Theranostics
13.3
Impact Factor
- Current Issue
- Advance Articles
- Volume 16; 2026
- Volume 15; 2025
- Volume 14; 2024
- Volume 13; 2023
- Volume 12; 2022
- Archive
- Cover Images
- Cover Suggestion
- Special Issues
Top
Introduction
Materials and Methods
Results
Discussion
Supplementary Material
Acknowledgements
References
Introduction
Materials and Methods
Results
Discussion
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2016; 6(8):1205-1219. doi:10.7150/thno.15083 This issue Cite
Research Paper
Gas6/Axl Axis Contributes to Chemoresistance and Metastasis in Breast Cancer through Akt/GSK-3β/β-catenin Signaling
Cun Wang1*, Haojie Jin1*, Ning Wang1*, Shaohua Fan2*, Yanyan Wang3, Yurong Zhang1, Lin Wei1, Xuemei Tao1, Dishui Gu1, Fangyu Zhao1, Jingyuan Fang1, Ming Yao1, Wenxin Qin1 ![]()
1. State Key Laboratory of Oncogenes and Related Genes, Shanghai Cancer Institute, Renji Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai, China;
2. Department of Biotechnology, School of Life Science, Jiangsu Normal University, Xuzhou, China;
3. Department of Oncology, The Affiliated City Hospital of Xuzhou Medical College, Xuzhou, China.
*These authors contribute equally to this work.
Received 2016-1-25; Accepted 2016-4-20; Published 2016-5-24
Citation:
Wang C, Jin H, Wang N, Fan S, Wang Y, Zhang Y, Wei L, Tao X, Gu D, Zhao F, Fang J, Yao M, Qin W. Gas6/Axl Axis Contributes to Chemoresistance and Metastasis in Breast Cancer through Akt/GSK-3β/β-catenin Signaling. Theranostics 2016; 6(8):1205-1219. doi:10.7150/thno.15083. https://www.thno.org/v06p1205.htm
Other stylesAbstract

Chemoresistance in breast cancer has been of great interest in past studies. However, the development of rational therapeutic strategies targeting chemoresistant cells is still a challenge in clinical oncology. By integrating data from global differences of gene expression and phospho-receptor tyrosine kinases between sensitive parental cells (MCF-7) and doxorubicin-resistant cells (MCF-7/ADR), we identified Axl as a potential target for chemoresistance and metastasis in multidrug resistant breast cancer cells. We analyzed Axl expression in 57 breast cancer cell lines and detected a dramatic increase in its expression level in mesenchymal breast cancer cell lines. Axl silencing suppressed invasive and metastatic potentials of chemoresistant breast cancer cells as well as increased elimination of cancer cells when combined with doxorubicin. Furthermore, in preclinical assays, an Axl inhibitor R428 showed increased cell death upon doxorubicin treatment. Additionally, using phospho-kinase array based proteomic analysis, we identified that Akt/GSK-3β/β-catenin cascade was responsible for Axl-induced cell invasion. Nuclear translocation of β-catenin then induced transcriptional upregulation of ZEB1, which in turn regulated DNA damage repair and doxorubicin-resistance in breast cancer cells. Most importantly, Axl was correlated with its downstream targets in tumor samples and was associated with poor prognosis in breast cancer patients. These results demonstrate that Gas6/Axl axis confers aggressiveness in breast cancer and may represent a therapeutic target for chemoresistance and metastasis.
Keywords: Axl, EMT, R428, drug-resistance, breast cancer.
Introduction
Breast cancer is the most frequently diagnosed malignancy and the first leading cause of cancer-related deaths among females, with an estimated 1.7 million annual diagnoses and half-million annual deaths worldwide [1]. Chemotherapeutics are important treatments for breast cancer. The commonly used chemotherapeutic drugs include 5-fluorouracil, doxorubicin (Dox), cyclophosphamide, paclitaxel, and docetaxel [2-4]. Although improvements in surgical operation and the combination of systemic treatment drugs contribute to decrease in mortality rates, a large number of breast cancer patients still inexorably experience multidrug resistance and develop the recurrent disease in the next several years.
The most recognized mechanism of multidrug resistance is the overexpression of cell surface efflux pumps including P-glycoprotein and MDR-associated proteins [5]. Accumulating evidence has indicated that acid tumor microenvironment caused by defective pH regulation effectively reduces the uptake of drugs and induces drug resistance [6, 7]. Our previous study showed that LASS2 could enhance chemosensitivity of breast cancer by counteracting acidic tumor microenvironment through inhibiting the activity of V-ATPase proton pump [8]. It has also been described that increased proportion of cells with cancer stem cell characteristics after prolonged selection of chemotherapy may contribute to the emergence of multidrug resistance [9]. Despite these advances, multidrug resistance remains a critical clinical challenge for successful chemotherapy.
Tyro3, Axl, and Mer belong to the TAM family of receptor tyrosine kinases (RTKs), which are the central regulators of various signaling pathways implicated in cancer [10]. Upon binding with its ligand, growth arrest-specific protein 6 (Gas6), Axl undergoes dimerization and autophosphorylation which subsequently activates downstream signaling pathways, including ERK, PI3K/Akt, p38, NF-ĸB, RHO family proteins, JAK-STAT and SRC family kinases [11]. It has been reported that Axl overexpression correlates with poor prognosis in a wide range of cancers [12-14]. Increasing evidence shows that Axl plays a critical role in metastasis in a variety of tumor types [13, 15, 16]. Kirane A et al found that Gas6-induced Axl signaling is a critical for pancreatic cancer progression and its inhibition with warfarin may improve outcome of the patients [17]. It has been also indicated that Axl is a potential therapeutic target for renal cell carcinoma and head and neck squamous cell carcinoma [13, 18]. In breast cancer, Axl represents a downstream effector of epithelial to mesenchymal transition (EMT), which is believed to be a requirement for cancer metastasis [19]. Antagonizing Axl signaling by pharmacologic inhibition or RNA interference suppresses pulmonary metastasis [20, 21]. Recently, it has been reported that Axl receptor mediates cancer cell resistance to multiple targeted drugs (ALK inhibitor [22], EGFR inhibitors [23-25], BRAF inhibitor [26], ERK inhibitor [26], PI3Kα inhibitor [27], or antiangiogenic therapy [28]). Axl also leads to chemoresistance in several cancer types [29, 30]. Targeting Axl pathway with specific antibody or small molecule inhibitor alone or in combination with other drugs can suppress Axl-mediated signaling pathways and improve therapeutic efficacy [31]. In breast cancer, Axl diversifies EGFR signaling and limits the response to EGFR-targeted inhibitors [32]. Activation of Axl has been identified as a mechanism of lapatinib resistance in HER2-positive breast cancer cells [33]. However, the functional attributes, downstream mechanisms, and potential therapeutic significance of Axl in acquired multidrug resistance in breast cancer remain unclear.
To elucidate novel mechanisms of chemoresistance in breast cancer, we performed microarray analysis of global gene expression and measured the activities of RTKs in MCF-7/ADR and parental MCF-7 cells. We report here a novel mechanism by which activation of Axl contributes to chemoresistance and EMT in breast cancer. Our findings establish a biological foundation for introducing inactivation of Axl to improve the activity of chemotherapeutic drugs. Our results potentially provide important translational implications to improve the efficiency of chemotherapy and clinical outcome in patients with breast cancer.
Materials and Methods
Cell culture
MCF-7 breast cancer cells (American Type Culture Collection, Manassas, VA, USA) and MCF-7/ADR cells were cultured in RPMI-1640 medium with 10% fetal calf serum (Sigma-Aldrich, St Louis, MO, USA), 100 IU/ml penicillin G, and 100 mg/ ml streptomycin sulfate (Gibco, Invitrogen, Carlsbad, CA, USA) at 37°C in a humidified 5% CO2 atmosphere. To maintain the resistance property, MCF-7/ADR cells were cultured in the presence of a low concentration of Dox (1 µg/ml) and passaged for 1 week in the drug-free medium before the experiments. The identities of the cell lines were confirmed by STR testing in 2013.
CCK8 assay
Cells were seeded in 96-well plates (4000 cells per well). Twenty-four hours after seeding, indicated concentrations of anti-cancer drugs were added to cells. Cells were then incubated for 24 h or 48 h with indicated anti-cancer drugs and cell viability was measured using Cell Counting Kit-8 assay (Dojindo Laboratories, Kumamoto, Japan) according to the manufacturer's instructions. Relative survival was normalized to the untreated controls after background subtraction.
Microarray analysis
For the analysis of gene expression profiles of MCF-7 and MCF-7/ADR cells, total RNA was prepared. Affymetrix Human U133 Plus 2.0 arrays were used according to the manufacturer's instructions. Gene expression levels of samples were normalized and analyzed with Microarray Suite, MicroDB, and Data Mining tool software (Affymetrix, Santa Clara, CA, USA).
Quantitative real-time PCR
Total RNA was extracted from cells using Trizol reagent (Invitrogen) and reversely transcribed using the PrimeScript™ RT Reagent Kit (TaKaRa Biotechnology). The real-time PCR was subsequently performed according to the manufacturer's instructions (TaKaRa Biotechnology). The expression levels were normalized against the internal reference gene GAPDH, and the relative expression levels were displayed using the 2-ΔΔCt method.
Immunofluorescent staining
Cells were grown on glass coverslips. After an attachment period of 24 h, cells were fixed in 4% paraformaldehyde for 30 min and permeabilized using 0.5% Triton X-100 for 10 min. After blocking with 10% donkey serum in PBS for 1 h, cells were incubated with the primary antibodies anti-E-cadherin (1:100 dilution, Proteintech), Keratin 19 (1:100 dilution, Abcam), N-cadherin (1:100 dilution, Proteintech), Vimentin (1:100 dilution, Proteintech), p-Axl (1:100, dilution, R&D Systems), β-catenin (1:200 dilution, Cell Signaling Technology), or γH2AX (1:200 dilution, Cell Signaling Technology) overnight at 4°C. After thorough washing, cells were then incubated with Alexa-Fluor 488 donkey anti-rabbit IgG (1:100 dilution, Life Technologies) for 30 min. Finally, cells were washed and stained with DAPI. Images were captured using an inverted fluorescent microscope (Olympus IX71).
In vitro migration and invasion assays
Cell migration was performed by the transwell assay (BD Biosciences, CA, USA). Briefly, 5 × 104 cells in serum-free RPMI-1640 were seeded on a membrane (8.0-μm pore size) inserted in the wells of a 24-well plate. RPMI-1640 containing 10% FBS was added to the lower chamber of each well. After 24 h, cells in the upper chamber were removed by cotton swab and the cells that had reached the underside of the membrane were fixed and stained with crystal violet (1% in methyl alcohol) for 10 min. The cells located on the underside of the filter (5 fields / filter) were counted. The cell invasion assay was carried out similarly, except that the matrigel (BD Biosciences, CA, USA) was added to each well 6 h before cells were seeded on the membrane. After 48 h, matrigel and any remaining cells in the upper chamber were removed by cotton swabs. Cells on the lower surface of the membrane were fixed and stained as described above.
Western blotting
Western blotting was performed as previously described [34]. Cell lysates were extracted with the T-PER tissue protein extraction reagent (Pierce, Rockford, IL) with a cocktail of proteinase inhibitors (Roche Applied Science, Switzerland) and a cocktail of phosphatase inhibitors (Roche Applied Science). Equal amounts of total proteins (20 μg) were separated by 10% SDS-PAGE and transferred onto PVDF membrane using a Bio-Rad SemiDry apparatus. After blocking for nonspecific binding, the membranes were incubated with anti-PARP (1:1000 dilution, Cell Signaling Technology), E-cadherin (1:1000 dilution, Proteintech), Keratin 19 (1:10000 dilution, Abcam), N-cadherin (1:1000 dilution, Proteintech), Vimentin (1:1000 dilution, Proteintech), p-Axl (1:200 dilution, R&D Systems), Axl (1:1000 dilution, Cell Signaling Technology), p-Akt (1:1000 dilution, Cell Signaling Technology), Akt (1:1000 dilution, Cell Signaling Technology), p-GSK3β (1:1000 dilution, Cell Signaling Technology), GSK3β (1:1000 dilution, Cell Signaling Technology), β-catenin (1:1000 dilution, Cell Signaling Technology), ZEB1 (1:1000 dilution, Cell Signaling Technology), γH2AX (1:1000 dilution, Cell Signaling Technology) or GAPDH (1:5000 dilution; Bioworld) overnight at 4°C, followed by HRP-conjugated secondary antibodies for 1 h at room temperature. After washing three times in TBST, protein bands were visualized using chemiluminescence detection.
Human Phospho-Receptor Tyrosine Kinase Array
Phospho-RTK analysis was performed using the human Phospho-RTK Arrays from R&D Systems (Minneapolis, USA). All steps were performed according to the manufacturer's instructions.
Bioinformatics analysis
Relative gene expression levels of Axl and EMT markers were acquired from the Cancer Cell Line Encyclopedia (CCLE) and The Cancer Genome Atlas (TCGA). The publicly available data were analyzed by Pearson's correlation analysis.
Gene knockdown using siRNA
Short interfering RNAs against Axl, Akt, β-catenin and ZEB1 (Biotend, Shanghai, China) were transfected into MCF-7/ADR or MCF-7 cells using Lipofectamine 2000 reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's instructions. The siRNA sequences used were listed in Supplementary Table 1.
Human phospho-kinase array
Phospho-kinase assays were utilized to analyze alterations of kinase signaling in response to Gas6 stimulation in MCF-7 cells according to the manufacturer's instructions (Proteome Profiler; R&D Systems, Minneapolis, MN, USA).
ChIP assays
ChIP assays were carried out using the EZ-ChIP kit (17-371, Millipore, Billerica, MA, USA) according to the manufacturer's instructions. Briefly, MCF-7/ADR cells were incubated for 10 min with 1% formaldehyde solution at room temperature, followed by incubation with glycine. The antibody or control IgG used for ChIP were as follows: rabbit anti-β-catenin (Cell Signaling Technology 8480), normal rabbit IgG (Cell Signaling Technology 2729). For the β-catenin/TCF binding site at position -161 of the human ZEB1 promoter, the primers amplifying the region between -325 and -101 were as follows: forward 5'-TTTACCTTTCCAACTCCGACAGC-3' and reverse 5-GGCTTTACGACATCACCTTCCTTAC-3. PCR products were analyzed on a 1.5% agarose gel by ethidium bromide staining.
Xenograft model and treatments
Six- to eight-week-old female BALB/c nude mice were housed under standard conditions and cared for according to protocols approved by the Shanghai Medical Experimental Animal Care Commission. 17β-estradiol pellets (NE-121, Innovative Research of America, Sarasota, FL, USA) were subcutaneously implanted into the back area of mice 3 days before tumor cell injection. For the tail vein metastasis models, MCF-7/ADR cells (2×106) stably expressing control or Axl shRNA were suspended in 200μl serum-free RPMI-1640 and injected into the tail vein of nude mice. After 30 days, lung metastasis was determined by examining serial sections of every lung tissue block by microscopy.
To assess the suppressive effects of the Axl inhibitor on metastasis, MCF-7/ADR cells (2×106) were suspended in 200μl serum-free RPMI-1640 and injected into the tail vein of nude mice. Then mice were randomly assigned to the vehicle-treated or R428 treated group (25 mg/kg, gavage, twice daily). After treatment for 30 days, all of the mice were sacrificed. The lung tissues were dissected and fixed in 10% formalin for at least 24 h. Lung tissues were subsequently analyzed by hematoxylin and eosin staining and lung metastasis was determined by examining serial sections of every lung tissue block by microscopy.
For the subcutaneously injected model, control or Axl-silenced MCF-7/ADR cells (5×106) were suspended in 100 μl serum-free RPMI-1640 and subcutaneously injected into the upper flank of each mouse. At day 10 after cell injection, mice were randomly divided into 4 groups: (1) sh-Control group, (2) sh-Axl group, (3) sh-Control + Dox-treated group, (4) sh-Axl + Dox-treated group. Mice in groups (3) and (4) were injected intraperitoneally with Dox (2mg/kg body weight; equivalent to a dose of 6.5 mg/m2 in patients). Mice were treated twice per week for 3 weeks. To assess the combined effects of Dox with Axl inhibitor in vivo, subcutaneous MCF-7/ADR xenografts were generated by injecting 5×106 cells into the upper flank of each mouse. At day 10 after cell injection, mice were randomly assigned to relative treatment groups consisting of vehicle-treated, Dox alone (Dox 2 mg/kg, i.p., twice weekly), R428 alone (25 mg/kg, gavage, twice daily) or the combination of Dox and R428. Tumor growth was measured every 5 days using Vernier calipers. Tumor volume was measured according to the formula: V = (a x b2)/2, where a and b are the maximal and minimal diameter in millimeters, respectively. At day 30 after cell injection, the mice were killed, and the tumors were weighed immediately after dissection. Immunohistochemical staining for p-Axl, Axl, p-Akt, p-GSK3β, β-catenin, ZEB1 and γH2AX was done using previously described methods [34]. Cell apoptosis was measured by TUNEL assay as previously described [34].
Clinical samples and immunohistochemistry staining
Fifty female patients, who had been diagnosed with breast cancer were treated at the Xuzhou First Renmin Hospital and received primary surgical treatment (either mastectomy or lumpectomy) without preoperative neoadjuvant therapy. After surgery, all the patients received four or more cycles of Dox-based adjuvant chemotherapy (cyclophosphamide / Dox / 5-FU; docetaxel / Dox / cyclophosphamide). Ethics approval was obtained from the local hospital's ethic committees and written consent was obtained from each patient before sample collection. The overall survival was defined as the length of time between the surgery and death. For surviving patients, the data were censored at the last follow-up. The disease-free survival was defined as the length of time between the date of the surgery and the date of detection of any tumor recurrence. Immunohistochemistry staining for p-Axl, β-catenin, and ZEB1 was done using previously described methods [35]. Immunohistochemical score was independently assessed by two pathologists without knowledge of patient characteristics.
Statistical analysis
Statistical analysis was carried out with SPSS software (Chicago, IL). Experimental data expressed as mean ± SD were analyzed by Student's t-test. All t-tests were conducted at the two-sided 0.05 level of significance. The relationship between expressions of indicated genes was analyzed by calculating Spearman's correlation coefficient.
Results
MCF-7/ADR cells show an EMT phenotype
The resistant property of MCF-7/ADR cells was confirmed by long term culture with Dox, cell viability, and PARP cleavage assays (Supplementary Figure 1A-C). Microarray analysis was performed to compare the global differences in gene expression between MCF-7 and MCF-7/ADR cells. This analysis revealed 3695 deregulated genes (fold change>2, P<0.01) in MCF-7/ADR cells, of which 51.5% were upregulated and 48.5% were downregulated (Figure 1A-B).
Figure 1
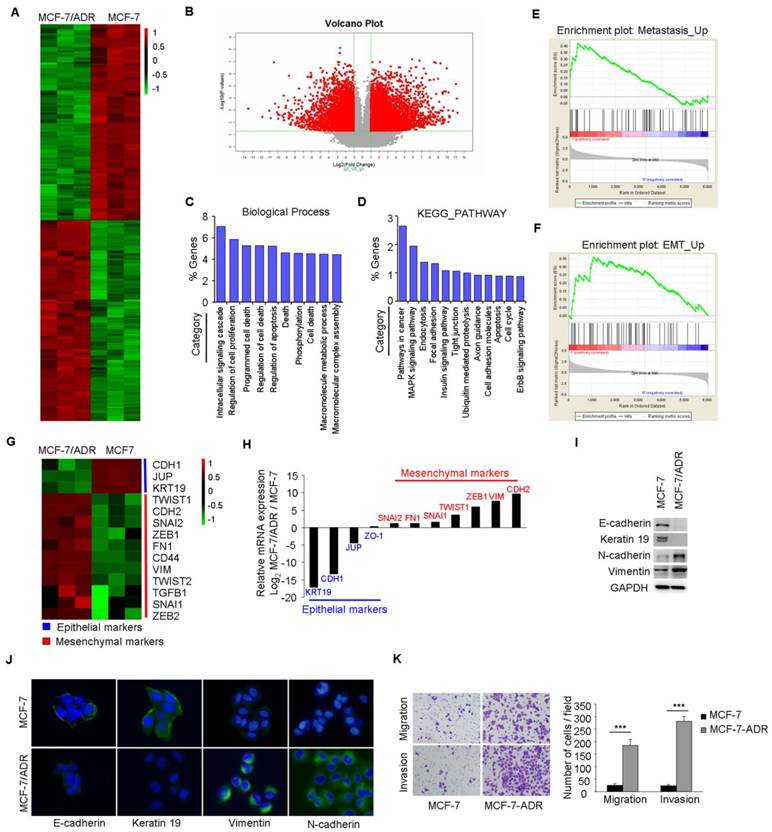
MCF-7/ADR cells exhibit an EMT phenotype. (A) Heatmap illustrated genes with at least 2-fold increase or decrease in transcript expression levels between parental and drug-resistant MCF-7 cells. (B) Volcano plot indicated the global differences of gene expression between MCF-7 and MCF-7/ADR cells. (C) Gene ontology categories of overlapping genes. (D) KEGG PATHWAY mapping showed the top 12 enriched pathways. (E-F) GSEA demonstrated a significant enrichment of gene signature associated with metastasis and EMT. (G) Heatmap illustrated the expression of EMT related genes in parental and Dox-resistant MCF-7 cells. (H) qRT-PCR indicated the downregulation of epithelial markers and the upregulation of mesenchymal genes in MCF-7/ADR cells. (I-J) The expression levels of epithelial markers (E-cadherin and Keratin 19) and mesenchymal markers (N-cadherin and Vimentin) in MCF-7 and MCF-7/ADR cells were compared by Western blotting and immunofluorescent staining. (K) Migratory and invasive behaviors were analyzed using migration and matrigel invasion assays for MCF-7 and MCF-7/ADR cells (magnification, ×200). Data are representative of results from three independent experiments. ***P<0.001.
Gene Ontology (GO) analysis of these deregulated genes indicated that, besides the expected changes in biological processes, such as cell death, cell proliferation, and apoptosis, other categories including intracellular signaling cascade and phosphorylation were significantly represented (Figure 1C). KEGG analysis also showed that several categories including pathways in cancer, cell adhesion, and apoptosis were deregulated in MCF-7/ADR cells (Figure 1D). A significant enrichment of gene signatures representative of metastasis and EMT were also observed in MCF-7/ADR cells (Figure 1E-F).
Microarray analysis showed significantly higher mRNA levels of mesenchymal markers (TWIST1, CDH2, SNAI2, ZEB1, FN1, CD44, VIM, TWIST2, TGFB1, SNAI1, ZEB2), but lower mRNA levels of epithelial markers (CDH1, KRT19, and JUP) in MCF-7/ADR cells than that in MCF-7 cells (Figure 1G). We subsequently used real-time RT-PCR to validate the expression profiles for 11 genes (KRT19, CDH1, JUP, ZO-1, SNAI2, FN1, SNAI2, TWIST1, ZEB1, VIM, and CDH2) associated with EMT. The trends of gene expression were similar between the microarray and real-time RT-PCR analysis (Figure 1H). Reduced expression levels of E-cadherin and Keratin 19 and increased levels of N-cadherin and Vimentin in MCF-7/ADR cells were further confirmed by Western blotting and immunofluorescent staining (Figure 1I-J). Furthermore, increased migratory and invasive potentials in MCF-7/ADR cells were observed using migration and matrigel-coated invasion assays (Figure 1K). In summary, MCF-7/ADR cells displayed phenotypes of EMT as well as increased invasive capacity.
Phenotypes of drug resistance and EMT are associated with activation of Axl
To gain further insight of the molecular mechanisms associated with acquired Dox resistance and phenotype of EMT in MCF-7/ADR cells, p-RTK arrays were used to simultaneously measure the activities of 42 RTKs between MCF-7 and MCF-7/ADR cells. This analysis revealed increased activities of Axl, Alk, and Ryk and decreased activities of IGF-1R and HGF-R in MCF-7/ADR cells (Figure 2A-B).
Figure 2
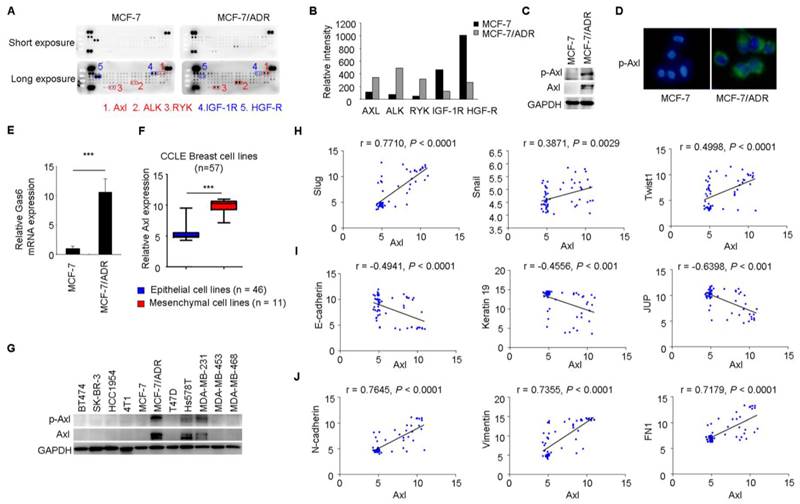
Axl activation is closely associated with EMT in breast cancer cells. (A-B) Whole-cell extracts from MCF-7 and MCF-7/ADR cells were processed with Human Phospho-Receptor Tyrosine Kinase Array Kit (R&D). The differential p-RTKs between MCF-7 and MCF-7/ADR cells were labeled and their relative activities of these RTKs between MCF-7 and MCF-7/ADR cells were shown graphically. (C) Western blot validation for phospho- and total Axl in MCF-7 and MCF-7/ADR cells. (D) Immunofluorescent staining for p-Axl in MCF-7 and MCF-7/ADR cells. (E) qRT-PCR indicated the upregulation of Gas6 mRNA in MCF-7/ADR cells. (F) Human breast cancer lines derived from mesenchymal group (n = 11) expressed high levels of Axl. (G) Levels of phospho- and total Axl in the indicated breast cancer cell lines by Western blotting. (H-J) Correlation between the level of Axl and EMT markers in a panel of 59 breast cancer cell lines. ***P<0.001.
Figure 3
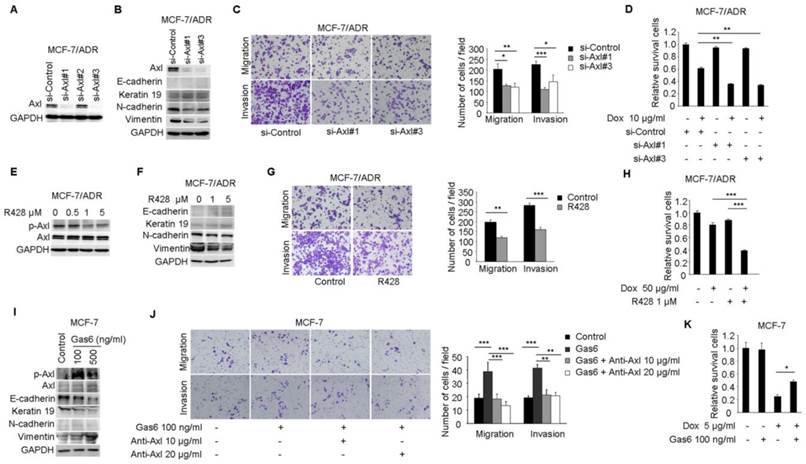
Axl activation is responsible for Dox resistance and EMT in breast cancer cells. (A) Western blot analysis for Axl expression in MCF-7/ADR cells transfected with three Axl si-RNAs or a control si-RNA. (B) Western blot analysis for epithelial markers (E-cadherin and Keratin 19) and mesenchymal markers (N-cadherin and Vimentin) was performed in Axl-silenced MCF-7/ADR cells. (C) Migration and matrigel invasion assays in Axl-silenced MCF-7/ADR cells and control cells (magnification, ×200). (D) MCF-7/ADR cells transfected with si-Axl or si-control were cultured in the absence or presence of 10 µg/ml Dox for 48 h. Relative cell survival was analyzed by CCK8 assays. (E) MCF-7/ADR cells were treated with increasing concentrations of R428 for 24 h, and activity of Axl was determined by Western blotting. (F) MCF-7/ADR cells were treated with indicated concentrations of R428 for 24 h, and Western blot analysis for epithelial (E-cadherin and Keratin 19) and mesenchymal markers (N-cadherin and Vimentin) was performed. (G) MCF-7/ADR cells were cultured in the absence or presence of 1 µM R428 for 24 h. Migratory and invasive behaviors were analyzed using transwell and matrigel invasion assays. (H) MCF-7/ADR cells were cultured with Dox (50 µg/ml), R428 (1 µM), or their combination for 24 h. Relative cell survival was analyzed by CCK8 assays. (I) MCF-7 cells were treated with increasing concentrations of Gas6 for 24 h, then the activity of Axl and the expression of epithelial (E-cadherin and Keratin 19) and mesenchymal markers (N-cadherin and Vimentin) were determined by Western blotting. (J) MCF-7 cells were cultured with Gas6 alone (100 ng/ml), or combined with neutralizing Axl antibody for 24h. Migratory and invasive behaviors were analyzed using transwell and matrigel invasion assays (magnification, ×200). (K) MCF-7 cells stimulated by Gas6 were cultured in the absence or presence of 5 µg/ml Dox for 24 h. Relative cell survival was analyzed by CCK8 assays. Data are representative of results from three independent experiments. *P<0.05, **P<0.01, ***P<0.001.
Increased levels of total Axl and p-Axl in MCF-7/ADR cells were validated by Western blotting and immunofluorescent staining (Figure 2C-D). Gas6 is the ligand of Axl. Similar with the result from microarray analysis, Gas6 mRNA level showed a 10-fold increase in MCF-7/ADR cells compared with that in MCF-7 cells through real-time RT-PCR analysis (Figure 2E).
Breast cancer cell lines (n = 57) were classified into two distinct groups epithelial group (n = 46) and mesenchymal group (n = 11) by hierarchical clustering in a previous study (Supplementary Table 2, two breast cancer cell lines in CCLE was not included in this reference) [36]. Then, we used publicly available data from CCLE to compare Axl expression between epithelial and mesenchymal cell lines. The expression of Axl was dramatically increased in mesenchymal cancer cell lines (Figure 2F). In addition, we validated the total Axl and p-Axl in a panel of breast cancer cell lines. We found that all three mesenchymal cell lines exhibited high level of total Axl and p-Axl (Figure 2G). Significant correlations between the level of Axl and EMT markers were also observed in 59 breast cancer cell lines by using gene expression data from the CCLE database (Figure 2H-J).
To investigate whether the activitity of Axl is critical for EMT and chemotherapeutic resistance in MCF-7/ADR cells, RNA interference was used to silence the expression of Axl in MCF-7/ADR cells (Figure 3A). According to microarray and real-time PCR analysis, we found that the expression of E-cadherin and Keratin 19 in MCF-7/ADR cells were very low. Following Axl knockdown, expression of Keratin 19 was slightly increased, while the expression of two mesenchymal markers (N-cadherin and Vimentin) was decreased (Figure 3B). Decreased expression of Axl also impaired migratory and invasive abilities of MCF-7/ADR cells (Figure 3C). We next evaluated whether Axl knockdown potentiated the effect of Dox. Our results indicated that Axl knockdown significantly increased the activity of Dox, reducing cell viability by an additional 40% compared with cells treated with control si-RNA and Dox (Figure 3D). Changes of survival/apoptotic factors (Bcl-xl, Bcl-2, MCL-1, Bad, Bax, and Bim) were analyzed. In Axl-silenced MCF-7/ADR cells, we observed obvious decreased level of antiapoptotic protein Bcl-xl, and significant upregulation of proapoptotic proteins Bim and Bad after Dox treatment. However, the levels of Bcl-2, MCL-1, and Bax were not changed (Supplementary Figure 2). These results indicate that Axl signaling pathway may be involved in cell survival and apoptosis, which is linked to expression levels of survival/apoptotic factors. Subsequently, we treated MCF-7/ADR cells with a selective small molecule Axl inhibitor R428. R428 significantly suppressed the activity of Axl in MCF-7/ADR cells at the concentration of 1 µM (Figure 3E). R428 treatment also suppressed the expression of N-cadherin and Vimentin (Figure 3F). A concomitant reduction of migratory and invasive potentials of MCF-7/ADR cells was observed after treatment with R428 (Figure 3G). In order to analyze the synergistic effects of Dox and R428 for MCF-7/ADR cells, we need to choose appropriate concentration and time point that Dox or R428 alone did not reduce cell viability obviously. And we found that if we treated MCF-7/ADR cells with R428 at the concentration of 1 µM for 48 h, a lot of dead cells will be occurred. We also found that Dox (10 µg/ml) for 24 h is not enough for synergistic effect. When we treated cells with either R428 (1 µM) or Dox (50 µg/ml) for 24 h, neither compound had a notable effect on cell viability. However, the combination of Dox and R428 resulted in a significant inhibition of cell viability (Figure 3H). Also, we observed that several chemotherapeutic drugs currently in clinical use (paclitaxel, vinorelbine bitartrate, epirubicin hydrochloride, and mitoxantrone) had a synergistic effect with R428 (Supplementary Figure 3A-F).
Stimulation of MCF-7 cells with Gas6 led to dose-dependent (ranging from 10-100 ng/ml) activation of Axl (Data not shown). Western blot analysis demonstrated that although N-cadherin was still almost undetectable, Gas6 treatment downregulated the levels of epithelial markers E-cadherin and Keratin 19 and upregulated the mesenchymal marker Vimentin (Figure 3I). Gas6 treatment also enhanced cell migration and invasion, while blocking antibody of Axl (10 or 20 μg/ml) completely inhibited migration and invasion of MCF-7 cells induced by Gas6 (Figure 3J). Furthermore, Gas6 pretreatment inhibited the effect of Dox, doubling cell viability compared with cells treated with Dox alone (Figure 3K). Taken together, these results indicate that activity of Axl plays an important role for Dox resistance and EMT in breast cancer cells.
Axl activates Akt/GSK3β/β-catenin signaling
Our studies suggested that Axl signaling was a critical regulator of acquired resistance and EMT in breast cancer cells. To identify signaling pathways which functioned as activated downstream of Axl, phospho-kinase arrays were utilized to analyze alterations of kinase signaling in response to Gas6 stimulation. In MCF-7 cells, p-GSK3β, p-Akt, and β-catenin were significantly increased upon Gas6 treatment compared with untreated cells (Figure 4A-B). Next, we validated Gas6-induced upregulation of Akt/GSK3β/β-catenin signaling in MCF-7 cells by Western blotting (Figure 4C). On the contrary, Axl knockdown significantly suppressed the activity of Akt/GSK3β/β-catenin signaling in MCF-7/ADR cells (Figure 4C). Furthermore, Gas6 stimulation resulted in nuclear translocation of β-catenin in MCF-7 cells, whereas Axl knockdown caused obvious decrease of nuclear β-catenin in MCF-7/ADR cells (Figure 4D). Overall, these data indicate that Axl activation can regulate the Akt/GSK3β/β-catenin signaling.
Akt/GSK3β/β-catenin signaling is responsible for Axl-induced cell invasion
We next analyzed whether Akt/GSK3β/β-catenin signaling is responsible for Axl-induced EMT and cell invasion. RNA interference was used to downregulate the expression of Akt or β-catenin in MCF-7/ADR cells (Figure 4E). Following Akt knockdown, the activity of Akt/GSK3β/β-catenin signaling was clearly suppressed while β-catenin knockdown effectively silenced the expression of β-catenin in MCF-7/ADR cells but had no influence on the activation of its upstream molecules (Figure 4F). si-Akt or si-β-catenin treatment resulted in decreased expression of mesenchymal markers N-cadherin and Vimentin, and caused a corresponding decrease in migration and invasion in MCF-7/ADR cells (Figure 4G-H). To further determine whether Axl exerts its function through β-catenin signaling, we ectopically expressed β-catenin in Axl-silenced MCF-7/ADR cells. Overexpression of β-catenin significantly restored cell migration and invasion, which were suppressed by Axl knockdown (Figure 4I). Similarly, Akt or β-catenin knockdown inhibited migration and invasion of MCF-7 cells induced by Gas6 stimulation (Supplementary Figure 4A-D). These observations suggest that Gas6/Axl pathway regulates cell motility and invasion through Akt/GSK3β/β-catenin signaling.
Figure 4
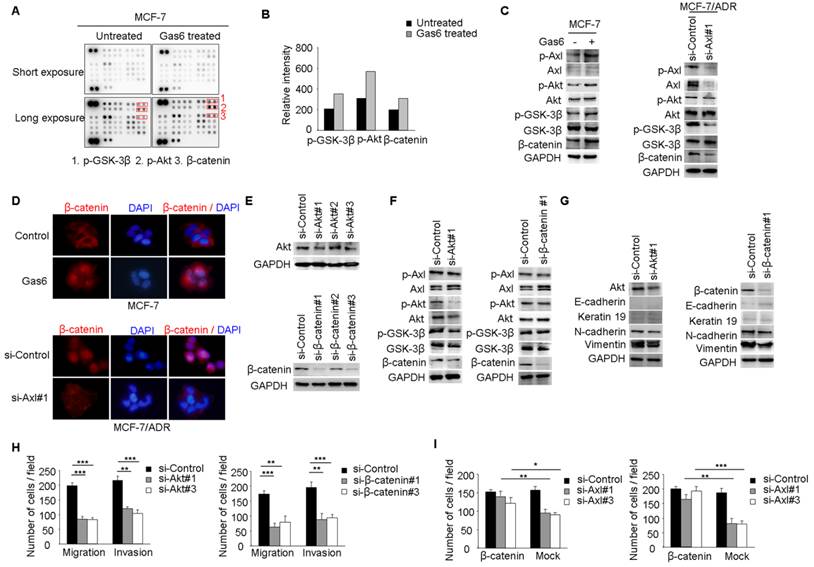
Axl regulates motility and invasion of MCF-7/ADR cells through Akt/GSK3β/β-catenin signaling. (A-B) Gas6 stimulation activated signaling pathways in MCF-7 cells. Results from phosphokinase array suggested that GSK3β, Akt, and β-catenin functioned as downstream signaling molecules of Axl. (C) MCF-7 cells were stimulated with 100 ng/ml Gas6 for 24 h. MCF-7/ADR cells were transfected by si-Axl. Whole-cell lysates were evaluated by Western blot analysis for phospho- and total Axl, Akt, GSK3β, and β-catenin. (D) Immunofluorescent staining for β-catenin was performed in Gas6 stimulated MCF-7 cells and Axl-silenced MCF-7/ADR cells. (E) Protein levels of Akt and β-catenin in MCF-7/ADR transfected with respective si-RNAs. (F) Western blot analysis of total and phospho-Axl, Akt, GSK3β, and β-catenin was performed in the indicated cells. (G) Western blot analysis of epithelial (E-cadherin and Keratin 19) and mesenchymal markers (N-cadherin and Vimentin) was performed in Akt-silenced or β-catenin-depleted MCF-7/ADR cells. (H) Migratory and invasive behaviors of Akt-silenced or β-catenin-silenced MCF-7/ADR cells were analyzed (magnification, ×200). (I) Ectopic expression of β-catenin significantly rescued cell migration and invasion in Axl-depleted cells. Data are representative of results from three independent experiments. **P<0.01, ***P<0.001.
Akt/GSK3β/β-catenin/ZEB1 signaling is responsible for Axl-mediated drug resistance
We performed a series of studies to clearly establish Akt/GSK3β/β-catenin pathway as the functional signaling in Axl-mediated drug resistance. Cell viability studies showed that Akt or β-catenin knockdown reduced the chemotherapy-resistant properties of MCF-7/ADR cells (Figure 5A). Furthermore, Akt or β-catenin knockdown at least partially counteracted the effects of Gas6 stimulation on cell survival in MCF-7 cells (Supplementary Figure 5A-D). Next, we transduced Axl-silenced MCF-7/ADR cells with control or β-catenin expression vector. Notably, cell viability studies showed that the upregulation of β-catenin significantly restored the biological defects instigated by Axl knockdown in the chemotherapy-resistant cells (Figure 5B-C).
ZEB1 is a transcription factor that plays an important role in therapeutic resistance [37, 38] and previous study found that β-catenin/TCF4 could activate ZEB1 transcription [39, 40]. We further determined whether ZEB1 was the functional mediator of Axl. Knockdown of endogenous Axl or β-catenin in MCF-7/ADR cells drastically downregulated ZEB1 expression at both mRNA and protein levels (Figure 5D-E). ChIP assay further confirmed that β-catenin could directly bind to the promoter region of ZEB1 in MCF/ADR cells (Figure 5F). We also used RNA interference to knockdown the expression of ZEB1 in MCF-7/ADR cells (Figure 5G). Cell viability studies showed that ZEB1 knockdown significantly enhanced the activity of Dox (Figure 5H). In ZEB1-silenced MCF-7/ADR cells but not in mock cells, we observed obvious accumulation of γH2AX after Dox treatment, suggesting that ZEB1-silenced cells were less able to repair DNA damage (Figure 5I-J). Similar with we observed in MCF-7/ADR cells, ZEB1 knockdown also partly reduced the protective effects of Gas6 stimulation on cell survival in MCF-7 cells (Supplementary Figure 5E-H). Collectively, these results suggest that Akt/GSK3β/β-catenin/ZEB1 signaling functions as downstream of Axl-mediated drug resistance.
Knockdown or inhibition of Axl suppresses metastasis and drug resistance in vivo
Having established the potential role of Axl-Akt/GSK3β/β-catenin axis in the aggressiveness of breast cancer, we next determined if Axl could be a therapeutic target in breast cancer. Compared with mice injected with the control MCF-7/ADR cells (metastasis incidence: 8/8, 100%), mice that received an intravenous injection of Axl-silenced MCF-7/ADR cells showed a 75% reduction in the incidence of metastasis (2/8, 25%). The number of metastatic nodules in the lung was also significantly decreased (Figure 6A-D). Subsequently, we inactivated Axl by using a small molecule inhibitor R428. We found that Axl inhibition, as compared to control group, significantly reduced metastasis incidence. Compared with control group (averaging 12.50 metastases per mouse), mice that received R428 showed an 81.6% reduction in the number of metastatic nodules in the lung, with an average of 2.30 metastases per mouse (Figure 6E-H). These results suggest that Axl has a great potential as a therapeutic target for breast cancer metastasis.
We also established subcutaneous tumors in nude mice using MCF-7/ADR cells. Compared with the control group, Axl knockdown or Dox treatment alone had no obvious anticancer effect. However, Axl knockdown strongly restored the efficacy of Dox by significantly inhibiting the growth of xenografts by about 85% for tumor volume and 83.3% for tumor weight (Figure 6I-6L). Immunohistochemical analysis also showed that Axl knockdown alone or in combination with Dox treatment suppressed the levels of Axl, p-Akt, p-GSK3β, β-catenin, and ZEB1 (Supplementary Figure 6A). Axl knockdown also dramatically rescued the efficacy of Dox on DNA damage and cell apoptosis in MCF-7/ADR subcutaneous tumors (Supplementary Figure 6B-E).
Figure 5
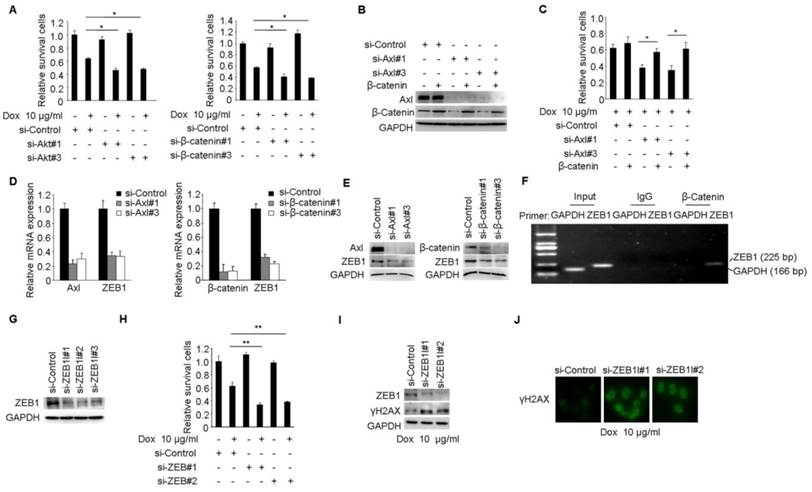
Axl mediates Dox resistance in MCF-7/ADR cells through Akt/GSK3β/β-catenin/ZEB1 cascade. (A) MCF-7/ADR cells transfected by si-Akt, si-β-catenin or si-control were cultured in the absence or presence of 10 µg/ml Dox for 48 h. Relative cell survival was analyzed by the CCK8 assay. (B-C) Ectopic expression of β-catenin significantly rescued cell survival in Axl-depleted cells. (D-E) Both mRNA and protein levels were detected in MCF-7/ADR cells transfected with si-Axl or si-β-catenin, respectively. (F) ChIP assay was performed with antibody against β-catenin or control IgG in MCF-7/ADR cells. The immunoprecipitated DNA was analyzed by PCR followed by agarose gel electrophoresis. (G) Western blot analysis of ZEB1 protein level in MCF-7/ADR cells transfected with ZEB1 si-RNAs or a control si-RNA. (H) MCF-7/ADR cells transfected with si-ZEB1 or si-control were cultured in the absence or presence of 10 µg/ml Dox for 48 h. Relative cell survival was analyzed by CCK8 assay. (I-J) MCF-7/ADR cells transfected by si-ZEB1 or si-control were cultured in the presence of 10 µg/ml Dox for 48 h. Western blot analysis and immunofluorescent staining for γH2AX were performed. *P<0.05, **P<0.01.
Figure 6
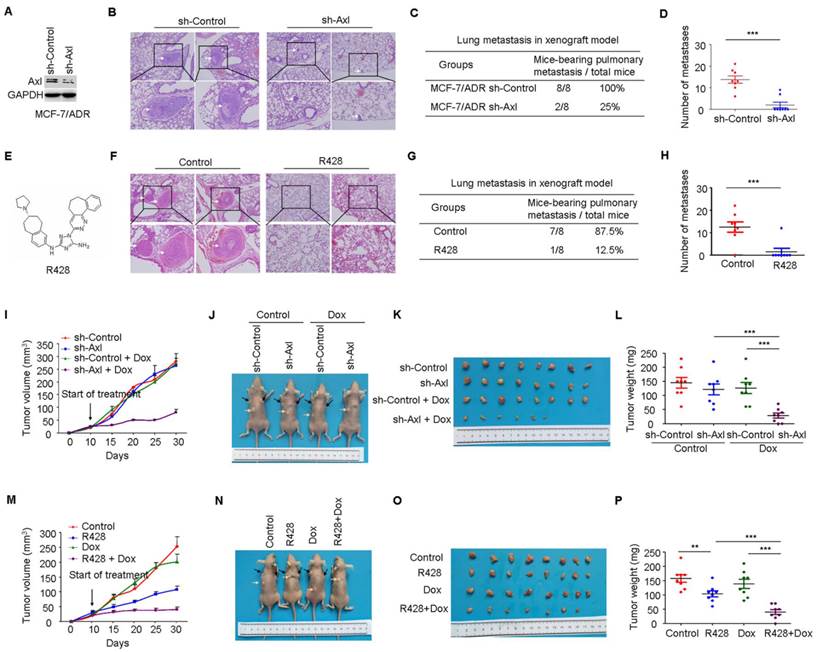
Axl knockdown or inhibition suppresses metastasis and restores the efficacy of chemotherapy in preclinical models. (A) Western blot analysis was used to validate the knockdown of Axl in MCF-7/ADR cells. (B) Representative images of H&E staining from indicated groups. (C-D) Metastasis incidence and the number of metastatic nodules in the lungs of mice with tail vein injection of Axl-silenced or mock-infected MCF-7/ADR cells at day 30 after implantation. (E) Structure of Axl inhibitor R428 used in the study. (F) Representative images of H&E staining from vehicle or R428 treatment groups. (G-H) Metastasis incidence and the number of metastatic nodules in the lungs of mice with tail vein injection of MCF-7/ADR after 30 days of therapy with vehicle or R428 (25 mg/kg orally twice daily). (I) Volumes of subcutaneous control or Axl-silenced MCF-7/ADR xenografts during 21 days of therapy with vehicle or Dox. (J-L) Representative tumor-bearing mice, tumors isolated from nude mice, and tumor weights of the subcutaneous tumor model from the indicated group. (M) Volumes of subcutaneous MCF-7/ADR xenografts during 21 days of therapy with vehicle, R428, Dox, or combination treatment. (N-P) Representative tumor-bearing mice, tumors isolated from nude mice, and tumor weights of MCF-7/ADR subcutaneous tumor model treated as indicated. Black arrows show the tumors and the yellow arrows show the 17β-estradiol pellets. **P<0.01, ***P<0.001.
Consistent with our in vitro data, R428 or Dox exhibited modest activity as single agent in vivo. In contrast, in combination, R428 significantly restored the efficacy of Dox, as evidenced by tumor volume, tumor weight, DNA damage, and cell apoptosis following 3 weeks of combination therapy (Figure 6M-P and Supplementary Figure 7B-E). Immunohistochemical analysis following treatment demonstrated that inhibition of p-Axl, p-Akt, p-GSK3β, β-catenin, and ZEB1 was similar between R428 and the combination treatment (Supplementary Figure 7A).
Clinical significance of Axl activation in human breast cancer
Finally, based on the above findings, the clinical significance of Axl activation was determined in human breast cancer patients. We analyzed whether there were correlations between the levels of p-Axl and its downstream targets (β-catenin and ZEB1) in the clinical specimens. As shown in Figure 7A-C, 66.6% (14 cases) and 61.9% (13 cases) exhibited high levels of β-catenin or ZEB1 in the specimens with high levels of p-Axl (21 cases), respectively. On the other hand, decreased expression of β-catenin and ZEB1 was observed in 69.0% (20 cases) and 75.9% (22 cases), respectively in a total of 29 samples with low levels of p-Axl (chi-square test, P<0.05). Using gene expression data from CCLE, we detected a significant positive correlation between Axl and ZEB1 mRNA levels in a panel of 59 breast cancer cell lines (r=0.7074, P<0.001; Figure 7D) and in a tumor data set comprising 1079 human breast cancer specimens from TCGA (r=0.6946, P<0.001, Figure 7E). Notably, significant associations between p-Axl level and shorter overall survival and disease-free survival were observed in our cohort (P=0.0279 and P=0.0087, respectively, Figure 7F-H). The expression level of ZEB1 was also analyzed in human breast cancer tissues. High expression of ZEB1 trended toward poorer survival of breast cancer patients, although the data did not meet statistical significance (Supplementary Figure 8A-C).
Taken together, our data indicate that Axl activation could stimulate Akt/GSK3β/β-catenin signaling, leading to nuclear translocation of β-catenin and transcriptional upregulation of ZEB1, which in turn regulate DNA damage repair and Dox-resistance in breast cancer cells. Furthermore, activation of Axl promotes EMT and cell invasion through Akt/GSK3β/β-catenin signaling cascade (Figure 7I). Our results demonstrate that Gas6/Axl Axis may represent a therapeutic target for chemoresistance and metastasis in breast cancer.
Figure 7
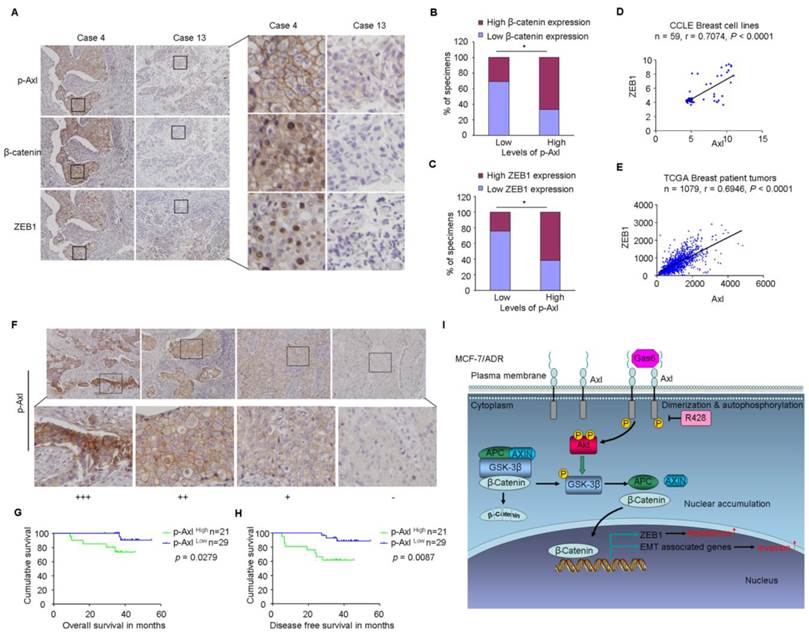
Clinical significance of Axl signaling in human breast cancer. (A) Levels of p-Axl were associated with the expression levels of β-catenin and ZEB1 in clinical breast cancer specimens. Two representative cases are shown. Original magnification, ×200. (B-C) Percentage of specimens showing low or high p-Axl levels in relation to the expression levels of β-catenin and ZEB1 (chi-square test, *P<0.05). (D-E) Positive correlation between Axl and ZEB1 mRNA levels in a panel of 59 breast cancer cell lines (Pearson's correlation, r=0.7074, P<0.0001) and 1079 human breast cancer specimens (Pearson's correlation, r=0.6946, P<0.0001). Relative gene expression levels of Axl and ZEB1 were acquired from the CCLE and TCGA databases. (F) Representative immunohistochemical staining patterns for p-Axl. (G-H) The impact of p-Axl level on overall survival and disease-free survival of breast cancer patients treated with Dox-based adjuvant chemotherapy. (I) Schematic representation of the contribution of axl activation to chemoresistance and EMT in breast cancer via Akt/GSK-3β/β-catenin signaling.
Discussion
Breast cancer is a widespread malignancy, which deserves intensive investigation. Using a cell model of multidrug resistance, we identified a Dox-resistant phenotype of MCF-7/ADR cells characterized by absence of epithelial markers and increased mesenchymal markers, as well as dramatically upregulated potentials of migration and invasion. It may be a potential reason a considerable proportion of breast cancer patients with adjuvant chemotherapy still experience distant metastasis. Consistent with our data, a previous study identified high scoring networks related to EMT in MCF-7/ADR cells through Ingenuity Pathway Analysis [9]. Recently, Axl has been implicated in EMT and metastasis in multiple tumor types [10]. Gjerdrum C et al reported that Axl was an important EMT-induced regulator of breast cancer metastasis [19]. Asiedu MK et al demonstrated that Axl could induce EMT and regulate the function of breast cancer stem cells [41]. In this study, we found that the activity of Axl was significantly increased in MCF-7/ADR cells. Besides in vitro and in vivo studies, we also observed an inverse correlation between Axl and epithelial markers and a positive correlation between Axl and mesenchymal markers in a panel of 59 breast cancer cell lines using mRNA gene expression data from CCLE. This suggests that the effect of Axl on epithelial and mesenchymal markers is a general mechanism conserved in heterogeneous breast cancer cell lines.
We used phospho-kinase arrays to analyze alterations in the downstream kinase signaling of Axl. Aberrant activation of Akt/GSK3β/β-catenin signaling was identified following stimulation with Gas6, the main ligand of Axl. We further validated that EMT in MCF-7/ADR cells was associated with the activation of Akt/GSK3β/β-catenin pathway, which suppressed the activity of GSK-3β and stabilized β-catenin. As an important driver of EMT, the accumulation and nuclear import of β-catenin could interact with TCF/LEF and induce the expression of genes responsible for the EMT process. Interestingly, canonical EMT inducing transcription factors Slug, Snail, and Twist were also upregulated in MCF-7/ADR cells. Several independent studies exhibited that these EMT inducing transcription factors could induce the expression of Axl, which further upregulated the expression of Twist, Snail, and Slug through a positive feedback loop [10]. Whether it is a general mechanism conserved in multidrug resistant breast cancers remains to be further investigated.
We also explored a novel mechanism for multidrug resistance of breast cancer, which involved increased activity of Axl in MCF-7/ADR cells. It has been observed that Axl activation is involved in resistance to multiple targeted drugs [10]. Like other RTKs, Axl activation subsequently induces autophosphorylation, which leads to the stimulation of a range of pathways. In our study, we validated that Akt/GSK3β/β-catenin signaling was responsible for Axl-mediated drug resistance. More importantly, we found the EMT inducing transcription factor ZEB1 was transcriptionally upregulated by β-catenin, which was nuclear accumulated induced by Gas6/Axl Axis. Consistent with previous studies [39, 40], we found that β-catenin could dirctely bind the promoter of ZEB1 and then induced the expression of ZEB1. Accumulating evidence indicates that ZEB1 has important implications in therapeutic resistance [37, 38, 42]. High expression of ZEB1 in glioblastoma patients was a predictor for shorter survival and poor temozolomide response [38]. Moreover, ZEB1 directly interacted with USP7 and increased its ability to deubiquinylate and stabilize CHK1, thereby promoting DNA repair and resistance to radiation [37]. Our findings revealed that ZEB1-silenced MCF-7/ADR cells were less able to repair DNA damage. Furthermore, ZEB1 was predictive for shorter overall survival and disease-free survival in breast cancer patients. It has also been shown that EMT significantly contributed to chemoresistance in several tumor types [43, 44]. Wilson C et al indicated that Axl inhibition was specifically synergistic with antimitotic agents in killing cancer cells that had undergone EMT [30]. Based on the important role of Axl in resistance and EMT, it is reasonable to conclude that there is a complex regulatory network involving Axl, EMT, and resistance in breast cancer. These findings reveal a novel combined treatment strategy for tumors displaying mesenchymal features [30]. Also, as an important EMT activator, the role of ZEB1 in crosstalk between EMT and drug resistance merits special attention in future studies.
Chemoresistance in breast cancer has been a great interest in past studies; however, the development of novel and rational therapeutic strategies that targeting chemoresistant cells is still a challenge for clinical oncology. Recently, several approaches designed to antagonize Axl signaling have been explored in preclinical and clinical studies. The Axl inhibitor used in our research is R428 (also known as BGB324), a selective small molecule inhibitor of Axl kinase, which is a first-in-class Axl kinase inhibitor, entered phase 1 clinical trials [45]. R428 exhibited preclinical anti-tumor potentials in a variety of tumour types. It enhanced tumor response to metformin in prostate cancer [46]. In head and neck squamous cell carcinoma, targeting Axl increased the sensitivity of HNSCC cell lines to erlotinib, cetuximab, and radiation [13, 23]. Furthermore, a robust induction of CLL B-cell apoptosis was observed in both time- and dose-dependent manners after R428 treatment [47]. We discovered that R428 alone was sufficient to reduce distant metastasis of MCF-7/ADR cells. Also, targeting Axl in combination with Dox significantly inhibited tumor growth in mouse xenografts. These observations suggested Axl inhibition as a promising therapeutic strategy to overcome acquired multidrug resistance in breast cancer.
In conclusion, we have presented evidence that Gas6/Axl axis confers aggressiveness in breast cancer through Akt/GSK-3β/β-catenin signaling. These data conclusively indicate that clinically translatable Axl inhibitors are potential therapeutics for breast cancer, especially for the breast cancer patients who have developed multidrug resistance.
Supplementary Material
Supplementary tables and figures.
Acknowledgements
This work was supported by grants from National Key Basic Research Program of China (973 Program: 2015CB553905), National Natural Science Foundation of China (81301818, 81402278, 81572311, 81421001), National Key Sci-Tech Special Project of China (2012ZX10002011-004), and projects of Special Research Fund for Healthy (201402003), Shanghai Jiao Tong University School of Medicine (YG2014MS44, YG2015QN34), State Key Laboratory of Oncogenes and Related Genes (SB16-04), and Key Discipline and Specialty Foundation of Shanghai Municipal Commission of Health and Family Planning.
Accession numbers
Microarray data have been deposited at GEO with the accession number GSE76540.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65:87-108
2. Del ML, De Placido S, Bruzzi P, De Laurentiis M, Boni C, Cavazzini G. et al. Fluorouracil and dose-dense chemotherapy in adjuvant treatment of patients with early-stage breast cancer: an open-label, 2 x 2 factorial, randomised phase 3 trial. Lancet. 2015;385:1863-72
3. Martin M, Ruiz A, Ruiz BM, Barnadas A, Gonzalez S, Calvo L. et al. Fluorouracil, doxorubicin, and cyclophosphamide (FAC) versus FAC followed by weekly paclitaxel as adjuvant therapy for high-risk, node-negative breast cancer: results from the GEICAM/2003-02 study. J Clin Oncol. 2013;31:2593-9
4. Mackey JR, Martin M, Pienkowski T, Rolski J, Guastalla JP, Sami A. et al. Adjuvant docetaxel, doxorubicin, and cyclophosphamide in node-positive breast cancer: 10-year follow-up of the phase 3 randomised BCIRG 001 trial. Lancet Oncol. 2013;14:72-80
5. Higgins CF. Multiple molecular mechanisms for multidrug resistance transporters. Nature. 2007;446:749-57
6. Tredan O, Galmarini CM, Patel K, Tannock IF. Drug resistance and the solid tumor microenvironment. J Natl Cancer Inst. 2007;99:1441-54
7. Gerweck LE, Seetharaman K. Cellular pH gradient in tumor versus normal tissue: potential exploitation for the treatment of cancer. Cancer Res. 1996;56:1194-8
8. Fan S, Niu Y, Tan N, Wu Z, Wang Y, You H. et al. LASS2 enhances chemosensitivity of breast cancer by counteracting acidic tumor microenvironment through inhibiting activity of V-ATPase proton pump. Oncogene. 2013;32:1682-90
9. Calcagno AM, Salcido CD, Gillet JP, Wu CP, Fostel JM, Mumau MD. et al. Prolonged drug selection of breast cancer cells and enrichment of cancer stem cell characteristics. J Natl Cancer Inst. 2010;102:1637-52
10. Graham DK, DeRyckere D, Davies KD, Earp HS. The TAM family: phosphatidylserine sensing receptor tyrosine kinases gone awry in cancer. Nat Rev Cancer. 2014;14:769-85
11. Linger RM, Keating AK, Earp HS, Graham DK. TAM receptor tyrosine kinases: biologic functions, signaling, and potential therapeutic targeting in human cancer. Adv Cancer Res. 2008;100:35-83
12. Reichl P, Dengler M, van Zijl F, Huber H, Fuhrlinger G, Reichel C. et al. Axl activates autocrine transforming growth factor-beta signaling in hepatocellular carcinoma. Hepatology. 2015;61:930-41
13. Brand TM, Iida M, Stein AP, Corrigan KL, Braverman CM, Coan JP. et al. AXL is a logical molecular target in head and neck squamous cell carcinoma. Clin Cancer Res. 2015;21:2601-12
14. Hattori S, Kikuchi E, Kosaka T, Miyazaki Y, Tanaka N, Miyajima A. et al. Relationship between increased expression of the Axl/Gas6 signal cascade and prognosis of patients with upper tract urothelial carcinoma. Ann Surg Oncol. 2016;23:663-70
15. Lee HJ, Jeng YM, Chen YL, Chung L, Yuan RH. Gas6/Axl pathway promotes tumor invasion through the transcriptional activation of Slug in hepatocellular carcinoma. Carcinogenesis. 2014;35:769-75
16. Dunne PD, McArt DG, Blayney JK, Kalimutho M, Greer S, Wang T. et al. AXL is a key regulator of inherent and chemotherapy-induced invasion and predicts a poor clinical outcome in early-stage colon cancer. Clin Cancer Res. 2014;20:164-75
17. Kirane A, Ludwig KF, Sorrelle N, Haaland G, Sandal T, Ranaweera R. et al. Warfarin blocks Gas6-mediated Axl activation required for pancreatic cancer epithelial plasticity and metastasis. Cancer Res. 2015;75:3699-705
18. Yu H, Liu R, Ma B, Li X, Yen HY, Zhou Y. et al. Axl receptor tyrosine kinase is a potential therapeutic target in renal cell carcinoma. Br J Cancer. 2015;113:616-25
19. Gjerdrum C, Tiron C, Hoiby T, Stefansson I, Haugen H, Sandal T. et al. Axl is an essential epithelial-to-mesenchymal transition-induced regulator of breast cancer metastasis and patient survival. Proc Natl Acad Sci U S A. 2010;107:1124-9
20. Holland SJ, Pan A, Franci C, Hu Y, Chang B, Li W. et al. R428, a selective small molecule inhibitor of Axl kinase, blocks tumor spread and prolongs survival in models of metastatic breast cancer. Cancer Res. 2010;70:1544-54
21. Zhang YX, Knyazev PG, Cheburkin YV, Sharma K, Knyazev YP, Orfi L. et al. AXL is a potential target for therapeutic intervention in breast cancer progression. Cancer Res. 2008;68:1905-15
22. Debruyne DN, Bhatnagar N, Sharma B, Luther W, Moore NF, Cheung NK. et al. ALK inhibitor resistance in ALK-driven neuroblastoma is associated with AXL activation and induction of EMT. Oncogene. 2015 doi: 10.1038/onc.2015.434
23. Giles KM, Kalinowski FC, Candy PA, Epis MR, Zhang PM, Redfern AD. et al. Axl mediates acquired resistance of head and neck cancer cells to the epidermal growth factor receptor inhibitor erlotinib. Mol Cancer Ther. 2013;12:2541-58
24. Brand TM, Iida M, Stein AP, Corrigan KL, Braverman CM, Luthar N. et al. AXL mediates resistance to cetuximab therapy. Cancer Res. 2014;74:5152-64
25. Zhang Z, Lee JC, Lin L, Olivas V, Au V, LaFramboise T. et al. Activation of the AXL kinase causes resistance to EGFR-targeted therapy in lung cancer. Nat Genet. 2012;44:852-60
26. Muller J, Krijgsman O, Tsoi J, Robert L, Hugo W, Song C. et al. Low MITF/AXL ratio predicts early resistance to multiple targeted drugs in melanoma. Nat Commun. 2014;5:5712
27. Elkabets M, Pazarentzos E, Juric D, Sheng Q, Pelossof RA, Brook S. et al. AXL mediates resistance to PI3Kalpha inhibition by activating the EGFR/PKC/mTOR axis in head and neck and esophageal squamous cell carcinomas. Cancer Cell. 2015;27:533-46
28. Zhou L, Liu XD, Sun M, Zhang X, German P, Bai S. et al. Targeting MET and AXL overcomes resistance to sunitinib therapy in renal cell carcinoma. Oncogene. 2015 doi: 10.1038/onc.2015.343
29. Li Y, Wang X, Bi S, Zhao K, Yu C. Inhibition of Mer and Axl receptor tyrosine kinases leads to increased apoptosis and improved chemosensitivity in human neuroblastoma. Biochem Biophys Res Commun. 2015;457:461-6
30. Wilson C, Ye X, Pham T, Lin E, Chan S, McNamara E. et al. AXL inhibition sensitizes mesenchymal cancer cells to antimitotic drugs. Cancer Res. 2014;74:5878-90
31. Scaltriti M, Elkabets M, Baselga J. Molecular Pathways: AXL, a membrane receptor mediator of resistance to therapy. Clin Cancer Res. 2016;22:1313-7
32. Meyer AS, Miller MA, Gertler FB, Lauffenburger DA. The receptor AXL diversifies EGFR signaling and limits the response to EGFR-targeted inhibitors in triple-negative breast cancer cells. Sci Signal. 2013;6:a66
33. Liu L, Greger J, Shi H, Liu Y, Greshock J, Annan R. et al. Novel mechanism of lapatinib resistance in HER2-positive breast tumor cells: activation of AXL. Cancer Res. 2009;69:6871-8
34. Wang C, Jiang K, Kang X, Gao D, Sun C, Li Y. et al. Tumor-derived secretory clusterin induces epithelial-mesenchymal transition and facilitates hepatocellular carcinoma metastasis. Int J Biochem Cell Biol. 2012;44:2308-20
35. Jin GZ, Yu WL, Dong H, Zhou WP, Gu YJ, Yu H. et al. SUOX is a promising diagnostic and prognostic biomarker for hepatocellular carcinoma. J Hepatol. 2013;59:510-7
36. Shaul YD, Freinkman E, Comb WC, Cantor JR, Tam WL, Thiru P. et al. Dihydropyrimidine accumulation is required for the epithelial-mesenchymal transition. Cell. 2014;158:1094-109
37. Zhang P, Wei Y, Wang L, Debeb BG, Yuan Y, Zhang J. et al. ATM-mediated stabilization of ZEB1 promotes DNA damage response and radioresistance through CHK1. Nat Cell Biol. 2014;16:864-75
38. Siebzehnrubl FA, Silver DJ, Tugertimur B, Deleyrolle LP, Siebzehnrubl D, Sarkisian MR. et al. The ZEB1 pathway links glioblastoma initiation, invasion and chemoresistance. EMBO Mol Med. 2013;5:1196-212
39. Wang Y, Bu F, Royer C, Serres S, Larkin JR, Soto MS. et al. ASPP2 controls epithelial plasticity and inhibits metastasis through beta-catenin-dependent regulation of ZEB1. Nat Cell Biol. 2014;16:1092-104
40. Sanchez-Tillo E, de Barrios O, Siles L, Cuatrecasas M, Castells A, Postigo A. beta-catenin/TCF4 complex induces the epithelial-to-mesenchymal transition (EMT)-activator ZEB1 to regulate tumor invasiveness. Proc Natl Acad Sci U S A. 2011;108:19204-9
41. Asiedu MK, Beauchamp-Perez FD, Ingle JN, Behrens MD, Radisky DC, Knutson KL. AXL induces epithelial-to-mesenchymal transition and regulates the function of breast cancer stem cells. Oncogene. 2014;33:1316-24
42. Meidhof S, Brabletz S, Lehmann W, Preca BT, Mock K, Ruh M. et al. ZEB1-associated drug resistance in cancer cells is reversed by the class I HDAC inhibitor mocetinostat. EMBO Mol Med. 2015;7:831-47
43. Fischer KR, Durrans A, Lee S, Sheng J, Li F, Wong ST. et al. Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature. 2015;527:472-6
44. Zheng X, Carstens JL, Kim J, Scheible M, Kaye J, Sugimoto H. et al. Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature. 2015;527:525-30
45. Sheridan C. First Axl inhibitor enters clinical trials. Nat Biotechnol. 2013;31:775-6
46. Bansal N, Mishra PJ, Stein M, DiPaola RS, Bertino JR. Axl receptor tyrosine kinase is up-regulated in metformin resistant prostate cancer cells. Oncotarget. 2015;6:15321-31
47. Ghosh AK, Secreto C, Boysen J, Sassoon T, Shanafelt TD, Mukhopadhyay D. et al. The novel receptor tyrosine kinase Axl is constitutively active in B-cell chronic lymphocytic leukemia and acts as a docking site of nonreceptor kinases: implications for therapy. Blood. 2011;117:1928-37
Author contact
![]() Corresponding author: Wenxin Qin; Mailing Address: State Key Laboratory of Oncogenes and Related Genes, Shanghai Cancer Institute, Renji Hospital, Shanghai Jiao Tong University School of Medicine, No. 25/Ln 2200 Xie-Tu Road, Shanghai 200032, China; Tel: +86-21-64436581; Fax: +86-21-64432142; Email: wxqinedu.cn.
Corresponding author: Wenxin Qin; Mailing Address: State Key Laboratory of Oncogenes and Related Genes, Shanghai Cancer Institute, Renji Hospital, Shanghai Jiao Tong University School of Medicine, No. 25/Ln 2200 Xie-Tu Road, Shanghai 200032, China; Tel: +86-21-64436581; Fax: +86-21-64432142; Email: wxqinedu.cn.
Citation styles
APA
Wang, C., Jin, H., Wang, N., Fan, S., Wang, Y., Zhang, Y., Wei, L., Tao, X., Gu, D., Zhao, F., Fang, J., Yao, M., Qin, W. (2016). Gas6/Axl Axis Contributes to Chemoresistance and Metastasis in Breast Cancer through Akt/GSK-3β/β-catenin Signaling. Theranostics, 6(8), 1205-1219. https://doi.org/10.7150/thno.15083.
ACS
Wang, C.; Jin, H.; Wang, N.; Fan, S.; Wang, Y.; Zhang, Y.; Wei, L.; Tao, X.; Gu, D.; Zhao, F.; Fang, J.; Yao, M.; Qin, W. Gas6/Axl Axis Contributes to Chemoresistance and Metastasis in Breast Cancer through Akt/GSK-3β/β-catenin Signaling. Theranostics 2016, 6 (8), 1205-1219. DOI: 10.7150/thno.15083.
NLM
Wang C, Jin H, Wang N, Fan S, Wang Y, Zhang Y, Wei L, Tao X, Gu D, Zhao F, Fang J, Yao M, Qin W. Gas6/Axl Axis Contributes to Chemoresistance and Metastasis in Breast Cancer through Akt/GSK-3β/β-catenin Signaling. Theranostics 2016; 6(8):1205-1219. doi:10.7150/thno.15083. https://www.thno.org/v06p1205.htm
CSE
Wang C, Jin H, Wang N, Fan S, Wang Y, Zhang Y, Wei L, Tao X, Gu D, Zhao F, Fang J, Yao M, Qin W. 2016. Gas6/Axl Axis Contributes to Chemoresistance and Metastasis in Breast Cancer through Akt/GSK-3β/β-catenin Signaling. Theranostics. 6(8):1205-1219.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) License. See http://ivyspring.com/terms for full terms and conditions.