Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and Methods
Results and Discussion
Conclusions
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2016; 6(9):1277-1292. doi:10.7150/thno.15133 This issue Cite
Research Paper
Green Tea Catechin-Based Complex Micelles Combined with Doxorubicin to Overcome Cardiotoxicity and Multidrug Resistance
Tangjian Cheng1, Jinjian Liu2, Jie Ren1, Fan Huang2, Hanlin Ou1, Yuxun Ding1, Yumin Zhang2, Rujiang Ma1, Yingli An1, Jianfeng Liu2 ![]() , Linqi Shi1, 3
, Linqi Shi1, 3 ![]()
1. State Key Laboratory of Medicinal Chemical Biology, Key Laboratory of Functional Polymer Materials of Ministry of Education, Institute of Polymer Chemistry, Nankai University, Tianjin 300071, P.R. China.
2. Tianjin Key Laboratory of Radiation Medicine and Molecular Nuclear Medicine, Institute of Radiation Medicine, Chinese Academy of Medical Science & Peking Union Medical College, Tianjin, 300192, P.R. China.
3. Collaborative Innovation Center of Chemical Science and Engineering (Tianjin), Nankai University, Tianjin 300071, P.R. China.
Received 2016-1-29; Accepted 2016-5-4; Published 2016-6-6
Abstract

Chemotherapy for cancer treatment has been demonstrated to cause some side effects on healthy tissues and multidrug resistance of the tumor cells, which greatly limits therapeutic efficacy. To address these limitations and achieve better therapeutic efficacy, combination therapy based on nanoparticle platforms provides a promising approach through delivering different agents simultaneously to the same destination with synergistic effect. In this study, a novel green tea catechin-based polyion complex (PIC) micelle loaded with doxorubicin (DOX) and (-)-Epigallocatechin-3-O-gallate (EGCG) was constructed through electrostatic interaction and phenylboronic acid-catechol interaction between poly(ethylene glycol)-block-poly(lysine-co-lysine-phenylboronic acid) (PEG-PLys/PBA) and EGCG. DOX was co-loaded in the PIC micelles through π-π stacking interaction with EGCG. The phenylboronic acid-catechol interaction endowed the PIC micelles with high stability under physiological condition. Moreover, acid cleavability of phenylboronic acid-catechol interaction in the micelle core has significant benefits for delivering EGCG and DOX to same destination with synergistic effects. In addition, benefiting from the oxygen free radicals scavenging activity of EGCG, combination therapy with EGCG and DOX in the micelle core could protect the cardiomyocytes from DOX-mediated cardiotoxicity according to the histopathologic analysis of hearts. Attributed to modulation of EGCG on P-glycoprotein (P-gp) activity, this kind of PIC micelles could effectively reverse multidrug resistance of cancer cells. These results suggested that EGCG based PIC micelles could effectively overcome DOX induced cardiotoxicity and multidrug resistance.
Keywords: combination therapy, catechin, cardiotoxicity, multidrug resistance.
Introduction
Doxorubicin (DOX), an anthracycline antibiotic, is a kind of chemotherapeutical agent widely used in solid tumors [1, 2]. However, various limitations that reduce the therapeutic efficacy are presented in the DOX based chemotherapy: low water solubility, cumulative and irreversible cardiotoxicity [3, 4], nonspecific delivery throughout the body [5] and DOX resistance of cancer cells [6, 7]. In recent decades, nanotechnology based nanomedicines provided solutions to some limitations, including encapsulating drug in the nanocarriers to improve solubility [8-12] and targeted strategies to increase the specificity delivery [13-16]. However, limited effectiveness in reduced cardiotoxicity and reversing multidrug resistance are still encountered in some nanomedicines [17-19].
(-)-Epigallocatechin-3-O-gallate (EGCG), a major component of green tea, has been reported to exhibit strong antioxidant activity and scavenging effects on active oxygen and free radicals because of the two triphenolic groups in its molecular structure [20-22]. It is well known that long-term chemotherapy with DOX is frequently accompanied by serious cardiac toxicity, severely damaging patients' health and causing additional suffering of patients [3]. A main mechanism for cardiotoxicity is the increasing oxygen free radicals generated via a redox reaction of DOX catalyzed by endogenous reductases during the metabolic activation of DOX [23-25]. Therefore, EGCG combined with DOX could be useful to scavenge intracellular oxygen free radicals and prevent DOX induced cardiotoxicity [26-28].
In addition, multidrug resistance (MDR) of tumor cells is an important limiting factor for effectiveness of chemotherapy [29]. P-glycoprotein (P-gp), a drug efflux pump on cell membrane, plays an important role to mediate efflux of the drugs from cancer cells [30-31]. Increased P-gp on cell membrane endows tumor cell with multidrug resistance. To overcome MDR, various strategies have been designed to inhibit or modulate the activity of P-gp in cancer cells, especially in pharmacological and biochemical fields [32-34]. One important approach is to use MDR inhibitors or modulators. MDR inhibitors or modulators with ability to reverse the resistance against anticancer drugs are co-administrated with drugs to block the efflux of drugs from cancer cells, leading to increased drugs accumulation in cancer cells and enhanced therapeutic efficacy [35-37]. However, low affinity of numerous MDR modulators with P-gp requires high doses to achieve desired effect. Unfortunately, high doses of MDR modulators will inevitably cause enhanced toxicity to normal tissues because of their intrinsic toxicity [36, 38]. Holl et al. reported that verapamil (VER), a weaker calcium channel blocker, showed inhibitory activity of P-gp [39]. However, to achieve effective inhibition of P-gp, the dose of VER significantly exceeded the limit of the clinical dose for arrhythmia and would induce cardiotoxicity [40, 41]. Therefore, a low toxicity and high affinity for P-gp MDR modulator combined with anticancer drugs could be more viable candidate for reversing MDR. Fortunately, EGCG was firstly found to inhibit the transport activity of P-gp in 2002 [42]. Since then, EGCG was suggested to be a low toxicity and effective P-gp modulator to chemo-sensitize the resistant tumor cells to DOX and increase the DOX accumulation in MDR cells [43, 44].
Based on the oxygen free radicals scavenging activity and drug resistance reversing of EGCG, combination therapy with EGCG and DOX could be the best strategy to overcome both DOX induced cardiotoxicity and multidrug resistance. Compared with monotherapy, combination therapy could be more effective in the enhancement of cancer therapy by achieving synergistic effects and reducing toxicity [26, 43, 45]. However, the combination therapy based on simply mixed administration of different drugs will lead to distinct pharmacokinetic profiles of different therapeutics and an inconsistent biodistribution in vivo, which would reduce the synergistic effect between different agents. On the contrary, nanoparticle-based combination therapy systems, such as vesicles, polymeric micelles and gold nanoparticles, are widely used to simultaneously release the different chemotherapeutic agents at the same destination and maximize synergistic effects [46-48]. Therefore, constructing a nanoparticle-based combination therapy encapsulating DOX and EGCG in the nanoparticle could achieve better therapeutic effects.
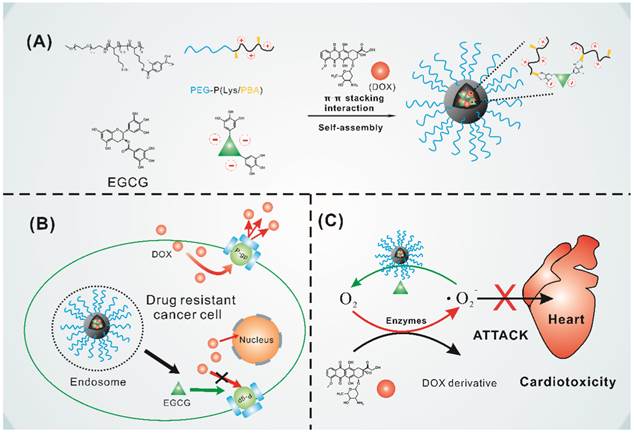
Because EGCG is hydrophilic and instable while DOX is hydrophobic under physiological environment, it is difficult to co-load DOX and EGCG in the hydrophobic nanoparticle core through hydrophobic interaction and maintain the stability of EGCG. Herein, we designed a type of green tea catechin-based polyion complex micelle with EGCG and DOX in the micelle core and PEG as the shell. EGCG based polyion complex micelle was firstly constructed through electrostatic interaction and phenylboronic acid-catechol interaction [49] between PEG-PLys/PBA and EGCG as shown in Figure 1A. With the binding property of EGCG with several biological molecules [50], DOX could be encapsulated in the micelle core through π-π stacking interaction with EGCG. For this system, the phenylboronic acid-catechol interaction between phenylboronic acid (PBA) and EGCG in micelle core achieved a cross-linked core which could obtain high stability of PIC micelles and EGCG under physiological environment [49]. More importantly, the boronic acid-catechol interaction was acid cleavable [51], endowed the micelles with effective and simultaneous DOX and EGCG release when internalized into cancer cells. Furthermore, as shown in Figure 1B, EGCG loaded in the micelles was expected to interact with P-gp and inhibit their transport activity, resulting in increased DOX accumulation in drug resistant cancer cells. Meanwhile, EGCG could timely and quickly scavenge oxygen free radicals generated by DOX in the heart, avoiding the cardiomyocytes damage caused by delayed oxygen free radicals scavenging as shown in Figure 1C.
(A) Schematic illustrating the formation of the core cross-linked micelle co-loaded with DOX. (B) Proposed mechanism to overcome multidrug resistance. (C) Mechanism to reduce cardiotoxicity.
Materials and Methods
Materials
Doxorubicin-HCl (DOX·HCl) was supplied by Jingyan Chemicals Corporation (Shanghai, China). EGCG was purchased from TCI. α-Methoxy-ω-aminopoly(ethylene glycol) (CH3O-PEG113-NH2; Mw = 5000; Mw/Mn = 1.05) was purchased from Aladdin and used after dried under vacuum. ε-(benzyloxycarbonyl)-L-lysine N-carboxyanhydride (Lys(Z)-NCA) were synthesized by the Fuchs-Farthing method using bis(trichloromethyl) carbonate (triphosgene) according to ref 52. 1-Ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride (EDC·HCl; >98%, Fluka), N-hydroxysuccinimide (NHS; >97%, Fluka), trifluoroacetic acid, hydrogen bromide (HBr; 45% in acetic acid) was 2′,7′-dichlorofluorescein diacetate (DCFH-DA) purchased from Sigma-Aldrich. 3-fluoro-4-carboxyphyenylboronic acid (FPBA) and 4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methylmorpholinium chloride n-hydrate (DMT-MM), nitro blue tetrazolium (NBT), xanthine, xanthine oxidase were purchased from J&K.
Synthesis of polymers
poly(ethylene glycol)-block-poly(ε-(benzyloxycarbonyl)-L-lysine) PEG-b-PLys(Z) was synthesized as shown in Figure S1 according to the previous report [11]. Briefly, Lys(Z)-NCA(1.96 g, 6.4 mmol) was dissolved in 30 ml of DMF and polymerized by addition of MeO-PEG113-NH2 (2.0 g, 0.4 mmol) with the terminal primary amino group as the initiator. The reaction mixture was stirred for 3 days at 35 °C under a dry argon atmosphere. Then solvent was evaporated under reduced pressure. The resulting product was dissolved in 25 ml of CHCl3 and then precipitated into excessive diethyl ether to obtain PEG-b-PLys(Z) (yield 90%).
Poly(ethylene glycol)-block-poly(L-lysine) (PEG-b-PLys) was prepared by deprotection of Z group in PEG-b-PLys(Z) as shown in Fig S1. In brief, PEG113-b-PLys(Z)13 (2.0 g) was dissolved in 20 ml of CF3COOH, followed by addition of HBr (33 wt.% in HOAc, 2 mL). The mixture was stirred at 0 °C for 2 h, then the reaction mixture was precipitated into excessive cold diethyl ether. The precipitation was dissolved in DMF and reprecipitated in excessive diethyl ether to remove the residual CF3COOH and obtain PEG-b-PLys (yield 60%). The product was dried at room temperature under vacuum.
PEG-b-PLys/PBA was synthesized as shown in Figure S1 according to previous report [53]. FPBA was conjugated by acylation of the pendent primary amino groups of PEG-b-PLys using DMT-MM as coupling agent. PEG113-b-PLys13 (100 mg, 0.18mmol) was dissolved in 10 ml of sodium bicarborate solution (50 mM, pH 8.5) in the presence of D-mannitol (100 mg, 0.55 mmol), followed by addition of the solution of FPBA in 1.7 ml of CH3OH. Then DMT-MM (254 mg, 0.92 mmol) was added and the reaction mixture was stirred at 25 °C for 12 h. The whole solution was transferred to a dialysis bag (MWCO 3500) and dialyzed against 0.01 N NaOH aq., 0.01N HCl aq., then deionized water. PEG-b-PLysPBA was obtained by lyophilisation.
Preparation of green tea catechin-based polyion complex micelles
The FPBA based core cross-linked micelle (CLM) was formed by mixing the aqueous solution of PEG-b-PLys/PBA and EGCG. Briefly, PEG-b-PLys/PBA (1.0 mg) was dissolved in 4 ml of deionized water, and then 75 μl of EGCG (1 mg/ml) in water was added. The polyion complex micelle was formed through the electrostatic interaction between PLys and EGCG. Based on boronic acid-catechol interaction, the cross-linked core was formed. With the same ratio of [Lys]/[EGCG], non-cross-linked micelle (NCLM) was prepared via the same method as CLM using PEG-b-PLys and EGCG.
As a control, micelle without EGCG (NP) was prepared via the nanoprecipitation technique. In brief, the solution of PEG-PLys(Z) (2 mg) in 1 ml of DMSO was added dropwise into 10 ml of PBS (pH 7.4, 10 mM). After stirring at room temperature for 4 h, NP was transferred into dialysis bag (MWCO 3500) and dialyzed against deionized water for 48 h.
Evaluation of stability of CLM and NCLM in vitro
The stability of CLM and NCLM was evaluated in 10 mM PBS (pH 7.4) at 37 °C. At predetermined time point, the required volume of CLM and NCM solution were taken and the mean size of micelles and light intensity were recorded by DLS.
Auto-oxidation of EGCG
EGCG (75 μg) was dissolved in 1 ml of PBS (10 mM, pH 7.4). Over time, the absorption peak of EGCG was measured by using UV-vis spectra.
pH responsiveness of EGCG based CLM
NCM core was cross-linked through boronic acid-catechol interaction. Under an acid environment, the interaction would be weakened and EGCG would release from micelle core.
The boronic acid-catechol interaction leads to the changed structure, which could be detected from the changes in the absorption peaks. UV-vis spectra of FPBA solution was recorded at pH 7.4, and then EGCG solution (75 μg/ml) was added into FPBA solution. UV-vis spectra of the mixture was recorded to detect changes in the characteristic peaks. Finally, pH value of the mixture was adjusted to 5.0, followed by monitoring the changes in the absorption peaks.
EGCG based micelles (CLM and NCLM) solution (1 mg/ml) were prepared in pH 7.4 PBS (10 mM), and then the absorption peaks of CLM and NCLM were detected by using UV-vis spectra at predetermined time point. pH value of CLM solution was adjusted to 5.0, and then the characteristic peak of CLM solution was detected to monitor the absorbance change.
1 ml of CLM solution (pH 5.0 and 7.4) (the content of EGCG was 100 μg) was transferred to dialysis bag (MWCO 3500), and then each dialysis bag was immersed in 40 ml of PBS solution with the same pH value and incubated in shaker (200 rpm, 37 °C). To simulate blood flow, at fixed time intervals, 4 ml of dialysis fluid was taken out for UV-vis measurement (at 276 nm) and an equal volume of fresh buffer was added in. The amount of released EGCG was determined by measuring the height of absorption peak (276 nm) using free EGCG as standard. The release experiments were conducted in triplicate, and the results presented are the average data.
Loading of DOX
DOX could be loaded into the CLM through π-π stacking interaction between DOX and EGCG. DOX-loaded CLM (CLM-DOX) was readily prepared by addition of DOX and EGCG (in DMSO) to the solution of PEG-PLys/PBA in PBS. Briefly, DOX·HCl (1 mg) was dissolved in DMSO with 5 μl of trimethylamine (TEA) for 2 h to obtain the doxorubicin base (DOX). And then a specific amount of DOX solution and EGCG solution in DMSO (weight ratio of DOX and EGCG was 0.9) were mixed firstly and added dropwise into 4 ml of PEG-PLys/PBA (1 mg) solution in 4 ml of PBS (pH 7.4, 10 mM). After stirring for 4 h, the DOX-loaded CLM was transferred into dialysis bag (MWCO 3500) and dialyzed against deionized water for 48 h. CLM-DOX was concentrated by ultrafiltration before use.
As a control, DOX-loaded micelle without EGCG (NP-DOX) was prepared via the nanoprecipitation technique. In brief, the solution of PEG-PLys(Z) (2 mg) and DOX (400 μg) in 1 ml of DMSO was added dropwise into 10 ml of PBS (pH 7.4, 10mM). After stirring at room temperature for 4 h, NP-DOX was transferred into dialysis bag (MWCO 3500) and dialyzed against deionized water for 48 h. NP-DOX was concentrated by ultrafiltration before use.
Drug loading content (DLC) and drug loading efficiency (DLE) of CLM-DOX and NP-DOX was measured by using a fluorescence spectrometer (excitation at 484 nm and emission at 590 nm). Briefly, the micelle solution was diluted into 50-fold volume of DMF and the DOX concentration was measured by calculating the height of emission peak at 590 nm according to the calibration curve of DOX. DLC and DLE were calculated according to the following formulas:
DLC (wt %) = (weight of loaded drug/total weight of polymer and loaded drug)×100 %
DLE (%) =(weight of loaded drug/weight of drug in feed) ×100 %
In vitro DOX release
The DOX release of CLM-DOX and NP-DOX was performed under different pH condition (pH 5.0, 6.5 and 7.4). Briefly, 2 ml of CLM-DOX and NP-DOX were transferred into dialysis bag (MWCO 3500) and immersed in 40 ml of PBS with different pH condition at 37 °C. At fixed time intervals (1, 2, 4, 6, 8, 10, 24 h), 4 mL of the solution outside the dialysis bag was taken out for fluorescent measurements (excitation at 484 nm) and an equal volume of fresh buffer was added in. The amount of released DOX was determined by measuring the height of emission peak (590 nm) using free DOX in PBS as standard. The release experiments were conducted in triplicate, and the results presented are the average data.
Superoxide scavenging activity (NBT assay)
Superoxide scavenging activity of EGCG based CLM was measured via NBT assay according to the previous report [54]. In this assay, xanthine-xanthine oxidase (X-XO) system was used to generate ·O2-. Reduced by ·O2-, NBT would be transformed into NBT formazan which would be detected in the absorbance at 560 nm. When EGCG based CLM was added into the system, ·O2- would be scavenged preferentially by CLM, leading to lower absorbance increase at 560 nm.
Xanthine (1 mg) and NBT (16 mg) were dissolved in 33 ml of PB (100 mM, pH 7.8) and incubated at 37 °C. Then each CLM solutions with different concentration was added into 2 ml of xanthine and NBT solution, respectively. XO (6 mU/ml) was added into the mixture to trigger the reaction. The increase of absorbance at 560 nm was recorded every 3 seconds by UV-vis spectra for 3 minutes to detect the production rate of NBT formazan at different CLM concentration. Each concentration generated a time-dependent curve, and the superoxide scavenging activity of CLM was calculated from the absorbance ratio of each micelle concentration to blank group at 3 min.
Cell culture
MCF-7 Cells and DOX resistant human breast carcinoma cells (MCF-7/Adr) were purchased from Nanjing Kaiji Biotech. Ltd. Co. (Nanjing, China). Cells were cultured in RPMI-1640 medium with 10% (v/v) fetal bovine serum (FBS) and 1% penicillin-streptomycin in a humidified atmosphere at 37 °C with 5% CO2. The cells were subcultured with 0.25% trypsin-EDTA when reaching 80-90% confluence.
Cellular uptake and cell viability in H9C2 cardiac muscle cells (H9C2 cells)
H9C2 cells were seeded into 96-well plates at a density of 0.6×104 cells per well in 100 µL RPMI-1640 medium/PBS. After an incubation of 24 hours, the free DOX, NP-DOX and CLM-DOX were added to each well. After 4 hours further incubation, the culture medium was removed and cells were washed three times with 500 μL PBS buffer. The cellular uptake of micelles was observed using confocal laser scanning microscope (TCS SP5).
The cytotoxicity of free DOX, NP-DOX and EGCG based CLM-DOX were determined against H9C2 cells by MTT assay. In the MTT assay, H9C2 cells were seeded into 96-well plates at a density of 4000 cells per well in 500 μL RPMI-1640 medium/PBS (pH 7.4). After 24 h incubation, free DOX, NP-DOX and CLM-DOX solution were added to each well with different DOX concentrations (0.01, 0.1, 1, 5, 20, 40 μg/mL). The saline solution was used as control. After 24 hours, 25 μL of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide (MTT) (5 mg/ml) was added to each well and the mixture was incubated for another 4 h, then 150 μL of DMSO was added to dissolve the obtained blue formazan crystals. The absorbance was measured at a wavelength of 570 nm and the viability was expressed as the percentage of the control.
Measurement of intracellular ROS generation
To measure the ROS generation in H9C2 cells, 5(6)-carboxy-2′,7′-dichlorofluorescein diacetate (DCFH-DA) was used as a cell-permeable probe which could be cleaved by intracellular esterase to non-fluorescent 2′,7′-dichlorofluorescin (DCFH) and oxidized by ROS to a fluorescent product dichlorofluorescein (DCF). The fluorescence intensity of DCF in H9C2 cells determines the level of intracellular ROS. H9C2 cells were seeded into 96-well plates at a density of 4000 cells per well in 500 μL RPMI-1640 medium/PBS (pH 7.4). After 24 h incubation, free DOX, NP-DOX and CLM-DOX solution were added to each well with same DOX concentrations. After incubation for 2 h, H9C2 cells were harvested and washed three times with 500 μL PBS buffer. Further incubation with 10 μM DCFH-DA for 20 min at 37 °C was conducted. The absorbance was measured at a wavelength of 488 nm and the ROS generation was expressed as the percentage of the control (PBS).
Histopathologic evaluation
The animal studies were performed in accordance with the Regulations for the Administration of Affairs Concerning Experimental Animals (Tianjin, revised in June2004) and adhered to the Guiding Principles in the Care and Use of Animals of the American Physiological Society. The BALB/c mice (n = 3) were injected intravenously via the tail vein with free DOX, NP-DOX, EGCG based CLM-DOX and saline. The dose of DOX was fixed at 5 mg/kg body weight. Free DOX, NP-DOX, CLM-DOX and saline were administrated for five times through tail vein every other day. To evaluate the DOX induced cardiotoxicity, mice were sacrificed and hearts were harvested immediately post dissection of these mice. The hearts were immediately fixed with 10% formalin for 1 day at room temperature, embedded in paraffin, and cut into 8-μm-thick sections for hematoxylin/eosin (H&E) observed using optical microscope.
Cellular uptake in MCF-7/Adr cells
MCF-7/Adr cells were seeded into 96-well plates at a density of 0.6×104 cells per well in 100 µL RPMI-1640 medium/PBS. After an incubation of 24 hours, CLM-DOX and NP-DOX were added to each well. After 12 hours further incubation, the cellular uptake of micelles was observed with an inverted fluorescence microscope (DMI6000B, Leica, Wetzlar, Germany).
Cell viability in MCF-7 cells and MCF-7/Adr cells
The cytotoxicity of different materials (CLM, NP, free DOX, NP-DOX and CLM-DOX) was determined against MCF-7 cells and MCF-7/Adr by MTT assay. In the MTT assay, MCF-7 and MCF-7/Adr cells were seeded into 96-well plates at a density of 2000 cells per well in 500 μL RPMI-1640 medium/PBS (pH 7.4). After 96 h incubation, CLM and NP solution were added to each well with different micelle concentration (0.5, 1, 5, 10, 100, 500, 1000μg/mL). Meanwhile free DOX, NP-DOX and CLM-DOX solution were added to each well with different DOX concentrations (0.1, 0.5, 1, 5, 20, 40 μg/mL). The saline solution was used as control. After 24 hours, 25 μL of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide (MTT) (5 mg/ml) was added to each well and the mixture was incubated for another 4 h, then 150 μL of DMSO was added to dissolve the obtained blue formazan crystals. The absorbance was measured at a wavelength of 570 nm and the viability was expressed as the percentage of the control.
In vivo biodistribution experiment
BALB/c mice were randomly divided into three groups and intravenously injected by and NP-DOX/Cy5 and CLM-DOX/Cy5 at same PEG-P(Lys-LysDox-LysCy5) (synthesized as described in the supporting information). At 1h, 6h, 24h post-injection, the mice were sacrificed, and then the heart as well as the major organs were harvested. ex vivo imaging was conducted by the Kodak IS in vivo FX imaging system.
Results and Discussion
Synthesis of polymers
PEG-PLys(Z) was synthesized by ring-opening polymerization. PEG-PLys was prepared by deprotection of benzyl group of PEG-b-PLys(Z). PEG-PLys/PBA was synthesized by conjugation of FPBA to the pendent amino of PLys using DMT-MM as coupling agent. Characterization of PEG-PLys(Z) and PEG-PLys was described in supporting information. PEG-b-PLys/PBA was characterized by 1H NMR. As shown in Figure S4, the peaks around 7.3 ppm which is the characteristic peaks of the phenylene protons of FPBA indicated that FPBA were conjugated to polymers successfully. The number of FPBA units introduced per polymer chain was estimated to be 6 by calculating the peak intensity ratio of the butylene protons of Lys units (1.2-1.8 ppm) to the phenylene protons of FPBA (7.2-7.5 ppm) as shown in Figure S4.
Formation of green tea catechin-based polyion complex micelles
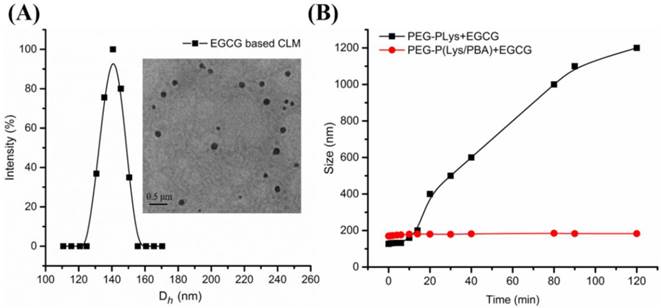
EGCG, the main polyphenol in green tea, has been demonstrated to possess anticancer effects [55], and antioxidant activity [20]. Here, inspired by binding property of EGCG with several biological molecules, polyion complex micelles (NCLM, CLM) encapsulated EGCG in the core were constructed for combining with hydrophobic DOX. Attributed to the negatively charged EGCG at pH 7.4, NCLM was formed through the electrostatic interaction between EGCG and PLys. However, polyion complex micelles (PIC micelles) suffer from instability under physiological condition. To improve the stability of green tea catechin based PIC micelles, FPBA was conjugated on the PLys. CLM was precisely constructed by self-assembly of PEG-Plys/PBA and EGCG through the electrostatic interaction between Plys and EGCG, and the cross-linked core of CLM was formed through phenylboronic acid-catechol interaction. NCLM and CLM were prepared with the same ratio of [Lys]/[EGCG] respectively. The drug loading content of EGCG was about 7.0 %. The hydrodynamic diameter distribution of CLM determined by DLS and TEM image of CLM were shown in Figure 2A. The average hydrodynamic diameters of CLM were measured to be around 140 nm (pH 7.4) with narrow size distributions (Figure 1A). The TEM image revealed that the obtained CLM was spherical and the sizes were around 130 nm (inset in Figure 2A).
(A) Hydrodynamic diameter distribution of CLM and TEM images of CLM at pH 7.4 in 10 mM phosphate buffer solution (inset). (B) Stability of CLM and NCLM. Size variation of CLM and NCLM in PBS buffer (10 mM, pH 7.4) at 37 °C.
Stability of EGCG in the micelles
Easily occurred auto-oxidation of EGCG under common experimental condition implicated the instability of EGCG [56]. To explore the auto-oxidation of EGCG, UV-vis spectra of EGCG dissolved in water exposed in the air was measured at room temperature as shown in Figure S6. EGCG solution exhibited the characteristic absorption peak at 276 nm. Moreover, the absorption peak at 326 nm which attributed to EGCG oxidation products (EGCG dimer) was observed, and the intensity of the absorption peak increased over the time, indicating the gradual auto-oxidation process of EGCG.
To evaluate the stability of EGCG in the micelle, the absorption peak of NCLM and CLM was measured by using UV-vis spectra. As shown in Figure S8, the absorption peak of EGCG oxidation products at 326 nm was obviously observed during the NCLM condition. Furthermore, the intensity of the absorption peak increased rapidly over time and the solution turned to be brown, indicating that EGCG was unstable in the NCLM core. While for the condition of CLM, the absorption peak of EGCG oxidation products at 326 nm were not detected for 4 h, and a new absorption peak at 312 nm appeared which was ascribed to the structure of boronate ester formed by phenylboronic acid and catechol (described below). Compared with the brown solution of NCLM at 4 h, the solution of CLM was colorless and transparent (inset in Figure S9). These results demonstrated that the movement of EGCG in the CLM core was restrained by cross-linked core, leading to high stability of EGCG in the CLM.
Stability of PIC micelles
The stability under physiological condition is of fundamental importance for drug delivery systems to achieve the high efficacy in controlled drug delivery. For PIC micelles, instability is a major obstacle in their therapeutic application. In recent decades, crosslinking in the micelle became the main strategy for PIC micelles to improve the stability under physiological condition [57]. In this study, CLM core was cross-linked based on phenylboronic acid-catechol interaction between FPBA and EGCG. To demonstrate the effect of core cross-linking on the stability of CLM, the size and intensity variation of CLM and NCLM were assessed in 10 mM PBS buffer by DLS. As shown in Figure 2B, the mean size of NCLM increased rapidly from 130 nm at 0 h to 1300 nm at 4 h, indicating the aggregation of micelles. This aggregation was also detected through measurement of relative light scattering intensity (I/I0) changes as functions of time. It was clearly shown that the relative light scattering intensity of NCLM increased to about 1100% in Figure S10, which was consistent with the mean size increase. The structure of NCLM was unstable because of electrostatic screening effect under physiological condition and EGCG existed in different micelle core tended to form EGCG dimer in the air, which together resulted in the aggregation of micelles. The ten-fold increase in mean size and relative light scattering intensity increase of NCLM suggested that NCLM exhibited disappointing stability under physiological condition. But for CLM, the mean size of CLM maintained unchanged at 170 nm for 2 h and the same result was confirmed by the little change of relative light scattering intensity. These results implicated that phenylboronic acid-catechol interaction endowed CLM with cross-linked core and greatly enhanced their stability under physiological condition.
pH responsiveness of CLM
The high stability of CLM under physiological condition lay the foundation for drug delivery system. However, when nanoparticles enter the tumor site, rapid and sufficient release of drug contained was vital for better therapeutic effect [58]. In this study, the pH cleavable boronate ester formed by EGCG and FPBA endowed CLM with rapid release of EGCG under an acid environment as illustrated in Figure 3A. To confirm this fact, the stability of boronate ester and EGCG release at different pH were investigated. The characteristic peak of phenylboronic-catechol interaction was determined by UV-vis spectra using small molecules FPBA and EGCG as standard. As shown in Figure 3B, the UV-spectra of EGCG and FPBA at pH 7.4 exhibited similar characteristic absorption peak around 276 nm. When the EGCG and FPBA solution were mixed, a new peak at 312 nm was detected. After that, pH value of the mixture was adjusted to 5.0, and then the peak at 312 nm was disappeared. These results indicated that the structure of boronate ester formed by phenylboronic acid and catechol could be detected by UV-vis spectra at 312 nm and phenylbonorate ester was cleavable under acid environment. Moreover, the formation of phenylbonorate ester in the CLM was confirmed with the same method. As shown in Figure 3C, the characteristic absorption peak of phenylbonorate ester at 312 nm was observed from the UV-spectra of PEG-PLys/PBA and EGCG mixed solution, and then disappearance of the peak was detected after adjusting pH value to 5.0. These results suggested that the absorption peak at 312 nm was belonged to the structure of phenylboronate ester formed by phenylboronic acid and EGCG, and phenylboronate ester was acid cleavable.
(A) Illustration of pH responsiveness of CLM. (B) UV-vis spectra of FPBA, EGCG, mixture of EGCG and FPBA at pH 7.4 and mixture of EGCG and FPBA at pH < 5.0. (C) UV-vis spectra of CLM over time under different pH condition.
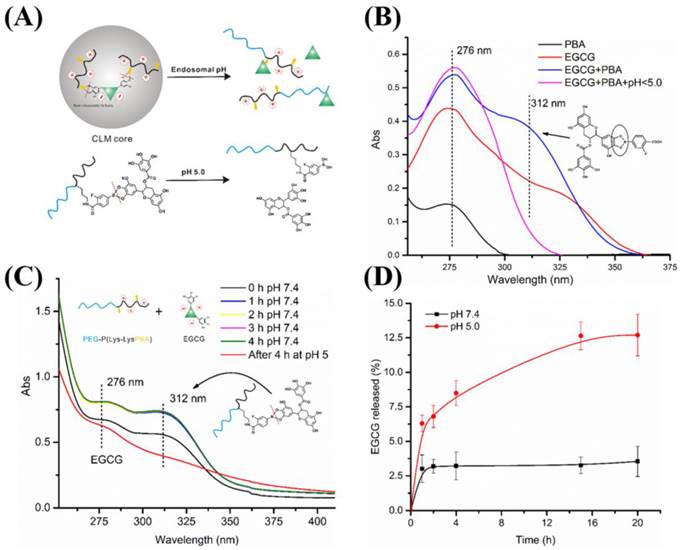
In vitro EGCG release from CLM was conducted under different pH condition. As shown in Figure 3D, under the physiological condition (pH 7.4), the cumulative release of EGCG was less than 4% in 20 h, exhibiting the high stability of CLM. While at pH 5.0 (endosomal pH), remarkably increased cumulative release to 12.5% in 20 h was achieved because of the rupture of phenylboronate ester. These results implied that CLM could selectively release EGCG at endosomal pH.
DOX loaded CLM and in vitro DOX release
Based on the binding property of EGCG with biological molecules, DOX loaded micelle, CLM-DOX was prepared through π-π stacking interaction between DOX and EGCG. As a control, DOX loaded NP-DOX was prepared through hydrophobic interaction between PLys(Z) and DOX without encapsulation of EGCG. As shown in Table 1. DLC of CLM-DOX and NP-DOX were 10.5% and 12.4%, respectively. The corresponding DLE were 50% and 55%. These result showed that DOX was encapsulated in the CLM successfully and the DOX encapsulation efficacy of CLM had little difference with NP.
Characterization of DOX loaded micelles.
| Micelles | DLC (wt %) | DLE (%) | Diameter (nm) | Zeta potential (mV) |
|---|---|---|---|---|
| CLM-DOX | 10.5 ± 2 | 50 ± 1 | 140± 10 | -2.4±1.0 |
| NP-DOX | 12.4 ± 3 | 55 ± 2 | 120±8 | -5.1±2.1 |
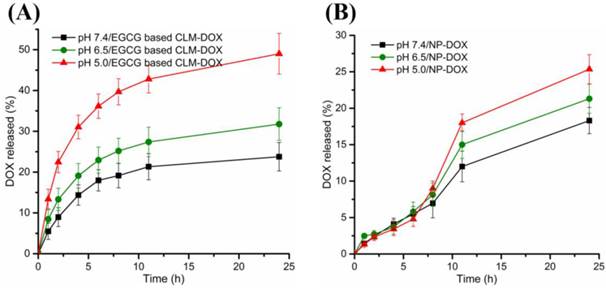
Because of pH responsiveness of CLM, DOX could rapidly release from CLM-DOX at acid pH. To confirm this fact, In vitro DOX release from CLM-DOX and NP-DOX was investigated under different pH condition (pH 7.4, 6.5 and 5.0). Under the physiological pH (7.4), the cumulative release of DOX was 22% for CLM-DOX, exhibiting similar release with NP-DOX about 18% as shown in Figure 4. However, the cumulative release of DOX from CLM-DOX increased significantly to 48% at pH 5.0, which was two-fold higher than that from NP-DOX (24%) at pH 5.0. It implicated that the acid cleavable phenylboronate ester endowed the CLM-DOX with selective release of DOX, which was excellent feature for drug delivery systems.
In vitro release profiles of DOX from (A) CLM-DOX and (B) NP-DOX under different pH conditions.
In vitro superoxide scavenging activity
Doxorubicin is an effective and widely used anticancer drug for various malignancies. However, oxygen free radicals (reactive oxygen species) generated by DOX in the heart induced cardiotoxicity, which limited the use of DOX for cancer treatment. In this study, CLM encapsulated with EGCG was prepared to prevent DOX induced cardiomyocyte toxicity because of the oxygen free radicals scavenging activity of EGCG. To simulate the production of reactive oxygen species (ROS) in cardiomyocyte, X-XO system was employed in vitro to generate ·O2-. And then NBT assay was performed to evaluate the superoxide scavenging activity of CLM at different micelle concentration. As illustrated in Figure 5A, NBT would be reduced by ·O2- (generated by X-XO system) to be a blue NBT formazan, exhibiting increased absorption at 560 nm from UV-vis experiment. After pre-addition of EGCG based CLM into the system, superoxide anion could be scavenged by CLM rapidly, which prevented NBT reduction, leading to the low UV absorbance at 560 nm. As shown in Figure 5B, UV absorbance increased rapidly to 0.35 at 3 h without addition of CLM solution, indicating the high content of superoxide anion generated by X-XO system in the whole solution. While increasing the concentration of CLM in the system from 40 to 300 μg/ml, decreased UV absorbance was followed spectrophotometrically, implicating that the NBT reduction was inhibited resulted from the superoxide scavenging activity of CLM. The superoxide scavenging activity for each concentration of micelle was calculated as follow: [Absorbance (0 μg/ml at 3min) - Absorbance at 3min)] *100% / Absorbance (0 μg/ml at 3min). As shown in Figure 5C, with the concentration of micelle increasing, the ability to scavenge superoxide was stronger for micelles. These results suggested that EGCG based CLM displayed outstanding superoxide scavenging activity and could be applied to reduce cardiotoxicity of DOX.
Superoxide scavenging activity of CLM. (A) Illustration of the role of CLM in NBT assay. (B) Effect of CLM on the inhibition of the NBT reduction under different micelle concentration. (C) Superoxide scavenging activity of CLM at 3 min under different micelle concentration.

Cellular uptake in cardiomyocytes
Mediated by intracellular enzymes, more DOX endocytosed into cardiomyocytes could generate more ROS and appears to induce cardiotoxicity. Therefore, reduced cellular uptake by cardiomyocytes would greatly decrease the cytotoxicity of DOX-loaded micelles. To evaluate the endocytosis capacity of free dox, NP-DOX and CLM-DOX in cardiomyocytes, H9C2 cells were incubated with free DOX, NP-DOX and CLM-DOX for 2 h. As shown in Figure 6, the fluorescence intensity of free DOX was high in nucleus, indicating that free DOX entered into nucleus easily. However, DOX-loaded micelles (NP-DOX and CLM-DOX) exhibited low fluorescence intensity in nucleus. And closed fluorescence intensity in the cytoplasm for NP-DOX and CLM-DOX revealed that these two DOX-loaded micelles possessed similar endocytosis capacity in H9C2 cells. These results suggested that micelles encapsulated with DOX reduced DOX content in cardiomyocyte nucleus compared with free DOX, which could effectively reduce DOX cardiotoxicity.
Inverted fluorescent microscopy images of H9C2 cells treated with free DOX, NP-DOX and EGCG based CLM-DOX after 4 h incubation at equivalent DOX concentration of 10 μg/mL. From left to right, the images show DOX fluorescence (red), cell nuclei stained by DAPI (blue), and the merge of the two images. Scale bars are 25 μm.
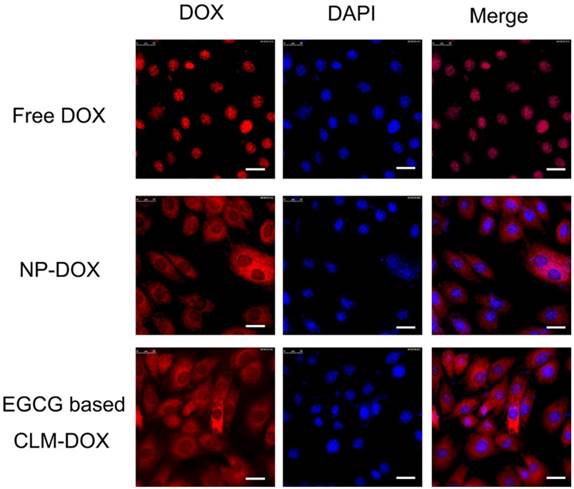
Reduced cellular uptake of CLM-DOX and NP-DOX by cardiomyocytes were demonstrated by flow cytometry as shown in Figure S13. The lower mean fluorescence intensity of H9C2 cells incubated with CLM-DOX and NP-DOX indicated that CLM-DOX and NP-DOX could effectively reduce the accumulation of DOX in the cardiomyocytes. This was consistent with the above results observed by the fluorescent microscopy images of cellular uptake.
ROS generation in cardiomyocytes and cytotoxicity assay against H9C2 cells
EGCG based CLM was demonstrated to exhibit outstanding superoxide scavenging activity from the NBT assay discussed above. To evaluate the effect of EGCG on intracellular ROS generation, free DOX, NP-DOX, CLM-DOX were incubated with H9C2 cells for 2 h using DCFH-DA as the ROS probe. ROS content in H9C2 cells was calculated through measuring the fluorescence intensity of DCF (the oxidation product of DCFH by intracellular ROS). Cells treated with PBS were used as control. As shown in Figure 7A, H9C2 cells treated with free DOX exhibited highest fluorescence intensity, indicating the highest ROS generation in H9C2 cells. And cells incubated with DOX-loaded micelles (NP-DOX and CLM-DOX) displayed decreased ROS generation, which was ascribed to reduced cellular uptake in H9C2 cells compared with free DOX. This result was consistent with inverted fluorescent microscopy images of H9C2 cells. Importantly, compared with NP-DOX treated cells, cells incubated with CLM-DOX exhibited lowest intracellular ROS generation. Because of similar endocytosis capacity of NP-DOX and CLM-DOX in H9C2 cells observed from cellular uptake results, the inhibition of ROS generation was mainly resulted from the ROS scavenging activity of EGCG encapsulated in CLM-DOX.
(A) ROS content in H9C2 cells treated with PBS, free DOX, NP-DOX and EGCG based CLM-DOX after 2 h incubation. (B) Cytotoxicity of free DOX, NP-DOX and EGCG based CLM-DOX on H9C2 cells.
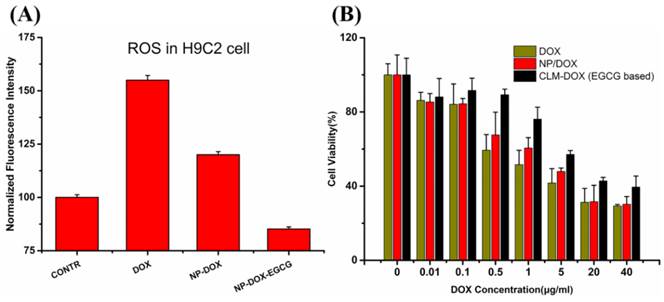
Based on inhibition of DOX induced ROS generation in cardiomyocytes by EGCG based CLM-DOX, it was expected that CLM-DOX could greatly decrease DOX induced injury of cardiac muscle cells. To confirm this fact, cytotoxicity of DOX, NP-DOX and CLM-DOX was investigated on H9C2 cells using MTT assay. Cells were treated with free DOX, NP-DOX and CLM-DOX containing different DOX concentration (0.01, 0.1, 0.5, 1, 5, 20, 40 μg/mL) at pH 7.4. As shown in Figure 7B, at each DOX concentration, free DOX caused highest cytotoxicity in H9C2 and EGCG based CLM-DOX displayed lowest cytotoxicity. These results indicated that EGCG in CLM-DOX scavenged ROS generated by DOX and prevented ROS attacking H9C2 cells. This was consistent with the ROS assay results.
Evaluation of cardiotoxicity
To investigate the cardiotoxicity of free DOX and different DOX loaded micelles, The BALB/c mice were injected intravenously via the tail vein with free DOX, NP-DOX, EGCG based CLM-DOX and saline. Histological observation of hearts slices were shown in Figure 8. Compared with DOX loaded micelles group, flake myocardial degeneration in cardiac ventricle, myocardial swelling, myofibril loose and nucleus degeneration were observed in free DOX treated group, indicating the severe cardiotoxicity of free DOX because of nonspecific tissue accumulation in hearts. However, the clear and compact structure in heart tissue was observed in CLM-DOX treated group, indicated the extremely low cardiac damage, which was attributed to reduced micelles accumulation in the heart and inhibition of DOX induced ROS generation by EGCG. These results suggested that nanoparticle based combination therapy with EGCG and DOX could effectively reduce cardiotoxicity by achieving the synergistic effect between different agents.
Histopathologic analysis of hearts after H&E staining, which were harvested from mice treated with free DOX, NP-DOX, CLM-DOX and saline. Scale bars are 50 μm.

Cellular uptake in MCF-7/Adr cells
Multidrug resistance of cancer cells hinders the effectiveness of chemotherapy. Increasing drug efflux pumps (P-gp) on the cell membrane is the main mechanism of multidrug resistance of cancer cells. However, EGCG could interact with P-gp and inhibit P-gp transport activity, leading to enhanced accumulation of DOX in multidrug resistant cancer cell according to the previous report [42]. To evaluate the effect of EGCG based CLM-DOX on the accumulation of DOX in multidrug resistant cells, MCF-7/Adr was incubated with NP-DOX and CLM-DOX at same DOX concentration for 4 h. As shown in Figure 9, strongest DOX fluorescence intensity for EGCG based CLM-DOX was observed in the cytoplasm compared with that for free DOX and NP-DOX without EGCG encapsulation, indicating that introduction of EGCG in DOX-loaded micelle could effectively inhibit DOX efflux mediated by P-gp.
Inverted fluorescent microscopy image of MCF-7/Adr cells after incubation with free DOX, NP-DOX and EGCG based CLM-DOX. From left to right, cell nucleus stained by DAPI (blue), the images show DOX fluorescence (red), and the merge of the two images.
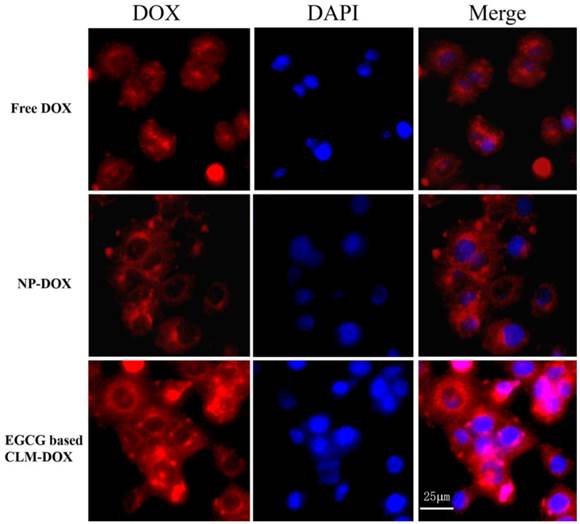
The enhanced DOX accumulation in MCF-7/ADR for EGCG based CLM-DOX was demonstrated by flow cytometry as shown in Figure S14. The highest mean fluorescence intensity of MCF-7/Adr cells incubated with CLM-DOX revealed that EGCG could effectively inhibit DOX efflux mediated by P-gp.
Cytotoxicity against MCF-7/Adr cells
The cytotoxicity of nanocarrier (NP and CLM) was investigated on MCF-7/Adr using MTT assay at different micelle concentration. As shown in Figure 10A, NP and CLM exhibited negligible toxicity assessed by cell viability assay with micelle concentration ranging from (0.5 - 1000 μg/ml). Because micelles consisted of biocompatible PEG and poly (amino acid), two kinds of micelles and EGCG were found to be nontoxic in MCF-7/Adr cells.
(A) Cytotoxicity of different micelle against resistant human breast cancer cells MCF-7/Adr. (B) Dose-response curves of DOX in CLM-DOX and NP-DOX against MCF-7/Adr. (Mean ±standard deviation, n=3).
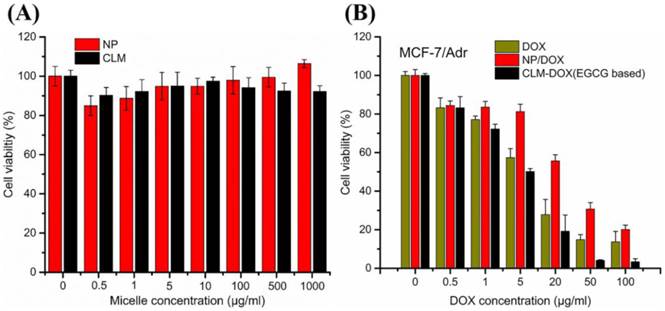
To investigate whether EGCG encapsulated in CLM-DOX could reverse DOX resistance, the cytotoxicity of free DOX, NP-DOX and CLM-DOX was evaluated on MCF-7/Adr cells at different DOX concentration. As shown in Figure 10B, at each concentration, CLM-DOX displayed highest toxicity against the DOX resistant cells compared with free DOX and NP-DOX. These results indicated that the combination of EGCG and DOX could sensitize MCF-7/Adr cells to DOX and increased DOX accumulation in DOX resistant cells.
Because of similar size and surface property of CLM-DOX and NP-DOX, they exhibited similar endocytosis capacity on each kind of cells (H9C2, MCF-7 and MCF-7/ADR cells). For H9C2 cells, the similar endocytosis capacity leaded to similar DOX content in cells. Because of the oxygen free radicals scavenging of EGCG, CLM-DOX exhibited slightly lower cytotoxicity on H9C2 than NP-DOX. But for MCF-7 and MCF-7/ADR cells where P-gp existed on the cell membrane, EGCG could inhibit P-gp transport activity, leading to reduced DOX efflux from cancer cells. In addition, EGCG could also inhibit the growth of cancer cells [59]. Attributed to these mechanism, CLM-DOX was more effective on MCF-7 and MCF-7/ADR compared with NP-DOX.
Biodistribution of micelles
The biodistribution of micelles are performed at the time points of 1 h, 6 h and 24 h as shown in Figure 11. NP-DOX and CLM-DOX presented strong fluorescence signals in the liver and kidney regions, which was consistent with Park's report that the majority of administered nanoparticles were accumulated in RES organs [60]. At the time point of 1 h and 6 h, Both NP-DOX and CLM-DOX accumulated in the heart at a low amount, which indicated that nanoparticle based therapy could effectively reduce the amount of DOX in the heart. However, it is well known that nanoparticle loaded with DOX is a kind of long-term therapy, which could gradually increase the amount of DOX in the heart. But for CLM-DOX co-loaded with EGCG, it could timely and quickly scavenge oxygen free radicals generated by DOX in the heart, avoiding the cardiomyocytes damage. In addition, a large amount of DOX accumulated in other tissues. Similar with the mechanism of reduced cardiotoxicity, CLM-DOX could also reduce these organs damage.
ex vivo fluorescence (from DOX) imaging of the heart and normal tissues harvested from BALB/c mice at 1 h, 6 h and 24 h post-injection. The numeric label for each organ is as follows: 1, heart; 2, liver; 3, spleen; 4, lung; 5, kidney.

Conclusions
In summary, we have successfully developed a novel green tea catechin-based PIC micelle, CLM combined with DOX, which could effectively overcome DOX induced cardiotoxicity and multidrug resistance. Based on the oxygen free radicals scavenging activity and drug resistance reversing of EGCG, combination therapy with EGCG and DOX in the micelles exhibited both reduced cardiotoxicity attributed to inhibition of ROS generation in cardiomyocytes and reversal of multidrug resistance induced by modulating the activity of P-gp in the cancer cells. This micelle could effectively address the limitations of chemotherapy including DOX induced cardiotoxicity and multidrug resistance.
Supplementary Material
Supplementary materials and methods, Supplementary figures.
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (Nos. 51390483, 91527306, 21274001, 81471727, and 51203189), the Natural Science Foundation of Tianjin, China (No. 15JCYBJC29700) and the PCSIRT (IRT1257).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Tewey K, Rowe T, Yang L, Halligan B, Liu L. Adriamycin-induced DNA damage mediated by mammalian DNA topoisomerase II. Science. 1984;226:466-8
2. Hortobágyi GN. Anthracyclines in the Treatment of Cancer. Drugs. 2012;54:1-7
3. Chatterjee K, Zhang J, Honbo N, Karliner JS. Doxorubicin Cardiomyopathy. Cardiology. 2010;115:155-62
4. Mason J. Anthracycline cardiotoxicity: recognition, management, and avoidance. Compr Ther. 1979;5:64
5. Primeau AJ, Rendon A, Hedley D, Lilge L, Tannock IF. The Distribution of the Anticancer Drug Doxorubicin in Relation to Blood Vessels in Solid Tumors. Clin Cancer Res. 2005;11:8782-8
6. Harker WG, Sikic BI. Multidrug (Pleiotropic) Resistance in Doxorubicin-selected Variants of the Human Sarcoma Cell Line MES-SA. Cancer Res. 1985;45:4091-6
7. Tacar O, Sriamornsak P, Dass CR. Doxorubicin: an update on anticancer molecular action, toxicity and novel drug delivery systems. J Pharm Pharmacol. 2013;65:157-70
8. Kataoka K, Harada A, Nagasaki Y. Block copolymer micelles for drug delivery: design, characterization and biological significance. Adv Drug Del Rev. 2001;47:113-31
9. Tyrrell ZL, Shen Y, Radosz M. Fabrication of micellar nanoparticles for drug delivery through the self-assembly of block copolymers. Prog Polym Sci. 2010;35:1128-43
10. Du J-Z, Du X-J, Mao C-Q, Wang J. Tailor-Made Dual pH-Sensitive Polymer-Doxorubicin Nanoparticles for Efficient Anticancer Drug Delivery. J Am Chem Soc. 2011;133:17560-3
11. Cheng T, Ma R, Zhang Y, Ding Y, Liu J, Ou H. et al. A surface-adaptive nanocarrier to prolong circulation time and enhance cellular uptake. Chem Commun. 2015;51:14985-8
12. Guan X, Li Y, Jiao Z, Chen J, Guo Z, Tian H. et al. A pH-sensitive charge-conversion system for doxorubicin delivery. Acta Biomater. 2013;9:7672-8
13. Brannon-Peppas L, Blanchette JO. Nanoparticle and targeted systems for cancer therapy. Adv Drug Del Rev. 2012;64(Supplement):206-12
14. Gao H, Cheng T, Liu J, Liu J, Yang C, Chu L. et al. Self-Regulated Multifunctional Collaboration of Targeted Nanocarriers for Enhanced Tumor Therapy. Biomacromolecules. 2014;15:3634-42
15. Yang L, Sajja HK, Cao Z, Qian W, Bender L, Marcus AI. et al. uPAR-targeted Optical Imaging Contrasts as Theranostic Agents for Tumor Margin Detection. Theranostics. 2014;4:106-18
16. Zhong Y, Meng F, Deng C, Zhong Z. Ligand-Directed Active Tumor-Targeting Polymeric Nanoparticles for Cancer Chemotherapy. Biomacromolecules. 2014;15:1955-69
17. Hu YP, Henry-Toulmé N, Robert J. Failure of liposomal encapsulation of doxorubicin to circumvent multidrug resistance in an in vitro model of rat glioblastoma cells. Eur J Cancer. 1995;31:389-94
18. Rivera E, Valero V, Esteva FJ, Syrewicz L, Cristofanilli M, Rahman Z. et al. Lack of activity of stealth liposomal doxorubicin in the treatment of patients with anthracycline-resistant breast cancer. Cancer Chemother Pharmacol. 2002;49:299-302
19. Pereverzeva E, Treschalin I, Bodyagin D, Maksimenko O, Kreuter J, Gelperina S. Intravenous tolerance of a nanoparticle-based formulation of doxorubicin in healthy rats. Toxicol Lett. 2008;178:9-19
20. Yen G-C, Chen H-Y. Antioxidant Activity of Various Tea Extracts in Relation to Their Antimutagenicity. J Agric Food Chem. 1995;43:27-32
21. Yang CS, Wang X, Lu G, Picinich SC. Cancer prevention by tea: animal studies, molecular mechanisms and human relevance. Nat Rev Cancer. 2009;9:429-39
22. Kim Y-J, Chung JE, Kurisawa M, Uyama H, Kobayashi S. Superoxide Anion Scavenging and Xanthine Oxidase Inhibition of (+)-Catechin-Aldehyde Polycondensates. Amplification of the Antioxidant Property of (+)-Catechin by Polycondensation with Aldehydes. Biomacromolecules. 2004;5:547-52
23. Olson RD, Mushlin PS. Doxorubicin cardiotoxicity: analysis of prevailing hypotheses. The FASEB Journal. 1990;4:3076-86
24. Fu Z, Guo J, Jing L, Li R, Zhang T, Peng S. Enhanced toxicity and ROS generation by doxorubicin in primary cultures of cardiomyocytes from neonatal metallothionein-I/II null mice. Toxicol In Vitro. 2010;24:1584-91
25. Brookins Danz ED, Skramsted J, Henry N, Bennett JA, Keller RS. Resveratrol prevents doxorubicin cardiotoxicity through mitochondrial stabilization and the Sirt1 pathway. Free Radic Biol Med. 2009;46:1589-97
26. Li W, Nie S, Xie M, Chen Y, Li C, Zhang H. A Major Green Tea Component, (-)-Epigallocatechin-3-gallate, Ameliorates Doxorubicin-Mediated Cardiotoxicity in Cardiomyocytes of Neonatal Rats. J Agric Food Chem. 2010;58:8977-82
27. Kozluca O, Olcay E, Sürücü S, Güran Z, Kulaksiz T, Üskent N. Prevention of doxorubicin induced cardiotoxicity by catechin. Cancer Lett. 1996;99:1-6
28. Saeed NM, El-Naga RN, El-Bakly WM, Abdel-Rahman HM, Salah ElDin RA, El-Demerdash E. Epigallocatechin-3-gallate pretreatment attenuates doxorubicin-induced cardiotoxicity in rats: A mechanistic study. Biochem Pharmacol. 2015;95:145-55
29. Stein WD, Bates SE, Fojo T. Intractable Cancers: The Many Faces of Multidrug Resistance and the Many Targets it Presents for Therapeutic Attack. Curr Drug Targets. 2004;5:333-46
30. Navarro G, Sawant RR, Biswas S, Essex S, Tros de Ilarduya C, Torchilin VP. P-glycoprotein silencing with siRNA delivered by DOPE-modified PEI overcomes doxorubicin resistance in breast cancer cells. Nanomedicine. 2011;7:65-78
31. Gottesman MM. Mechanisms of Cancer Drug Resistance. Annu Rev Med. 2002;53:615-27
32. Creixell M, Peppas NA. Co-delivery of siRNA and therapeutic agents using nanocarriers to overcome cancer resistance. Nano Today. 2012;7:367-79
33. Lukianova-Hleb EY, Ren X, Zasadzinski JA, Wu X, Lapotko DO. Plasmonic Nanobubbles Enhance Efficacy and Selectivity of Chemotherapy Against Drug-Resistant Cancer Cells. Adv Mater. 2012;24:3831-7
34. Wang J, Sun J, Chen Q, Gao Y, Li L, Li H. et al. Star-shape copolymer of lysine-linked di-tocopherol polyethylene glycol 2000 succinate for doxorubicin delivery with reversal of multidrug resistance. Biomaterials. 2012;33:6877-88
35. Krishna R, Mayer LD. Multidrug resistance (MDR) in cancer: Mechanisms, reversal using modulators of MDR and the role of MDR modulators in influencing the pharmacokinetics of anticancer drugs. Eur J Pharm Sci. 2000;11:265-83
36. Ullah MF. Cancer multidrug resistance (MDR): a major impediment to effective chemotherapy. Asian Pac J Cancer Prev. 2008;9:1-6
37. Ford JM, Hait WN. Pharmacology of drugs that alter multidrug resistance in cancer. Pharmacol Rev. 1990;42:155-99
38. Lampidis TJ, Krishan A, Planas L, Tapiero H. Reversal of Intrinsic Resistance to Adriamycin in Normal Cells by Verapamil. Cancer Drug Deliv. 1986;3:251-9
39. Höll V, Kouba M, Dietel M, Vogt G. Stereoisomers of calcium antagonists which differ markedly in their potencies as calcium blockers are equally effective in modulating drug transport by P-glycoprotein. Biochem Pharmacol. 1992;43:2601-8
40. Pennock GD, Dalton WS, Roeske WR, Appleton CP, Mosley K, Plezia P. et al. Systemic Toxic Effects Associated With High-Dose Verapamil Infusion and Chemotherapy Administration. J Natl Cancer Inst. 1991;83:105-10
41. Wu J, Lu Y, Lee A, Pan X, Yang X, Zhao X. et al. Reversal of multidrug resistance by transferrin-conjugated liposomes co-encapsulating doxorubicin and verapamil. J Pharm Pharm Sci. 2007;10:350-7
42. Jodoin J, Demeule M, Béliveau R. Inhibition of the multidrug resistance P-glycoprotein activity by green tea polyphenols. Biochim Biophys Acta. 2002;1542:149-59
43. Zhang Q, Wei D, Liu J. In vivo reversal of doxorubicin resistance by (-)-epigallocatechin gallate in a solid human carcinoma xenograft. Cancer Lett. 2004;208:179-86
44. Liang G, Tang A, Lin X, Li L, Zhang S, Huang Z. et al. Green tea catechins augment the antitumor activity of doxorubicin in an in vivo mouse model for chemoresistant liver cancer. Int J Oncol. 2010;37:111-23
45. Gerber H-P, Ferrara N. Pharmacology and Pharmacodynamics of Bevacizumab as Monotherapy or in Combination with Cytotoxic Therapy in Preclinical Studies. Cancer Res. 2005;65:671-80
46. Hu C-MJ, Aryal S, Zhang L. Nanoparticle-assisted combination therapies for effective cancer treatment. Ther Deliv. 2010;1:323-34
47. Song W, Tang Z, Li M, Lv S, Sun H, Deng M. et al. Polypeptide-based combination of paclitaxel and cisplatin for enhanced chemotherapy efficacy and reduced side-effects. Acta Biomater. 2014;10:1392-402
48. Jiang T, Mo R, Bellotti A, Zhou J, Gu Z. Gel-Liposome-Mediated Co-Delivery of Anticancer Membrane-Associated Proteins and Small-Molecule Drugs for Enhanced Therapeutic Efficacy. Adv Funct Mater. 2014;24:2295-304
49. Ren J, Zhang Y, Zhang J, Gao H, Liu G, Ma R. et al. pH/Sugar Dual Responsive Core-Cross-Linked PIC Micelles for Enhanced Intracellular Protein Delivery. Biomacromolecules. 2013;14:3434-43
50. Chung JE, Tan S, Gao SJ, Yongvongsoontorn N, Kim SH, Lee JH. et al. Self-assembled micellar nanocomplexes comprising green tea catechin derivatives and protein drugs for cancer therapy. Nat Nano. 2014;9:907-12
51. Springsteen G, Wang B. A detailed examination of boronic acid-diol complexation. Tetrahedron. 2002;58:5291-300
52. Daly WH, Poché D. The preparation of N-carboxyanhydrides of α-amino acids using bis(trichloromethyl)carbonate. Tetrahedron Lett. 1988;29:5859-62
53. Naito M, Ishii T, Matsumoto A, Miyata K, Miyahara Y, Kataoka K. A Phenylboronate-Functionalized Polyion Complex Micelle for ATP-Triggered Release of siRNA. Angew Chem Int Ed. 2012;51:10751-5
54. Fernandes AS, Gaspar J, Cabral MF, Caneiras C, Guedes R, Rueff J. et al. Macrocyclic copper(II) complexes: Superoxide scavenging activity, structural studies and cytotoxicity evaluation. J Inorg Biochem. 2007;101:849-58
55. Jankun J, Selman SH, Swiercz R, Skrzypczak-Jankun E. Why drinking green tea could prevent cancer. Nature. 1997;387:561 -
56. Sang S, Lee M-J, Hou Z, Ho C-T, Yang CS. Stability of Tea Polyphenol (-)-Epigallocatechin-3-gallate and Formation of Dimers and Epimers under Common Experimental Conditions. J Agric Food Chem. 2005;53:9478-84
57. Matsumoto S, Christie RJ, Nishiyama N, Miyata K, Ishii A, Oba M. et al. Environment-Responsive Block Copolymer Micelles with a Disulfide Cross-Linked Core for Enhanced siRNA Delivery. Biomacromolecules. 2009;10:119-27
58. Wei H, Zhuo R-X, Zhang X-Z. Design and development of polymeric micelles with cleavable links for intracellular drug delivery. Prog Polym Sci. 2013;38:503-35
59. Zhu A, Wang X, Guo Z. Study of tea polyphenol as a reversal agent for carcinoma cell lines' multidrug resistance (study of TP as a MDR reversal agent). Nucl Med Biol. 2001;28:735-40
60. Bae YH, Park K. Targeted drug delivery to tumors: Myths, reality and possibility. J Control Release. 2011;153:198-205
Author contact
![]() Corresponding authors: Linqi Shi, State Key Laboratory of Medicinal Chemical Biology, Key Laboratory of Functional Polymer Materials of Ministry of Education, Institute of Polymer Chemistry, Collaborative Innovation Center of Chemical Science and Engineering (Tianjin), Nankai University, Tianjin 300071, P.R. China, E-mail: shilinqiedu.cn. Fax: +86 22 23503510. Or Jianfeng Liu, Tianjin Key Laboratory of Radiation Medicine and Molecular Nuclear Medicine, Institute of Radiation Medicine, Chinese Academy of Medical Science & Peking Union Medical College, Tianjin, 300192, P.R. China, E-mail address: lewis78com.
Corresponding authors: Linqi Shi, State Key Laboratory of Medicinal Chemical Biology, Key Laboratory of Functional Polymer Materials of Ministry of Education, Institute of Polymer Chemistry, Collaborative Innovation Center of Chemical Science and Engineering (Tianjin), Nankai University, Tianjin 300071, P.R. China, E-mail: shilinqiedu.cn. Fax: +86 22 23503510. Or Jianfeng Liu, Tianjin Key Laboratory of Radiation Medicine and Molecular Nuclear Medicine, Institute of Radiation Medicine, Chinese Academy of Medical Science & Peking Union Medical College, Tianjin, 300192, P.R. China, E-mail address: lewis78com.