Impact Factor
 Global reach, higher impact
Global reach, higher impact
Theranostics 2016; 6(7):1004-1011. doi:10.7150/thno.14471 This issue Cite
Research Paper
Safe and Immunocompatible Nanocarriers Cloaked in RBC Membranes for Drug Delivery to Treat Solid Tumors
Brian T. Luk1, Ronnie H. Fang1, Che-Ming J. Hu2, Jonathan A. Copp1, Soracha Thamphiwatana1, Diana Dehaini1, Weiwei Gao1, Kang Zhang1,3, Shulin Li4, Liangfang Zhang1 ![]()
1. Department of NanoEngineering and Moores Cancer Center, University of California, San Diego, La Jolla, CA 92093, U.S.A.
2. Institute of Biomedical Sciences, Academia Sinica, Taipei, Taiwan.
3. Shiley Eye Institute, University of California, San Diego, La Jolla, CA 92093, U.S.A.
4. Department of Pediatric Research, MD Anderson Cancer Center, Houston, TX 77030, U.S.A.
Received 2015-11-18; Accepted 2015-12-11; Published 2016-4-28
Abstract

The therapeutic potential of nanoparticle-based drug carriers depends largely on their ability to evade the host immune system while delivering their cargo safely to the site of action. Of particular interest are simple strategies for the functionalization of nanoparticle surfaces that are both inherently safe and can also bestow immunoevasive properties, allowing for extended blood circulation times. Here, we evaluated a recently reported cell membrane-coated nanoparticle platform as a drug delivery vehicle for the treatment of a murine model of lymphoma. These biomimetic nanoparticles, consisting of a biodegradable polymeric material cloaked with natural red blood cell membrane, were shown to efficiently deliver a model chemotherapeutic, doxorubicin, to solid tumor sites for significantly increased tumor growth inhibition compared with conventional free drug treatment. Importantly, the nanoparticles also showed excellent immunocompatibility as well as an advantageous safety profile compared with the free drug, making them attractive for potential translation. This study demonstrates the promise of using a biomembrane-coating approach as the basis for the design of functional, safe, and immunocompatible nanocarriers for cancer drug delivery.
Keywords: nanomedicine, biomimetic nanoparticle, immunocompatible nanocarrier, drug delivery, lymphoma treatment.
Introduction
Nanoparticle-based drug delivery platforms are often tasked with navigating complex biological environments, and their performance can ultimately be governed by their ability to avoid nonspecific interactions while exhibiting a high degree of target selectivity [1-3]. As such, the facile incorporation of complex functionalities onto nanoparticle surfaces has long been desirable [4-7]. Recently, the direct use of naturally derived biomembrane as a coating material represents an emerging strategy for nanoparticle functionalization [8-10]. The faithful, right-side-out translocation of all membrane-bound moieties from the cell surface onto the surface of a nanoparticle can naturally bestow the nanoparticle with desirable properties such as long circulation, immune evasion, and targeting affinity without the need to explicitly engineer these functionalities from the bottom-up [11-13]. This cell membrane cloaking strategy has demonstrated utility for a variety of purposes, including biodetoxification [14-16], antibacterial vaccination [17, 18], antibiotic delivery [19], photothermal therapy [20], and cancer immunotherapy [13, 21]. Using this approach, an endless number of applications can be envisioned, as it should be possible to combine the membrane from any cell type with a variety of different nanoparticle core materials [22-25].
One cell type that has been widely explored as a source for membrane coating is the red blood cell (RBC), which represents nature's own long circulating carrier. RBCs express a variety of immunomodulatory markers that enable the body to recognize them as self [26, 27], and functionalization of nanoparticles with RBC membrane has been proven to promote immune evasion and significantly enhance circulation residence time [10, 22]. These properties make RBC membrane-coated nanocarriers a truly appealing candidate for cancer drug delivery, which is a field that has long benefited from the use of long-circulating, lowly immunogenic nanocarriers [28, 29]. As more nanoparticle-based chemotherapies are being investigated in the clinic, a great deal of emphasis has been placed on safety and immunocompatibility [30, 31]. To this end, RBC membrane-coated polymeric nanoparticles (denoted RBC-NP), consisting of a poly(lactic-co-glycolic acid) (PLGA) core and an RBC membrane shell, represent a promising delivery system due to their combination of high drug carrying capacity along with an inherently biocompatible membrane coating [32]. Here, we demonstrate that RBC-NP can be effectively used to deliver a model chemotherapeutic drug, doxorubicin (DOX), in a mouse model of lymphoma. We study the ability of the drug-loaded nanoparticles to control tumor growth while concurrently assessing their ability to eliminate the toxicities commonly associated with free drug administration. Further, the short- and long-term immune effects of the RBC-NP upon systemic administration are studied as well.
Materials and Methods
Preparation of RBC-NP and RBC-NP(DOX): Whole blood was collected from C57BL/6 mice (Harlan Sprague Dawley) and collected by centrifugation at 500 × g for 10 min. RBC membrane vesicles were then prepared using a sonication approach. PLGA polymeric cores loaded with DOX were prepared with carboxy-terminated 50:50 PLGA polymer (LACTEL Absorbable Polymers) using a double emulsion process. DOX was dissolved in 25 μL of 500 mM Tris-HCl at pH 8 as the inner phase, and sonicated with 500 μL of PLGA in dichloromethane (DCM) at 10 mg/mL. The solution was then added to 5 mL of 10 mM Tris-HCl at pH 8 and sonicated again. This final solution was then added to an additional 10 mL of 10 mM Tris-HCl at pH 8 and allowed to evaporate for at least 4 h with stirring. Empty PLGA cores were prepared in the same fashion, but without DOX in the inner phase. RBC-NPs were prepared by fusing RBC membrane vesicles on preformed PLGA cores using a previously established protocol [15]. The size and zeta potential of the RBC-NP were obtained from three dynamic light scattering (DLS) measurements using a Malvern ZEN 3600 Zetasizer. Transmission electron microscopy (TEM) was used to characterize the morphology of RBC-NP. Briefly, a drop of RBC-NP solution (1 mg/mL) was deposited onto a glow-discharged carbon-coated TEM grid, followed by washing with 10 drops of distilled water and staining with 1 wt% uranyl acetate. An FEI Sphera Microscope operating at 200 kV was used to image the sample. The DOX loading was evaluated by measuring its fluorescence (excitation at 480 nm; emission at 580 nm). Drug release was studied by dialyzing samples against phosphate buffered saline (PBS) buffer (1 X, pH = 7.4) using Slide-A-Lyzer MINI Dialysis Cups (Thermo Scientific) with a molecular weight cut-off of 10 kDa.
In Vitro Cytotoxicity and Uptake: EL4 cells (American Type Culture Collection) were plated at 5,000 cells per well. Free DOX and RBC-NP(DOX) at varying drug concentrations were incubated with the cells for 72 h at 37 °C and 5% CO2, after which an XTT Cell Proliferation Kit (Roche Diagnostics) was used to assess cell viability. DOX uptake was assessed by incubating 100,000 EL4 cells with varying concentrations of free DOX or RBC-NP(DOX) for 1 h at 37 °C. After 1 h, the samples were washed and fixed with 10% formalin for analysis. Flow cytometry was used to measure the DOX signal in the cells using a Becton Dickinson FACSCanto II.
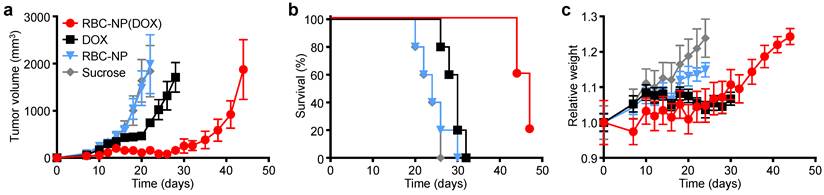
In Vivo Antitumor Efficacy: 75,000 EL4 cells were implanted subcutaneously into the right flank of 6-week-old male C57BL/6 mice. The tumors were allowed to grow for 9 days. RBC-NP(DOX), free DOX, empty RBC-NP(without DOX), or sucrose was administered every other day starting from day 9 post-implantation of the tumor cells for 2 weeks (n = 5 per group). Where applicable, 200 μL of each respective formulation was administered intravenously via tail vein injection at a concentration equivalent to 3 mg/kg DOX, which was found to be the maximum tolerated dose of the drug. Tumor dimensions and mouse weights were measured every other day beginning on day 8 post-implantation and every 3 days beginning on day 35. Tumor volume was calculated using the equation 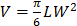 , where V is volume, L is length, and W is width. Survival was pre-defined as tumor size < 2000 mm3 prior to the initiation of the study.
, where V is volume, L is length, and W is width. Survival was pre-defined as tumor size < 2000 mm3 prior to the initiation of the study.
In Vivo Safety Studies: To examine the effect of RBC-NP(DOX) on normal physiological parameters, 200 μL of sucrose, RBC-NP(DOX), or free DOX at 3 mg/kg of drug was injected intravenously into the tail vein of C57BL/6 mice (n = 3 per group). Whole blood was collected into heparinized tubes before and 24 h after injection. Hematological parameters (RBC count, platelet count, hemoglobin, hematocrit, white blood cell count, neutrophil count, lymphocyte count, and monocyte count) were evaluated using a Drew Scientific Hemavet 950 FS Multi-Species Hematology System. To evaluate serum chemistry, blood was collected and allowed to clot for 4 h at room temperature. Samples were then centrifuged at 7000 × g, and 300 μL of serum was collected. Serum chemistry components were measured using the SEAL AutoAnalyzer 3 HR.
In Vivo Immunogenicity Studies: To examine the safety of the RBC-NP platform, 200 μL of RBC-NP at a particle dosing of 30 mg/kg was injected intravenously into the tail vein of C57BL/6 mice; additional mice were administered isotonic sucrose solution or a mixture of lipopolysaccharide (10 μg/kg) and D-galactosamine (100 mg/kg) (n = 3 per group). Blood was collected 6 h post-injection and the plasma was separated. An IL-6 ELISA kit (Biolegend) was used to measure the levels of IL-6 following the manufacturer's protocol. To study the anti-RBC IgG and IgM titers, blood was collected on day 30 post-challenge from tumor-bearing mice administered with RBC-NP in the above antitumor efficacy study (n = 5). In the study, mice were injected with RBC-NP at a particle dosing of 30 mg/kg every other day for 2 weeks starting from day 9 post-challenge. Plasma was separated from whole blood. To measure anti-RBC titers, RBCs in PBS were coated onto Costar 96 well plates (Corning) at 106 RBCs per well. The collected plasma was used as the primary immunostain. Goat anti-mouse IgG-HRP (Biolegend) or goat anti-mouse IgM-HRP (Santa Cruz Biotechnology) was used as the secondary antibody for detecting the presence of autoantibodies against RBCs, and TMB substrate (Thermo Scientific) was used to develop the plate.
Results and Discussion
Preparation of DOX-Loaded RBC-NP and Physicochemical Characterization. Empty or DOX-loaded PLGA nanoparticles were prepared using a double emulsion method. RBC membrane derived from the blood of C57BL/6 mice was coated onto the polymeric cores using a sonication approach as previously described [15]. The general structure of the resulting nanoparticles is depicted in Figure 1a with the DOX loaded inside the PLGA core and the RBC membrane coating, with all its associated proteins, forming the outer layer. Drug loading into the PLGA core could be controlled by varying the initial input concentration of DOX (Figure 1b). By increasing the input of DOX, loading of the drug was also increased, and a saturation level was reached at approximately 40 wt% (DOX weight/PLGA weight) drug input, corresponding to approximately 10 wt% loading. As the drug input concentration was increased, the encapsulation efficiency decreased markedly, dropping from 50% efficiency at an input of 10 wt% down to 20% efficiency at the maximal tested input of 50 wt%. A formulation approaching the saturation loading level of 10 wt% DOX was used for subsequent in vitro and in vivo studies.
Physicochemical characterization and drug loading of red blood cell membrane-cloaked nanoparticle (RBC-NP). (a) Schematic of doxorubicin (DOX)-loaded RBC-NP, denoted “RBC-NP(DOX)”. (b) Loading yield and encapsulation efficiency of DOX into PLGA nanoparticles at various initial drug inputs. (c) Dynamic light scattering (DLS) measurements of the size and surface zeta potential of bare PLGA core, RBC-NP, and RBC-NP(DOX) with a drug loading yield of 10 wt%. (d) Transmission electron microscopy (TEM) visualization of RBC-NP(DOX) with uranyl acetate negative staining (scale bar = 100 nm). (e) Cumulative release profile of DOX from RBC-NP(DOX) with 10 wt% DOX loading yield over a period of 7 days.
Coating of 140 nm empty or DOX-loaded PLGA cores with RBC membranes resulted in a size increase to approximately 155 nm and an increase in zeta potential of approximately 15 mV, from ~-45 mV to -30 mV (Figure 1c). This is consistent with previous findings, as the membrane layer adds to the hydrodynamic size, while the membrane coating, which is less negatively charged than the core, shields the highly negative carboxyl groups present on the surface of the core [13]. The core-shell structure of the drug-loaded RBC-NP, herein denoted RBC-NP(DOX), was confirmed by visualization using transmission electron microscopy (TEM) with uranyl acetate negative staining (Figure 1d). Morphologically, the drug-loaded RBC-NP is similar in appearance to unloaded PLGA coated with the same membrane, suggesting that drug-loaded RBC-NP of varying sizes can easily be fabricated to meet the requirements of future applications [12]. The resulting RBC-NP(DOX) demonstrated sustained release over time (Figure 1e); approximately 80% of the encapsulated drug was released over the course of 7 days, with the majority of the release occurring within the first 72 h. This observed prolonged release is due in part to the RBC membrane coating acting as a diffusional barrier for DOX release, a phenomenon that has been previously reported [32, 33].
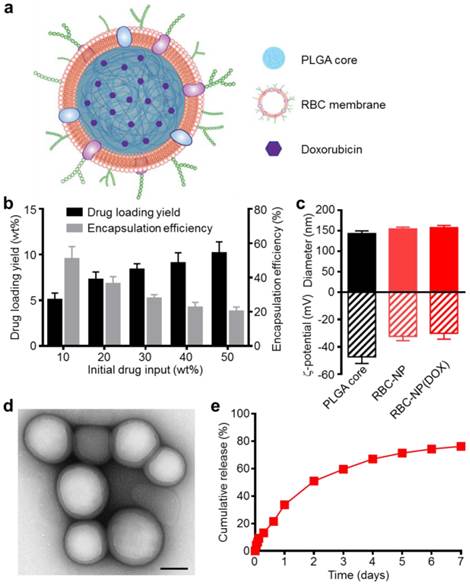
In Vitro Cytotoxicity and Cellular Uptake of RBC-NP(DOX). To evaluate whether the drug encapsulated within the RBC-NP(DOX) could retain its tumor killing activity, an in vitro cytotoxicity test was conducted. It was shown that RBC-NP(DOX) exhibited cytotoxicity when incubated together with EL4 mouse lymphoma cells for 72 h in vitro (Figure 2a). Under the conditions tested, the nanoparticle formulation had an apparent drug IC50 of 5.6 ng/mL. Free DOX demonstrated slightly better efficacy against EL4 tumor cells in vitro compared with RBC-NP(DOX) with an IC50 of 1.4 ng/mL. This difference can be rationalized by the fact that, at the 72 h conclusion of the experiment, there was still incomplete release of drug from RBC-NP(DOX). Additionally, free DOX can easily diffuse into the cancer cells, making it extremely potent over extended incubation periods, especially given that the EL4 cell line is not inherently DOX-resistant [34]. While RBC-NP(DOX) and free DOX showed similar activity over longer incubation periods, encapsulated DOX showed enhanced uptake by EL4 cells after a short, 1 h incubation (Figure 2b). The observed difference in intracellular localization is likely due to the fact that nanoparticles are taken up via active mechanisms such as endocytosis [35], enabling a higher capacity for transport across the cellular membrane compared with pure diffusion. This effect can be further enhanced with active targeting ligands, which can be introduced onto the surface of RBC-NP [36]. These observations indicate that encapsulating DOX into RBC-NP has the potential to facilitate quick uptake of the drug by cancer cells and slow release in a sustained manner over time.
In vitro cellular toxicity and uptake. (a) In vitro cytotoxicity of RBC-NP(DOX) in comparison with free DOX against EL4 murine lymphoma cells after 72 h of incubation. (b) Uptake of DOX by EL4 cells after 1 h incubation with either RBC-NP(DOX) or free DOX.
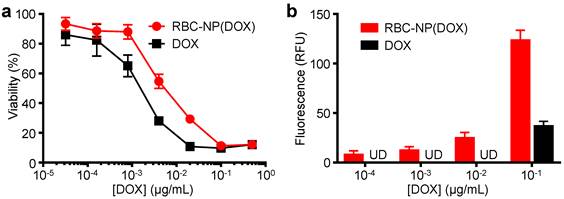
In Vivo Therapeutic Efficacy. To determine the ability of drug-loaded RBC-NP to function as effective therapy against tumor growth in vivo, we analyzed long-term tumor burden in a murine lymphoma model. EL4 cells were implanted subcutaneously into the right flank of C57BL/6 mice and were first allowed to develop for 9 days, after which tumor-bearing mice were treated every other day with RBC-NP(DOX), free DOX, empty RBC-NP, or isotonic sucrose at a drug dosage of 3 mg of DOX per kg of body weight (3 mg/kg) (Figure 3a). The dosing was determined to be just under the maximum tolerated dose for 6-week-old male C57BL/6 mice, designated as the dose at which the mouse body weight decreases 10% (data not shown). Isotonic sucrose and RBC-NP treatment alone did not affect tumor growth. Mice treated with free DOX exhibited marginal control of tumor growth, extending median survival by 6 days compared with no treatment. The RBC-NP(DOX) treatment group showed the most significant efficacy in terms of tumor growth inhibition, with the median survival nearly doubling from 24 days for the control group to 47 days for the treated group (Figure 3b). Though not an actively targeted formulation, it is believed that RBC-NP(DOX) was able to accumulate at the tumor site via the enhanced permeation and retention (EPR) effect [37, 38], thereby substantially increasing the local drug concentration at the tumor site. It should be noted that it was not until treatment ceased on day 23 post-implantation that the tumor growth kinetics started to accelerate. During this period, mice treated by RBC-NP(DOX) showed no appreciable decrease in weight, a global parameter of formulation safety (Figure 3c). It should also be noted that, for the free DOX treatment group, the observed steady weight kinetics reflects an increase in tumor burden, which indicates an appreciable toxic effect. Mice treated with RBC-NP(DOX) continued to increase in weight despite minimal tumor burden during the treatment period, suggesting that, in future explorations, the nanoparticle formulation can be further increased to exert an even more potent antitumor effect.
In vivo treatment of solid tumors. (a) Tumor growth inhibition in mice treated with RBC-NP(DOX), free DOX, RBC-NP, or isotonic sucrose by tail vein injection (n = 5). Treatment was initiated 9 days post-challenge, and mice were administered formulations every other day for 2 weeks. (b) Survival of mice treated with RBC-NP(DOX), free DOX, RBC-NP, or isotonic sucrose. (c) Relative weights of tumor-bearing mice treated with RBC-NP(DOX), free DOX, RBC-NP, or isotonic sucrose. Values were normalized within each group using the measurements on day 0.
In Vivo Safety and Immunogenicity of the RBC-NP Platform. Next, we evaluated the safety profile of RBC-NP(DOX) in order to assess the formulation's potential as a clinically translatable drug delivery platform. Isotonic sucrose, RBC-NP(DOX), or free DOX was administered at a drug dosage of 3 mg/kg, and whole blood was collected prior to injection and 24 h post-injection. Hematological parameters largely showed no difference between the sucrose, RBC-NP(DOX), and free DOX treatment groups (Figure 4a-d).
In vivo safety studies of RBC-NP(DOX). (a) RBC count, (b) platelet count, (c) hemoglobin quantification, (d) hematocrit, (e) white blood cell count, (f) neutrophil count, (g) lymphocyte count, and (h) monocyte count in mice before and 24 h after injection of sucrose, RBC-NP(DOX) or free DOX. (i) Comprehensive serum chemistry panel conducted on mice intravenously injected with isotonic sucrose, RBC-NP(DOX), or free DOX. Serum was collected 24 h post-injection (n = 3 for all experiments; *P ≤ 0.05; ***P ≤ 0.001).
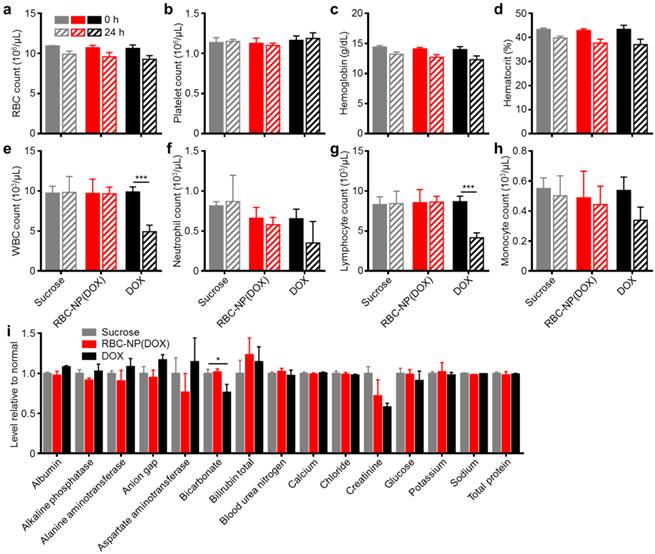
RBC, platelet, hemoglobin, and hematocrit quantifications were all normal 24 h after injection. Free DOX, however, is known to have myelosuppressing effects, which can lead to severe complications in the clinic such as neutropenic fever, infections, hemorrhage, and even death [39]. This was reflected in the white blood cell (WBC) quantifications (Figure 4e-h). When free DOX was administered, the mice suffered a significant decrease in WBC count. This decrease in overall WBC count was seen across different leukocyte subsets, with the sharpest reduction occurring in the number of lymphocytes. The RBC-NP(DOX) formulation was able to stably sequester the drug, delivering it for potent tumor control with no observable myelosuppression, which is often the dose-limiting toxic side effect of DOX in a clinical setting.
Additionally, RBC-NP(DOX) did not elicit any adverse physiological effects based on a comprehensive chemistry panel of mouse serum (Figure 4i). The creatinine levels for mice treated with free DOX were significantly decreased, possibly indicating increased activity of the kidneys to remove excess free drug. Of potential note are the decrease in bicarbonate and concurrent increase in anion gap for mice treated with free DOX only. These results, when taken together, can indicate a shift towards acidosis, which can be an early sign of kidney failure. Furthermore, RBC-NP(DOX) treatment did not result in any increase in serum alanine aminotransferase (ALT) or aspartate aminotransferase (AST) levels, suggesting that RBC-NP(DOX) administration does not induce liver injury. As is the case with most nanoparticle formulations, RBC-NPs are cleared by the liver [22], so it is important that the DOX-loaded RBC-NPs do not induce any acute hepatic damage. Compared with free DOX, RBC-NP(DOX) demonstrated a lack of acute systemic abnormalities after administration of a therapeutic dose, thereby underscoring the biocompatibility and safety of RBC-NP as a drug carrier.
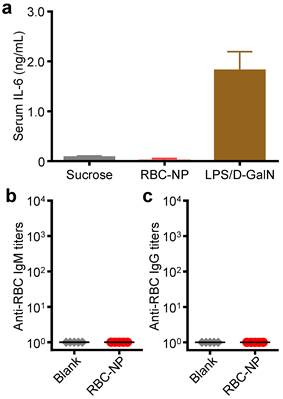
To further evaluate the translatability of RBC-NP as a cancer drug carrier, we investigated the immunological implications of RBC-NP administration. RBC-NP did not induce elevated serum levels of interleukin-6 (IL-6), which indicates lack of an acute systemic inflammatory response against the nanocarrier (Figure 5a). Nanoscale drug delivery vehicles must also not induce any long-term immune responses, the lack of which helps to preserve functionality of the particles upon repeated injections [40]. Multiple administrations of RBC-NP induced no detectable serum IgM or IgG titers against RBCs after 30 days (Figure 5b,c), indicating that RBC-NPs can be used in vivo repeatedly without reduction in their abilities to function as a drug delivery vehicle. Additionally, the lack of antibody titers against RBCs reveals that no autoimmunity against self-RBCs is developed as a result of repeated RBC-NP injections. Taken together, these results demonstrate that RBC-NP themselves produce no significant acute or long-term immunological responses as a delivery vehicle, highlighting the “self” nature of the RBC-NP platform. Thus, RBC-NP can serve as an effective option for drug delivery to overcome the limitations of traditional chemotherapy.
In vivo immunogenicity of RBC-NP. (a) Serum analysis of interleukin-6 (IL-6) production in mice administered with isotonic sucrose, RBC-NP, or lipopolysaccharide with D-galactosamine (LPS/D-GalN) as a positive control (n = 3). (b) anti-RBC IgM and (c) anti-RBC IgG titers produced in mice receiving repeated dosing of RBC-NP (n = 5). Tumor-bearing mice were administered with RBC-NP every other day starting from day 9 post-implantation of the tumor cells for 2 weeks and blood was collected on day 30 post-implantation for titer analyses.
The focus of these studies as a whole was to evaluate a biocompatible nanoparticle drug delivery agent with clinical translation potential to treat solid tumors with enhanced efficacy. Therapeutic nanocarriers must be safe for use in vivo. Previous nanoparticle studies on bare materials such as carbon nanotubes [41, 42], iron nanoparticles [43], and titanium dioxide [44], have shown adverse responses indicative of material toxicity. RBC-NP(DOX), employing a highly generalizable membrane cloaking approach, was able to deliver a toxic chemotherapy drug payload to the tumor site and significantly prolong survival without any acute increases in myelosuppression, systemic immune response, or abnormal blood chemistry parameters. Furthermore, early mortality and toxicity from high DOX dosages were not observed during long-term and repetitive treatment with RBC-NP(DOX).
Conclusion
The studies described here indicate that the RBC-NP platform is able to unite several important drug delivery properties into a single system. Of great importance, RBC-NP with its natural membrane coating, demonstrated good biocompatibility and did not elicit immune reactions. Furthermore, the versatility of the inner polymeric core provides opportunities to deliver a wide variety of therapeutics (i.e. hydrophilic/hydrophobic small molecules, proteins, nucleic acids, etc.) [45]. Although translating RBC-NP to human use remains to be investigated, large-scale synthesis of the particles should be feasible given the existing infrastructure for blood collection and transfusion as well as the maturing development of polymeric therapeutics [46]. Ultimately, the safety and biocompatibility of RBC-NP paired with its efficacy against solid tumors reveal that this platform possesses many of the requisite characteristics for a clinically translatable drug delivery system. The results presented here provide a promising foundation for continued development and future clinical tests of the RBC-NP platform.
Acknowledgements
This work is supported by the National Science Foundation Grant DMR-1505699 and the National Institute of Diabetes and Digestive and Kidney Diseases of the National Institutes of Health under Award Number R01DK095168. B. L. is supported by a National Institutes of Health 5F31CA186392 training grant from the National Cancer Institute. R. F. is supported by a National Institutes of Health R25CA153915 training grant from the National Cancer Institute.
Competing Interests
The authors declare no competing financial interests.
References
1. Desai N. Challenges in development of nanoparticle-based therapeutics. AAPS J. 2012;14:282-95
2. Mitragotri S, Burke PA, Langer R. Overcoming the challenges in administering biopharmaceuticals: formulation and delivery strategies. Nat Rev Drug Discov. 2014;13:655-72
3. De Jong WH, Borm PJ. Drug delivery and nanoparticles: applications and hazards. Int J Nanomedicine. 2008;3:133-49
4. Jiang S, Win KY, Liu S, Teng CP, Zheng Y, Han MY. Surface-functionalized nanoparticles for biosensing and imaging-guided therapeutics. Nanoscale. 2013;5:3127-48
5. Mout R, Moyano DF, Rana S, Rotello VM. Surface functionalization of nanoparticles for nanomedicine. Chem Soc Rev. 2012;41:2539-44
6. Sperling RA, Parak WJ. Surface modification, functionalization and bioconjugation of colloidal inorganic nanoparticles. Philos Trans A Math Phys Eng Sci. 2010;368:1333-83
7. Hu CM, Fang RH, Luk BT, Zhang L. Polymeric nanotherapeutics: clinical development and advances in stealth functionalization strategies. Nanoscale. 2014;6:65-75
8. Yoo JW, Irvine DJ, Discher DE, Mitragotri S. Bio-inspired, bioengineered and biomimetic drug delivery carriers. Nat Rev Drug Discov. 2011;10:521-35
9. Fang RH, Hu CM, Zhang L. Nanoparticles disguised as red blood cells to evade the immune system. Expert Opin Biol Ther. 2012;12:385-9
10. Hu CM, Fang RH, Zhang L. Erythrocyte-inspired delivery systems. Adv Healthc Mater. 2012;1:537-47
11. Hu CM, Fang RH, Luk BT, Chen KN, Carpenter C, Gao W. et al. 'Marker-of-self' functionalization of nanoscale particles through a top-down cellular membrane coating approach. Nanoscale. 2013;5:2664-8
12. Luk BT, Hu CM, Fang RH, Dehaini D, Carpenter C, Gao W. et al. Interfacial interactions between natural RBC membranes and synthetic polymeric nanoparticles. Nanoscale. 2014;6:2730-7
13. Fang RH, Hu CM, Luk BT, Gao W, Copp JA, Tai Y. et al. Cancer cell membrane-coated nanoparticles for anticancer vaccination and drug delivery. Nano Lett. 2014;14:2181-8
14. Hu CM, Fang RH, Copp J, Luk BT, Zhang L. A biomimetic nanosponge that absorbs pore-forming toxins. Nat Nanotechnol. 2013;8:336-40
15. Copp JA, Fang RH, Luk BT, Hu CM, Gao W, Zhang K. et al. Clearance of pathological antibodies using biomimetic nanoparticles. Proc Natl Acad Sci USA. 2014;111:13481-6
16. Pang Z, Hu CM, Fang RH, Luk BT, Gao W, Wang F. et al. Detoxification of organophosphate poisoning using nanoparticle bioscavengers. ACS Nano. 2015;9:6450-8
17. Gao W, Fang RH, Thamphiwatana S, Luk BT, Li J, Angsantikul P. et al. Modulating antibacterial immunity via bacterial membrane-coated nanoparticles. Nano Lett. 2015;15:1403-9
18. Hu CM, Fang RH, Luk BT, Zhang L. Nanoparticle-detained toxins for safe and effective vaccination. Nat Nanotechnol. 2013;8:933-8
19. Hu CM, Fang RH, Wang KC, Luk BT, Thamphiwatana S, Dehaini D. et al. Nanoparticle biointerfacing by platelet membrane cloaking. Nature. 2015;526:118-21
20. Piao JG, Wang L, Gao F, You YZ, Xiong Y, Yang L. Erythrocyte membrane is an alternative coating to polyethylene glycol for prolonging the circulation lifetime of gold nanocages for photothermal therapy. ACS Nano. 2014;8:10414-25
21. Guo Y, Wang D, Song Q, Wu T, Zhuang X, Bao Y. et al. Erythrocyte membrane-enveloped polymeric nanoparticles as nanovaccine for induction of antitumor immunity against melanoma. ACS Nano. 2015;9:6918-33
22. Hu CM, Zhang L, Aryal S, Cheung C, Fang RH, Zhang L. Erythrocyte membrane-camouflaged polymeric nanoparticles as a biomimetic delivery platform. Proc Natl Acad Sci USA. 2011;108:10980-5
23. Gao W, Hu CM, Fang RH, Luk BT, Su J, Zhang L. Surface functionalization of gold nanoparticles with red blood cell membranes. Adv Mater. 2013;25:3549-53
24. Zhang J, Gao W, Fang RH, Dong A, Zhang L. Synthesis of nanogels via cell membrane-templated polymerization. Small. 2015;11:4309-13
25. Parodi A, Quattrocchi N, van de Ven AL, Chiappini C, Evangelopoulos M, Martinez JO. et al. Synthetic nanoparticles functionalized with biomimetic leukocyte membranes possess cell-like functions. Nat Nanotechnol. 2013;8:61-8
26. Oldenborg PA, Zheleznyak A, Fang YF, Lagenaur CF, Gresham HD, Lindberg FP. Role of CD47 as a marker of self on red blood cells. Science. 2000;288:2051-4
27. Rodriguez PL, Harada T, Christian DA, Pantano DA, Tsai RK, Discher DE. Minimal "self" peptides that inhibit phagocytic clearance and enhance delivery of nanoparticles. Science. 2013;339:971-5
28. Davis ME, Chen ZG, Shin DM. Nanoparticle therapeutics: an emerging treatment modality for cancer. Nat Rev Drug Discov. 2008;7:771-82
29. Peer D, Karp JM, Hong S, Farokhzad OC, Margalit R, Langer R. Nanocarriers as an emerging platform for cancer therapy. Nat Nanotechnol. 2007;2:751-60
30. Ishida T, Maeda R, Ichihara M, Irimura K, Kiwada H. Accelerated clearance of PEGylated liposomes in rats after repeated injections. J Control Release. 2003;88:35-42
31. Knop K, Hoogenboom R, Fischer D, Schubert US. Poly(ethylene glycol) in drug delivery: pros and cons as well as potential alternatives. Angew Chem Int Ed Engl. 2010;49:6288-308
32. Aryal S, Hu CM, Fang RH, Dehaini D, Carpenter C, Zhang DE. et al. Erythrocyte membrane-cloaked polymeric nanoparticles for controlled drug loading and release. Nanomedicine (Lond). 2013;8:1271-80
33. Zhang L, Chan JM, Gu FX, Rhee JW, Wang AZ, Radovic-Moreno AF. et al. Self-assembled lipid-polymer hybrid nanoparticles: a robust drug delivery platform. ACS Nano. 2008;2:1696-702
34. Maccubbin DL, Wing KR, Mace KF, Ho RL, Ehrke MJ, Mihich E. Adriamycin-induced modulation of host defenses in tumor-bearing mice. Cancer Res. 1992;52:3572-6
35. Iversen TG, Skotland T, Sandvig K. Endocytosis and intracellular transport of nanoparticles: present knowledge and need for future studies. Nano Today. 2011;6:176-85
36. Fang RH, Hu CM, Chen KN, Luk BT, Carpenter CW, Gao W. et al. Lipid-insertion enables targeting functionalization of erythrocyte membrane-cloaked nanoparticles. Nanoscale. 2013;5:8884-8
37. Matsumura Y, Maeda H. A new concept for macromolecular therapeutics in cancer chemotherapy: mechanism of tumoritropic accumulation of proteins and antitumor agent smancs. Cancer Res. 1986;46:6387-92
38. Maeda H. Macromulecular therapeutics in cancer treatment: the EPR effect and beyond. J Control Release. 2012;164:138-44
39. Rostad ME. Current strategies for managing myelosuppression in patients with cancer. Oncol Nurs Forum. 1991;18:7-15
40. Rao L, Bu LL, Xu JH, Cai B, Yu GT, Yu X. et al. Red blood cell membrane as a biomimetic nanocoating for prolonged circulation time and reduced accelerated blood clearance. Small. 2015;11:6225-36
41. Kam NW, O'Connell M, Wisdom JA, Dai H. Carbon nanotubes as multifunctional biological transporters and near-infrared agents for selective cancer cell destruction. Proc Natl Acad Sci USA. 2005;102:11600-5
42. Monteiro-Riviere NA, Nemanich RJ, Inman AO, Wang YY, Riviere JE. Multi-walled carbon nanotube interactions with human epidermal keratinocytes. Toxicol Lett. 2005;155:377-84
43. Pisanic TRI, Blackwell JD, Shubayev VI, Finones RR, Jin S. Nanotoxicity of iron oxide nanoparticle internalization in growing neurons. Biomaterials. 2007;28:2572-81
44. Warheit DB, Webb TR, Reed KL, Frerichs S, Sayes CM. Pulmonary toxicity study in rats with three forms of ultrafine-TiO2 particles: differential responses related to surface properties. Toxicology. 2007;230:90-104
45. Luk BT, Zhang L. Current advances in polymer-based nanotheranostics for cancer treatment and diagnosis. ACS Appl Mater Interfaces. 2014;6:21859-73
46. Hrkach J, Von Hoff D, Mukkaram Ali M, Andrianova E, Auer J, Campbell T. et al. Preclinical development and clinical translation of a PSMA-targeted docetaxel nanoparticle with a differentiated pharmacological profile. Sci Transl Med. 2012;4:128ra39
Author contact
![]() Corresponding author: Tel: 858-246-0999, Email: zhangedu.
Corresponding author: Tel: 858-246-0999, Email: zhangedu.