Impact Factor
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Results and Discussion
Materials and Methods
Conclusions
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2016; 6(9):1293-1305. doi:10.7150/thno.15081 This issue Cite
Research Paper
Supramolecular PEGylated Dendritic Systems as pH/Redox Dual-Responsive Theranostic Nanoplatforms for Platinum Drug Delivery and NIR Imaging
Yunkun Li#, Yachao Li#, Xiao Zhang, Xianghui Xu ![]() , Zhijun Zhang, Cheng Hu, Yiyan He, Zhongwei Gu
, Zhijun Zhang, Cheng Hu, Yiyan He, Zhongwei Gu ![]()
National Engineering Research Center for Biomaterials, Sichuan University, Chengdu 610064, Sichuan, China.
#These authors contributed equally in this work.
Received 2016-1-25; Accepted 2016-4-27; Published 2016-6-5
Abstract

Recently, self-assembling small dendrimers into supramolecular dendritic systems offers an alternative strategy to develop multifunctional nanoplatforms for biomedical applications. We herein report a dual-responsive supramolecular PEGylated dendritic system for efficient platinum-based drug delivery and near-infrared (NIR) tracking. With a refined molecular/supramolecular engineering, supramolecular dendritic systems were stabilized by bioreducible disulfide bonds and endowed with NIR fluorescence probes, and PEGylated platinum derivatives coordinated onto the abundant peripheral groups of supramolecular dendritic templates to generate pH/redox dual-responsive theranostic supramolecular PEGylated dendritic systems (TSPDSs). TSPDSs markedly improved the pharmacokinetics and biodistribution of platinum-based drugs, owing to their stable nanostructures and PEGylated shells during the blood circulation. Tumor intracellular environment (low pH value and high glutathione concentration) could trigger the rapid disintegration of TSPDSs due to acid-labile coordination bonds and redox-cleavable disulfide linkages, and then platinum-based drugs were delivered into the nuclei to exert antitumor activity. In vivo antitumor treatments indicated TSPDSs not only provided high antitumor efficiency which was comparable to clinical cisplatin, but also reduced renal toxicity of platinum-based drugs. Moreover, NIR fluorescence of TSPDSs successfully visualized in vitro and in vivo fate of nanoplatforms and disclosed the intracellular platinum delivery and pharmacokinetics. These results confirm tailor-made supramolecular dendritic system with sophisticated nanostructure and excellent performance is a promising candidate as smart theranostic nanoplatforms.
Keywords: supramolecular dendritic systems, theranostic nanoplatform, pH and redox sensitivity, platinum delivery, near-infrared imaging
Introduction
In recent years, more and more attentions in nanomedicine domain have focused on the development of theranostic nanoplatforms which are perfect combinations of diagnostic and therapeutic agents in a single formulation, since the success of theranostic protocols largely depends on advanced nanomaterials [1-3]. Numerous animal trials and clinical treatments have proven that nanoparticulate therapeutics (e.g., Doxil® and Abraxane®) indeed increase drug efficacy and decrease systemic toxicity [4, 5]. Meanwhile, modern nanocarriers are urgently endowed with diagnostic potentials to monitor real-time pharmacokinetics, detect drug localization and personalize intervention program [1, 2]. To this end, broad-spectrum anticancer drugs (e.g., doxorubicin, paclitaxel and cisplatin) are very hopeful to acquire more efficient treatment through well-designed theranostic nanoplatforms [6, 7]. Noticeably, platinum-based anticancer drugs play a pivotal role in chemotherapy of several solid tumors including testicular, ovarian and lung cancers, but platinum-based therapeutics are often accompanied by severe systemic toxicity (e.g., nephrotoxicity and neurotoxicity) due to poor tumor specificity [8, 9]. Although a variety of nanovehicles have been engineered for efficient treatment and minimal untoward effects, there are as yet only a few theranostic nanoplatforms explored for platinum-based drug delivery [10-12]. As a result, the development of appropriate theranostic nanoplatforms is of great value for promoting therapeutic index of platinum-based drugs and monitoring treatment process.
Owing to the available biocompatibility, biodegradability and multiple functionality of natural and synthetic macromolecules, polymeric nanomaterials become the primary choice for engineering theranostic nanoplatforms [13-15]. Dendrimers, as the latest generation of artificial macromolecules, are inherent nanoplatforms for diagnostic and therapeutic agent delivery, due to their precise molecular structures, highly-branched architectures and monodispersity [16-19]. For these merits, some dendrimer-based nanocarriers have been designed for improving platinum-based drug metabolism [20-22], as well as diagnostic accuracy [19, 23]. However, traditional dendrimer-based nanocarriers (e.g., poly(amidoamine) dendrimers) have exposed some shortcomings in clinical trials, including disappeared advantages (e.g., precise structure and good water-solubility) after conjugation and encapsulation of theranostic agents, significant toxicity, complicated manufacture and low input-output ratio [24, 25]. To address these problems, self-assembling small dendrimers into supramolecular dendritic systems (SDSs) emerges as a facile approach to develop a new generation of biocompatible and efficient nanoplatforms for nanomedicine [26, 27]. Recently, we also have demonstrated a series of strategies to fabricate multifunctional supramolecular dendritic systems for improving drug and gene delivery [28-34]. Consequently, supramolecular dendritic system is an excellent candidate for developing desired theranostic nanoplatforms.
To perfectly integrate diagnostics and therapeutics, more considerations including robust nanostructures, long-circulation time, high tumor accumulation, site-specific delivery and efficient biological imaging, should take into account for engineering supramolecular dendritic platforms. First of all, poor drug bioavailability which is due to nanostructural instability, premature release and elimination by the reticuloendothelial system (RES) during blood circulation, is a major limitation of current nanocarriers in clinical uses [1, 35]. To elevate drug availability and weaken side effects, dynamic covalent bonds could be utilized to fix the supramolecular dendritic systems, which are expected to have more stable nanostructures than our previous completely non-covalent SDSs [36, 37]; and PEGylation of SDSs could avoid RES capture and alter drug plasma concentration profile [38]. Second, stimuli-responsive engineering of dendritic systems can facilitate site-specific controlled release in the targeted tumor tissues and cells for enhancing anticancer efficacy [39, 40]. In the meantime, stimuli-triggered rapid drug release at the target site is promising to reduce drug resistance in cancer cells [41]. Last but not least, diagnostic and therapeutic agents could be introduced into one nanoplatform by a facile and reliable fabrication, which is very essential for future application and clinical translation [24, 42].
To confirm our concept, a novel type of theranostic supramolecular dendritic systems is customized to satisfy tumor therapy and biological tracking with the following features (Figure 1): (i) lipoic acid (LA)-functionalized amphiphilic dendrons and PEG derivatives are designed as biocompatible and readily available building blocks for hierarchical supramolecular fabrication [28, 30, 34]; (ii) tailor-made near-infrared (NIR) fluorescent probe as initiator precursor can be introduced into supramolecular dendritic systems after polymerization, rendering sensitive and specific imaging in deep tissue [43, 44]; (iii) supramolecular dendritic systems are able to be stabilized by disulfide polymerization with abundant terminal groups and firm architectures [36, 37]; (iv) PEGylated platinum derivatives will coordinate with peripheral carboxyl of dendritic systems for antitumor therapy, (v) the immediate formation of PEG shells can prolong drug circulation time in blood and optimize in vivo drug distribution [38]; (vi) redox-sensitive disulfide linkages and acid-cleavable coordination can synergistically trigger the site-specific release of platinum drug to carry on the anti-tumor treatment [39, 40].
Schematic illustrations for molecular and supramolecular engineering of theranostic supramolecular PEGylated dendritic systems, including self-assembly of amphiphilic dendrons, introduction of NIR probe, core crosslinking of dendritic systems, platinum-drug coordination and supramolecular PEGylation.
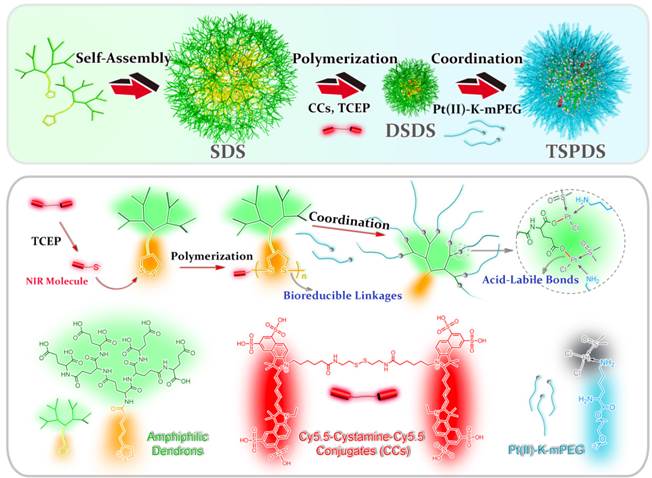
Results and Discussion
LA-functionalized dendrons were first synthesized through a divergent approach, and the matrix-assisted laser desorption/ionization time-of-flight mass spectrometry result confirmed the accurate molecular weight of peptide-based dendrons ([M+H]+=1110.07) which well agreed with theoretical value of 1110.34 (Figure S9, for details on the syntheses and characterizations in Supporting Information). In an aqueous solution, LA moieties would aggregate as inner cores of SDSs, and hydrophilic dendritic fragments self-assembled as outer spheres. A critical aggregation concentration (CAC) of 43.6 μg mL-1 was determined using pyrene as a polar fluorescence probe (Figure S14). As shown in Figure 2A, the transmission electron microscopy (TEM) image represented that the amphiphilic dendrons self-assembled into spherical nanoparticles with an average size of 160.9 ± 2.4 nm by dynamic light scattering (DLS) measurement. The loose nanostructures of SDSs should be attributed to relatively weak hydrophobic forces from LA moieties, and it is predicted that disulfide polymerization of LA moieties could make supramolecular dendritic systems more compact and stable [45, 46]. Thereby, we customized the novel NIR molecules (Cy5.5-cystamine-Cy5.5 conjugates, CCs) as initiator precursors, which can be in situ reduced into the active Cy5.5-SH for initiating polymerization of LA-functionalized dendrons (Figure 1) [45, 47]. UV-Vis spectra showed that SDSs had a characteristic absorption peak of lipoyl rings at 330 nm, while the polymerization of peptide dendrons induced the disappearance of absorbance at 330 nm and the appearance of Cy5.5 absorbance at 679 nm (Figure S15), suggesting the ring-opening polymerization was successful [48]. On the other hand, the enhanced viscosity also indicated successful polymerization with increasing molecular weight (Figure S16). After core crosslinked polymerization by disulfide linkages, the supramolecular dendritic systems were bundled into the stable nanostructures having abundant peripheral residues of glutamic acid (-COOH) and NIR initiator-contained cores. As speculated, these disulfide-stabilized supramolecular dendritic systems (DSDSs) exhibited more tight nanostructures and a smaller average size (67.6 ± 5.7 nm) as compared with un-crosslinked supramolecular dendritic systems (Figure 2B).
Hierarchical nanostructures of supramolecular dendritic systems. DLS results (in aqueous solutions) and TEM images for (A) SDSs, (B) DSDSs and (C) TSPDSs.
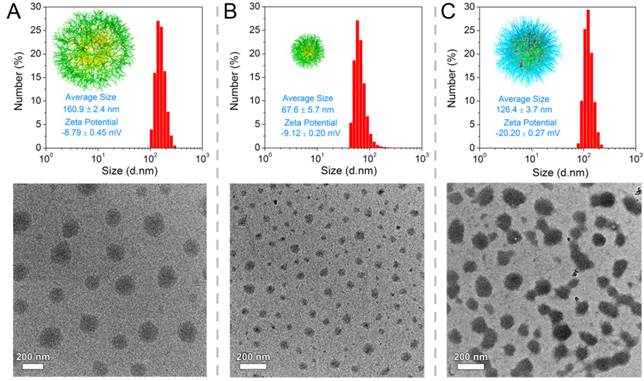
Next, methoxypolyethylene glycol (mPEG) was modified with lysine (K) to obtain platinum ligands (mPEG-K). With a facile procedure, Pt(DMSO)2Cl2 would coordinate with amino groups of mPEG-K to generate PEGylated platinum derivatives (Pt(II)-K-mPEG) in methanol. The detailed syntheses and characterizations of above-mentioned compounds can be found in the Supporting Information. After removal of chloride ion in Pt(II)-K-mPEG, PEGylated platinum precursors would coordinate with carboxylate ions on DSDSs to form theranostic supramolecular PEGylated dendritic systems (TSPDSs), resulting in moderate growth in nanoparticle size (Figure 2C). And the zeta potential of TSPDSs was about -20.20 mV, allowing for protein adsorption resistance [49].
To illustrate the key features of TSPDSs for tumor theranostics, we characterized them via energy dispersive spectroscopy (EDS) analysis, which indicated that the PEGylated platinum derivatives were successfully incorporated into TSPDSs after hierarchical supramolecular self-assembly (Figure 3A). Moreover, X-ray photoelectron spectroscopy (XPS) result at Pt4f region showed two distinct peaks at 72.3 eV (Pt4f7/2) and 75.8 eV (Pt4f5/2) for the binding energy of platinum(II) in the coordination sites [Pt(II)-O-C(O)], which implied platinum-based drug maintained antitumor potentials after supramolecular fabrication (Figure 3B) [50].
Physicochemical properties of the theranostic supramolecular PEGylated dendritic systems. (A) EDS spectrum and (B) XPS spectrum of TSPDSs. (C) TGA curves of TSPDSs and organic components (dendrimers and mPEG-K complex) under flowing air with a ramp rate of 10 ºC/min. (D) Fluorescent excitation spectra (Ex, hollow dot) and emission spectra (Em, solid dot) of SDSs, DSDSs and TSPDSs in aqueous solutions.
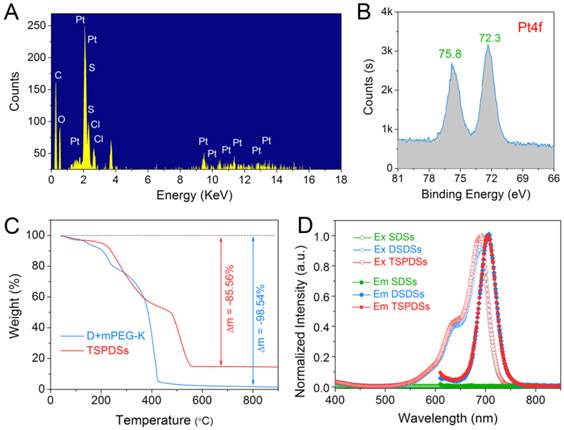
The thermo gravimetric analysis (TGA) manifested 85.56% weight loss for the organic substances in TSPDSs (Figure 3C). As compared with 98.54% weight loss of organic complex (dendrimers and mPEG-K), the platinum loading capacity in TSPDSs was calculated to be 12.98%, which was consistent with inductively coupled plasma-mass spectrometry (ICP-MS) result of 12.09%. At the same time, near-infrared characters of TSPDSs was confirmed by fluorescence spectra, with obvious excitation at 691 nm and emission at 705 nm (Figure 3D).
To verify the dual-responsive property, we displayed the size variations of TSPDSs under different biomimetic conditions. In the phosphate buffered solution (PBS, pH 7.4) similar to normal physiological condition, TSPDSs can keep stable size around 120 nm for 12.0 h (Figure 4A). The nice dimensional stability of TSPDSs should be ascribed to disulfide-stabilized dendritic templates and strong coordination interactions to firm multicomponent assemblies. Nevertheless, PBS (pH 5.0) simulated intracellular pH condition can induce the decrease of TSPDSs size from 120 nm to 60 nm (Figure 4B), which implied acidic condition triggered the shedding of Pt(II)-K-mPEG from the disulfide-stabilized supramolecular dendritic systems. Mimicking tumor intracellular reductive environment by 10 mM DTT, the size of TSPDSs markedly enlarged due to the destruction of disulfide immobilization (Figure 4C). In addition, TSPDSs showed favorable stability at pH 6.8 and 10 μM DTT, which indicated TSPDSs could bear tumor microenvironment to keep their performances (Figure S18).
Stimuli-sensitivity, in vitro drug release and cytotoxicity of TSPDSs. Size distributions of TSPDSs at (A) pH 7.4, (B) pH 5.0 and (C) 10 mM DTT with different incubation time (0 h, 0.5 h, 1.0 h, 6.0 h and 12.0 h). (D) Platinum release profiles of TSPDSs at the special conditions mimicking normal physiological environment (pH 7.4, no DTT), tumor microenvironment (pH 6.8, 10 μM DTT) and tumor intracellular environment (pH 5.0, 10 mM DTT) for 48 h at 37 °C, respectively (means ± standard deviation (SD), n = 3). The platinum amount was determined by ICP-MS. (E) The cell viability of A549 tumor cells after incubation with dendrimers, mPEG-K, D+mPEG-K complex and TSPDSs for 48 h at various platinum concentrations (means ± SD, n = 6; *p < 0.05, **p < 0.01, ***p < 0.001, TSPDSs compared with D+mPEG-K).
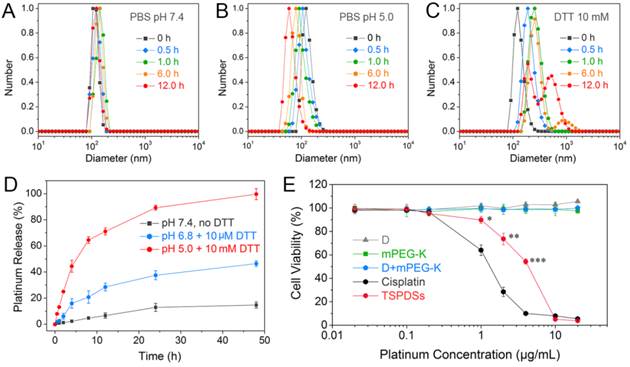
Then we determined the in vitro release profiles of platinum drug from TSPDSs under various biomimetic conditions by ICP-MS analysis. As shown in Figure 4D, a very slow platinum release was observed in normal physiological environment, and the cumulative release of platinum just reached about 10 % until 48 h. At pH 6.8 with 10 μM DTT mimicking the tumor microenvironment, the platinum release rate from TSPDSs was a little faster than pH 7.4 due to acid-activated platinum release. In the line with expectations, pH 5.0 with 10 mM DTT, analogous to the tumor intracellular condition, triggered the burst release of platinum; and the amount of platinum release was more than 60% within 12 h. Comparison of platinum release at pH 5.0 with DTT or not (Figure S19 in Supporting Information) inferred that (a) the acid-activated platinum release was a main factor for drug delivery, because Pt(II)-O-C(O) coordinate bonds were more cleavable under an acid condition [51]; (b) the redox-induced disintegration of TSPDSs could accelerate platinum release to a certain extent.
Once the physicochemical requirements of TSPDSs were confirmed, we turned to evaluate their in vitro antitumor effects against A549 tumor cell line by CCK-8 assay (Figure 4E). First, organic components including dendrimers, mPEG-K and their complex (with the same amount as platinum-loaded TSPDSs) were found no obvious toxicity to A549 tumor cell line. When the platinum(II) concentration in TSPDSs reached to 1.0 μg mL-1, TSPDSs revealed antitumor activity to A549 tumor cells; and 4.0 μg mL-1 platinum(II) inhibited the cell viability of A549 tumor cells around 50%. As a consequence, above-mentioned features of TSPDSs on dimensional stability, stimuli-responsive platinum release and considerable antitumor activity were beneficial to in vivo drug transportation, tumor accumulation and site-specific drug delivery.
Based on the inherent NIR fluorescence of theranostic nanoplatform, visualizations for intracellular fate of TSPDSs were carried out with fluorescent subcellular location. Confocal laser scanning microscopy (CLSM) images showed that TSPDSs were fast internalized into A549 tumor cells within 1 h (Figure 5A, TSPDSs with red signals and DiO-labeled cell membrane with green signals). After exposure to TSPDSs for 3 h, the CLSM images and fluorescence-activated cell sorting (FACS) analysis consistently indicated stronger NIR signals can be detected in more A549 tumor cells. To clarify the endocytosis of supramolecular dendritic systems (TSPDSs), we investigated the effects of various endocytosis inhibitors on the internalization of TSPDSs. Quantitative results on the internalization of TSPDSs showed that low temperature of 4 ºC and energy suppression by NaN3 remarkably lowered cellular uptake of TSPDSs, implying the internalization of TSPDSs was through an active endocytic mechanism (Figure S22). In the meantime, distinguishing from other inhibitors (e.g., cytochalasin D, nystatin and M-β-CD), chlorpromazine resulted in the decrease of 16.4% internalization of TSPDSs indicating clathrin-mediated endocytosis (Table S1).
Biological tracking of intracellular platinum delivery and antitumor utility to A549 tumor cell line. (A) CLSM images for cellular uptake of TSPDSs including Cy5.5 channel for TSPDSs (red), DiO-stained cell membrane (green) and overlay; and FACS analysis for quantitative internalization of TSPDSs with different incubating time points. (B) CLSM images of A549 tumor cells after incubation with TSPDSs for 3 h and 6 h, and GSH-OEt pretreated A549 tumor cells after incubation with TSPDSs for 3 h, including Cy5.5 channel for TSPDSs (red), LysoTracker-stained lysosome (blue) and overlay. (C) Quantitative internalization and intracellular distribution of platinum in A549 tumor cells after culture in the cisplatin solution and TSPDSs solution with the same platinum concentration of 1.5 μg mL-1 for 24 h. (D) Cell cycle distribution of A549 tumor cells after incubation with cisplatin and TSPDSs (1.5 μg mL-1 platinum) for 24 h, respectively (means ± SD, n = 3).
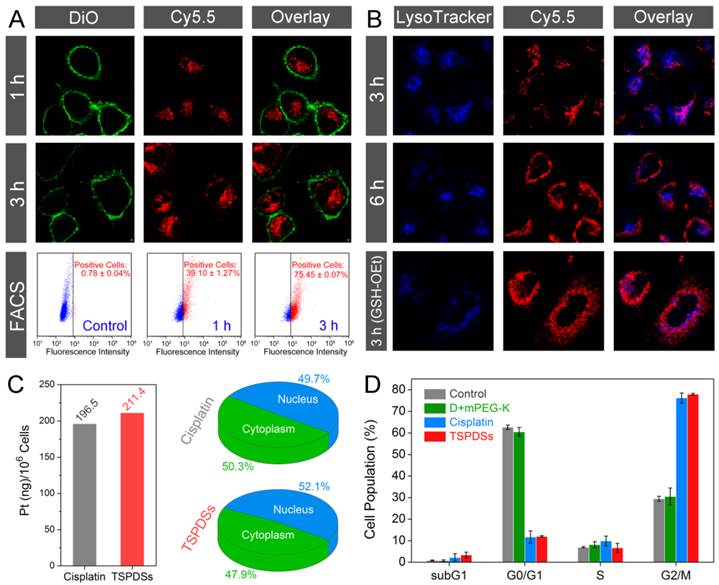
In the CLSM images, most of the red fluorescence (TSPDSs) overlapped with the acidic organelles (LysoTracker-stained lysosome with blue signals) at 3 h; with incubation time last for 6 h, a part of TSPDSs could escape from lysosome (Figure 5B). However, in the GSH-OEt pretreated A549 tumor cells, wide distribution of red signals in cytoplasm indicated endosomal escape of TSPDSs could be promoted by the redox-responsive degradation of disulfide-stabilized cores.
To further explore the antitumor mechanism of TSPDSs, we carefully quantified the intracellular distribution of platinum and their influences on cell cycles. A clinical formulation of cisplatin for cancer patients was selected as a positive control group. Figure 5C illustrated that the internalization of platinum(II) from TSPDSs was slightly more than the clinical cisplatin. Furthermore, 52.1% platinum(II) from TSPDSs was transported into nucleus to exert antitumor activity. Interestingly, little red fluorescence could be observed in the nucleus with a long incubation time of 24 h (Figure S23), suggesting that intracellular lower pH value and higher GSH concentration could trigger the disassembly of TSPDSs in cytoplasm and effectively deliver platinum(II) into nucleus. According to the previous researches, cisplatin chemotherapy could largely trapped cell cycle of tumor cells in G2/M phase, and ultimately induced apoptotic cell death. As shown in Figure 5D, TSPDSs offered the same utility for cell cycle arrest as the clinical cisplatin. In general, this strategy for theranostic nanoplatform succeeded in generating high antitumor activity of platinum-based drug and tracing delivery pathway, owing to their stable nanostructures for tumor cellular uptake, dual stimuli-responsive feature for site-specific delivery, and NIR fluorescence for biological tracking.
One of the major purposes of this theranostic nanoplatform is to improve antitumor efficiency of platinum(II) and overcome adverse reactions. The in vivo antitumor effects of TSPDSs were evaluated in nude mice bearing A549 tumor xenografts using intravenous injection (3.25 mg platinum kg-1). After a treatment course of 21 d, the relative tumor volume in the saline-administered group prominently increased to 3.5-fold than that of the TSPDSs-administered group (**p < 0.01, Figure 6A). However, the organic components (dendrimers and mPEG-K complex) showed no obvious effect on tumor suppression. Encouragingly, in vivo tumor inhibition of TSPDSs was comparable to the positive control group of the clinical cisplatin for tumor patients, and tumor inhibitory rates of TSPDSs and clinical cisplatin were 71.7% and 68.1%, respectively.
In vivo therapeutic efficacy against nude mice bearing A549 xenograft. (A) Tumor volume changes of A549 solid tumor administered with saline, organic complex, cisplatin and TSPDSs by intravenous injection (3.25 mg platinum kg-1, means ± SD, n = 6; *p < 0.05, **p < 0.01, TSPDSs compared with saline treated group). (B) Histological and immunohistochemical analysis of H&E, CD31, Ki-67, and TUNEL assays for A549 tumor xenografts. CD31-positive vessels, Ki-67-positive cells and TUNEL-positive cells are stained brown. (C) Body weight changes of nude mice with different administrations over 21 d (n = 6, *p < 0.005, TSPDSs compared with saline treated group). (D) H&E and TUNEL assays for kidney tissue.
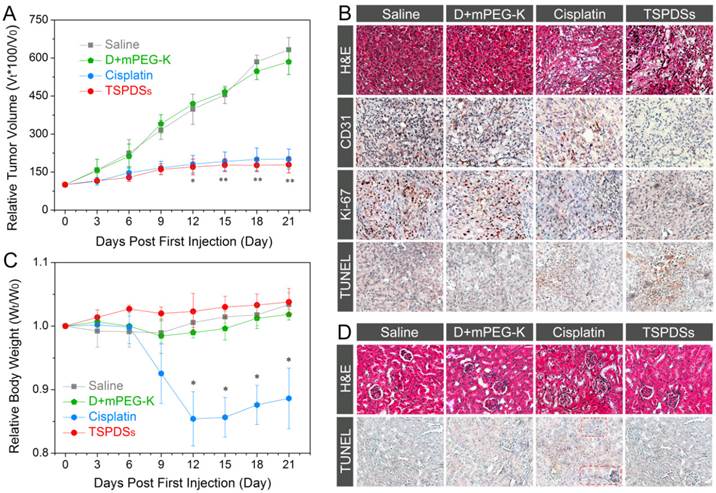
Afterwards, in vivo antitumor efficacy of TSPDSs was determined by histological and immunohistochemical studies (Figure 6B and Figure S24). Haematoxylin and eosin (H&E) staining of tumor slices displayed that systemic administration of TSPDSs made tumor tissue become loose and irregular, unlike superabundant and compact tumor cells in the saline-administered group. Meanwhile, CD31-positive vessels and proliferating Ki-67-positive tumor cells were effectively inhibited after TSPDSs treatment, whose inhibitory effect was even slightly better than clinical cisplatin. More importantly, the administration of TSPDSs caused the most apoptotic A549 tumor cells (TUNEL-positive cells) at tumor site, which was about 16-fold higher than that of the saline-administered group (Figure S25).
After nude mice finished the course of chemotherapy, an obvious reduction of their body weight (more than 10%) was observed in the cisplatin-treated group (Figure 6C). In contrast, a modest increase of body weight emerged in the TSPDSs-administered group. Furthermore, histological slices showed that the cellular morphologies at renal tissue were deformed after administration with the clinical cisplatin, and glomeruli became swollen and fuzzy (Figure 6D). Besides, more apoptotic cells (TUNEL-positive cells) were found in renal tissue. Additionally, no obvious histological lesion or abnormality was exhibited in other major organs, such as heart, liver, spleen and lung in TSPDSs-administered group compared with saline group (Figure S26). These results indicated TSPDSs did provide high therapeutic effects equal to the clinical medication of cisplatin, but also successfully resolved some drawbacks on adverse effects.
Subsequently, diagnostic potentials by the native NIR fluorescence were utilized to monitor in vivo real-time distribution of TSPDSs. After intravenous injection of the theranostic nanoplatforms, NIR signals dramatically appeared on tumor site, suggesting TSPDSs were mainly enriched in tumor tissue by passive targeting (Figure 7A). More importantly, along with the extending of time, more and more TSPDSs accumulated at solid tumor region to combat with tumor cells. The ex vivo NIR imaging for different organs and tumors also revealed that tumor tissue was a predominant site for TSPDSs accumulation (Figure 7B), and the diagnostic function should be attributed to enhanced permeability and retention (EPR) effects at tumor site.
(A) In vivo NIR imaging of nude mice bearing A549 tumor xenografts before and after intravenous injection of TSPDSs (3.25 mg platinum kg-1). (B) Ex vivo NIR imaging of solid tumors and organs (heart, liver, spleen, lung and kidney) after 24 h intravenous injection of saline (control) and TSPDSs. (C) Pharmacokinetic profiles of TSPDSs and cisplatin (3.25 mg platinum kg-1) after intravenous injection, and the amount of platinum in blood was determined by ICP-MS (means ± SD, n = 3). (D) Distribution of platinum in kidney and tumor after intravenous injection of TSPDSs and cisplatin for 2 h and 12 h, respectively (means ± SD, n = 3; *p < 0.05, **p < 0.01). (E) Blood urea nitrogen (BUN) and creatinine level with different administrations after a treatment course of 21 d (means ± SD, n = 3).
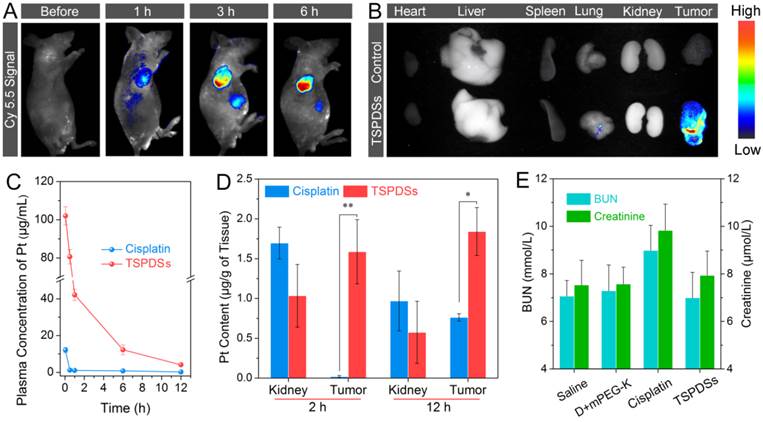
The advantages of this theranostic nanoplatform for systemic administration would be highlighted by pharmacokinetics and platinum distribution. After exposure to bovine serum albumin solution, little proteins adsorbed onto TSPDSs due to the good protein resistance of PEGylated shells (Figure S27), which was equivalent to the resistance ability of clinical cisplatin. The plasma platinum concentration versus time profiles after intravenous injection of TSPDSs and clinical cisplatin were monitored and recorded in Figure 7C. As expected, the maximum plasma concentration (Cmax) of platinum in the TSPDSs-administered group was 8.4-fold higher than that of clinical cisplatin; and compared with the clinical cisplatin having area under the curve (AUC, 12.10 mg/L·h), half-life (t1/2, 1.00 h) and blood clearance (CL, 0.80 L/h), TSPDSs showed many merits on pharmacokinetics including increased AUC of 262.26 mg/L·h (21.67-fold), enlarged t1/2 of 3.17 h (3.17-fold) and reduced CL of 0.04 L/h (20.00-fold) as shown in Table S2. For another, quantitative platinum distribution manifested 6.83% platinum accumulated in tumor tissue (1.84 μg platinum per gram of tumor tissue). TSPDSs realized relatively lower distribution at kidney and higher accumulation at tumor tissue as compared with clinical cisplatin (Figure 7D), confirming TSPDSs could minimize nephrotoxicity and enhance antitumor therapy. To further evaluate the nephrotoxicity, the serum levels of blood urea nitrogen (BUN) and creatinine were measured after treatment in nude mice (Figure 7E). It was found that administration of cisplatin increased BUN and creatinine in plasma because of nephrotoxicity, while TSPDSs had no influence on the biochemical criterions. The results of NIR imaging, pharmacokinetics and biological distribution were well agreed with in vivo tumor suppression and slice analysis.
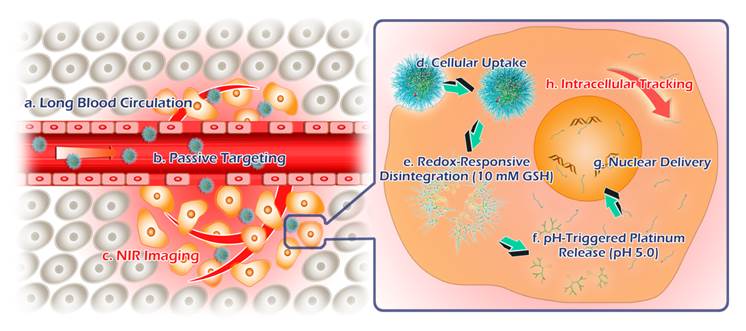
Taken together, this molecular and supramolecular strategy on TSPDSs fabrication successfully provide some benefits for tumor theranostics as follows (Figure 8): (a) disulfide-stabilized cores to maintain their stable nanostructures for blood circulation and passive targeting, achieve their intracellular disintegration for site-specific drug delivery, and provide hydrophobic pockets for hydrophobic antitumor drug encapsulation (Figure S28), (b) coordination interactions for platinum(II) drug loading and pH-dependent release, (c) supramolecular PEGylated outer shells to improve pharmacokinetics and distribution of platinum-based drug, (d) polymerization-introduced NIR probes to trace the fate of the theranostic nanoplatforms and monitor real-time drug metabolism in vitro and in vivo.
Schematic illustrations for tumor theranostics by TSPDSs, including blood transportation, tumor accumulation, intracellular delivery and tracking.
Materials and Methods
Materials
H-Glu(OtBu)-OtBu, Boc-Lys(Boc)-OH, 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride (EDC.HCl) and benzotriazol-1-yl-1-hydroxybenzotriazole hydrate (HOBT) were purchased from GL Biochem (Shanghai, China). Cystamine dihydrochloride, glutathione reduced ethyl ester (GSH-OEt), AgNO3 and K2PtCl4 were obtained from Sigma Aldrich (USA). Lipoic acid (LA) and methoxypolyethylene glycol (mPEG, Mw=1000) were obtained from Aladdin (Shanghai, China). Dithiothreitol (DTT) and tris(2-carboxyethyl)phosphine hydrochloride (TCEP) were obtained from TCI Development Co., Ltd. (Shanghai, China). Cy5.5 hydroxysuccinimide ester (Cy5.5-NHS) was obtained from Fanbo Biochemicals (Beijing, China). N,N-diisopropylethylamine (DIPEA) and trifluoroacetic acid (TFA) were purchased from Asta Tech Pharmaceutical (Chengdu, China). All the above reagents were used as received. All the organic solvents were purified using the standard method and distilled before use. Clinical cisplatin for tumor patients as a positive control was produced by Qilu Pharmaceutical Co., Ltd (Shandong, China). RPMI 1640 medium, fetal bovine serum (FBS) and NE-PER nuclear and cytoplasmic extraction reagent were purchased from Thermo Fisher Scientific (USA). Penicillin and streptomycin were purchased from Hyclone (USA). LysoTracker Blue DND-22 was purchased from Invitrogen (Carlsbad, CA, USA). Cell counter kit-8 (CCK-8) was purchased from Dojindo Laboratories (Kumamoto, Japan). Cell cycle and apoptosis analysis kit and 3,3'-dioctadecyloxacarbocyanine perchlorate (DiO) were purchased from Beyotime Institute of Biotechnology (Beijing, China). BALB/c mice and BALB/c nude mice were purchased from Dashuo Experimental Animal Center (Chengdu, China).
Molecular synthesis of supramolecular components
Synthesis of LA-functionalized dendrons
Briefly, LA-functionalized dendrons were synthesized using a divergent approach [30, 52]. The condensation of LA (1.0 equiv) and H-Glu(OtBu)-OtBu (1.2 equiv) was catalyzed by EDC.HCl (1.2 equiv), HOBT (1.2 equiv) and DIPEA (4.0 equiv) in CH2Cl2 for 24 h under a nitrogen atmosphere. After removal of tert-butoxy (OtBu) groups by TFA, all carboxyl groups were reacted with H-Glu(OtBu)-OtBu by condensation reaction to obtain Generation 2 dendrons with protecting groups. Repeating the above steps would generate LA-functionalized Generation 3 dendrons. Each crude product was purified by washing and column chromatography to obtain pure compound. Carboxyl groups in LA-functionalized dendrons were exposed after treatment with TFA.
Synthesis of NIR initiator
Cystamine dihydrochloride (1.0 equiv) was reacted with Cy5.5-NHS (2.5 equiv) in an aqueous solution for 24 h under dark condition. The Cy5.5-cystamine-Cy5.5 conjugates (CCs) were dialyzed against Millipore water to remove the free NIR dye (MWCO 1000) in low temperature (4 °C) and dark condition. Then freeze-dried CCs were obtained as NIR initiators.
Synthesis of PEGylated platinum derivatives
mPEG was conjugated with Boc-Lys(Boc)-OH, and mPEG-K was obtained after removal of N-tert-butoxycarbonyl (Boc) groups. Meanwhile, K2PtCl4 (1.0 equiv) was reacted with DMSO (5.0 equiv) to harvest Pt(DMSO)2Cl2 according to previous reports [51, 53]. Pt(DMSO)2Cl2 (1.0 equiv) would coordinate with mPEG-K (1.0 equiv) by the sufficient stirring for 24 h to form PEGylated platinum derivatives (Pt(II)-K-mPEG) [51, 54], which was obtained after filtration and concentration.
Chemical characterizations of all above-mentioned compounds could be found in Supporting Information.
Supramolecular engineering of theranostic nanoplatforms
Amphiphilic LA-functionalized dendrons would self-assemble into core-shell nanostructures with lipoic acids as inner core and abundant peripheral carboxyl groups in aqueous solution. A catalytic amount of CCs were reduced by TCEP in deionized water for 45 min to generate thiol-NIR (HS-Cy5.5), and then thiol-NIR molecules could induce the polymerization of the LA-functionalized dendrons to immobilize the inner core of supramolecular dendritic systems at room temperature for 24 h. The carboxyl groups (-COOH) on the disulfide-stabilized supramolecular dendritic systems (DSDSs) were ionized into -COO- by NaOH. Subsequently, Pt(II)-K-mPEG pretreated with AgNO3 would coordinate onto DSDSs as theranostic supramolecular PEGylated dendritic systems (TSPDSs) [55].
Characterizations of supramolecular nanoplatforms
Sizes and zeta potentials of supramolecular nanoplatforms were determined by dynamic light scattering (DLS, Malvern Zetasizer Nano ZS, UK) in aqueous solutions at a concentration of 100 μg mL-1. Their nanostructures were performed by transmission electron microscopy (TEM, Tecnai GF20S-TWIN, USA) on the copper grids. Element compositions of TSPDSs were detected by energy-dispersive spectroscopy (EDS, JSM-5900LV, Japan). The chemical nature of platinum in TSPDSs was observed by X-ray photoelectron spectroscopy (XPS, XSAM800, UK) with C1s peak at 284.6 eV for energy calibration.
The platinum amount in TSPDSs was determined by thermo gravimetric analysis (TGA, NETZSCH STA 449C, Germany) over a temperature range of 40 ºC to 900 ºC in air with a programmed temperature increase of 10 ºC/min. The platinum amount was also directly measured by inductively coupled plasma mass spectrometry (ICP-MS, Perkin ELAN DRC-e, Canada).
In vitro drug release
TSPDSs were dissolved with a concentration of 1.0 mg mL-1 in different PBS, which simulated the normal physiological conditions (pH 7.4, no DTT), tumor microenvironment (pH 6.8, 10 μM DTT) and tumor intracellular conditions (pH 5.0, 10 mM DTT), respectively. The samples (1.0 mL) were sealed into dialysis tubes (MWCO 2000) with three parallel samples. The dialysis tubes were placed into the corrresponding PBS of 20 mL at 37 ºC with gentle shaking. 0.5 mL of outside buffer solution was taken for platinum concentration analysis at monitoring time points, and 0.5 mL fresh buffer solution was supplemented. The release amount of platinum was measured by ICP-MS.
Cytotoxicity to A549 tumor cell line
Human lung epithelial tumor cell line A549 was cultured in RPMI 1640 medium supplemented with 10% (v/v) FBS and 1% (v/v) penicillin/streptomycin at 37 ºC in a humidified 5% CO2 atmosphere.
To assess the cytotoxicity of TSPDSs to A549 tumor cells, cells were cultured in 96-well plate at a density of 8×103/well for 24 h. After incubation with TSPDSs and other components at the different concentrations for 48 h, the cell viability of A549 cell was detected using CCK-8 reagent.
Intracellular tracking of drug delivery
Fluorescence activated cell sorting (FACS, Beckman Coulter Cytomics FC-500, USA) was used to quantify the internalization of TSPDSs. A549 tumor cells were seeded in 6-well plates with a cell density of 2×105 cells/well and cultured for 24 h. After removal of culture media, TSPDSs-contained culture media were added to each well for various incubating time. Then, the cells were washed three times with PBS and harvested for FACS analysis.
A549 tumor cells were seeded in glass bottom dishes at a density of 4×103 cells/well and cultured for 24 h. After incubation with TSPDSs for 1 h and 3 h, A549 tumor cells were fixed with 4% paraformaldehyde, and cell membranes were stained by DiO. To monitor endosomal escape of TSPDSs, lysosomes of A549 tumor cells were stained by LysoTracker Blue after incubation with certain time. To strengthen intracellular reducing condition, A549 tumor cells were pretreated with GSH-OEt for 2 h. Intracellular tracking of TSPDSs was performed by confocal laser scanning microscopy (CLSM, Leica TCP SP5, Germany).
Intracellular platinum distribution
A549 tumor cells were seeded into 6-well plates and incubated with cisplatin and TSPDSs (1.5 μg mL-1 platinum) for 24 h. First, subcellular fractionation of nucleus and cytoplasm was carried out with NE-PER nuclear and cytoplasmic extraction reagent in accordance with the manufacturer's handbook [56], and more concrete operation can be found in the Supporting Information. The cytoplasmic extract and nucleus extract were digested by H2O2 and HNO3, and platinum amount was measured by ICP-MS.
Cell cycle analysis
After treatment with D+mPEG-K complex, clinical cisplatin and TSPDSs (1.5 μg mL-1 platinum), the A549 tumor cells were fixed with pre-cooled 70% ethanol at 4 ºC for 2 h. Then A549 tumor cells were stained with propidium iodide (PI) at 37 ºC for 0.5 h. Cell cycle distribution was analyzed by flow cytometry.
In vivo antitumor efficacy of TSPDSs
All animal experiments were conducted in accordance with the ethics committee of Sichuan University. A549 tumor xenografts were established by subcutaneous injection of 2×106 cells into the right flanks of nude mice. When the tumor volumes reached about 100 mm3, nude mice were randomly divided into four groups for antitumor treatment, including normal saline, D+mPEG-K, clinical cisplatin and TSPDSs group. The xenograft-bearing mice were administred with a dose of 3.25 mg platinum kg-1 body weight using tail intravenous injection at a 3-day interval for four times. The xenograft volume and body weight were monitored at a 3-day interval for 21 days. The tumor volumes were measured using a caliper and calculated by the formula of V = AB2/2 mm3, A and B presented the length and width of the solid tumor. The tumor volume and body weight were analyzed by t test to evaluate significant difference.
Histological and immunohistochemical evaluation
After the course of antitumor treatment for 21 days, xenografts and major organs were dissected from the mice and fixed in 10 % (v/v) formalin saline for 24 h. The tissues were embedded in paraffin and cut at 5 μm thickness. The xenograft sections stained by hematoxylin and eosin (H&E), platelet endothelial cell adhesion molecule-1 (CD31), nuclear nonhistone protein Ki-67 and terminal dUTP nick-end labeling (TUNEL) for histopathological and immunohistochemistry analysis. Kidney tissue slices stained by H&E and TUNEL were carried out for toxicological analysis.
In vivo and ex vivo NIR imaging
Xenograft-bearing mice were administrated with TSPDSs (3.25 mg platinum kg-1 by tail intravenous injection. Then, the NIR fluorescence signals were collected by Maestro in vivo fluorescence imaging system (CRi Maestro EX, USA) with excitation at 675 nm and emission at 695 nm at the monitoring time points. When the mice were sacrificed at time point of 24 h, the xenograft and major organs were excised for fluorescence imaging at the same field of view.
Pharmacokinetics and biodistribution
The pharmacokinetics of TSPDSs and cisplatin were carried out with a single intravenous injection at a dose of 3.25 mg platinum kg-1 using 4-week male mice. At the preset time points, the blood was taken by removalling eyeball and digested with H2O2 and HNO3. The concentrations of platinum were detected by ICP-MS.
The biodistribution of platinum in A549-tumor bearing mice was conducted with intravenous administration of TSPDSs and cisplatin (3.25 mg platinum kg-1). At the preset time points (2 h and 12 h), mice were sacrificed to separate kidneys and xenografts. After tissues were weighed and digested with H2O2 and HNO3, platinum contents were determined by ICP-MS.
Peripheral Blood Analysis
After the course of antitumor treatment, the mice were sacrificed to harvest 1 mL blood in a heparinized tube. After centrifugation at 3000 rpm for 15 min, serum was separated for clinical biochemical analysis, including blood urea nitrogen (BUN) and creatinine.
Conclusions
In summary, we demonstrated a specifically-made molecular and supramolecular strategy for developing the dual-responsive theranostic nanoplatforms for platinum drug delivery and NIR imaging. The theranostic supramolecular PEGylated dendritic systems provided good dimensional stability at the normal physiological condition owing to disulfide-stabilized cores and coordination-connected PEG-shells, which were particularly suited to resist protein adsorption, prolong drug circulation time and achieve passive tumor targeting. Intracellular pH and redox dual-responsive disintegration of TSPDSs created the necessary conditions for tumor site-specific platinum release and nuclear delivery, as well as for cell cycle arrest and tumor cell growth inhibition. In vivo animal results proved that TSPDSs not only provided high antitumor efficiency comparable to clinical cisplatin, but also avoided some side effects (e.g., nephrotoxicity) after chemotherapy. More importantly, polymerization-introduced NIR fluorescence of TSPDSs visualized in vitro and in vivo fate of theranostic nanoplatforms, which contributed to the disclosure of intracellular platinum release and pharmacokinetics. As a result, theranostic supramolecular PEGylated dendritic systems successfully realized high in vivo antitumor activity comparable to clinical cisplatin and reduced adverse effects, as well as intracellular tracking and tumor location. We hope that this work will offer an alternative strategy to develop supramolecular dendritic theranostic nanoplatforms.
Supplementary Material
Materials and methods, experimental details, additional data, figures (Figures S1-S28) and table (Table S1-S2).
Acknowledgements
This work was supported by National Natural Science Foundation of China (NSFC, 51133004, 81361140343 and 51503128), Joint Sino-German Research Project (GZ756 and GZ905), Scientific Research Foundation for Talent Introduction (YJ201463) and Youth Scholars (2015SCU11037) from Sichuan University.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Lim E-K, Kim T, Paik S, Haam S, Huh Y-M, Lee K. Nanomaterials for Theranostics: Recent Advances and Future Challenges. Chem Rev. 2014;115:327-94
2. Muthu MS, Leong DT, Mei L, Feng S-S. Nanotheranostics˗ application and further development of nanomedicine strategies for advanced theranostics. Theranostics. 2014;4:660-77
3. Xie J, Lee S, Chen X. Nanoparticle-based theranostic agents. Adv Drug Delivery Rev. 2010;62:1064-79
4. Barenholz YC. Doxil®—the first FDA-approved nano-drug: lessons learned. J Controlled Release. 2012;160:117-34
5. Bourzac K. Nanotechnology: carrying drugs. Nature. 2012;491:S58-S60
6. Yang E, Qian W, Cao Z, Wang L, Bozeman EN, Ward C. et al. Theranostic Nanoparticles Carrying Doxorubicin Attenuate Targeting Ligand Specific Antibody Responses Following Systemic Delivery. Theranostics. 2015;5:43-61
7. Wang H, Wu Y, Zhao R, Nie G. Engineering the assemblies of biomaterial nanocarriers for delivery of multiple theranostic agents with enhanced antitumor efficacy. Adv Mater. 2013;25:1616-22
8. Wang D, Lippard SJ. Cellular processing of platinum anticancer drugs. Nat Rev Drug Discovery. 2005;4:307-20
9. Wong E, Giandomenico CM. Current status of platinum-based antitumor drugs. Chem Rev. 1999;99:2451-66
10. Dai Y, Xiao H, Liu J, Yuan Q, Ma Pa, Yang D. et al. In vivo multimodality imaging and cancer therapy by near-infrared light-triggered trans-platinum pro-drug-conjugated upconverison nanoparticles. J Am Chem Soc. 2013;135:18920-9
11. Min Y, Li J, Liu F, Yeow EK, Xing B. Near-Infrared Light-Mediated Photoactivation of a Platinum Antitumor Prodrug and Simultaneous Cellular Apoptosis Imaging by Upconversion-Luminescent Nanoparticles. Angew Chem Int Ed. 2014;53:1012-6
12. Zhu Z, Wang X, Li T, Aime S, Sadler PJ, Guo Z. Platinum (II)-Gadolinium (III) Complexes as Potential Single-Molecular Theranostic Agents for Cancer Treatment. Angew Chem Int Ed. 2014;53:13225-8
13. Luk BT, Fang RH, Zhang L. Lipid-and polymer-based nanostructures for cancer theranostics. Theranostics. 2012;2:1117-26
14. Wang Z, Niu G, Chen X. Polymeric materials for theranostic applications. Pharm Res. 2014;31:1358-76
15. Krasia-Christoforou T, Georgiou TK. Polymeric theranostics: using polymer-based systems for simultaneous imaging and therapy. J Mater Chem B. 2013;1:3002-25
16. Cheng Y, Zhao L, Li Y, Xu T. Design of biocompatible dendrimers for cancer diagnosis and therapy: current status and future perspectives. Chem Soc Rev. 2011;40:2673-703
17. Stiriba SE, Frey H, Haag R. Dendritic polymers in biomedical applications: from potential to clinical use in diagnostics and therapy. Angew Chem Int Ed. 2002;41:1329-34
18. Wolinsky JB, Grinstaff MW. Therapeutic and diagnostic applications of dendrimers for cancer treatment. Adv Drug Delivery Rev. 2008;60:1037-55
19. Lo S-T, Kumar A, Hsieh J-T, Sun X. Dendrimer Nanoscaffolds for Potential Theranostics of Prostate Cancer with a Focus on Radiochemistry. Mol Pharm. 2013;10:793-812
20. Malik N, Evagorou EG, Duncan R. Dendrimer-platinate: a novel approach to cancer chemotherapy. Anti-Cancer Drugs. 1999;10:767-76
21. Chen G, Kumar J, Gregory A, Stenzel MH. Efficient synthesis of dendrimers via a thiol-yne and esterification process and their potential application in the delivery of platinum anti-cancer drugs. Chem Commun. 2009:6291-3
22. Kapp T, Dullin A, Gust R. Platinum (II)- dendrimer conjugates: synthesis and investigations on cytotoxicity, cellular distribution, platinum release, DNA, and protein binding. Bioconjugate Chem. 2010;21:328-37
23. Nwe K, Bryant LH, Brechbiel MW. Poly(amidoamine) Dendrimer Based MRI Contrast Agents Exhibiting Enhanced Relaxivities Derived via Metal Preligation Techniques. Bioconjugate Chem. 2010;21:1014-7
24. Svenson S. The dendrimer paradox-high medical expectations but poor clinical translation. Chem Soc Rev. 2015;44:4131-44
25. Helms B, Meijer E. Dendrimers at Work. Science. 2006;313:929-30
26. Al-Jamal KT, Ramaswamy C, Florence AT. Supramolecular structures from dendrons and dendrimers. Adv Drug Delivery Rev. 2005;57:2238-70
27. Sun H-J, Zhang S, Percec V. From structure to function via complex supramolecular dendrimer systems. Chem Soc Rev. 2015;44:3900-23
28. Xu X, Yuan H, Chang J, He B, Gu Z. Cooperative Hierarchical Self-Assembly of Peptide Dendrimers and Linear Polypeptides into Nanoarchitectures Mimicking Viral Capsids. Angew Chem Int Ed. 2012;51:3130-3
29. Li H, Xu X, Li Y, Geng Y, He B, Gu Z. Design and self-assembly of amphiphilic peptide dendron-jacketed polysaccharide polymers into available nanomaterials. Polym Chem. 2013;4:2235-8
30. Xu X, Jian Y, Li Y, Zhang X, Tu Z, Gu Z. Bio-Inspired Supramolecular Hybrid Dendrimers Self-Assembled from Low-Generation Peptide Dendrons for Highly Efficient Gene Delivery and Biological Tracking. ACS Nano. 2014;8:9255-64
31. Xu X, Li Y, Li H, Liu R, Sheng M, He B. et al. Smart Nanovehicles Based on pH-Triggered Disassembly of Supramolecular Peptide-Amphiphiles for Efficient Intracellular Drug Delivery. Small. 2014;10:1133-40
32. Li Y, Lai Y, Xu X, Zhang X, Wu Y, Hu C. et al. Capsid-like supramolecular dendritic systems as pH-responsive nanocarriers for drug penetration and site-specific delivery. Nanomedicine NBM. 2016;12:355-64
33. Xu X, Jiang Q, Zhang X, Nie Y, Zhang Z, Li Y. et al. Virus-inspired mimics: self-assembly of dendritic lipopeptides into arginine-rich nanovectors for improving gene delivery. J Mater Chem B. 2015;3:7006-10
34. Zhang Z, Zhang X, Xu X, Li Y, Li Y, Zhong D. et al. Virus-Inspired Mimics Based on Dendritic Lipopeptides for Efficient Tumor-Specific Infection and Systemic Drug Delivery. Adv Funct Mater. 2015;25:5250-60
35. Janib SM, Moses AS, MacKay JA. Imaging and drug delivery using theranostic nanoparticles. Adv Drug Delivery Rev. 2010;62:1052-63
36. Bowman CN, Kloxin CJ. Covalent adaptable networks: reversible bond structures incorporated in polymer networks. Angew Chem Int Ed. 2012;51:4272-4
37. Nyström AM, Wooley KL. The importance of chemistry in creating well-defined nanoscopic embedded therapeutics: devices capable of the dual functions of imaging and therapy. Acc Chem Res. 2011;44:969-78
38. Gref R, Domb A, Quellec P, Blunk T, Müller RH, Verbavatz JM. et al. The controlled intravenous delivery of drugs using PEG-coated sterically stabilized nanospheres. Adv Drug Delivery Rev. 2012;64(Supplement):316-26
39. Cheng R, Meng F, Deng C, Klok H-A, Zhong Z. Dual and multi-stimuli responsive polymeric nanoparticles for programmed site-specific drug delivery. Biomaterials. 2013;34:3647-57
40. Ge Z, Liu S. Functional block copolymer assemblies responsive to tumor and intracellular microenvironments for site-specific drug delivery and enhanced imaging performance. Chem Soc Rev. 2013;42:7289-325
41. Yin Q, Shen J, Zhang Z, Yu H, Li Y. Reversal of multidrug resistance by stimuli-responsive drug delivery systems for therapy of tumor. Adv Drug Delivery Rev. 2013;65:1699-715
42. Svenson S. Theranostics: Are we there yet? Mol Pharm. 2013;10:848-56
43. Ye Y, Chen X. Integrin targeting for tumor optical imaging. Theranostics. 2011;1:102-26
44. Kim J, Piao Y, Hyeon T. Multifunctional nanostructured materials for multimodal imaging, and simultaneous imaging and therapy. Chem Soc Rev. 2009;38:372-90
45. Li YL, Zhu L, Liu Z, Cheng R, Meng F, Cui JH. et al. Reversibly stabilized multifunctional dextran nanoparticles efficiently deliver doxorubicin into the nuclei of cancer cells. Angew Chem Int Ed. 2009;48:9914-8
46. Won YW, Yoon SM, Lim KS, Kim YH. Self-Assembled Nanoparticles with Dual Effects of Passive Tumor Targeting and Cancer-Selective Anticancer Effects. Adv Funct Mater. 2012;22:1199-208
47. Bang E-K, Gasparini G, Molinard G, Roux A, Sakai N, Matile S. Substrate-initiated synthesis of cell-penetrating poly (disulfide) s. J Am Chem Soc. 2013;135:2088-91
48. Gasparini G, Bang E-K, Molinard G, Tulumello DV, Ward S, Kelley SO. et al. Cellular uptake of substrate-initiated cell-penetrating poly (disulfide) s. J Am Chem Soc. 2014;136:6069-74
49. Veronese FM, Pasut G. PEGylation, successful approach to drug delivery. Drug Discov Today. 2005;10:1451-8
50. Xing B, Zhu H, Shi Y, Tang W. In vitro binding of an orally active platinum antitumor drug, JM216 to metallothionein. BioMetals. 2001;14:51-7
51. Binauld S, Scarano W, Stenzel MH. pH-Triggered release of platinum drugs conjugated to micelles via an acid-cleavable linker. Macromolecules. 2012;45:6989-99
52. Zhang X, Zhang Z, Xu X, Li Y, Li Y, Jian Y. et al. Bioinspired Therapeutic Dendrimers as Efficient Peptide Drugs Based on Supramolecular Interactions for Tumor Inhibition. Angew Chem Int Ed. 2015;54:4289-94
53. Price JH, Birk JP, Wayland BB. Thermal and photochemical cis-trans isomerization of PtL2Cl2 (L= dialkyl sulfoxide) complexes. Kinetics and mechanisms for thermal isomerization. Inorg Chem. 1978;17:2245-50
54. Karim KJA, Binauld S, Scarano W, Stenzel MH. Macromolecular platinum-drugs based on statistical and block copolymer structures and their DNA binding ability. Polym Chem. 2013;4:5542-54
55. Cabral H, Matsumoto Y, Mizuno K, Chen Q, Murakami M, Kimura M. et al. Accumulation of sub-100 nm polymeric micelles in poorly permeable tumours depends on size. Nat Nanotechnol. 2011;6:815-23
56. Hung J-H, Lu Y-S, Wang Y-C, Ma Y-H, Wang D-S, Kulp SK. et al. FTY720 induces apoptosis in hepatocellular carcinoma cells through activation of protein kinase C δ signaling. Cancer Res. 2008;68:1204-12
Author contact
![]() Corresponding authors: xianghui.xucom (X. Xu); zwguedu.cn (Z. Gu)
Corresponding authors: xianghui.xucom (X. Xu); zwguedu.cn (Z. Gu)