Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and methods
Results
Discussion
Supplementary Material
Abbreviations
Acknowledgements
Conflicts of Interest
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2016; 6(8):1160-1175. doi:10.7150/thno.14848 This issue Cite
Research Paper
Redox Regulation of Stem-like Cells Though the CD44v-xCT Axis in Colorectal Cancer: Mechanisms and Therapeutic Implications
Huai-Qiang Ju1,2*, Yun-Xin Lu1*, Dong-Liang Chen1*, Tian Tian1, Hai-Yu Mo1, Xiao-Li Wei1, Jian-Wei Liao1, Feng Wang1, Zhao-Lei Zeng1, Helene Pelicano3, Mitzi Aguilar2, Wei-Hua Jia1, Rui-Hua Xu1 ![]()
1. Sun Yat-sen University Cancer Center, State Key Laboratory of Oncology in South China, Collaborative Innovation Center for Cancer Medicine, Guangzhou, 510060, China.
2. Department of Molecular and Cellular Oncology, The University of Texas MD Anderson Cancer Center, Houston, TX 77030, USA.
3. Department of Translational Molecular Pathology, The University of Texas MD Anderson Cancer Center, Houston, TX 77030, USA.
*These three authors contributed equally to this work.
Received 2015-12-30; Accepted 2016-4-25; Published 2016-5-23
Abstract

Colorectal cancer (CRC) is a common neoplastic disease and a frequent cause of death. Drug resistance is a major challenge to CRC treatment and stem-like side-population (SP) cells may play a key role in this resistance. Although it has been recognized that cancer stem cells may be affected by redox status, the underlying mechanisms for this effect and the roles of celllular redox adaptation and antioxidant capacity in CRC remain elusive. Our study shows that CRC SP cells are highly dependent on cellular GSH to maintain ROS levels below those of non-SP cells. Exposing CRC cells to H2O2 produced a significant decrease in the percentage of SP cells, which was rescued by adding N-acetylcysteine. Mechanistically, CD44v interacts with and stabilizes xCT and thereby promotes the uptake of cysteine for GSH synthesis and stimulates SP cell enrichment. Additionally, miR-1297 levels were inversely correlated with the expression of xCT; thus, reduced miR-1297 contributes to SP cell enrichment in CRC tumors, which results in tumor aggressiveness and poor clinical outcomes. Importantly, redox modification by PEITC significantly reduces CRC SP cells in vitro and impairs tumors growth in vivo. The combination of 5FU and PEITC led to synergistic cytotoxic effects against CRC cells in vitro and in vivo. Taken together, our data suggest that a GSH-mediated reduction in cellular ROS levels is an essential regulator of CRC SP cells mediated by the CD44v-xCT axis, and disrupting the redox status may eliminate the chemotherapy-resistant CRC SP cells with potentially significant benefits for cancer treatment.
Keywords: Colorectal cancer, Side population, Redox status, CD44v-xCT, miR-1297.
Introduction
Colorectal cancer (CRC) is a common neoplastic disease and a frequent cause of death in humans. It is the third most common cancer and the third leading cause of cancer-related deaths in men and women in the United States [1, 2]. Also, the incidence of CRC has continued to rapidly rise in China over the past decade [3]. Despite significant improvements have been made in clinical therapy in recent years, most patients still die of this disease as a result of distant metastasis, therapeutic resistance and recurrence. An increasing number of studies suggest that tumors in various tissues, including colorectal tumors, include a small subpopulation of cells with stem-like characteristics, often referred to as cancer stem cells (CSCs) or tumor-initiating cells. These cells are believed to be responsible for tumor initiation, growth and metastasis [4]. The recurrence of CRC after surgery, chemotherapy or radiation therapy may be due to the persistence of CSCs. A subset of cells that highly express ABC transporters and have CSC-like characteristics, such as the capacity for self-renewal and tumorigenicity, are called side population (SP) cells [5]. In addition, the percentage of SP cells in CRC tumors has been shown to be correlated with patient prognosis and tumor grade [6]. It is therefore important to carry out quantitative analyse of CRC SP cells and to study the factors that affect SP cells. Although certain key molecules and signaling pathways that affect the stemness of CSCs and their ability to differentiate have been identified, the redox status of CSCs remain largely to be investigated.
Even though cancer cells have higher levels of reactive oxygen species (ROS) in general, which are essential for the initiation and progression of malignancy, CSCs seem to harbor only low levels of ROS [7, 8]. Elevated ROS levels have been observed to induce the differentiation of embryonic stem cells into cardiovascular and mesendodermal cells [9, 10]. In addition, breast cancer stem cells might have a high ROS-scavenging capacity and lower cellular ROS levels than the corresponding non-tumorigenic cells [11]. Our recent study also showed that serum could activate mitochondrial respiration and promote the generation of mitochondrial ROS, leading to an apparent loss of certain stem cell markers and a reduce ability to form neurospheres [12]. Although it has been recognized that CSCs may be affected by redox status, the underlying mechanisms for this effect and the roles of cellular redox adaptations and antioxidant capacity in CRC remain elusive.
Based on the observation that redox status plays a significant role in determining the fates of cancer stem cells, we reasoned that CRC CSCs might have a high antioxidant capacity to maintain moderate levels of cellular ROS and retain both the stemness and the cancer-forming capabilities of the cells. CD44 is known as an adhesion molecule that is expressed in CRC stem-like cells [13] . A recent study has shown that a CD44 variant (CD44v) interacts with xCT, a glutamate-cysteine transporter, and controls the intracellular level of reduced glutathione (GSH) in colorectal cancer cells [13]. The findings discussed above led us to evaluate the impact of redox status and the role of the CD44v-xCT axis on CRC SP cells, to investigate the underlying molecular mechanisms, and to develop new therapeutic strategies for killing chemotherapy-resistant CRC CSC cells.
Materials and methods
Tissue specimens and patient information
A total of 119 archived colorectal cancer specimens and 48 adjacent normal tissue samples were obtained immediately after surgery and were either stored at -80°C or paraffin-embedded at the Sun Yat-sen University Cancer Center (Guangzhou, China) between January 2010 and July 2013. Prior to sample collection, the donors' written consents and approvals from the Institutional Research Ethics Committee were obtained.
Cell lines and cell culture
Colorectal normal and cancer cell lines (LoVo, SW 620, SW 1116, SW 480 and RKO) and immortalized colon epithelial cell lines (CCD-112Con and CCD 841) were obtained from the American Type Culture Collection (Manassas, VA, USA) and were cultured under the conditions specified by the manufacturer. All cell lines were authenticated based on short tandem repeat (STR) fingerprinting before use at the Medicine Laboratory of the Forensic Medicine Department of Sun Yat-sen University (Guangzhou, China).
Reagents and antibodies
Hoechst 33342, PEITC, 5FU, NAC, H2O2, verapamil and MTT were obtained from Sigma-Aldrich (St. Louis, MO, USA). The following antibodies were used for Immunoblotting analyses: GAPDH (Millipore, Billerica, USA); Oct4, xCT, (Abcam, Cambridge, USA); CD44, CD133, ABCG2 and SOX2 (Cell Signaling Technology, Beverly, USA).
Measurement of cellular ROS and GSH
CM-H2DCF-DA (Invitrogen, used for H2O2 detection) was used to measure cellular ROS levels via flow cytometry as previously described [14]. Total cellular GSH contents were measured using a GSH assay kit (Cayman Chemical, Arbor, USA) according to a protocol provided by the manufacturer.
Flow cytometric sorting of SP and non-SP cells
Cells were detached via trypsinization, washed, and resuspended in pre-warmed RPMI 1640 medium containing 2% FBS at a density of 1×106 cells/ml. Cell staining was performed using the method described by Goodell et al [15], with the following modifications. The cells were incubated with Hoechst 33342 at a nontoxic concentration (5 mg/ml) in the presence or absence of the ABC transporter inhibitor verapamil (50 mM) for 90 min at 37 °C in darkness with intermittent shaking. The cells were then washed with cold PBS, resuspended in PBS, and kept at 4 °C for flow cytometry and sorting. Cell analysis and sorting were performed using a MoFlo XDP Cell Sorter (Beckman Coulter, Brea, USA).
Sphere-forming and colony formation assays
A sphere-forming assay was preformed according to published protocol with minor modifications. Briefly, single-cell suspensions were plated in six-well ultralow attachment plates (Corning Inc. Corning, USA) at a density of 1.0x103 cells/well Sorted SP and non-SP cells were cultured in serum-free DMEM/F12 (Invitrogen) supplemented with 20 ng/ml of epidermal growth factor (Miltenyi Biotec, Auburn, USA), 20 ng/ml of basic fibroblast growth factor (Miltenyi Biotec), and 2 mM L-glutamine (Mediatech Inc.). The number and size of tumor spheres formed were evaluated via light microscopy after 7 days. For the colony formation assay, the cells were plated in 6-well plates (500 cells/well) and cultured for 14 days. The colonies were fixed with 10% formaldehyde for 10 min and then stained with 1% crystal violet for 15min.
RNA extraction and qPCR analysis
Total miRNA was extracted using the mirVana miRNA Isolation Kit (Ambion, Carlsbad, USA) according to the manufacturer's instructions. miRNA was reverse-transcribed and the expression levels of miRNA were quantified using the All-in-One™ miRNA qRT-PCR Detection Kit (GeneCopoeia, Rockville, USA). Human U6 snRNA was used as an endogenous control for the quantitation of miRNAs. Total RNA was extracted using Trizol reagent (Life Technologies, Carlsbad, USA) according to the manufacturer's instructions. The mRNA expression level was analyzed by qPCR analysis as previously described [16]. Specific primers were synthesized by Sigma-Aldrich, and the sequences are described in the supplementary material and methods section.
Immunoblotting and immunohistochemical analysis
Cells were harvested from 10-cm dishes, lysed in a radioimmunoprecipitation assay protein lysis buffer, and 50 mg of each protein extract was used for the analysis. The Immunoblotting analysis was performed as previously described [17]. Immunohistochemical analyses were conducted according to standard procedures as previously described [16]. The slides were blocked and then incubated with an anti-xCT antibody (1:100 dilution, Abcam, #ab111822) or CD44 (1:100 dilution, Cell Signaling Technology, #3570) at 4°C overnight.
Cell transfection and luciferase assay
Small interfering RNA (siRNA) targets for xCT (#1: ATGCCCAGATATGCATCGT; #2: GGAAGAGATTCAAGTATTA) and CD44 (#1: GCAAGGCTTTCAATAGCAC; #2: CCGCTTTGCAGGTGTATTC) were synthesized by RiboBio (Guangzou, China). siRNA#1-resistant expression vectors for CD44 and xCT with same-sense mutations (CD44-R: TGTAAAGCATTTAACTCAACC, xCT-R: AAATGTCCTGACATGCACAGG) were synthesized by GeneCopoeia (Rockville, USA). Cells (~70% confluent) were transfected with 20 nM siRNA or NC using DharmaFECT transfection reagent (GE Dharmacon, Lafayette, USA) according to the manufacturer's instructions. miR-1297 and miR-1297-mut mimics were purchased from RiboBio (GuangZhou, China). DNA fragments from 3'UTR of xCT containing the predicted complementary sites of miR-1297 were cloned into a pGL3-basic vector (Addgene, Cambridge, USA). Ten thousand cells were seeded in triplicate in 48-well plates and allowed to settle for 24 h. Then, the pGL3-xCT-3'UTR reporter plasmids (100 ng) plus 5 ng of pRL-TK renilla plasmid (Promega, Madison, USA), and increasing amounts (10 and 50 nM) of negative control (NC), miR-1297 or miR-1297-mut mimic were co-transfected into CRC cells using Lipofectamine LTX reagent (Invitrogen, Carlsbad, USA) according to the manufacturer's recommendation. Luciferase and renilla signals were measured 24 h after transfection using the Dual Luciferase Reporter Assay Kit (Promega, Madison, USA) according to a protocol provided by the manufacturer.
Evaluation of in vivo tumorigenicity
To test the effects of 5FU and PEITC on tumor-initiating capacity, LoVo cells were treated with each drug at their respective IC50 concentrations for 24 h in vitro, washed, and cultured in fresh medium (without either drug) for another 48 h. The cells were then harvested and injected subcutaneously into the flanks of athymic nude mice at 1.0×105 or 2.0×105 cells per injection site, with 10 mice per group. The presence or absence of a visible or palpable tumor was evaluated and tumor growth was monitored every 2-3 days. Tumor volume was measured using a caliper and was calculated using the formula V=length×width2/2. All animal studies were conducted with the approval of the Sun Yat-sen University Institutional Animal Care and Use Committee.
In vivo therapeutic study
LoVo cells (2×106) were injected subcutaneously into the flanks of Balb/c nude mice (4-5 weeks old). When palpable tumors reached approximately 100 mm3 (after 2 weeks), mice were randomly assigned (n=6 per group) to receive the following treatments on a weekly schedule: group 1 served as a control and received no treatment; group 2 received 25 mg/kg of intraperitoneal 5FU once a week; group 3 was treated with 25 mg/kg of intraperitoneal PEITC for 3 consecutive days per week; and group 4 was treated with 25 mg/kg of 5FU once a week, followed by 25 mg/kg of PEITC for the next 3 days. Treatments were continued for 4 weeks. Tumor volumes were measured and calculated as described above. Body weights were also recorded. On the day after the end of treatment, the mice were euthanized via carbon dioxide inhalation and the tumors were excised and weighed.
Statistical analysis
All data are presented as the mean ± SD. For comparisons of the statistical differences among more than two groups, one-way ANOVAs and Newman-keuls multiple comparison tests were used. All other statistical differences were evaluated via the Student's unpaired t tests using Prism GraphPad software (San Diego, CA, USA). A P value of < 0.05 was considered statistically significant. The cytotoxic effect of PEITC combined with 5FU was calculated using CalcuSyn Biosoft software (Ferguson, MO, USA).
Results
Increased cellular antioxidant activity decreases ROS level to sustain the SP cell fraction in CRC cells
To assess the redox status in CRC CSCs, a fluorescence-activated cell sorting (FACS) technique that can exclude Hoechst 33342 dye was used to isolation CRC SP cells [15]. The cell lines LoVo and SW620 contained 5.18% and 6.52% SP cells, respectively, while the other tested CRC cellscontained less than 1% SP cells in the population, as shown in the distinct dim 'tail' in the flow cytometry plots (Figure 1A, S1A). The ABC transporter inhibitor verapamil (Vera, 50 mM), which effectively blocks the export of the Hoechst dye and thus leads to the disappearance of the SP subpopulation, was used as a control (Figure 1A). To determine whether CRC SP cells were enriched as CSCs, the ability of the CRC SP cells to form colonies, or spheres was studied. Figure 1B shows that CRC SP cells had a greater ability to form colonies than the non-SP cells and the unsorted cells after 15 days of culturing. Similarly, the sphere formation assay confirmed that SP cells formed a greater number of spheres than the non-SP cells and the unsorted cells. A limiting dilution-type in vivo assay also showed that the frequency of tumor initiation following inoculation with small numbers (1x104 or 1x105) of CRC SP cells was higher than the frequency following inoculations with non-SP cells (P<0.05, Figure S1B). To verify these observations, PCR and an immunoblotting assay were performed to analyze the expression of molecules that are either CSC surface markers or are highly expressed in CSCs, including CD44, CD133, Oct4, SOX2 and ABCG2. The results shows that the mRNA and protein expression levels of the analyzed molecules were higher in the CRC SP cells than in the non-SP cells (Figure 1C, S1C). These data suggested that LoVo and SW620 cells in human CRC have a stem-like side population.
Increased cellular antioxidant activity decreases ROS level to sustain the SP cell fraction in CRC cells. (A) The percentages of SP cells in LoVo and SW620 cells were compared using Hoechst 33342 staining followed by flow cytometry. (B) Images and quantification of colony or sphere formation by SP, non-SP or unsorted cells. (C) The stem cell markers CD44, CD133, Oct4, ABCG2 and Sox2 were analyzed using immunoblotting in both SP and non-SP cells. (D) Representative histograms of significant decreases in the ROS contents of SP, non-SP or unsorted cells as detected by the fluorescent probe DCF-DA. (E) Cellular GSH levels in SP, non-SP or unsorted cells were measured via spectrophotometric analysis. (F) Reversion of H2O2-induced decreases in SP cells following NAC treatment. LoVo or SW620 cells were treated with 3 mM NAC for 2 h, followed by 100 μM H2O2 for 24 h. Representative dot plots of three separate experiments are shown. Data in B and E are presented as the mean ± SD (n=3). *P < 0.05 versus control cells.
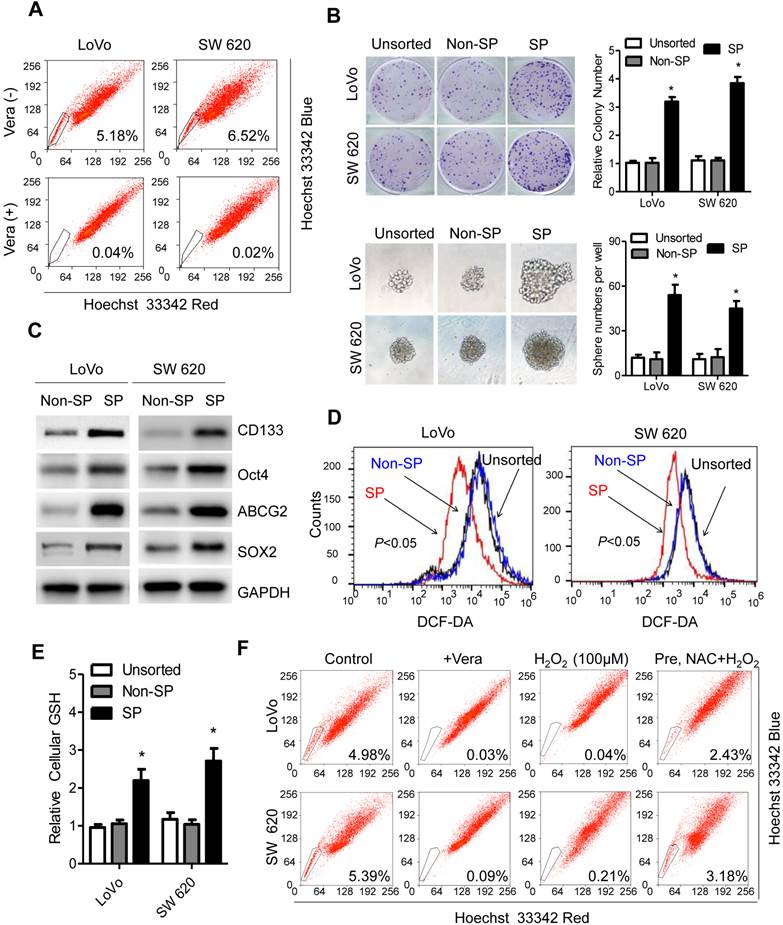
To study the redox status in the CRC SP fractions, we first compared the ROS levels between the SP, non-SP and unsorted cells. CRC SP cells exhibited a significant decrease in their basal ROS content, which was quantified via flow cytometry using CM-H2DCF-DA as a fluorescent probe (Figure 1D). Further analyses revealed that the reduced ROS levels were due to an increase in antioxidant capacity, as evidenced by the elevated GSH content in CRC SP cells relative to that in non-SP cells or in unsorted cells (Figure 1E). These results suggest that SP cells might be highly dependent on GSH for maintaining lower ROS levels in CRC cells. We next analyzed how exogenous H2O2 affected the percentage of SP cells. As shown in Figure 1F, the SP population was greatly decreased in LoVo cells (from 4.98% to 0.04%) treated with 100 μM H2O2 for 24 h. In contrast, treatment with 3 mM N-acetylcysteine (NAC, a GSH precursor) 2 h before adding H2O2 moderately rescued LoVo SP cells from the injury caused by H2O2, as demonstrated by the fact that the percentage of SP cells in the population was increased to 2.43%. Similar results were also observed in SW620 cells (Figure 1F). The results of our study indicate that CRC SP cells have an increased antioxidant capacity that maintains cellular ROS at a low level, as well as maintaining long-term self-renewal and stemness capabilities.
The CD44v-xCT axis contributes to ROS defense and to CRC SP cell enrichment
The above findings led us to further investigate the molecular mechanisms controlling the redox status and cellular GSH synthesis in CRC SP cells. Studies have recently shown that the expression of xCT, which is a subunit of a glutamate-cysteine transporter, on the surface of cancer cells is regulated by CD44v8-10 (an isoform including the sequence by variant exons v8-10). CD44v8-10 interacts with and stabilizes xCT within the plasma membrane, resulting in the promotion of cysteine uptake for GSH synthesis [13]. CD44 has recently been shown to be a cell surface marker associated with CSCs in many tumors, including CRCs [18, 19]. We tested whether the CD44v-xCT axis contributes to the ROS defense and to SP enrichment in colorectal cancer cells. RT-PCR analysis showed that the abundance of CD44v8-10 mRNA was greater in CRC SP cells than non-SP cells (Figure 2A). Consistently, we also found that the xCT protein was highly expressed on the membranes of LoVo and SW620 SP cells (Figure 2A).
The CD44v-xCT axis contributes to the defense against ROS and to SP cell enrichment. (A) mRNA expression of CD44v8-10 and xCT protein expressions in SP and non-SP cells were compared using RT-PCR and immunoblotting, respectively. (B) RT-PCR and immunoblotting analysis of the mRNA expression of CD44v8-10 and protein expression of xCT, respectively, in LoVo and SW620 cells transfected with two independent CD44 siRNAs or with scrambled control siRNA (sc). Na+/K+ ATPase was used as the membrane marker in A and B. (C) The expression of xCT and GAPDH in LoVo and SW620 cells transfected with two independent xCT siRNAs or scrambled control siRNA (sc) was analyzed via immunoblotting. (D) Intracellular GSH content and ROS level of LoVo cells after the siRNA depletion of CD44 or xCT. (E) ROS levels were compared in SW620 cells after the siRNA depletion of CD44 or xCT. (F) Comparison of the % of SP cells in LoVo and SW620 cell populations after the siRNA depletion of CD44 or xCT. Representative dot plots of three separate experiments are shown. Data in D and E are presented as the mean ± SD (n=3). *P < 0.05 versus control cells.
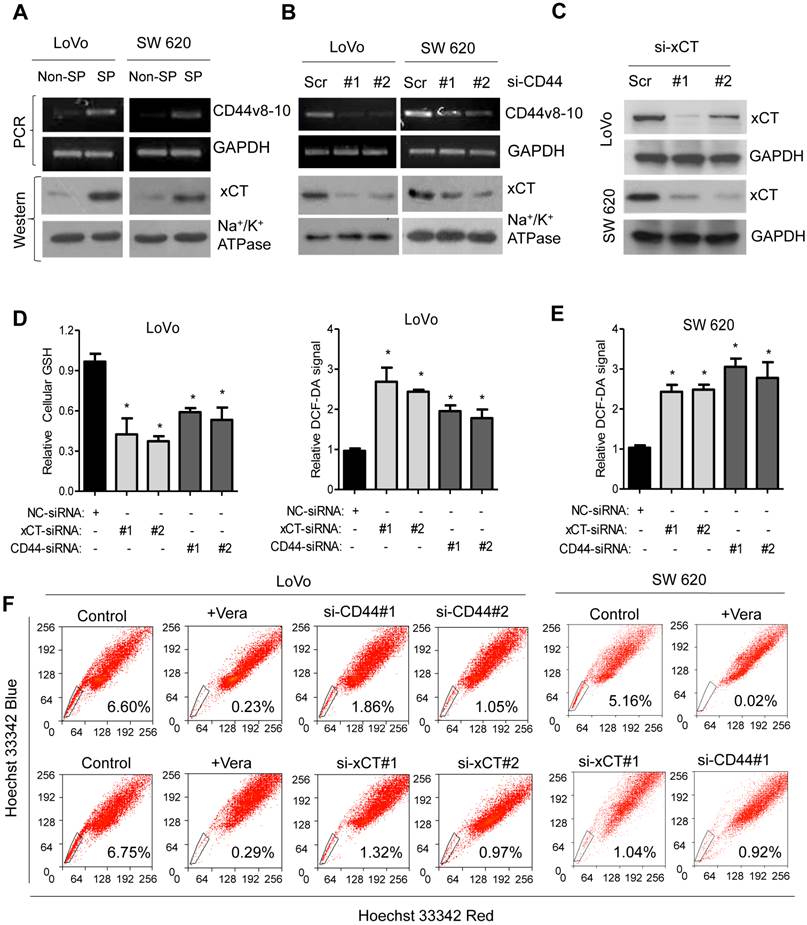
Two different siRNAs that target different sequences of CD44 mRNA decreased the expression of CD44v8-10 and the cell surface expression of xCT (Figure 2B). This suggests that CD44v stabilizes localization of the xCT protein on the surface of the membrane, as reported previously [13]. In addition, the RNAi-mediated ablation of CD44v resulted in a reduction in the intracellular GSH content and significantly increased the cellular ROS level in LoVo cells (Figure 2D). The siRNA depletion of CD44 for 48 h produced a rapid decrease in SP cells, from 6.60% to 1.86% or 1.05% in LoVo cells according to flow cytometric analysis (Figure 2F). In addition, we used the RNAi ablation strategy to knock down xCT expression in LoVo cells and tested the effect this had on SP cells in LoVo cells (Figure 2C, S2A). The results show that the RNAi-mediated ablation of xCT significantly depleted cellular GSH and increased ROS levels in CRC cells (Figure 2D), which led to a rapid reduction of the SP cell population (Figure 2F). Also, the decreased proportion of SP cells was significantly rescued in LoVo cells co-transfected with siRNA#1 and siRNA#1-resistant expression vectors for CD44 and xCT (Figure S2C). In agreement with the finding from the LoVo cells, similar results were also observed in SW620 cells after the siRNA depletion of CD44 or xCT, as evidenced by the reduced cellular GSH contents, elevated cellular ROS levels (Figure 2E, S2B) and decreased SP cell population (Figure 2F). Taken together, these results indicate that CD44v interacts with and stabilizes xCT and thereby promotes the uptake of cysteine for GSH synthesis and SP enrichment in colorectal cancer cells.
xCT is upregulated and is a target for miR-1297 in CRC cells
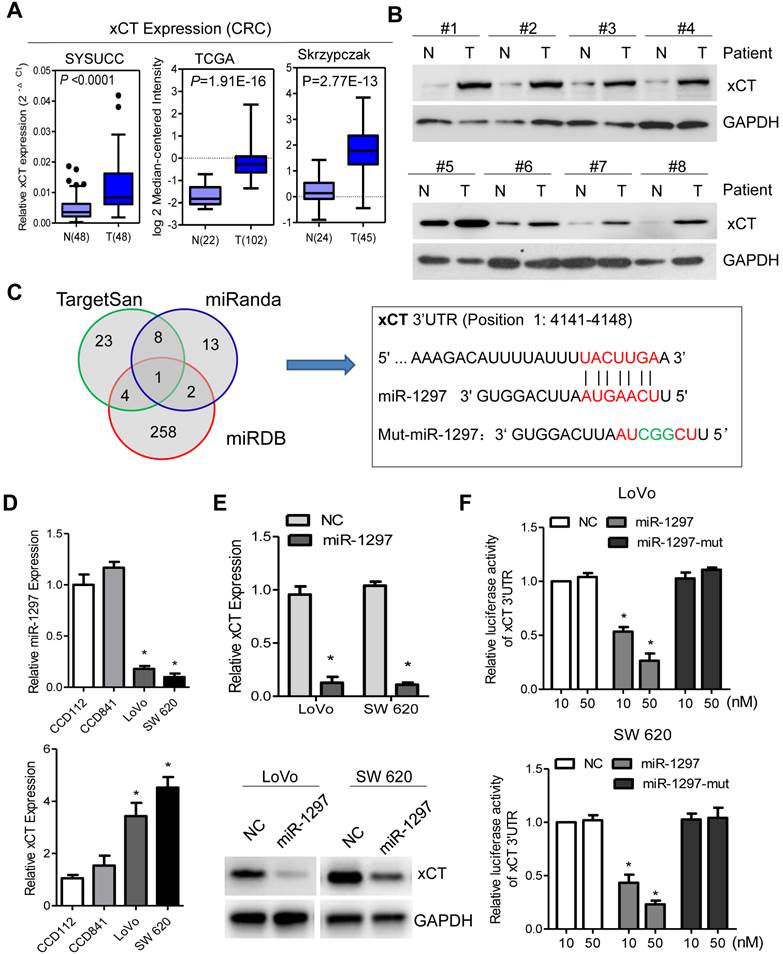
The significant role of the CD44v-xCT axis in ROS defense and in CRC SP cell enrichment prompted us to further investigate its regulation and role in CRC cells. The expression of xCT was analyzed with qPCR, and it was shown to be significantly higher in 48 CRC tumor tissues relative to their matched adjacent normal tissues. These results are also supported by the TCGA and Skrzypczak data sets available from Oncomine (Figure 3A). Also, the expression of xCT was differentially increased compared to its expression in paired normal adjacent colorectal tissues in 8 primary CRC tissue samples (Figure 3B). These results indicate that xCT is upregulated in CRC. MicroRNAs (miRNAs) are small non-coding RNAs that have been shown to be master regulators of gene expression in many cancer-related signaling pathways and these miRNAs can be used as targets and biomarkers for molecular diagnostics [20]. Analysis using publicly available algorithms (TargetScan, miRanda and miRDB) showed that xCT is the predicted target of miR-1297 (Figure 3C), a miRNA that has been found to be substantially decreased in lung and colorectal cancer [21, 22]. To explore whether xCT expression is negatively regulated by miR-1297 in the CRC cellular environment, we measured the expression level of miR-1297 and xCT in LoVo and SW620 cells by qPCR. The results show that miR-1297 expression levels were significantly decreased, while, xCT was significantly increased in LoVo and SW620 cells relative to levels in colon epithelial CCD112 and CCD841 cells (Figure 3D). In determining whether miR-1297 affects xCT expression, we found that the mRNA and protein levels of xCT were drastically decreased in CRC cells transfected with miR-1297 mimics (Figure 3E). Moreover, a luciferase reporter assay showed that the overexpression of miR-1297 repressed the luciferase activity of xCT-3'UTR (P<0.05 for both; Figure 3F). However, mutational mimics ectopically expressing miR-1297 showed no inhibition of xCT-3'UTR luciferase activity (Figure 3F), and no difference in luciferase activity was observed in cells cotransfected with xCT-mt-3'UTR and miR-1297 mimics (Figure S3A). Therefore, these results demonstrate that xCT is a target of miR-1297 and that it may be a significant redox modifier that provides SP enrichment in CRC cells.
xCT is upregulated and is a target for miR-1297 in CRC cells. (A) xCT expression in patient samples from our hospital and in other cancer microarray data sets (TCGA and Skrzypczak) available from Oncomine (https://www.oncomine.com//). (B) xCT expression according to immunoblotting analysis in 10 pairs of human CRC tissues (T) and in matched adjacent non-tumor tissues (N) from the same patient. The loading control was GAPDH. (C) Venn diagrams showing the number of potential miRNAs targeting the 3'UTR of xCT, as predicted by three databases: TargetScan, miRanda and miRDB. Sequences of the indicated miR-1297 and its potential binding site at the 3'UTR of xCT are shown. (D) Results from the qPCR analysis of xCT and miR-1297 expression, with GAPDH and U6 expression used to normalize transcript levels, respectively. (E) Expression levels of xCT according to qPCR and immunoblotting analysis in cells transfected with either the negative control (NC) or the miR-1297 mimic. The loading control was GAPDH. (F) Luciferase activity results from reporters containing the 3'UTR end of xCT in cells transfected with the miR-1297 mimic, miR-1297 mutation mimic (miR-1297-mut) and negative control (NC). Means ± SD (n=3) are shown in D, E and F. *P < 0.05 versus control cells
Reduced miR-1297 levels in CRC cells contribute to SP cell enrichment and correlate with patient prognoses
In previous studies, miR-1297 has been shown to be capable of inhibiting human CRC cell proliferation, migration and invasion in vitro and tumorigenesis in vivo [22]. We postulated that miR-1297 may serve as a tumor suppressor and that it might have a significant impact on SP cells through redox modification and xCT regulation in CRC cells. To test these possibilities, we assessed the percent of SP cells in LoVo cells and SW620 cells that had been transfected with miR-1297 mimics. As shown in Figure 4A, the overexpression of miR-1297 caused a significant decrease in the cellular GSH contents and increased the ROS levels in LoVo and SW620 cells. Additionally, the proportion of SP cells was markedly reduced in LoVo (from 5.26% to 1.35%) and SW620 (from 6.36% to 2.61%) cells transfected with miR-1297 mimics (Figure 4B). Meanwhile, miR-1297 overexpression significantly reduced the mRNA expression levels of multiple CSCs markers (Figure 4C). To further confirm the above finding, CRC cells were pretreated with 3 mM N-acetylcysteine (NAC, a GSH precursor) for 2 h before miR-1297 mimic transfection. Our results show that treatment with NAC before miR-1297 mimic transfection resulted in the rescue of ROS levels and SP proportions in these cells, demonstrating that NAC suppresses the effects of miR-1297 on SP cells through redox modifications (Figure S3B).
Reduced miR-1297 levels in CRC contribute to SP cell enrichment and correlate with patient prognoses. (A) ROS levels and the intracellular GSH content were assessed in cells transfected with miR-1297 mimic. (B) CRC SP cell quantification in cells transfected with negative control (NC) or miR-1297 mimic. (C) qPCR analysis of the expression of CSC markers in the indicated cells. (D) The expression level of miR-1297 was examined in CRC specimens, and Kaplan-Meier curves of patients with CRCs with low versus high expression levels of miR-1297 are shown (n=119; p< 0.001, log-rank test). (E) Correlation of miR-1297 expression with xCT mRNA expression in CRC patient samples (n=119). (F) Representative IHC staining indicates that xCT protein expression was upregulated and that miR-1297 levels were inversely associated with xCT in 119 primary human CRC specimens. Two representative cases are shown (scale bar: 100 µm). The percentages of CRC tissues that had low or high miR-1297 expression levels relative to xCT expression levels. Data in A and C are presented as the mean ± SD (n=3). *P < 0.05 versus control cells.
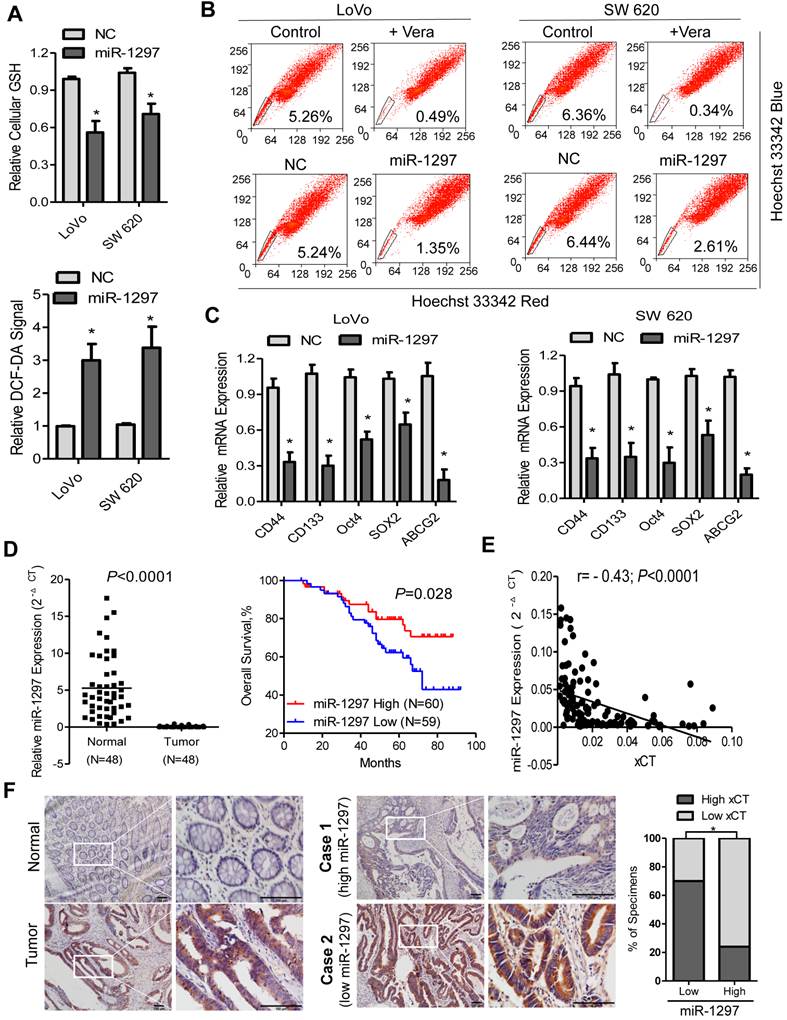
Furthermore, a qPCR analysis revealed that miR-1297 was downregulated in 48 CRC samples compared with the matched adjacent non-tumor tissues (Figure 4D). To evaluate whether the down-regulation of miR-1297 correlates with the clinical progression of CRC, we examined miR-1297 expression in 119 archived human CRC specimens. Patients with high miR-1297 levels had more favorable clinical outcomes, as shown in the Kaplan-Meier survival curves, while, patients with lower miR-1297 expression levels demonstrated shorter survival times (P=0.028; Figure 4D). Of note, a multivariate analysis showed that miR-1297 expression levels, metastasis and lymph node invasion were independent prognostic factors for CRC patients (P<0.001, P=0.007 and P=0.001, respectively; Table 1). In addition, the miR-1297 levels in the 119 CRC samples were inversely correlated with the expression of xCT mRNA (r = -0.43; P<0.0001), further confirming the inhibitory effects of miR-1297 on xCT in colorectal cancer (Figure 4E). To further investigate the correlation of miR-1297 and xCT in CRC, xCT expression was examined in 119 cases of CRC using IHC. As illustrated, xCT was markedly upregulated in CRC samples, but was only marginally detectable in normal colorectal tissues (Figure 4F). Statistical analyses revealed that miR-1297 levels were inversely correlated with the expression of xCT (Figure 4F), which is consistent with the qPCR analysis. These observations further support the notion that miR-1297 contributes to SP cells enrichment in CRC, which results in tumor aggressiveness and poorer clinical outcome.
Univariate and Multivariate Analyses of Various Potential Prognostic Factors in 119 CRC Patients.
| Univariate Analysis | Multivariate Analysis | |||
|---|---|---|---|---|
| Factors | HR (95% CI) | P value | HR (95% CI) | P value |
| Age | 2.31 (1.19-4.50) | 0.014 | 2.20(1.11-4.35) | 0.023 |
| Sex | 0.48 (0.24-0.96) | 0.038 | 0.45(0.21-0.94) | 0.035 |
| Histological grade | 1.50 (0.90-2.50) | 0.121 | - | - |
| Tumor size | 1.16 (0.63-2.16) | 0.634 | - | - |
| Tumor depth | 3.06 (1.28-7.30) | 0.012 | - | - |
| Lymph node invasion | 4.31 (2.11-8.34) | <0.001 | 3.85 (1.85-8.03) | <0.001 |
| Metastasis | 4.95 (2.47-9.91) | <0.001 | 3.59(1.73-7.48) | 0.001 |
| TNM stage | 4.95 (2.27-10.79) | <0.001 | - | - |
| miR-1297 Expression | 0.49 (0.26-0.95) | 0.028 | 0.39 (0.19-0.77) | 0.007 |
*P < 0.05. HR, hazard ratio; CI, confidence interval.
In vivo inhibition of tumor formation with pretreatment of CRC cells with PEITC through redox modulation in vitro
CSCs have been shown to be intrinsically resistant to many anticancer agents, which mean that new strategies are required to eliminate these cells. Because SP cells are highly dependent on GSH levels, we speculated that this subpopulation might be sensitive to redox changes. PEITC is a compound that effectively conjugates with GSH to export it from cells [14, 23]. Whether it could kill CRC SP cells by altering their redox status was assessed. As shown, the incubation of CRC cells with 5 μM PEITC for 6 h significantly depleted cellular GSH content, which led to an increase in the cellular ROS level (Figure 5A). Significantly, treating LoVo cells with equally toxic concentrations of PEITC and the conventional chemotherapeutic agent 5FU showed a striking difference in their impact on CRC SP cells. As shown in Figure 5B, PEITC at its IC50 concentration (5.83±0.27μM, Figure S4A) significantly reduced the proportion of SP cells from 6.11% to 1.57% in 48 h. An equally toxic concentration of 5FU (IC50=4.01±0.23 μM, Figure S4B) caused a substantial increase in the proportion of SP cells (from 6.11% to 11.58%), which indicated that 5FU killed non-SP cells and enriched CRC SP cells. Similar results were also observed in SW620 cells. Moreover, a qPCR analysis showed that the treatment of LoVo and SW620 cells with PEITC caused a significant decrease in the mRNA expression levels of multiple CSCs markers, which is consistent with the ability of PEITC to decrease the proportion of CRC SP cells (Figure 5C). However, the 5FU treatment showed the opposite results (Figure S4C). Our results also show that the proportion of SP cells was significantly increased in the 5FU-treated CRC cells, but decreased in PEITC-treated CRC cells after the 48h drug-free incubation for cell death (Figure S4D), indicating that 5FU firstly kills non-SP cells and PEITC are more effective in killing SP cells. Sulfasalazine has been widely reported as a potent inhibitor of xCT [24]. Consistent with this, our results showed that incubation with SSZ (100 μM or 300 μM) resulted in a significant depletion of GSH contents and an accumulation of ROS levels in SW 620 and LoVo cells (Figure S5A). Also, the percentage of SP cells was significantly reduced in these cells after SSZ treatment (Figure S5B). These finding indicate that xCT is a candidate for use as a therapeutic target in reducing CRC SP cells.
Pre-treatment of CRC cells with PEITC in vitro caused inhibition of tumor formation in vivo. (A) Depletion of cellular GSH and increased ROS levels in response to PEITC in both LoVo and SW620 cells as measured by spectrophotometric and flow cytometric analysis, respectively. (B) CRC cells were pretreated with the IC50 concentrations of PEITC and 5FU for 48 h. They were then washed and cultured in fresh medium without either drug for 48 h to allow cell death to occur. Viable cells were harvested and then stained with Hoechst 33342 to identify SP cells. (C) CD44, CD133, Oct4, ABCG2 and SOX2 expression levels were analyzed via qPCR in the indicated cells. (D) Experimental design as described in B. Equal numbers of control and drug-treated cells were inoculated subcutaneously into the flanks of athymic mice using 1.0x105 cells/injection site (Group 1) or 2.0x105 cells/injection site (Group 2). (E) Images of tumors from representative mice in each group on day 14. Measurement of tumor volumes was carried out on the indicated days. The right panel shows the mean tumor weights in each group on day 50. Data in A and C are presented as the mean±SD (n=3). *P < 0.05 versus control cells.
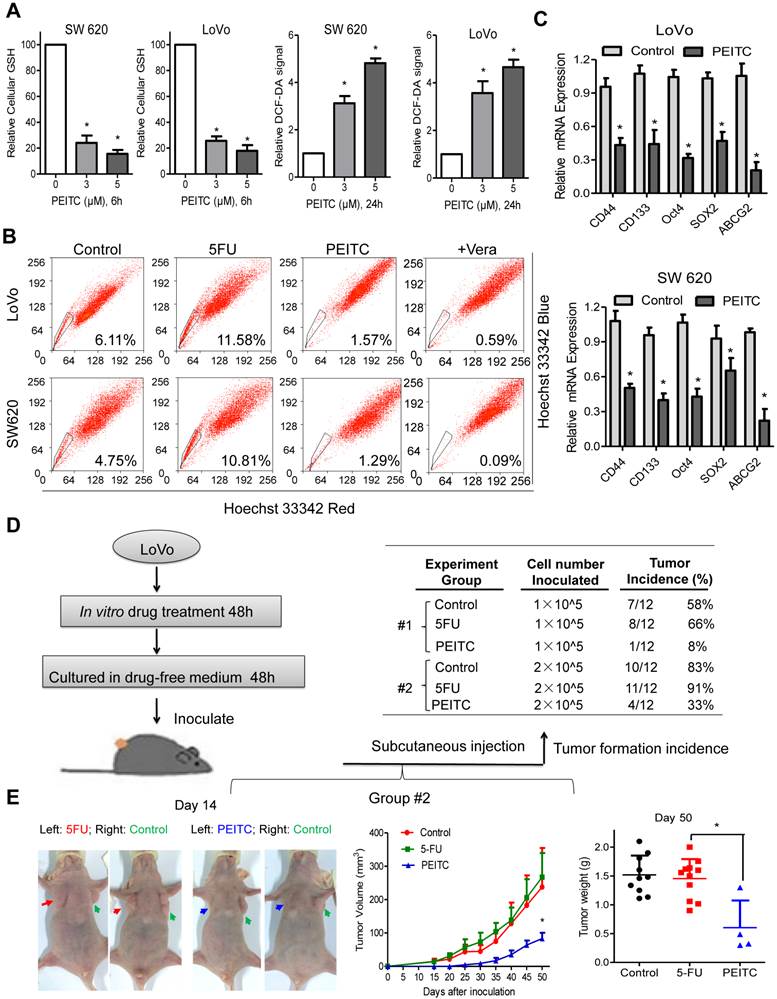
Because PEITC reduced CRC SP cell populations and 5FU increased them, we assessed the effect of PEITC and 5FU on LoVo cells forming tumors in vivo. As shown in Figure 5D, equal numbers of viable cells were subcutaneously inoculated into the flanks of athymic mice. Observation of tumor formation in these mice were performed without further drug treatment. The results show that about half of the mice in the control and 5FU-treated groups developed tumors when the inoculated cell number was 1x105 cells (Group #1). PEITC pre-treatment reduced the tumor incidence to 8%. Most of the mice in the control group and 5FU-treated group developed tumors when the mice were inoculated with 2x105 cells (Group #2). The tumor incidence in the PEITC-treated group was dramatically reduced to 30% (Figure 5D). Consistently, tumor growth was retarded substantially and tumor weight was decreased significantly by pre-treatment with PEITC. In contrast, since 5FU primarily kills non-SP cells, the remaining cells were mainly SP cells and showed a higher tumor incidence when equal numbers of viable cells were inoculated (Figure 5E). LoVo cells were treated with 5FU or PEITC at equal cytotoxic concentrations, however, the decrease in tumor formation that was observed in the PEITC-treated group suggests that PEITC was effective in killing CRC SP cells.
PEITC enhanced the 5FU-induced cytotoxic effects in CRC cell lines and in a xenograft mouse model
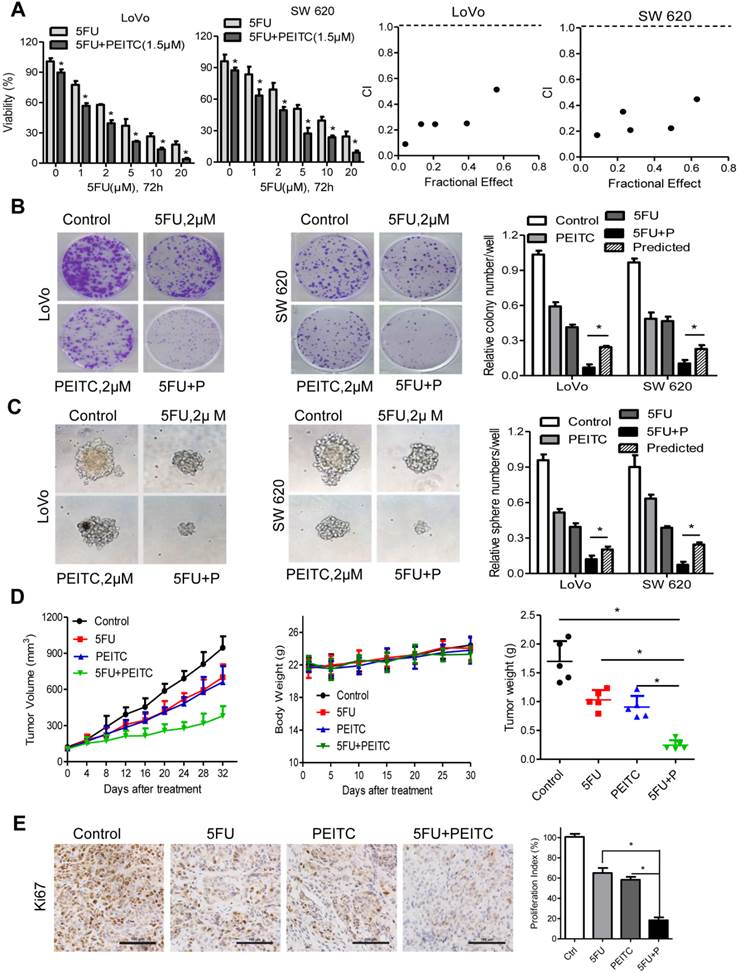
To test our hypothesis, based on the above findings that 5FU administered in combination with PEITC may be a useful therapeutic strategy for treating colorectal cancer, we examined the antitumor capability of PEITC plus 5FU in CRC cell lines and in a xenograft mouse model. MTS analysis demonstrated that PEITC significantly enhanced 5FU's cytotoxicity in CRC cells. The results show a synergistic interaction between 5FU and PEITC, as demonstrated by a consistent CI value of less than 1.0 (Figure 6A, S4A). A colony formation assay was also used to test the effect of this combination on CRC cell growth. Either agent alone caused some delays in cell growth in LoVo and SW620 cells, but together 5FU and PEITC showed a synergistic effect in inhibiting colony formation compared with the calculated/predicted additive value [25, 26] (Figure 6B). Similarly, the sphere formation assay also confirmed that the combination of 5FU and PEITC led to synergistic cytotoxic effects on CRC SP cells (Figure 6C). Finally, we determined whether PEITC plus 5FU at clinically relevant doses can inhibit the growth of LoVo cells in a xenograft nude mouse model. Twenty mice were subcutaneously injected with LoVo cells and randomly assigned to one of four groups (n = 5 per group). Treatment with 5FU, PEITC, both agents, or a vehicle control began 2 weeks after the injection and lasted for 4 weeks. Compared with the control group, after 4 weeks of treatment, the groups treated with 5FU or PEITC alone showed moderate effects. However, the mice treated with both agents experienced a large reduction in tumor burden, which was evident by the redused tumor growth curve and tumor weight (Figure 6D). No significant weight loss or other signs of acute or delayed toxicity were observed in any mouse during the treatment period (Figure 6D). Also, the combination of 5FU and PEITC resulted in the greater decrease in cell proliferation indices (Ki67-positive) when compared with the effects of treatment with 5FU or PEITC alone, as detected via IHC staining (Figure 6D). The IHC analysis also showed that the expression level of CD44 was markedly elevated in 5FU-treated tumors but was decreased in the tumors treated with PEITC alone or combined with 5FU (Figure S5C). Overall, the combination of PEITC and 5FU exhibited a highly synergistic therapeutic activity against the colorectal cancer cells in vitro and in vivo.
PEITC enhanced the 5FU-induced cytotoxic effects in CRC cell lines and in xenograft mouse models. (A) LoVo or SW 620 cells were incubated with various concentrations of 5FU and with or without PEITC (1.5 μM) for 72 h. The effect on cell viability was determined via MTS assay. A combination index (CI) of 5FU and PEITC treatment in CRC cells was analyzed using a median dose-effect method with CalcuSyn software (Biosoft). CI = 1 indicates an additive effect, CI < 1 indicates a synergistic effect and CI > 1 indicates an antagonist effect. (B) LoVo and SW620 cells were incubated with 5FU, PEITC, or both for 2 weeks, and the cell colonies were fixed in formalin, stained with crystal violet, and counted. (C) CRC cells were plated in 6-well plates and cultured in CSC medium with 5FU, PEITC, or both for 7 days, and the numbers of spheres formed were quantified. The predicted value was calculated by multiplying the relative colony/tumor sphere numbe in the 5FU-treated and PEITC-treated sample. The combination effect is considered additive when the observed value is equal to the predicted value. When observed value is less than the predicted value, the combination effect is considered as synergistic. (D) The volume of the tumors and the weight of mice were measured and recorded, and a tumor growth curve was created for each group. (E) Paraffin-embedded tumor sections were stained with anti-Ki67 antibody (scale bar: 100 μm), the proliferation index was quantified. Data in A, B and C are presented as the mean±SD (n=3). *P < 0.05 versus control cells.
Discussion
It has been known for a long time that cancer cells exhibit increased ROS levels and have an altered redox status, which has been associated with active metabolism and oncogenic stimulation. Increased levels of ROS may play an important role in maintaining cancer cell phenotypes and are considered unfavorable because of their stimulatory effects on genetic instability, cell proliferation and metastasis. Recent studies have suggested that CSCs may actually have lower ROS levels than non-tumorigenic cells and that ROS levels might affect CSCs [11, 27]. However, the redox status and its regulation in CRC CSCs remains largely unknown. Consistent with previous findings, our current study shows that CRC SP cells have a high antioxidant capacity that can keep ROS at moderate levels and maintain both the stemness and cancer-forming capabilities of CSCs, which might contribute to CSC survival and drug resistance. We also used multiple analyses to investigate the underlying mechanisms in which GSH plays a major role in maintaining the CSC phenotype through the miR-1297-xCT-CD44v axis in CRC cells (Figure 7). Based on these finding, we suggest a novel therapeutic strategy to kill CRC SP cells by disrupting their redox status using the natural compound PEITC.
Proposed model for the regulation of the redox status in SP cells through the CD44v-xCT axis in CRC and its therapeutic implications. The GSH-mediated reduction incellular ROS levels is an essential regulator of CRC SP cells mediated by the CD44v-xCT axis, and redox modifications caused by the natural compound PEITC significantly reduced the chemotherapy-resistant CRC SP cells. The combination of PEITC and 5FU exhibited highly synergistic cytotoxic effects in vitro and in vivo, which merits further testing in pre-clinical and clinical trials.
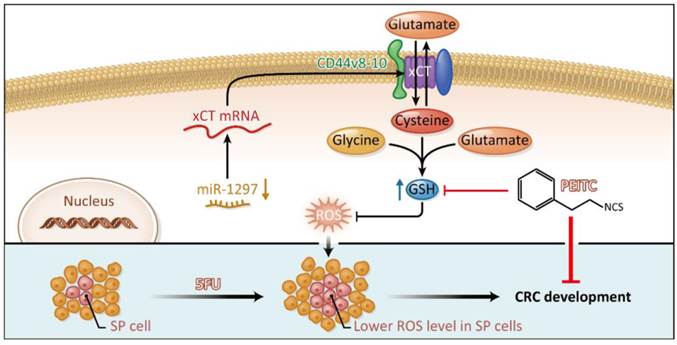
Cancer cell redox adaptations contribute to cell survival under the conditions of persistent oxidative stress that result from their intrinsically active metabolism and from extrinsic factors in the microenvironment. These adaptations also result in resistance to some anticancer agents. Our study shows that redox adaptations in CRC SP cells may be potentiated because of the enhanced antioxidant capacity related to GSH content. GSH is biosynthesized from the amino acids cysteine, glycine and glutamate, which are transported by LAT1, ASCT2 and xCT [28]. Cysteine acts as a rate-limiting factor in GSH biosynthesis in cells because the sulfhydryl group in cysteine acts as a proton donor, which is responsible for its biological activity [29]. Consistently, in the current study, we proved that xCT-mediated cysteine uptake plays a significant role in GSH synthesis and CRC SP cell enrichment. Furthermore, we found xCT was mainly regulated by miR-1297 and CD44v at the mRNA and protein levels, respectively. The role of glutamate, glycine and their transporters in CRC SP cells, and the underlying regulatory mechanisms involved, however, still need further clarification.
CD44 and its variant isoforms have been implicated in multiple steps required for cancer metastasis, including matrix invasion, extravasation and colonization [30]. Previous study has shown that CD44 v8-10 stabilizes the xCT subunit of system xc(-) in the plasma membrane and thereby promotes tumor growth in CRC cells [13]. CD44v8-10 expression levels increase xCT-dependent ROS defenses and thereby allow cancer cells to evade metastatic stress [13]. ESRP-regulated alternative splicing of CD44 mRNA results in CD44v expression in metastatic cancer cells [31]. xCT is overexpressed in CRC cells and tissues, and its expression level correlates with disease recurrence [32, 33]. CSCs are thought to be responsible for tumor initiation and metastasis [4]. Yet, as far as we know, the effect of the CD44-xCT axis on CRC stem-like SP cells has not been clarified. We showed that both CD44v8-10 mRNA and xCT protein levels were increased in CRC SP cells than non-SP cells. This resulted in the promotion of GSH synthesis and the enrichment of CRC SP cells. In addition, the RNAi-mediated destruction of CD44v reduced the cell surface expression of xCT, thus, depleting the GSH content and decreasing the proportion of SP cells present. Therefore, targeting the CD44v-xCT system may impair the ability of CRC CSCs to defend against ROS. This may sensitize them to currently available treatments.
Many studies have revealed that miRNAs can influence colorectal tumor growth, angiogenesis, and metastasis and can effect diagnosis and prognosis of CRC [20, 22, 34]. Our previous study indicated that the down-regulation of miR-214 led to the up-regulation of its target FGFR1, which led to CRC proliferation and metastasis [35]. In this study, we also investigated the post-transcriptional regulation of xCT beyond the role of CD44v in stabilizing its protein level in the plasma membrane. It has been reported that miR-1297 acts as a tumor suppressor in lung cancer [21]. MiR-1297 also inhibits CRC cellular proliferation, migration, and invasion in vitro, as well as tumorigenesis in vivo, by targeting Cox-2 [22]. Here, we report that xCT is a direct target of miR-1297. Also, reduced miR-1297 expression contributes to CRC SP cell enrichment through redox modification, which results in tumor aggressiveness and poor clinical outcomes. ATF4 and Nrf2 have been reported to enhance the expression of xCT in cancer cells [36]. However, our further study demonstrated that miR-1297 expression was independent of ATF4- and Nrf2-mediated signaling pathways (data not shown). Therefore, we believe that miR-1297 might be useful as a diagnostic biomarker to assess the stage of CRC or as a predictive biomarker to determine the outcome of CRC. It may also serve as a therapy tool to neutralize xCT overexpression, thereby terminating malignant tumor proliferation.
CSCs are not only critical in the initiation, development, metastasis, and recurrence of tumors, but they also play roles in the ineffectiveness of conventional cancer therapies [37, 38]. In agreement with previous reports, our data show that the classic chemotherapeutic agent 5FU was able to eradicate the bulk of the CRC cells, but not the CSCs, which may make CRC resistant to this drug. Based on the redox status and regulation in CRC SP cells, we reasoned that modulating the redox regulatory mechanisms by reducing the GSH pool may affect CRC SP cell survival. The natural compound PEITC, which has been reported to deplete GSH and subsequently to elevate oxidative stress [35, 39], can not only eliminate the drug-resistant cell population, but can also kill CSCs [40]. The rapid depletion of GSH with PEITC was effective in killing CRC SP cells, which is in agreement with the above studies. Also, redox modifications caused by PEITC significantly reduced the CRC SP cells in vitro and impaired their ability to form tumors in vivo. The combination of PEITC and 5FU exhibited highly synergistic cytotoxic effects on CRC cell lines and in a xenograft mouse model. Based on our results, it seems logical to combine standard chemotherapeutic agents with redox-modifying compounds such as PEITC to simultaneously eliminate the bulk of the cancer cells along with the SP cells. This novel therapeutic strategy could potentially improve the outcomes of patients undergoing cancer treatments, and it merits further testing in pre-clinical and clinical trials.
Supplementary Material
Supplementary method and figures.
Abbreviations
CRC, colorectal cancer; SP, side population; CSC, cancer stem cells; ROS, reactive oxygen species; GSH, glutathione; PEITC, β-phenylethyl isothiocyanate; CD44v, CD44 variant; NAC, N-acetylcysteine; Vera, verapamil.
Acknowledgements
This research was supported by the National Natural Science Foundation of China (No.81372570, 81572392), the Natural Science Foundation of Guangdong Province (No.2014A030312015) and by grants from the China Postdoctoral Science Foundation (No.2015M570746).
Conflicts of Interest
No potential conflicts of interest were disclosed.
References
1. Siegel R, Desantis C, Jemal A. Colorectal cancer statistics, 2014. CA Cancer J Clin. 2014;64:104-17
2. Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin. 2014;64:9-29
3. Zhou Q, Li K, Lin GZ, Shen JC, Dong H, Gu YT. et al. Incidence trends and age distribution of colorectal cancer by subsite in Guangzhou, 2000-2011. Chin J Cancer. 2015;34:34
4. Clarke MF, Dick JE, Dirks PB, Eaves CJ, Jamieson CH, Jones DL. et al. Cancer stem cells-perspectives on current status and future directions: AACR Workshop on cancer stem cells. Cancer Res. 2006;66:9339-44
5. Wu C, Alman BA. Side population cells in human cancers. Cancer Lett. 2008;268:1-9
6. Wu C, Wei Q, Utomo V, Nadesan P, Whetstone H, Kandel R. et al. Side population cells isolated from mesenchymal neoplasms have tumor initiating potential. Cancer Res. 2007;67:8216-22
7. Pervaiz S, Taneja R, Ghaffari S. Oxidative stress regulation of stem and progenitor cells. Antioxid Redox Signal. 2009;11:2777-89
8. Zhou D, Shao L, Spitz DR. Reactive oxygen species in normal and tumor stem cells. Advances in cancer research. 2014;122:1-67
9. Sauer H, Wartenberg M. Reactive oxygen species as signaling molecules in cardiovascular differentiation of embryonic stem cells and tumor-induced angiogenesis. Antioxid Redox Signal. 2005;7:1423-34
10. Ji AR, Ku SY, Cho MS, Kim YY, Kim YJ, Oh SK. et al. Reactive oxygen species enhance differentiation of human embryonic stem cells into mesendodermal lineage. Exp Mol Med. 2010;42:175-86
11. Diehn M, Cho RW, Lobo NA, Kalisky T, Dorie MJ, Kulp AN. et al. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature. 2009;458:780-3
12. Yuan S, Lu Y, Yang J, Chen G, Kim S, Feng L. et al. Metabolic activation of mitochondria in glioma stem cells promotes cancer development through a reactive oxygen species-mediated mechanism. Stem Cell Res Ther. 2015;6:198
13. Ishimoto T, Nagano O, Yae T, Tamada M, Motohara T, Oshima H. et al. CD44 variant regulates redox status in cancer cells by stabilizing the xCT subunit of system xc(-) and thereby promotes tumor growth. Cancer Cell. 2011;19:387-400
14. Ju HQ, Gocho T, Aguilar M, Wu M, Zhuang ZN, Fu J. et al. Mechanisms of Overcoming Intrinsic Resistance to Gemcitabine in Pancreatic Ductal Adenocarcinoma through the Redox Modulation. Mol Cancer Ther. 2015;14:788-98
15. Goodell MA, Brose K, Paradis G, Conner AS, Mulligan RC. Isolation and functional properties of murine hematopoietic stem cells that are replicating in vivo. J Exp Med. 1996;183:1797-806
16. Chang Z, Ju H, Ling J, Zhuang Z, Li Z, Wang H. et al. Cooperativity of oncogenic K-ras and downregulated p16/INK4A in human pancreatic tumorigenesis. PLoS One. 2014;9:e101452
17. Ju HQ, Wang SX, Xiang YF, Liu Z, Liu JY, Chen ZP. et al. BJ-B11, a novel Hsp90 inhibitor, induces apoptosis in human chronic myeloid leukemia K562 cells through the mitochondria-dependent pathway. Eur J Pharmacol. 2011;666:26-34
18. Collins AT, Berry PA, Hyde C, Stower MJ, Maitland NJ. Prospective identification of tumorigenic prostate cancer stem cells. Cancer Res. 2005;65:10946-51
19. Dalerba P, Dylla SJ, Park IK, Liu R, Wang X, Cho RW. et al. Phenotypic characterization of human colorectal cancer stem cells. Proc Natl Acad Sci U S A. 2007;104:10158-63
20. Hur K, Toiyama Y, Schetter AJ, Okugawa Y, Harris CC, Boland CR. et al. Identification of a metastasis-specific MicroRNA signature in human colorectal cancer. J Natl Cancer Inst. 2015:107
21. Zhang C, Chi YL, Wang PY, Wang YQ, Zhang YX, Deng J. et al. miR-511 and miR-1297 inhibit human lung adenocarcinoma cell proliferation by targeting oncogene TRIB2. PLoS One. 2012;7:e46090
22. Chen P, Wang BL, Pan BS, Guo W. MiR-1297 regulates the growth, migration and invasion of colorectal cancer cells by targeting cyclo-oxygenase-2. Asian Pac J Cancer Prev. 2014;15:9185-90
23. Trachootham D, Zhou Y, Zhang H, Demizu Y, Chen Z, Pelicano H. et al. Selective killing of oncogenically transformed cells through a ROS-mediated mechanism by beta-phenylethyl isothiocyanate. Cancer Cell. 2006;10:241-52
24. Gout PW, Buckley AR, Simms CR, Bruchovsky N. Sulfasalazine, a potent suppressor of lymphoma growth by inhibition of the x(c)- cystine transporter: a new action for an old drug. Leukemia. 2001;15:1633-40
25. Hu Y, Lu W, Chen G, Zhang H, Jia Y, Wei Y. et al. Overcoming resistance to histone deacetylase inhibitors in human leukemia with the redox modulating compound beta-phenylethyl isothiocyanate. Blood. 2010;116:2732-41
26. Wu WJ, Zhang Y, Zeng ZL, Li XB, Hu KS, Luo HY. et al. beta-phenylethyl isothiocyanate reverses platinum resistance by a GSH-dependent mechanism in cancer cells with epithelial-mesenchymal transition phenotype. Biochem Pharmacol. 2013;85:486-96
27. Sato A, Okada M, Shibuya K, Watanabe E, Seino S, Narita Y. et al. Pivotal role for ROS activation of p38 MAPK in the control of differentiation and tumor-initiating capacity of glioma-initiating cells. Stem Cell Res. 2013;12:119-31
28. Pompella A, Visvikis A, Paolicchi A, De Tata V, Casini AF. The changing faces of glutathione, a cellular protagonist. Biochem Pharmacol. 2003;66:1499-503
29. Conrad M, Sato H. The oxidative stress-inducible cystine/glutamate antiporter, system x (c) (-): cystine supplier and beyond. Amino Acids. 2012;42:231-46
30. Ponta H, Sherman L, Herrlich PA. CD44: from adhesion molecules to signalling regulators. Nat Rev Mol Cell Biol. 2003;4:33-45
31. Yae T, Tsuchihashi K, Ishimoto T, Motohara T, Yoshikawa M, Yoshida GJ. et al. Alternative splicing of CD44 mRNA by ESRP1 enhances lung colonization of metastatic cancer cell. Nat Commun. 2012;3:883
32. Sugano K, Maeda K, Ohtani H, Nagahara H, Shibutani M, Hirakawa K. Expression of xCT as a predictor of disease recurrence in patients with colorectal cancer. Anticancer Res. 2015;35:677-82
33. Ma MZ, Chen G, Wang P, Lu WH, Zhu CF, Song M. et al. Xc inhibitor sulfasalazine sensitizes colorectal cancer to cisplatin by a GSH-dependent mechanism. Cancer Lett. 2015
34. Verma AM, Patel M, Aslam MI, Jameson J, Pringle JH, Wurm P. et al. Circulating plasma microRNAs as a screening method for detection of colorectal adenomas. Lancet. 2015;385(Suppl 1):S100
35. Chen DL, Wang ZQ, Zeng ZL, Wu WJ, Zhang DS, Luo HY. et al. Identification of microRNA-214 as a negative regulator of colorectal cancer liver metastasis by way of regulation of fibroblast growth factor receptor 1 expression. Hepatology. 2014;60:598-609
36. Ye P, Mimura J, Okada T, Sato H, Liu T, Maruyama A. et al. Nrf2- and ATF4-dependent upregulation of xCT modulates the sensitivity of T24 bladder carcinoma cells to proteasome inhibition. Mol Cell Biol. 2014;34:3421-34
37. Wilson BJ, Schatton T, Frank MH, Frank NY. Colorectal Cancer Stem Cells: Biology and Therapeutic Implications. Curr Colorectal Cancer Rep. 2011;7:128-35
38. Liao K, Xia B, Zhuang QY, Hou MJ, Zhang YJ, Luo B. et al. Parthenolide inhibits cancer stem-like side population of nasopharyngeal carcinoma cells via suppression of the NF-kappaB/COX-2 pathway. Theranostics. 2015;5:302-21
39. Zhang H, Trachootham D, Lu W, Carew J, Giles FJ, Keating MJ. et al. Effective killing of Gleevec-resistant CML cells with T315I mutation by a natural compound PEITC through redox-mediated mechanism. Leukemia. 2008;22:1191-9
40. Wang D, Upadhyaya B, Liu Y, Knudsen D, Dey M. Phenethyl isothiocyanate upregulates death receptors 4 and 5 and inhibits proliferation in human cancer stem-like cells. BMC Cancer. 2014;14:591
Author contact
![]() Corresponding author: Prof. Rui-Hua Xu, M.D. Ph.D.; Address: 651 Dongfeng East Road, Guangzhou 510060, China; Tel: +86-20-8734-3228; Fax: +86-20-8734-3392; E-mail: xurhorg.cn.
Corresponding author: Prof. Rui-Hua Xu, M.D. Ph.D.; Address: 651 Dongfeng East Road, Guangzhou 510060, China; Tel: +86-20-8734-3228; Fax: +86-20-8734-3392; E-mail: xurhorg.cn.