Impact Factor
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and Methods
Results and discussion
Conclusions
Supplementary Material
Abbreviations
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2016; 6(8):1261-1273. doi:10.7150/thno.14302 This issue Cite
Research Paper
HER2 Targeting Peptides Screening and Applications in Tumor Imaging and Drug Delivery
Lingling Geng1,3*, Zihua Wang1,2*, Xiangqian Jia1,4, Qiuju Han1,4, Zhichu Xiang1,2, Dan Li1,2, Xiaoliang Yang1,2, Di Zhang1, Xiangli Bu1,2, Weizhi Wang1,2 ![]() , Zhiyuan Hu1,2
, Zhiyuan Hu1,2 ![]() , Qiaojun Fang1,2
, Qiaojun Fang1,2 ![]()
1. CAS Key Laboratory for Biomedical Effects of Nanomaterials & Nanosafety, National Center for Nanoscience and Technology, Beijing 100190, China;
2. CAS Center for Excellence in Nanoscience, National Center for Nanoscience and Technology, Beijing 100190, China;
3. National Laboratory of Biomacromolecules, Institute of Biophysics, Chinese Academy of Sciences, Beijing 100101, China;
4. Pharmacy College, Liaoning Medical University, Jinzhou, Liaoning 121001, China.
*These authors contributed equally to this work.
Received 2015-11-3; Accepted 2016-4-14; Published 2016-5-28
Abstract

Herein, computational-aided one-bead-one-compound (OBOC) peptide library design combined with in situ single-bead sequencing microarray methods were successfully applied in screening peptides targeting at human epidermal growth factor receptor-2 (HER2), a biomarker of human breast cancer. As a result, 72 novel peptides clustered into three sequence motifs which are PYL***NP, YYL***NP and PPL***NP were acquired. Particularly one of the peptides, P51, has nanomolar affinity and high specificity for HER2 in ex vivo and in vivo tests. Moreover, doxorubicin (DOX)-loaded liposome nanoparticles were modified with peptide P51 or P25 and demonstrated to improve the targeted delivery against HER2 positive cells. Our study provides an efficient peptide screening method with a combination of techniques and the novel screened peptides with a clear binding site on HER2 can be used as probes for tumor imaging and targeted drug delivery.
Keywords: HER2 targeting peptide, tumor imaging, drug delivery, breast cancer, MD simulation.
Introduction
The human epidermal growth factor receptor (EGFR/ERBB) family which includes four members: HER1 (EGFR, ERBB1), HER2 (NEU, ERBB2), HER3 (ERBB3), and HER4 (ERBB4) has been associated with many cancers. HER2 is a unique receptor molecule due to its lacking of a ligand but functions as a co-receptor to form homodimers and heterodimers with the other three HER (1, 3 and 4) family proteins [1, 2]. Amplification and/or high expression of the HER2 occurs in approximately 20‑25% of invasive breast cancers classified as HER2 positive breast cancer [3, 4], which has worse prognosis, higher mortality in early-stage disease, increased incidence of metastases, and reduced time to relapse comparing with HER2-normal breast cancer [3, 5, 6]. Therefore, HER2 has become an important validated therapeutic target in breast cancer.
Computational methods including: molecular dynamics (MD) modeling, molecular mechanics/generalized born surface area (MM/GBSA) binding free energy calculations [7-9], and binding free energy decomposition analysis [10-12] are widely used in peptide probes or drug agents design and screening these years [13-17]. Computational aided virtual peptide library screening can provide peptides with a clear structure information of binding site and mechanism [18], but the efficiency of screening is limited when compared to high throughput screening. OBOC library screening [19-21] is efficient and has been applied in discovery of novel peptides targeting at cancer and other diseases. In addition, OBOC library combining in situ mass spectrometry (MS) sequencing and identification by using a microfluidic chip [22-24] with magnetic trapping and sheath flow sorting functions have been successfully employed in sorting a large number of peptide beads library in our previous works [25]. However, the screening efficiency of this method can be further improved by applying computational simulation in designing the library to limit mutations of residues in peptide for favorite interactions rather than aimless random mutations.
In this study, computational simulation was performed to design the OBOC peptide library for screening, Combined with in situ mass spectrometry (MS) sequencing using a microfluidic chip, 72 positive peptides were obtained as candidates for targeting HER2 protein. Moreover, the following Surface Plasmon Resonance imaging (SPRi) analysis indicated that the KD value of P51 is as low as 18.6 nmol/L. The nude mice imaging further verified that two novel peptides (P51 and P25) have strong affinity and high specificity for HER2 in ex vivo and in vivo test. The molecular dynamic (MD) simulation leaded OBOC peptide library design is successfully applied in screening for novel targeting peptides efficiently. And the results of simulation also indicate that the binding sites of the peptides remain the same as their starting model. These peptides can be used as novel probes in HER2 positive breast cancer imaging and targeted drug delivery, as demonstrated in our study of the improved cytotoxicity of peptide modified liposome nanoparticles loaded with DOX.
Materials and Methods
Peptide screening strategy
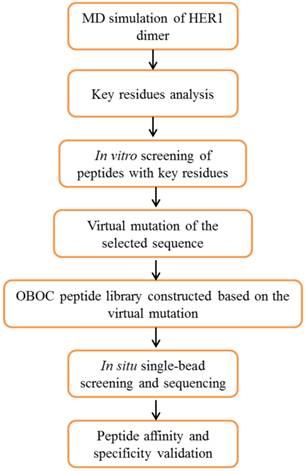
The whole design strategy of this study is shown in Figure 1. There is currently no reports of the crystal structures of human epidermal growth factor receptor-2 (HER2) homodimer or heterodimers with three other HER (1, 3 and 4) family members, therefore the crystal structure of homodimer of HER1 extracellular domains (PDB entry: 1IVO) [26] was used as the starting structure for simulation. Molecular dynamics (MD) simulations followed by binding free energy calculation and decomposition provide the key residues in the four members of HER family proteins forming homodimers or heterodimers with HER2. An initial screening using experimental analysis in vitro showed that WP1 peptide has the highest affinity among eight peptides. It was chosen for the starting sequence for peptide library design. Virtual single mutation for OBOC peptide library was constructed based on preferred amino acids for each residue in WP1 and the properties of their interacting residues of HER2 receptor. The positive peptide beads were selected and sequenced using in situ microarray technique. Finally the candidate peptides were synthesized and validated for their affinity and specificity of targeting HER2 by in vitro and in vivo assays.
Overview of the procedures for screening HER2-targeting peptides using a strategy combining computational modeling, OBOC in situ single-bead screening and sequencing.
MD Simulations
There is currently no reports of the crystal structures of HER2 homodimers or heterodimers with three other HER (1, 3 and 4) family members, therefore the crystal structure of homodimer of HER1 extracellular domains (PDB entry: 1IVO) [26] was used as the starting structure for simulation. Models of HER1/PS1 and HER1/WP1 were constructed from 1IVO by keeping the whole structure of one monomer and that of Met243-Asn255 and Asp237-Met252 respectively. Models of HER1/PS2, HER1/PS3, HER1/PS4, HER1/WP2, HER1/WP3 and HER1/WP4 were constructed based on the structure of HER1/PS1 and HER1/WP1 by changing sidechains based on the sequences. Then models of HER2/PS1, HER2/PS2, HER2/PS3, HER2/PS4, HER2/WP1, HER2/WP2, HER2/WP3 and HER2/WP4 were constructed by aligning and merging the crystal structure of HER2 (PDB entry: 3MZW) [27] with HER1 in the HER1/ligand complexes, the ligand Z(HER2:342) in 3MZW and HER1 were deleted. The single mutants of WP1 and HER2 complexes were acquired by mutating the amino acids based on ligand sequences.
Primary sequences of PS1, PS2, PS3, PS4, WP1, WP2, WP3, WP4 and residues 237-255 in HER1, HER2, HER3 and HER4 were aligned by using ClustalW program available on the web of EMBnet [28].
The AMBER03 force field was used to establish the potentials of the proteins in the following molecular mechanics (MM) minimizations and MD simulations [29]. The MD simulations of all the complexes were carried out with AMBER12 software package. The binding free energy of each system was calculated using MM/GBSA method [18, 30, 31]. And the MM/GBSA free energy decomposition was performed by the mm_pbsa program in AMBER12. More details of the MD simulation methods are provided in Supplementary Materials and Methods.
The photo labile OBOC peptide library synthesis and in situ MALDI-TOF identification of the peptides
The photo labile OBOC library and all positive peptides were synthesized using Fmoc strategy SPPS (solid phase peptide synthesis). Tentagel resin (loading: 0.53 mmol/g) was used as the solid phase support. The OBOC library synthesis process and in situ MALDI-TOF sequencing and identification was performed following the literature [25]. Briefly, positive peptides were trapped by magnetic field on a microchip. Positive peptide beads will be surrounded by the magnetic beads through the interaction bridge of peptide-HER2-biotin-streptavidin, while native beads will remain naked and cannot be trapped. The detail of the OBOC screening process was shown in Section I. Supplementary Materials and Methods of Supporting Information. The synthesized peptides were purified by using HPLC system (L-7100, Japan) on a TSK gel ODS-100 V column (150 mm × 4.6 mm) with a flow rate of 2.0 mL min-1 with a 25 minutes gradient of 5 to 80% acetonitrile containing 0.1% TFA. MALDI-TOF MS analysis was carried out on a Bruker ULTRAFLEXTREME mass spectrometer (Bruker Daltonics, Germany).
9-Fluorenylmethoxycarbonyl (Fmoc)-protected amino acids and 2-(1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HBTU) were purchased from GL Biochem (China). Trifluoroacetic acid (TFA), and fluorescein 5-isothiocyanate were from Sigma-Aldrich (USA). N-Methyl morpholine (NMM) and N,N-dimethylformamide (DMF) were from Beijing chemical plant (China).
Cell assays
Human breast cancer cell line SKBR3 with high expression of HER2, human breast cancer cell line MDA-MB-468 with high expression of HER1 and no expression of HER2 and human embryonic kidney cell line 293A (HER2 negative) cells were used in this study [32]. SKBR3 cells were cultured in in RPMI 1640 medium (Hyclone) supplemented with 10% fetal bovine serum (Gibco). 468 and 293A cells were cultured in DMEM/High glucose (Hyclone) medium, with 10% fetal bovine serum.
For flow cytometry analysis, all cells were cultured overnight and approximately 100 μL of 1×106 mL-1 cells were used for FITC-labeled peptides and antibody (ebioscience) tests. Specifically, FITC-labeled peptides were dissolved into a concentration of 50 μM with cell culture mediums and the antibody was used with the recommended dilution of 1:20. Then cells were incubated with FITC-labeled peptides on ice for 20 minutes. The non-bound peptides were rinsed away with PBS for three times. The whole process was performed on ice and with minimal light exposure. A Beckman Quanta SC flow cytometry was used for the test.
For confocal fluorescence imaging analysis approximately 1×105 cells (1 mL) were seeded into culture dishes and cultured overnight. 200 uL FITC-labeled peptide at 50 μM and 1 mM Hoechst 33342 was dissolved in cell culture medium, and added into the culture dishes. Then cells were incubated in the solution for 20 minutes at 4℃. Finally, the cells were washed three times with cold PBS. For the colocalization analysis, the cells were treated with phycoerythrin (PE) conjugated anti-human HER2 antibody (LS-C213848, 1:20 dilution). FITC-labeled peptide (50μM) and 1 mM Hoechst 33342 were added simultaneously. Cellular binding and internalization of the liposomes, LS-DOX, P51-LS-DOX and P25-LS-DOX, were assessed by confocal microscopy. SKBR3 cells were seeded onto culture dishes at a density of 1×105 cells (1 mL) and cultured overnight. Then cells were incubated with LS-DOX, P51-LS-DOX and P25-LS-DOX at a DOX concentration of 2 μg/mL (200μL) for 5, 10, 15, 30, 60 and 120 minutes, at 37°C, respectively, followed by cell nuclei staining with 1 mM Hoechst 33342 for another 15 minutes. Finally, the cells were washed with cold PBS three times.
An Olympus FV1000-IX81 confocal-laser scanning microscope was used for confocal fluorescence imaging. For FITC and DOX, A FV5-LAMAR 488 nm laser was the excitation source, and emission was collected between 520-620 nm. Hoechst 33342 was excited by a FV5- LD405-2 405 nm laser and collected within the range of 422-472 nm. PE was excited by a FV10-LD559 559 nm laser with emission maximum of 573 nm. All parameters of the microscope were set to be the same for different samples to allow comparisons of the binding ability of different peptides.
SPRi assay
SPRi chip used in this work is a Plexera® Nanocapture® Chip with a layer of 47.5 nm thickness of bare gold. For detection, all peptides were linked a cysteine (Cys) in the amino terminal for interacting with bare gold, since the thiol side chain of Cys can form Au-S bond with Au [33]. 0.5 uL of 1mg/mL peptides were pointed on the gold surface of the chip and incubated at 4 oC overnight in water box. Then, the SPRi chip was washed with PBS and water before 5% non-fat milk was applied to block the chip overnight at 4 oC. After the chip was washed with PBS and water, it was dried with nitrogen for use. The purified HER2 protein purchased from Sino Biological Inc (China) was used as the mobile phase. HER2 protein was diluted to 20 μg/mL in PBST (PBS with 0.1% Tween-20) and further diluted to 10 μg/mL, 5 μg/mL, 2.5 μg/mL and 1.25 μg/mL. The SPRi analysis procedure follows the following cycle of injections: running buffer (PBST, baseline stabilization); sample (one of the five concentrations of protein) for binding assay; running buffer (PBST) for washing; and 0.5% (vol/vol) H3PO4 in deionized water for regeneration. After one cycle was finished for a sample, the next sample will start on the same chip until all were completed. Real-time binding signal were recorded and analyzed by PlexArray HT system (Plexera LLC, Bothell, WA). The dissociation constant was calculated by fitting the association-dissociation curves.
In vivo and ex vivo fluorescence imaging
All animal experiments were carried out in compliance with the guide for the care and use of laboratory animals of Beijing University Animal Study Committee's requirements. The Beijing University Animal Study Committee approved the experiments. 1 × 107 SKBR3 cells were injected by subcutaneously (s.c.) into the right flank of the 5-7 week-old female BALB/c nude mice to establish xenografted tumors. Tumor size was measured periodically using calipers, and the tumors were allowed to grow to 6-8 mm in diameter. Cy5.5-NHS purchased from Lumiprobe was used to labeled peptides. The conjugation of peptides and Cy5.5-NHS was carried out following the protocol provided by Lumiprobe. Chemical reaction happens with Cy5.5-NHS and -NH2 group of peptide in PBS buffer (pH 7.4) at room temperature overnight and the C terminal of peptide keeps carboxylic acid. The crude product was purified by semi-preparative reverse phase HPLC. Cy5.5-peptides (1 μM, 200 μL) were injected into tumor-bearing nude mice via the tail vein and the tumor images were obtained using the small animal in vivo imaging system (CRI Maestro 2). Near-infrared fluorescence (NIRF) images of nude mice bearing subcutaneous tumors were acquired after vein injection of Cy5.5-P51 and Cy5.5-P25, while the control tumor bearing nude mouse was intravenously injected with Cy5.5 (1 μM, 200 μL). Half an hour after the injection, the mice were anesthetized and placed into the imaging system. For each peptide and control, at least three mice were used. Near-infrared fluorescence (NIRF) imaging of nude mice bearing subcutaneous tumors were taken with an exposure time of 50 ms using the Cy5.5 filter sets (excitation: 673 nm, emission: 707 nm) and the intensities were quantified using the CRI Maestro software. Then the nude mice were sacrificed and tumors as well as the main organs were harvested and the NIRF images were taken individually.
Peptide modified DOX-loaded liposome preparation
DSPE-PEG2000-NHS and peptide were mixed in newly distilled DMF at 1:1 molar ratio, and pH was adjusted to 8.0-8.5 with N-methyl morpholine under moderate stirring at room temperature. After 72 h incubation, the reaction mixture was put into a dialysis bag with cut off molecular weight (MW) of 3500 Da and dialyzed against deionized water for 48 h to remove free ligands. Finally DSPE-PEG2000-peptide was formed. The DSPE-PEG2000-peptide solution was lyophilized and stored at -20 °C. The conjugation efficiency was determined by using a Hitachi HPLC system (L-7100, Japan) on a TSK gel ODS-100V column (150 mm×4.6 mm) at a flow rate of 2 mL min-1 with 25 min gradient using 5-80% acetonitrile containing 0.1% Trifluoroacetic acid (TFA) solution. Then the product was characterized by MALDI-TOF MS (Bruker Daltonics). Finally, DSPE-PEG2000-peptide, soybean phosphatidylcholine (SPC), cholesterol (CHOL) and doxorubicin (DOX) were mixed to produce peptide modified DOX-loaded liposome (P51-LS-DOX or P25-LS-DOX) by conventional thin lipid film method. Briefly, lipids were dissolved in chloroform and dried until thin lipid film formed on a rotary evaporator. The dried lipid film was hydrated with PBS and sonicated with a bath type sonicator [34]. DOX was remote-loaded via the ammonium sulfate gradient method [35, 36]. As control, DOX-loaded liposome without peptide modification (LS-DOX) was also prepared. Free DOX was removed by Sepharose CL-4B column. DOX (doxorubicin) was from Sigma-Aldrich (USA); DSPE-PEG2000-NHS, (1, 2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[poly(ethyleneglycol)-2000]-N-hydroxysuccinimidyl) were purchased from Nanosoft Biotechnology LLC (USA); CHOL (cholesterol) and SPC (soybean phosphatidylcholine) were purchased from A.V.T. Pharmaceutical Co; NMM (N-methyl morpholine) and DMF (N,N-Dimethylformamide) were from Beijing chemical plant (China); LS (liposome), LS-DOX, DOX-loaded liposome; P51-LS-DOX, P51 modified DOX-loaded liposome; P25-LS-DOX, P25 modified DOX-loaded liposome.
Transmission electron microscopy (TEM)
The morphology and size of liposomes were observed by TEM (Tecnai G2 20 S-TWIN, FEI, USA). Briefly, the liposome dispersion was applied to a copper grid and after 5-minute incubation at room temperature, the grid was stained with 1% uranyl acetate and the image was taken after drying.
MTT assay
P51 and P25 modified DOX-loaded liposomes were tested on SKBR3 cells by MTT assay for cell viability to compare the toxicity of liposomes in the absence or presence of peptides. Cells resuspended at a concentration of 3 × 104 cells/mL in fresh cell culture medium were seeded into 96-well plates at 3 × 103 cells per well and cultured for 24 h with 5% CO2 at 37 °C. Then, the medium was discarded and replaced with the fresh medium containing P51-LS-DOX or P25-LS-DOX or LS-DOX at the final concentration of 0, 5, 10, 50, 100, 200 and 500 μg/mL and cultured for 20 minutes. After replacing with fresh culture medium, cells continue to grow for 24 hours. The MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide, obtained from Sigma USA) solution was added to each well and the plates were incubated at 37°C for 3 h. Afterwards, the MTT solution was removed and 150 µL of dimethyl sulfoxide (DMSO) was added to each well. The optical density (OD) of each well was measured by using an ELISA reader at 570 nm after 10 minutes of vibration mixing.
Results and discussion
Key binding residues analysis based on the MD simulation
The structure of HER1 dimer (from PDB entry: 1IVO) is shown in Figure 2A. In order to find the key residues for the dimer interaction, MD simulation was carried out. The results show that the predicted binding free energy between two monomers in HER1 complex is -222.56 kcal/mol. The van der Waals (ΔEvdw) contribution (-220.71 kcal/mol) is the main component (Table S1). Root-Mean-Square Displacement (RMSD) values of backbone atoms of the simulated structures and their corresponding starting structure for each monomer and complex are stable after ∼1000 ps (Figure S1).
Structure of HER1 dimer (A) and key residues in HER1 dimer binding interface (B-E).
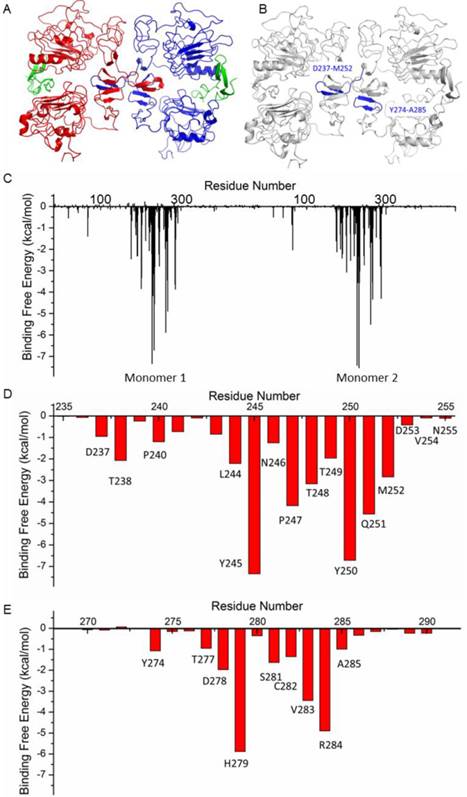
The following energy decomposition analyses based on MD simulation results illustrate that residues Gln193-Lys302 in HER1 complex are the main residues for the binding between monomers (Figures 2B and 2C). In detail, residues Asp237-Met252 and Tyr274-Ala285 show significant contributions (Figures 2B-E), especially from Leu244 to Met252. By examining the structure, the 13-mer fragment from Met243 to Asn255 of HER1 forms a hairpin loop, which was selected as the template in the following study. These key residues and their corresponding ones in HER2, HER3 and HER4 should have similar type of interactions in the formation of homo- or heterodimers because of structural and sequence similarity (Figure 3A). Therefore, aiming to selecting HER2 targeting peptides, 13-mer peptides PS1 (Met243-Asn255HER1), PS2 (Val247-Asn259HER2), PS3 (Leu238-Asn250HER3) and PS4 (Phe241-Asn253HER4), and 16-mer peptides WP1 (Asp237-Met252HER1), WP2 (Leu241-Ser256HER2), WP3 (Pro232-Leu247HER3) and WP4 (Thr235-Leu250HER4) were derived from the four HER family proteins after structural alignment (Figures 3B and 3C).
Sequences alignment of key binding residues in HER1, HER2, HER3 and HER4 (A) and cellular analyses of peptides affinity for HER2 binding. Alignment of the key binding residues, the sequences of 13-mer and 16-mer peptides selected to synthesize (B, C). Immunocytochemistry analysis of peptides PS1, PS2, PS3, PS4, WP1, WP2, WP3 and WP4 (labeled with FITC, green) in HER2 positive SKBR3 cells (D and E), colocalization analysis of WP1 (labeled with FITC, green) and anti-HER2 antibody (labeled with PE, red) in HER2 positive SKBR3 cells (F). Hoechst 33342-stained cell nuclei are in blue. (G, H) Flow cytometry analysis of WP1, WP2, WP3, WP4 and phosphate buffer saline (PBS) control for SKBR3 cell line and the binding percentages are plotted as bars in panel h, respectively (n = 3).
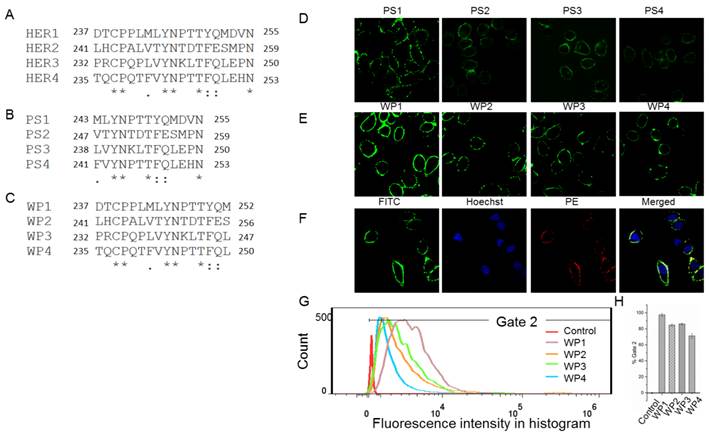
As a preliminary screening to select the best peptide from these eight ones for the following virtual mutation and screening, confocal fluorescence imaging was first carried out to estimate which peptide binds best to HER2 protein. All peptides labeled with FITC bind to the extracellular part of HER2 in SKBR3 cells which has a high expression of HER2 (Figures 3D, 3E and S2) and weakly in the HER2 negative 293A cells (Figure S3). The fluorescence intensity shows that PS1 and WP series peptides bind better than others. In particular, WP1 has the highest intensity, indicating the strongest binding to HER2 on SKBR3 cells among these 8 peptides. In addition, colocalization analysis indicates that fluorescence signals of WP1 (labeled with FITC) and anti-HER2 antibody (labeled with PE) overlap on the cell surface of HER2 positive cells (Figure 3F), since the binding site of anti-HER2 antibody is at 30-59 of human HER2 (LS-C213848), which is different from the binding site of WP1. The flow cytometry analysis was also performed on WP series peptides using SKBR3 cells. Figures 3G and 3H show WP1 has the most fluorescence intensity among all peptides. These results further confirm that WP1 binds to HER2 protein specifically and with high affinity. Moreover, MD simulations of eight complexes HER2/PS1, HER2/PS2, HER2/PS3, HER2/PS4, HER2/WP1, HER2/WP2, HER2/WP3 and HER2/WP4 were performed, RMSD values of the backbone atoms of these eight complexes show an acceptable level of stabilization in all MD trajectories (Figures S4 and S5).The binding free energy calculation shown in Table 1 indicates that WP1 has the lowest binding energy with HER2, which is consistent with the above confocal fluorescence imaging and flow cytometry results. The binding free energy decomposition results (Figure S6) suggest that the last three residues in the 13-mer peptides PS1-PS4 are unfavorable for HER2 binding and residues 2, 9 and 14 in the 16-mer peptides of WP1-WP4 contribute most, while residues 6 and 10 show less contribution (Figure S7).
Binding free energies and individual energy terms for HER2 and peptides complexes calculated by MM/GBSA (kcal/mol).
| Complexes | ΔEvdw | ΔEele | ΔGGB | ΔGSA | ΔGtot |
|---|---|---|---|---|---|
| HER2/PS1 | -59.60±4.81 | 12.16±2.32 | -5.36±2.64 | -8.42±0.55 | -61.21±4.47 |
| HER2/PS2 | -62.30±5.13 | 32.14±6.88 | -21.19±6.19 | -9.54±0.55 | -60.90±5.72 |
| HER2/PS3 | -52.43±3.75 | -7.00±2.67 | 15.51±2.53 | -7.76±0.51 | -51.68±3.76 |
| HER2/PS4 | -52.09±3.80 | 17.34±2.41 | -8.18±2.39 | -7.41±0.49 | -50.34±3.85 |
| HER2/WP1 | -71.11±5.14 | 1.57±3.68 | 6.93±2.91 | -11.21±0.50 | -73.83±5.10 |
| HER2/WP2 | -70.97±5.09 | 21.83±6.22 | -11.35±5.64 | -12.02±0.70 | -72.51±5.48 |
| HER2/WP3 | -65.45±6.87 | -74.53±9.45 | 82.28±9.26 | -10.49±1.01 | -68.19±7.35 |
| HER2/WP4 | -68.66±4.63 | -23.92±3.01 | 32.91±2.52 | -11.63±0.49 | -71.30±4.61 |
ΔEvdw, van der Waals contribution; ΔEele, electrostatic contribution; ΔGGB, the polar contribution of desolvation; ΔGSA, nonpolar contribution of desolvation; ΔGtot, the calculated total binding free energy.
In summary, cellular experiments and the MD simulation indicate that 16-mer peptide WP1 has the highest binding affinity toward HER2. Therefore, WP1 is selected as the starting sequence for the following screening to further improve the affinity.
Virtual screening by single mutation of WP1
The binding free energy decomposition results (Figure S7) indicate that residues 6 and 10 in the 16-mer peptides WP1-WP4 contribute slightly to HER2 binding. Residue 11 is a proline which is usually highly conserved at the turn region. Therefore residues 6, 10 and 11 were fixed, 27 single mutations of WP1 were made for the rest residues based on the properties of amino acids in both HER2 and peptide, as well as the space among the interactions. Basic rules are that mutations should favor electrostatic and van der Waals interactions, without causing steric overlap. Then interactions of single mutated peptides and HER2 complexes were simulated by MD simulation, and MM/GBSA binding free energy was calculated to look for peptides with lower binding free energies than WP1. The sequences and modeling data were shown in the Supporting Information (Table S2). Except two mutations, the binding free energies of most mutants (25) are lower than the wide type WP1 and expected to have a tight binding to HER2. The highly successful mutation rate demonstrates that we have a good understanding of the interactions in the structure complex. This result provides the mutation basis for the OBOC library construction.
OBOC peptides library was constructed based on the above results. As Au-S bond formation is required in the SPRi detection, a cysteine was added at the N-terminal of the library peptides (see Methods). Mutations of residues in the peptide library were shown in Figure 4A, i.e., two or three amino acids selected by virtual screening were used to build the peptide library. As shown in Figure 4A, in the OBOC library, the peptide sequences on each bead were randomly distributed, so that the capacity of the peptide library was 2 × 3 × 2 × 2 × 2 × 2 × 3 × 2 × 2 × 2 × 2 × 3 × 3 = 41 472 and the redundancy of the library was five. Therefore, the total peptide beads for screening could be ∼ 2 × 105. Normally, for a 16-mer peptide, a library with blind mutations is much larger. However, the computational simulation and virtual screening has greatly reduced the library size in this case.
2 × 105 peptide beads library construct strategy (A), 72 verified positive sequences sorted to be three classes (B-D), peptides with PYL***NP (B), peptides with YYL***NP (C), and peptides with PPL***NP (D).
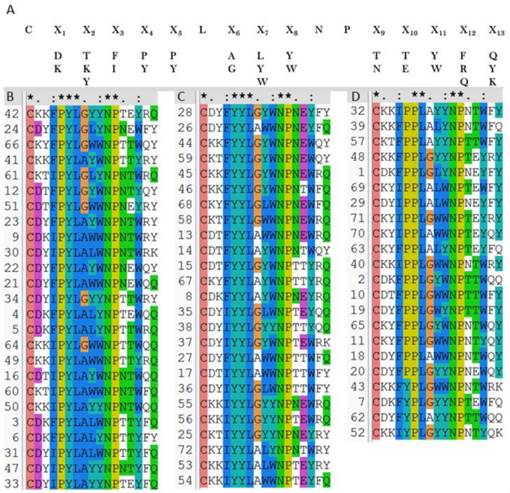
Then the OBOC peptides library was screened and sequenced using MALDI-TOF MS combined in situ microarray technique (Figure S8 and Methods). Sequences of 72 positive peptides were acquired. All sequences were aligned using clustalW program and we found the peptides are clustered into three categories with sequence characterized as: PYL***NP, YYL***NP and PPL***NP, respectively (Figures 4B-D).
Dissociation constants (KD) of positive peptides measured by SPRi
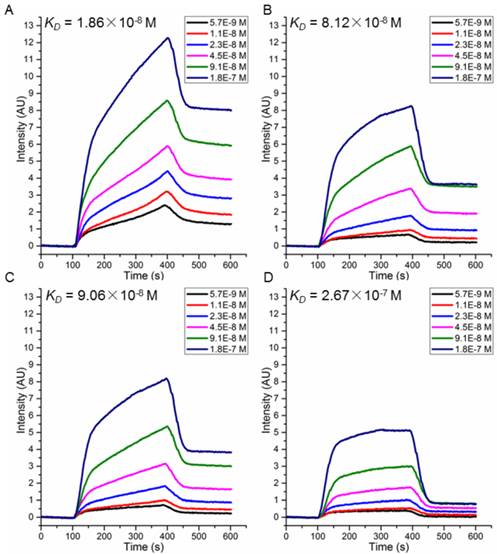
To choose candidate peptides for the following tests by experiments, 72 positive peptides and HER2 complexes were simulated by MD modeling and the binding free energies were also calculated (data not shown). Based on our previous study, we have observed a high correlation between the binding free energies calculated by MD simulation and actual binding affinity when the changes of interactions in peptide-protein complex is small [18]. Finally, two peptides with the lowest binding free energy (P51: CDTFPYLGWWNPNEYRY and P25: CKTIYYLGYYNPNEYRY) and two with the highest (P47: CDYIPYLAYYNPNTYFQ and P40: CKKIPPLGWWNPNTWRY) were chosen (Table 2) for further validation. Peptides were synthesized and their affinity to HER2 protein measured by SPRi method as described in the Methods. The dissociation constant was calculated from kinetic constants obtained by curve-fitting association and dissociation rates to real-time binding and washing data. All four peptides P51, P25, P47 and P40 have high affinity against HER2 protein (Figure 5), while no binding signals for human serum albumin (HSA) (Figure S9). The dissociation constants (KD) of the peptides P51, P25, P47, P40 and HER2 protein are 18.6, 81.2, 90.6 and 267 nmol/L, respectively. SPRi detection of the relative binding signals of these four peptides and WP1 toward HER2 protein at concentration of 9.1×10-8 M/L were also performed, indicating that four peptides except P40 bind stronger than WP1 to HER2 (Figure S10). The SPRi results are consistent with the computational simulation results shown in Table 2, with P51 having the lowest binding free energy and P40 having a binding energy closest to WP1.
Binding free energies and individual energy terms of complexes for HER2 and WP1 or the four selected peptides calculated by MM/GBSA (kcal/mol).
| Complexes | ΔEvdw | ΔEele | ΔGGB | ΔGSA | ΔGtot |
|---|---|---|---|---|---|
| HER2/WP1 | -71.11±5.14 | 1.57±3.68 | 6.93±2.91 | -11.21±0.50 | -73.83±5.10 |
| HER2/P51 | -111.00±5.29 | -1.10±7.03 | 14.37±6.06 | -15.64±0.59 | -113.36±5.24 |
| HER2/P25 | -96.14±6.15 | -80.27±10.68 | 89.78±9.89 | -14.71±0.78 | -101.35±6.18 |
| HER2/P47 | -82.96±6.89 | 10.14±3.93 | 1.38±4.16 | -12.24±1.17 | -83.68±7.25 |
| HER2/P40 | -77.18±5.98 | -22.31±2.93 | 32.44±2.77 | -12.38±0.69 | -79.43±5.85 |
SPRi detection of the binding affinity of peptides P51 (A), P25 (B), P47 (C) and P40 (D) toward HER2.
Confirmation of affinity and specificity of peptides to HER2 positive cells in vitro
Flow cytometry analyses for peptides P51 and P25 were performed to verify the affinity and specificity of the screened peptides on HER2 positive human breast cancer cell line SKBR3 and HER2 negative human embryonic kidney cell line 293A [32] (Figures 6A-D). 99.7% and 98% SKBR3 cells treated by fluorescein isothiocyanate (FITC) labeled peptides P51 and P25 respectively show significant fluorescence signals, indicating high affinities of P51 and P25 toward HER2 protein. However, for 293A cells, less than 3% cells are detected with fluorescence. The results show a high specificity of P51 and P25 at the cellular level.
Flow cytometry analysis shows P51 and P25 have high affinity and specificity for SKBR3 (HER2 high expression) cells. The fluorescence intensities of cells bound with peptides or antibody for SKBR3 and 293A cell lines are shown in A and C, respectively, and the binding percentages in two cell lines are plotted as bars in panel B and D, respectively (n = 3). Panel E shows the colocalization analysis of peptides and antibodies in HER2 or HER1 high expression cell lines, both peptides showed significant fluorescence signals and overlap in SKBR3 cell surface, but very weak signals are detected in 468 cells. HER2 high expression SKBR3 cells were treated with peptides (labeled with FITC, green) and anti-HER2 antibody (labeled with PE, red), and HER2 low expression but HER1 high expression 468 cells were treated with peptides (labeled with FITC, green) and anti-HER1 antibody (labeled with AlexaFluor555, red).
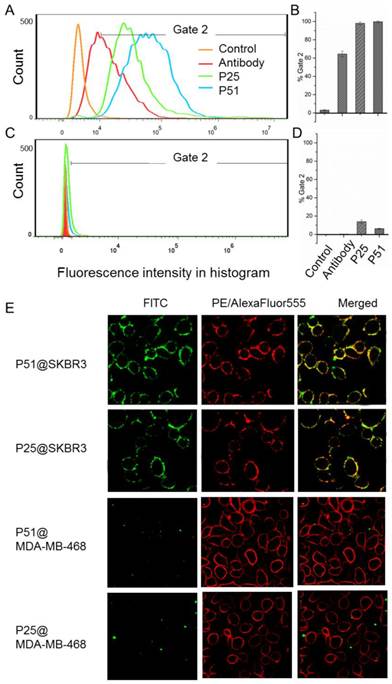
Confocal fluorescence imaging analyses further confirm the high affinities and specificities of peptides toward HER2 (Figure 6E) using HER2 positive SKBR3 cells and HER1 positive but HER2 negative 468 cells. The colocalization analysis shows peptides and the anti-HER2 antibody can overlap on the surface of SKBR3 cells, while no fluorescence signal of peptides is detected on 468 cells, confirming that both P51 and P25 target at HER2, but not HER1. Moreover, confocal fluorescence imaging analysis of peptides P51 and P25 binding to cell lines with medium expression of HER2 (MCF-7 and MDA-MB-231) and low expression of HER2 (293A) were also performed. Both peptides show weak signals in MCF-7 and MDA-MB-231 cells and no signals in 293A cells (Figure S11).
Confirmation of the ability of peptides to target tumors in vivo and ex vivo
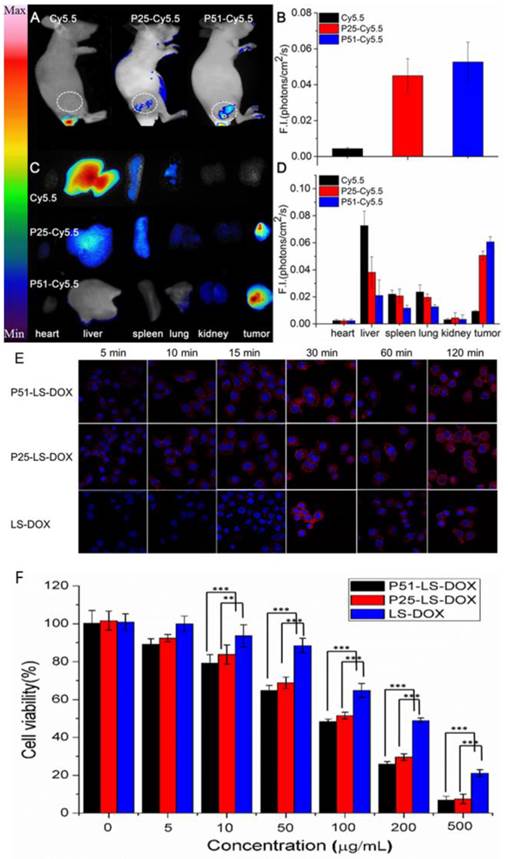
To investigate the affinity and specificity of peptides to HER2 positive tumors, in vivo and ex vivo tests were conducted. MALDI-TOF-MS data of crude products of Cy5.5 labeled peptides (P51 and P25) are shown in Figure S12, and the molecular weights are consistent with the theoretical values. Nude mice xenografted with human breast tumors were generated by inoculation of SKBR3 cells into nude mice. When tumors size was around 6-8 mm in diameter, Cy5.5 labeled peptides (Cy5.5-P51, Cy5.5-P25) and control Cy5.5 were injected into mice via the tail vein. Half an hour after the injection, in vivo tumor detection and imaging were carried out. Near-infrared fluorescence (NIRF) images of nude mice were acquired using the small animal in vivo imaging system (CRI Maestro 2) as described in Methods. There are clear differences between the intensity of signals in tumor with Cy5.5-P51 or Cy5.5-P25 injected and those of the Cy5.5 control (Figure 7A). MDA-MB-468 cells with high expression of HER1 and no expression of HER2 were also used to generate tumor-bearing mice models as the negative control. In vivo imaging shows that these tumors do not have fluorescence signals and therefore excludes the possibility of tumor nonspecific uptake of peptides (Figure S13). Intensities of signals increase 12.16-fold for Cy5.5-P51 and 10.39-fold for Cy5.5-P25 as compared to the control (Figure 7B). After in vivo imaging, the nude mice were sacrificed and tumors as well as the main organs were harvested and the NIRF images were individually taken. Figures 7C and 7D show that tumors targeted with P51 and P25 have higher fluorescence signal than control and other main organs. The results are consistent with the in vitro results that P51 and P25 have a high specificity and affinity for HER2 positive cells and tumors. Both peptides can be used as probes for imaging HER2 positive breast cancer.
Peptides applications in tumor imaging and drug delivery. In vivo and ex vivo imaging of tumor targeting by P51-Cy5.5 and P25-Cy5.5 (A-D): (A), in vivo fluorescence imaging of P51-Cy5.5 and P25-Cy5.5 to tumor and quantification of fluorescence signals in bar charts (B); (C), ex vivo fluorescence imaging of tumor accumulation and biodistribution, and quantification of fluorescence signals in bar charts (D). Fluorescence intensity was measured in terms of counts/energy/area and is presented as an average (n = 3) in all bar charts. Confocal microscopy images of P51-LS-DOX, P25-LS-DOX and LS-DOX treated SKBR3 cells and MTT assay of cytotoxicity (E-F): (E), Confocal microscopy observation of liposomes modified with or without peptides in SKBR3 cells for cellular uptake and distribution study. Liposomes modified with peptides bind quickly within the first 5 minutes, while LS-DOX within 15 minutes. (Hoechst 33342-stained cell nuclei are in blue, DOX in red). (F), MTT assay of SKBR3 cell viability with P51 and P25 peptides modified DOX-loaded liposomes treatment. All data represent mean ± s.d. n = 10. *P<0.05, **P<0.01, ***P<0.001.
Peptide-modified liposomes for doxorubicin loading
P51 and P25 modified liposomes loaded with doxorubicin (P51-LS-DOX and P25-LS-DOX) and liposome loaded with doxorubicin without peptide modification (LS-DOX) were prepared and detected by transmission electron microscope (TEM). Figure S14 illustrates the particle sizes of P51-LS-DOX, P25-LS-DOX and LS-DOX are all around 100 nm.
To check the targeting effect, cellular uptake and distribution of liposomes with or without peptides modified were observed by confocal microscopy. As shown in Figure 7E, peptide modified liposomes quickly bind to cell membranes within the first 5 minutes, while this phenomenon does not happen in LS-DOX treated cells until 15 mins later. With time going on, liposomes may enter the cells and then further into the cell nuclei as shown by DOX fluorescence, which is found to be partly overlapped with the Hoechst 33342-stained cell nuclei at 2 h for all formulations. The results suggest that targeting effect of peptides happens at the early stage of binding.
We then performed MTT assay to check the cell viability of SKBR3 cells treated with P51 and P25 modified DOX-loaded liposomes. As shown in Figure 7F, after exposure to drug for 20 minutes, the cell viability decreases approximately 10% for P51-LS-DOX and P25-LS-DOX at 5 μg/mL, respectively, and 20% for P51-LS-DOX at 10 μg/mL. With the concentration of DOX increases, the difference in cell viability decreases. The cytotoxicity of P25-LS-DOX and P51-LS-DOX is significantly higher than that of LS-DOX when DOX concentration is higher than 50 μg/mL, indicating that the cytotoxicity is higher for targeted liposome than for non-targeted ones when the drug concentration increases. This is consistent with results shown in Figure 7E that in the early stage of drug exposure, targeted liposomes enter the cells more efficiently; therefore a higher concentration will result in a more significant difference of drug concentration inside cells and hence higher cytotoxicity. The increasing efficiency may due to that peptides modified liposomes enter the cells by receptor-meditated and nonspecific endocytosis, while LS-DOX liposomes by nonspecific pathway only. The increased drug cytotoxicity due to targeting is comparable to other reports [34, 37]. Finally, the stability of the peptides in plasma was measure and both peptides have the half-life of approximately 20 minutes (Figure S15). Although the half-life is short, as shown in Figure 7E, the binding of targeted-liposomes to the cell membrane happens in the first 5 minutes, much faster than the degradation of the peptides. With proper modifications to improve stability, these peptides can be used in drug delivery for cancer therapy.
Conclusions
With computational aided analysis of the crystal structure of HER1 dimer and its homologous HER2, we identified a 16-mer peptide WP1 to have the ability to target HER2 protein. By combining molecular dynamics modeling, MM/GBSA binding free energy calculations, and binding free energy decomposition analysis, a series of WP1 single mutants were screened virtually for the construction of an OBOC peptide library. Finally, 72 positive peptides were selected and dissociation constants (KD) of four selected peptides P51, P25, P47 and P40 toward HER2 protein were measured with the best affinity (P51) at 18.6 nmol/L. Peptides P51 and P25 showed high affinity and specificity to the extracellular domain of HER2 proteins by confocal fluorescence imaging and flow cytometry analysis in vitro. Furthermore, in vivo and ex vivo imaging showed both peptides have strong affinity and high specificity for HER2 positive tumors. The introduction of computational modeling in the design of OBOC library not only improves the screening efficiency by reducing the size of library, but also limits the number of mutations so that the selected peptides remain the same binding site as their original form. The consistency of calculated binding energy with SPRi measured KD for all peptides confirms that their interacting sites with HER2 are the same as the starting model. These novel designed peptides may provide alternative probes to improve HER2 positive breast cancer early detection, diagnosis, and targeted therapy. These peptides can also be used in drug loading for breast cancer therapy as shown in the peptide modified DOX-loaded liposomes in this study. In summary, we have provided an efficient method that integrates computational modeling, OBOC peptide library screening coupled with in situ single-bead sequencing microarray methods and have successfully applied in screening of HER2 targeting peptides. While most peptides screened from a library are unknown for their binding sites to the targets, our method provides a way to screen peptides with clear binding information, which can help design multi-targeted probes with other peptides or antibodies [18], or be used at the same time for molecular imaging as a tracer [38, 39] in the presence of therapeutic monoclonal antibodies.
Statistical analysis
Tests of statistical significance were performed using a two-tailed Student's t-test. Standard deviation was used to determine the error bars shown in the figures.
Supplementary Material
Supplementary tables and figures.
Abbreviations
HER1: human epidermal growth factor receptor 1; HER2: human epidermal growth factor receptor 2; OBOC: one-bead-one-compound; MD: molecular dynamics; SPRi: Surface Plasmon Resonance Imaging; KD: dissociation constants; EGFR/ERBB: epidermal growth factor receptor; RTK: receptor tyrosine kinases; MM: molecular mechanics; PME: particle mesh ewald; ∆EvdW: van der Waals energy; ∆Eele: electrostatic energy; GB: generalized born; SASA: solvent accessible surface area; ∆GGB: polar contribution of desolvation free energy; ∆GSA: nonpolar solvation contribution; RMSD: root-mean-square displacement; SPPS: solid phase peptide synthesis; HBTU: 2-(1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate; TFA: Trifluoroacetic acid; FITC: fluorescein 5-isothiocyanate; DSPE-PEG2000-NHS: (1, 2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[poly(ethyleneglycol)-2000]-N-hydroxysuccinimidyl); CHOL: cholesterol; SPC: soybean phosphatidylcholine; NMM: N-Methyl morpholine; DMF: N,N-dimethylformamide; PBS: phosphate buffer saline; DOX: doxorubicin; PE: phycoerythrin; MTT: 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide; DMSO: dimethyl sulfoxide; OD: optical density; s.c.: subcutaneously; NIR: Near-infrared. TEM: transmission electron microscope.
Acknowledgements
This work is supported by “Strategic Priority Research Program” of Chinese Academy of Sciences (XDA09040300), Chinese Academy of Science (100 Talents Program of the Chinese Academy of Sciences: Y4362911ZX), Beijing Natural Science Foundation Beijing Natural Science Foundation (5142021) and National Natural Science Foundation of China (No. 31270875, 31470049).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Olayioye MA, Neve RM, Lane HA, Hynes NE. The ErbB signaling network: receptor heterodimerization in development and cancer. Embo J. 2000;19:3159-67
2. Yarden Y, Sliwkowski MX. Untangling the ErbB signalling network. Nat Rev Mol Cell Biol. 2001;2:127-37
3. Slamon DJ, Clark GM, Wong SG, Levin WJ, Ullrich A, McGuire WL. Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science. 1987;235:177-82
4. Slamon DJ, Godolphin W, Jones LA, Holt JA, Wong SG, Keith DE. et al. Studies of the HER-2/neu proto-oncogene in human breast and ovarian cancer. Science. 1989;244:707-12
5. Seshadri R, Firgaira FA, Horsfall DJ, McCaul K, Setlur V, Kitchen P. Clinical significance of HER-2/neu oncogene amplification in primary breast cancer. The South Australian Breast Cancer Study Group. J Clin Oncol. 1993;11:1936-42
6. Gabos Z, Sinha R, Hanson J, Chauhan N, Hugh J, Mackey JR. et al. Prognostic Significance of Human Epidermal Growth Factor Receptor Positivity for the Development of Brain Metastasis After Newly Diagnosed Breast Cancer. J Clin Oncol. 2006;24:5658-63
7. Wang J, Hou T, Xu X. Recent Advances in Free Energy Calculations with a Combination of Molecular Mechanics and Continuum Models. Curr Comput-Aided Drug Design. 2006;2:287-306
8. Kollman PA, Massova I, Reyes C, Kuhn B, Huo S, Chong L. et al. Calculating Structures and Free Energies of Complex Molecules: Combining Molecular Mechanics and Continuum Models. Accounts Chem Res. 2000;33:889-97
9. Homeyer N, Gohlke H. Free Energy Calculations by the Molecular Mechanics Poisson-Boltzmann Surface Area Method. Mol Inform. 2012;31:114-22
10. Gohlke H, Kiel C, Case DA. Insights into protein-protein binding by binding free energy calculation and free energy decomposition for the Ras-Raf and Ras-RalGDS complexes. J Mol Biol. 2003;330:891-913
11. Hou T, Xu Z, Zhang W, McLaughlin WA, Case DA, Xu Y. et al. Characterization of domain-peptide interaction interface: a generic structure-based model to decipher the binding specificity of SH3 domains. Mol Cell Proteomics. 2009;8:639-49
12. Hou T, Zhang W, Case DA, Wang W. Characterization of domain-peptide interaction interface: a case study on the amphiphysin-1 SH3 domain. J Mol Biol. 2008;376:1201-14
13. Jiang Y, Lu J, Wang Y, Zeng F, Wang H, Peng H. et al. Molecular-dynamics-simulation-driven design of a protease-responsive probe for in-vivo tumor imaging. Adv Mater. 2014;26:8174-8
14. Razavi AM, Wuest WM, Voelz VA. Computational screening and selection of cyclic peptide hairpin mimetics by molecular simulation and kinetic network models. J Chem Inf Model. 2014;54:1425-32
15. Hwang S, Thangapandian S, Lee Y, Sakkiah S, John S, Lee KW. Discovery and evaluation of potential sonic hedgehog signaling pathway inhibitors using pharmacophore modeling and molecular dynamics simulations. J Bioinform Comput Biol. 2011;1:15-35
16. Frederix PW, Ulijn RV, Hunt NT, Tuttle T. Virtual Screening for Dipeptide Aggregation: Toward Predictive Tools for Peptide Self-Assembly. J Phys Chem Lett. 2011;2:2380-4
17. Parikesit AA, Kinanty, Tambunan US. Screening of commercial cyclic peptides as inhibitor envelope protein dengue virus (DENV) through molecular docking and molecular dynamics. Pak J Biol Sci. 2013;16:1836-48
18. Geng L, Wang Z, Yang X, Li D, Lian W, Xiang Z. et al. Structure-based Design of Peptides with High Affinity and Specificity to HER2 Positive Tumors. Theranostics. 2015;5:1154-65
19. Drews J. Drug discovery: a historical perspective. Science. 2000;287:1960-4
20. Fodor SP, Read JL, Pirrung MC, Stryer L, Lu AT, Solas D. Light-directed, spatially addressable parallel chemical synthesis. Science. 1991;251:767-73
21. Moulin E, Cormos G, Giuseppone N. Dynamic combinatorial chemistry as a tool for the design of functional materials and devices. Chem Soc Rev. 2012;41:1031-49
22. Zheng H, Wang W, Li X, Wang Z, Hood L, Lausted C. et al. An automated Teflon microfluidic peptide synthesizer. Lab Chip. 2013;13:3347-50
23. Wang W, Huang Y, Liu J, Xie Y, Zhao R, Xiong S. et al. Integrated SPPS on continuous-flow radial microfluidic chip. Lab Chip. 2011;11:929-35
24. Wang W, Li M, Wei Z, Wang Z, Bu X, Lai W. et al. Bimodal imprint chips for peptide screening: integration of high-throughput sequencing by MS and affinity analyses by surface plasmon resonance imaging. Anal Chem. 2014Apr15;86(8):3703-7 doi: 10.1021/ac500465e
25. Wang W, Wei Z, Zhang D, Ma H, Wang Z, Bu X. et al. Rapid Screening of Peptide Probes through In Situ Single-Bead Sequencing Microarray. Anal Chem. 2014;86:11854-9
26. Ogiso H, Ishitani R, Nureki O, Fukai S, Yamanaka M, Kim JH. et al. Crystal structure of the complex of human epidermal growth factor and receptor extracellular domains. Cell. 2002;110:775-87
27. Eigenbrot C, Ultsch M, Dubnovitsky A, Abrahmsén L, Härd T. Structural basis for high-affinity HER2 receptor binding by an engineered protein. Proc Natl Acad Sci U S A. 2010;107:15039-44
28. Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H. et al. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947-8
29. Duan Y, Wu C, Chowdhury S, Lee MC, Xiong G, Zhang W. et al. A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. J Comput Chem. 2003;24:1999-2012
30. Wei C, Desheng L, Jian G, Fang L, Lingling G, Mingjuan J. Molecular dynamics and free energy studies of chirality specificity effects on aminobenzo[a]quinolizine inhibitors binding to DPP-IV. J Mol Model. 2013;19:1167-77
31. Geng L, Gao J, Cui W, Tang Y, Ji M, Chen B. Computational insights into the selectivity mechanism of APP-IP over matrix metalloproteinases. J Comput Aided Mol Des. 2012;26:1327-42
32. Neve RM, Chin K, Fridlyand J, Yeh J, Baehner FL, Fevr T. et al. A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell. 2006;10:515-27
33. Schuck P. Use of surface plasmon resonance to probe the equilibrium and dynamic aspects of interactions between biological macromolecules. Annu Rev Biophys Biomol Struct. 1997;26:541-66
34. Yu X, Liang L, Xueqing W, Jiancheng W, Xuan Z, Qiang Z. Chloride channel-mediated brain glioma targeting of chlorotoxin-modified doxorubicine-loaded liposomes. J Control Release. 2011;152:402-10
35. Haran G, Cohen R, Bar LK, Barenholz Y. Transmembrane ammonium sulfate gradients in liposomes produce efficient and stable entrapment of amphipathic weak bases. BBA - Biomembranes. 1993;1151:201-15
36. Zhou X, Zhang M, Yung B, Li H, Zhou C, Lee LJ. et al. Lactosylated liposomes for targeted delivery of doxorubicin to hepatocellular carcinoma. Int J Nanomed. 2012;7:5465-74
37. Elbayoumi TA, Torchilin VP. Enhanced Cytotoxicity of Monoclonal Anticancer Antibody 2C5-Modified Doxorubicin-Loaded PEGylated Liposomes against Various Tumor Cell Lines. Eur J Pharm Sci. 2007;32:159-68
38. Tolmachev V, Orlova A, Pehrson R, Galli J, Baastrup B, Andersson K. et al. Radionuclide therapy of HER2-positive microxenografts using a 177Lu-labeled HER2-specific Affibody molecule. Cancer Res. 2007;67:2773-82
39. Baum RP, Prasad V, Muller D, Schuchardt C, Orlova A, Wennborg A. et al. Molecular imaging of HER2-expressing malignant tumors in breast cancer patients using synthetic 111In- or 68Ga-labeled affibody molecules. J Nucl Med. 2010;51:892-7
Author contact
![]() Corresponding authors: Qiaojun Fang, National Center for Nanoscience and Technology, Beijing 100190, China. Phone: +86-10-82545562; Fax: +86-10-82545643; E-mail: fangqjcn. Zhiyuan Hu, National Center for Nanoscience and Technology, Beijing 100190, China. Phone: +86-10-82545643; Fax: +86-10-82545643; E-mail: huzycn. Weizhi Wang, National Center for Nanoscience and Technology, Beijing 100190, China. Phone: +86-10-82545643; Fax: +86-10-82545643; E-mail: wangwzcn.
Corresponding authors: Qiaojun Fang, National Center for Nanoscience and Technology, Beijing 100190, China. Phone: +86-10-82545562; Fax: +86-10-82545643; E-mail: fangqjcn. Zhiyuan Hu, National Center for Nanoscience and Technology, Beijing 100190, China. Phone: +86-10-82545643; Fax: +86-10-82545643; E-mail: huzycn. Weizhi Wang, National Center for Nanoscience and Technology, Beijing 100190, China. Phone: +86-10-82545643; Fax: +86-10-82545643; E-mail: wangwzcn.