Impact Factor
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Results and Discussion
Conclusion
Material and Methods
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2016; 6(10):1506-1513. doi:10.7150/thno.15871 This issue Cite
Research Paper
Simple, Sensitive and Accurate Multiplex Detection of Clinically Important Melanoma DNA Mutations in Circulating Tumour DNA with SERS Nanotags
Eugene J.H. Wee1, #, ![]() , Yuling Wang1, #,
, Yuling Wang1, #, ![]() , Simon Chang-Hao Tsao3, 4, Matt Trau1, 2,
, Simon Chang-Hao Tsao3, 4, Matt Trau1, 2, ![]()
1. Center for Personalized Nanomedicine. Australian Institute for Bioengineering and Nanotechnology, The University of Queensland, Queensland, Australia
2. School of Chemistry and Molecular Biosciences, The University of Queensland, Queensland, Australia
3. Olivia Newton-John Cancer Research Institute, Heidelberg, Victoria, Australia
4. Department of Surgery, University of Melbourne, Austin Health. Heidelberg, Victoria, Australia
# Authors contributed equally
Received 2016-4-17; Accepted 2016-5-31; Published 2016-6-17
Abstract

Sensitive and accurate identification of specific DNA mutations can influence clinical decisions. However accurate diagnosis from limiting samples such as circulating tumour DNA (ctDNA) is challenging. Current approaches based on fluorescence such as quantitative PCR (qPCR) and more recently, droplet digital PCR (ddPCR) have limitations in multiplex detection, sensitivity and the need for expensive specialized equipment. Herein we describe an assay capitalizing on the multiplexing and sensitivity benefits of surface-enhanced Raman spectroscopy (SERS) with the simplicity of standard PCR to address the limitations of current approaches. This proof-of-concept method could reproducibly detect as few as 0.1% (10 copies, CV < 9%) of target sequences thus demonstrating the high sensitivity of the method. The method was then applied to specifically detect three important melanoma mutations in multiplex. Finally, the PCR/SERS assay was used to genotype cell lines and ctDNA from serum samples where results subsequently validated with ddPCR. With ddPCR-like sensitivity and accuracy yet at the convenience of standard PCR, we believe this multiplex PCR/SERS method could find wide applications in both diagnostics and research.
Keywords: SERS, multiplex PCR, melanoma, BRAF, cKIT, NRAS, ctDNA
Introduction
Aberrant DNA base changes are hallmarks of cancer. [1-3] However, sensitive, accurate and timely identification of some DNA mutations can influence clinical decisions thus potentially leading to better prognosis and in some cases, remission. Currently in practise, multiplexed PCR-based assays used to detect such mutations rely on probe-based quantitative strategies (qPCR) or some form of targeted sequencing.[4] More recently, droplet digital PCR have been demonstrated to accurately quantify mutant copies from limiting DNA input e.g. circulating tumour DNA (ctDNA).[5] While highly accurate, these fluorescence-based approaches require expensive specialized equipment, have limited sensitivity and multiplexing capability. As such, novel approaches to address the limitations of current gold-standards are still in demand.
Herein, we describe a novel multiplex PCR/SERS detection method using surface-enhanced Raman spectroscopy (SERS) nanotags to address the limitations of fluorescence-based detection. [6-15] SERS is a physical phenomenon that occurs on metal nanoparticles surfaces upon laser excitation resulting in enhanced Raman scattering that are characteristic of the adsorbed molecules.[6] With potentially higher sensitivity to fluorescence probes,[16] SERS is also better suited for multiplexing due to the benefits of unique and narrow spectral peaks. [6-15] This multiplexing advantage is potentially very useful for maximizing the use of limited samples, such as detecting multiple mutations in ctDNA. In addition, only a single laser source is needed for excitation hence a SERS-based assay could also potentially lead to a simpler, lower-cost instrumentation. Thus a SERS-based approach could be an alternative strategy to fluorescence for sensitive multiplexed detection of DNA.[17-22]. Currently most PCR-based SERS applications use labelled DNA probes that hybridize within PCR amplicons followed by salt induced nanoparticle aggregation to detect a positive amplification.[23-27] While effective, these approaches have not been demonstrated for translational applications in detecting single DNA base changes from limiting inputs (e.g. ctDNA) which requires exquisite specificity and sensitivity. Therefore, there is still an urgent need to develop novel multiplexed PCR/SERS detection strategies.
Our proposed multiplex PCR/SERS detection method was designed, as a proof-of-concept, for the sensitive and specific identification of 3 clinically important DNA point mutations in melanoma (BRAF V600E, c-Kit L576P and NRAS Q61K) which could potentially direct clinical decisions.[28-36] The combination of biochemical (PCR) and physical (SERS) amplification allowed the assay to detect as few as 10 mutant alleles from a background of 10,000 wild type sequences (0.1%). From less than 5 ng of genomic DNA, the assay could accurately genotype cell lines and serum-derived ctDNA of melanoma patients. Results were also validated by droplet digital PCR (ddPCR). In addition to having the convenience of a single-tube multiplex assay, this method is potentially as sensitive as ddPCR, has assay times comparable to current qPCR-based methods, while potentially achieving higher multiplexing than standard fluorescence-based strategies. We believe the translational potential of our method could have wide applications in both diagnostics and research.
Results and Discussion
Multiplex PCR/SERS assay
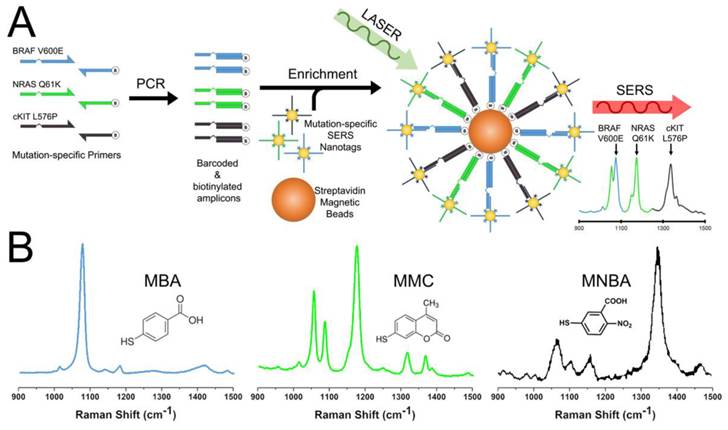
Melanoma lesions with single base changes resulting in the BRAF V600E mutation and c-Kit L576P mutations have potent corresponding targeted drug therapies that can significantly improve prognosis.[28-34] While NRAS mutations, such as Q61K, have novel drug therapies that are currently being evaluated in clinical trials.[35, 36] Hence, detecting such DNA anomalies in multiplex in clinic is extremely useful. To this end, as a proof-of-concept, we coupled SERS with end-point PCR for a simple 3-plex assay to simultaneously amplify DNA sequences that resulted in BRAF V600E (c.1799T>A), c-Kit L576P (c.1727T>C) and NRAS Q61K (c.181C>A) mutations (Figure 1A). Each forward primer had a 15 nt unique barcode sequence followed by an internal carbon spacer upstream of the mutation-specific sequence. This effectively created a 5' overhang after PCR as DNA polymerases cannot extend beyond the carbon spacer on the reverse strand. The reverse primers contained a 5'-biotin molecule which later allowed for convenient enrichment by streptavidin coated magnetic beads (SMB). If a mutation was present, the resulting PCR amplicon would have a biotin handle on one end and a 5' overhang barcode on the other. SERS-nanotags were gold nanoparticles (AuNPs) modified with Raman reporters and DNA probes that were complementary to barcodes sequences on the amplicons thus resulting in a SERS/DNA/biotin molecule. After enrichment with SMB to remove excess SERS nanotags, the identity of the remaining SERS nanotags was ascertained with a Raman spectrometer, which in turn, reflected the presence of a particular mutation. Raman reporters used in this study were 4-Mercaptobenzoic acid (MBA) for BRAF V600E at Raman shift of 1076 cm-1, 2,7-mercapto-4-methylcoumarin (MMC) for NRAS Q61K at 1174 cm-1 and 4-mercapto-3-nitrobenzoic acid (MNBA) for c-Kit L576P at 1334 cm-1 (Figure 1B). To the best of our knowledge, this is the first demonstration of a nanotag-based PCR/SERS assay for multiplexed detection of clinically important point mutations.
Conceptual schematic of the multiplex PCR/SERS assay and SERS nanotags. (A) Multiplex mutation specific primers were used to amplify tumour DNA. Amplicons were then tagged with mutation-specific SERS nanotags and enriched using magnetic beads. Mutation status was then evaluated using Raman spectroscopy where unique spectral peaks indicate the presence of the mutation of interest; (B) Molecular structure of Raman reporters and the corresponding SERS spectra of the SERS nanotags.
Specific multiplex assay
Amplification specificity is crucial for multiplexed point mutation detection. To this end, we challenged the 3-plex PCR assay with BRAF V600E, c-Kit L576P and NRAS Q61K synthetic targets individually and with a 3-plex mixture of SERS nanotags (Figure 2). The rational of this experiment was to demonstrate that when only specific products were amplified, as determined by the input target, only the corresponding SERS signal representing the target mutation would be detected. As expected, only a single SERS spectrum corresponding to the input was observed (MBA at 1076 cm-1 for BRAF V600E; MMC at 1174 cm-1 for NRAS Q61K and MNBA at 1334 cm-1 for c-Kit L576P).
Specific multiplex detection by SERS. Specific detection of individual mutant (MT) targets: (A) BRAF V600E, (B) cKIT L576P and (C) NRAS Q61K. (D) 3-plex detection of 3 mutant targets. (E) Negligible signal from wild type (WT) sequences. NoT (dotted lines) is the no template control. (F) Gel electrophoresis verification of specific amplification.
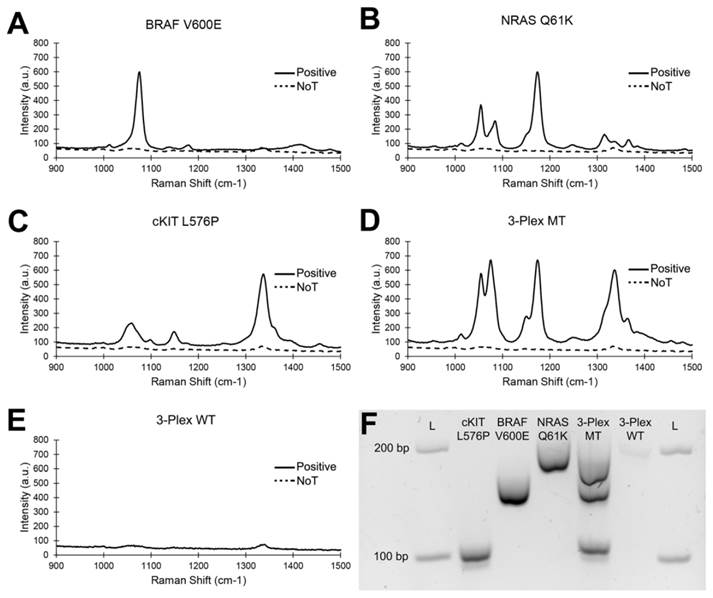
Clinically, BRAF, c-Kit and NRAS mutations are mutually exclusive, i.e., only in extremely rare cases will a tumour contain more than one mutation.[37-39] Nonetheless, to further demonstrate the specificity of the assay in a hypothetical “multi-mutant” tumour, a 1:1:1 mix of all three targets was challenged. As expected, only three distinct SERS spectra of similar intensity were observed. In contrast, no SERS signal was detected for wild type (WT) sequences, indicating that the assay was specific only to mutant sequences. Specific PCR amplification was also verified with standard gel electrophoresis, where the presence of amplicons associated with their respective SERS peaks. Similar results were also obtained with WT-specific primers where only WT sequences amplified (Fig 4). Together, the data indicated the high specificity and unbiased detection of target mutations in multiplex. To evaluate the potential interference/overlap between the 1063 cm-1 peak of MMC with the main 1076 cm-1 peak of MBA, we performed a deconvolution of the 3-plex spectra (Fig. S2). Due to highly similar profiles of the raw and deconvoluted spectra, we concluded that genotyping could be easily done without post-processing.
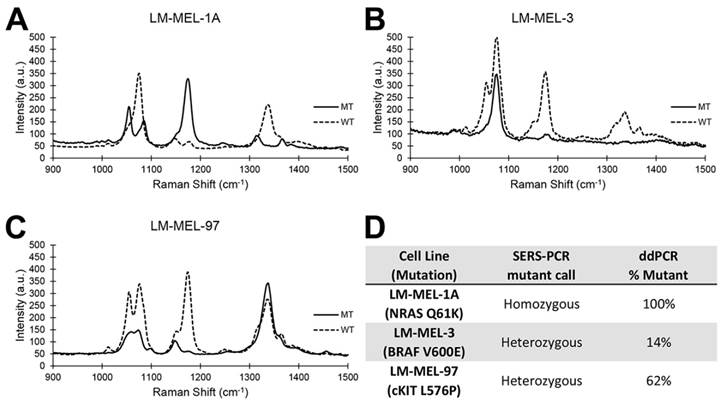
Accurate genotyping of melanoma cell lines. (A-C): typical SERS spectra for three melanoma cell lines. Solid lines represent MT. Broken lines represent WT. (D): Genotype assessment by PCR/SERS compared to ddPCR.
While we have only demonstrated a 3-plex system, the method could easily be multiplexed to a higher capacity to fully exploit the discrete spectral separation which was only possible with SERS.[6-15] Nonetheless, compared to current commercial assays, e.g. the Roche Cobas systems[4] and ddPCR,[5] which required multi-tube/reactions qPCR assays to evaluate multiple mutations, PCR/SERS assay described herein could interrogate at least 3 targets per tube/reaction. In the future, higher multiplex could be achieved and thus lead to reduced reagent/sample requirements.
Sensitive mutation detection
High sensitivity assays are required for early disease detection and for detecting low copy numbers of targets e.g. tumour DNA in a background of normal DNA. Hence to evaluate the performance of our proposed PCR/SERS method, we titrated known copies of the BRAF V600E sequence in a background of WT BRAF sequences (10,000 copies in total). To simulate the complexity of a typical biopsy-derived sample, 5 ng of salmon sperm DNA was included to further increase the complexity of the system. As little as 0.1% mutant sequence (10 copies) could be detected reproducibly (t-test p < 0.01) over the no input control (Figure 3). The inter- and intra- assay coefficient of variability (CV) was found to be 8.7% (n = 3) and 8.8% (n = 6) respectively, indicating good assay reproducibility.
Detecting low levels of mutation load. (A) Typical raw Raman spectra and (B) bar graph of average SERS intensities at 1076 cm-1 over a range of mutation loads for 10,000 input copies. NoT is the no template control. Error bars represent SD of 3 independent experiments. Inserts are magnification of data at 1%, 0.1% and 0% mutation loading and the NoT control.
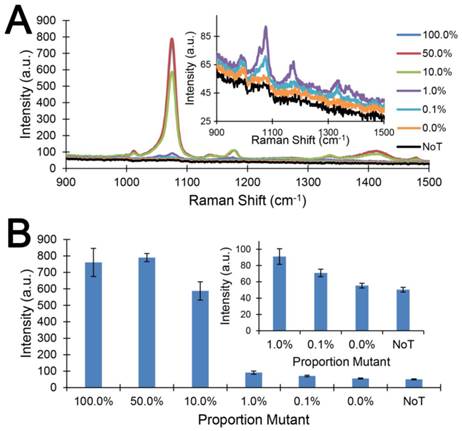
This level of sensitivity (0.1%) in detecting low levels of targets as achieved by our method (Figure 3) was 10 fold higher than that described for commercial PCR-based assays (1%), e.g. Roche Cobias 4800 BRAF assay [4]. Our method was, therefore, a potentially more sensitive yet specific alternative assay for detecting BRAF mutations (Table S1). In addition, compared to a recent SERS nanotag assay[14] for single DNA base changes via the ligase chain reaction (10% sensitivity), the current approach was 100 times more sensitive in detecting low abundance targets.
Accurate genotyping in cells and circulating serum DNA
Next, we evaluated the accuracy of genotyping melanoma cell lines for BRAF V600E, c-Kit L576P and NRAS Q61K mutations and compared our results with that obtained using ddPCR (Figure 4). Separate PCR/SERS assays for the mutant (MT) and wild type (WT) sequences were also performed concurrently. As expected, the PCR/SERS assay was as accurate as ddPCR at correctly genotyping the cell lines. For instance, LM-MEL-1a was homozygous for NRAS Q61K mutations but WT for cKIT and BRAF. So, in the MT profile, only the peak for NRAS was observed. Conversely, only peaks for cKIT and BRAF was seen in the WT assay. Likewise, inferring from the profiles of the MT and WT assays, LM-MEL-3 was genotyped to be heterozygous for bothV600E and WT BRAF but WT for cKIT and NRAS while LM-MEL-97 was heterozygous for L576P and WT cKIT but WT for BRAF and NRAS. These genotyping results were also consistent with that in literature. [40]
Finally, we performed the PCR/SERS assay on ctDNA derived from the plasma of melanoma patients (Figure 5). As samples were limiting, only the MT assay was performed on ctDNA samples. Again, BRAF V600E mutation calls concurred with that of ddPCR. Of note, Patient 4 was negative for the BRAF V600E mutation but positive for the V600K and thus was not detected by our assay, further underscoring high assay specificity.
Accurate genotyping of ctDNA samples. (A-E): Typical SERS spectra for 5 ctDNA samples. (F): Genotype assessment by PCR/SERS compared to ddPCR.
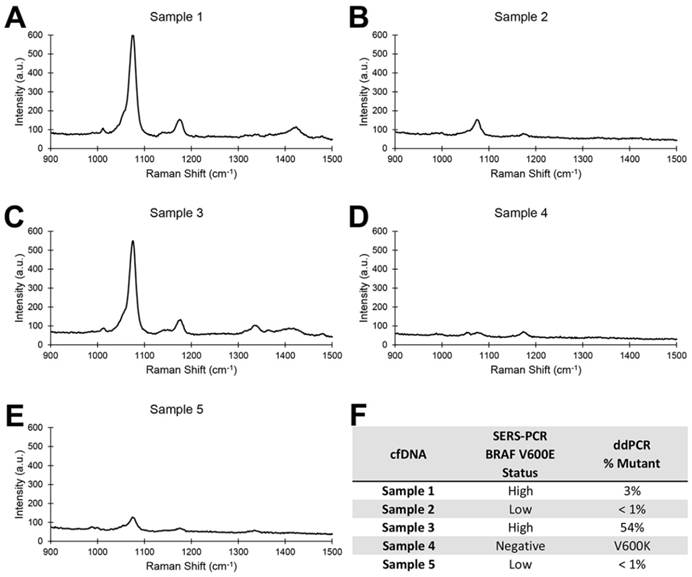
The current iteration of the method was not able to quantify the levels of tumour DNA due to the nature of an “end point” PCR assay. This limitation was evident in the rapid saturation in SERS signal as target loading increased above 10% (Figure 3). This could also explain in part the high SERS signal for Patient 1 while ddPCR measured only a 3% ctDNA loading. Perhaps, compartmentalization of the PCR assay similar to BEAMing digital PCR[41] may resolve this limitation by enabling an absolute quantitation scheme in the future. Nonetheless, the method in its current form was able to accurately identify mutation carriers which may be sufficient for clinicians to make treatment decisions based on the presence of targetable mutation(s).
Conclusion
In conclusion, we have demonstrated, as a proof-of-concept, a multiplex PCR/SERS assay for detecting clinically important BRAF V600E, c-Kit L576P and NRAS Q61K resulting DNA anomalies in ctDNA. The method was successfully applied to cell lines and serum derived DNA where results were subsequently validated with ddPCR. With its specificity and sensitivity approaching that of ddPCR yet at the simplicity of standard PCR techniques (Table S1), we believe this PCR/SERS approach could have wide applications as a tool for developing novel multiplex assays in both diagnostics and research.
Material and Methods
Cell line DNA sample preparation
Melanoma cell lines were obtained from Ludwig Institute for Cancer Research, Melbourne. They were established, typed and cultured as previously described.[40] Genomic DNA (gDNA) was purified using the DNeasy Blood and Tissue kit (Qiagen) as instructed by the manufacturer. gDNA concentrations were determined using spectrometry (NanoDrop, Thermo Scientific) and 5 ng were used as input for the PCR/SERS assay.
Patient serum ctDNA samples
Patients provided their written informed consent for the samples collected for the research study protocol, which was approved by the Human Research Ethics Committee of the Austin Hospital, Melbourne (H2012/04446) and The University of Queensland Human Research Ethics Committee (2011001315). Five patients with known BRAF V600E and V600K mutation statues previously determined by sequencing and ddPCR[5] were used in this study. ctDNA was isolated from 1 mL frozen aliquots of plasma using the QIAamp Circulating Nucleic Acid kit (Qiagen). gDNA amounts were then determined using the Qubit dsDNA High Sensitivity kit (Thermo Scientific). Generally, 0.2 - 1 ng/µL of ctDNA was obtained. Two microliters was then used in the PCR/SERS assay.
Preparation of SERS nanotags
SERS nanotags were prepared according to our previous report.[14] The gold nanoparticles (AuNPs) were synthesized by citrate reduction of HAuCl4.[42] SERS nanotags were synthesized by the coating of Raman reporters and DNA probe on the AuNPs surface. Briefly, 1mL AuNPs were mixed with 10 μL 50 μM TCEP treated thiolated DNA oligonucleotides (IDT) at RT for 12 hours. Then, 100 μL of 1 mM Raman reporters, 4-Mercaptobenzoic acid (MBA), 2,7-mercapto-4-methylcoumarin (MMC) or 4-mercapto-3-nitrobenzoic acid (MNBA) were added to the AuNPs and incubated at RT for overnight. Then 0.6 M NaCl in 1 mM PBS was used to age the SERS nanotags at RT for 12 hours before being centrifuged and resuspended into 10 mM PBS solution prior to use on the SERS detection assay. Successful functionalization of SERS nanotags were confirmed with UV-Vis (Fig S1A). The enhancement factor of gold nanoparticles to the Raman reporter was estimated to be 106-108, consistent with previously reported values.[43] SERS nanotag concentrations were then normalized by diluting in PBS such that a 1:1:1 SERS signal was obtained for a 1:1:1 target mix. Final concentration of the SERS nanotag mix was 0.5 nM as determined by the UV extinction spectra for AuNPs. DNA sequences are provided in Table 1.
Oligonucleotides used in this study. Modifications are as indicated.
| Oligonucleotide | 5'-Sequence-3' |
|---|---|
| BRAF V600E F | CAGATCGTCATGTTC/iSpC3/ATAGGTGATTTTGGTCTAGCTACTGA |
| BRAF WT F | CAGATCGTCATGTTC/iSpC3/ATAGGTGATTTTGGTCTAGCTACTGT |
| BRAF R | /5BiosG/AGTAACTCAGCAGCATCTCAGG |
| BRAF nanotag | GAACATGACGATCTGTTTTT/3ThioMC3-D/ |
| cKIT L576P R | TCTGCACCAATGTAC/iSpC3/ACTCCCATTTGTGATCATAAGCAG |
| cKIT WT R | TCTGCACCAATGTAC/iSpC3/ACTCCCATTTGTGATCATAAGCAA |
| cKIT F | /5BiosG/TTTGTTCTCTCTCCAGAGTGCT |
| cKIT nanotag | GTACATTGGTGCAGATTTTT/3ThioMC3-D/ |
| NRAS Q61K F | AGTCTGATGGCAGCA/iSpC3/GGACATACTGGATACAGCTGTAA |
| NRAS WT F | AGTCTGATGGCAGCA/iSpC3/GGACATACTGGATACAGCTGTAC |
| NRAS R | /5BiosG/TGATGGCAAATACACAGAGGA |
| NRAS nanotag | TGCTGCCATCAGACTTTTTT/3ThioMC3-D/ |
PCR/SERS assay
Multiplex PCR was performed using the Robust2G Hotstart PCR kit (KapaBiosistems) with major modifications. Each 10 µL reaction contained 1.25x Buffer A, 3.125 mM MgCl2, 1.8 M ethylene glycol, 800 ng BSA, 166 nM of each primer, 200 nM of dNTP and 0.2 U of polymerase. Thermal cycling conditions were 95°C for 2 min followed by 32 cycles of 95°C for 30 s, 55°C for 30s and 72°C for 30s. After PCR, 5 µL were then evaluated by gel electrophoresis to confirm amplification. The remaining 5 µL was then used for SERS detection. DNA sequences are provided in Table 1. For cell line experiments, WT and MT specific assays were performed separately. For ctDNA experiments, there were only sufficient material for 3 replicates of MUT-specific assay. For all other experiments, 3 independent experiments were performed with 3 technical replicates each.
To prepare PCR samples for SERS, 1µL SERS nanotag mix was added to each sample and incubated at 35°C for 15 mins. Subsequently 5 µL of streptavidin coated magnetic beads (SMB) was added to the PCR/SERS mix and left to incubate for another 10 mins at room temperature. The PCR/SERS/SMB complex was then isolated with a magnet and washed 3 times by resuspending the pellet with 0.25x PBS supplemented with 0.01% Tween 20. After the final wash, the pellet was then resuspended in 60 µL 2x PBS and transferred to a quartz cuvette prior to SERS measurement on the IM-52 portable Raman microscope (Snowy Range Instruments). SERS spectra were obtained from five 2-second acquisitions using a 785 nm excitation laser at 70 mW. The binding of the SERS nanotags on magnetic beads was confirmed by Transmission Electron Microscopy (TEM), in which SERS nanotags were clearly observed on the bead surface only in the presence of target DNA (Fig S1B-C).
Supplementary Material
Supplementary tables and figures.
Acknowledgements
The work acknowledges funding support from UQ ECR (2014002940) to Y.W. Although not directly funding this work, we also acknowledge funding received from the National Breast Cancer Foundation of Australia (CG-08-07 and CG-12-07). These grants have significantly contributed in creating the environment to enable the research described here. We also thank Yadveer Grewal and Kevin Koo for facilitating TEM imaging.
Author Contributions
All authors were involved in conceiving the study and drafting the manuscript. EJHW and YL designed the method and perform all experiments. SCHT collected all cell lines and patient samples, isolated and quantified their DNA and provided ddPCR data.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Pleasance ED, Cheetham RK, Stephens PJ, McBride DJ, Humphray SJ, Greenman CD. et al. A comprehensive catalogue of somatic mutations from a human cancer genome. Nature. 2010;463:191-6
2. Komar AA. Genetics. SNPs, silent but not invisible. Science. 2007;315:466-7
3. Greenman C, Stephens P, Smith R, Dalgliesh GL, Hunter C, Bignell G. et al. Patterns of somatic mutation in human cancer genomes. Nature. 2007;446:153-8
4. Lopez-Rios F, Angulo B, Gomez B, Mair D, Martinez R, Conde E. et al. Comparison of Testing Methods for the Detection of BRAF V600E Mutations in Malignant Melanoma: Pre-Approval Validation Study of the Companion Diagnostic Test for Vemurafenib. PLoS ONE. 2013;8:e53733
5. Chang-Hao Tsao S, Weiss J, Hudson C, Christophi C, Cebon J, Behren A. et al. Monitoring response to therapy in melanoma by quantifying circulating tumour DNA with droplet digital PCR for BRAF and NRAS mutations. Scientific Reports. 2015;5:11198
6. Schlucker S. Surface-Enhanced Raman Spectroscopy: Concepts and Chemical Applications. Angew Chem Int Edit. 2014;53:4756-95
7. Moskovits M. SPECTROSCOPY Expanding versatility. Nature. 2010;464:357 -
8. Anker JN, Hall WP, Lyandres O, Shah NC, Zhao J, Van Duyne RP. Biosensing with plasmonic nanosensors. Nat Mater. 2008;7:442-53
9. Bantz KC, Meyer AF, Wittenberg NJ, Im H, Kurtulus O, Lee SH. et al. Recent progress in SERS biosensing. Phys Chem Chem Phys. 2011;13:11551-67
10. Sharma B, Frontiera RR, Henry AI, Ringe E, Van Duyne RP. SERS: Materials, applications, and the future. Mater Today. 2012;15:16-25
11. Schlucker S. SERS Microscopy: Nanoparticle Probes and Biomedical Applications. Chemphyschem. 2009;10:1344-54
12. Wang YL, Schlucker S. Rational design and synthesis of SERS labels. Analyst. 2013;138:2224-38
13. Lai YM, Sun SQ, He T, Schlucker S, Wang YL. Raman-encoded microbeads for spectral multiplexing with SERS detection. Rsc Adv. 2015;5:13762-7
14. Wang YL, Wee EJH, Trau M. Highly sensitive DNA methylation analysis at CpG resolution by surface-enhanced Raman scattering via ligase chain reaction. Chem Commun. 2015;51:10953-6
15. Maiti KK, Samanta A, Vendrell M, Soh KS, Olivo M, Chang YT. Multiplex cancer cell detection by SERS nanotags with cyanine and triphenylmethine Raman reporters. Chem Commun. 2011;47:3514-6
16. Faulds K, Barbagallo RP, Keer JT, Smith WE, Graham D. SERRS as a more sensitive technique for the detection of labelled oligonucleotides compared to fluorescence. Analyst. 2004;129:567-8
17. Cao YC, Jin R, Mirkin CA. Nanoparticles with Raman spectroscopic fingerprints for DNA and RNA detection. Science. 2002;297:1536-40
18. Papadopoulou E, Bell SE. Label-free detection of single-base mismatches in DNA by surface-enhanced Raman spectroscopy. Angewandte Chemie. 2011;50:9058-61
19. Johnson RP, Richardson JA, Brown T, Bartlett PN. A label-free, electrochemical SERS-based assay for detection of DNA hybridization and discrimination of mutations. Journal of the American Chemical Society. 2012;134:14099-107
20. Gracie K, Correa E, Mabbott S, Dougan JA, Graham D, Goodacre R. et al. Simultaneous detection and quantification of three bacterial meningitis pathogens by SERS. Chemical Science. 2014;5:1030-40
21. Shanmukh S, Jones L, Driskell J, Zhao Y, Dluhy R, Tripp RA. Rapid and sensitive detection of respiratory virus molecular signatures using a silver nanorod array SERS substrate. Nano letters. 2006;6:2630-6
22. van Lierop D, Larmour IA, Faulds K, Graham D. SERS Primers and Their Mode of Action for Pathogen DNA Detection. Anal Chem. 2013;85:1408-14
23. Zhou H, Lin S, Nie Y, Yang D, Wang Q, Chen W. et al. Surface-enhanced resonance Raman scattering (SERRS) simulates PCR for sensitive DNA detection. Analyst. 2015;140:7518-21
24. Hibbitts S, White PL, Green J, McNay G, Graham D, Stevenson R. Human papilloma virus genotyping by surface-enhanced Raman scattering. Analytical Methods. 2014;6:1288-90
25. Hoppmann EP, Yu WW, White IM. Detection of Deoxyribonucleic Acid (DNA) Targets Using Polymerase Chain Reaction (PCR) and Paper Surface-Enhanced Raman Spectroscopy (SERS) Chromatography. Applied Spectroscopy. 2014;68:909-15
26. White PL, Hibbitts SJ, Perry MD, Green J, Stirling E, Woodford L. et al. Evaluation of a commercially developed semiautomated PCR-surface-enhanced raman scattering assay for diagnosis of invasive fungal disease. Journal of clinical microbiology. 2014;52:3536-43
27. Strelau KK, Brinker A, Schnee C, Weber K, Möller R, Popp J. Detection of PCR products amplified from DNA of epizootic pathogens using magnetic nanoparticles and SERS. Journal of Raman Spectroscopy. 2011;42:243-50
28. Sensi M, Nicolini G, Petti C, Bersani I, Lozupone F, Molla A. et al. Mutually exclusive NRASQ61R and BRAFV600E mutations at the single-cell level in the same human melanoma. Oncogene. 2006;25:3357-64
29. Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S. et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949-54
30. Ascierto PA, Minor D, Ribas A, Lebbe C, O'Hagan A, Arya N. et al. Phase II trial (BREAK-2) of the BRAF inhibitor dabrafenib (GSK2118436) in patients with metastatic melanoma. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2013;31:3205-11
31. Beadling C, Jacobson-Dunlop E, Hodi FS, Le C, Warrick A, Patterson J. et al. KIT gene mutations and copy number in melanoma subtypes. Clinical cancer research: an official journal of the American Association for Cancer Research. 2008;14:6821-8
32. Carvajal RD, Antonescu CR, Wolchok JD, Chapman PB, Roman RA, Teitcher J. et al. KIT as a therapeutic target in metastatic melanoma. Jama. 2011;305:2327-34
33. Hodi FS, Friedlander P, Corless CL, Heinrich MC, Mac Rae S, Kruse A. et al. Major response to imatinib mesylate in KIT-mutated melanoma. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2008;26:2046-51
34. Antonescu CR, Busam KJ, Francone TD, Wong GC, Guo T, Agaram NP. et al. L576P KIT mutation in anal melanomas correlates with KIT protein expression and is sensitive to specific kinase inhibition. International journal of cancer Journal international du cancer. 2007;121:257-64
35. Ascierto PA, Schadendorf D, Berking C, Agarwala SS, van Herpen CM, Queirolo P. et al. MEK162 for patients with advanced melanoma harbouring NRAS or Val600 BRAF mutations: a non-randomised, open-label phase 2 study. The Lancet Oncology. 2013;14:249-56
36. Johnson DB, Smalley KS, Sosman JA. Molecular pathways: targeting NRAS in melanoma and acute myelogenous leukemia. Clinical cancer research: an official journal of the American Association for Cancer Research. 2014;20:4186-92
37. Beadling C, Jacobson-Dunlop E, Hodi FS, Le C, Warrick A, Patterson J. et al. KIT Gene Mutations and Copy Number in Melanoma Subtypes. Clinical Cancer Research. 2008;14:6821-8
38. Chernoff KA, Bordone L, Horst B, Simon K, Twadell W, Lee K. et al. GAB2 Amplifications Refine Molecular Classification of Melanoma. Clinical Cancer Research. 2009;15:4288-91
39. Davies MA, Samuels Y. Analysis of the genome to personalize therapy for melanoma. Oncogene. 2010;29:5545-55
40. Behren A, Anaka M, Lo PH, Vella LJ, Davis ID, Catimel J. et al. The Ludwig institute for cancer research Melbourne melanoma cell line panel. Pigment Cell Melanoma Res. 2013;26:597-600
41. Dressman D, Yan H, Traverso G, Kinzler KW, Vogelstein B. Transforming single DNA molecules into fluorescent magnetic particles for detection and enumeration of genetic variations. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:8817-22
42. Frens G. Controlled Nucleation for Regulation of Particle-Size in Monodisperse Gold Suspensions. Nature-Phys Sci. 1973;241:20-2
43. Orendorff CJ, Gole A, Sau TK, Murphy CJ. Surface-enhanced Raman spectroscopy of self-assembled monolayers: sandwich architecture and nanoparticle shape dependence. Analytical chemistry. 2005;77:3261-6
Author contact
![]() Corresponding authors: m.trauedu.au, j.weeedu.au, y.wang27edu.au
Corresponding authors: m.trauedu.au, j.weeedu.au, y.wang27edu.au