Impact Factor
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2016; 6(8):1244-1260. doi:10.7150/thno.13804 This issue Cite
Research Paper
Mitochondrial Transplantation Attenuates Airway Hyperresponsiveness by Inhibition of Cholinergic Hyperactivity
Yuan Su1, 2, †, Liping Zhu1, †, Xiangyuan Yu1, †, Lei Cai1, †, Yankai Lu1, Jiwei Zhang1, 3, Tongfei Li4, Jiansha Li1, 5, Jingyan Xia6, Feng Xu7, ![]() , Qinghua Hu1, 8,
, Qinghua Hu1, 8, ![]()
1. Key Laboratory of Pulmonary Diseases of Ministry of Health and Department of Pathophysiology, School of Basic Medicine, Tongji Medical College, Huazhong University of Science and Technology (HUST), Wuhan 430030, People's Republic of China (PRC);
2. Department of Respiratory Medicine, Union Hospital, Tongji Medical College, HUST, Wuhan 430022, PRC;
3. Department of Pathology, Union Hospital, Tongji Medical College, HUST, Wuhan 430030, PRC;
4. Department of Pathology, School of Basic Medical Sciences, Hubei University of Medicine, Shiyan 442000, PRC;
5. Department of Pathology, Tongji Hospital, Tongji Medical College, HUST, Wuhan 430030, PRC;
6. Department of Radiation Oncology, Second Affiliated Hospital, Zhejiang University School of Medicine (ZUSM), Hangzhou 310009, PRC;
7. Department of Infectious Diseases, Second Affiliated Hospital, ZUSM, Hangzhou 310009, PRC;
8. Key Laboratory of Molecular Biophysics of the Ministry of Education, HUST, PRC.
† Equal contributor
Received 2015-9-9; Accepted 2016-4-26; Published 2016-5-24
Abstract

Increased cholinergic activity has been highlighted in the pathogenesis of airway hyperresponsiveness, and alternations of mitochondrial structure and function appear to be involved in many lung diseases including airway hyperresponsiveness. It is crucial to clarify the cause-effect association between mitochondrial dysfunction and cholinergic hyperactivity in the pathogenesis of airway hyperresponsiveness. Male SD rats and cultured airway epithelial cells were exposed to cigarette smoke plus lipopolysaccharide administration; mitochondria isolated from airway epithelium were delivered into epithelial cells in vitro and in vivo. Both the cigarette smoke plus lipopolysaccharide-induced cholinergic hyperactivity in vitro and the airway hyperresponsiveness to acetylcholine in vivo were reversed by the transplantation of exogenous mitochondria. The rescue effects of exogenous mitochondria were imitated by the elimination of excessive reactive oxygen species or blockage of muscarinic M3 receptor, but inhibited by M receptor enhancer. Mitochondrial transplantation effectively attenuates cigarette smoke plus lipopolysaccharide-stimulated airway hyperresponsiveness through the inhibition of ROS-enhanced epithelial cholinergic hyperactivity.
Keywords: airway hyperresponsiveness, mitochondrial transplantation, cholinergic hyperactivity
Introduction
The airway hyperresponsiveness (AHR) is a state or phenotype of increased responsiveness of the airway to a variety of nonspecific triggers such as the cholinergic receptors agonist, acetylcholine (Ach) [1;2]. Rather than the consequence of chronic obstructive pulmonary diseases (COPD), AHR may serve as a contributing mechanism to the development of this disease [3]. About two third of patients with COPD have demonstrated AHR [4]. The aggravated airway smooth muscle contraction, which is an important feature of AHR plays a vital role for COPD. The successful application of anti-cholinergic medications in COPD therapy highlights the significance of increased cholinergic activity in the pathogenesis [2]. Additionally, the mortality from COPD is correlated with the severity of AHR and the trend is more pronounced in smokers than non-smokers [5].
Cigarette smoke is the most predominate reason for COPD and can be used in animal models to replicate COPD. Previous studies have indicated that cigarette smoking not only results in airway inflammation, but also increases airway responsiveness [4;6]. COPD patients experience periodic exacerbations that mostly caused by respiratory tract bacterial infection [7;8]. Furthermore, it has also been reported that cigarette smoke increased the non-neuronal cholinergic activity of lung cells [9]. However, the exact role and mechanisms of AHR and cholinergic hyperactivity induced by the pathogenic factors such as smoking in the pathogenesis and pathophysiology of COPD have not been understood yet.
Mitochondria play multiple and critical roles in cell structural organization and functions. Alternations of the ultrastructure or functions of mitochondria themselves are considered to be involved in a variety of pathological process including airway chronic inflammation and hyperresponsiveness [10]. Although mitochondrial dysfunction has been demonstrated in airway epithelial cells exposed to cigarette smoke extracts [11;12], the cause-effect association between mitochondria alternations and cholinergic hyperactivity has never been revealed. Herein we explore the involvement of mitochondria in the regulation of cholinergic activity of airway epithelium as well as their mechanistic significance underlying AHR upon cigarette smoke plus lipopolysaccharide (LPS) exposure.
We show that exogenous mitochondria transplantation reverses the cigarette smoke plus lipopolysaccharide-stimulated cholinergic hyperactivity in vitro as well as the AHR to Ach in vivo, which is probably due to the limitation of reactive oxygen species production in airway epithelial cells.
Materials and Methods
Animals
All experiments involving Sprague-Dawley rats obtained approval from the Institutional Animal Care and Use Committee of the Tongji Medical College, and performed in keeping with the Guide for the Care and Use of Laboratory Animals of National Institutes of Health. The male adult SD rats aged of 6 weeks were randomly allocated to cigarette smoke plus LPS (Sigma) exposed (CS/LPS) and room air plus the vehicle for LPS (control) groups. The rats were caged in a smoking box (0.25 m3, 8 rats per box), and exposed to fresh air or a 12:1 mixture of air and smoke of 9 cigarettes (Hong Shuangxi Brand, Wuhan, China; tar 16 mg, nicotine 1.0 mg in each), delivered at 5 ml puff every 2 seconds within 1.5 hours per day for 28 consecutive days. The CO level at the time of smoke was 1147.19 ± 65.09 mg/m3 (917.75 ± 52.08 ppm, n=4). On the first and the 14th day of smoke exposure, rats were anesthetized by chloral hydrate (0.35g/Kg, intraperitoneally), and vehicle or LPS (1mg/mL, 200 μL/animal/time) were instilled into the trachea through an intubation needle [13;14]. For exogenous mitochondria treatment, after 28 days of CS/LPS exposure or air/vehicle, 100 μl of mitochondria suspension (3.75×108 /ml in PBS) were intratracheally introduced into rats once every other day for additional 6 days. The rats were sacrificed after measurement of airway responsiveness on day 35. For the evaluation of leukocyte accumulation, the lungs of rats with or without treatment were lavaged for the collection of bronchoalveolar lavage fluid. For detecting of inflammatory mediators of LTB4 and IL-8, the lungs of rats with or without treatment were immediately preserved in the liquid nitrogen. For histological study, the lungs were fixed in 4% paraformaldehyde. For electron microscopy, the airway epithelium tissues freshly stripped from the rats were fixed with 2.5% glutaraldehyde.
Cell culture
Rat airway epithelial cells (RAECs) were cultured from male SD rats (80-100 g) as previously described [15]. The tracheae were removed from sacrificed rats from the thyroid cartilage to the level of the bifurcation. After carefully removing the surrounding tissue(s), the tracheae were cut lengthwise and rinsed at least 3 times with Hanks' balanced salt solution supplemented with penicillin/streptomycin. After they were sliced into thin rings between the cartilaginous rings with a blade, the tracheal rings were transferred into 12-well plate. After standing in a small amount of bronchial epithelial media (BEM) at 37℃ in 10% CO2 for 24 hours, then the medium was exchanged. BEM was prepared from DEMEM/F12 by supplementing fetal bovine serum (10%), EGF (10 ng/ml), insulin (5 μg/ml), transferrin (10 μg/ml), tretinoin (0.5 μg/ml) and BSA (0.5 mg/ml). The culture was maintained for another 5 to 7 days, and the medium was exchanged every other day. From 3 to 4 days after the primary culture, numerous ciliated epithelial cells were crawling from the tracheal ring, and were employed in experiments. The purity of airway epithelial cells was identified to be > 95% by the expression of cytokeratins-14 (Figure S1), the marker for upper airway epithelial cells [16;17].
For exogenous mitochondria delivery, RAECs were incubated with 3.75×108/ml mitochondria isolated from rat airway epithelium for 24 hours, then treated with stimulus.
Cigarette smoke extract (CSE) Preparation
The cigarette smoke extract (CSE) used in the current study was obtained following the protocol reported before [18]. Briefly, a medium containing warm (37ºC) PBS was connected between cigarette and a vacuum pump. Commercial cigarettes (Hong Shuangxi Brand, Wuhan, China; tar 16 mg, nicotine 1.0 mg in each) were continuously smoked with a vacuum pump. Mainstream smoke was drowned and dissolved into 25 mL PBS. The smoking lasted for 5 min for each cigarette, and 6 cigarettes in total were smoked to prepare each 25 mL of CSE. The cigarette burning setting was 337.5 ± 75.01 mg/m3 (270 ± 60 ppm, n=4) for carbon monoxide (CO), 25.96 ± 5.77 mg/m3 (n=3) for nicotine and an estimated tar level of ~ 415 mg/m3, yielding the CSE with the nicotine content of 87.62 ± 5.18 μg/ml (n=3). CSE was frozen in aliquots and stored at - 80℃. 10% CSE solution was freshly prepared before use by diluting the aliquot CSE into DMEM/F12 medium.
Liver of Wilson's disease rat model
The Wilson's disease model was developed by copper-loaded diet in rats [19], which were fed with food containing 1g/kg copper sulfate and water containing 0.185% copper sulfate for 8 weeks. The mitochondria in the liver of the above Wilson's disease rats were characteristic in ultrastructure including unclear and greatly swelling cristae, increased density of matrix, discrete inclusions as well as shape alterations, distinguishable in morphology from mitochondria of airway epithelial cells in normal rats and thus employed in the present study as one tracer to track the localization of exogenous mitochondria.
Mitochondrial labeling and preparations from airway epithelial cells
To label mitochondria, DsRed-Mito or ascorbate peroxidase (APEX)-Mito plasmid (Clontech) was transfected into RAECs using Lipofectamine 2000 (Invitrogen) as we reported before [20]. Briefly, RAECs monolayers grown in 60-mm dishes were used for transfection with 250 µl opti-MEM containing 4 µg plasmid and 10 µl Lipofectamine 2000 at 37oC and 5% CO2. After 6 hours, culture medium was changed into DMEM containing 10% FBS.
Mitochondria were isolated from cultured RAECs by a modification of the previously described method [21] as follows: Exponentially growing RAECs were collected by trypsinization, rinsed 3 times by Tris-HCl (1.0 mM, pH 7.0), NaCl (0.13 M), KCl (5.0 mM), MgCl2 (7.5 mM), then fragmented with one half of the packed cell volume (PCV) of Tris-HCl (3.5 mM, pH 7.8), NaCl (2.0 mM) and MgCl2 (0.5 mM) with the aid of a homogenizer. The homogenate was promptly admixed with one ninth of PCV of Tris-HCl (0.35 M, pH 7.8), NaCl (0.2 M), MgCl2 (50 mM) and centrifuged for 5 min at 1800 g. The recovered supernatant was spun again using the above parameters. The supernatant was finally partitioned among Eppendorf tubes and spun at approximately 13,000 g in an Eppendorf microcentrifuge for 2 min. The final pellets of mitochondria were rinsed serially by Tris-HCl (35 mM, pH 7.8), NaCl (20 mM), MgCl2 (5 mM), then three times with Tris-HCl (10 mM, pH 7.4), EDTA (1 mM), sucrose (0.32 M), and re-suspended finally with the appropriate incubation solution. All the above purification procedures were conducted at 4 oC.
Mitochondria isolated from rat airway epithelium were prepared following Patel's method [22]. Freshly isolated tracheal epithelium from the control rat was immersed within 1 mL ice-cold isolation solution containing mannitol (215 mM), sucrose (75 mM), bovine serum albumin (BSA, 0.1%), HEPES (20 mM), EGTA (1 mM), trypsin (0.25 mg/mL) and a protease inhibitor cocktail (Sigma-Aldrich, P2714-1BTL), pH 7.2 (adjusted with KOH), and the tissue(s) were repeatedly snipped using scissors and homogenized for 15 min on ice. The homogenates were spun at 800g for 6 min at 4°C, and the recovered supernatant was spun again at 7000g for 15 min at 4°C. The final pellets of mitochondria were resuspended in ice cold PBS, and the number was counted under contract microscope with the staining of Janus green B. The purity of tracheal epithelium was verified under electronic microscope containing epithelial cells, however no fibroblasts or smooth muscle cells (Figure S2).
The respiratory functions of the naked mitochondria isolated from cultured RAECs and from fresh airway epithelium tissue were evaluated for the respiratory control ratio (RCR) and the NAD+/NADH (Figure S3A-B) and the integrity of isolated, naked mitochondria was examined under electronic microscope showing intact ultra-structures including double-layer membrane and clear cristae (Figure S3C-D).
Transplantation of isolated mitochondria into cultured RAECs
The cultured RAECs were incubated with DsRed- or APEX-labeled, exogenous mitochondria or with DsRed or APEX protein at 37oC and 5% CO2 respectively, then the RAECs were completely washed by PBS for DsRed fluorescent examination under confocal microscope, or prepared for electronic microscopy identification of APEX.
Separation and recovery of transplanted mitochondria
To separate and recover exogenous mitochondria after their transplantation into rat airway epithelia cells (RAECs), the endogenous mitochondria of RAECs were pre-labeled with GFP and then the RAECs were incubated with 3.75×108/ml DsRed-labeled exogenous mitochondria at 37ºC for 24 hours. Then mitochondria were isolated from ~ 2.5×106 RACEs, suspended in 500 µl Tris-buffer containing sucrose (320 mM), EDTA (1 mM), Tris-Base (10 mM), pH 7.4, BSA (0.1% wt/vol). 300 µl of the mitochondrial suspension were processed on a FACS flow cytometer (BD FACSAria II, Becton, Dickinson and Company, USA) and subsequently analyzed using BD FACSDiva software. The unlabeled mitochondria were used a negative control to set up voltages of forward scatter (FSC) and side scatter (SSC) at 220 and 346v, respectively. The wavelengths for detecting GFP and DsRed fluorescence were 488 nm and 530 ± 30 nm, or 554 nm and 585 ± 15 nm for excitation and emission, respectively.
Assessment of mitochondrial respiratory function
The oxygen consumption in RACEs was determined with a Clark-type oxygen electrode system (Oxytherm, Hansatech, UK) in a sealed glass chamber. 1×106/ml RACEs suspended in 2 ml of DMEM culture medium were placed in the chamber with a magnetic stirrer at 37°C. The oxygen consumption was defined as nanomoles O2 consumed per minute per 106 cells. The respiratory control ratio (RCR) in mitochondrial suspension was monitored with the Clark-type oxygen electrode system and a Mitochondria RCR Assay Kit (Genmed. Arlington, MA, USA) following the manufactures' procedures. Briefly, the chamber was filled with 2 ml medium containing KCl (100 mM), K2HPO4 (10 mM), MgCl2 (10 mM), EDTA (1 mM), glucose (20 mM), MOPSO (50 mM) and glutamate (5 mM), sealed and incubated for 1 min, then 20 µl isolated mitochondria (0.1 mg protein/µl) were introduced into the chamber through a connected microinjector and incubated for 1 min. The respiration of mitochondria was triggered by supplying the substrates of pyruvate (1.25 mM), malate (1.25 mM) and an inhibitor of ATP synthase, oligomycin (2 µM) through the microinjector, and the reaction was allowed for 2 min for the determination of the State IV respiration rate. The State III respiration rate was determined before and after adding an uncoupler, carbonyl cyanide m-chlorophenylhydrazone (CCCP, 3 µM) and 0.375 mM ADP. RCR was defined as the ratio of State III against State IV respiration rate. Triplicate measurements were performed for each separate experiment.
Estimation of cellular NAD+
Cellular NAD+ levels were measured using the colorimetric method and a NAD/NADH Quantification Colorimetric Kit (BioVision, USA). The pellet of 2×105 cells for each assay was collected by centrifuging at 2000 rpm for 5 min in a miro-centrifuge tube. The cells were broken by repeated freeze (20 min on dry-ice) and thaw (10 min at room temperature) using 400 μl of NADH/NAD Extraction Buffer. The cellular homogenates were vortexed for 10 sec, then spun down at 15000 rpm for 10 min. The extracted NADH/NAD supernatant was recovered. To decompose NADH, 0.2 mL of the supernatant was warmed for 30 min at 60°C, and 0.05 mL of NAD decomposed samples were added in a 96-well plate each by triplicates. 0.1 mL of NAD cycling enzyme and buffer mixture was mixed with each sample and incubated for 10 min at room temperature to convert NAD into NADH. 0.01 mL of NADH developer was supplied into each reaction and incubated for 5 hours at room temperature before the final measurement of OD450nm. The standard curve was generated following the manufacturer's protocol. And the ratio of NAD/NADH was determined by the formula of NADt (NAD and NADH) - NADH/NADH.
Measurement of airway responsiveness
Rats were anesthetized with urethane (1g/Kg, intraperitoneally). Following tracheal intubation, the animals were mechanically ventilated using a rat ventilator with the settings of a tidal volume at 6ml per Kg and a frequency of 76 breaths per min under muscle paralysis (suxamethonium chloride, 10 mg/kg, i.m). The whole airway resistance (RRS) were assessed by a differential pressure transducer (AD instruments, MLI 1050/D) and measured continuously. Following 1 min's nebulization of normal saline to record the baseline, rats were aerosol challenged successively with increasing concentration of Ach (0.3-100 mg/ ml in PBS) through an atomizer (Yuehua Model: WH-2000) with a circuit of 1 min's challenge and 5 min's interval, or alternatively with the gradient concentrations of NaCl at 1.8%, 2.7%, 3.6%, 4.5%, 5%, 5.4% and 6.3% each for 5 min's challenge and 5 min's interval. The atomizer was set at the working frequency of 1.7 MHz, which atomized more than 2 ml liquid in one minute and produced particles of 1~5 μm in their diameters. RRS was recorded after each challenge.
Measurement of Ach
Both the rat bronchoalveolar lavage fluid (BALF) and the supernatant of cultured RAECs were collected to quantify Ach level by commercial kit (Biovision, K615-100) [9]. The standard choline and samples were in placed into a 96 well plate. The Ach in the BALF and RAECs supernatant were converted to choline by acetyl cholinesterase added to the reaction. The amounts of choline including the total and free in each of the samples were calibrated by applying the fluorescence intensities to the standard curve.
Determination of ciliary beating frequency
The ciliary beating frequency (CBF) in both the freshly isolated tracheal ring and cultured RAECs were evaluated. Tracheal rings were obtained by slicing between the cartilaginous rings with a blade from the distal part of the intubated trachea of anesthetized rats. Approximately 2-3 rings, 0.5 mm in thickness were obtained from each trachea. The rings were prepared as described described [23], and placed in a small amount of 37℃ PBS on glass slides. The preparation was viewed at 200× using phase contrast with an inverted microscope (Olympus IX71). A small area of the ciliated epithelium was observed, images of ciliary beating were recorded by a high speed CCD (iCube NS1300cu) with a speed of 62 frames per second, digital images (640×480 pixels) were directly recorded. The image areas with beating cilia at the edge of the tracheal epithelium and RAECs were analyzed with the PC software HCImage. The HCImage software seized the history of all motion images within each of the analysis grids for the entire duration of the video recorded and expressed the variation of grayscale intensity as the function of time. This process tracking allowed the accurate measurement of CBF. The CBF was recorded for 5 seconds at 5 randomly selected sites of each ring.
Estimation of reactive oxygen species (ROS) and mitochondrial membrane potential
The ROS monitoring and quantitative estimation using DCFDA (invitrogen) in RAECs were performed as we recently described in detail [20] and also detailed in supplement. Alternatively, the superoxide production from RAECs was also evaluated using Dihydroethidium (DHE, Beyotime Biotechnology) and MitoSOX (Molecular Probes). After incubation with 5 μM DHE or 5 μM MitoSOX at 37℃ for 30min and wash three times, the red fluorescence from RAECs was detected under fluorescent microscope with excitation and emission wavelength of 535 nm and 610 nm, or 510 nm and 580 nm for DHE or MitoSOX, respectively.
To evaluate RAECs mitochondrial function, we performed the JC-1 assay to detect mitochondrial membrane potential (MMP). At low MMP, JC-1 is predominantly a monomer that yields green fluorescence with emission of 530 ± 15 nm. At high MMP, the dye aggregates yielding a red colored emission (590 ± 17.5 nm). Thus the depolarization of MMP was defined as a decline in the ratio of red against green fluorescence intensity. RAECs were loaded with JC-1 (10 μg/ml, invitrogen) at 37°C for 20 min and excited with wavelengths of 514 and 585 nm respectively, the alterations of the red (590 nm) against the green (530 nm) emission fluorescence intensity ratio were observed on confocal microscope. CCCP was used as a positive control for the induction of mitochondrial depolarization.
Measurement of IL-8 and LTB4 by ELISA
Rat IL-8 ELISA kit and rat LTB4 ELISA kit (both from R&D Systems) were used to measure IL-8 and LTB4 in the samples of rat lung tissue and the supernatant of cultured RAECs following the manufacturer's instructions.
BALF cell count
Immediately after the rats were sacrificed, the rat lungs were lavaged four times with PBS (2 ml per lavage) from a tracheal cannula and the bronchoalveolar lavage fluid (BALF) samples were collected and combined. The BALF was centrifuged (1500 rpm, 10 min) and the precipitation was resuspended with 1 ml PBS, total leukocytes in the BALF fluid were determined with a hemocytometer.
Histological study
The Paraformaldehyde fixed lung lobe was embedded in paraffin and sliced into slides with thickness of 4 μm. Following HE staining, the lung histology was observed under light microscope.
Electron microscopy
After being washed by PBS three times, the epithelium layer was peeled off carefully from the rat bronchus. Firstly, the tracheal epithelium, cultured RAECs or liver tissue was fixed with a phosphate-buffered glutaraldehyde (3%), then post-fixed with a phosphate-buffered osmium tetroxide (OsO4, 2%) [19;24]. The thin tissue sections were cut and prepared using an ultramicrotome, and finally examined under a transmission electron microscope (FEI Tecnai G2 20 TWIN) using an Olympus CCD (Cantega G2) and software (Cantega G2). To visualize ascorbate peroxidase (APEX)-labeled mitochondria [25;26], the samples were overlaid with H2O2 and 3,3'-diaminobenzidine (DAB) to allow APEX-catalyzed polymerization for 25 minutes before staining with OsO4.
Statistical Analysis
Results were presented as Mean ± SEM. The analysis of Student's t-test or One-Way ANOVA was performed for comparisons between 2 or ≥ 3 groups, respectively. Probability values < 0.05 were considered significant.
Results
Increased airway responsiveness to Ach and mitochondrial morphological changes upon cigarette smoke plus LPS exposure
To evaluate the changes of airway responsiveness in rats chronically exposed to cigarette smoke with infections, the animals were co-administrated with cigarette smoke and LPS for 4 weeks (CS/LPS treated rats), then airway responsiveness was measured after aerosolized challenge with increasing concentrations of Ach (0-100 mg/ml). As shown in Fig. 1A, nebulization of Ach elicited a concentration-dependent increase in the resistance of the whole respiratory system (RRS). The dose-response curve upon CS/LPS exposure moved upward, indicating increased airway responsiveness (p < 0.01 for each dosage of Ach). Of note, the airway resistance of CS/LPS rats was also higher than control in the absence of Ach stimulation, indicating the increased airway resistance at the basal level in rats after exposure to cigarette smoke plus LBS (Fig. 1A).
Changes of airway responsiveness and mitochondrial morphology in epithelium of rats upon cigarette smoke plus LPS exposure. A: Cigarette smoke plus LPS (CS/LPS) increased Ach-induced contractile response of rats bronchia (n=13) as compared to that of control (n=17). * P < 0.05. B: Electron micrographs showing long spindle mitochondria with clear cristae in airway epithelium in control rats (left, B), round mitochondria with swelling, unclear cristae in airway epithelium in CS/LPS rats (right, B).
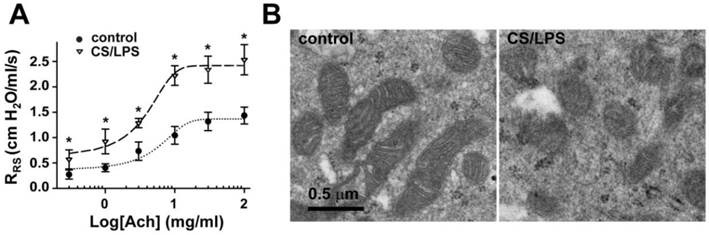
To further explore the possible underlying mechanisms of increased airway responsiveness in CS/LPS treated rats, ultrastructures of mitochondria in airway epithelium cells were observed (Fig. 1B). In epithelial cells of bronchus isolated from control rats, mitochondria were long spindles and their matrices were dense with well-developed cristae (Fig. 1B, left). In contrast, the percentage of damaged mitochondria represented as lower dense matrix and loss of missing cristae in CS/LPS treated group reached ~ 45%, dramatically higher than control of < 1% (Fig. 1B, right; and Fig. 3C).The increased quantity, size of the ribosome and their further aggregation along enlarged endoplasmic reticulum were also noted in the rats exposed to CS/LPS (Fig. 1B).
Mitochondria in the regulation of airway responsiveness in rats. A. The airway responsiveness in control, CS/LPS exposed and CS/LPS exposed rats delivered with exogenous mitochondria prepared from control rats (n=11-17, * p < 0.05 vs. control, some data in Fig 1A were re-presented for comparison). B-C Electron micrographs showing long spindle mitochondria with clear cristae in ciliated airway epithelium in control rats (left, B, control) and in normal rats for mitochondrial preparation (middle, B, mito), round mitochondria with swelling, unclear cristae in airway epithelium in CS/LPS exposed rats (middle, B, CS/LPS ), and mixture of two types of mitochondria with distinct morphology in ciliated airway epithelium in CS/LPS exposed rats after delivery of exogenous mitochondria prepared from control rats (right, B, CS/LPS+mito) and quantitative comparisons of two shapes of mitochondria (C). Quantitative analysis was obtained from 204, 197, 194 and 221 mitochondria of 11-17 separate ciliated airway epithelial cells from 5, 4, 6 and 7 rats, respectively for control, mito, CS/LPS and CS/LPS+mito rats respectively. The mitochondria in each cell were counted from six fields randomly chosen each with an area of 5.25 μm2. D-F The tissue of tracheal epithelium was freshly stripped from the airway of rats after an administration of APEX-labeled mitochondria. The subsequent electronic microscopy examination identified the localization of the APEX-labeled, exogenous mitochondria mainly within the ciliated epithelial cells (D), occasionally within the goblet cells (E) and basal cells (F). The results were consistent in 4 tissues from 3 individual rats. endo mito: endogenous mitochondria (white arrow); exo mito: exogenous mitochondria (black arrow). white star filled: ciliated epithelial cells; white star open: goblet cells; black star: basal cells.
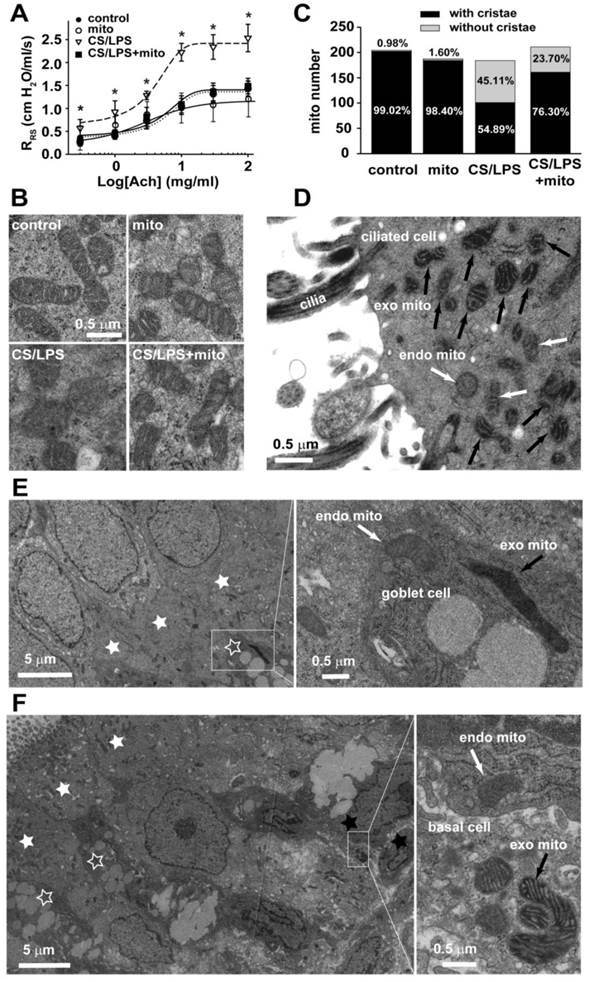
However, the above observation can not reveal if the changes of mitochondria were the reason or the consequence of increased airway responsiveness, so we delivered normal epithelial mitochondria into airway epithelium in vitro and in vivo, to explore whether mitochondrial delivery can reverse the AHR induced by cigarette smoke and LPS co-exposure.
Transplantation of exogenous mitochondria into airway epithelial cells in vitro and in vivo
To determine if exogenous mitochondria can be delivered into rat airway epithelial cells (RAECs), mitochondria labeled with DsRed (Fig. 2A) or prepared from Wilson's disease rat liver (Fig. 2H-J) were employed respectively. DsRed-labeled mitochondria were incubated with RAECs in a series of mitochondrial quantity or concentration. After different periods of incubation respectively and washout of mitochondria-containing medium, DsRed fluorescence was identified by live cell confocal imaging to localize within RAECs after incubation with DsRed-labeled mitochondria (Fig. 2A, upper), not in those after incubation with DsRed protein or blank control (Fig. 2A, middle and lower), indicating the successful transplantation of exogenous mitochondria into RAECs.
Transplantation of mitochondria into airway epithelial cells. A: Live cell confocal imagings of rat airway epithelial cells (RAECs) in culture at 37℃ and 5% CO2 after 24 h incubation with DsRed-labeled mitochondria (upper), DsRed (middle) or vehicle (lower, n=3 for each), and staining with DAPI and Dio for localization of nucleus and cell membrane, respectively. B-G: Dynamics of intracellular transplantation of DsRed labeled mitochondria in RAECs showing the calibration cure between the numbers of isolated mitochondria (mito) and DsRed fluorescent intensities (B), the dependence of intracellular DsRed-mitochondrial quantity (D) and efficiency of DsRed-mitochondria transplanted cells (C) on exogenous mitochondrial concentrations after a 24 hour incubation, the relative distributions of intracellular DsRed-mitochondrial quantity after a 24 hours incubation of 3.75×108/ml mitochondria (G), the time-dependence of intracellular DsRed-mitochondrial quantity (* p < 0.05 vs. 6h, F) and efficiency of DsRed-mitochondria_transplanted cells (* p < 0.05 vs. 6h, E) after incubating with 3.75×108/ml mitochondria (n=3 for B-G). H-K: Electron micrographs showing long spindle mitochondria with clear cristae in RAECs (H), round mitochondria with swelling, unclear cristae in liver cells from Wilson's rats (LC, H), mixture of two types of mitochondria with distinct morphology in RAECs after intratracheally instilled rats with mitochondria prepared from liver of Wilson's rats (RAEC/LC, H) and quantitative comparisons of two shapes of mitochondria (I) as well as the ratio of elongated mitochondria to round ones in cells (* p < 0.05 vs. LC, J). Quantitative analysis was obtained from 204, 142 and 164 mitochondria of 17, 11 and 12 separate cells for RAEC, LC and RAEC/LC respectively. K Representative electronic microscopy images showing endogenous mitochondria (white arrow) in control RAECs without any incubation of exogenous mitochondria (left), endogenous mitochondria (white arrow) in RAECs after incubation with APEX protein (middle), and the intracellular localization of APEX-labeled mitochondria (black arrow) together with endogenous ones (white arrow) within RAECs after incubation with APEX-labeled, exogenous mitochondria (right). L-O: The sorting and recovery of GFP-labeled, endogenous mitochondria and Ds-Red-labeled, exogenous mitochondria from RAECs without (L) or with transplantation of exogenous mitochondria (M), and the subsequent evaluation of mitochondrial function indexed by NAD+/NADH (N) and RCR (O) in endogenous (endo mito) and exogenous mitochondria (exo mito).
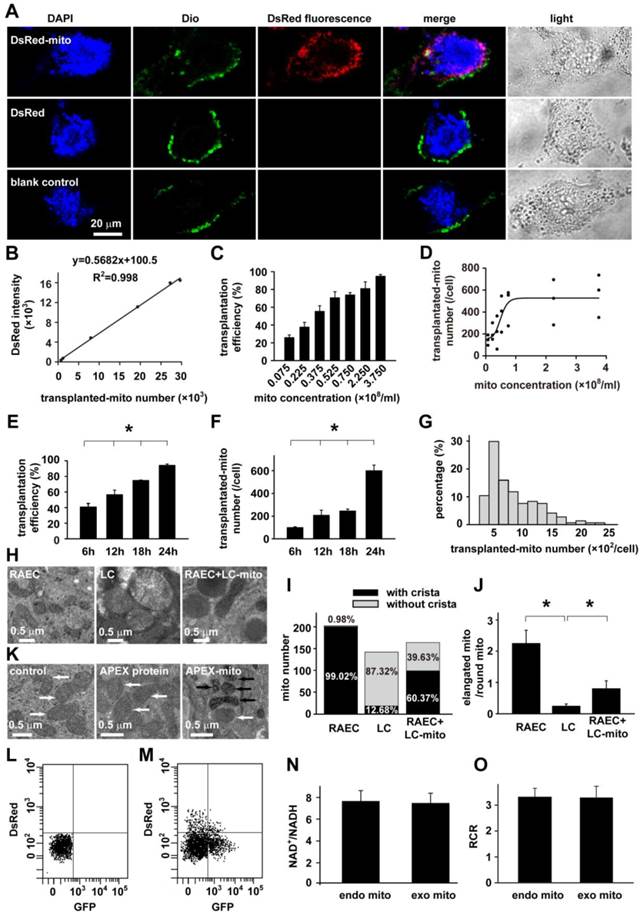
To verify the intracellular localization of the DsRed-labeled exogenous mitochondria, the final wash elution was collected for DsRed fluorescence detection. The OD value of elution was almost similar with PBS and obviously lower than the incubation medium (Figure S4), indicating the complete washout of the DsRed-labeled exogenous mitochondria. Furthermore, RAECs were also incubated with DAPI and DIO, the marker for nucleus and cytoplasmic membrane respectively. As also shown in the Fig 2A, the DAPI, DIO and DsRed were co-existed at the same layer under confocal microscope, clearly illustrating the intracellular localization of the exogenous mitochondria in RACEs.
Quantitative analysis clearly demonstrated a concentration- (Fig. 2B-D) and time-dependent transplantation of exogenous mitochondria into RAECs in culture (Fig. 2E-G). Hereafter, when RAECs were incubated with 3.75×108/ml DsRed-labeled mitochondria for 24 hours, the transplantation efficiency reached ~ 93% and the average amount of intracellularly-transplanted mitochondria reached ~ 600 for each cell (Fig. 2E-F).
To verify the successful transplantation of exogenous mitochondria (RAECs), mitochondria of Wilson's liver with characteristic ultrastructure were employed in the study as an alternative method. Compared with the long spindle mitochondria with clear cristae in RAECs (Fig. 2H, left), mitochondria in liver cells of Wilson's disease rat exhibited distinct ultrastructure since they were big and round with less dense matrices and unclear or swelling cristae (Fig. 2H, middle). These features were exploited to distinguish them from endogenous mitochondria in RAECs and to confirm the feasibility of intracellular delivery of exogenous mitochondria in vivo. For this purpose, rats were intratracheally instilled with mitochondria prepared from Wilson's disease rat liver. 24 hours later, airway epithelium were isolated and prepared for electron microscopy. As shown in the electron micrograph, there was a mixture of two types of mitochondria with different morphology (Fig. 2H, right) in administrated epithelium, long spindle endogenous mitochondria with clear cristae from the epithelial cell itself, and big round exogenous mitochondria with unclear cristae. Quantitative evaluation of the mitochondria by their cristae morphology (Fig. 2I) as well as their shape (Fig. 2J) among the three types of cells also proved the intracellular transplantation of liver cell mitochondria.
We then further confirmed the intracellular delivery of exogenous mitochondria in normal, healthy status in contrast to the above liver mitochondrial from cancer cells. The mitochondria in RAECs were pre-labeled with mitochondria-targeted ascorbate peroxidase (APEX), a genetic tag for mitochondrial tracking under electron microscopy [25;26]. After incubation with the APEX-labeled mitochondria prepared from RAECs, the subsequent electronic microscopy examination successfully identified the intracellular localization of the exogenous mitochondria (black arrow) within RAECs (Fig. 2K).
In order to determine if transplanted mitochondria still remain functional, endogenous mitochondria of RAECs were pre-labeled with GFP and then the RAECs were transplanted with DsRed- labeled exogenous mitochondria. The mitochondria were finally isolated, sorted and recovered from the RAECs by flow cytometry for functional evaluation (Fig. 2 L and M). As shown in Fig. 2, the ratio of NAD+/NADH in endogenous and exogenous mitochondria was almost equal (7.73 ± 1.03 vs. 7.56 ± 0.89, p > 0.05, Fig. 2N). Consistently, there was no statistical difference in respiratory control ratio (RCR) between endogenous and exogenous mitochondria (3.31 ± 0.35 vs. 3.29 ± 0.44, p > 0.05, Fig. 2O). These results suggest that transplanted exogenous mitochondria were functional in host cell.
To experimentally address the mechanisms underlying how the cells take the naked mitochondria, we explored several pathways or factors involved in pinocytosis, a process describing endocytosis of particle. Chlorpromazine, the inhibitor of clathrin did not affect the internalization of DsRed-labeled exogenous mitochondria. By contrast, the inhibitor of sodium-hydrogen exchanger, ethylisopropylamiloride (EIPA); the inhibitor of microtubule polymerization, nocodazole; and the inhibitor of actin interaction/polymerization, cytochalasin D, significantly inhibited the internalization of DsRed-labeled exogenous mitochondria, respectively (Figure S5). The above results suggest that the internalization of exogenous mitochondria in RAECs involves macropinocytosis, but does not depend on clathrin. The above findings are generally consistent with a recent study in uterine endometrial gland-derived mesenchymal cells [27].
Delivery of exogenous mitochondria on airway hyperresponsiveness (AHR)
To explore any potential effect of exogenous mitochondria on AHR, mitochondria extracted from the freshly isolated airway epithelium of control rats were introduced intratracheally into CS/LPS-treated rats at the concentration of 3.75×108 /ml for an additional week after cigarette smoke plus LPS treatment. The transplantation of exogenous mitochondria significantly attenuated AHR caused by 4 weeks treatment of CS/LPS intervention (p < 0.05, Fig. 3A). However, transplanted mitochondria had no effects on airway responsiveness in control rats (p > 0.05). Meanwhile, the transplantation or delivery of exogenous mitochondria substantially restored the mitochondrial structural changes in airway epithelium (Fig. 3B). As shown in Fig. 3C, the quantitative estimations of the mitochondria with or without clear cristae in epithelium of among the three types of animals further confirmed the restoration of mitochondrial morphology by transplantation of exogenous mitochondria to CS/LPS treated rats, e.g. the abnormal or damaged mitochondria as characterized by the absence of clear crista in CS/LPS-exposed rats after mitochondrial transplantation obviously decreased to ~24% as compared to ~45% in the CS/LPS-exposed rats without mitochondrial transplantation (p < 0.05). To reveal what type(s) of epithelial cells were transplanted, we performed electronic microscopy examination on our endothelium tissues stripped from rats after an administration of APEX-labeled mitochondria. As shown in the Fig. 3D-F, the exogenous mitochondria were identified within the ciliated epithelial cells, the goblet and basal cells and the majority of them appeared with the ciliated epithelial cells.
The above results demonstrated that exogenous mitochondrial transplantation rectified the changes of endogenous mitochondrial in airway epithelium and reversed the AHR induced by CS/LPS exposure.
Mitochondria and cholinergic activity in the regulation of CBF
Effective ciliary beating is an important function for mucus clearance, and the ciliary beating frequency (CBF) at a sufficiently high speed is necessary for the superior ciliary function. Interestingly and by accident, we found that the challenge of Ach, in the process of detecting airway responsiveness, depressed the CBF of tracheal epithelium in both control and CS/LPS treated rats (Fig. 4A). Similar with previous reports, the baseline CBF of CS/LPS treated rats was apparently lower than that of control subjects which represented CBF level at physiological status (6.12 ± 1.05 vs. 8.25 ± 1.65 Hz, p < 0.05). In addition, after 5 min administration of 30 mg/ml Ach, the CBF was significantly decreased in both CS/LPS (4.83 ± 0.97 Hz) and control groups (6.51 ± 1.58 Hz). Ipratropium bromide (IB), a muscarinic M3 receptor blocker, remarkably increased the baseline CBF of CS/LPS treated rats, while it showed no obvious effect on that of control groups (Fig. 4A and supplementary movie: Tissue1-6), further indicating the enhancement or activation of muscarinic receptor activity in down-regulating CBF. Of note, IB at the concentration of 75 µg/ml was chosen in the above experiments. The 75 µg/ml was the highest concentration without interfering CBF by IB itself according to our preliminary examination in vitro on the effects of IB on CBF in cultured RAECs (Figure S6).
Mitochondria in the regulation of CBF. A: The effects of M receptor inhibitor, ipratropium bromide (IB) on CBF in rats without any treatment (blank) and acetylcholine (Ach)-induced decline in CBF in rat right inferior lobar bronchus in control and CS/LPS exposed rats (n=10-13, * p < 0.05). The waveform graph of cilia beating last for 1000 millisecond was drawn and columns represented mean±SD of CBF. B: The quantitative analysis of Ach level in BALF of CS/LPS-exposed rats (n=6 or 4 for control or CS/LPS, respectively, p > 0.05). C: The quantitative analysis of Ach in supernatant of cultured RAECs treated by CSE/LPS (n=5 for each, p > 0.05). D: Time-dependent decline in CBF of cultured RAECs exposed to LPS plus cigarette smoking extract (CSE/LPS) (n=5 for each, * p < 0.05). E: IB and mitochondrial transplantation on 3 hour exposure of CSE/LPS-induced decline in CBF of cultured RAECs and the effects of M receptor enhancer, win62577 (WIN) (n=4-8, * p < 0.05).
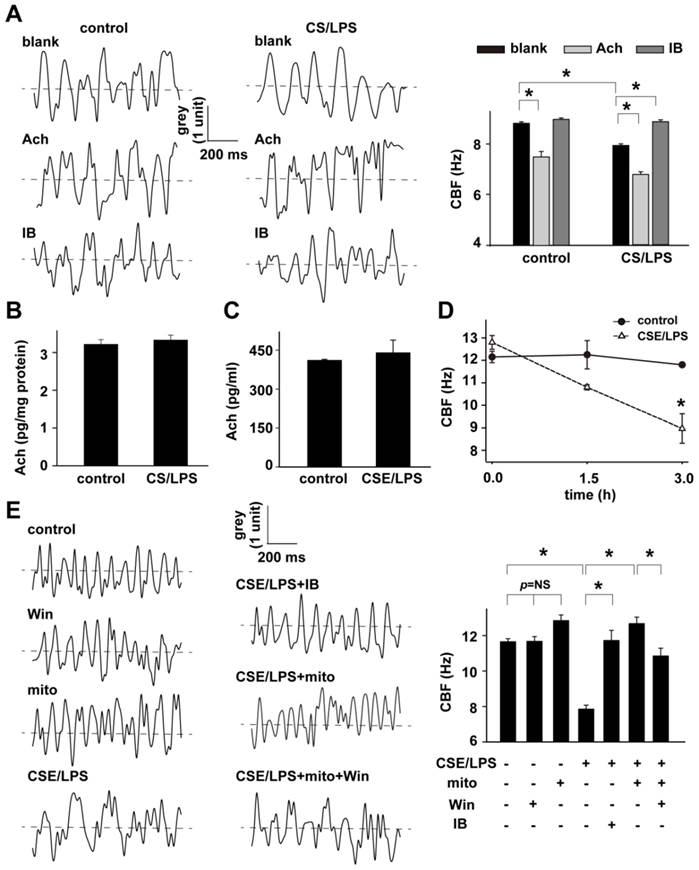
Influence of CS(E)/LPS on Ach level in the BALF or supernatant of cultured RAECs
Although IB significantly restored CBF of CS/LPS-treated rats, the Ach levels in BALF and supernatant of cultured RAECs were all similar with controls (Fig. 4B and C).
Role of mitochondria in augmented cholinergic activity
To further investigate the potential role of mitochondria in the down-regulated CBF upon cigarette smoke and LPS exposure, a cell model was adopted by exposing RAECs to 10% cigarette smoking extract plus 1mg/ml lipopolysaccharide (CSE/LPS) [28]. As shown in Fig. 4D, the CBF of control RAECs maintained stable around 12 Hz during 3 hours' observation, but showed a time-dependent decline in RAECs stimulated with CSE/LPS, and there was a significant decrease by the exposure for 3 hours. Thus we chose 3 hours as the exposure time period to carry out the subsequent experiments to examine the effects of exogenous mitochondria on down-regulated CBF by CSE/LPS exposure and its correlations with cholinergic activity. The transplantation of exogenous mitochondria slightly modulated the baseline CBF level in control cells (mito, p = NS vs. control, Fig. 4E), but significantly increased CBF in CSE/LPS-exposed cells (CSE/LPS + mito, * p < 0.05 vs. CSE/LPS, Fig. 4E). A similar effect was also observed by blocking muscarinic M3 receptor with IB administration (CSE/LPS + IB, * p < 0.05 vs. CSE/LPS, 4E). However, there was no extra increase CBF in CSE/LPS-exposed treated with mitochondria plus IB. The Win62577 was originally defined as a neurokinin-1 receptor antagonist but also an allosteric enhancer of muscarinic M3 receptor sensitivity [29;30]. Win62577 at the concentration of 3 µg/mL, which did not affect CBF by itself (Figure S7), significantly attenuated the protective role of mitochondrial transplantation (CSE/LPS + mito + Win, p < 0.05 vs. CSE/LPS + mito, 4E and supplementary movie: Cell1-7).
These data indicate the possibility of the existence of increased cholinergic activity in RAECs upon CSE/LPS stimulation, and the delivery of exogenous mitochondria effectively attenuated the increased cholinergic activity, which in turn reversed the depressed ciliary beating. It is known that the cholinergic activity is determined by two factors, the quantity of produced Ach and the sensitivity of muscarinic receptors. Similar to a previous study [9], 3 hours' CSE/LPS stimulation didn't change the level of Ach in the cultured supernatant of RAECs (Fig. 4C), excluding the possibility of over produced or secreted Ach in enhancing the cholinergic activity. These results strongly indicate that the increased cholinergic activity induced by CSE/LPS stimulation is mainly determined by the increased receptor activity, and that exogenous mitochondria regulated the CBF of airway epithelium, possibly through affecting the cholinergic sensitivity.
Mitochondria affect epithelium cholinergic sensitivity by regulating ROS production
Non-neuronal cholinergic system has been recently suggested to be critical for the mobilization of airway epithelium and the releasing of chemotactic agents mediating neutrophil activation and accumulation within the airways of COPD patients [31]. Leukotriene B4 (LTB4) is an important inflammatory mediator in the pathogenesis of COPD, which was very recently demonstrated to be released from human airway epithelial cells by the stimulation of Ach with a concentration dependent manner and via M receptors [9]. Thereafter, LTB4 released in the supernatant of cultured RAECs with different stimulations was tested to evaluate/represent the non-neuronal cholinergic activity of airway epithelial cells in our study.
Considering that the CSE/LPS stimulation had no obvious effect on Ach production of RAECs, we then used the LTB4 level in the culture supernatant to quantitatively evaluate the non-neuronal cholinergic sensitivity of RAECs. As shown in Fig. 5A-B, there was a trace amount of LTB4 baseline in the supernatant of control RAECs, which was increased in the presence of M receptor enhancer win62577 (Win vs. control, p<0.05. Fig. 5A), but not significantly affected by IB administration (Fig. 5A, IB) or exogenous mitochondrial delivery (Fig. 5B, mito). CSE/LPS stimulation significantly increased the LTB4 secretion (CSE/LPS vs. control, p<0.05, Fig. 5A-B), suggesting increased cholinergic receptor sensitivity. However, this increasing was remarkably reversed by exogenous mitochondrial transplantation (CSE/LPS + mito vs. CSE/LPS, p < 0.05, Fig. 5B), as well as the treatment of IB (CSE/LPS + IB vs. CSE/LPS, p < 0.05, Fig. 5A). These data indicated that mitochondrial transplantation suppressed the enhancement, not the baseline, of cholinergic receptor sensitivity caused by CSE/LPS stimulation.
Mitochondria in the regulation cholinergic sensitivity and oxidant stress in cultured RAECs. A: Evaluation of cholinergic sensitivity by secreted level of LTB4 from RAECs and the effects of CSE/LPS exposure. B: Mitochondria transplantation on CSE/LPS-enhanced cholinergic sensitivity in RAECs. C: Mitochondrial transplantation on CSE/LPS-stimulated oxidant stress in RAECs. D. Oxidant stress on the regulation of CSE/LPS-enhanced cholinergic sensitivity in RAECs. E-F: Representative fluorescence of JC-1 showing CSE/LPS-induced alterations in mitochondria membrane potential and ratio of red against green fluorescence (n=4, * p < 0.05 for each).
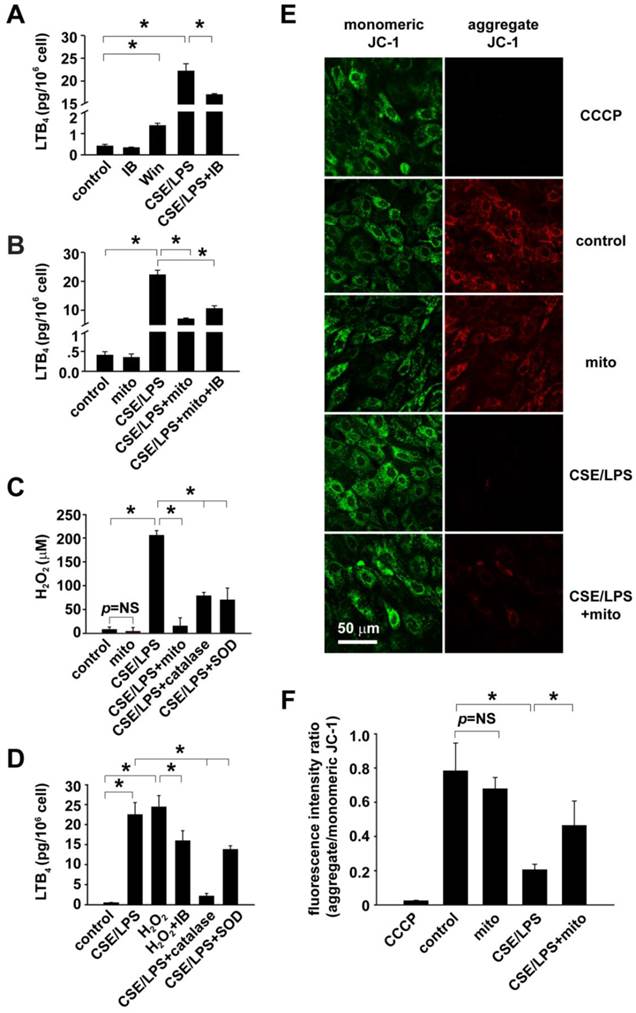
In order to estimate the mechanistic importance of ROS in the regulation of epithelial cholinergic sensitivity by mitochondria, ROS production in RAECs was detected and calibrated using DCFDA (Fig. 5C). In CSE/LPS-stimulated RAECs, the ROS production was equivalent to 198.72 ± 17.16 μM exogenous H2O2; catalase and SOD effectively decreased the CSE/LPS-induced ROS production, respectively. In mitochondria-delivered RAECs, the CSE/LPS-enhanced ROS production was also significantly inhibited (CSE/LPS + mito vs. CSE/LPS, p < 0.05, Fig. 5C). The CSE/LPS-stimulated ROS production and the rescue effect of mitochondrial transplantation were also verified using DHE/MitoSOX to examine the superoxide level (Figure S8). Then 200 μM H2O2, the equivalent amount of ROS production in CSE/LPS-stimulated RAECs, was applied to RAECs to mimic oxidative status induced by CSE/LPS, and LTB4 secretion was detected for cholinergic receptor sensitivity. We observed that under this condition, the RAECs exhibited a cholinergic activity similar to that of CSE/LPS stimulation, which was decreased by co-treatment with IB (H2O2 + IB vs. H2O2, p < 0.05, Fig. 5D). Meanwhile, the increased LTB4 secretion induced by CSE/LPS was significantly suppressed by catalase or SOD, and catalase appeared to have more significant effects (Fig. 5D). These results suggested that CSE/LPS stimulation enhanced the cholinergic receptor sensitivity of RAECs by ROS production, and H2O2 was the principal ROS.
To further determine the functional alternations of mitochondria after CSE/LPS stimulation or exogenous mitochondrial delivery, the changes of mitochondrial membrane potential was assessed in RAECs with the staining of JC-1. Significantly decreased potential was found in CSE/LPS treated cells (CSE/LPS vs. control, p < 0.05, Fig. 5F). Mitochondrial delivery attenuated the membrane potential decline caused by CSE/LPS stimulation (CSE/LPS + mito vs. CSE/LPS, p < 0.05, Fig. 5F), while it had no obvious influence on the control (mito vs. control, p = NS, Fig. 5F). Taken together, these results indicate that CSE plus LPS stimulation induced mitochondrial dysfunction in RAECs, leading to the increased production of ROS, which subsequently resulted in augmented cholinergic sensitivity of RAECs.
Mitochondrial transplantation attenuates airway remodeling and inflammation in CS/LPS-exposed rats
Chronic inflammation in peripheral airways and lung parenchyma and subsequent bronchial remodeling are important features of COPD. To prove our findings in vivo, we next investigated the effect of exogenous mitochondrial transplantation on histopathology changes of the CS/LPS treated rat model. In the rat model after 4 weeks cigarette smoke exposure plus 2 times of LPS intratracheally instillation, characteristic pathological alternations of COPD were observed in the lung as expected. The histologic slice showed various degrees of enlarged airspace and alveolar wall destruction in the lung, and the significantly decreased mean linear intercept (MLI) (CS/LPS vs. control, p < 0.05, Fig. 6A) indicated the occurrence of emphysema. The bronchiolar wall appeared apparently thickened (CS/LPS vs. control, p < 0.05, Fig. 6B) for proliferation of mucus cell and infiltration of inflammatory cells. In addition, the images of electron microscopy showed incomplete and less cilium on the surface of trachea epithelium of CS/LPS treated rats compared with control (CS/LPS vs. control, p < 0.05, Fig. 6C), and the measurement of CBF also revealed a decline of the cilia beating (CS/LPS vs. control, p < 0.05, Fig. 6D).
Mitochondrial transplantation on CS/LPS exposure-induced bronchial remodeling and chronic airway inflammation. A: pulmonary alveoli structure and quantification of mean linear intercept (MLI). B: bronchial remodeling and quantification of bronchial wall thickness. C: cilia ultrastructure and quantification of cilia number. D: CBF of airway epithelium in vivo. E: leukocyte accumulation in bronchoalveolar lavage fluid. F: LTB4 levels in pulmonary tissue homogenate. G: IL-8 levels in pulmonary tissue homogenate. * p < 0.05 for each. Shown in each bar is the total rat number of the group from at least 3 independent sets of experiments.
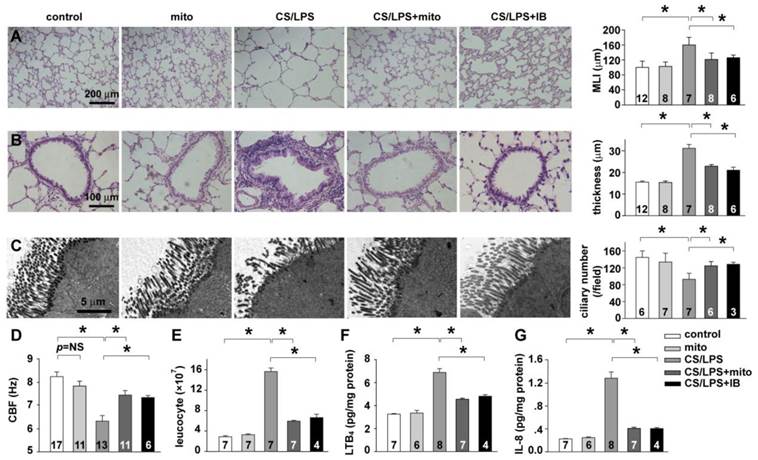
These results indicated an impaired cilia structure and function in the CS/LPS treated rat model. To further evaluate the lung inflammation in CS/LPS treated rats, leukocytes accumulation and neutrophil chemoattractants expression were measured. The significant increase of leukocytes in bronchoalveolar lavage fluid (CS/LPS vs. control, p < 0.05, Fig. 6E) as well as the elevated level of LTB4 and IL-8 in pulmonary tissue homogenate (CS/LPS vs. control, p < 0.05, Fig. 6F-G) consistently suggested an augmented inflammation in CS/LPS-treated rats. Intriguingly, delivery of exogenous mitochondria intratracheally significantly rescued the above pathological alternations of COPD, which similar to the effects of IB (CS/LPS + mito vs. CS/LPS, p < 0.05, Fig. 6A-G).
Discussion
We established COPD rat with AHR by cigarette smoke and LPS intratracheal instillation [13;14;32], with expectation to better mimic the clinical progression of COPD usually caused and or exacerbated by cigarette smoke and repetitive respiratory infection [33]. The release of Ach in the airway can lead to airway contraction and mucus secretion through the muscarinic receptors and thus contribute to the development of AHR and COPD [34]. As an inhibitor of muscarinic receptor, IB at the concentration of 75 μg/ml that by itself did not affect CBF partially restored CBF in CS/LPS-treated rat and cultured RAECs (Fig. 4), suggesting the possible role of hypercholinergic activity in CS/LPS-induced decline in CBF. This experimental finding is in consistence with a recent report showing that tiotropium bromide, a long-acting anti-cholinergic medication, caused a persistent increase in CBF in nasal cilia from COPD patients [35]. Although there was evidence indicating an increased Ach release in AHR [34], CS/LPS did not alter the level of Ach either in BALF of rats or in the cultured supernatant of RAECs in the current study (Fig. 4), hence, our results were more inclined to suggest that it is the increased muscarinic receptor sensitivity which is involved in the CBF decline associated with COPD and AHR upon cigarette smoke and LPS exposure. The hypercholinergic activity appeared to contribute to the development of AHR by > 60% as quantitatively evaluated in airway responsiveness to hypertonic NaCl (Fig. S9).
The synthesis and release of Ach are involving not only parasympathetic nerves but also several other cell types such as epithelial cell, fibroblasts, mast cells, lymphocytes and alveolar macrophages, named the non-neuronal cholinergic system [36;37]. In non-neuronal cholinergic system, the airway epithelium is anatomically the primary cell type first exposed to smoke and may therefore be the original site for smoke to initiate inflammation and the subsequent downstream events. The epithelial Ach regulates airway inflammation and mucociliary clearance through a local auto/paracrine pathway [37]. Since Ach stimulated LTB4 secretion with a concentration-dependent manner via M receptors [9], the change of LTB4 level was measured to indirectly reflect the cholinergic activity in CSE/LPS-treated RAECs. Consistently, CSE/LPS significantly augmented the LTB4 secretion (Fig. 5), but had no effect on the secreted level of Ach in the cultured supernatant of RAECs as mentioned above. These results further suggested the existence of increased cholinergic receptor sensitivity in airway epithelial cells upon cigarette smoke and LPS stimulation. Of note, the AHR is the complex phenotype in several kinds of respiratory disease such as COPD, asthma and cystic fibrosis [1], and AHR cannot be mimicked in vitro approaches. The experimental findings from some in vitro studies may be limited for understanding the exact mechanisms in vivo.
Mitochondrial dysfunction and morphological alternations have been observed in a wide array of pathological process including airway inflammation and AHR [10-12]. In electron microscopic examination of the airway tissues, we demonstrated that mitochondria in airway epithelial cells tended to swell and lose normal cristae in rats that had been treated with cigarette smoke and LPS (Fig. 1), suggesting mitochondrial injury and even predictable dysfunction occurred during CS/LPS exposure. This result is in accordance with previous reports showing that mitochondria in the airway epithelial cells from COPD patients tended to fragment and deplete in cristae, while the same morphological change was noted in human bronchial epithelial cell lines that experienced prolonged CSE exposure for 6 months [12]. In sample preparation for electron microscopic examination in the current study, the bronchus epithelial layer was stripped and washed by PBS. We thus were unable to observe mitochondrial changes in airway smooth muscle cell or macrophagocyte. But in several studies, mitochondrial damage or dysfunction were also found in airway smooth muscle cell of COPD patient or cultured airway smooth muscle cell after exposure to CSE [38;39].
To further clarify the correlation of mitochondrial alternation and the AHR to Ach in CS/LPS treated rats, a technique for the delivery exogenous mitochondrial into airway epithelial cells was established. The successful intracellular delivery of exogenous mitochondria was verified by tracking the DsRed fluorescence-labeled exogenous mitochondria in vitro and by ultrastructural identification of the unique morphology of exogenous mitochondria prepared from Wilson's liver in vivo. The genetic tag of APEX labeling and tracking the normal, healthy mitochondria prepared from RAECs further confirmed the feasibility of mitochondrial transplantation for the subsequent studies in evaluation of their biological role(s). The AHR and the increased LTB4 secretion induced by CSE/LPS stimulation was remarkably reversed (Fig. 3 and Fig. 5), indicating a critical role of mitochondria in regulating airway responsiveness and also cholinergic receptor sensitivity of airway epithelial cells.
Interestingly, we found that CBF in separated tracheal epithelium (Fig. 4) were slightly lower than that of cultured RAECs (Fig. 4). This difference may be relevant to the intrinsic characteristic of separated tissue and cultured cell. However, the exact reason was unclear. Depressed cilia function and impaired cilia mucus clearance are important characteristics and may play critical roles in the pathogenesis of various airway inflammatory diseases including COPD and asthma [39;40]. In the current study, we demonstrated a consistent decline of CBF in the airway epithelium of CS/LPS treated rats as well as the CSE/LPS-stimulated RAECs (Fig. 4). These results are in accordance with previous reports, one showing a time dependent decrease of CBF baseline in mice trachea epithelium upon months of cigarette exposure [41], and the other demonstrating a decline of CBF from ~12Hz to ~8Hz in primary airway epithelial cells by 3 hours treatment of 5% CSE and 100 mmol/L ethanol [42]. CBF reflects the speed of cilia beating and mainly depends on its function, not its number. However, cilia loss in RAECs was also a remarkable characteristic of airway inflammation induced by CS/LPS. Thus, slower CBF together with and reduced cilia number resulted in the mucus clearance inhibition facilitating the development of inflammation. In our study, exogenous mitochondria transplantation not only increased CBF (Fig 4 and Fig 6) but also cilia number (Fig 6).
Moreover, this protective role of mitochondria was significantly inhibited by win62577, an enhancer of M3 receptor. Additionally, the level of Ach was not elevated in CSE/LPS treated RAECs and rats, all these results suggest that transplanted mitochondria decreased M3 receptor activity. Till the present, there were only very limited studies exploring the possible mechanism underlying how mitochondria are involved in ciliary dysfunction [43;44]. Manzanares proposed a reactive oxygen species (ROS)-dependent mechanism of ciliary regulation [45], which may suggest, to some degree, a clue connecting mitochondria and the mechanisms of ciliary beating regulation.
Mitochondria are primary sites of ROS production inside eukaryotic cells. While low level of ROS serves as signaling messengers and participates in multiple phisiological functions, when their production overwhelms the innate scavenging mechanisms, the resultant oxidative stress is harmful to the cell [46]. In the lung, several studies have shown that mitochondria- derived ROS can trigger expression and release of proinflammatory cytokines and promote the pulmonary inflammation [47]. In the current study, we observed a considerable increase of ROS production as well as increased LTB4 secretion in RAECs after CSE/LPS stimulation. We further found that exogenous mitochondrial transplantation attentuated this potentially deleterious ROS increase, and the elevated cholinergic activity (Fig. 5). These results further clarified the functional change of mitochondria upon cigarette and LPS exposure besides the mophologial alternation we have disscussed above. Furthermore, it indicated that ROS may be involved in the regulation of epithelial cholinegic activity. Next, we demonstrated that when ROS scanveger catalase was supplied, the increased epithelial cholinegic activity induced by CSE/LPS stimulation was significantly decreased (Fig. 5), logistically proving the role of ROS, especially the H2O2 derived from mitochondria, in epithelial cholinergic regulation.
Although the mechanism underlying oxidant stress or H2O2-enhanced activity of the muscarinic receptors is beyond the scope of this paper, it may be related with the oxidation of cysteine residues, the formation of disulfide bonds and the subsequent conformational change of the receptors. It is also noted that the above in vitro findings trying to tie ROS and cholinergic activity are indrect proof, we can only speculate that cigarette smoke plus infection stimulation can cause mitochondria injury in airway epithelium, and then lead to the over production of mitochondrial derived ROS, which subsequently augments the non-neuronal epithelial cholinergical activity, and finally results in increased airway inflammation and decreased cialiary clerance. In this line, mitochondria are possibly the primary trigger in initiating the downstream events. To verify our speculation, we transplanted noraml epithelial mitochondria into the rat airway, and demonstrated that exogenous mitochondria delivery rescued most of the chronic obstructive pathological alternations, including the AHR (Fig. 3), the airway remodeling, the ciliary morphology and the inflammatory cytokine expression in the airway of rats treated with cigarette smoke and LPS for 4 weeks (Fig. 6).
Supplementary Material
Supplementary Methods, References, Figure S1-S9 and Legends.
Acknowledgements
Funding: The work was supported by the grants from Natural Science Foundation of China (81170048, 81330001, 81400027, 30971162, 81470252, 81170049, 31270031, 31400990, 81572274, 30900545 and 81370176), State Key Laboratory of Respiratory Disease (SKLRD2016OP001), Ministry of Education of China (NCET-12-0484), Health Department of Zhejiang Province (201342966) and Natural Science Foundation of Zhejiang Province (LR12H01003).
Author contributions: Q Hu originated the hypothesis; Q Hu, F Xu, Y Su, X Yu, L Cai, L Zhu, J Li and J Xia designed the experiments; Q Hu, F Xu, Y Su, X Yu, L Cai, T Li, Y Lu, J Xia, L Zhu, J Zhang and J Li carried out the data analyses and interpretation; Y Su, X Yu, L Cai, T Li, Y Lu, L Zhu, J Zhang and J Li performed the data acquisition; Q Hu, F Xu, Y Su and L Cai wrote the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Scichilone N, Battaglia S, La Sala A, Bellia V. Clinical implications of airway hyperresponsiveness in COPD. Int J Chron Obstruct Pulmon Dis. 2006;1:49-60
2. Prakash A, Babu KS, Morjaria JB. Novel anti-cholinergics in COPD. Drug Discov Today. 2013;18:1117-1126
3. Celli BR, MacNee W. Standards for the diagnosis and treatment of patients with COPD: a summary of the ATS/ERS position paper. Eur Respir J. 2004;23:932-946
4. Tashkin DP, Altose MD, Bleecker ER, Connett JE, Kanner RE, Lee WW. et al. The lung health study: airway responsiveness to inhaled methacholine in smokers with mild to moderate airflow limitation. The Lung Health Study Research Group. Am Rev Respir Dis. 1992;145:301-310
5. Vestbo J, Hansen EF. Airway hyperresponsiveness and COPD mortality. Thorax. 2001;56(Suppl2):ii11-14
6. Tamimi A, Serdarevic D, Hanania NA. The effects of cigarette smoke on airway inflammation in asthma and COPD: therapeutic implications. Respir Med. 2012;106:319-328
7. Mallia P, Footitt J, Sotero R, Jepson A, Contoli M, Trujillo-Torralbo MB. et al. Rhinovirus infection induces degradation of antimicrobial peptides and secondary bacterial infection in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2012;186:1117-1124
8. Sethi S, Murphy TF. Infection in the pathogenesis and course of chronic obstructive pulmonary disease. N Engl J Med. 2008;359:2355-2365
9. Profita M, Bonanno A, Montalbano AM, Ferraro M, Siena L, Bruno A. et al. Cigarette smoke extract activates human bronchial epithelial cells affecting non-neuronal cholinergic system signalling in vitro. Life Sci. 2011;89:36-43
10. Aravamudan B, Thompson MA, Pabelick CM, Prakash YS. Mitochondria in lung diseases. Expert Rev Respir Med. 2013;7:631-646
11. Van der Toorn M, Rezayat D, Kauffman HF, Bakker SJ, Gans RO, Koëter GH. et al. Lipid-soluble components in cigarette smoke induce mitochondrial production of reactive oxygen species in lung epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2009;297:L109-114
12. Hoffmann RF, Zarrintan S, Brandenburg SM, Kol A, de Bruin HG, Jafari S. et al. Prolonged cigarette smoke exposure alters mitochondrial structure and function in airway epithelial cells. Respir Res. 2013;14:97
13. Nie YC, Wu H, Li PB, Luo YL, Zhang CC, Shen JG. et al. Characteristic comparison of three rat models induced by cigarette smoke or combined with LPS: to establish a suitable model for study of airway mucus hypersecretion in chronic obstructive pulmonary disease. Pulm Pharmacol Ther. 2012;25:349-356
14. Hardaker EL, Freeman MS, Dale N, Bahra P, Raza F, Banner KH. et al. Exposing rodents to a combination of tobacco smoke and lipopolysaccharide resultsin an exaggerated inflammatory response in the lung. Br J Pharmacol. 2010;160:1985-1996
15. Zhang L, Sanderson MJ. Oscillations in ciliary beat frequency and intracellular calcium concentration in rabbit tracheal epithelial cells induced by ATP. J Physiol. 2003;546:733-49
16. Nakajima M, Kawanami O, Jin E, Ghazizadeh M, Honda M, Asano G. et al. Immunohistochemical and ultrastructural studies of basal cells, Clara cells and bronchiolar cuboidal cells in normal human airways. Pathol Int. 1998;48:944-953
17. Yunus MH, Siang KC, Hashim NI, Zhi NP, Zamani NF, Sabri PP. et al. The effects of human serum to the morphology, proliferation and gene expression level of the respiratory epithelium in vitro. Tissue Cell. 2014;46:233-240
18. Luppi F, Aarbiou J, van Wetering S, Rahman I, de Boer WI, Rabe KF. et al. Effects of cigarette smoke condensate on proliferation and wound closure of bronchial epithelial cells in vitro: role of glutathione. Respir Res. 2005;6:140
19. Zischka H, Lichtmannegger J, Schmitt S, Jägemann N, Schulz S, Wartini D. et al. Liver mitochondrial membrane crosslinking and destruction in a rat model of Wilson disease. J Clin Invest. 2011;121:1508-1518
20. Zhang J, Zhou J, Cai L, Lu Y, Wang T, Zhu L. et al. Extracellular calcium-sensing receptor is critical in hypoxic pulmonary vasoconstriction. Antioxid Redox Signal. 2012;17:471-484
21. King MP, Attardi G. Injection of mitochondria into human cells leads to a rapid replacement of the endogenous mitochondrial DNA. Cell. 1988;52:811-819
22. Patel SP, Gamboa JL, McMullen CA, Rabchevsky A, Andrade FH. Lower respiratory capacity in extraocular muscle mitochondria: evidence for intrinsic differences in mitochondrial composition and function. Invest Ophthalmol Vis Sci. 2009;50:180-186
23. Burman WJ, Martin WJ 2nd. Oxidant-mediated ciliary dysfunction. Possible role in airway disease. Chest. 1986;89:410-413
24. Hayes JM, Kim SK, Abitua PB, Park TJ, Herrington ER, Kitayama A. et al. Identification of novel ciliogenesis factors using a new in vivo model for mucociliary epithelial development. Dev Biol. 2007;312:115-130
25. Martell JD, Deerinck TJ, Sancak Y, Poulos TL, Mootha VK, Sosinsky GE. et al. Engineered ascorbate peroxidase as a genetically encoded reporter for electron microscopy. Nat. Biotechnol. 2012;30:1143-1148
26. Rhee HW, Zou P, Udeshi ND, Martell JD, Mootha VK, Carr SA. et al. Proteomic mapping of mitochondria in living cells via spatially restricted enzymatic tagging. Science. 2013;339:1328-1331
27. Kitani T, Kami D, Matoba S, Gojo S. Internalization of isolated functional mitochondria: involvement of macropinocytosis. J Cell Mol Med. 2014;18:1694-1703
28. Victoni T, Gleonnec F, Lanzetti M, Tenor H, Valença S, Porto LC. et al. Roflumilast N-oxide prevents cytokine secretion induced by cigarette smoke combined with LPS through JAK/STAT and ERK1/2 inhibition in airway epithelial cells. PLoS One. 2014;9:e85243
29. Lazareno S, Popham A, Birdsall NJ. Analogs of WIN 62,577 define a second allosteric site on muscarinic receptors. Mol Pharmacol. 2002;62:1492-1505
30. Eglen RM. Muscarinic receptor subtype: pharmacology and physiology. Prog Med Chem. 2005;43:105-136
31. Profita M, Giorgi RD, Sala A, Bonanno A, Riccobono L, Mirabella F. et al. Muscarinic receptors, leukotriene B4 production and neutrophilic inflammation in COPD patients. Allergy. 2005;60:1361-1369
32. Zhou Y, Tan X, Kuang W, Liu L, Wan L. Erythromycin ameliorates cigarette-smoke-induced emphysema and inflammation in rats. Transl Res. 2012;159:464-472
33. Monsó E, Ruiz J, Rosell A, Menterola J, Fiz J, Morera J. et al. Bacterial infection in chronic obstructive pulmonary disease. A study of stable and exacerbated outpatients using the protected specimen brush. Am J Respir Crit Care Med. 1995;152:1316-1320
34. Coulson FR, Fryer AD. Muscarinic acetylcholine receptors and airway diseases. Pharmacol Ther. 2003;98:59-69
35. Yaghi A, Zaman A, Cox G, Dolovich MB. Ciliary beating is depressed in nasal cilia from chronic obstructive pulmonary disease subjects. Respir Med. 2012;106:1139-1147
36. Proskocil BJ, Sekhon HS, Jia Y, Savchenko V, Blakely RD, Lindstrom J. et al. Acetylcholine is an autocrine or paracrine hormone synthesized and secreted by airway bronchial epithelial cells. Endocrinology. 2004;145:2498-2506
37. Lips KS, Lührmann A, Tschernig T, Stoeger T, Alessandrini F, Grau V. et al. Down-regulation of the non-neuronal acetylcholine synthesis and release machinery in acute allergic airway inflammation of rat and mouse. Life Sci. 2007;80:2263-2269
38. Wiegman CH, Michaeloudes C, Haji G, Narang P, Clarke CJ, Russell KE. et al. Oxidative stress-induced mitochondrial dysfunction drives inflammation and airway smooth muscle remodeling in patients with chronic obstructive pulmonary disease. J Allergy Clin Immunol. 2015;136:769-780
39. Ahmad T, Sundar IK, Lerner CA, Gerloff J, Tormos AM, Yao H. et al. Impaired mitophagy leads to cigarette smoke stress-induced cellular senescence: implications for chronic obstructive pulmonary disease. FASEB J. 2015;29:2912-2929
40. Thomas B, Rutman A, Hirst RA, Haldar P, Wardlaw AJ, Bankart J. et al. Ciliary dysfunction and ultrastructural abnormalities are features of severe asthma. J Allergy Clin Immunol. 2010;126:722-729
41. Simet SM, Sisson JH, Pavlik JA, Devasure JM, Boyer C, Liu X. et al. Long-term cigarette smoke exposure in a mouse model of ciliated epithelial cell function. Am J Respir Cell Mol Biol. 2010;43:635-640
42. Wyatt TA, Sisson JH, Allen-Gipson DS, McCaskill ML, Boten JA, DeVasure JM. et al. Co-exposure to cigarette smoke and alcohol decreases airway epithelial cell cilia beating in a protein kinase Cepsilon-dependent manner. Am J Pathol. 2012;181:431-440
43. Fliegauf M, Benzing T, Omran H. When cilia go bad: cilia defects and ciliopathies. Nat Rev Mol Cell Biol. 2007;8:880-893
44. Hu J, Barr MM. ATP-2 interacts with the PLAT domain of LOV-1 and is involved in Caenorhabditis elegans polycystin signaling. Mol Biol Cell. 2005;16:458-469
45. Manzanares D, Monzon ME, Savani RC, Salathe M. et al. Apical oxidative hyaluronan degradation stimulates airway ciliary beating via RHAMM and RON. Am J Respir Cell Mol Biol. 2007;37:160-168
46. Birben E, Sahiner UM, Sackesen C, Erzurum S, Kalayci O. Oxidative stress and antioxidant defense. World Allergy Organ J. 2012;5:9-19
47. Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2010;469:221-225
Author contact
![]() Corresponding authors: Qinghua Hu, Department of Pathophysiology, School of Basic Medicine, Tongji Medical College, Huazhong University of Science and Technology, Wuhan 430030, PRC. Tel: (27) 6331-4909, Fax: (027) 8369-2608, email: qinghuaatjmu.edu.cn; or Feng Xu, Department of Infectious Diseases, Second Affiliated Hospital, Zhejian University School of Medicine, Hangzhou 310009, Tel: (571) 8778-4583, Fax: (571) 8778-4571, email: xufeng99com.
Corresponding authors: Qinghua Hu, Department of Pathophysiology, School of Basic Medicine, Tongji Medical College, Huazhong University of Science and Technology, Wuhan 430030, PRC. Tel: (27) 6331-4909, Fax: (027) 8369-2608, email: qinghuaatjmu.edu.cn; or Feng Xu, Department of Infectious Diseases, Second Affiliated Hospital, Zhejian University School of Medicine, Hangzhou 310009, Tel: (571) 8778-4583, Fax: (571) 8778-4571, email: xufeng99com.