of established orthotopic brain tumors initiated from patient-derived GBM-SCs
The efficacy of AC133 mAb-based NIR-PIT in models of established orthotopic NCH421k tumors might be reduced by several different factors compared to s.c. tumors: (i) the invasive growth pattern; (ii) the blood-brain and blood-tumor barriers, which may hinder the penetration of systematically injected mAb tracers into brain tumors; and (iii) since we had decided to apply the light on the intact skull, insufficient light might reach the intracerebrally located tumor cells.
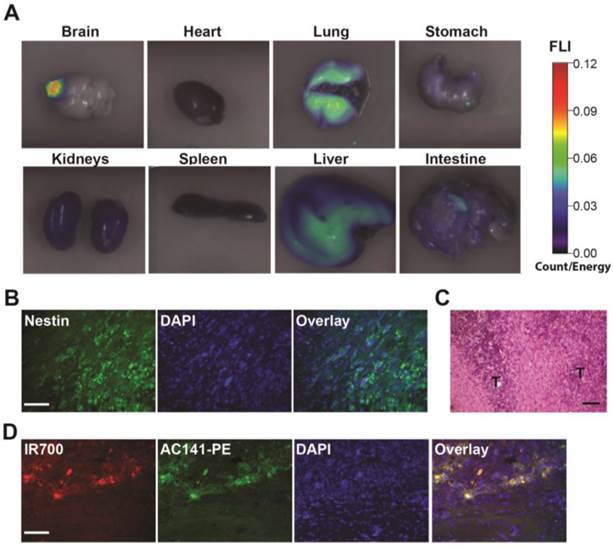
We first examined whether the i.v. injected AC133-IR700 conjugate accumulated in brain tumors, using the CD133-OE U251 glioma model. At 24 h after injection, the IR700-conjugated AC133 mAb indeed accumulated in CD133-OE U251 gliomas growing orthotopically in the brain of nude mice, indicating the ability of the AC133-IR700 tracer to cross the blood-brain/blood-tumor barriers (Fig. 5A). Unlike CD133-OE U251 gliomas, brain tumors initiated from NCH421k GBM-SCs exhibit an invasive growth pattern (Fig. 5B and C, and [19, 21]). Therefore, we next examined whether the i.v. injected mAb conjugate reached AC133/CD133+ tumor cells at the invasive front of orthotopically growing NCH421k brain tumors, which was indeed the case (Fig. 5D).
Biodistribution of the i.v. injected AC133 mAb conjugate in mice bearing orthotopic AC133+ brain tumors. (A) Mice with CD133-OE U251 brain tumors were injected i.v. with AC133-IR700; 24 h later, organs were excised and imaged using the FMT-1500 system. (B) Confocal microscopic images of brain sections encompassing the NCH421k tumor periphery. The sections were stained with DAPI to visualize nuclei and with Alexa Fluor® 488-labeled Nestin mAb. Note the infiltrative growth pattern of the NCH421k cells. Scale bar, 50 μm. (C) H&E staining of NCH421 brain tumor sections also showing irregular tumor margins (T: tumor). Scale bar, 50 μm. (D) Immunohistological detection of the i.v. injected AC133-IR700 conjugate at the margins of orthotopically growing NCH421k brain tumors. The brain tumor sections were stained with DAPI to visualize nuclei and PE-labeled AC141 mAb to visualize CD133 expression. Scale bar, 50 μm. Pictures shown are representative of three independent experiments.
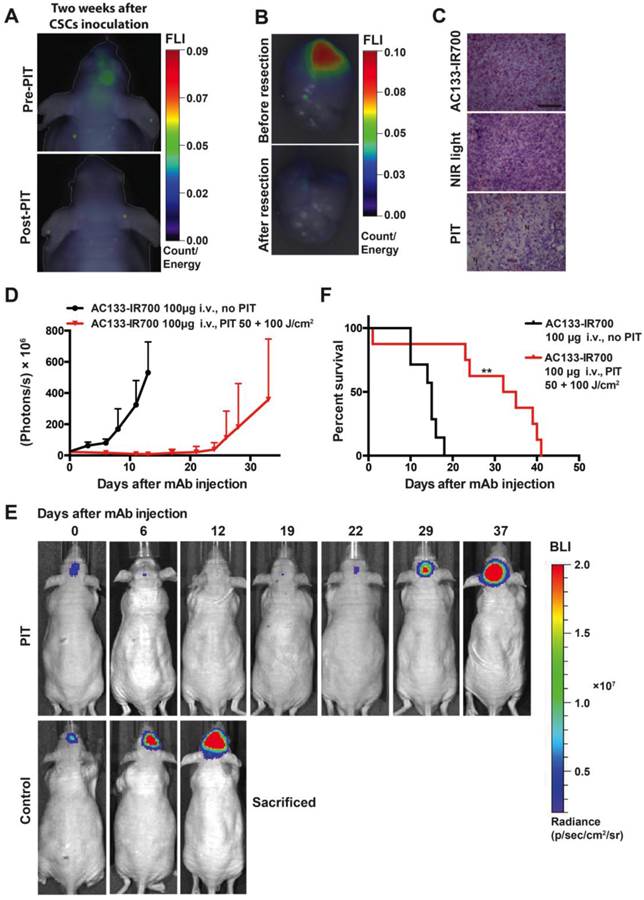
PIT experiments in mice with orthotopically growing luciferase-expressing NCH421k tumors were conducted after approx. 2 weeks of tumor growth when a BLI signal of approx. 2 × 107 photons/s was reached. Mice were then randomized into two groups, both of which received 100 μg of AC133-IR700 i.v.; whereas one group was not exposed to NIR light, the other one was irradiated with a dose of 50 J/cm2 NIR light on day 1 and a dose of 100 J/cm2 on day 3 after AC133-IR700 injection. At 24 h after injection of the AC133-IR700 conjugate, IR700 fluorescence was detected by non-invasive NIR-FMT in the brain area (Fig. 6A, upper). Ex vivo NIR-FMT imaging of brains harbouring NCH421k GBM xenografts before and after excision of the brain tumor demonstrated that AC133-IR700 penetrated also into orthotopic NCH421k tumors (Fig. 6B). A histopathological analysis of the tumor area revealed necrosis and only few viable cells 3 days after the second NIR light irradiation (Fig. 6C). Accordingly, the signals of non-invasive FMT and BLI decreased significantly after NIR light exposure (Fig. 6A lower and 6E). The strongly decreased BLI signal did not show any increase for about 3 weeks thereafter, while the BLI signal in control mice, which received the AC133-IR700 injection but were not exposed to NIR light, increased by more than 20 times within 14 days, by which time the control animals had to be sacrificed (Fig. 6D, E). The BLI data were confirmed by evaluating the overall survival. Whereas non-irradiated control mice had a median survival time of 15 days, mice treated with only one round of NIR-PIT had a median survival of 33.5 days after treatment (Fig. 6F).
Theranostic imaging and efficient PIT-mediated eradication of NCH421k GBM-SCs in 2-week established orthotopic brain tumors. (A) Non-invasive detection of AC133-IR700 by NIR-FMT before and after NIR light irradiation. 100 μg of AC133-IR700 was injected i.v. 24 h earlier. Representative images are shown. (B) NIR-FMT imaging of brains harbouring NCH421k GBM xenografts 24 h after injection of AC133-IR700 before (upper) and after (lower) ex vivo excision of the brain tumor. (C) Histological evaluation of NCH421 brain tumors revealed necrosis and a lower tumor cell density after NIR-PIT. Note that there was no significant necrosis in mice exposed to either AC133-IR700 injected i.v. or NIR light alone. All sections were stained with H&E. Scale bar, 100 μm. Representative images are shown. (D) BLI assessing the growth of the NCH421k brain tumors treated (red) or not (black) by PIT. All mice received AC133-IR700 i.v.; whereas the control mice were not irradiated, the mice in the PIT group were irradiated with 50 and 100 J/cm2 of NIR light 24 and 72 h after tracer injection (n = 7 mice per group). (E) Representative BLI images of a control and an irradiated mouse at different time points after treatment. (F) Survival of the mice according to Kaplan-Meier (n = 7 mice in each group, **p < 0.01 for mice receiving NIR irradiation compared to the control group using a log-rank test).
Discussion
Here, we report for the first time the successful eradication of patient-derived CSCs by PIT [2, 36] and the successful PIT of orthotopic brain tumor xenografts in mice. The anti-tumor effects of PIT based on an AC133-IR700 conjugate were very strong in a model of invasive, established brain tumors initiated from patient-derived GBM-SCs. Already a single mAb tracer injection followed by a short hypofractionated application of harmless NIR light led to an extension of the post-treatment survival of the mice by more than a factor of 2. It should also be noted that the treatment was highly efficient in spite of the fact that the NIR light was applied through the skull bone of the mice. In addition, we were able to visualize the accumulation of the IR700-conjugated AC133 mAb in the orthotopically growing brain tumors non-invasively by NIR-FMT.
Although AC133 mAb-based NIR-PIT was highly efficient in both the s.c. and orthotopic models after a single and a short split-dose application, respectively, it is likely that the treatment efficacy could be further increased by repeated mAb/NIR light treatment cycles or by using cocktails of mAb-IR700 conjugates targeting different antigens [4], particularly when the tumors are heterogeneous in terms of surface antigen expression on the tumor (stem) cells, or by combining PIT with other treatment modalities [3, 37, 38].
We proved that the i.v. injected IR700-conjugated mAb accumulated in the brain tumor xenografts and there bound to CD133+ tumor cells including those at the invasive tumor front. Since NIR light penetrates 1-2 cm of tissue [5], the method, e.g. when applied post-resection, should also allow the killing of tumor cells that have evaded into the surrounding normal brain tissue while sparing normal brain cells.
We have also demonstrated here the possibility of non-invasive imaging of the AC133+ CSCs in brain tumors by NIR imaging. Although this may not be feasible in humans because of the thicker skull bone and the larger brain dimensions, in the future, NIR fluorescence imaging after systemic injection or local application of NIR fluorescence-labeled mAbs might well be used in patients for fluorescence-guided tumor resection and histopathological determination of tumor margins [12, 39-43]. This could be followed by NIR-PIT-mediated ablation of left-over tumor cells within approx. 1 cm of tissue lining the resection cavity by exposing the surgery field to light.
Although PIT had strong therapeutic effects on the brain tumor xenografts, we observed the development of cerebral edema several hours after treatment, which was probably caused by the necrotic cell death induced by PIT. Therefore, we developed the split-dose schedule for the treatment of mice with brain tumors [44]. If used in brain tumor patients, it might be necessary to restrict PIT to small tumor masses or residual tumor tissue after surgical debulking. However, with intensive care, edema caused by PIT or PDT might be better controlled in patients [45].
Promising results in the treatment of brain tumors including GBM have also been reported for non-targeted conventional PDT both in the preclinical and the clinical setting [45, 46]. In comparison to PIT, PDT is less specific and therefore can kill tumor cells independent of target antigen expression but does also kill normal tissue cells including normal brain cells [47]. Potential resistance factors of CSCs against conventional PDT include the induction of autophagy [24], the efflux of conventional photosensitizers from the CSCs due to high expression of drug efflux pumps in CSCs [48] and low oxygen levels in hypoxic CSC niches as well as strong anti-reactive oxygen species (ROS) defense systems typical of normal stem cells and CSCs [9, 49, 50]. An alternative approach to selective PDT targeting CD133+ or other CSCs is the recently described photochemical internalization (PCI) of immunotoxins, which has been shown to cause growth delay in s.c. tumor models using stem-like cells sorted from tumor cell lines [49, 51]. In comparison to NIR-PIT, NIR light-induced PCI requires endocytosis of the macromolecular PCI conjugate, it may more heavily rely on ROS than PIT, and it may engender more difficulties in avoiding off-target toxicity of the toxin conjugate compared to the mAb-dye conjugate used for NIR-PIT. Avoidance of off-target toxicity is important because, in humans, stem cell-specific CD133 epitopes are also expressed on some normal-tissue stem cells, e.g., a subpopulation of hematopoietic stem cells [25]. Another alternative NIR light-induced approach is CD133-targeted NIR light-induced thermoablation of CSCs, but this approach has not yet been evaluated in established or orthotopic tumor models [52].
Taken together, we applied AC133-specific NIR-PIT in mouse tumor models based on patient-derived GBM-SCs. We demonstrate that AC133-IR700 conjugates bind specifically to AC133+ CSCs in vitro and in vivo. Furthermore, this theranostic tracer facilitated the non-invasive imaging of AC133+ GBM-SCs in both s.c. and orthotopic brain tumor models. Importantly, one split PIT dose after a single injection of the theranostic tracer dramatically prolonged the survival time of mice bearing CSC-derived orthotopic brain tumors.
Materials and methods
Reagents
The water-soluble silicon-phthalocyanine derivative IRDye 700DX NHS ester (IR700; C74H96N12Na4O27S6Si3) was purchased from LI-COR Bioscience. The AC133 mAb was purified from supernatant of AC133.1 hybridoma cells (ATCC HB-12346) according to standard methods. The IgG1 isotype control mAb was purchased from BioXcell (West Lebanon).
Cell lines
The establishment of the CD133-OE U251 glioma cell line and the patient-derived NCH421k GBM-SC line has been described previously [21, 53]. NCH421k cells were cultured under stem cell culture conditions as previously described [19]. All three cell lines were transduced with a lentivirus coding for firefly luciferase (L1-mCherry-IRES-FFneo or L1-FF-IRESneo) constructed in our laboratory according to standard procedures.
Antibody conjugation with IR700
Antibody (1 mg, 6.8 nmol) was incubated with IR700 (66.8 μg, 34.2 nmol) in 0.1 M Na2HPO4 (pH 8.5) at room temperature for 2 h. The mixture was then purified over a Sephadex G50 column (PD-10; GE Healthcare) and concentrated with an Amicon Ultra 4 centrifugation filter (Millipore). Thereafter, the protein concentration was determined with a Coomassie Plus protein assay kit (Pierce Biotechnology) by measuring the absorption at 595 nm. The concentration of IR700 was measured by fluorescence spectroscopy to determine the number of fluorophore molecules conjugated to each antibody molecule. The number of IR700 molecules per antibody molecule was approximately three. The purity of the AC133-IR700 conjugates was confirmed by SDS polyacrylamide gel electrophoresis using gradient (4-20%) polyacrylamide gels (Invitrogen) under non-reducing conditions. Just after separating the proteins, the fluorescence intensity was analyzed with a Fujifilm FLA-5100 fluorescence scanner with an internal laser of 670 nm for excitation and a 705-nm long-pass filter for emission. The gels were then stained with a Colloidal Blue Staining Kit (Invitrogen) and digitally scanned. The potential formation of protein aggregates was examined by performing size exclusion chromatography on an Enrich™ SEC 650 column (Bio-Rad).
Titration of labeled antibody
Preservation of binding activity upon labelling with IR700 was certified through side-by-side flow-cytometric titration analysis of labeled and unlabeled mAbs. Serially diluted antibody was incubated with 5 × 105 HCT116 colon carcinoma cells (which are AC133 positive) and 5 × 105 p53-deficient HCT116 colon carcinoma cells (which are AC133 negative) suspended in 100 μl FACS buffer (PBS with 0.5% BSA and 2 mM EDTA) for 15 min. After washing, the cells were incubated with 1.5 μg anti-mouse PE-conjugated F(ab')2 fragment (Dianova) in 100 μl FACS buffer for 20 min. Samples were then washed twice and analyzed on a BD FACSVerseTM flow cytometer (BD Bioscience). Statistical analyses were performed using GraphPad Prism 6.0 statistical software (GraphPad Software Inc.).
Flow cytometry
CD133-OE U251, WT U251 and NCH421k cells were incubated with AC133-IR700 for 30 min at 37 °C, at AC133-IR700 concentrations of 20 and 40 μg/ml. IR700 fluorescence was measured thereafter by NIR flow cytometry using a Fortessa flow cytometer (BD Biosciences).
Fluorescence microscopy
CD133-OE U251 cells or WT U251 cells were seeded on cover glass-bottomed dishes and incubated for 24 h at 37 °C. In the blocking setting, cells were pre-exposed to 400 μg/ml AC133 mAb for 1 h. AC133-IR700 was added to the culture medium at 20 μg/ml and incubated for 6 h at 37 °C. The cells were washed with PBS and stained with a PE-labeled AC141 mAb (Miltenyi Biotec). After washing and nuclear staining with 4',6-diamidino-2-phenylindole (DAPI), the slides were mounted and analyzed with an Olympus ScanR microscope.
In vitro PIT
U251 cells were seeded in 96-well plates and incubated for 24 h at 37 °C. The culture medium was refreshed and AC133-IR700 or isotype control IgG-IR700 was administered at 20 μg/ml. After incubation at 37 °C for 6 h, the cells were washed and irradiated with a red light-emitting diode (LED), which emits light at wavelengths of 670-710 nm (L690-66-60; Marubeni America Co.) at a dose of 12 J/cm2. Power densities were measured with an optical power meter (PM 100, Thorlabs). Cell viability was analyzed by Trypan blue exclusion.
For NCH421k cells, AC133-IR700 or isotype control IgG-IR700 was administered at 40 μg/ml. The cells were incubated at 37 °C for 14 h, washed once with PBS and irradiated with the red LED at a dose of 18 J/cm2. Cell viability was analyzed by Trypan blue exclusion.
Sphere-forming assay
NCH421k cells (2.5 × 104) were seeded into 24-well plates and cultured under stem cell culture conditions. AC133-IR700 or isotype control IgG-IR700 was administered at 40 μg/ml and PIT was performed on single-cell suspensions under the conditions described above for NCH421k cells. The sphere-forming ability of control and PIT-treated cells was assessed by photographing the cultures 3 days later using an inverted microscope (IX51, Olympus).
Serum stability assay
The AC133-IR700 conjugate in PBS was mixed with mouse serum (Sigma-Aldrich) at a 1:5 volume ratio for 24, 48, and 72 h at 37 °C in a CO2 incubator. Control sample was prepared by diluting AC133-IR700 in mouse serum (1:5) immediately before the PIT assay.
Animal and tumor models
All animal experiments were performed in accordance with the German Animal License Regulations and were approved by the animal care committee of the Regierungspräsidium Freiburg (registration numbers: G-10/64, G-13/12 and G-13/101).
For s.c. tumor models, 5 × 106 U251 (CD133-OE or WT) or NCH421k cells were implanted into the left and right flanks of 6-8-week-old immunocompromised nude mice (Harlan). The growth of the xenografts was monitored by caliper measurement, and the tumor volume was calculated using the formula: length × width2 × 0.5. For orthotopic brain tumor models, 2.5 × 105 luciferase-transfected NCH421k cells were implanted free-handedly into the brains of 6-8-week-old nude mice 3 mm anterior and 3 mm to the right of the bregma. The cells were injected at a depth of 3 mm with a Hamilton syringe. The syringe was held in position for 5 min. After the injection, the surface was cleaned with a sterile cotton swab. Thereafter, the tumor growth was monitored non-invasively using in vivo BLI on an IVIS spectrum imaging system (Perkin Elmer).
In vivo PIT
For assessing any impairment of the tumor initiation capacity of CSCs, 5 × 106 NCH421k cells were pre-incubated with 40 μg/ml AC133-IR700 in vitro for 14 h, washed twice with PBS and then injected into the left and right flanks of 6-8-week-old nude mice. After determining the AC133-IR700 signal of the cells by FMT and the luciferase signal of the cells by BLI, one flank was irradiated with NIR light (100 J/cm2) while the other flank was covered with aluminium foil.
Treatment of established s.c. tumors: Approximately 17 days after injection of CD133-OE U251 cells or 40 days after injection of NCH421k cells, the tumor volume reached approximately 50 mm3 or 100 mm3, respectively. AC133-IR700 (100 μg) was then injected i.v. into tumor-bearing mice. After 24 h, one tumor was irradiated with 100 J/cm2 NIR light while the other tumor on the same mouse was covered with aluminium foil.
Treatment of established, orthotopic NCH421k-derived brain tumors: At 2 weeks after NCH421k cell implantation, the BLI signal usually reached an intensity of 2 × 107 photons/s. The mice were then randomized into two groups: (1) 100 μg of AC133-IR700 was injected i.v. but no NIR light was applied; (2) 100 μg of AC133-IR700 was injected i.v. and 50 J/cm2 NIR light was applied on day 1 (lasting 14 min at a light intensity of 60 mW/cm2) and 100 J/cm2 on day 3 (lasting 28 min at a light intensity of 60 mW/cm2) after AC133-IR700 injection. Non-invasive FMT and BLI imaging was performed over time with an FMT1500 system (Perkin Elmer) and an IVIS spectrum imaging system, respectively.
Ex Vivo Biodistribution
At 24 h after AC133-IR700 injection, the animals were euthanized by cervical dislocation under anesthesia. The selected organs were excised and ex vivo FMT imaging was performed.
Western Blot Analyses
Tumor lysates were prepared in RIPA buffer supplemented with a protease inhibitor mixture (Roche) and the phosphatase inhibitors NaF and Na3VO4 (Sigma). Then, 40 µg each of the cell lysates was separated by SDS-PAGE and blotted onto nitrocellulose membranes. The blots were probed with the indicated antibodies and developed by enhanced chemiluminescence (Amersham Biosciences). The following antibodies were used: sex-determining region Y (SRY)-box 2 (Sox2) (R&D Systems), Nestin (Millipore), Musashi (Millipore), GFAP (DAKO), Actin (Santa Cruz Biotechnology), and HRP-conjugated secondary antibodies (Dianova).
MRI imaging
MRI experiments were performed 2 days before PIT and 14 days post PIT. Nude mice bearing NCH421k s.c. tumors were anesthetized with isoflurane (1.0-1.5%) in O2 (1.2 l/min) applied with a face mask. The animal body temperature was kept at 37 °C with a circulating water pad, and respiration (spontaneous breathing) was continuously monitored. The mice were held in supine position. A 9.4-T animal scanner (BioSpec 94/20, Bruker Biospin) with a birdcage resonator was used. T2-weighted multislice RARE scans were acquired with the following parameters: 30 axial slices, slice thickness 1.0 mm, field of view 30 mm × 30 mm, matrix size 192 × 192, in-plane resolution 0.16 mm × 0.16 mm, TR/TEeffective = 4.4 s/33 ms, RARE factor 8, 2 averages.
Histopathology
PFA (4%)-fixed brains were cut in a vibratome (Leica VT-1000S, Leica Microsystems AG) into horizontal sections 50 μm apart. Hematoxylin and eosin (H&E) staining was performed according to standard procedures. For analysis of CD133 expression in tumor tissue, the sections were stained using a PE-labeled mAb against human AC141 (Miltenyi Biotec) for 30 min. For Nestin staining, the primary antibody was added overnight, followed by 1 h of secondary antibody incubation. Sections were then stained with DAPI to identify the nuclei, mounted and analyzed with an Olympus ScanR microscope.
Statistical analysis
Results are presented as mean ± SE. Statistical analyzes were carried out using GraphPad Prism 6.0 statistical software (GraphPad Software Inc.). A p-value < 0.05 was considered significant (*p < 0.05, **p < 0.01, ***p < 0.001 and ****p < 0.0001).
Abbreviations
BLI: bioluminescence imaging; CSC: cancer stem cell; DAPI: 4',6-diamidino-2-phenylindole; FMT: fluorescence molecular tomography; GBM-SC: glioblastoma stem cell; H&E: hematoxylin and eosin; i.v.: intravenous; IR700: IRDye 700DX NHS ester; LED: light-emitting diode; mAb: monoclonal antibody; MRI: magnetic resonance imaging; NIR-PIT: near-infrared photoimmunotherapy; OE: overexpressing; PCI: photochemical internalization; PDT: photodynamic therapy; PE: phycoerythrin; ROS: reactive oxygen species; s.c.: subcutaneous; SE: standard error; WT: wild type.
Supplementary Material
Supplementary figures.
Acknowledgements
We wish to thank Dr. Marcia Machein for her advice regarding immunofluorescence stainings. We also thank Dr. Martin Holzer for his help with DLS and zeta measurements.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Mitsunaga M, Ogawa M, Kosaka N, Rosenblum LT, Choyke PL, Kobayashi H. Cancer cell-selective in vivo near infrared photoimmunotherapy targeting specific membrane molecules. Nat Med. 2011;17:1685-91
2. Warram JM, de Boer E, Sorace AG, Chung TK, Kim H, Pleijhuis RG. et al. Antibody-based imaging strategies for cancer. Cancer Metastasis Rev. 2014;33:809-22
3. Sano K, Nakajima T, Choyke PL, Kobayashi H. The effect of photoimmunotherapy followed by liposomal daunorubicin in a mixed tumor model: a demonstration of the super-enhanced permeability and retention effect after photoimmunotherapy. Mol Cancer Ther. 2014;13:426-32
4. Nakajima T, Sano K, Choyke PL, Kobayashi H. Improving the efficacy of Photoimmunotherapy (PIT) using a cocktail of antibody conjugates in a multiple antigen tumor model. Theranostics. 2013;3:357-65
5. Richards-Kortum R, Sevick-Muraca E. Quantitative optical spectroscopy for tissue diagnosis. Annu Rev Phys Chem. 1996;47:555-606
6. Sato K, Hanaoka H, Watanabe R, Nakajima T, Choyke PL, Kobayashi H. Near infrared photoimmunotherapy in the treatment of disseminated peritoneal ovarian cancer. Mol Cancer Ther. 2015;14:141-50
7. Sato K, Nagaya T, Choyke PL, Kobayashi H. Near Infrared Photoimmunotherapy in the Treatment of Pleural Disseminated NSCLC: Preclinical Experience. Theranostics. 2015;5:698-709
8. Nguyen LV, Vanner R, Dirks P, Eaves CJ. Cancer stem cells: an evolving concept. Nat Rev Cancer. 2012;12:133-43
9. Yan K, Yang K, Rich JN. The evolving landscape of glioblastoma stem cells. Curr Opin Neurol. 2013;26:701-7
10. Hermann PC, Bhaskar S, Cioffi M, Heeschen C. Cancer stem cells in solid tumors. Semin Cancer Biol. 2010;20:77-84
11. Yano S, Miwa S, Kishimoto H, Toneri M, Hiroshima Y, Yamamoto M. et al. Experimental curative fluorescence-guided surgery of highly invasive glioblastoma multiforme selectively labeled with a killer-reporter adenovirus. Mol Ther. 2015
12. Stummer W, Pichlmeier U, Meinel T, Wiestler OD, Zanella F, Reulen HJ. et al. Fluorescence-guided surgery with 5-aminolevulinic acid for resection of malignant glioma: a randomised controlled multicentre phase III trial. Lancet Oncol. 2006;7:392-401
13. Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ. et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987-96
14. Ortensi B, Setti M, Osti D, Pelicci G. Cancer stem cell contribution to glioblastoma invasiveness. Stem Cell Res Ther. 2013;4:18
15. Chen R, Nishimura MC, Bumbaca SM, Kharbanda S, Forrest WF, Kasman IM. et al. A hierarchy of self-renewing tumor-initiating cell types in glioblastoma. Cancer Cell. 2010;17:362-75
16. Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T. et al. Identification of human brain tumour initiating cells. Nature. 2004;432:396-401
17. Liu S, Cong Y, Wang D, Sun Y, Deng L, Liu Y. et al. Breast cancer stem cells transition between epithelial and mesenchymal states reflective of their normal counterparts. Stem Cell Reports. 2014;2:78-91
18. Hermann PC, Huber SL, Herrler T, Aicher A, Ellwart JW, Guba M. et al. Distinct populations of cancer stem cells determine tumor growth and metastatic activity in human pancreatic cancer. Cell Stem Cell. 2007;1:313-23
19. Gaedicke S, Braun F, Prasad S, Machein M, Firat E, Hettich M. et al. Noninvasive positron emission tomography and fluorescence imaging of CD133+ tumor stem cells. Proc Natl Acad Sci U S A. 2014;111:E692-701
20. Piccirillo SG, Reynolds BA, Zanetti N, Lamorte G, Binda E, Broggi G. et al. Bone morphogenetic proteins inhibit the tumorigenic potential of human brain tumour-initiating cells. Nature. 2006;444:761-5
21. Campos B, Wan F, Farhadi M, Ernst A, Zeppernick F, Tagscherer KE. et al. Differentiation therapy exerts antitumor effects on stem-like glioma cells. Clin Cancer Res. 2010;16:2715-28
22. Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB. et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444:756-60
23. Colak S, Medema JP. Cancer stem cells-important players in tumor therapy resistance. FEBS J. 2014;281:4779-91
24. Wei MF, Chen MW, Chen KC, Lou PJ, Lin SY, Hung SC. et al. Autophagy promotes resistance to photodynamic therapy-induced apoptosis selectively in colorectal cancer stem-like cells. Autophagy. 2014;10:1179-92
25. Yin AH, Miraglia S, Zanjani ED, Almeida-Porada G, Ogawa M, Leary AG. et al. AC133, a novel marker for human hematopoietic stem and progenitor cells. Blood. 1997;90:5002-12
26. Kemper K, Sprick MR, de Bree M, Scopelliti A, Vermeulen L, Hoek M. et al. The AC133 epitope, but not the CD133 protein, is lost upon cancer stem cell differentiation. Cancer Res. 2010;70:719-29
27. Zeppernick F, Ahmadi R, Campos B, Dictus C, Helmke BM, Becker N. et al. Stem cell marker CD133 affects clinical outcome in glioma patients. Clin Cancer Res. 2008;14:123-9
28. O'Brien CA, Pollett A, Gallinger S, Dick JE. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature. 2007;445:106-10
29. Puisieux A, Brabletz T, Caramel J. Oncogenic roles of EMT-inducing transcription factors. Nat Cell Biol. 2014;16:488-94
30. Lampson LA. Monoclonal antibodies in neuro-oncology: Getting past the blood-brain barrier. MAbs. 2011;3:153-60
31. Dijkers EC, Oude Munnink TH, Kosterink JG, Brouwers AH, Jager PL, de Jong JR. et al. Biodistribution of 89Zr-trastuzumab and PET imaging of HER2-positive lesions in patients with metastatic breast cancer. Clin Pharmacol Ther. 2010;87:586-92
32. Veeravagu A, Liu Z, Niu G, Chen K, Jia B, Cai W. et al. Integrin alphavbeta3-targeted radioimmunotherapy of glioblastoma multiforme. Clin Cancer Res. 2008;14:7330-9
33. Weissleder R, Pittet MJ. Imaging in the era of molecular oncology. Nature. 2008;452:580-9
34. Prasad S, Gaedicke S, Machein M, Mittler G, Braun F, Hettich M. et al. Effective Eradication of Glioblastoma Stem Cells by Local Application of an AC133/CD133-Specific T-cell-Engaging Antibody and CD8 T Cells. Cancer Res. 2015;75:2166-76
35. Pollard SM, Yoshikawa K, Clarke ID, Danovi D, Stricker S, Russell R. et al. Glioma stem cell lines expanded in adherent culture have tumor-specific phenotypes and are suitable for chemical and genetic screens. Cell Stem Cell. 2009;4:568-80
36. Goff BA, Bamberg M, Hasan T. Photoimmunotherapy of human ovarian carcinoma cells ex vivo. Cancer Res. 1991;51:4762-7
37. Sano K, Nakajima T, Choyke PL, Kobayashi H. Markedly enhanced permeability and retention effects induced by photo-immunotherapy of tumors. ACS Nano. 2013;7:717-24
38. Maawy AA, Hiroshima Y, Zhang Y, Garcia-Guzman M, Luiken GA, Kobayashi H. et al. Photoimmunotherapy lowers recurrence after pancreatic cancer surgery in orthotopic nude mouse models. J Surg Res. 2015
39. Huang R, Vider J, Kovar JL, Olive DM, Mellinghoff IK, Mayer-Kuckuk P. et al. Integrin alphavbeta3-targeted IRDye 800CW near-infrared imaging of glioblastoma. Clin Cancer Res. 2012;18:5731-40
40. Bouvet M, Hoffman RM. Glowing tumors make for better detection and resection. Sci Transl Med. 2011;3:110fs10
41. Chi C, Du Y, Ye J, Kou D, Qiu J, Wang J. et al. Intraoperative imaging-guided cancer surgery: from current fluorescence molecular imaging methods to future multi-modality imaging technology. Theranostics. 2014;4:1072-84
42. Nguyen QT, Tsien RY. Fluorescence-guided surgery with live molecular navigation-a new cutting edge. Nat Rev Cancer. 2013;13:653-62
43. van Dam GM, Themelis G, Crane LM, Harlaar NJ, Pleijhuis RG, Kelder W. et al. Intraoperative tumor-specific fluorescence imaging in ovarian cancer by folate receptor-alpha targeting: first in-human results. Nat Med. 2011;17:1315-9
44. Mitsunaga M, Nakajima T, Sano K, Choyke PL, Kobayashi H. Near-infrared theranostic photoimmunotherapy (PIT): repeated exposure of light enhances the effect of immunoconjugate. Bioconjug Chem. 2012;23:604-9
45. Stylli SS, Kaye AH, MacGregor L, Howes M, Rajendra P. Photodynamic therapy of high grade glioma - long term survival. J Clin Neurosci. 2005;12:389-98
46. Stummer W, Beck T, Beyer W, Mehrkens JH, Obermeier A, Etminan N. et al. Long-sustaining response in a patient with non-resectable, distant recurrence of glioblastoma multiforme treated by interstitial photodynamic therapy using 5-ALA: case report. J Neurooncol. 2008;87:103-9
47. Celli JP, Spring BQ, Rizvi I, Evans CL, Samkoe KS, Verma S. et al. Imaging and photodynamic therapy: mechanisms, monitoring, and optimization. Chem Rev. 2010;110:2795-838
48. Ishikawa T, Kajimoto Y, Inoue Y, Ikegami Y, Kuroiwa T. Critical role of ABCG2 in ALA-photodynamic diagnosis and therapy of human brain tumor. Adv Cancer Res. 2015;125:197-216
49. Selbo PK, Bostad M, Olsen CE, Edwards VT, Hogset A, Weyergang A. et al. Photochemical internalisation, a minimally invasive strategy for light-controlled endosomal escape of cancer stem cell-targeting therapeutics. Photochem Photobiol Sci. 2015
50. Catalano V, Turdo A, Di Franco S, Dieli F, Todaro M, Stassi G. Tumor and its microenvironment: a synergistic interplay. Semin Cancer Biol. 2013;23:522-32
51. Bostad M, Olsen CE, Peng Q, Berg K, Hogset A, Selbo PK. Light-controlled endosomal escape of the novel CD133-targeting immunotoxin AC133-saporin by photochemical internalization - A minimally invasive cancer stem cell-targeting strategy. J Control Release. 2015;206:37-48
52. Wang CH, Chiou SH, Chou CP, Chen YC, Huang YJ, Peng CA. Photothermolysis of glioblastoma stem-like cells targeted by carbon nanotubes conjugated with CD133 monoclonal antibody. Nanomedicine. 2011;7:69-79
53. Tsurumi C, Esser N, Firat E, Gaedicke S, Follo M, Behe M. et al. Non-invasive in vivo imaging of tumor-associated CD133/prominin. PLoS One. 2010;5:e15605
Author contact
![]() Corresponding author: Gabriele Niedermann; gabriele.niedermannde
Corresponding author: Gabriele Niedermann; gabriele.niedermannde