Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Results
Discussion
Conclusion
Materials and Methods
Supplementary Material
Abbreviations
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2016; 6(10):1477-1490. doi:10.7150/thno.14158 This issue Cite
Research Paper
Dihydropyrimidine Dehydrogenase Is a Prognostic Marker for Mesenchymal Stem Cell-Mediated Cytosine Deaminase Gene and 5-Fluorocytosine Prodrug Therapy for the Treatment of Recurrent Gliomas
Taemoon Chung1,2,3,4*, Juri Na1,2,3,4*, Young-il Kim1,5, Da-Young Chang7,8, Young Il Kim4,6, Hyeonjin Kim2,4,6, Ho Eun Moon9, Keon Wook Kang1,2,3, Dong Soo Lee1,5, June-Key Chung1,2,3 ![]() , Sung-Soo Kim8, Haeyoung Suh-Kim8, Sun Ha Paek9, Hyewon Youn1,3,6
, Sung-Soo Kim8, Haeyoung Suh-Kim8, Sun Ha Paek9, Hyewon Youn1,3,6 ![]()
1. Department of Nuclear Medicine, Seoul National University College of Medicine, Seoul, Korea.
2. Department of Biomedical Sciences, Seoul National University College of Medicine, Seoul, Korea.
3. Cancer Research Institute, Seoul National University College of Medicine, Seoul, Korea.
4. Institute of Radiation Medicine, Seoul National University Medical Research Center, Seoul, Korea.
5. Department of Molecular Medicine and Biopharmaceutical Sciences, Seoul National University, Seoul, Korea,
6. Cancer Imaging Center, Seoul National University Hospital, Seoul, Korea.
7. Department of Radiology, Seoul National University College of Medicine, Seoul, Korea.
8. Department of Anatomy, Ajou University School of Medicine, Suwon, Korea.
9. Department of Neurosurgery, Seoul National University College of Medicine, Seoul, Korea.
*These authors contributed equally to this research.
Received 2015-10-9; Accepted 2016-5-6; Published 2016-6-17
Abstract

We investigated a therapeutic strategy for recurrent malignant gliomas using mesenchymal stem cells (MSC), expressing cytosine deaminase (CD), and prodrug 5-Fluorocytosine (5-FC) as a more specific and less toxic option. MSCs are emerging as a novel cell therapeutic agent with a cancer-targeting property, and CD is considered a promising enzyme in cancer gene therapy which can convert non-toxic 5-FC to toxic 5-Fluorouracil (5-FU). Therefore, use of prodrug 5-FC can minimize normal cell toxicity. Analyses of microarrays revealed that targeting DNA damage and its repair is a selectable option for gliomas after the standard chemo/radio-therapy. 5-FU is the most frequently used anti-cancer drug, which induces DNA breaks. Because dihydropyrimidine dehydrogenase (DPD) was reported to be involved in 5-FU metabolism to block DNA damage, we compared the survival rate with 5-FU treatment and the level of DPD expression in 15 different glioma cell lines. DPD-deficient cells showed higher sensitivity to 5-FU, and the regulation of DPD level by either siRNA or overexpression was directly related to the 5-FU sensitivity. For MSC/CD with 5-FC therapy, DPD-deficient cells such as U87MG, GBM28, and GBM37 showed higher sensitivity compared to DPD-high U373 cells. Effective inhibition of tumor growth was also observed in an orthotopic mouse model using DPD- deficient U87MG, indicating that DPD gene expression is indeed closely related to the efficacy of MSC/CD-mediated 5-FC therapy. Our results suggested that DPD can be used as a biomarker for selecting glioma patients who may possibly benefit from this therapy.
Keywords: Dihydropyrimidine dehydrogenase (DPD), 5-Fluorouracil (5-FU), 5-Fluorocytosine (5-FC), mesenchymal stem cells (MSC), cytosine deaminase (CD), gene therapy.
Introduction
Glioma is a malignant tumor of the brain and spinal cord (most commonly the brain) which shows a high rate of recurrence even after standard anti-cancer therapy including surgery, radiotherapy, and chemotherapy. Though various advanced therapies have been suggested in addition to standard therapy, these have shown only a modest therapeutic effect. Therefore, glioma remains a malignancy with a high mortality rate, and one that is not easily cured. [1, 2] In this regard, new strategies for glioma therapy are eagerly awaited. [3, 4] Most failures in glioma treatment are related to the invasive nature of glioma, inefficient drug-delivery and/or dose-limiting toxicities. [5, 6] To reduce the side effects from the treatment, the use of 'suicide' (apoptosis-causing) genes combined with a prodrug have been suggested as an alternative option. [7, 8]
Cytosine deaminase (CD), which is expressed in bacteria and fungi but is not expressed in humans, can de-aminate non-toxic 5-Fluorocytosine (5-FC) into the toxic 5-Fluorouracil (5-FU) frequently used in anti-cancer therapy. Compared to other enzyme/prodrug systems, CD/5-FC is an attractive suicide gene for cancer therapy, because freely diffusible 5-FC has strong anti-cancer effects. [10, 11] Recently, tumor-specific delivery of the CD gene as well as tumor tropism of stem cells have been extensively studied [12, 13].
Unlike viral vector-mediated conventional suicide gene therapy, stem cell-mediated suicide gene therapy can ablate therapeutic stem cells after treatment with prodrug. Therefore, any potential safety risks from insertional mutation caused by the genetically modified therapeutic stem cells may be diminished by cell self-destruction. Since the cell membrane is highly permeable to 5-FC and 5-FU by simple diffusion, cytotoxic effect can easily propagate to the neighboring tumor cells. This 'bystander' effect targets tumor cells present in the vicinity of the therapeutic stem cells. Aboody et al. has shown preclinical results from combination therapy with neural stem cells-mediated CD gene and 5-FC, which led to the approval of a human trial for the treatment of recurrent high grade gliomas [14, 15]. Instead of neuronal stem cells (NSCs), Suh-Kim et al. have suggested that mesenchymal stem cells (MSCs) could be used as a vehicle to deliver CD gene to brain tumors [16, 17]. Compared to NSCs, MSCs can be easily isolated from bone marrow and adipose tissues of patients, are highly proliferative, and therefore can be expanded in vitro suggesting the possibility of MSCs' production at an industrial scale as a cell therapeutic agent. Furthermore, the lack of human leukocyte antigen, antigen D related (HLA-DR) on MSCs reduces their immunogenic property contributing to a reduced immune rejection response after allogeneic injection of therapeutic MSCs [17, 18].
General regimens for treating GBM patients with chemo-radiotherapy do not take into consideration inter-patient variability in the expression of specific target genes [19, 20]. Considering the invasive nature of GBMs and their almost inevitable recurrence, application of MSC/CD therapy to recurrent gliomas appears to be a plausible strategy. However, cell therapy is costly and carries with it the possible risk of other side effects; hence, the candidate recurrent glioma patients to receive this advanced therapy should be carefully selected. Identification of a prognostic biomarker would therefore be critical in tailoring MSC/CD with 5-FC therapy to specific patients who are likely to benefit from it. To search for a possible prognostic marker for MSC/CD with 5-FC therapy, we investigated the correlation between the therapeutic effect of MSC/CD with 5-FC and the expression levels of genes related to 5-FU metabolism in gliomas.
In this study, we evaluated sensitivity and therapeutic efficacy of MSC/CD with 5-FC treatment in established glioblastoma cell lines as well as in primary cells from GBMs acquired from patients. We then selected the most effective cell line that displayed a low expression level of dihydropyrimidine dehydrogenase (DPD). By taking advantage of multimodal imaging systems including Positron Emission Tomography (PET), Magnetic Resonance Imaging (MRI), Magnetic Resonance Spectroscopy (MRS) and Bioluminescence Imaging (BLI), we also evaluated the therapeutic effect of MSC/CD with 5-FC treatment using an orthotopic mouse model to prove its feasibility.
Results
Changes in gene expression in gliomas after standard chemo/radio-therapy
The standard therapy for malignant gliomas comprises radiation and temozolomide (TMZ) treatment following surgical resection. We performed microarray analysis to investigate the molecular alterations in gliomas that ensue in response to this conventional therapy. Gliobastoma cell lines, U87MG and U373 were chosen because they have been most frequently used to show a therapeutic benefit from the combination therapy of TMZ and radiation [20]. We also included primary cells from a GBM patient before (GBM28) and after (GBM37) standard therapy. Microarray results demonstrated higher expression of genes related to DNA damage repair in all three cases, i.e. in U87MG, U373 and GBM37 (Figure 1A).
Analysis of gene expression and 5-FU sensitivity of glioblastoma cell lines and primary cells: (A) Expression levels of genes related to DNA damage repair by MA-Agilent Human 44K v1 microarray: (B) In vitro cytotoxicity of 5-FU on gliomas; viable luciferase-expressing glioma cells were imaged with IVIS 100. (C) Cytotoxicity of 5-FU evaluated by bioluminescence imaging (D) Expression levels of genes related to 5-FU metabolism by microarray analysis.
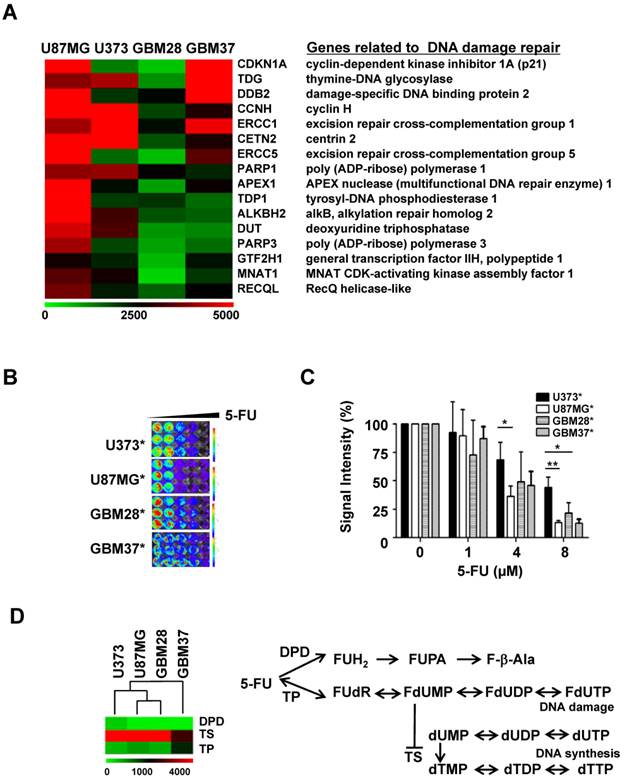
5-FU cytotoxicity- and metabolism-related alterations in gene expression in glioma cells
To explore the possibility of using 5-FU as a plausible agent targeting DNA damage in recurrent glioma treatment, as was evident by the microarray analysis, we evaluated the anti-cancer effect of 5-FU in four glioma cell lines and primary glioma cells. Reporters-expressing glioma cells (Figure S1) were used for visualizing the cytotoxicity of 5-FU on glioma cells. Though the viability of all four glioma cells decreased in a concentration-dependent manner, 5-FU was more toxic to U87MG than to U373 or to the two primary glioma cells, GBM28 and GBM37 (Figure 1B & 1C).
DNA break-inducing 5-FU toxicity is caused by the collapse of stalled replication forks and is closely related to its metabolism. Using microarray analysis, we compared the expression level of each enzyme known to be involved in the metabolism of 5-FU. Among these, the expression of 5-dihydropyrimidine dehydrogenase (DPD) showed an inverse relationship with 5-FU sensitivity (Figure 1D). U373 cells, which are less sensitive to the therapeutic effect of 5-FU, showed a relatively higher gene expression of DPD than other glioma cells. There was no change in the expression of thymidylate synthase (TS) and thymidine phosphorylase (TP) genes, which are also involved in the metabolism of 5-FU.
5-FU cytotoxicity and DPD gene expressions in various glioma cells
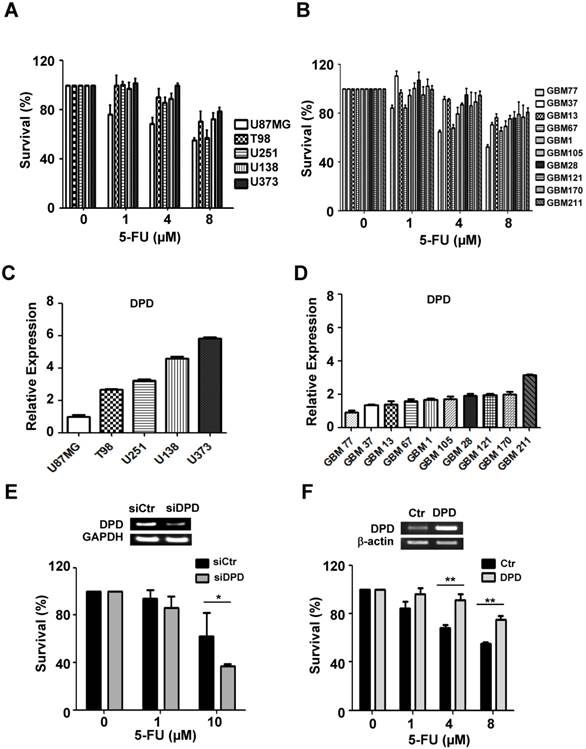
To evaluate the variable sensitivity of glioma cells to 5-FU, we performed the CCK-8 assay in a larger panel of five established glioma cell lines and 10 primary glioma cells derived from patients (Figure 2A&B). In particular, we investigated the relationship between 5-FU sensitivity and the expression level of DPD by performing the real-time quantitative PCR (Figure 2C & D). Our results indicate that DPD-deficient glioma cells were more sensitive to 5-FU treatment than DPD-high U373 cells, indicating that increased expression of DPD might be the underlying mechanism for inhibiting the 5-FU toxicity. To support this hypothesis, we tested whether inhibition of DPD by siRNA might increase 5-FU toxicity in U373 cells. Upon treatment with DPD siRNA, U373 cells became sensitive to 5-FU (Figure 2E). In addition, we evaluated the effect of DPD over-expression on 5-FU sensitivity in DPD-deficient U87MG. As shown in Figure 2F, over-expression of DPD increased the survival rate of U87MG after 5-FU treatment. These results implied (or strongly suggested) that 5-FU is an efficient chemotherapeutic agent in DPD-deficient gliomas.
5-FU sensitivity and DPD expression in gliomas: (A) 5-FU sensitivity in 5 established glioma cell lines and (B) 10 primary glioma cells from patients; CCK-8 assay was performed to measure cell survival following treatment with different concentrations of 5-FU. (C) Relative DPD expression in 5 established glioma cell lines and (D) 10 primary cells from patients. Real-time PCR was performed to measure DPD expression. The relative level of DPD expression of each glioma was compared with U87MG. (E) Effect of down-regulation of DPD on 5-FU sensitivity in U373 cells (high DPD-expressing cells). GAPDH was used as loading control. (F) Effect of up-regulation of DPD on 5-FU sensitivity in U87MG cells (low DPD-expressing cells). β-actin was used as loading control (*, P<0.05; **, P<0.01; N=3).
Characterization of MSC/CDs
Since we anticipated that suicide gene therapy using CD-expressing MSCs with prodrug 5-FC may be a better option for the gliomas resistant to conventional therapy, we introduced MSC/CDs [16, 17] in two glioblastoma cell lines and cells derived from a GBM patient (U87MG, U373, GBM28, GBM37). Our strategy was to use the tumor targeting property of MSC/CDs as well as the bystander effect generated by MSC/CDs by converting the non-toxic prodrug 5-FC to toxic 5-FU (Figure 3A).
Therapeutic strategy and characterization of MSC/CD: (A) Strategy of MSC-mediated cytosine deaminase with prodrug therapy. (B) Cytosine deaminase expression of MSCs was validated by RT-PCR and immunoblot assay. β-actin was used as loading control. (C) Fluorescence-activated cell sorting analysis (FACS) was performed for characterization of stem cell marker expression on naive MSC and gene-manipulated MSCs. * indicates luciferase reporter-expressing cells for bioluminescence imaging. Stem cell characteristics were evaluated by FACS analysis.
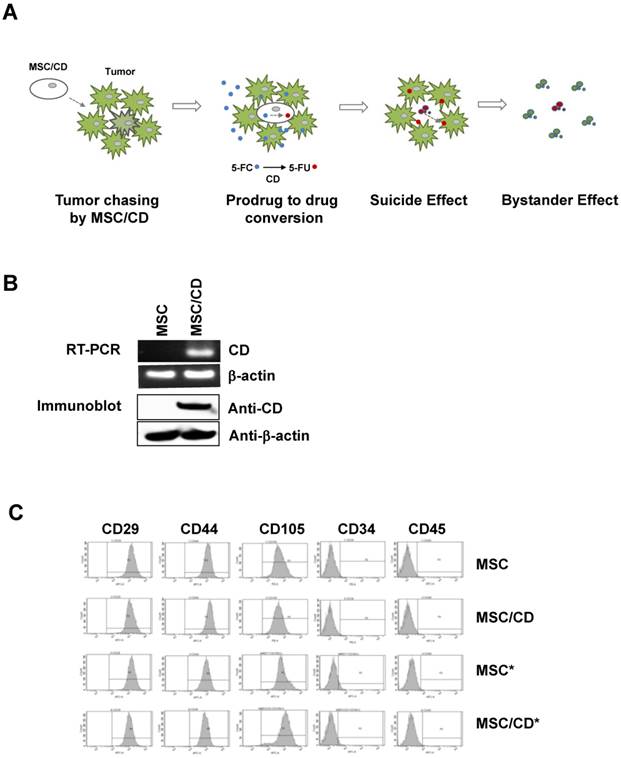
Both mRNA and protein levels of CD were monitored in MSC/CD cells. Normal (i.e. naïve) MSCs do not express CD, but the enzyme was expressed in gene-transduced MSCs (MSC/CDs) (Figure 3B). Since we introduced reporters in stem cells for visualization (Figure S2), we could validate that stem cell characteristics of MSC/CDs were unchanged (Figure 3C). Preservation of stem cell characteristics in the CD-transduced MSCs was described in an earlier report [17]. Stem Cell characteristics were evaluated by monitoring the expression of CD29 (Integrin protein), CD44 (hyaluronate receptor), CD105 (endoglin protein), CD34 (hematopoietic progenitor cell marker), and CD45 (leukocyte common antigen) markers.
The ability of MSC/CDs to convert 5-FC to 5-FU was evaluated using magnetic resonance spectroscopy (19F-MRS). The chemical shift from 5-FC to 5-FU was only observed in MSC/CD culture media, indicating that MSC/CD could convert prodrug 5-FC into 5-FU in vitro (Figure 4A).
Therapeutic effect of MSC/CDs by conversion of 5-FC to 5-FU and their tumor targeting property: (A) In vitro 5-FC to 5-FU conversion by MSCs and MSC/CDs. Chemical shift from 5-FC to 5-FU was analyzed with 19F-MRS. (B) 5-FC-dependent suicide effect in MSCs and MSC/CDs. Viable MSC and MSC/CD cells were measured by bioluminescence. (C) 5-FC-dependent anti-cancer effect of MSC/CDs on gliomas. (D) MSC/CDs number-dependent anticancer effect on gliomas. Different ratios of MSC/CDs to glioma cells were co-cultured with 100 µM of 5-FC. (E) In vivo 5-FC to 5-FU conversion by MSC/CDs. MSC/CDs transplanted regions of 5-FC and 5-FU were analyzed by 19F-MRS. (F) The tumor targeting property of MSC/CDs in a mouse orthotopic glioma model. Luciferase-expressing-MSC/CDs were introduced into the left striatum of mice 1 week after transplantation of U87MG cells (right striatum). Tumor development and localization of MSCs were monitored using MRI and BLI.
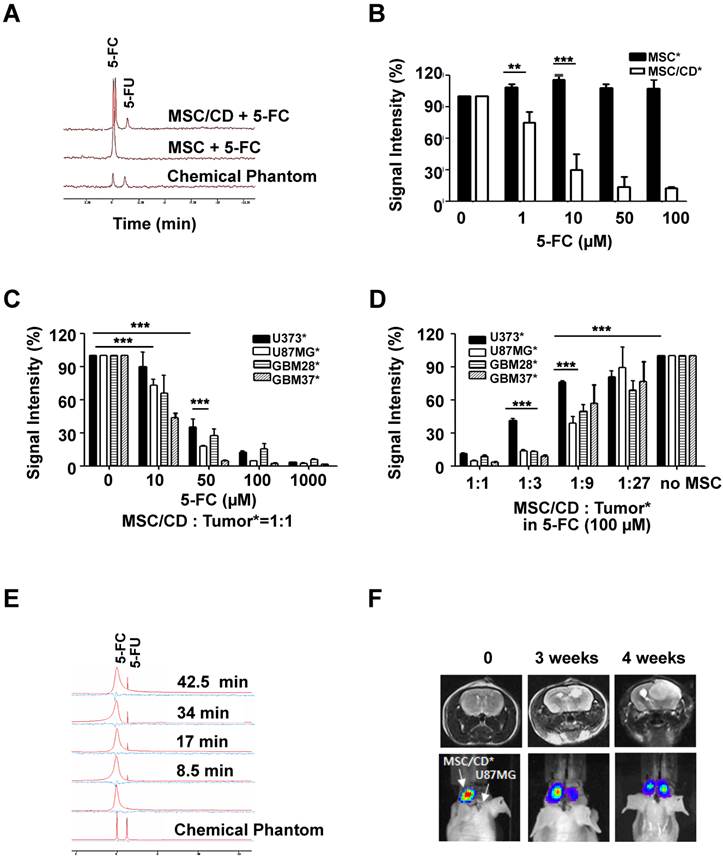
Next, we evaluated the suicide effect of the MSC/CD cells after treatment with prodrug 5-FC. As shown in Figure 4B, the viability of MSC/CD cells decreased with increasing 5-FC concentration. In contrast, treatment with 5-FC had no suicide effect on naïve MSCs (without CD expression). This result confirms that prodrug 5-FC was cytotoxic only to CD-expressing cells.
To determine the bystander effect of MSC/CD, therapeutic MSC/CD cells were co-cultured with the same number of luciferase reporter-expressing glioma cells with different 5-FC concentrations. The viable glioma cells were measured by luminescence intensity. Figure 4C shows that the survival rate of glioma cells co-cultured with MSC/CDs decreased in a 5-FC concentration-dependent manner. At a concentration of 10 µM of 5-FC, DPD-deficient U87MG, GBM28, GBM37 showed significant reduction of their survival compared to U373. Moreover, the survival rate between DPD-high U373 cells (34.9%) and DPD-deficient U87MG cells (17.5%) was significantly different when they were exposed to 50 µM of 5-FC. At a concentration of 100 µM of 5-FC, less than 10% of all three DPD-deficient glioma cells survived in all of the gliomas. This result supports the conclusion that DPD-deficient U87MG is more susceptible to MSC/CD therapy than DPD-high U373.
To determine the optimal number of MSC/CDs needed for therapeutic efficacy, various ratios of MSC/CD to reporter-expressing glioma cells were co-cultured. The survival of glioma cells was monitored by luminescence (Figure S2). At a 1:1 ratio of MSC/CDs to glioma cells, MSC/CD therapy showed an anti-cancer effect on all glioma cells when 100 µM of 5-FC was used. At a 1:3 ratio of MSC/CDs to glioma cells, less than 20% of DPD-deficient glioma cells (U87MG, GBM28, and GBM37) survived. On the other hand, U373 cells showed 41% survival. At a 1:9 ratio of MSC/CDs to glioma cells, half of the DPD-deficient U87MG cells were killed when 100 µM of 5-FC treatment was used (Figure 4D). As expected, co-cultured MSC/CDs with various glioma cells showed a variable degree of therapeutic effect dependent on the level of DPD expression. DPD-deficient U87MG cells were most sensitive to MSC/CD therapy with 5-FC treatment.
Conversion of 5-FC to-5-FU in the in vivo mouse model was also confirmed with 19F-MRS. The 5-FU peak gradually increased within 10 min following 500 mg/kg of 5-FC administration by intraperitoneal injection (Figure 4E). After U87MG cells had been transplanted into the right cranium of mice, luciferase-expressing MSC/CD cells were inoculated into the left side of the mice brain to test the tumor-targeting property of MSC/CDs. Four weeks later, 55.4% of MSC/CD cells had migrated to the tumor region (Figure 4F), indicating that MSC/CDs have the ability to target the tumor.
Therapeutic effect of MSC/CD and 5-FC on the orthotopic glioma model
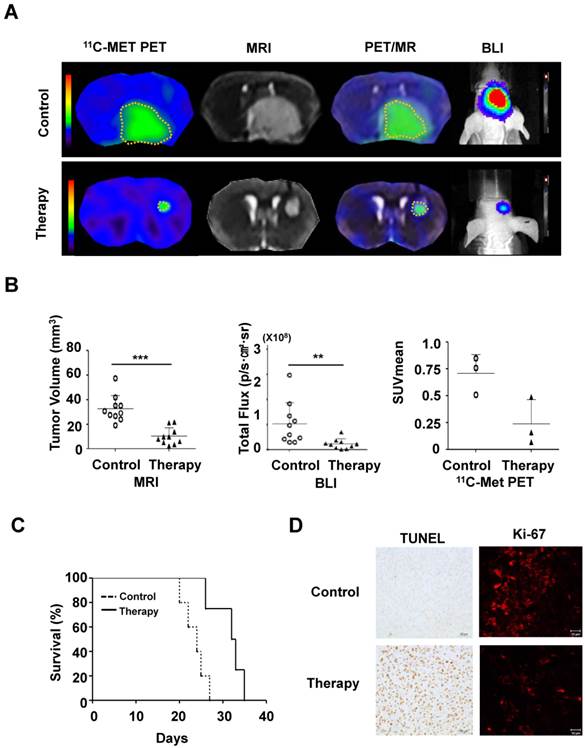
To evaluate the therapeutic effect of MSC/CD with 5-FC on pre-existing glioma, we transplanted MSC/CD cells 4 days after U87 tumor cell inoculation. As visualized with PET (11C-MET), MRI, and BLI imaging, the growth of glioma was dramatically suppressed by MSC/CD with 5-FC therapy (Figure 5A). MRI and PET fused images provided a combined biological and anatomical picture of the glioma. Tumor development was measured by changes in tumor volume, bioluminescent intensity (total flux) and Standardized Uptake Value (SUVmean). The results showed that MSC/CD with 5-FC therapy successfully suppressed tumor growth (Figure 5B). The size of the tumors in the control group gradually increased after 3 weeks, but tumor growth in MSC/CD therapy group was dramatically suppressed until after 4 weeks (Figure S3). Kaplan-Meier survival analysis revealed prolonged survival in the MSC/CD therapy group compared with the control group (Figure 5C). Histological analysis of brain tissue showed that the proliferation, as represented by Ki-67 tumor marker, was greatly increased in the control group compared to the MSC/CD therapy group. TUNEL assay revealed that more apoptosis had occurred in the MSC/CD therapy group compared to the control group (Figure 5D). Taken together, these results indicated that MSC/CD with 5-FC therapy had effectively suppressed the U87MG tumor growth.
Therapeutic effect of MSC/CDs with 5-FC on the glioma orthotopic model: (A) Tumor growth was visualized following therapy using MSC/CDs with 5-FC and monitored by PET (11C-MET), MRI and BLI. (B) Quantification of tumor volume (MRI), bioluminescent intensity (BLI) and SUV mean (PET) for each group, N=10 (C) Kaplan-Meier survival curves of mice in control and therapy groups. (D) Immunohistochemistry images of TUNEL (apoptosis) and Ki67 (proliferation) staining of each group (**, P<0.01; ***, P<0.001; N=3).
Discussion
GBM belongs to a group of the most malignant brain tumors. Standard therapy for malignant gliomas includes radiation and chemotherapy with the alkylating/methylating agent TMZ following surgical resection. The therapeutic efficacy depends on radiation sensitivity and o-6-methyguanine-DNA methyltransferase (MGMT) gene expression in the target gliomas. However, inter-patient variability is not generally considered for glioma treatment. Most importantly, there is no other treatment option for recurrent gliomas. Primary factors contributing to the failure of treatment are the invasive nature of the tumor, inefficient drug delivery, and the dose-limiting toxicity of treatment. Development of a more effective strategy is therefore desired to improve drug delivery and to reduce toxicity.
In an effort to develop new strategies for recurrent gliomas, we performed microarray analysis of GBMs following the conventional chemo-radiation therapy. Our analysis showed increased gene expression related to DNA damage repair, suggesting a requirement of DNA-targeting strategy for recurrent gliomas. Based upon these observations, we proposed stem cell-mediated enzyme/prodrug therapy for gliomas which would be more efficient in a subset of tumors with enhanced DNA damage repair capability. Our proposed approach would also overcome the delivery and toxicity issues associated with the conventional treatment of gliomas.
Our strategy is based upon the conversion of non-toxic 5-FC to cytotoxic 5-FU by the CD- expressing MSCs. Since it was first synthesized in 1957, 5-FU has become one of the most frequently used first-line drugs in the treatment of various cancers. Even though the enzymatic conversion of 5-FU from 5-FC and its metabolites are well-known, the actual mechanism(s) of 5-FU cellular toxicity remain poorly understood. It has been suggested that 5-FU inhibits thymidylate synthase (TS) to induce the stalling of the replication fork and increased mis-incorporation of nucleotides. Subsequently, excision of mis-incorporated nucleotides from DNA takes place leading to DNA breaks. Among the 5-FU metabolism-related enzymes, DPD and TP are recognized as important [32]. Our data showed that the expression of DPD, which depletes 5-FU by metabolizing 5-FU to α-fluoro-β-alanine (F-β-Ala), was inversely related to 5-FU toxicity (Figure 1 & 2). We tested 15 different glioma cells, 10 from patients and 5 from established glioma cell lines, by real-time PCR to evaluate the level of DPD expression. Though the viability of all glioma cells decreased by 5-FU in a concentration-dependent manner, 5-FU was shown to be more toxic to DPD-deficient U87MG than to other glioma cells. On the contrary, U373 cells with high DPD expression showed the least therapeutic response to MSC/CD prodrug therapy.
Over the last few decades, the cell-based approach to cancer therapy has been extensively studied using therapeutic genes such as HSV-TK, TRAIL, and others [21-24]. However, in their clinical application, one of the problems of these strategies is that the genetically-manipulated cells remain in the patient after therapy. In this study, we showed that MSC/CDs mediate the cytotoxicity via 5-FC conversion to FU to influence not only the MSC/CDs themselves (i.e. the suicide effect) but also the surrounding glioma cells through the bystander effect (Figure 4B & 4D). This indicates that the risk of using genetically-manipulated cells in clinical applications can be avoided by using cytosine deaminase, as the therapeutic cells could otherwise be depleted by the suicide effect. For safety issues, our previous study demonstrated that retroviral-infected MSCs maintain their original properties as well as genetic stability [17]. In addition, we also monitored tumorigenesis of retroviral-infected MSC cells (MSC/CD). During 24 weeks, there was no sign of tumorigenesis from MSC/CDs (Figure S4).
Our therapeutic strategy is to inhibit tumor recurrence after conventional therapy. Implanted MSC/CDs would localize to residual and invasive brain tumor foci after the tumor resection and convert prodrug 5-FC to 5-FU at the tumor site. Thus, the undesirable side effect can be minimized. Besides the observation of neural progenitor cell migration to tumor lesion across the corpus callous [36], there is growing evidence that MSCs can migrate to the tumor and the injury site [25-27]. Molecular factors behind MSC migration to the tumor have been revealed as the stromal cell derived factor-1(SDF-1), the platelet-derived growth factor-AB (PDGF-AB), members of the CCR family, and others [28-31]. Since the tumor environment provides sufficient chemo-attractants, tumor tropism of MSC/CDs was clearly observed in an orhtotopic mouse model (Figure 4F). Because 5-FC is a promising prodrug with limited toxicity only in the CD expressing cells, our therapeutic MSC/CDs, which have effective tumor-targeting ability, could greatly reduce normal brain damage.
Though MSC/CD with prodrug therapy has a great potential in the treatment of gliomas, the treatment is very costly, and there is also a possible risk of side effects or complications. Furthermore, to establish more effective therapeutic strategies, it is important to identify those glioma patients who may possibly benefit from a particular treatment. For example, identification of potential prognostic markers for MSC/CD with 5-FC therapy would be critical for selecting glioma patients who may benefit from this treatment strategy. Our results have identified DPD as a potential biomarker to predict the therapeutic efficacy of MSC/CD with 5-FC therapy. Considering the ability of DPD to deplete 5-FU, it is reasonable to expect that DPD-deficient tumors would be more sensitive to the MSC/CD with 5-FC therapy and patients with DPD-deficient gliomas may show a better response to MSC/CD therapy with 5-FC. To select patients who would possibly benefit from MSC/CDs with 5-FC treatment, the DPD levels should be measured with real-time PCR following the surgical biopsy. Further optimization of the treatment regimen, for instance, 5-FC dose or frequency of treatment with the therapeutic MSCs and 5-FC, could enhance the therapeutic effect.
Conclusion
In this study, we proposed MSC-mediated prodrug therapy to target DNA damage for the treatment of recurrent gliomas after conventional therapy. We showed that therapeutic MSC/CD cells have the ability to convert prodrug 5-FC to toxic 5-FU and successfully target gliomas to suppress tumor growth. Since the higher therapeutic efficacy of MSC/CDs with 5-FC treatment was observed in DPD-deficient gliomas, DPD can be a useful biomarker to select the group of patients who may potentially benefit from MSC/CD with 5-FC therapy.
Materials and Methods
Cells
Human glioma cell lines (U373, U87MG, T98, U251, and U138) were purchased from ATCC (American Type Culture Collection, Manassas, VA, USA) and Sigma-Aldrich Korea (Youngin, Korea). The glioma cells were maintained in Minimum Essential Medium (Welgene, Worcester, MA, USA) containing 10% fetal bovine serum and 1% antibiotic-antimycotic solution (Gibco, Gran Island, NY, USA). Primary glioma cells (GBM1, GBM13, GBM28, GBM37, GBM67, GBM77, GBM105, GBM121, GBM170, and GBM211) were obtained from patients after surgery and expanded with the approval of the Institutional Review Board of Seoul National University Hospital (IRB No. 1009-025-331). Primary glioma cells were maintained in Dulbecco's modified Eagle's medium (Welgene, Worcester, MA, USA) containing 10% fetal bovine serum and 1% antibiotic-antimycotic solution (Gibco, Grand Island, NY, USA). All cells were incubated at 37℃ in a humidified atmosphere containing 5% CO2.
For microarray analysis, U373 and U87MG cells received TMZ in a dose of 50 mM for 2 hours and were exposed to 2Gy radiation using a 137Cs irradiator at 4 Gy/min (CIS Bio International, Cedex, France). GBM28 cells were isolated from a surgical biopsy from a patient, and GBM37 cells were isolated from recurrent glioma from the same patient after standard therapy of TMZ (150mg/ m2/d, oral for 5d, 4 week cycle) and gamma knife radiation (1.8Gy, 5d/w for 7 weeks).
Primary human MSCs obtained from the iliac crest bone marrow of healthy donors were isolated with the approval of the Institutional Review Board of Ajou University Medical Center (IRB No. 00002500) and cultured as previously reported [16, 17]. The CD gene from E.coli K12 MG1655 (KRIBB, Daejon, Korea) [17] was stably transfected with the retroviral vector into the human MSCs. The transfected cells were cultured in Dulbecco's modified Eagle's medium (Welgene, Worcester, MA, USA) containing 10% fetal bovine serum and 1% antibiotic-antimycotic solution (Gibco, Grand Island, NY, USA). All experiments were performed at cell passage <9.
For optical imaging, glioma cells and MSCs were stably transfected with pMSCV-Luc-IRES-eGFP (luciferase bioluminescence/green fluorescence) or pMSCV-Luc-IRES-tdTomato (luciferase bioluminescence/red fluorescence) reporter constructs.
Microarray analysis
Total RNA was extracted form glioma cells (U8MG, U373, GBM28, and GBM37) with TRIzol reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's protocol. The transcriptional profiles of glioma cells were characterized by oligonucleotide microarray analysis, using MA-human Agilent 44K (Agilent Technology, Santa Clara, CA, USA). Global median normalization was performed to analyze one channel microarray data by GeneSpring GX 7.3 (EBIOGEN, Seoul, Korea) [33].
To define DNA expression patterns of glioma cells, four glioma cells (U373, U87MG, GBM28, and GBM37) were clustered together by DNA repair-related genes and 5-FU metabolism-related genes such as dihydropyrimidine dehydrogenase (DPD), thymidine phosphorylase (TP), and thymidylate synthase (TS) using a TIGR MultiExperiment viewer (Mev4.8.1, http://tm4.org/mev/). A heat-map represented the expression levels of genes in 4 different glioma cells. Hierarchical clustering was performed grouping clusters according to their similarities in gene function representation using the MeV software program. Each cluster was determined by hierarchical clustering algorithms with Pearson correlation as a similarity measurement for each cluster.
Bioluminescence imaging
Luciferase-expressing glioma cells (U87MG*, U373*, GBM28*, and GBM37*) and MSC (MSC* and MSC/CD*) cells were plated in a 24-well plate at concentrations ranging from 0.9 to 30 x 104 cells. After addition of D-luciferin (30 µg, Caliper Life Sciences, Hopkinton, MA, USA) to the media, bioluminescence images were captured by IVIS 100 system (Caliper Life Sciences, Hopkinton, MA, USA). Correlations between cell numbers per well and bioluminescence intensity were measured by Living Image, version 2.50.1 (Xenogen, Alameda, CA) and IGOR, version 4.09A (WaveMetrics, Portland, OR) image analysis software.
Confocal microscopy
GFP expression of glioma cells and tdTomato expression of MSCs were detected by confocal microscopy using an LSM510 META confocal microscope (Carl Zeiss Inc., Oberkochen, Germany) with a 40x magnification. Excitation light was generated by diode and a helium-neon laser. Cells were fixed for 10 min with 3.7% paraformaldehyde (USB, Cleveland, OH, USA), washed with PBS, and mounted with ProLong Gold (Invitrogen, Carlsbad, CA, USA) anti-fading reagent.
5-FU cytotoxicity assay
Glioma cells were seeded in a 96-well plate at a concentration of 5000 cells/well. After 24 hours, the medium was replaced with fresh medium with or without 5-FU (0, 1, 4, 8 μM). The cells were further incubated for 72 hours following which 5-FU toxicity was examined using a Cell Counting Kit (CCK-8, Dojindo Molecular Technologies, Kumamoto, Japan) according to the manufacturer's protocol. Pre-warmed CCK-8 solution (10μl) was added to each well and incubated for 4 hours. The absorbance at 450 nm was measured using a microplate reader. Six wells were counted for each 5-FU concentration (N=3).
RT-PCR and Real-Time PCR
To determine the mRNA expression of CD and 5-FU metabolism-related genes in the cells, RT-PCR and real-time PCR were performed. Total RNA was extracted with TRIzol reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's protocol. cDNA was synthesized using 1 µg of total RNA and a cDNA synthesis master kit (Genedepot, Barker, TX, USA). PCR conditions were: 30 sec at 95℃ for denaturation, 40 sec at 58℃ for primer annealing, and 30 sec at 72℃ for primer extension. Primer pairs for RT-PCR were as follows: (CD forward - CAGTTGATGGGCTACGGG; CD reverse - TGGCTTCTGGCTGCTCC; DPD forward AGGACGCAAGGAGGGTTTG; DPD reverse GTCCGCCGAGTCCTTACTGA; GAPDH forward-TGAAGGTCGGAGTCAACGGATTTGGT; GAPDH reverse - CATGTGGGCCATGAGGTCCACCAC; β-actin forward TGACGGGGTCACCCAACTGTGCCCATCTA; β-actin reverse CTAGAAGCATTTGCGGTGGACGATGGAGGG). The PCR products were analyzed by 1.2 % agarose gel electrophoresis. Gene expression of DPD was measured by quantitative Real-time PCR using a 7700 ABI PRISM sequence detector system (Applied Biosystems, Foster City, CA, USA). PCR primers and fluorogenic probes for the DPD gene (Hs00559279_ml) and endogenous controls (Hs99999905_ml) were purchased as Assays-On-Demand (Applied Biosystems, Foster City, CA, USA).
Regulation of DPD expression
To reduce DPD expression, non-targeting control siRNA and DPD-targeted siRNA were purchased from Santa Cruz Biotechnology (Santa Cruz, Dallas, TX, USA). The U373 cells (1x104 /well in 12-well plates) were transfected with the 5nM siRNA for control (siCtr) or DPD (siDPD) using lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA). After incubating for 72 hours, cells were washed and used for subsequent experiments.
For over-expression of DPD, pCMV6-DPD-GFP clones were purchased from Origene (Rockville, MD, USA). The U87MG cells (5x104 /well for 12-well plates) were cultured overnight and then transfected with 0.5 μg of vectors with lipofectamine 2000. Cells were washed after 48 hours of incubation and used for subsequent experiments.
Immunoblot assay
Cells were lysed in RIPA buffer (Sigma-Aldrich, St Louis, MI, USA) with protease inhibitors (Qiagen, Hilden, Germany). Proteins (20 µg) were separated by SDS-PAGE gel electrophoresis and transferred to polyvinylidene difluoride membrane (Millipore, Billeroca, MA, USA). The membranes were incubated with antibodies specific to CD (obtained from Dr. Suh-Kim, Aju University, Korea), DPD (Cell Signaling, Beverly, MA, USA), and β-actin (Sigma-Aldrich, St Louis, MO, USA). Membranes were then treated with HRP conjugated anti-rabbit (Cell Signaling, Beverly, MA, USA) or anti-mouse (Invitrogen, Carlsbad, CA, USA) secondary antibody. The specific proteins were visualized by chemo-luminescence (Roche, Indianapolis, IN, USA) according to the manufacturer's protocol and detected with LAS-3000 (Fuji Film, Stockholm, Sweden).
FACS analysis
Naïve MSCs and gene-manipulated MSCs (MSC/CD, MSC/CD*) were washed with 2% FBS/phosphate-buffered saline (PBS) and incubated at 4℃ for 30 minutes with 5 µl of monoclonal antibody specific for CD 29, CD44, CD105, CD34, and CD45 (BD Biosciences, San Jose, CA, USA). Unstained cells were used as controls. FACS Canto II (BD Biosciences, San Jose, CA, USA) was used for cytometry analysis.
5-FC cytotoxicity assay
To validate the suicide effect of MSC/CD with 5-FC, luciferase-expressing MSC* and MSC/CD* cells were seeded at a density of 1x104 cells/well in 24 well plates with increasing concentrations of 5-FC (Kolon Life Science, Gyeonggi, Korea).
To validate the bystander effect, luciferase-expressing glioma cells (U373*, U87MG*, GBM28*, and GBM37*) were seeded in 24 well plates at a density of 1x104 cells/well. Same number of MSC/CDs (1x104 cells/well) were co-cultured in the media with increasing doses of 5-FC (0, 10, 50, 100, 100 nM).
To evaluate the effect of the tumor to MSC/CDs ratio, 1x104 cells/well of glioma cells were mixed with different numbers of MSC/CDs (1, 1/3, 1/9, 1/27 of 1x104 cells) and each glioma-MSC/CD mixture was seeded in a 24 well plate. 5-FC was added to a final concentration of 100 µM.
To measure the number of viable cells, 30 µg of D-Luciferin (Caliper Life Sciences, Hopkinton, MA, USA) was added to each well. Bioluminescence signal from viable cells was acquired for 5 seconds with IVIS 100 system (Caliper Life Sciences, Hopkinton, MA, USA) and analyzed with Living Image, version 2.50.1 (Xenogen, Alameda, CA) software.
19F-MRS measurement
For in vitro 19F-MRS experiments, MSC and MSC/CD cells (1 x 105) were grown in 6 well plates. Cells were then incubated in growth medium containing 100 μM of 5-FC (Kolon Life Science, Gyeonggi, Korea). After 24 hours, conditioned media were collected and analyzed using 9T (375.567 MHz) Agilent NMR spectrometer (Agilent Technologies, Santa Clara, CA, USA). Chemical phantom samples were constructed by diluting 5-FC (Kolon Life Science, Gyeonggi, Korea) and 5-FU (Sigma-Aldrich, MI, USA) to 200 uM each in 5ml of DMEM with 10% FBS. Spectra were acquired with a single pulse sequence whole region excitation with 11 min scan time (average 512). The ratio of peak intensities was measured using VnmrJ 3.2 version software program (Agilent Technologies, Santa Clara, CA, USA).
For in vivo 19F-MRS experiments, MSC/CD cells (1x106) were subcutaneously injected into the right flank of mice. Serial 19F-NMR spectra were acquired every 8.5 min during 0 to 1.5 h (total repetition time, 500 ms; the number of averages, 1024; spectral width, 25 kHz; acquisition size, 2048 points) after administration of 5-FC (500 mg/kg, intraperitoneally). The chemical shift of the 5-FC resonance was set to 0 ppm and the 5-FU signal was observed at 1.2 ppm. The signal positions were verified with phantom samples of 5-FC and 5-FU in the culture medium which were described above.
Animal modeling
BALB/c nude mice (male, 6 to 8 weeks old) were used in accordance with the Institutional Animal Care and Use Committee of Seoul National University Hospital guidelines (IACUC No.; 10-0212). Mice were anesthetized with 1.5% isoflurane gas at a flow rate of 1.5L/min.
The intracranial tumor model in mouse brain was constructed as described previously [16]. U87MG cells (3x105 in 4 μl PBS) were transplanted into the right striatum (anteroposterior, +0.5; mediolateral, +1.8; dorsoventral, 3.0) with a Hamilton syringe (10 μl) at a rate of 0.25 ul/min using a stereotactic device (Stoelting, IL, USA).
To evaluate the tumor tropism, MSC/CD* cells (3x105 in 4 μl PBS) were inoculated into the left striatum 1 week after tumor inoculation. Migration of MSC/CD* cells was monitored once a week with the bioluminescence imaging (BLI) and the tumor was monitored with MRI.
To evaluate the therapeutic effect of MSC/CD and 5-FC, MSC/CD cells (3x105) were inoculated into the tumor lesion (in right striatum, intracranial tumor model described above) 4 days after U87MG inoculation. 5-FC (500 mg/kg) was injected intraperitoneally for 5 times a week. For the control group, only 5-FC was administrated in the tumor-bearing mice without MSC/CD inoculation (N=10 each, control and treatment group). The tumor growth of the orthotopic model was monitored and evaluated using BLI, MRI, and PET imaging.
Animal bioluminescence imaging
Before image acquisition, 3 mg of D-luciferin (Caliper Life Sciences, Hopkinton, MA, USA) was administrated by intraperitoneal injection (100 μl per mouse, 30 mg/ml with saline stock). Bioluminescence signals were collected at 10-30 min with maximum intensity using IVIS100 (Caliper Life Sciences, Hopkinton, MA, USA). The signals emitted from the animals were presented as pseudo-color spectra, ranging from red (maximum) to blue (minimum) based on their intensity. Gray-scale photographs of the mouse and corresponding pseudo-color images were superimposed with Living Image, version 2.12 (Xenogen, Alameda, CA, USA) and IGOR, version 1.24 (WaveMetrics, Portland, OR, USA) image analysis software. Signals emitted by regions of interest (ROIs) were measured and expressed as photon flux [photons/sec/cm2/sr], which refers to the photons emitted from a unit solid angle of a sphere. The background signal intensity was subtracted electronically for normalization both from images and from the measurements of photon flux.
Animal PET/CT
A small animal PET/CT scanner eXplore Vista-CT (GE Healthcare, Buckinghamshire, United Kingdom) was used for image acquisition. 11C-Methionine (11C-MET) was purchased from Seoul National University Cancer Hospital (Seoul, Korea) and injected intravenously at a dose ranging from 14 to 22.2 MBq per mice. Glioma images with 11C-MET as an amino acid tracer showed high sensitivity and specificity [34]. Static images were acquired for 10 min after tracer injection and CT was also performed for attenuation correction. Acquired images were reconstructed using an ordered subset expectation maximization (OSEM) algorithm. The value of standard uptake (SUVmean) was used to evaluate tumor growth [35] by the eXplore Vista/CT software program (GE Healthcare, Buckinghamshire, United Kingdom).
Animal MRI
MRIs were acquired by SIMENS 3-tesler scanner with animal 6 channel coil (SIMENS, Berlin, Germany). T2-weighted turbo spin-echo sequences were collected for each mouse (TR = 3000 ms; TE = 100 ms; FOV = 35 x 35 mm2; thickness = 0.8 mm). These images were used to calculate tumor volume using Osirix 5.5 version software program (http://www.osirix-viewer.com/). The margins of the tumor were drawn in MRI images. The longest and shortest axes were measured to calculate area, and this number was multiplied by the thickness (0.8 mm) of the slides. The volume of each slice was added to generate total tumor volume. We fused the PET images with MR images using the OsiriX image processing software program.
Immunohistochemistry
Glioma-bearing mice were sacrificed 4 weeks after tumor cell inoculation for histologic analysis. The brain was fixed in 10% formalin for 48 hours and embedded in paraffin. Tissue slices were stained with hematoxylin and eosin (H&E), Ki-67 (Invitrogen, Carlsbad, CA, USA) for cell proliferation, and terminal deoxyribonucleotidyl transferase-mediated dUTP nick end labeling (TUNEL assay kit, Millipore, Billeroca, MA, USA) for apoptotic activity.
Statistical Analysis
All data were expressed as means ± standard deviation (SD). Statistically significant differences of all results were determined using the Student t test. Differences were considered statistically significant when P was <0.05 (*, P<0.05; **, P<0.01; ***, P<0.001).
Supplementary Material
Supplementary figures.
Abbreviations
MSC: mesenchymal stem cells; CD: cytosine deaminase; DPD: Dihydropyrimidine dehydrogenase; TP: thymidine phosphorylase; TS: thymidylate synthase; 5-FC: 5-Fluorocytosine; 5-FU: 5-Fluorouracil; FUH2: 5-fluoro-5,6-dihydrouracil; F-β-Ala: α-fluoro-β-alanine; FUdR: 5-fluorodeoxyuridine; FURP: 5-fluorouridine; FdUMP: 5-fluorodeoxyuridine monophosphate; FUMP: 5-fluorouridine monophosphate; FdUDP: 5-fluorodeoxyuridine diphosphate; FUDP: 5-fluorouridine diphosphate; FdUTP: Fluoro-deoxyuridine triphosphate; FUTP: 5-fluorouridine triphosphate; dTMP: deoxythymidine monophosphate; dTDP: deoxythymidine diphosphate; dTTP: deoxythymidine triphosphate.
Acknowledgements
This work was supported by grants from the Cancer Research Institute of Seoul National University, Seoul National University Hospital (No. 0420111150), the Korea Health Technology R & D Project through the Korea Health Industry Development Institute (KHIDI) funded by the Korea Ministry of Health & Welfare (A101446, HI14C1072), and the National Research Foundation of Korea (NRF) grant for the Global Core Research Center (GCRC) funded by the Korea MSIP (Ministry of Science, ICT & Future Planning) (No. NRF-2011-0030001).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Wen PY, Kesari S. Malignant gliomas in adults. N Engl J Med. 2008;359:492-507
2. Westphal M, Lamszus K. The neurobiology of gliomas: from cell biology to the development of therapeutic approaches. Nat Rev Neurosci. 2011;12:495-508
3. Galldiks N, Ullrich R, Schroeter M. et al. Imaging biological activity of a glioblastoma treated with an individual patient-tailored, experimental therapy regimen. J Neurooncol. 2009;93:425-30
4. Viel T, Talasila KM, Monfared P. et al. Analysis of the growth dynamics of angiogenesis-dependent and -independent experimental glioblastomas by multimodal small-animal PET and MRI. J Nucl Med. 2012;53:1135-45
5. Neuwelt EA, Bauer B, Fahlke C. et al. Engaging neuroscience to advance translational research in brain barrier biology. Nat Rev Neurosci. 2011;12:169-82
6. Norden AD, Young GS, Setayesh K. et al. Bevacizumab for recurrent malignant gliomas: efficacy, toxicity, and patterns of recurrence. Neurology. 2008;70:779-87
7. Greco O, Dachs GU. Gene directed enzyme/prodrug therapy of cancer: historical appraisal and future prospectives. J Cell Physiol. 2001;187:22-36
8. Okura H, Smith CA, Rutka JT. Gene therapy for malignant glioma. Mol Cell Ther. 2014;2:21
9. Rainov NG. A phase III clinical evaluation of herpes simplex virus type 1 thymidine kinase and ganciclovir gene therapy as an adjuvant to surgical resection and radiation in adults with previously untreated glioblastoma multiforme. Hum Gene Ther. 2000;11:2389-401
10. Trinh QT, Austin EA, Murray DM. et al. Enzyme/prodrug gene therapy: comparison of cytosine deaminase/5-fluorocytosine versus thymidine kinase/ganciclovir enzyme/prodrug systems in a human colorectal carcinoma cell line. Cancer Res. 1995;55:4808-12
11. Ramnaraine M, Pan W, Goblirsch M. et al. Direct and bystander killing of sarcomas by novel cytosine deaminase fusion gene. Cancer Res. 2003;63:6847-54
12. Parekkadan B, Milwid JM. Mesenchymal stem cells as therapeutics. Annu Rev Biomed Eng. 2010;12:87-117
13. Corsten MF, Shah K. Therapeutic stem-cells for cancer treatment: hopes and hurdles in tactical warfare. Lancet Oncol. 2008;9:376-84
14. Aboody KS, Brown A, Rainov NG. et al. Neural stem cells display extensive tropism for pathology in adult brain. Proc Natl Acad Sci U S A. 2000;97:12846-51
15. Aboody KS, Najbauer J, Metz MZ. et al. Neural stem cell-mediated enzyme/prodrug therapy for glioma. Sci Transl Med. 2013;5:184ra59
16. Chang DY, Yoo SW, Hong Y. et al. The growth of brain tumors can be suppressed by multiple transplantation of mesenchymal stem cells expressing cytosine deaminase. Int J Cancer. 2010;127:1975-83
17. Park JS, Chang DY, Kim JH. et al. Retrovirus-mediated transduction of a cytosine deaminase gene preserves the stemness of mesenchymal stem cells. Exp Mol Med. 2013;45:e10
18. Koppula PR, Chelluri LK, Polisetti N. et al. Histocompatibility testing of cultivated human bone marrow stromal cells - a promising step towards pre-clinical screening for allogeneic stem cell therapy. Cell Immunol. 2009;259:61-5
19. van Rijn J, Heimans JJ, van den Berg J. et al. Survival of human glioma cells treated with various combination of temozolomide and X-rays. Int J Radiat Oncol Biol Phys. 2000;47:779-84
20. Barazzuol L, Jena R, Burnet NG. et al. In vitro evaluation of combined temozolomide and radiotherapy using X rays and high-linear energy transfer radiation for glioblastoma. Radiat Res. 2012;177:651-62
21. Sampath D, Rao VA, Plunkett W. Mechanisms of apoptosis induction by nucleoside analogs. Oncogene. 2003;22:9063-74
22. Mesnil M, Yamasaki H. Bystander effect in herpes simplex virus-thymidine kinase/ganciclovir cancer gene therapy: role of gap-junctional intercellular communication. Cancer Res. 2000;60:3989-99
23. Gambhir SS, Barrio JR, Phelps ME. et al. Imaging adenoviral-directed reporter gene expression in living animals with positron emission tomography. Proc Natl Acad Sci U S A. 1999;96:2333-8
24. Kim SM, Woo JS, Jeong CH. et al. Effective combination therapy for malignant glioma with TRAIL-secreting mesenchymal stem cells and lipoxygenase inhibitor MK886. Cancer Res. 2012;72:4807-17
25. Sasportas LS, Kasmieh R, Wakimoto H. et al. Assessment of therapeutic efficacy and fate of engineered human mesenchymal stem cells for cancer therapy. Proc Natl Acad Sci U S A. 2009;106:4822-7
26. Ji JF, He BP, Dheen ST. et al. Interactions of chemokines and chemokine receptors mediate the migration of mesenchymal stem cells to the impaired site in the brain after hypoglossal nerve injury. Stem Cells. 2004;22:415-27
27. Klopp AH, Spaeth EL, Dembinski JL. et al. Tumor irradiation increases the recruitment of circulating mesenchymal stem cells into the tumor microenvironment. Cancer Res. 2007;67:11687-95
28. Satake K, Lou J, Lenke LG. Migration of mesenchymal stem cells through cerebrospinal fluid into injured spinal cord tissue. Spine. 2004;29:1971-9
29. Studeny M, Marini FC, Champlin RE. et al. Bone marrow-derived mesenchymal stem cells as vehicles for interferon-beta delivery into tumors. Cancer Res. 2002;62:3603-8
30. Gao H, Priebe W, Glod J. et al. Activation of signal transducers and activators of transcription 3 and focal adhesion kinase by stromal cell-derived factor 1 is required for migration of human mesenchymal stem cells in response to tumor cell-conditioned medium. Stem Cells. 2009;27:857-65
31. Ponte AL, Marais E, Gallay N. et al. The in vitro migration capacity of human bone marrow mesenchymal stem cells: comparison of chemokine and growth factor chemotactic activities. Stem Cells. 2007;25:1737-45
32. Ando T, Ishiguro H, Kuwabara Y. et al. Relationship between expression of 5-fluorouracil metabolic enzymes and 5-fluorouracil sensitivity in esophageal carcinoma cell lines. Dis. Esophagus. 2008;21:15-20
33. Yang YH, Dudoit S, Luu P. et al. Normalization for cDNA microarray data: a robust composite method addressing single and multiple slide systematic variation. Nucleic Acids Res. 2002;30:e15
34. Nariai T, Tanaka Y, Wakimoto H. et al. Usefulness of L-[methyl-11C] methionine-positron emission tomography as a biological monitoring tool in the treatment of glioma. J Neurosurg. 2005;103:498-507
35. Lucignani G. SUV and segmentation: pressing challenges in tumour assessment and treatment. Eur J Nucl Med Mol Imaging. 2009;36:715-20
36. Tang Y, Shah K, Messerli SM, Snyder E. et al. In vivo tracking of neural progenitor cell migration to glioblastoma. Gene Ther. 2003;14:1247-1254
Author contact
![]() Corresponding author: Hyewon Youn, Ph.D. Cancer Imaging Center, Seoul National University Hospital, #207-1, Samsung Cancer Research Building, 103 Daehak-ro, Jongno-gu, Seoul 110-799, Korea E-mail: hwyounac.kr; Tel: +82-2-3668-7026; Fax : +82-2-766-4477. Or June-Key Chung, M.D., Ph.D. Department of Biomedical Sciences, Seoul National University College of Medicine, 101 Daehak-ro, Jongno-gu, Seoul 110-744, Korea. E-mail: jkchungac.kr; Tel:+82-2-2072-2706; Fax : +82-2-766-4477.
Corresponding author: Hyewon Youn, Ph.D. Cancer Imaging Center, Seoul National University Hospital, #207-1, Samsung Cancer Research Building, 103 Daehak-ro, Jongno-gu, Seoul 110-799, Korea E-mail: hwyounac.kr; Tel: +82-2-3668-7026; Fax : +82-2-766-4477. Or June-Key Chung, M.D., Ph.D. Department of Biomedical Sciences, Seoul National University College of Medicine, 101 Daehak-ro, Jongno-gu, Seoul 110-744, Korea. E-mail: jkchungac.kr; Tel:+82-2-2072-2706; Fax : +82-2-766-4477.