Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Supplementary Material
Author Contributions
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2016; 6(10):1629-1640. doi:10.7150/thno.15253 This issue Cite
Research Paper
High-Resolution PET Imaging with Therapeutic Antibody-based PD-1/PD-L1 Checkpoint Tracers
Michael Hettich1,3, Friederike Braun2,3, Mark D. Bartholomä2, Reinhold Schirmbeck4, Gabriele Niedermann1,5,6 ![]()
1. Department of Radiation Oncology, Medical Center - University of Freiburg, D-79106 Freiburg, Germany.
2. Department of Nuclear Medicine, Medical Center - University of Freiburg, D-79106 Freiburg, Germany.
3. Faculty of Biology, University of Freiburg, D-79104 Freiburg, Germany.
4. Department of Internal Medicine I, Medical Center - University of Ulm, D-89081 Ulm, Germany.
5. German Cancer Consortium (DKTK), D-79106 Freiburg, Germany.
6. German Cancer Research Center (DKFZ), D-69120 Heidelberg, Germany.
Received 2016-2-12; Accepted 2016-4-29; Published 2016-6-18
Abstract

Checkpoint-blocking antibodies like those targeting the PD-1/PD-L1 pathway have revolutionized oncology. We developed radiotracers based on therapeutic checkpoint-blocking antibodies permitting sensitive and high-resolution PET imaging of both PD-1 and PD-L1 in immunocompetent mice. ImmunoPET of naive mice revealed similar overall expression patterns for PD-1 and PD-L1 in secondary lymphoid organs (spleen and lymph nodes). Interestingly, PD-L1 was also detected in brown adipose tissue (BAT), confirming the notion that BAT is immunologically relevant. Under pathophysiological conditions, strong expression of the receptor/ligand pair was also found in non-lymphoid tissues. Both were specifically detected in malignant tumors. PD-1 was readily detected after combined immunoradiotherapy causing massive tumor infiltration by PD-1+ lymphocytes. PD-L1 tracer uptake was reduced in PD-L1 knockout tumors. Moreover, monitoring the expression changes of PD-L1 in response to its main inducer, the effector T cell cytokine IFN-γ, revealed robust upregulation in the lung. This suggests that T cell responses in the lung, a vital organ continuously exposed to a variety of antigens, are strongly restrained by the PD-1 checkpoint. In turn, this could explain the association of PD-1 checkpoint inhibition with potentially fatal immune-mediated pneumonitis and partially also its efficacy in lung cancer.
Keywords: non-invasive imaging, PET, PD-1/PD-L1 checkpoint, antibody imaging.
Introduction
Immune checkpoints are crucial for maintaining self-tolerance and modulating the duration and amplitude of immune responses. In tumors, they constitute a major mechanism of immune resistance, particularly against tumor antigen-specific T cells. Immune checkpoints are usually triggered by the interaction of negative regulatory lymphocyte co-receptors and their ligands, and can thus be blocked by antagonistic antibodies to either the receptor or the ligand [1, 2].
Programmed cell death-1 (PD-1) is a critical negative co-receptor expressed on activated lymphocytes [3]. On cells from solid tumors, the major PD-1 ligand expressed is PD-L1 (B7-H1) [4]. The PD pathway [4, 5] can be blocked with antagonistic antibodies to either PD-1 or PD-L1, resulting in greatly enhanced antitumor immune responses [6].
After the preclinical demonstration of immune checkpoint blockade-induced tumor regression in mice, checkpoint antibodies have been developed for clinical use [2, 7]. Following initial reports of clinical benefits in advanced melanoma patients treated with the CTLA-4 checkpoint blocker ipilimumab, trials testing anti-PD-1/PD-L1 checkpoint antibodies have demonstrated better responses compared to standard therapy in advanced melanoma, as well as in lung and kidney cancer, leading to initial FDA approvals. Based on these encouraging results, these antibodies are currently evaluated either alone or in combination with CTLA-4 antibodies or other therapies in several dozen clinical trials for the treatment of multiple tumor types [8].
Successful PD checkpoint blockade requires the expression of both PD-1 and PD-L1 in the tumor. Thus, the response rates are often higher for PD-L1-positive tumors than for tumors that appear to be PD-L1 negative by immunohistochemistry (IHC), which led to great efforts to develop reliable assays for the assessment of intratumoral PD-L1 and PD-1 expression by IHC [9-11]. However, IHC requires invasive methods such as surgical resection or biopsy to obtain tumor material. Moreover, checkpoint molecule expression can be highly dynamic, varying during the time course of the malignant disease or post-treatment [12]. Indeed, expression can be heterogeneous in a given tumor or in different metastases [10]. Clinically relevant methods permitting the whole-body non-invasive detection of PD-1 and PD-L1 at high sensitivity and resolution could thus be highly useful in patient selection and treatment monitoring [11]. Non-invasive imaging in mice of either PD-L1 [13-16] or PD-1 [17] has recently been reported. PD-L1 imaging has been performed in immunocompromised mice with xenograft tumors expressing human PD-L1 [13, 14, 16]. One SPECT study with an anti-murine PD-L1-tracer visualized tumor and spleen in immunocompetent mice [15]. Tumor and spleen were also clearly visualized with a 64Cu-DOTA-anti-murine PD-1 mAb tracer in immunocompetent mice.
Here, we developed immunoPET tracers [18] for imaging of both PD-1 and PD-L1 using anti-murine checkpoint-blocking antibodies with proven antitumoral activity [19, 20]. We used the chelator 1,4,7-triazacyclononane-1,4,7-triacetic acid (NOTA) which forms stable complexes with the radioisotope 64Cu (t1/2 = 12.7 h) [21-23]. These tracers permitted the non-invasive determination of PD-1 and PD-L1 expression in malignant tumors and the biodistribution monitoring of the checkpoint-blocking antibodies. In addition, they allowed the monitoring of PD-1 and PD-L1 expression in naive immunocompetent mice with high sensitivity and resolution, yielding novel insights into the physiological expression patterns of this important receptor/ligand pair. In addition, this is the first study, which unequivocally demonstrates imaging specificity by using PD-1 and PD-L1 knockout mice. Moreover, we visualized body-wide PD-L1 expression changes in response to its major inducer, the cytokine IFN-γ [5, 24].
Materials and Methods
Mice, cell lines and tumor experiments
All animal experiments were performed in accordance with the German Animal License Regulations and were approved by the animal care committee of the Regierungspräsidium Freiburg (registration number: G-13/101). Animals were kept under standard pathogen-free conditions. C57BL/6N mice were purchased from Janvier Labs. PD-1-deficient (PD-1-/-) [25] and PD-L1-deficient (B7-H1-/-/PD-L1-/-) [26] mice were kindly provided by Dr. Tasuku Honjo (Kyoto University) and Dr. Lieping Chen (Yale University), respectively. All experiments were performed with mice 8-12 weeks of age. For immunoPET studies of PD-1 tracer uptake in tumors, mice were subcutaneously (s.c.) injected with 2×105 CD133-expressing B16F10 melanoma cells into the right flank. Evaluation of PD-L1 tracer uptake was performed in mice injected with 2×105 CD133-expressing B16F10 cells into the left flank along with 8×105 PD-L1-deficient B16F10 melanoma cells into the right flank.
Generation of PD-L1-deficient B16F10 melanoma cells
CRISPR/Cas9 technology [27] was used to generate PD-L1 knockout (PD-L1 KO) B16F10 melanoma cells. Cells were simultaneously transfected with pGS-CMV-hCAS9 and three different guideRNA vectors (pGS-U6-gRNA; GenScript) with the following PD-L1-specific sequences: TAGAAAACATCATTCGCTG, CTTCCCACTCACGGGTTGG, TATGGCAGCAACGTCACGA. Transfection was performed with Lipofectamine 2000 (Invitrogen) following the manufacturer's protocol. KO efficacy was tested via cultivation of transduced and antibiotic-selected cells in the presence of (50 IU/ml) IFN-γ (BioLegend) for 24 h and subsequent analysis of PD-L1 surface expression. Transduced cells were FACS-sorted for PD-L1 deficiency.
Immunoradiotherapy
Tumor growth of mice bearing subcutaneous B16F10 melanoma tumors was followed by caliper measurement. When tumors reached 500 mm3, local tumor radiation therapy was started and consisted of 2 × 12 Gy on consecutive days. Irradiation was performed with a RadSource RS2000 γ-irradiator operated at 160 kV with 0.3 mm copper filtration and a dose rate of 3.93 Gy min-1. Only the subcutaneous tumors were irradiated while the rest of the body was protected by 3 mm lead shielding. Mice were turned upside down for the second irradiation to ensure dose homogeneity. The second tumor irradiation was followed by the injection of anti-PD-L1 and anti-CTLA-4 checkpoint-blocking antibodies (BioXCell; 200 µg, intraperitoneal). Five days later, the tumor-bearing mice were injected with 64Cu-NOTA-PD-1 mAb and imaged 24 h thereafter.
IFN-γ treatment of mice and preparation of the lungs for FACS analysis
Recombinant mouse IFN-γ (5×104 IU; BioLegend) was injected intravenous (i.v.) daily for 4 days. IFN-γ-treated animals were then subjected to PET/CT imaging and ex vivo biodistribution or lung tissue analysis by FACS. For FACS analysis, the lungs of treated mice were extracted, perfused via the right ventricle and rinsed with PBS to remove blood cells. Single-cell suspensions were generated with a Miltenyi lung dissociation kit according to the manufacturer's instructions. Cells were further stained with fluorochrome-conjugated mAbs, and PD-L1 expression was analyzed on a BD FACSVerse flow cytometer.
Preparation of brown fat (BAT) for FACS analysis
BAT was dissociated with a mouse/rat adipose tissue dissociation kit (Miltenyi) according to the manufacturer's instructions.
Preperation of radiolabeled antibodies
α-PD-1 (RMP1-14) and α-PD-L1 (10F.9G2) were purchased from BioXCell and conjugated with the metal chelator S-2-(4-isothiocyanatobenzyl)-1,4,7-triazacyclononane-1,4,7-triacetic acid (p-SCN-Bn-NOTA), as described before [28]. Radioactive 64Cu (Eberhard Karls University, Tübingen, Germany) was used for mAb loading as described before [28]. In vivo antibody binding specificity was tested with the same mAbs labeled with AlexaFluor 680 (AF680-SAIVI Kit, Invitrogen) according to the manufacturer's instructions.
ImmunoPET and CT imaging
ImmunoPET was performed 24 h after i.v. injection of either 1.5 µg 64Cu-NOTA-PD-1 or 20 μg 64Cu-NOTA-PD-L1 mAb using a microPET Focus 120 (Concorde), immediately followed by CT imaging in two-bed positions with a tube voltage of 40 keV and a tube current of 1 mA (microCT scanner; CT Imaging). Mice were anesthetized using 2% isoflurane/O2 during PET/CT imaging. The scanning duration for immunoPET was dependent on the injected activity and elapsed time since tracer injection. PD-1 immunoPET signals were recorded for 45-55 minutes. PD-L1 immunoPET scans were acquired for 15 - 20 minutes. For all scans, a total count of at least 4,5 Mio was recorded.
PET/CT image analysis
A routine 2D ordered subset expectation maximization (OSEM2D) algorithm provided by the scanner software was used to reconstruct PET images with a resolution of 1.5 mm. CT scans were reconstructed with a resolution of 120 µm and a T30 kernel, using the software provided by the manufacturer. Fusion of the PET and CT images was performed with AMIDE software 1.0.5 [29]. Quantitative analysis of radiotracer uptake and image display was performed with AMIDE. The reported values represent the mean activity concentration expressed as percent injected activity per cubic centimeter of tissue (% IA/cm3).
Ex vivo biodistribution
After PET/CT imaging, mice were euthanized, organs and blood collected, flushed and weighed, and the activity was measured with a Wizard2 gamma counter (PerkinElmer). All values were decay- and background corrected and expressed as percent of injected activity per gram tissue (% IA/g) using a standard of 100% of the injected dose. Left- and right-side lymph nodes were pooled for biodistribution analysis.
Flow cytometry
Flow cytometric analysis of single-cell suspensions from lymph nodes, spleen, tumor-infiltrating lymphocytes (TILs), and tumor cells was performed using α-PD-1, α-PD-L1, α-CD45 (all BioLegend), α-CD3 (BD Pharmingen), α-CD4, α-CD8, α-CD19 (all eBioscience), α-PD-1-AF680, and α-PD-L1-AF680. Adipocytes were identified after BAT dissociation (as described above) and subsequent staining with α-PAT-2 (mouse IgM; Santa Cruz) and α-mouse IgM (eBioscience) as secondary antibodies. Cells were analyzed using a BD FACSVerse flow cytometer with FACSuite software (Becton Dickinson).
Statistics
Results are presented as means ± SD. Data were compared using the unpaired two-tailed Student's t-test or an ordinary one-way ANOVA with Tukey's multiple comparison test when comparing more than two groups. A P-value <0.05 was considered significant. All analyses were performed using Prism 6.0 (GraphPad software).
Results
Tracer development and characterization
After conjugation of the PD-1 and PD-L1 mAbs with the chelator NOTA, which resulted in 2,5 +/- 0,5 and 1,4 +/- 0,1 chelators per molecule of antibody, respectively (Fig. S1), we radiolabeled the conjugates with 64Cu. The radiochemical purity of both tracers exceeded 94%, with specific activities of 108.7 ± 31.9 MBq/nmol for the 64Cu-NOTA-PD-1 mAb and 81.6 ± 30.2 MBq/nmol for the 64Cu-NOTA-PD-L1 mAb. Conjugation of the chelator to the antibodies did not impair binding to their respective epitopes, revealed through side-by-side flow cytometric titration of both the NOTA-PD-1 and NOTA-PD-L1 mAbs against the corresponding unmodified antibodies (Fig. S2).
Detection of PD-1 in naive mice
The most important prerequisite for effective immunoPET imaging is the cell surface expression of the target molecules. PD-1 is expressed on activated but not on resting T, B, and myeloid cells [3]. Thus, in the absence of pathogenic stimuli, only few cells express PD-1. Accordingly, we detected PD-1 only on a small percentage of cells in secondary lymphoid organs (spleen and lymph nodes) of healthy untreated mice (Fig. 1A and B, left panels), most of which were CD3+ T cells (Fig. S3A). The expression level per cell was low (Fig. 1C). The absolute number of PD-1-positive CD4 T cells was highest compared to CD8 T cells, CD19 B cells, or CD45+ non-lymphocytes (Fig. 1A and B, right panels). Immunosuppressive regulatory T cells (CD4+ CD25+ FoxP3+) constituted approximately half of the PD-1+ cells among the CD4+ cells (Fig. S3B and C). The surrogate PD-1 checkpoint-blocking antibody used in this study indeed bound specifically to its target cells in vivo, as demonstrated by injecting a fluorescently labeled version into C57BL/6 mice followed by ex vivo co-staining of lymph node and splenic single-cell suspensions with a mAb detecting an alternative PD-1 epitope. PD-1-deficient mice [25] served as additional specificity control (Fig. 1C).
PD-1 immunoPET/CT in naive mice. Frequency of PD-1+ and PD-1- cells in CD45+ leukocytes and the absolute cell count of PD-1+ and PD-1- cells among CD4 and CD8 T cells, CD19 B cells, and other leukocytes in spleen (A) and lymph nodes (B); n = 3 each. (C) Representative density dot plots of splenic CD4/CD8 T cells from C57BL/6 and PD-1-/- mice i.v. injected with α-PD-1-AF680 (RMP1-14), ex vivo counterstained with α-PD-1-PE (29F.1A12). (D) Representative coronal (C) and transverse (T) immunoPET/CT sections from WT, antigen-blocked, and PD-1-/- mice. White ticks in the C-sections indicate the position of the T-section. Scanning duration was ca. 45-50 min. (E) Uptake of 64Cu-NOTA-PD-1 mAb as measured by microPET in various organs of WT, antigen-blocked, and PD-1-/- mice 24 h p.i. of 1.5 µg tracer (1.13 ± 0.31 MBq). Antigen-blocked mice were pretreated with unlabeled α-PD-1 (1 mg, i.v.) 24 h before; n = 6 (WT), 5 (Block), and 3 (PD-1-/-). (F) Ex vivo biodistribution of radiotracer uptake 48 h p.i.; n = 14 (WT), 11 (Block), and 3 (KO). Abbreviations: cLN - cervical lymph node, aLN - axillary lymph node, iLN - inguinal lymph node, mLN - mesenteric lymph node, Sp - spleen, Thym - thymus, Panc - pancreas, Stom - stomach, Intest - intestines, Blad - bladder, BAT - brown adipose tissue.
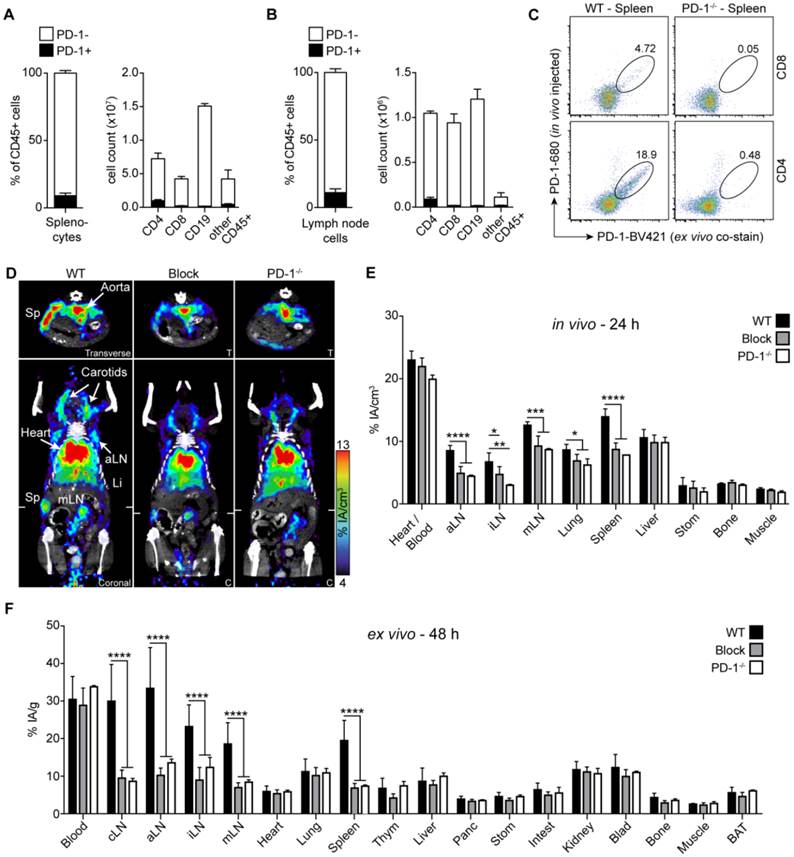
Despite the sparse expression of PD-1 in healthy mice, PET imaging with 64Cu-NOTA-PD-1 mAb enabled specific detection of the spleen and individual lymph nodes 24 h post injection (p.i.) (Fig. 1D and E; Movie S1 in Additional File 2). To confirm immunoPET specificity, we performed blocking experiments by pretreatment with cold anti-PD-1. As shown in Fig. 1D and E, the spleen and lymph node signals were strongly reduced in antigen-blocked animals. However, such blocking experiments cannot unequivocally prove the specificity of a given antibody. Thus, we also performed PD-1 PET imaging in PD-1-deficient mice. As shown in Fig. 1D, secondary lymphoid organs were not detectable in PD-1-/- mice; only strong signals for the blood pool (heart, carotids, and aorta) and a moderate one for the liver (the organ where antibodies are mainly metabolized) could be detected. These observations were confirmed by ex vivo biodistribution analysis 48 h p.i., where specific tracer uptake was only found in lymph nodes and the spleen (Fig. 1F).
Detection of PD-L1 in naive mice
PD-L1 is more broadly expressed than PD-1 both on hematopoietic and non-hematopoietic cells even under physiologic conditions [30]. Reports on its constitutive expression in non-hematopoietic tissues have been somewhat controversial [6, 24, 30]. While mRNA expression has been found in a variety of organs, protein expression has been reported for cardiac endothelium, lung macrophages, and selected cell types at immune-privileged sites including the placenta, pancreas islets, and the retina.
Our flow cytometry analyses detected PD-L1 on virtually all cells of secondary lymphoid organs in naive mice (Fig. 2A). Similar to PD-1, the constitutive per cell expression levels were low (Fig. 2B). ImmunoPET scans obtained 24 h after 64Cu-NOTA-PD-L1 mAb injection nicely visualized the spleen and lymph nodes (Fig. 2C and D, Fig. S4, Movie S2 in Additional File 3), which perfectly reflects the uniform receptor expression detected by FACS in these organs.
PD-L1 immunoPET/CT in naive mice. (A) Frequency of PD-L1+ cells among CD45+ leukocytes in spleen and lymph nodes of healthy (untreated) mice; n = 5. (B) Representative FACS histograms for PD-L1 expression on splenic or lymph node leukocytes. (C) Representative 24-h coronal (C), transverse (T), and sagittal (S) immunoPET/CT sections of WT, antigen-blocked, and PD-L1-/- mice injected with 64Cu-NOTA-PD-L1 mAb (20 µg; 6.38 ± 0.35 MBq). Antigen-blocked mice were pretreated with 1 mg unlabeled α-PD-L1 (i.v.) 24 h before. White ticks in the C-sections indicate the positions of the T- and S-sections. Scanning duration was ca. 15-20 min. (D) Uptake of 64Cu-NOTA-PD-L1 mAb as measured by microPET in various organs; n = 5 (WT and antigen-blocked), 3 (PD-L1-/-). (E) Ex vivo biodistribution of radiotracer uptake 48 h p.i.; n = 10 (WT and antigen-blocked), 3 (PD-L1-/-). (F) Upper panel: PD-L1 expression levels in BAT cell populations (CD45-positive leukocytes, CD45-negative adipocytes and non-adipocytes) from C57BL/6 and PD-L1-/- mice, calculated against values from PD-L1-/- mice. Lower panel: Comparison of CD3+ T cells and macrophages; n = 5. Abbreviations: cLN - cervical lymph node, aLN - axillary lymph node, iLN - inguinal lymph node, mLN - mesenteric lymph node, Li - liver, Sp - spleen, Thym - thymus, Panc - pancreas, Stom - stomach, Intest - intestines, Blad - bladder, BAT - brown adipose tissue.
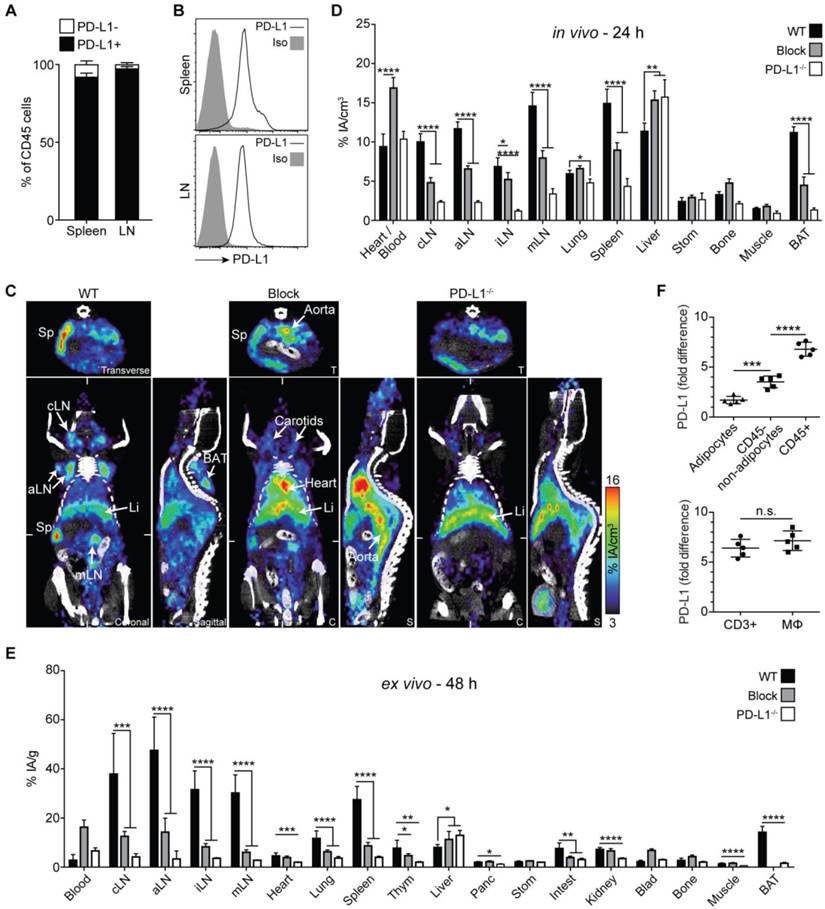
Surprisingly, we also detected relatively strong tracer uptake in the upper dorsal region (Fig. 2C, sagittal section of the wild-type [WT] panel; Movie S2). Closer inspection revealed that this tissue had the shape of a butterfly and was located between the scapulae, typical of BAT (Fig. S5; Movie S3 in Additional File 4), the main function of which is heat production [31, 32]. The tracer uptake was subsequently confirmed by ex vivo biodistribution analysis (Fig. 2E).
Antigen-blocked and PD-L1-deficient mice showed markedly reduced or completely absent uptake in lymphatic tissue and BAT, confirming the specificity of PET-based detection of PD-L1 mAb tissue uptake. Moreover, blood pool and liver uptake was higher in these animals, presumably caused by elevated levels of circulating unbound 64Cu-NOTA-PD-L1 tracer in the absence of specific binding sites (Fig. 2C-E). Besides a strong uptake in secondary lymphatic organs and BAT, ex vivo biodistribution studies revealed slightly higher PD-L1 signals in the lungs of WT compared to PD-L1-deficient mice (Fig. 2E). Additionally, slight but significant differences were found for the heart, thymus, intestines, pancreas, and kidneys.
To find out which cell type in BAT expresses PD-L1, we isolated CD45-positive leukocytes, CD45-negative adipocytes, and other CD45-negative cells. Flow cytometric analyses of BAT from WT and PD-L1-deficient mice revealed the highest PD-L1 expression for CD45+ leukocytes and only a very weak one for adipocytes. Among the leukocytes, the PD-L1 expression levels per cell were similar for macrophages and T cells (Fig. 2F).
PD-1 immunoPET detects PD-1+ TILs after combined immunoradiotherapy
Combined immunoradiotherapy consisting of high-dose hypofractionated tumor γ-irradiation together with checkpoint-blocking mAbs, as depicted in Fig. 3A, can strongly induce TILs ([33, 34]; Hettich et al., in revision). A few days after treatment start, the majority of induced TILs are CD8+ effector cytotoxic T lymphocytes (CTLs) which uniformly express PD-1 on their cell surface (Fig. 3B, Fig. S6). TILs from untreated and immunoradiotherapy-treated mice show considerably higher PD-1 expression compared with T cells in secondary lymphoid organs of healthy mice (Fig. 3B). However, after immunoradiotherapy, there is a strong increase in the numbers and the CD8/CD4 T cell ratio (data not shown).
PD-1 immunoPET/CT in tumor-bearing mice. (A) Treatment scheme for analyzed mice. Subcutaneous flank tumors were locally irradiated with 2×12 Gy when their volume reached 500 mm3, and the mice were treated with α-PD-L1/CTLA-4 as indicated. Five days later, mice received 1.5 µg 64Cu-NOTA-PD-1 mAb (ca. 1.44 ± 0.32 MBq) and PET images were acquired 24 h p.i. Antigen-blocked mice were pretreated with unlabeled α-PD-1 (1 mg) 24 h before tracer injection. Scanning duration was ca. 45-55 min. (B) Representative FACS histogram overlays acquired 5 days post treatment start comparing PD-1 expression from lymph node and tumor-infiltrating T cells. (C) Representative coronal (C), transverse (T), and sagittal (S) PD-1 immunoPET/CT sections. White ticks in the C-sections indicate the positions of the T- and S-sections. (D) Uptake of 64Cu-NOTA-PD-1 mAb as measured by microPET in various organs. n = 7 (WT) and 5 (antigen-blocked). Abbreviations: RT - radiotherapy, Sp - spleen, Tu - tumor, td iLN - tumor-draining inguinal lymph node.
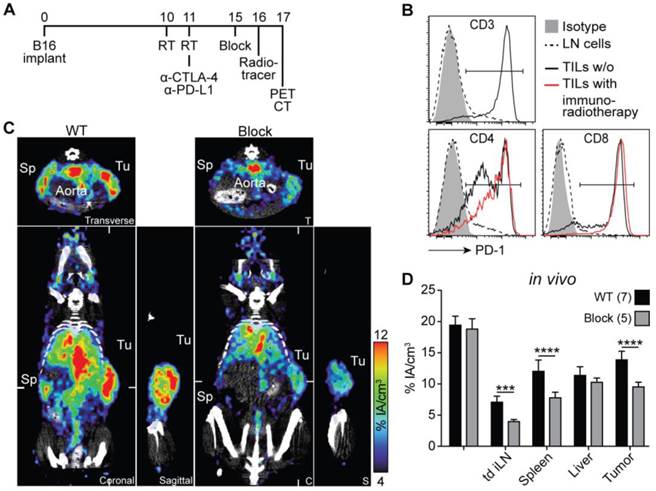
In this study, mice that were treated with immunoradiotherapy were injected with 64Cu-NOTA-PD-1 mAb and imaged 24 h thereafter (Fig. 3A). As shown in Fig. 3C and Movie S4 in Additional File 5, homogeneous and high tracer uptake was found in the tumor, which was significantly reduced in antigen-blocked animals. In these blocking experiments, not only the uptake into the tumor but also into lymph nodes (including tumor-draining lymph nodes) and the spleen was lower, whereas the blood pool and liver uptakes were unaltered (Fig. 3C and D).
PD-L1 immunoPET detects differences in intratumoral PD-L1 expression
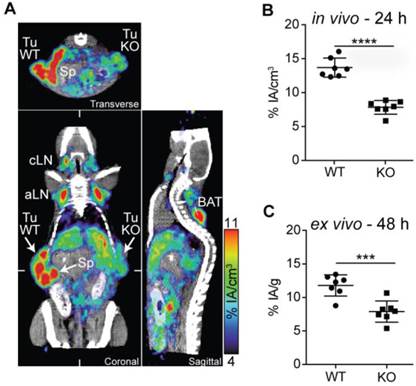
Within tumors, PD-L1 can be expressed on malignant and stromal cells including infiltrating hematopoietic cells [9, 10]. To determine if the PD-L1 radiotracer enabled the detection of differences in PD-L1 expression, we performed PD-L1 immunoPET/CT in mice bearing WT and PD-L1-deficient B16F10 melanomas on opposing flanks. PD-L1-deficient B16F10 melanoma cells were generated using the CRISPR/Cas9 technology [27]. While WT B16F10 cells exhibited basal PD-L1 expression augmented upon IFN-γ exposure, both were completely abrogated in the PD-L1 KO cells (Fig. S7). In vivo and ex vivo analysis of 64Cu-NOTA-PD-L1 tracer uptake showed a dramatically reduced signal in the PD-L1 KO tumors (Fig. 4A-C; Movie S5). Additionally, the spleen, various lymph nodes, and the BAT could again be detected with high tumor-to-background contrast (Fig. 4A; Movie S5).
PD-L1 immunoPET/CT in mice with WT and PD-L1 KO tumors. (A) C57BL/6 mice were s.c. injected with 2×105 B16F10 CD133 (left) and 8×105 B16F10 CD133 PD-L1 KO tumor cells (right). Ten days later, mice were injected with 64Cu-NOTA-PD-L1 mAb (20 µg; 11.65 ± 0.13 MBq), and 24 h later, representative coronal (C), transverse (T) and sagittal (S) immunoPET/CT scans were acquired. White ticks in the C-sections indicate the positions of the T- and S-sections. Scanning duration was ca. 15 min. (B) Uptake of 64Cu-NOTA-PD-L1 mAb as measured by microPET in various organs and (C) ex vivo biodistribution of tracer uptake into WT and PD-L1 KO tumors. n = 7. Abbreviations: Sp - spleen, Tu WT - wild-type tumor, Tu KO - PD-L1 KO tumor, cLN - cervical lymph node, aLN - axillary lymph node, BAT - brown adipose tissue.
PD-L1 immunoPET identifies the lung as an organ with pronounced IFN-γ-inducible PD-L1 expression
IFN-γ secreted by activated antiviral, antitumoral, auto- or alloreactive T lymphocytes is an important inducer of PD-L1 [5, 24]. Non-invasive PD-L1 immunoPET should facilitate the body-wide assessment of IFN-γ-inducible changes in PD-L1 expression, which could give novel insights into the regulation of PD-L1 in response to this important cytokine.
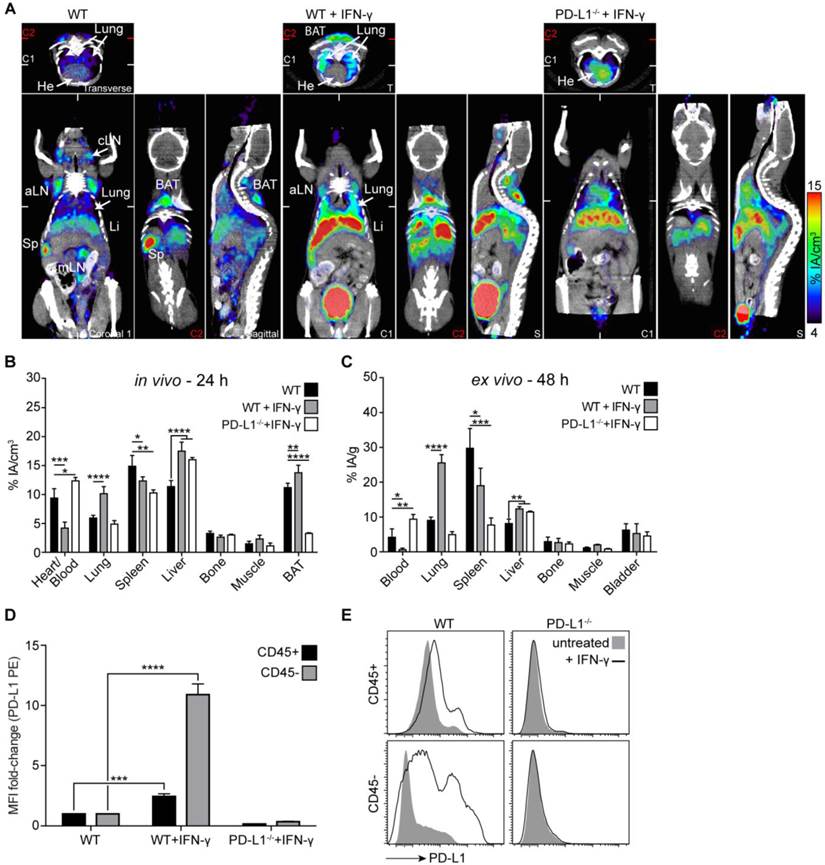
Before PET imaging with 64Cu-NOTA-PD-L1 mAb, we treated both WT and PD-L1-deficient mice systemically with IFN-γ for 4 days. As shown in Fig. 5A-C, particularly strong, specific induction of PD-L1 expression was detected in the lungs. To find out to what extent leukocytes and non-leukocytes contribute to this upregulation, we separated CD45-positive and -negative cells from single-cell suspensions generated from lung tissue. As shown in Fig. 5D and E, CD45-negative cells (i.e., non-leukocytes) exhibited much stronger PD-L1 upregulation in response to IFN-γ. The IFN-γ-inducible upregulation of PD-L1 was so strong that the tracer concentration in the blood and secondary lymphoid organs was reduced, which likely indicates that the amount of tracer was not sufficient under these conditions to saturate all binding sites in the mice (Fig. 5A-C). In vivo bladder signals were not regularly increased in IFN-γ-treated mice (Fig. 5A) and increased bladder signals, which were sometimes also detected in non-treated WT and in PD-L1 knockout mice, are likely due to radioactivity in the urine. There was no significant increase in bladder wall uptake in the ex vivo analyses (Fig. 5C).
PD-L1 immunoPET/CT after systemic IFN-γ treatment revealed the strongest PD-L1 upregulation in the lung. (A) Representative coronal (C), transverse (T), and sagittal (S) immunoPET sections of untreated WT and IFN-γ-treated (4×50,000 IU, i.v.) WT and PD-L1-/- mice injected with 64Cu-NOTA-PD-L1 mAb (20 µg; 6.49 ± 0.30 MBq). White ticks in the C-sections indicate the positions of the T- and S-sections. Two C-sections are shown with positions indicated by white and red ticks in the T-sections. Scanning duration was ca. 15-20 min. (B) Uptake of 64Cu-NOTA-PD-L1 mAb as measured by microPET in various organs and (C) ex vivo biodistribution of tracer uptake in indicated organs/tissues. n = 5 (WT and WT + IFN-γ) or 3 (PD-L1-/- + IFN-γ). (D) Ex vivo lung tissue FACS analysis of PD-L1 expression. Fold change of the mean fluorescence intensity (MFI) on CD45+ and CD45- lung cells before and after IFN-γ treatment. n = 4 (WT + IFN-γ) and 3 (WT and KO + IFN-γ). (E) Representative FACS histograms of PD-L1. Abbreviations: He - heart, Sp - spleen, cLN - cervical lymph node, aLN - axillary lymph node, mLN - mesenteric lymph node, Li - liver, BAT - brown adipose tissue.
Discussion
Here, we report on the non-invasive in vivo imaging of the receptor/ligand pair of the important PD-1/PD-L1 immune checkpoint by PET using 64Cu-NOTA-conjugated surrogate checkpoint-blocking antibodies in immunocompetent mice. These tracers permitted the imaging of PD-1 and PD-L1 expression in healthy mice, tumor-bearing mice, and mice exposed to the pathophysiologically important cytokine IFN-γ, with high sensitivity and resolution. They also permitted non-invasive imaging of the biodistribution of the two surrogate checkpoint-blocking antibodies. The results suggest grossly similar expression patterns for PD-1 and PD-L1 in healthy mice, mainly in secondary lymphoid organs (spleen and lymph nodes). Furthermore, PD-L1 immunoPET revealed rather high ligand expression in BAT. Under pathophysiological conditions, both PD-1 and PD-L1 were also detected in non-lymphoid organs and tissues. Both could be specifically detected in malignant tumors where PD-1 was strongly expressed on treatment-induced TILs, while PD-L1 immunoPET detected differences in PD-L1 expression in untreated tumors. Moreover, we found particularly pronounced PD-L1 induction in the lung in response to its major inducer IFN-γ [5, 24]. Subsequent FACS analyses assigned the highest increase to non-hematopoietic cells, suggesting that the lung, which is a vital organ continuously exposed to a variety of antigens [35], is strongly protected against immune attacks by PD-L1 upregulation.
The PD-1/PD-L1 immune checkpoint is often discussed to mainly operate in extralymphatic organs [1]. However, in naive mice, we non-invasively detected PD-1 and PD-L1 mainly in secondary lymphoid organs. This may reflect the less frequently discussed function of the PD-1/PD-L1 checkpoint in preventing the priming of autoreactive T cells (in the induction of peripheral T cell self-tolerance) and in limiting effector T cell induction [36-38]. On the other hand, the upregulation in extralymphatic tissues (including tumors) under inflammatory conditions reflects its function in inducing T cell exhaustion. T cell exhaustion limits effector T cell activity, expansion, and survival, and also protects peripheral tissue (including tumors) against immune-mediated destruction [1, 3, 6]. Our data on PD-1 and PD-L1 expression on T cells (including Tregs), B cells and non-lymphoid leukocytes in naive mice, as well as on TILs are very similar to published data [1, 3, 24, 30, 38].
Recently, SPECT and microPET imaging of PD-L1 have been reported in mice. Two studies using human-specific probes visualized tumor xenografts expressing human PD-L1 in immunodeficient mice [13, 14]. However, given the nature of these probes, information on endogenous expression of PD-L1 could not be provided. In one SPECT study with radiolabeled anti-murine PD-L1 in immunocompetent mice, PD-L1 was specifically detected in tumor and spleen [15]. Contrary to our results, neither lymph nodes nor BAT were detected, presumably because of the relatively low spatial resolution and sensitivity of SPECT. Chatterjee et al. [16], using a human/mouse cross-reactive PD-L1 SPECT tracer, detected tumors and BAT in NSG mice, but endogenous PD-L1 expression in lymphatic organs cannot be properly assessed in these severely immunodeficient mice. Another study reported that 64Cu-DOTA-anti-mouse PD-1 visualized PD-1 in tumor and spleen of immunocompetent mice [17]; however, lymph nodes were not detected and a dominant liver signal prevented further evaluation in large parts of the abdominal region.
Instead of DOTA, we used the chelator NOTA which forms more stable radiometal complexes [22, 23], resulting in less trans-chelation and thus lower unspecific liver signals [22, 23, 39]. In addition, we titrated the dose of the 64Cu-NOTA-PD-1 tracer, revealing that specific PD-1 imaging was only feasible with extremely low amounts of mAb tracer (approx. 1.5 μg). This is similar to what Heskamp et al. [13] reported for PD-L1 imaging in a tumor xenograft model in immunodeficient mice, and is likely caused by the low number of receptors in the mice under these conditions. It is also important to mention that we used PD-1- and PD-L1-deficient animals [25, 26], unequivocally proving the specificity of our imaging experiments in immunocompetent mice, not previously reported in other studies. Moreover, this is the first study reporting the non-invasive imaging of PD-1 and PD-L1 side-by-side.
Ours is also the first study to non-invasively visualize both PD-1 and PD-L1 in lymph nodes. The detection of lymph nodes with the PD-1 mAb tracer is particularly remarkable not only because the lymph nodes are small but also because they contain only few, small PD-1-positive lymphocytes with low receptor expression. The PD-L1 mAb tracer more readily detected lymph nodes, presumably because PD-L1 is expressed on virtually all cells in secondary lymphoid organs even under physiologic conditions. As mentioned above, PD-L1 expression has also been reported for non-lymphoid organs under physiologic conditions, but mostly only for minor cell populations [6, 24, 30], which could explain why we did not detect clear PD-L1 signals on whole-body scans for these organs. However, our ex vivo biodistribution analyses showed low but specific tracer uptake in heart, lung, thymus, pancreas, intestines, and kidney compared with PD-L1-/- mice.
PD-L1 expression in BAT, which was clearly visible on immunoPET images of the interscapular region, has very recently also been reported by one other group [16]. Our study unequivocally proves specific tracer uptake in BAT, because BAT was not detected in PD-L1 knockout mice. Moreover, our FACS analyses revealed that PD-L1 is mainly expressed on CD45+ leukocytes such as adipose-tissue macrophages and T lymphocytes. The noticeable PD-L1 expression detected in BAT contributes to the notion of BAT as an immunologically relevant tissue [40-42].
As IFN-γ secreted by activated T lymphocytes is the main inducer of PD-L1 [5, 24], and thus responsible for adaptive PD-L1-mediated immune resistance [9, 10], we were interested in elucidating body-wide changes in ligand expression in response to this important cytokine. Interestingly, the lung turned out to be the organ with the strongest specific PD-L1 upregulation, which teleologically appears meaningful since the lung is a vital organ that is continuously exposed to a high level of antigens that can potentially induce IFN-γ-secreting lymphocytes [35, 43, 44]. In contrast, BAT is not continuously exposed to microorganisms. Therefore, protection of brown fat cells against effector T cells may not be essential. Why BAT contains an astonishingly high number of leukocytes (that are PD-L1+) is currently not clear. The particularly strong PD-L1 induction on non-leukocytes in the lung indicates that anti-lung T cell responses are strongly restrained by the PD immune checkpoint. This may also apply to T cell responses against lung cancers and could partially explain why PD-1/PD-L1 checkpoint inhibition is quite effective in lung cancer patients [45-49]. Further, it could explain the association with immune-mediated pneumonitis, a specific immune-related adverse effect of PD checkpoint blockade, which only occurs in few treated patients but can be fatal [45, 46, 50].
Traditionally, lung cancer was considered non-immunogenic. However, recently, anti-PD-1/PD-L1 responses have been reported in advanced lung cancer patients; they seem to correlate with the mutational load/antigenic neoepitope formation [51], and in some [45-47] but not all [48] studies, with tumor PD-L1 expression. If our finding of pronounced IFN-γ-mediated PD-L1 induction holds true for lung malignancies, these observations together suggest that, in lung cancer, adaptive immune resistance via the PD-1 checkpoint is crucial. Thus, its blockade might be particularly important to boost otherwise ineffective CTL responses against lung cancer.
The importance of PD-L1 expression as a predictive biomarker is currently one of the most extensively discussed topics in cancer immunotherapy research [9-11]. PD checkpoint inhibitors can be effective in tumors appearing PD-L1 negative by biopsy. However, the usually higher response rates for PD-L1-positive tumors have prompted extensive efforts towards the development of IHC assays to assess tumoral PD-L1 expression. Although controversial, the aim is to predict which patients may respond to checkpoint blockade, which is still expensive and, although comparatively well tolerated, is not without side effects. PD-1/PD-L1 immunoPET imaging has several potential advantages over IHC: It is non-invasive and could assess receptor expression in all metastases including those difficult to reach by biopsy or throughout larger tumors which cannot be covered completely [10]; it may also be useful for non-invasive evaluation of marker expression in a large variety of chronic inflammatory disease states. Additionally, it could be easily repeated, which appears important because of the dynamic changes in PD-1/PD-L1 expression. Clinical trials assessing non-invasive PD-L1 PET in tumor patients are already ongoing (NCT02453984, NCT02478099). These trials will show if the PD-L1 expression patterns revealed by non-invasive imaging in mice are conserved in humans. Here, we performed PD-1 imaging after combined immunoradiotherapy. Future studies will show if PD-1 and PD-L1 PET can reflect changes in PD-1/PD-L1 expression levels prior to and after therapy.
Non-invasive imaging of therapeutically effective PD-1 and PD-L1 antibodies is also of high interest for preclinical and potentially also for the clinical development of these drugs, as it provides an elegant opportunity to obtain quantitative and kinetic information on the whole-body biodistribution of these antibodies, including parameters such as tumor accumulation and blood half-life.
Our study demonstrates that high-sensitivity and high-resolution whole-body PET imaging using therapeutic antibody-based tracers can yield novel insights into the biology and pathophysiological importance of immune checkpoints. In addition, it suggests that it could also be exploited for the preclinical and clinical development of checkpoint-blocking drugs and of non-invasive diagnostic assays for patient selection and treatment monitoring.
Supplementary Material
Additional File 1Figures S1-S7.
Movie S1.
Movie S2.
Movie S3.
Movie S4.
Movie S5.
Author Contributions
The project was conceptualized and supervised by G.N. M.H., F.B., R.S., and G.N. designed the experiments. M.H. performed most experiments and data analyses. F.B. performed part of the immunoPET and ex vivo biodistribution analyses. M.D.B. performed radiolabeling and isotype dilution experiments. R.S. contributed valuable reagents/tools. M.H. and G.N. wrote the manuscript and all authors commented on it.
Acknowledgements
We thank S. Gaedicke for performing the NOTA conjugation and for excellent technical assistance, and R. Tönnesmann and J. Lahoti for their help with mouse dissections. We also thank T. Honjo (Kyoto University) for PD-1-/- mice and L. Chen (Yale School of Medicine) for PD-L1-/- (B7-H1-/-) mice. Further, we thank Dr. E. Firat for critical discussion of the manuscript and Prof. A. Grosu and Prof. P. T. Meyer for their generous support.
Conflict of Interest
The authors declare that they have no conflict of interest.
References
1. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12:252-64
2. Allison JP. Immune Checkpoint Blockade in Cancer Therapy: The 2015 Lasker-DeBakey Clinical Medical Research Award. JAMA. 2015;314:1113-4
3. Okazaki T, Chikuma S, Iwai Y, Fagarasan S, Honjo T. A rheostat for immune responses: the unique properties of PD-1 and their advantages for clinical application. Nat Immunol. 2013;14:1212-8
4. Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB. et al. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med. 2002;8:793-800
5. Freeman GJ, Long AJ, Iwai Y, Bourque K, Chernova T, Nishimura H. et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med. 2000;192:1027-34
6. Chen L, Han X. Anti-PD-1/PD-L1 therapy of human cancer: past, present, and future. J Clin Invest. 2015;125:3384-91
7. Berman D, Korman A, Peck R, Feltquate D, Lonberg N, Canetta R. The development of immunomodulatory monoclonal antibodies as a new therapeutic modality for cancer: the Bristol-Myers Squibb experience. Pharmacol Ther. 2015;148:132-53
8. Lesokhin AM, Callahan MK, Postow MA, Wolchok JD. On being less tolerant: enhanced cancer immunosurveillance enabled by targeting checkpoints and agonists of T cell activation. Sci Transl Med. 2015;7:280sr1
9. Lipson EJ, Forde PM, Hammers HJ, Emens LA, Taube JM, Topalian SL. Antagonists of PD-1 and PD-L1 in Cancer Treatment. Semin Oncol. 2015;42:587-600
10. Taube JM, Anders RA, Young GD, Xu H, Sharma R, McMiller TL. et al. Colocalization of inflammatory response with B7-h1 expression in human melanocytic lesions supports an adaptive resistance mechanism of immune escape. Sci Transl Med. 2012;4:127ra37
11. Teng MW, Ngiow SF, Ribas A, Smyth MJ. Classifying Cancers Based on T-cell Infiltration and PD-L1. Cancer Res. 2015;75:2139-45
12. Sharma P, Allison JP. The future of immune checkpoint therapy. Science. 2015;348:56-61
13. Heskamp S, Hobo W, Molkenboer-Kuenen JD, Olive D, Oyen WJ, Dolstra H. et al. Noninvasive Imaging of Tumor PD-L1 Expression Using Radiolabeled Anti-PD-L1 Antibodies. Cancer Res. 2015;75:2928-36
14. Maute RL, Gordon SR, Mayer AT, McCracken MN, Natarajan A, Ring NG. et al. Engineering high-affinity PD-1 variants for optimized immunotherapy and immuno-PET imaging. Proc Natl Acad Sci U S A. 2015;112:E6506-14
15. Josefsson A, Nedrow JR, Park S, Banerjee SR, Rittenbach A, Jammes F. et al. Imaging, Biodistribution, and Dosimetry of Radionuclide-Labeled PD-L1 Antibody in an Immunocompetent Mouse Model of Breast Cancer. Cancer Res. 2015;76:472-9
16. Chatterjee S, Lesniak WG, Gabrielson M, Lisok A, Wharram B, Sysa-Shah P. et al. A humanized antibody for imaging immune checkpoint ligand PD-L1 expression in tumors. Oncotarget. 2016
17. Natarajan A, Mayer AT, Xu L, Reeves RE, Gano J, Gambhir SS. Novel Radiotracer for ImmunoPET Imaging of PD-1 Checkpoint Expression on Tumor Infiltrating Lymphocytes. Bioconjug Chem. 2015;26:2062-9
18. Freise AC, Wu AM. In vivo imaging with antibodies and engineered fragments. Mol Immunol. 2015;67:142-52
19. Nomi T, Sho M, Akahori T, Hamada K, Kubo A, Kanehiro H. et al. Clinical significance and therapeutic potential of the programmed death-1 ligand/programmed death-1 pathway in human pancreatic cancer. Clin Cancer Res. 2007;13:2151-7
20. Eppihimer MJ, Gunn J, Freeman GJ, Greenfield EA, Chernova T, Erickson J. et al. Expression and regulation of the PD-L1 immunoinhibitory molecule on microvascular endothelial cells. Microcirculation. 2002;9:133-45
21. Anderson CJ, Ferdani R. Copper-64 radiopharmaceuticals for PET imaging of cancer: advances in preclinical and clinical research. Cancer Biother Radiopharm. 2009;24:379-93
22. Prasanphanich AF, Nanda PK, Rold TL, Ma L, Lewis MR, Garrison JC. et al. [64Cu-NOTA-8-Aoc-BBN(7-14)NH2] targeting vector for positron-emission tomography imaging of gastrin-releasing peptide receptor-expressing tissues. Proc Natl Acad Sci U S A. 2007;104:12462-7
23. Zhang Y, Hong H, Engle JW, Bean J, Yang Y, Leigh BR. et al. Positron emission tomography imaging of CD105 expression with a 64Cu-labeled monoclonal antibody: NOTA is superior to DOTA. PLoS One. 2011;6:e28005
24. Chen L. Co-inhibitory molecules of the B7-CD28 family in the control of T-cell immunity. Nat Rev Immunol. 2004;4:336-47
25. Nishimura H, Minato N, Nakano T, Honjo T. Immunological studies on PD-1 deficient mice: implication of PD-1 as a negative regulator for B cell responses. Int Immunol. 1998;10:1563-72
26. Dong H, Zhu G, Tamada K, Flies DB, van Deursen JM, Chen L. B7-H1 determines accumulation and deletion of intrahepatic CD8(+) T lymphocytes. Immunity. 2004;20:327-36
27. Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816-21
28. Gaedicke S, Braun F, Prasad S, Machein M, Firat E, Hettich M. et al. Noninvasive positron emission tomography and fluorescence imaging of CD133+ tumor stem cells. Proc Natl Acad Sci U S A. 2014;111:E692-701
29. Loening AM, Gambhir SS. AMIDE: a free software tool for multimodality medical image analysis. Mol Imaging. 2003;2:131-7
30. Francisco LM, Sage PT, Sharpe AH. The PD-1 pathway in tolerance and autoimmunity. Immunol Rev. 2010;236:219-42
31. Bartelt A, Heeren J. Adipose tissue browning and metabolic health. Nat Rev Endocrinol. 2014;10:24-36
32. Azhdarinia A, Daquinag AC, Tseng C, Ghosh SC, Ghosh P, Amaya-Manzanares F. et al. A peptide probe for targeted brown adipose tissue imaging. Nat Commun. 2013;4:2472
33. Demaria S, Formenti SC. Radiotherapy effects on anti-tumor immunity: implications for cancer treatment. Front Oncol. 2013;3:128
34. Deng L, Liang H, Fu S, Weichselbaum RR, Fu YX. From DNA Damage to Nucleic Acid Sensing: A Strategy to Enhance Radiation Therapy. Clin Cancer Res. 2016;22:20-5
35. The lungs at the frontlines of immunity. Nat Immunol. 2015;16:17
36. Probst HC, McCoy K, Okazaki T, Honjo T, van den Broek M. Resting dendritic cells induce peripheral CD8+ T cell tolerance through PD-1 and CTLA-4. Nat Immunol. 2005;6:280-6
37. Tsushima F, Yao S, Shin T, Flies A, Flies S, Xu H. et al. Interaction between B7-H1 and PD-1 determines initiation and reversal of T-cell anergy. Blood. 2007;110:180-5
38. Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol. 2008;26:677-704
39. Bass LA, Wang M, Welch MJ, Anderson CJ. In vivo transchelation of copper-64 from TETA-octreotide to superoxide dismutase in rat liver. Bioconjug Chem. 2000;11:527-32
40. DiSpirito JR, Mathis D. Immunological contributions to adipose tissue homeostasis. Semin Immunol. 2015;27:315-21
41. Vijgen G, van Marken Lichtenbelt W. Brown adipose tissue: clinical impact of a re-discovered thermogenic organ. Front Biosci (Elite Ed). 2013;5:823-33
42. Nyamdorj D, Dagdanbazar B, Uurtuya S, Amgalanbaatar D, Munkhtulga L, Enebish S. Experimental study detection of CD4+ and CD8+ T Cells in the brown adipose tissue. AJCEM. 2014;2:4-8
43. Gollwitzer ES, Saglani S, Trompette A, Yadava K, Sherburn R, McCoy KD. et al. Lung microbiota promotes tolerance to allergens in neonates via PD-L1. Nat Med. 2014;20:642-7
44. Chen HD, Fraire AE, Joris I, Brehm MA, Welsh RM, Selin LK. Memory CD8+ T cells in heterologous antiviral immunity and immunopathology in the lung. Nat Immunol. 2001;2:1067-76
45. Garon EB, Rizvi NA, Hui R, Leighl N, Balmanoukian AS, Eder JP. et al. Pembrolizumab for the treatment of non-small-cell lung cancer. N Engl J Med. 2015;372:2018-28
46. Herbst RS, Baas P, Kim DW, Felip E, Perez-Gracia JL, Han JY. et al. Pembrolizumab versus docetaxel for previously treated, PD-L1-positive, advanced non-small-cell lung cancer (KEYNOTE-010): a randomised controlled trial. Lancet. 2015
47. Borghaei H, Paz-Ares L, Horn L, Spigel DR, Steins M, Ready NE. et al. Nivolumab versus Docetaxel in Advanced Nonsquamous Non-Small-Cell Lung Cancer. N Engl J Med. 2015;373:1627-39
48. Brahmer J, Reckamp KL, Baas P, Crino L, Eberhardt WE, Poddubskaya E. et al. Nivolumab versus Docetaxel in Advanced Squamous-Cell Non-Small-Cell Lung Cancer. N Engl J Med. 2015;373:123-35
49. Reck M, Paz-Ares L. Immunologic checkpoint blockade in lung cancer. Semin Oncol. 2015;42:402-17
50. Nishino M, Sholl LM, Hodi FS, Hatabu H, Ramaiya NH. Anti-PD-1-Related Pneumonitis during Cancer Immunotherapy. N Engl J Med. 2015;373:288-90
51. Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ. et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015;348:124-8
Author contact
![]() Corresponding author: Prof. Dr. Gabriele Niedermann, Department of Radiation Oncology, Medical Center - University of Freiburg, Robert-Koch-Str. 3, D-79106 Freiburg. Phone: 0049 761 270 95140; Fax: 0049 761 270 95130; E-Mail: gabriele.niedermannde
Corresponding author: Prof. Dr. Gabriele Niedermann, Department of Radiation Oncology, Medical Center - University of Freiburg, Robert-Koch-Str. 3, D-79106 Freiburg. Phone: 0049 761 270 95140; Fax: 0049 761 270 95130; E-Mail: gabriele.niedermannde