Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and methods
Results and Discussion
Conclusion
Supplementary Material
Abbreviations
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2016; 6(10):1573-1587. doi:10.7150/thno.15164 This issue Cite
Research Paper
Ultrasound-Mediated Microbubble Destruction (UMMD) Facilitates the Delivery of CA19-9 Targeted and Paclitaxel Loaded mPEG-PLGA-PLL Nanoparticles in Pancreatic Cancer
Lingxi Xing1*, Qiusheng Shi1*, Kailiang Zheng2, Ming Shen3, Jing Ma1, Fan Li1, Yang Liu1, Lizhou Lin1, Wenzhi Tu4, Yourong Duan3 ![]() , Lianfang Du1
, Lianfang Du1 ![]()
1. Department of Ultrasound, Shanghai General Hospital, Shanghai Jiaotong University School of Medicine, Shanghai 200080, P. R. China
2. Engineering Department, Crop Science Division of Bayer, Institute, WV, 25112, USA
3. State Key Laboratory of Oncogenes and Related Genes, Shanghai Cancer Institute, Renji Hospital, Shanghai Jiaotong University School of Medicine, Shanghai 200032, P. R. China
4. The Comprehensive Cancer Center, Shanghai General Hospital, Shanghai Jiaotong University School of Medicine, Shanghai 200080, P. R. China
*Co-first Authors
Received 2016-2-1; Accepted 2016-5-1; Published 2016-6-18
Abstract

Pancreatic cancer, one of the most lethal human malignancies with dismal prognosis, is refractory to existing radio-chemotherapeutic treatment modalities. There is a critical unmet need to develop effective approaches, especially for targeted pancreatic cancer drug delivery. Targeted and drug-loaded nanoparticles (NPs) combined with ultrasound-mediated microbubble destruction (UMMD) have been shown to significantly increase the cellular uptake in vitro and drug retention in vivo, suggesting a promising strategy for cancer therapy. In this study, we synthesized pancreatic cancer-targeting organic NPs that were modified with anti CA19-9 antibody and encapsulated paclitaxol (PTX). The three-block copolymer methoxy polyethylene glycol-polylacticco-glycolic acid-polylysine (mPEG-PLGA-PLL) constituted the skeleton of the NPs. We speculated that the PTX-NPs-anti CA19-9 would circulate long-term in vivo, "actively target" pancreatic cancer cells, and sustainably release the loaded PTX while UMMD would "passively target" the irradiated tumor and effectively increase the permeability of cell membrane and capillary gaps. Our results demonstrated that the combination of PTX-NPs-anti CA19-9 with UMMD achieved a low IC50, significant cell cycle arrest, and cell apoptosis in vitro. In mouse pancreatic tumor xenografts, the combined application of PTX-NP-anti CA19-9 NPs with UMMD attained the highest tumor inhibition rate, promoted the pharmacokinetic profile by increasing AUC, t1/2, and mean residence time (MRT), and decreased clearance. Consequently, the survival of the tumor-bearing nude mice was prolonged without obvious toxicity. The dynamic change in cellular uptake, targeted real-time imaging, and the concentration of PTX in the plasma and tumor were all closely associated with the treatment efficacy both in vitro and in vivo. Our study suggests that PTX-NP-anti CA19-9 NPs combined with UMMD is a promising strategy for the treatment of pancreatic cancer.
Keywords: Ultrasound-mediated microbubble destruction, nanoparticles, controlled release, tumor target, pancreatic cancer.
Introduction
Pancreatic cancer is a malignancy with a poor prognosis and short median survival time [1]. Chemotherapy alone, or combined with surgical management, remains the mainstream treatment for pancreatic cancer. However, due to chemo-resistance, the 5-year survival rate remains less than 5% [2]. The dense stroma and deficient vascular network in the microenvironment of pancreatic cancer are believed to be the underlying reasons for inefficient delivery of chemotherapeutic drugs [3-5]. Therefore, there is an urgent need for developing a novel drug delivery system to improve drug concentration in the pancreatic tumor microenvironment [6].
Nanoparticles (NPs) modified with specific agents have been proven to be a promising carrier to efficiently deliver therapeutic drugs in a targeted manner to cells and tissues [7-11]. Our previous studies have demonstrated that NPs composed of a three-block copolymer of mPEG-PLGA-PLL have unique properties to deliver agents for cancer imaging and therapy. Among the three components of the NPs, mPEG protects the NPs from the reticuloendothelial system (RES) and maintains long half-lives of the NPs in circulation, PLGA generates a sustained release of the loaded drugs, and PLL facilitates the targeted modification [12-14]. However, the potential application of mPEG-PLGA-PLL NPs for the targeted imaging and therapy of pancreatic cancer remains to be determined.
In this study, we loaded mPEG-PLGA-PLL NPs, organic nanoparticles with a high affinity for hydrophobic drugs, with paclitaxol (PTX), which is a widely used anticancer drug for the treatment of a variety of cancers. As the targeting agent, we selected the anti CA19-9 antibody since CA19-9 is a well-studied, highly specific biomarker and is the only one approved by Food and Drug Administration for pancreatic ductal carcinoma [15-19]. We hypothesized that the "active targeting" of PTX-NP-antiCA19-9 would specifically deliver the loaded PTX to the pancreatic cancer cells.
Ultrasound mediated microbubble destruction (UMMD) is an effective method to enhance the cellular uptake of drugs with the help of auxiliary gas-filled microbubbles under proper sound pressure. It results in the cavitation of microbubbles and causes shear stress, micro-streaming, and jetting, which help to create transient pores on the cell membrane and enlarge the capillary gaps [20, 21]. Mounting evidence has shown that UMMD is a powerful tool to increase the permeability of cell membranes in vitro and promote the enhanced permeability and retention (EPR) effect in vivo [22-27]. It has further been reported that UMMD can enhance clathrin-mediated endocytosis [28-30], which is the most important internalization mechanism for NPs [31-34]. We postulated that the “passive targeting” process of UMMD would significantly enhance the "active targeting" by PTX-NP-anti CA19-9 at the irradiated tumor site and may significantly increase the cellular uptake and accumulation of PTX in pancreatic tumors (Fig. 1).
Schematic illustration of the enhanced cellular uptake and tumor accumulation by the combination of UMMD and PTX-NP-anti-CA19-9.
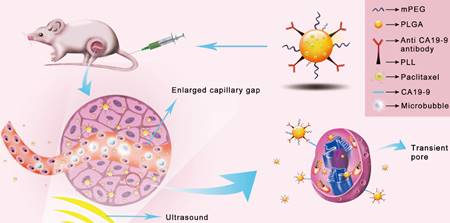
The objective of this study was to explore the treatment efficacy and toxicity of PTX-NP-anti CA19-9 delivery combined with UMMD for pancreatic cancer. In addition, we investigated the transport, distribution, and elimination of PTX-NP-anti CA19-9 when used with UMMD, thereby assessing the dynamic impact of the combination of "active targeting" and "passive targeting".
Materials and methods
Materials
Various chemicals and reagents and antibodies were acquired from the following sources: Paclitaxel (PTX) from J&K Scientific (Beijing, China), Taxol from Sino American Shanghai Squibb Pharmaceutical (Shanghai, China), Rhodamine B (RB) from Sigma-Aldrich (Shanghai, China) 1,1'-dioctadecyl-3,3,3',3'-tetramethylindotricarbocyanine iodide (DIR) was from Biotium (Hayward, CA, USA), Pluronic F68 from BASF (Shanghai, China), 4',6-diamino-2-phenylindole (DAPI) from Beijing Solarbio Science & Technology (Beijing, China), Anti-CA19-9 and anti-Ki67 monoclonal antibodies from Shanghai Linc-Bio Science (Shanghai, China), and the commercial SonoVue from Bracco (Milan, Italy).
Capan-1 cells were purchased from Shanghai Cancer Institute (Shanghai, China) and cultured in Dulbecco's Modified Eagle Medium (DMEM; Hyclone, USA) supplemented with 10% fetal bovine serum (Gibco, USA) in a humidified incubator at 37℃ with 5% CO2. The Cell Cycle Detection Kit and Annexin V-FITC/PI Cell Apoptosis Analysis Kit were purchased from Life Technologies (USA). Cell Counting Kit 8 (CCK-8) was obtained from Dojindo Molecular Technology (Japan) and the terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL) Kit was purchased from F. Hoffmann-La Roche (Basel, Switzerland).
Male BALB/c nude mice aged 4-6-weeks were obtained from Shanghai Laboratory Animal Center (Shanghai, China). All animal procedures were carried out in accordance with the protocols approved by the Animal Care and Use Committee at Shanghai General Hospital.
Preparation and characterization of PTX-mPEG-PLGA-PLL-anti-CA19-9 NPs
The PTX-mPEG-PLGA-PLL-anti-CA19-9 NPs were prepared at the Shanghai Cancer Institute by the emulsion-evaporation method as described previously [12, 14]. Briefly, 10mg mPEG-PLGA-PLL (mPEG MW = 2000, 10%) was added into 0.5ml methylene dichloride solution of PTX (0.5mg/ml) and the mixture was emulsified with an additional 40μl of pure water by a JY92-II ultrasonic processor (Ningbo Scientz Biotechnology, Ningbo, China) for 200s at 200W. Subsequently, 5ml of 0.5% F-68 was rapidly added and sonicated for 200s at 200W, followed by stirring at room temperature to remove the organic solvents. Next, 0.5mg anti-CA19-9 antibody, 1mg N-(3-Dimethylaminopropyl)-N'-ethylcarbodiimide hydrochloride (EDC) and 1mg N-hydroxysuccinimide (NHS) were dissolved in the mixture, sealed up, stirred for 1h and then dialyzed by distilled water rapidly (MWCO=14000) for 1h. The resultant emulsions were evaporated and lyophilized by a lyophilizer (EPSILON 2-6D; Martin Christ, Osterodeam Harz, Germany) to obtain the PTX-NPs-anti-CA19-9. The RB and DIR loaded NPs were prepared using the same method as above.
The size distribution and ζ potential were detected by Nicomp-380ZLS dynamic light scattering (Malvern, USA). The morphology of NPs was examined using a H-800 transmission electron microscope (Hitachi, Japan) and a Nanoscope® Multimode V atomic force microscope (AFM; Veeco, USA). The encapsulation efficiency was presented as the percentage of PTX encapsulated in the NPs from the total amount of PTX initially used. The drug-loading efficiency was defined as the percentage of PTX encapsulated in the NPs versus the total amount of NPs. Fourier transform infrared spectroscopy (FTIR) was used to identify the anti-CA19-9 antibody on the copolymer using a Nicolet NEXUS-670 Fourier Transform Infrared Spectrometer.
The drug release was determined by dialysis method. Briefly, 4ml solution of PTX loaded NPs was dissolved in 16ml salicylic acid (1M) and the mixture was transferred to a sealed dialysis bag (MWCO=3500). After horizontal shaking (50 rpm/min) at 37℃ for 1, 2, 4, 8, 12, 24 and 48hr, 0.2ml dialysate was collected and equal volume of solvent was added to the bag. The concentration of PTX was detected by high performance liquid chromatography (HPLC).
Experimental grouping
The experimental groups both in vitro and in vivo were as follows: Control, free PTX (PTX), PTX-NP (PN), PTX-NP-anti-CA19-9 (PN-Ab), PTX-NP-anti-CA19-9 plus anti-CA19-9 (PN-Ab+Ab), PTX-NP plus UMMD (PN+U), PTX-NP-anti-CA19-9 plus UMMD (PN-Ab+U). To confirm the targeted delivery of PN-Ab, in the group of PN-Ab+Ab, extra anti-CA19-9 antibody was added to the cell culture medium or injected to the tumor bearing nude mice prior to PTX-NP-anti-CA19-9 treatment to competitively bind the CA19-9 antigen on the cell membrane. The superiority of UMMD over ultrasound/microbubble alone in the enhanced drug delivery and therapeutic effect was shown in Fig. S1.
Dynamic cellular uptake analysis
Capan-1 cells, selected for their high expression of CA19-9 (Fig. S2), were seeded into 6-well plates at a density of 5×105cells/well for flow cytometry and into 35 mm glass bottom dishes with 10 mm micro-well (In Vitro Scientific, Sunnyvale, CA, USA) at a density of 5×105cells/dish for confocal microscopy and cultured overnight. RB instead of PTX was encapsulated in the NPs to serve as a fluorescence probe. RB and RB loaded NPs (RN) were added into the wells/dishes at an equivalent dose of RB (5μg/ml). Right before RB loaded NPs were added, UMMD was carried out in the groups of Rb-NP+UMMD (RN+U) and RN-NP-anti-CA19-9 (RN-Ab+U) by a Topteam161 Physioson-Basic therapeutic US machine (Physioson Elektromedizin AG, Laipersdorf, Germany). SF6 (59 mg) was dissolved in 5ml saline and the condition of UMMD (1.2W/cm2, 30sec, duty cycle 20%, 20% microbubble) was determined according to orthogonal design in the pre-experiment (Fig. S3 and S4). We performed UMMD before drug administration because as per our previous study, the simultaneous application of targeted NPs and UMMD decreased the cellular uptake and treatment effect in vitro. This was likely due to the fact that the powerful force of UMMD interfered with the binding of targeted NPs to cells [35]. Then at different time intervals (0.5h, 1h, 2h, 4h, 6h, 8h, 12h, 24h, 48h), cells in 6-well plates were collected, washed in PBS three times, and subjected to flow cytometry by an Accuri C6 Flow cytometer (BD Biosciences, CA, USA). Cells were fixed with 4% paraformaldehyde for 20min, incubated with DAPI for 2min at room temperature, washed in PBS three times, and imaged using a confocal laser scanning microscope (Leica Microsystems, Mannheim, Germany). The quantitative results of flow cytometry recorded the dynamic changes of cellular uptake, and the area under curve (AUC) was integrated by origin 8.
Cytotoxicity and IC50 assessment in vitro
Cytotoxicity was assessed by CCK-8 assay. Capan-1 cells were seeded into 96-well plates at 2000 cells/well and cultured overnight, then incubated with different formations of PTX with concentration of 0.625, 1.25, 2.5, 5.0, 10.0, 20.0, 40.0, 80.0, 160.0, 320.0μg/ml. UMMD was performed before drug administration in the groups of PN+U and PN-Ab+U with the same condition(1.2W/cm2, 30sec, duty cycle 20%, 20% microbubble) applied as in the cellular uptake assay. After 24h and 48h, 10μl CCK-8 agent was added to each well and incubated for 2h at 37℃. The OD value was measured at 450nm by a Spectra Max 190 microplate reader (BIO-RAD; Hercules, CA). The IC50 of 24h and 48h in different groups was evaluated by SPSS software 19.0.
Cell cycle analysis
Capan-1 cells were seeded into 6-well plates at 5×105 cells/well overnight. Different formulations of PTX were administered at the dose of 5.0μg PTX/ml and incubated for 24h and 48h. The cells were collected, rinsed in cold PBS three times, and fixed in cold 70% ethanol for 24 h at 4℃. After washing with PBS, cells were treated with 0.1% RNase A for 1h at 37℃, followed by propidium iodide (PI) staining for 15min at 4℃ in the dark. Samples were analyzed using the FACScan flow cytometry.
Cell apoptosis analysis
Cells were treated as described above for cell cycle analysis. Cells were collected and resuspended in 500μl binding buffer, followed by addition of 5μl Annexin V-FITC and 5μl PI, and incubated for 15 min at room temperature. Samples were analyzed for Annexin V staining by FAC Scan flow cytometry. For TUNEL staining, cells were seeded on slides before drug administration, then fixed with 4% paraformaldehyde for 20 min, and incubated with the TUNEL reaction cocktail for 5 min at room temperature.
Tumor mouse xenograft model
The pancreatic tumor mouse xenograft model was established by injecting 5×106 capan-1 cells into the subcutaneous space at the dorsum above the right leg in male 4-6-week-old BALB/c nude mice. Tumor volume was calculated through the equation: V=ab2/2, 'a' represents the length and 'b' represents the width [13, 14, 36]. Treatment began when the subcutaneous tumor reached 50-100mm3.
Targeted imaging and dynamic fluorescence biodistribution
To investigate the effect of targeted delivery and enhanced retention of targeted NPs combined with UMMD, DIR was encapsulated into the NPs instead of PTX. Different formulations of DIR were injected through the tail vein at an equivalent DIR dose of 1mg/Kg. For the groups of DIR-NP plus UMMD (DN+U) and DIR-NP-anti-CA19-9 plus UMMD (DN-Ab+U), the mice received a simultaneous injection of 200μl microbubbles (5.9 mg SonoVue dissolved in 5ml saline) from the retroorbital venous plexus and UMMD was conducted at the tumor site under preset conditions (1MHz, 2W/cm2, 2min, 20% duty cycle) which were determined in the pre-experiment (Fig. S4). The real-time imaging was conducted by a small animal in vivo fluorescence imaging system (LuminaXRMS; PerkinElmer, Walther, MA, USA) at the time intervals of 0.5h, 1h, 2h, 4h, 6h, 8h, 12h, 24h, 48h, 72h, 96h, 120h and 144h. The quantitative fluorescence intensity of the tumor was acquired using the onboard software. The dynamic change of tumor fluorescence intensity was recorded in a line chart and the AUC that partly reflects the total accumulation was calculated by origin 8.
Pharmacokinetics and PTX accumulation in the tumor
For pharmacokinetics and distribution investigations, mice with a subcutaneous tumor of approximately 200mm3 in the six groups of Taxol, PN, PN-Ab, PN-Ab+Ab, PN+U and PN-Ab+U were given the same PTX dose of 2mg PTX/Kg through the vena caudalis. At 0.5, 1, 2, 4, 8, 12 and 24h after drug injection, blood samples from the Taxol, PN and PN-Ab groups and tumor samples from all groups were collected. The samples were further processed into plasma and tissue homogenate respectively, and the PTX concentrations were subsequently detected by liquid chromatography/mass spectrometry (LC/MS) (Thermo Scientific TSQ Quantum Ultra; Massachusetts, USA). The pharmacokinetics analysis was operated by WinNonLin 6.3 software with non-compartmental modeling and the parameters were calculated as AUC, t1/2 (half time), MRT (mean residence time) and CL (clearance). The AUC of the concentration-time curve of PTX levels in the tumor was also calculated by WinNonLin 6.3 software and represented the total drug accumulated in the tumor within 24h.
Therapeutic effect in vivo and systemic toxicity
A mouse model was used to study the tumor inhibition rate and the serum indexes after treatment. The same dose of 2mg PTX/Kg for each treatment group was injected via the tail vein once a day for three weeks. The control group received saline injections. Body weight and tumor size were recorded twice a week. PTX treatment started at day 10 and ended at day 31 after tumor cell injections. After 31 days, all mice were sacrificed and blood was collected to detect the serum markers including albumin, lactate dehydrogenase (LDH), creatine kinase (CK), creatine kinase MB (CK-MB), aspartate aminotransferase (AST), alanine aminotransferase (ALT) and blood urea (BU). Unlike in the in vitro study, UMMD was performed simultaneously with the NP injection for the following reasons. First, although the NPs have a life greater than 5 days, the blood concentration dropped rapidly in the first several hours. And second, it was not clear how long the increased capillary permeability caused by UMMD lasted. At the end of the treatment course, a contrast-enhanced ultrasonography (CEUS) was conducted to detect the intra-tumoral vascularity by an iU-Elite ultrasonic diagnostic apparatus (Philips, Eindhoven, Holland), with a bolus injection of SonoVue through caudal vein at the dose of 10μl/g. The perfusion process was recorded and the images at peak intensity were captured. The tumor specimens were fixed in 10% Faure Marin and stained with Ki-67 to assess cell proliferation following treatments. An additional batch of mice was treated as above and used to observe the daily body weight changes and other symptoms based on the predetermined euthanasia criteria, including ascites, dyspnea, cyanosis, polypnea, apathy and no self-feeding;the survival time was recorded everyday (n=10).
Results and Discussion
Preparation and characterization of mPEG-PLGA-PLL NPs
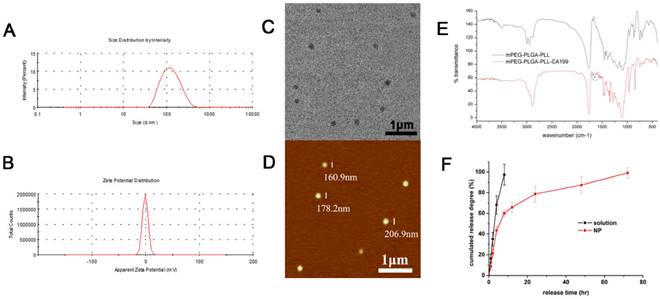
The mean size and ζ potential of the PTX-NPs-antiCA19-9 were approximately 184 nm and -0.907 mV (Fig. 2A and 2B), respectively. The spherical NPs dispersed uniformly with no aggregation (Fig. 2C and 2D). The presence of an absorption peak at 1650cm-1 represented a characteristic peak of the amide bond in proteins and indicated the attachment of anti CA19-9 antibody to the NPs (Fig. 2E). The encapsulation and drug loading efficiency was 81.5% and 2.04%, respectively. There was an initial burst release of PTX-NPs with about 60% of PTX released within 12h. However, it took 3 days to release the entire amount of the loaded PTX in PTX-NPs-antiCA19-9 (Fig. 2F). Thus, PTX-NPs-antiCA19-9 demonstrated a continuous release of PTX. By comparison, the free PTX was completely cleared within 8h.
Characterization of PTX-NP-anti-CA19-9: (A) size distribution and (B) zeta potential by dynamic light scattering; (C) morphology of NPs as detected by transmission electron microscopy and (D) atomic force microscopy; (E) FTIR of mPEG-PLGA-PLL and mPEG-PLGA-PLL-antiCA19-9 NPs;(F) drug release profiles of free PTX and PTX-NPs.
Targeted delivery of RB-NPs-anti CA19-9 and enhanced uptake by UMMD in pancreatic cancer cells
We determined the cellular uptake efficiency of free RB and NPs loaded with RB quantitatively by confocal laser scanning microscopy (Fig. 3A) and flow cytometry (Fig. 3B and 3C). The uptake of free RB started quickly and peaked at 8h post-administration. However, the cellular uptake of RB-loaded NPs (RN) was slower than that of free RB, likely due to the fact that RNs were internalized by cells through endocytosis, whereas entry of free RB into the cells was facilitated by diffusion. The uptake of RN-Ab was evidently greater than RN because the targeted NPs with the anti-CA19-9 antibody could specifically bind to the capan-1 cells and increase the local concentration of RB. The uptake of RN-Ab was similar to that of free RB before 8 h but kept increasing until 48 h. These results were consistent with the previous observations that the RB delivered via NPs was internalized by endocytosis, accumulated inside the cells, and could avoid degradation [37-40].
Cellular uptake of RB, RN, RN-Ab, RN-Ab+Ab, RN+U, and RB-NP- RN-Ab+U at different time intervals:)Analysis was performed by (A) confocal laser scanning microscope and (B) flow cytometry; (C) cellular uptake percentages over time (mean±SEM, n=3).
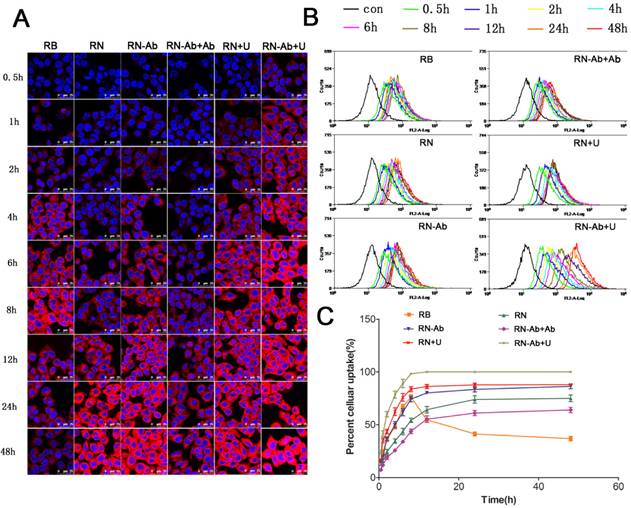
It has been reported that the cellular uptake of NPs was time-dependent and would increase until equilibrium was reached [37, 41]. The uptake had almost reached equilibrium at 24 h and increased slightly from 24 h to 48 h in the RN, RN-Ab and RN-Ab+Ab groups. The free RB reached equilibrium at 8 h and the uptake gradually began decreasing thereafter. Thus, the NPs, especially targeted NPs, could increase the time required to reach the equilibrium level. Furthermore, the targeted binding of RN-Ab to its ligand improved the cellular uptake. The uptake of RN-Ab+Ab, on the other hand, was significantly less than that of RN-Ab. It is possible that the added free anti CA19-9 antibody bound competitively to CA19-9 on the surface of cells, thereby decreasing the targeted internalization of RN-Ab. Our results were congruent with the earlier findings [42-45] that the efficacy of the targeted receptors diminished when the receptors on the cell membrane were saturated. Our previous studies demonstrated that UMMD could instantly create transient and repairable pores in the cell membrane by sonoporation that would greatly enhance the permeability of the membrane and increase the delivery of NPs to cells [22, 46, 47]. We had also reported earlier that UMMD increased the expression and accumulation of clathrin over a few hours thus enhancing clathrin-mediated endocytosis [28-30]. In the present study, in the RN+U and RN-Ab+U groups, the uptake efficiency of NPs increased rapidly in the first few hours, reached an equilibrium at 8h similar to the free RB group, and kept at a high level until 48h. The increased uptake of NPs is probably a consequence of UMMD causing an increase in the number of pores and clathrin.
It has been well documented that the antitumor effect of PTX mainly results from its intracellular accumulation over a period of time. We calculated the total uptake of RB in the six groups at 24 h and 48 h (Table 1). For both time points, The order of total uptake among the various groups was : RN-Ab+Ab<RN-Ab, RN<RN-Ab<RN-Ab+U, RN<RN+U. The total uptake at 48 h was significantly higher than that at 24 h in each group. Significant statistical difference existed between the total uptake of RN and RB at 48 h but not at 24 h, indicating continuous uptake of nanoparticles. The total uptake was higher in the RN-Ab group than in the RN group. Taken together, these data demonstrated a targeted delivery of RN-Ab and showed that UMMD significantly enhanced the total uptake of RN as well as RN-Ab.
Total uptake of RB and the IC50 of PTX formulations.
| Total uptake (24h) | Total uptake (48h) | IC50 (24h) | IC50 (48h) | |
|---|---|---|---|---|
| RB/PTX | 1221.23±39.48 | 2160.43±78.91*** | 25.076 | 19.532 |
| RN/PN | 1326.97±96.48*** | 3115.77±236.09** | 30.830 | 21.316 |
| RN-Ab/PN-Ab | 1661.82±83.81 | 3697.42±175.29 | 12.425 | 6.408 |
| RN-Ab+Ab/PN-Ab+Ab | 1099.16±62.69*** | 2599.56±163.12*** | 37.943 | 26.581 |
| RN+U/PN+U | 1838.53±23.75*** | 3945.33±27.45*** | 8.770 | 5.448 |
| RN-Ab+U/PN-Ab+U | 2157.02±34.24*** | 4557.02±34.24*** | 4.479 | 3.219 |
One-way ANOVA and Tukey HSD for statistical tests (n=3), * p<0.05, ** p<0.01, *** p<0.001 between groups of RN-Ab and RN/RN-Ab+Ab, RN and RN+U, and RN-Ab and RN-Ab+U for total uptake of 24 hand between groups of RB and RN, RN and RN-Ab, RN-Ab and RN-Ab+Ab, RN and RN+U, RN-Ab and RN-Ab+U for total uptake of 48 h. Data are presented as mean±SD.
Cytotoxicity and IC50 in vitro
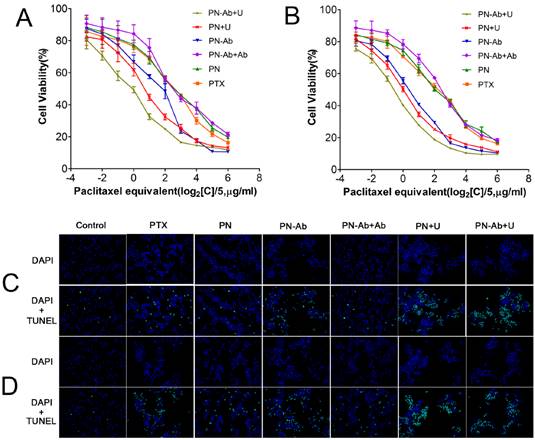
Previous studies have demonstrated that drug-loaded NPs work in a time- and concentration-dependent manner [37, 41]. The blank NPs and targeted NPs have been proven to be non-toxic (Fig. S6). The CCK8 assay indicated that the inhibitory effects of different formulations of PTX on cell viability were improved with increase in PTX concentration and treatment time (Fig. 4A and 4B). The cytotoxicity and cell apoptosis, as assessed by Hoechst 33342 staining (Fig. S7) and TUNEL assay (Fig. 4C), respectively, were more prominent with PN-Ab than with PN, and were further enhanced when combined with UMMD at 24 h and 48 h. These effects were further confirmed by the half maximal inhibitory concentration (IC50) values of different formulations of PTX for 24h and 48h (Table 1), the order of which was as follows: PN-Ab+Ab>PN-Ab, PN>PN-Ab>PN-Ab+U, PN>PN+U.The IC50 at 48 h was lower than that at 24 h in each group. These data indicated that the targeting properties of NPs and the additional physical processing of UMMD significantly improved the cell growth inhibitory effect of PTX. Interestingly, the cytotoxicity of PTX was higher than PN. This might be due to the fact that the free PTX could be easily internalized by passive diffusion, whereas PTX in the NPs is gradually released into the cytoplasm and therefore the cytotoxicity is cumulative over time in vitro [48-50]. However, when applied in vivo, free PTX may cause serious side effect, whereas the targeted NPs would deliver PTX to the tumor site specifically [50, 51]. Surprisingly, the IC50 was found to be consistent with the total uptake not only for 24 h but also for 48 h, where a lower IC50 matched a higher total uptake.
Concentration-dependent cytotoxicity as detected by CCK-8 assay: (A) 24 h; (B) 48 h (mean±SEM, n=3).Cell apoptosis as assessed by TUNEL staining in groups of PTX, PN, PN-Ab, PN-Ab+Ab, PN+U, and PN-Ab+U (C) 24 h and (D) 48 h.
Cell cycle distribution analysis by flow cytometry in groups of PTX, PN, PN-Ab, PN-Ab+Ab, PN+U and PN-Ab+U: (A and C) 24 h; (B and D) 48 h. Data are shown as mean±SEM (n=3), *p<0.05, **p<0.01, ***p<0.001.
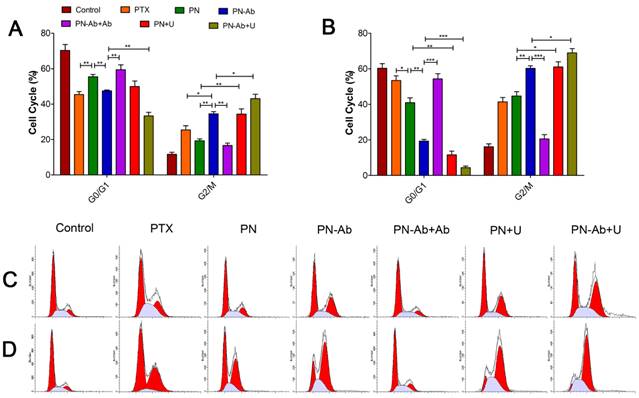
Cell cycle analysis
It is well established that PTX inhibits spindle formation and arrests cell cycle progression at the G2/M phase [52, 53]. Based on our cell cycle analysis by flow cytometry, the number of cells in G2/M phase subpopulationsin in different PTX formulations for 24 h (5A and 5C) and 48 h (5B and 5D) was as follows: PN-Ab+Ab<PN-Ab, PN<PN-Ab<PN-Ab+U, PN<PN+U. The arrest of cells at the G2/M phase and decreased number of cells at the G0/G1 phase disrupted the normal cell cycle transition and subsequently induced apoptosis [54, 55]. The higher percentage of cells in G2/M phase indicated higher cytotoxicity and lower IC50. Thus, PTX-NPs-anti-CA19-9 nanoparticles significantly induced cell cycle arrest at the G2/M phase in pancreatic cancer cells, and this effect was enhanced by UMMD.
Cell apoptosis analysis
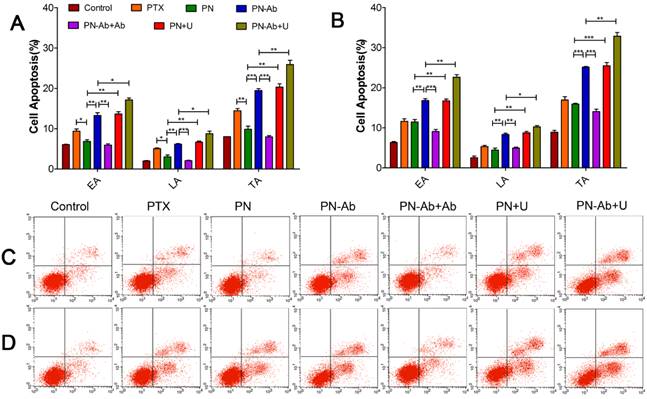
Following administration of different PTX formulations in Capan-1 cells, apoptosis was assessed by annexin-V-FITC/PI double staining assay. The extent of early apoptosis (EA), late apoptosis (LA) and total apoptosis (TA) in various groups in an ascending order was as follows: PN-Ab+Ab<PN-Ab, PN<PN-Ab<PN-Ab+U, PN<PN+U. Apoptosis at 48 h was significantly higher than that at 24 h in all groups with EA constituting the main proportion. This trend was consistent with those observed for the cell cycle distribution and IC50 (Fig. 6). The induction of apoptosis by PN-Ab was significantly higher than that by PN, and was further enhanced by UMMD. Taken together, these comprehensive analyses of cellular uptake, IC50, cell cycle, and cell apoptosis clearly demonstrated that mPEG-PLGA-PLL NPs were excellent carriers for PTX. In addition, anti-CA19-9 antibody modification on the surface of NPs improved the targeted binding and further enhanced the cellular uptake of PTX-NPs-anti CA19-9 nanoparticles.
Cell apoptosis analysis by flow cytometry in groups of PTX, PN, PN-Ab, PN-Ab+Ab, PN+U and PN-Ab+U: (A and C) 24 h; (B and D) 48 h. Data are shown as mean±SEM (n=3), *p<0.05, **p<0.01, ***p<0.001.
Targeted imaging and dynamic fluorescence biodistribution in mouse pancreatic tumor xenografts
The expression of CA19-9 in pancreatic tumor xenografts was detected by immunohistochemistry (Fig. S8). A small animal in vivo fluorescence imaging system was used to detect the accumulation of DIR in the subcutaneous tumor model in BALB/c nude mice. During the first 2 h post-injection, the fluorescence of DIR was mainly concentrated in the liver and/or brain. The signal intensity of the DIR fluorescence was high in DN+U and DN-Ab+U groups. The accumulation of fluorescent signals in tumors reached peak levels at 8 h in groups with NPs, and then decreased gradually (Fig. 7A and 7B). On the other hand, the intensity decreased immediately in DN-Ab+Ab because the additional antibody saturated the CA19-9 on the tumor cells. In the free DIR group, the fluorescence was barely observed in the tumor at 24 h and became very weak to be detected in the whole body at 72 h. However, in other groups of NPs, the fluorescence in the tumor lasted until 120 h (Fig. 7B). Also, the peak intensity of free DIR in vivo was lower than that of other groups of NPs. Thus, both the retention time and peak intensity of free DIR were inferior to that of NPs in vivo. On the contrary, the total cellular uptake of free RB was higher than that of RN and RN-Ab+Ab in in vitro experiments. The potential reason for this discrepancy might be due to the reason that the mPEG-PLGA-PLL NPs could escape from the reticuloendothelial system and stay longer in circulation. Also, the sustained release of NPs cargo would help DIR to accumulate in the tumor tissue over a longer time period.
Tumor-targeted real-time imaging using small animal in vivo fluorescence imaging system: Imaging was carried out at time intervals of 0.5h, 1h, 2h, 4h, 6h, 8h, 12h, 24h, 48h, 72h, 96h, 120h and 144h after administration of free DIR, DN, DN-Ab, DN-Ab+Ab, DN+U and DN-Ab+U. (A) the average quantitative fluorescence intensity in tumors over time. (B) representative images of the tumor-targeted real-time imaging using different DIR formulations.
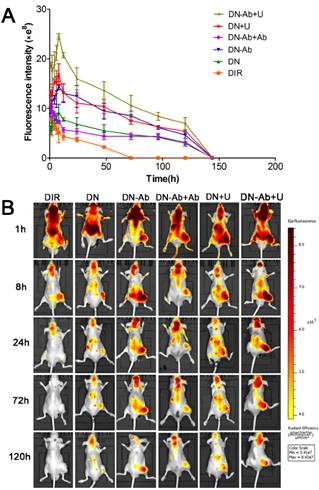
The peak intensities and AUC in various groups in the ascending order were as follows: DN-Ab+Ab<DN-Ab, DN<DN-Ab<DN-Ab+U, DN<DN+U. The AUC roughly reflected the total amount of DIR accumulation (Table 2). Compared with DN, DN-Ab aggregated specifically in the tumor site, with a 59.8% and 68.3% increase in peak intensity and AUC, respectively. Furthermore, with UMMD, the peak intensity and AUC were increased by 84.6% and 71.0%, respectively in the DP group and 68.4% and 40.5%, respectively, in the DN-Ab group. It has been reported that UMMD could transiently increase the capillary permeability of normal and tumor tissues [46, 47, 56-58]. Generally, tumor vessel walls are leaky due to the endothelial gaps ranging from hundreds of nanometers to a few micrometers [59-61] which would theoretically allow NPs to pass through the pores. Pancreatic cancers, on the other hand, are composed of over 80% of stroma and less than 20% of tumor cells [5, 62, 63]. Similar to the cellular uptake in vitro, the fluorescent signals increased immediately after UMMD in vivo, and then decreased gradually. The enhanced effect in the initial period both in vitro and in vivo possibly resulted from the fact that the pores on the membranes and the increased capillary permeability created by UMMD are transient [56-58]. Together, these results demonstrated that the accumulation of the loaded imaging agents in tumors was significantly increased by mPEG-PLGA-PLL-anti CA19-9 NPs and further enhanced by UMMD in mouse pancreatic tumor xenografts.
Drug accumulation in tumors, mean tumor inhibition rate and median survival.
| AUC of targeted imaging | AUC of PTX in the tumor | Mean tumor inhibition rate | median survival | |
|---|---|---|---|---|
| Control | / | / | / | 41±4.74 |
| DIR/PTX | 210.70±39.98** | 982.04±168.84 | 16.02% | 43±3.95 |
| DN/PN | 673.33±108.76 | 2488.18±568.92 | 62.24% | 51±4.65** |
| DN-Ab/PN-Ab | 1133.09±217.13 | 3689.42±1131.25* | 82.99% | 63±3.10* |
| DN-Ab+Ab /PN-Ab+Ab | 605.00±126.75* | 2992.34±669.73 | 68.38% | 53±3.16 |
| DN+U/PN+U | 1151.54±177.39 | 5317.92±1038.74* | 82.76% | 58±5.53 |
| DN-Ab+U /PN-Ab+U | 1591.72±307.71 | 6777.79±1369.98* | 90.51% | 75±7.12* |
One-way ANOVA and Tukey HSD for statistical tests in AUC of targeted imaging and PTX in tumors (n=3), * p<0.05, ** p<0.01, *** p<0.001 between group of DIR and DN, DN and DN-Ab, DN-Ab and DN-Ab+Ab, DN and DN+U, and DN-Ab and DN-Ab+U and in AUC of targeted imaging between groups of PN-Ab and PTX, PN and PN+U, and PN-Ab and PN-Ab+U. Log-rank test was used for median survival (n=10), ** p<0.01 between groups of PN and control, * p<0.05 between groups of PN and PN-Ab, and PN-Ab and PN-Ab+U. Data are presented as mean±SD.
Pharmacokinetics and tumor accumulation of PTX
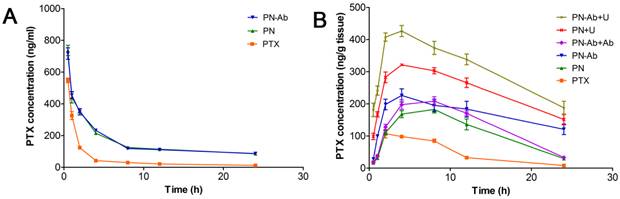
We further investigated the pharmacokinetics and accumulation of various PTX formulations in mouse pancreatic tumor xenografts. The PTX concentrations in plasma and tumor tissues are shown in Figure 8, and the pharmacokinetic parameters are listed in Table 3. Compared with PTX alone, PN and PN-Ab improved the pharmacokinetic profile of PTX by significantly increasing AUC, t1/2, MRT, and decreasing clearance. This excellent performance may be attributed to the ability of mPEG-PLGA-PLL NPs to have longer circulation time by evading the RES. Moreover, the NPs may protect their loaded PTX from elimination and provide enough time to penetrate into the tumor tissue. At 8 h post drug injection, the blood concentration of PTX in all three groups of free PTX, PN and PN-Ab groups was low. At the same time, the fluorescence in the tumors of DN and DN-Ab remained high while it significantly decreased in the free DIR group, indicating that DN and DN-Ab remained in the tumor, while free DIR was excreted. Considering that PTX accumulation in the tumor mesenchyme is ultimately responsible for the antitumor effect, we tested the PTX concentration at different time intervals following the administration of various PTX formulations with and without UMMD. The controlled release of NPs prolonged the residence time of the loaded PTX in the tumor, and the targeted binding of anti CA19-9 to its ligand on the cell membrane allowed increased accumulation of NPs in the tumor thereby increasing its concentration. When combined with UMMD, the concentration of PTX rose sharply and reached a higher level earlier than that with NPs alone. These results were predictable because the cavitation effect by UMMD facilitates the delivery of NPs by opening the capillary gaps. The AUC of the concentration-time curve in the tumor represented the total effective PTX with the following ascending order: PN-Ab+Ab<PN-Ab, PN<PN-Ab<PN-Ab+U, PN<PN+U (Table 2). The trend and profile of the concentration-time curve were generally in agreement with the results obtained by targeted imaging, which confirmed the EPR effect of targeted NPs and UMMD.
Pharmacokinetic parameters of PTX, PN, and PN-Ab in non-compartmental modeling by WinNonLin 6.3.
| PTX | PN | PN-Ab | |
|---|---|---|---|
| T1/2 (hr) | 12.82±0.64 | 29.20±1.95** | 36.10±6.64** |
| Cmax (ng/ml) | 547.60±25.10 | 736.91±55.51** | 716.23±61.60* |
| AUC0-t (hr*ng/ml) | 1434.25±131.92 | 4132.40±221.86*** | 4108.98±209.35*** |
| AUC0-∞ (hr*ng/ml) | 1665.11±158.75 | 7769.69±390.25*** | 8688.52±794.42*** |
| CL0-∞ (ml/hr/kg) | 1208.10±109.79 | 257.85±13.25*** | 231.53±22.00*** |
| MRT0-t (hr) | 4.25±0.16 | 7.67±0.21*** | 7.70±0.28*** |
| MRT0-∞ (hr) | 9.55±0.35 | 35.06±2.97** | 43.87±9.32** |
T1/2, half-life; Cmax, maximal concentration; AUC, area under the curve; CL, clearance; MRT, mean residence time. One-way ANOVA and Tukey HSD for statistical tests (n=3), * p<0.05, ** p<0.01, *** p<0.001 between groups of PTX and PN/PN-Ab. No statistical difference exists between groups of PN and PN-Ab. Data are presented as mean±SD.
Blood indeces indicating the functions of liver, kidney and heart.
| Albumin | AST | ALT | LDH | BU | CK | CK-MB | |
|---|---|---|---|---|---|---|---|
| Control | 16.9 ±5.4 | 271.2 ±87.9 | 40.5 ±11.6 | 3189.8±837.1 | 7.1±1.8 | 927.4±805.4 | 525.0±204.8 |
| PTX | 16.1 ±2.4 | 293.8 ±50.5 | 45.2 ±12.2 | 4062.8±622.4 | 6.6±0.8 | 1093.2±309.2 | 550.6±50.4 |
| PN | 18.2 ±2.7 | 203.4 ±47.7 | 35.6 ±9.8 | 3712.8±284.0 | 6.5±0.7 | 1065.4±457.8 | 516.6±39.5 |
| PN-Ab | 19.1 ±2.6 | 182.2 ±90.9 | 35.2 ±7.5 | 2565.6±832.7 | 7.1±0.7 | 835.8±506.4 | 424.3±137.2 |
| PN-Ab+Ab | 20.8 ±0.5 | 255.6 ±76.6 | 41.0 ±13.2 | 3023.6±1099.3 | 6.9±0.4 | 1416.8±558.7 | 494.9±166.7 |
| PN+U | 23.6 ±2.6* | 208.6 ±44.5 | 41.6 ±28.2 | 3561.2±646.3 | 7.5±0.8 | 923.2±213.3 | 499.6±54.3 |
| PN-Ab+U | 22.0 ±3.0 | 246.0 ±171.9 | 42.8 ±19.9 | 3799.0±1080.7 | 7.4±0.6 | 958.0±450.2 | 492.4±176.5 |
One-way ANOVA and Tukey HSD for statistical test in Albumin (n=4-5), Albumin * p<0.05 between groups of PN+U and control/PTX. Data are presented as mean±SD.
Pharmacokinetics of PTX after a single intravenous injection of PTX, PN, PN-Ab, PN-Ab+Ab, PN+U, and PN-Ab+U: Concentration of PTX in (A) plasma and (B) tumors. Data are presented as mean±SEM (n=3).
Therapeutic effect in vivo and systemic toxicity
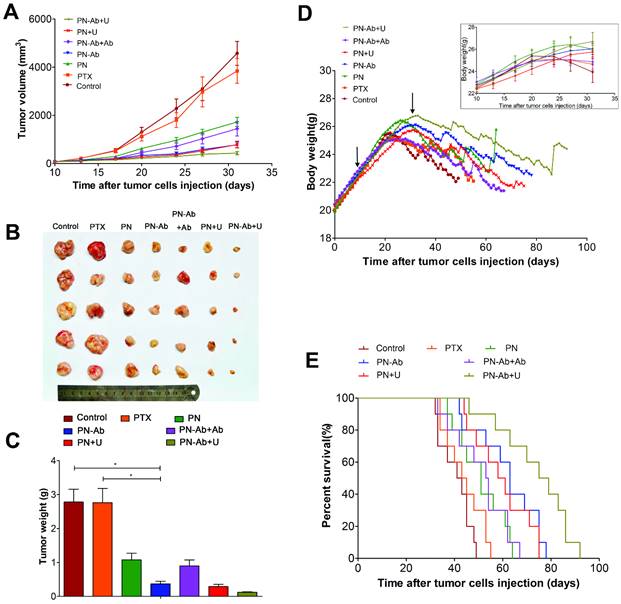
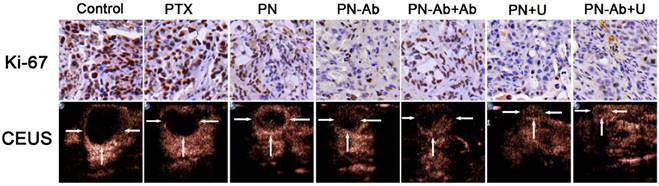
Finally, we tested the in vivo therapeutic effect and systemic toxicity of different formulations of PTX in mouse pancreatic tumor xenografts. The sequence of growth rate, final tumor volume, and weight were as follows: PN-Ab+Ab>PN-Ab, PN>PN-Ab>PN-Ab+U, PN>PN+U (Fig. 9A, 9B and 9C). The treatment effect of PN-Ab was significantly higher than PN. When PN-Ab and PN were applied simultaneously with UMMD, there was a dramatic increase in therapeutic efficacy resulting in 82.76% and 90.51% tumor inhibition, respectively. Ki-67 staining revealed that the tumor progression was slower after various treatments (Fig. 10), especially in the PN-Ab+U group. TUNEL staining also showed significant apoptosis in the PN-Ab+U group (Fig. S9). Contrast-enhanced ultrasonography (CEUS) was applied to evaluate the vascularity in the tumors after treatment. The time parameters in the CEUS course including the arrival time, peak time and washout time were almost similar in all seven groups. Based upon the peak intensities, the most effective treatment group was of PN-Ab+U in which the tumor was the smallest with the most abundant intratumoral blood flow (Fig. 10). The intratumoral blood flow detected by CEUS was suggested to be a predictor for prognosis in recent studies, and abundant blood flow might indicate betterr prognosis [64, 65]. As observed in the case of cellular uptake of nanoparticles and cytotoxicity analyses in vitro, the antitumor effect of PN-Ab was superior to that of PN-Ab+Ab, demonstrating a targeted drug delivery of PN-Ab. Thus, the targeting properties of PN-Ab and the application of UMMD greatly improved the therapy of subcutaneous pancreatic cancer in nude mice.
Therapeutic effect and systemic toxicity of saline, PTX, PN, PN-Ab, PN-Ab+Ab, PN+U, and PN-Ab+U in vivo: (A) tumor volumes over the treatment course in various groups; (B) subcutaneous tumor specimens at the end of the treatment; (C) average weight (n=5) of tumors; (D) change in body weight following tumor cells injection during the treatment course (top-right corner), arrows point to the beginning and end of treatment; (E) Kaplan-Meier survival analysis of mice with tumors(E) (n=10). Results were shown as mean±SEM, *p<0.05, for the comparison of average tumor weight by one-way ANOVA and Dunnett T3.
Ki67 immunohistochemical staining and contrast-enhanced ultrasonography of various treatment groups.
The median survival analyzed by Kaplan-Meier analysis determined for all groups is shown in Table 2 and was as follows: PN-Ab+Ab<PN-Ab, PN<PN-Ab<PN-Ab+U, PN<PN+U. The log-rank test showed statistically significant differences among the groups (p<0.001). At the 45th day after tumor cell injection, all mice in the PN-Ab+U group were alive while 80%, 60%, 40%, 30%, 20% and 20% of mice died in the groups of saline, PTX, PN, PN-Ab+Ab, PN-Ab and PN+U, respectively (Fig. 9E). Multiple comparisons analysis by log-rank test showed significant differences between the PN and PN-Ab groups and between the PN-Ab and PN-Ab+U groups (p<0.05). Importantly, when all the mice in the PN group died, 40% of mice in the PN-Ab group were still alive and when all the mice in the PN-Ab group died, 50% of mice in the PN-Ab+U group were alive; the longest surviving mouse was alive for 92 days. These results revealed that the targeted property and the employment of UMMD could significantly prolong the survival of mice bearing pancreatic cancer. However, no significant difference was observed between PTX and PN groups, or between PN and PN+U groups, further supporting that targeted delivery of PTX by PN-Ab is a key factor for the successful therapy.
To confirm the effect of various PTX formulations on systemic toxicity and the metabolism, we recorded the daily weights of mice after tumor cells injection and tested the serum markers. Figure 9D shows the daily weight changes throughout the course of treatment. There was no statistically significant difference in the weights of mice in various groups at any time during the treatment, indicating that the targeted NPs and UMMD caused no toxicity. However, the PN+U group had the highest albumin, and the PTX group had the lowest albumin but the highest AST, ALT, LDH, and CK-MB with significant statistical differences in the albumin. These results clearly indicate that PTX, when loaded in NPs, is less toxic than the free PTX. In other words, the mPEG-PLGA-PLL carrier could reduce the side effect of PTX. Furthermore, the serum albumin of mice in NPs groups and NPs combined with UMMD groups were higher than that in control group, suggesting that the effective therapy may relieve the systemic cachexia of malignancy. These results suggest that PN-Ab combined with UMMD is a promising strategy for targeted delivery of PTX to pancreatic cancer, significantly enhancing the treatment efficiency with no serious side effects.
Conclusion
The PTX-loaded mPEG-PLGA-PLL-anti CA19-9 NPs could target and deliver PTX to capan-1 cells in vitro and to subcutaneous pancreatic tumors in BABL/c nude mice in vivo. Furthermore, administration of UMMD greatly increased the cellular uptake in vitro and enhanced the EPR effect in vivo. Importantly, the enhanced antitumor effect of targeted and drug-loaded NPs with and without UMMD was closely associated with their transport, distribution, metabolism, and elimination both in vitro and in vivo. In particular, the total cellular uptake and PTX accumulation in subcutaneous tumors were consistent with the treatment efficacy. PTX-NP-anti-CA19-9 combined with UMMD significantly improved PTX uptake, cell cycle arrest and cell apoptosis in vitro while reducing the IC50. In vivo, the combined application of PTX-NP-antiCA19-9 with UMMD attained the highest tumor inhibition, prolonged the survival, and improved the pharmacokinetic profile as well as the general condition of the tumor-bearing nude mice without obvious toxicity. Interestingly, PTX-NP was inferior to PTX in vitro while superior in vivo. This might be due to the fact that the advantages of mPEG-PLGA-PLL NPs including RES escape, long circulation time, and sustained release could only be fully displayed in vivo. Our study demonstrated the superiority of PTX-NP-anti-CA19-9 combined with UMMD in pancreatic cancer therapy.
Supplementary Material
Supplementary figures S1-S9.
Abbreviations
mPEG-PLGA-PLL: methoxy polyethylene glycol-polylacticco-glycolic acid-polylysine
NPs: Nanoparticles
UMMD: Ultrasound mediated microbubble destruction
PTX: Paclitaxol
EPR effect: The enhanced permeability and retention effect
IC50: Half maximal inhibitory concentration
EDC: N-(3-Dimethylaminopropyl)-N'-ethylcarbodiimide hydrochloride
NHS: N-hydroxysuccinimide
FTIR: Fourier transform infrared spectroscopy
HPLC: High performance liquid chromatogram
PTX: free PTX; PN: PTX-NP; PN-Ab: PTX-NP-antiCA19-9; PN-Ab+Ab: PTX-NP-anti CA19-9 plus anti CA19-9; PN+U: PTX-NP plus UMMD; PN-Ab+U: PTX-NP-anti CA19-9 plus UMMD.
TUNEL: Terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling
AUC: Area under curve; t1/2: Half time; MRT: Mean residence time; CL: Clearance
LDH: Lactate dehydrogenase; CK: Creatine kinase; CK-MB: Creatine kinase MB; AST: Aspartate aminotransferase; ALT: Alanine aminotransferase; BU: Blood urea.
CEUS: contrast enhanced ultrasonography.
Acknowledgements
This study was supported by the National Natural Science Foundation of China (No. 81171352, 81271596, 81571677 and 81571679).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA: a cancer journal for clinicians. 2014;64:9-29
2. Sun C, Ansari D, Andersson R, Wu DQ. Does gemcitabine-based combination therapy improve the prognosis of unresectable pancreatic cancer? World journal of gastroenterology: WJG. 2012;18:4944-58
3. Fokas E, O'Neill E, Gordon-Weeks A, Mukherjee S, McKenna WG, Muschel RJ. Pancreatic ductal adenocarcinoma: From genetics to biology to radiobiology to oncoimmunology and all the way back to the clinic. Biochimica et biophysica acta. 2015;1855:61-82
4. Schober M, Jesenofsky R, Faissner R, Weidenauer C, Hagmann W, Michl P. et al. Desmoplasia and chemoresistance in pancreatic cancer. Cancers. 2014;6:2137-54
5. Erkan M, Hausmann S, Michalski CW, Fingerle AA, Dobritz M, Kleeff J. et al. The role of stroma in pancreatic cancer: diagnostic and therapeutic implications. Nature reviews Gastroenterology & hepatology. 2012;9:454-67
6. Durymanov MO, Rosenkranz AA, Sobolev AS. Current Approaches for Improving Intratumoral Accumulation and Distribution of Nanomedicines. Theranostics. 2015;5:1007-20
7. Sun T, Zhang YS, Pang B, Hyun DC, Yang M, Xia Y. Engineered nanoparticles for drug delivery in cancer therapy. Angewandte Chemie. 2014;53:12320-64
8. Du JZ, Mao CQ, Yuan YY, Yang XZ, Wang J. Tumor extracellular acidity-activated nanoparticles as drug delivery systems for enhanced cancer therapy. Biotechnology advances. 2014;32:789-803
9. Muthu MS, Leong DT, Mei L, Feng SS. Nanotheranostics - application and further development of nanomedicine strategies for advanced theranostics. Theranostics. 2014;4:660-77
10. Yang T, Zhao P, Rong Z, Li B, Xue H, You J. et al. Anti-tumor Efficiency of Lipid-coated Cisplatin Nanoparticles Co-loaded with MicroRNA-375. Theranostics. 2016;6:142-54
11. Hong CA, Nam YS. Functional nanostructures for effective delivery of small interfering RNA therapeutics. Theranostics. 2014;4:1211-32
12. Liu P, Yu H, Sun Y, Zhu M, Duan Y. A mPEG-PLGA-b-PLL copolymer carrier for adriamycin and siRNA delivery. Biomaterials. 2012;33:4403-12
13. Wang Y, Liu P, Qiu L, Sun Y, Zhu M, Gu L. et al. Toxicity and therapy of cisplatin-loaded EGF modified mPEG-PLGA-PLL nanoparticles for SKOV3 cancer in mice. Biomaterials. 2013;34:4068-77
14. Liu P, Qin L, Wang Q, Sun Y, Zhu M, Shen M. et al. cRGD-functionalized mPEG-PLGA-PLL nanoparticles for imaging and therapy of breast cancer. Biomaterials. 2012;33:6739-47
15. Poruk KE, Gay DZ, Brown K, Mulvihill JD, Boucher KM, Scaife CL. et al. The clinical utility of CA 19-9 in pancreatic adenocarcinoma: diagnostic and prognostic updates. Current molecular medicine. 2013;13:340-51
16. Zhang Y, Yang J, Li H, Wu Y, Zhang H, Chen W. Tumor markers CA19-9, CA242 and CEA in the diagnosis of pancreatic cancer: a meta-analysis. International journal of clinical and experimental medicine. 2015;8:11683-91
17. Fong ZV, Winter JM. Biomarkers in pancreatic cancer: diagnostic, prognostic, and predictive. Cancer journal. 2012;18:530-8
18. Tan S, Wu T, Zhang D, Zhang Z. Cell or cell membrane-based drug delivery systems. Theranostics. 2015;5:863-81
19. Zhu J, Huang H, Dong S, Ge L, Zhang Y. Progress in aptamer-mediated drug delivery vehicles for cancer targeting and its implications in addressing chemotherapeutic challenges. Theranostics. 2014;4:931-44
20. Kooiman K, Vos HJ, Versluis M, de Jong N. Acoustic behavior of microbubbles and implications for drug delivery. Advanced drug delivery reviews. 2014;72:28-48
21. Faez T, Emmer M, Kooiman K, Versluis M, van der Steen A, de Jong N. 20 years of ultrasound contrast agent modeling. IEEE transactions on ultrasonics, ferroelectrics, and frequency control. 2013;60:7-20
22. Zhao YZ, Tian XQ, Zhang M, Cai L, Ru A, Shen XT. et al. Functional and pathological improvements of the hearts in diabetes model by the combined therapy of bFGF-loaded nanoparticles with ultrasound-targeted microbubble destruction. Journal of controlled release: official journal of the Controlled Release Society. 2014;186:22-31
23. Carson AR, McTiernan CF, Lavery L, Grata M, Leng X, Wang J. et al. Ultrasound-targeted microbubble destruction to deliver siRNA cancer therapy. Cancer research. 2012;72:6191-9
24. Noble ML, Kuhr CS, Graves SS, Loeb KR, Sun SS, Keilman GW. et al. Ultrasound-targeted microbubble destruction-mediated gene delivery into canine livers. Molecular therapy: the journal of the American Society of Gene Therapy. 2013;21:1687-94
25. Kopechek JA, Carson AR, McTiernan CF, Chen X, Hasjim B, Lavery L. et al. Ultrasound Targeted Microbubble Destruction-Mediated Delivery of a Transcription Factor Decoy Inhibits STAT3 Signaling and Tumor Growth. Theranostics. 2015;5:1378-87
26. Liu HL, Fan CH, Ting CY, Yeh CK. Combining microbubbles and ultrasound for drug delivery to brain tumors: current progress and overview. Theranostics. 2014;4:432-44
27. Sirsi SR, Borden MA. Advances in ultrasound mediated gene therapy using microbubble contrast agents. Theranostics. 2012;2:1208-22
28. Jin LF, Li F, Wang HP, Wei F, Qin P, Du LF. Ultrasound targeted microbubble destruction stimulates cellular endocytosis in facilitation of adeno-associated virus delivery. International journal of molecular sciences. 2013;14:9737-50
29. Delalande A, Leduc C, Midoux P, Postema M, Pichon C. Efficient Gene Delivery by Sonoporation Is Associated with Microbubble Entry into Cells and the Clathrin-Dependent Endocytosis Pathway. Ultrasound in medicine & biology. 2015;41:1913-26
30. Meijering BD, Juffermans LJ, van Wamel A, Henning RH, Zuhorn IS, Emmer M. et al. Ultrasound and microbubble-targeted delivery of macromolecules is regulated by induction of endocytosis and pore formation. Circulation research. 2009;104:679-87
31. Syed A, Chan WC. How nanoparticles interact with cancer cells. Cancer treatment and research. 2015;166:227-44
32. Pearce TR, Shroff K, Kokkoli E. Peptide targeted lipid nanoparticles for anticancer drug delivery. Advanced materials. 2012;24:3803-22 710
33. Pae J, Pooga M. Peptide-mediated delivery: an overview of pathways for efficient internalization. Therapeutic delivery. 2014;5:1203-22
34. Oh N, Park JH. Endocytosis and exocytosis of nanoparticles in mammalian cells. International journal of nanomedicine. 2014;9(Suppl 1):51-63
35. Shi Q, Liu P, Sun Y, Zhang H, Du J, Li F. et al. siRNA delivery mediated by copolymer nanoparticles, phospholipid stabilized sulphur hexafluoride microbubbles and ultrasound. Journal of biomedical nanotechnology. 2014;10:436-44
36. Yin P, Wang Y, Qiu Y, Hou L, Liu X, Qin J. et al. Bufalin-loaded mPEG-PLGA-PLL-cRGD nanoparticles: preparation, cellular uptake, tissue distribution, and anticancer activity. International journal of nanomedicine. 2012;7:3961-9
37. Liu P, Sun Y, Wang Q, Sun Y, Li H, Duan Y. Intracellular trafficking and cellular uptake mechanism of mPEG-PLGA-PLL and mPEG-PLGA-PLL-Gal nanoparticles for targeted delivery to hepatomas. Biomaterials. 2014;35:760-70
38. Akita H, Enoto K, Tanaka H, Harashima H. Particle tracking analysis for the intracellular trafficking of nanoparticles modified with African swine fever virus protein p54-derived peptide. Molecular therapy: the journal of the American Society of Gene Therapy. 2013;21:309-17
39. Chen L, She X, Wang T, He L, Shigdar S, Duan W. et al. Overcoming acquired drug resistance in colorectal cancer cells by targeted delivery of 5-FU with EGF grafted hollow mesoporous silica nanoparticles. Nanoscale. 2015;7:14080-92
40. Yang C, Hu T, Cao H, Zhang L, Zhou P, He G. et al. Facile Construction of Chloroquine Containing PLGA-Based pDNA Delivery System for Efficient Tumor and Pancreatitis Targeting in Vitro and in Vivo. Molecular pharmaceutics. 2015;12:2167-79
41. Alqahtani S, Simon L, Astete CE, Alayoubi A, Sylvester PW, Nazzal S. et al. Cellular uptake, antioxidant and antiproliferative activity of entrapped alpha-tocopherol and gamma-tocotrienol in poly (lactic-co-glycolic) acid (PLGA) and chitosan covered PLGA nanoparticles (PLGA-Chi). Journal of colloid and interface science. 2015;445:243-51
42. Tomasina J, Poulain L, Abeilard E, Giffard F, Brotin E, Carduner L. et al. Rapid and soft formulation of folate-functionalized nanoparticles for the targeted delivery of tripentone in ovarian carcinoma. International journal of pharmaceutics. 2013;458:197-207
43. Liu C, Yu W, Chen Z, Zhang J, Zhang N. Enhanced gene transfection efficiency in CD13-positive vascular endothelial cells with targeted poly(lactic acid)-poly(ethylene glycol) nanoparticles through caveolae-mediated endocytosis. Journal of controlled release: official journal of the Controlled Release Society. 2011;151:162-75
44. Jiang X, Xin H, Ren Q, Gu J, Zhu L, Du F. et al. Nanoparticles of 2-deoxy-D-glucose functionalized poly(ethylene glycol)-co-poly(trimethylene carbonate) for dual-targeted drug delivery in glioma treatment. Biomaterials. 2014;35:518-29
45. Benezra M, Phillips E, Overholtzer M, Zanzonico PB, Tuominen E, Wiesner U. et al. Ultrasmall integrin-targeted silica nanoparticles modulate signaling events and cellular processes in a concentration-dependent manner. Small. 2015;11:1721-32
46. Hernot S, Klibanov AL. Microbubbles in ultrasound-triggered drug and gene delivery. Advanced drug delivery reviews. 2008;60:1153-66
47. Chen H, Hwang JH. Ultrasound-targeted microbubble destruction for chemotherapeutic drug delivery to solid tumors. Journal of therapeutic ultrasound. 2013;1:10
48. Zhang L, Zhu D, Dong X, Sun H, Song C, Wang C. et al. Folate-modified lipid-polymer hybrid nanoparticles for targeted paclitaxel delivery. International journal of nanomedicine. 2015;10:2101-14
49. Shi Y, van der Meel R, Theek B, Oude Blenke E, Pieters EH, Fens MH. et al. Complete Regression of Xenograft Tumors upon Targeted Delivery of Paclitaxel via Pi-Pi Stacking Stabilized Polymeric Micelles. ACS nano. 2015;9:3740-52
50. He X, Li L, Su H, Zhou D, Song H, Wang L. et al. Poly(ethylene glycol)-block-poly(epsilon-caprolactone)-and phospholipid-based stealth nanoparticles with enhanced therapeutic efficacy on murine breast cancer by improved intracellular drug delivery. International journal of nanomedicine. 2015;10:1791-804
51. Ma P, Zhang X, Ni L, Li J, Zhang F, Wang Z. et al. Targeted delivery of polyamidoamine-paclitaxel conjugate functionalized with anti-human epidermal growth factor receptor 2 trastuzumab. International journal of nanomedicine. 2015;10:2173-90
52. Horwitz SB. Taxol (paclitaxel): mechanisms of action. Annals of oncology: official journal of the European Society for Medical Oncology / ESMO. 1994;5(Suppl 6):S3-6
53. Arbuck SG. Taxol (paclitaxel): future directions. Annals of oncology: official journal of the European Society for Medical Oncology / ESMO. 1994;5(Suppl 6):S59-62
54. Tian H, Gao Z, Li H, Zhang B, Wang G, Zhang Q. et al. DNA damage response-a double-edged sword in cancer prevention and cancer therapy. Cancer letters. 2015;358:8-16
55. Lobrich M, Jeggo PA. The impact of a negligent G2/M checkpoint on genomic instability and cancer induction. Nature reviews Cancer. 2007;7:861-9
56. Bekeredjian R, Kroll RD, Fein E, Tinkov S, Coester C, Winter G. et al. Ultrasound targeted microbubble destruction increases capillary permeability in hepatomas. Ultrasound in medicine & biology. 2007;33:1592-8
57. Yang D, Tan KB, Gao YH, Liu H, Yang WX. Effects of diagnostic ultrasound-targeted microbubble destruction on permeability of normal liver in rats. Ultrasonics. 2012;52:1065-71
58. Wang G, Zhuo Z, Xia H, Zhang Y, He Y, Tan W. et al. Investigation into the impact of diagnostic ultrasound with microbubbles on the capillary permeability of rat hepatomas. Ultrasound in medicine & biology. 2013;39:628-37
59. Adiseshaiah PP, Hall JB, McNeil SE. Nanomaterial standards for efficacy and toxicity assessment. Wiley interdisciplinary reviews Nanomedicine and nanobiotechnology. 2010;2:99-112
60. Prabhakarpandian B, Shen MC, Nichols JB, Garson CJ, Mills IR, Matar MM. et al. Synthetic tumor networks for screening drug delivery systems. Journal of controlled release: official journal of the Controlled Release Society. 2015;201:49-55
61. Kobayashi H, Watanabe R, Choyke PL. Improving conventional enhanced permeability and retention (EPR) effects; what is the appropriate target? Theranostics. 2013;4:81-9
62. Cohen R, Neuzillet C, Tijeras-Raballand A, Faivre S, de Gramont A, Raymond E. Targeting cancer cell metabolism in pancreatic adenocarcinoma. Oncotarget. 2015;6:16832-47
63. Feig C, Gopinathan A, Neesse A, Chan DS, Cook N, Tuveson DA. The pancreas cancer microenvironment. Clinical cancer research: an official journal of the American Association for Cancer Research. 2012;18:4266-76
64. Yamashita Y, Ueda K, Itonaga M, Yoshida T, Maeda H, Maekita T. et al. Tumor vessel depiction with contrast-enhanced endoscopic ultrasonography predicts efficacy of chemotherapy in pancreatic cancer. Pancreas. 2013;42:990-5
65. Akasu G, Kawahara R, Yasumoto M, Sakai T, Goto Y, Sato T. et al. Clinicopathological analysis of contrast-enhanced ultrasonography using perflubutane in pancreatic adenocarcinoma. The Kurume medical journal. 2012;59:45-52
Author contact
![]() Corresponding authors: Dr. Lianfang Du, Department of Ultrasound, Shanghai General Hospital Affiliated to Shanghai Jiao Tong University School of Medicine, 85 Wu Jin Road, Shanghai 200080, China. Tel: +86-13386259562; Fax: (0086) 021-37798276; E-mail: du_lianfang2013com or Dr. Yourong Duan (yrduanorg).
Corresponding authors: Dr. Lianfang Du, Department of Ultrasound, Shanghai General Hospital Affiliated to Shanghai Jiao Tong University School of Medicine, 85 Wu Jin Road, Shanghai 200080, China. Tel: +86-13386259562; Fax: (0086) 021-37798276; E-mail: du_lianfang2013com or Dr. Yourong Duan (yrduanorg).