Theranostics
13.3
Impact Factor
- Current Issue
- Advance Articles
- Volume 16; 2026
- Volume 15; 2025
- Volume 14; 2024
- Volume 13; 2023
- Volume 12; 2022
- Archive
- Cover Images
- Cover Suggestion
- Special Issues
Top
Introduction
Results
Discussions
Conclusion
Materials and Methods
Supplementary Material
Acknowledgements
References
Introduction
Results
Discussions
Conclusion
Materials and Methods
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2016; 6(13):2295-2305. doi:10.7150/thno.16141 This issue Cite
Research Paper
X-Ray Induced Photodynamic Therapy: A Combination of Radiotherapy and Photodynamic Therapy
Geoffrey D. Wang1*, Ha T. Nguyen2*, Hongmin Chen1 ![]() , Phillip B. Cox3, Lianchun Wang4, Koichi Nagata5, Zhonglin Hao6, Andrew Wang7, Zibo Li8, Jin Xie1
, Phillip B. Cox3, Lianchun Wang4, Koichi Nagata5, Zhonglin Hao6, Andrew Wang7, Zibo Li8, Jin Xie1 ![]()
1. Department of Chemistry, Bio-Imaging Research Center, the University of Georgia, Athens, Georgia 30602, USA;
2. National Exposure Research Lab, US Environmental Protection Agency, Athens, Georgia 30605, USA;
3. Department of Cellular Biology, the University of Georgia, Athens, Georgia 30602, USA;
4. Department of Biochemistry and Molecular Biology, the University of Georgia, Athens, Georgia 30602, USA;
5. College of Veterinary Medicine, The University of Georgia, Athens, GA 30602, USA;
6. Section of Hematology and Oncology, Georgia Cancer Center, Medical College of Georgia, Augusta University, Augusta, Georgia 30912, USA;
7. Laboratory of Nano- and Translational Medicine, Department of Radiation Oncology, Lineberger Comprehensive Cancer Center, Carolina Center for Cancer Nanotechnology Excellence, University of North Carolina at Chapel Hill, Chapel Hill, USA;
8. Department of Radiology and Biomedical Research Imaging Center, University of North Carolina at Chapel Hill, Chapel Hill, NC, USA.
*Equal contribution
Received 2016-5-12; Accepted 2016-6-20; Published 2016-10-1
Citation:
Wang GD, Nguyen HT, Chen H, Cox PB, Wang L, Nagata K, Hao Z, Wang A, Li Z, Xie J. X-Ray Induced Photodynamic Therapy: A Combination of Radiotherapy and Photodynamic Therapy. Theranostics 2016; 6(13):2295-2305. doi:10.7150/thno.16141. https://www.thno.org/v06p2295.htm
Other stylesAbstract

Conventional photodynamic therapy (PDT)'s clinical application is limited by depth of penetration by light. To address the issue, we have recently developed X-ray induced photodynamic therapy (X-PDT) which utilizes X-ray as an energy source to activate a PDT process. In addition to breaking the shallow tissue penetration dogma, our studies found more efficient tumor cell killing with X-PDT than with radiotherapy (RT) alone. The mechanisms behind the cytotoxicity, however, have not been elucidated. In the present study, we investigate the mechanisms of action of X-PDT on cancer cells. Our results demonstrate that X-PDT is more than just a PDT derivative but is essentially a PDT and RT combination. The two modalities target different cellular components (cell membrane and DNA, respectively), leading to enhanced therapy effects. As a result, X-PDT not only reduces short-term viability of cancer cells but also their clonogenecity in the long-run. From this perspective, X-PDT can also be viewed as a unique radiosensitizing method, and as such it affords clear advantages over RT in tumor therapy, especially for radioresistant cells. This is demonstrated not only in vitro but also in vivo with H1299 tumors that were either subcutaneously inoculated or implanted into the lung of mice. These findings and advances are of great importance to the developments of X-PDT as a novel treatment modality against cancer.
Keywords: photodynamic therapy, radiotherapy, lung cancer, clonogenecity, nanoparticles.
Introduction
PDT is a relatively new and minimally invasive cancer therapy approach [1-3]. PDT utilizes photosensitizers that are activated by light in the presence of oxygen, producing reactive oxygen species (ROS) that are cytotoxic [3]. PDT can kill cancer cells directly and or damage tumor microvessels, leading to tissue ischemia [2, 4, 5]. It has been utilized in the clinic for treatments of different cancer types, including esophageal cancer, non-small cell lung cancer, bladder cancer, and head and neck cancer [6-10]. Despite of the promises, however, PDT suffers from the shallow tissue penetration of light, especially in the visible spectrum window [11]. There has been progress of developing near-infrared (NIR) photosensitizers, for instance Lumin, Motexafin lutetium, and TOOKAD (absorption peaks at 770 nm, 732 nm and 753 nm) [12, 13]. However, even in the NIR region, light can travel less than 1 cm in tissues. This restriction has largely limited the applications of PDT in the clinic.
Recently, we and others have developed a new PDT derivative called X-ray induced PDT, or X-PDT [14-17]. The goal is to break the shallow penetration restriction by using X-ray as an energy source [18]. The idea of utilizing X-ray to overcome the shallow penetration of PDT was first raised by Chen et al. in 2006 [19], but it was not until very recently that we and the Chen group demonstrated its feasibility in vivo [14-17]. In particular, we showed in a recent study that SrAl2O4:Eu2+ nanoparticle (SAO:Eu, a scintillator which convert X-ray photons to visible photons) and MC540 (a photosensitizer with matching excitation wavelength) co-loaded mesoporous silica nanoparticles (MC540-SAO:Eu@mSiO2) can produce singlet oxygen (1O2) under X-ray radiation, leading to efficient cancer cell death, even when the cells are beneath thick tissues [14].
Figure 1
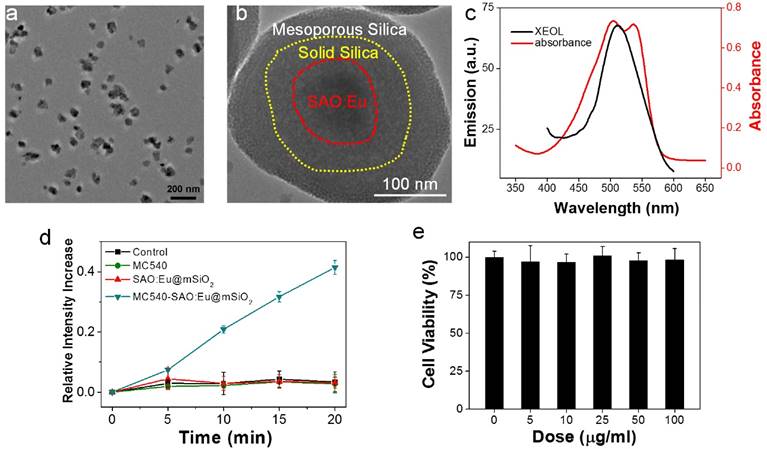
Characterizations of SAO:Eu@mSiO2 nanoparticles. (a) TEM image of SAO:Eu nanoparticles. (b) TEM image of a representative SAO:Eu@mSiO2 nanoparticle. (c) XEOL spectrum of SAO:Eu@mSiO2 nanoparticles and the absorption spectrum of MC540. The excitation wavelength of photosensitizers and the emission wavelength of SAO:Eu match well. (d) Singlet oxygen generation under X-ray irradiation. (e) Cytotoxicity of SAO:Eu@mSiO2 nanoparticles. Little toxicity was observed even at high nanoparticle concentrations.
It is clear that during X-PDT, not all X-ray energy, but a portion of it, is converted to visible photons to activate PDT. Despite of the studies by us and others observed enhanced treatment efficacy with X-PDT relative to radiation therapy (RT) alone at the same radiation doses [14-17], this phenomenon is intriguing, indicating that there is more to X-PDT than a mere PDT process. In the current study, we tap into the mechanisms behind X-PDT-induced cell death. Specifically, we conducted comprehensive studies to examine the impacts of X-PDT on cell viability, clonogenicity, apoptosis, necrosis, DNA damage, and membrane lipid damage. Our studies showed that X-PDT contains a RT component, and hence it is essentially a RT and PDT combination. The two modalities interplay to attack both cell membrane and DNA, leading to lethal damage that is beyond the repairs of cells. The synergy explains the better cell killing efficacy of X-PDT than RT, even for cells that are refractory to radiotherapy. In particular, we found that X-PDT can efficiently kill H1299 cells, which are radioresistant non-small cell lung cancer (NSCLC) cells. The enhanced efficacy was observed not only in vitro but also in vivo with a subcutaneous tumor model or when H1299 cells were percutaneously implanted into the lung. These findings are of great value to our understanding of X-PDT as a novel treatment modality and its further transformation for eventual clinical translation.
Results
Preparation of MC540-SAO:Eu@mSiO2 nanoparticles
The MC540-SAO:Eu@mSiO2 nanoparticles were prepared by following our published protocol [14]. Briefly, SrCO3, Al2O3, Eu2O3 and graphite powders were mixed and heated in a tube furnace at 1450 °C for 2 h under an argon flow. The pressure was maintained at 5 Torr. The as-synthesized bulk SAO:Eu was subject to mechanical grinding, followed by sedimentation, filtration and centrifugation, to yield nanoscale nanoparticles (73.5 ± 26.9 nm, Figure 1a&b). X-ray diffraction (XRD) analysis found that the composition of the material was monoclinic SrAl2O4 (JCPDS #74-0794, Figure S1). Inductively coupled plasma (ICP) confirmed the chemical composition and found that Eu accounts for ~1% of the total mass. As described in the previous study, SrAl2O4:Eu can be stimulated by both Uv-vis and X-ray to emit green photoluminescence (centered at ~520 nm, Figure 1c), which is attributed to 4f65d1→4f7 transition of Eu2+ ions [15].
SrAl2O4:Eu is a highly hydrolytic material and is quickly reduced to constituent ions in an aqueous solution [15]. For bio-applications, we coated SrAl2O4:Eu nanoparticles with two layers of silica (Figure 1b). These include an inner, solid silica layer that prevents direct contact with the aqueous surroundings, and an outer, mesoporous silica layer that provides a docking place for photosensitizers [14]. Into the resulting SrAl2O4:Eu@mSiO2 nanoparticles, MC540 (Figure S2), a common photosensitizer that has been investigated in both pre-clinical and clinical studies [20], was loaded. The absorbance wavelengths of MC540 well match the emission wavelengths of SAO:Eu (Figure 1c, Figure S3). Using singlet oxygen sensor green (SOSG) assays, we studied the ability of X-PDT to producing 1O2. It was found that X-ray alone (1-4 Gy) was not able to produce 1O2, either with PBS, MC540, or SrAl2O4:Eu nanoparticles (Figure 1d). As a comparison, when both SrAl2O4:Eu@mSiO2 (0.05 mg/mL) and X-ray were applied, efficient 1O2 production was observed (Figure 1d).
Figure 2
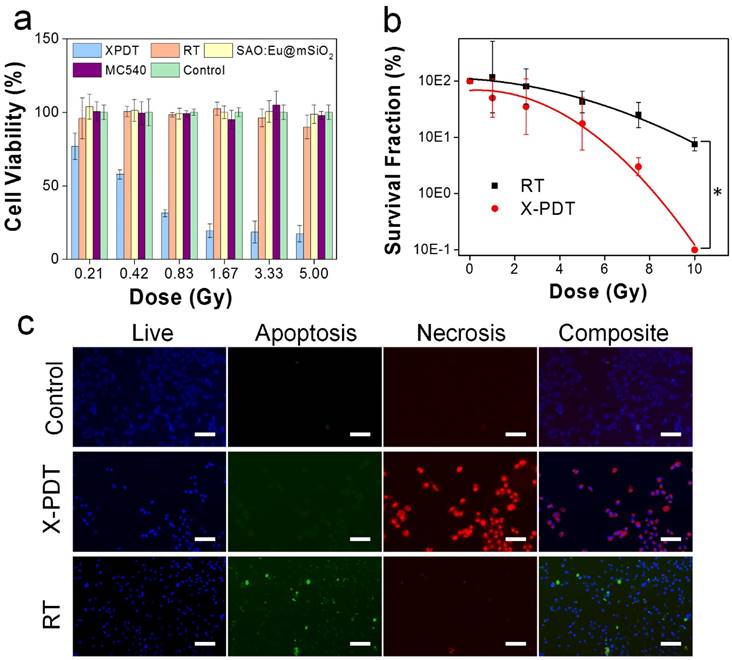
X-PDT induced cell death. (a) Cell viability, measured by MTT assays performed 24 h after X-PDT. In control groups, including RT alone and nanoparticles alone, there was no significant drop of viability. (b) Cell reproductive capacity, measured by clonogenic assays taken 14 days after X-PDT (* p-value < 0.01). (c) Apoptosis and necrosis assays, performed 24 h after X-PDT. Scale bars: 100 µm.
Impact of X-PDT on cell viability and clonogenecity
We first investigated the impact of X-PDT on cell viability by MTT assays. Radioresistant NSCLC H1299 cells were used in the studies [21-23]. In the absence of radiation, SAO:Eu and SAO:Eu@mSiO2 nanoparticles were not toxic to H1299 cells (Figures 1e, 2a). The low toxicity of SAO:Eu@mSiO2 was also observed in our previous studies with other cell lines [24] and is not surprising because all the constituents of the nanoparticles, including MC540, SAO:Eu, and SiO2, are not toxic in the dark [14]. Meanwhile, RT alone (0-5 Gy) did not induce significant cell death at 24 hours either (Figure 2a). As a comparison, when X-ray irradiation was applied after MC540-SAO:Eu@mSiO2 (50 µg/mL) incubation (i.e. X-PDT), there was significant cell viability drop (Figure 2a). Specifically, when irradiation of 0.83, 1.67, 3.33, and 5 Gy was applied, the 24-hour cell viability was reduced to 31.4 ± 2.3%, 19.6 ± 4.6%, 18.6 ± 7.4%, and 17.5 ± 5.6%, respectively.
While MTT assay is adequate to measures short-term cell viability, it is suboptimal in assessing the reproductive capacity of cancer cells. For RT, however, reproductive capacity is a more relevant ending point. This is because ionizing radiation mainly targets DNA, most importantly double-strand DNA break [25]. This is often manifested not as immediate cell death but reduced clonogenicity in days or weeks [26]. To investigate, we performed clonogenic assays with H1299 cells that received RT alone (0, 1, 2.5, 5, 7.5 and 10 Gy) or RT plus MC540-SAO:Eu@mSiO2 (50 μg/mL). X-PDT was much more effective than RT alone at all doses (Figure 2b). Taking 5 Gy for instance, X-PDT was able to reduce the survival fraction (SF) to 17.5%, compared to that of 42.5% for RT alone. The dose enhancement factor (DEF) was calculated to be 1.67 (Methods Section).
Impact of X-PDT on DNA and lipid membranes
To further investigate the cause of X-PDT induced cell death, we performed Apoptotic/Necrotic/Healthy assay (PromoCell, Heidelberg, Germany), again with H1299 cells. With RT only (5 Gy), cells showed a mediocre level of apoptosis at 24 h but no detectable necrosis (Figure 2c). On the contrary, when cells were treated with MC540-SAO:Eu@mSiO2 (50 µg/mL) plus radiation, there manifests extensive cell necrosis (Figure 2c). This pattern resembles membrane-targeted PDT, which causes oxidative degradation of unsaturated lipids and surface proteins [27]. To confirm, we conducted lipid peroxidation assay (Lipid Peroxidation Kit, Life Technologies, Carlsbad, CA). Compared to the control (PBS only), the lipid peroxidation level was increased by 90.5 ± 23.0% after RT (5 Gy); meanwhile, X-PDT under the same irradiation dose led to an increase of 201.5 ± 34.0% (Figure 3a). Such lipid peroxidation is attributed mainly to the PDT component of X-PDT and is responsible for the short-term cell necrosis and viability drop.
Figure 3
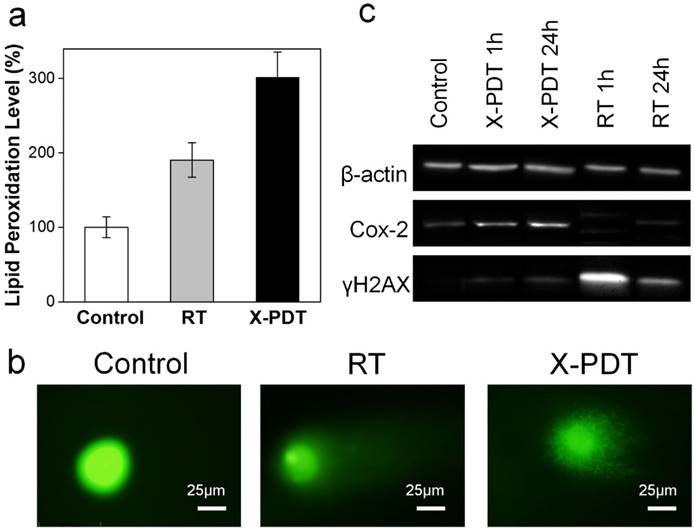
Impacts of X-PDT on cellular compartments. (a) Lipid damage assessment, measured by lipid peroxidation assays. (b) DNA damage, assessed by single cell electrophoresis assays. (c) Western blot assays, which further confirms the impact of X-PDT on DNA and membrane lipids.
Meanwhile, X-PDT also causes DNA damage. This was confirmed by comet assays in (also known as single cell gel electrophoresis assay, Figure 3b). Compared to the control, cells received RT alone (5 Gy) displayed a relatively long and intense tail, suggesting extensive DNA double-strand break [28]. Such a tail was also observed with X-PDT treated cells, but was shorter, less intense, and more discrete, likely associated with the extensive necrosis [28]. This suggests that there remains a RT component in X-PDT, which explains the reduced cell survival in clonogenic assays.
To further assess the cellular impacts of X-PDT, we analyzed expression levels of histone H2AX and Cox-2 in X-PDT treated H1299 cells with Western blot (Figure 3c). Histone H2AX plays a critical role in recruiting repair- or damage-signaling factors to the sites of DNA damage [29, 30] and is thus an indicator of RT-induced DNA breakage. Cox-2, on the other hand, is involved in lipid peroxidation, and is often up-regulated after PDT-induced membrane damage [31, 32]. We found that X-ray radiation (5 Gy) alone induced expression of H2AX but minimally affected the level of Cox-2 (Figure 3c). With X-PDT, on the other hand, both Cox-2 and H2AX expressions were increased, although the H2AX level was lower than that after RT (Figure 3c). These results corroborate with the preceding studies, again confirming that X-PDT is essentially a combination of RT and PDT. It is postulated that because the two modalities target different cellular compartments (DNA and unsaturated membrane lipids, respectively), a synergy in treatment occurs that leads to much more efficient cancer cell killing.
Impact of tissue depth on the efficacy of X-PDT
We next stdied the impact of tissue thickness on the treatment efficacy of X-PDT in vivo. This was first studied with mouse subcutaneous tumor models established with H1299 cells. Briefly, we intratumorally injected MC540-SAO:Eu@mSiO2 nanoparticles (4.25 mg/kg) to tumor bearing animals; we then irradiated the tumors (5 Gy, with the rest of the body lead-shielded), with pork tissues of 1- or 2-cm thickness lain on top. In control group, animals were injected with PBS and received no radiation.
Tumors in the control group grew very rapidly, and either died or reached an end point (tumor diameter > 1.7 cm) within 10 days (Figure 4a).
Figure 4
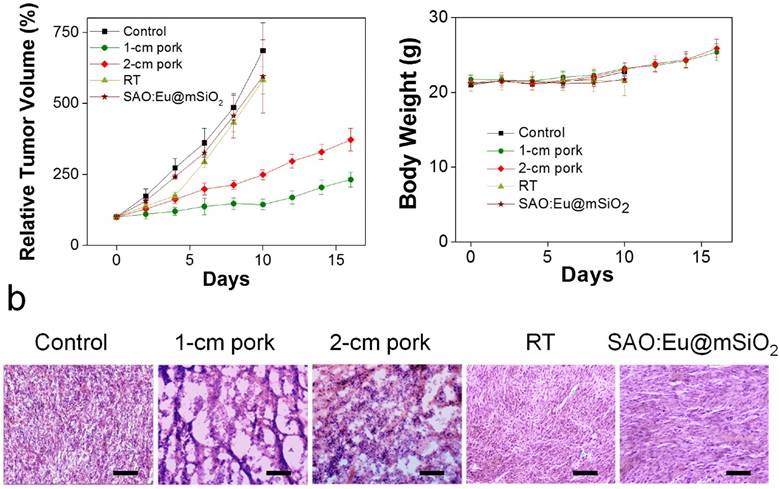
X-PDT to treat subcutaneously implanted tumors from above thick tissues. (a) Tumor growth curves. Despite of using thick pork as tissue blocks, X-PDT can efficiently suppress tumor growth. (b) H&E staining with tumor tissues. X-PDT caused extensive cancer cell death in H1299 tumors. Scale bars, 100 µm. (c) Body weight curves. X-PDT did not cause significant changes to the animal body weights.
Figure 5
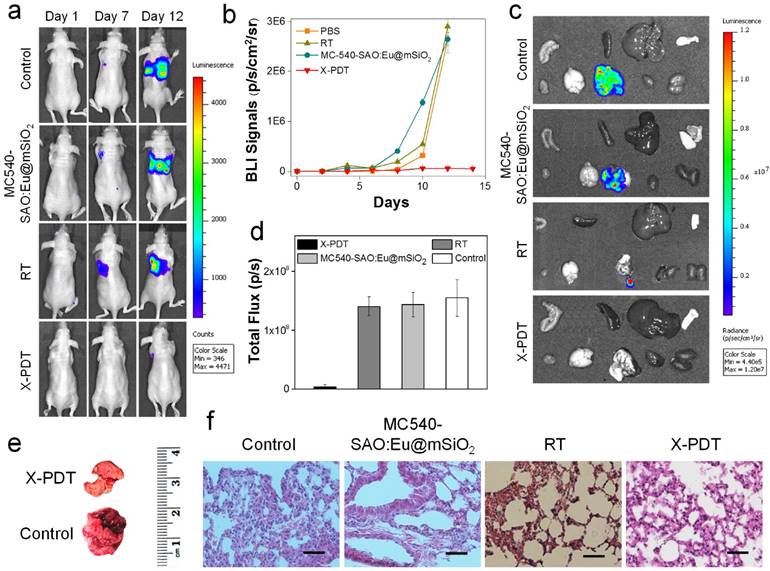
In vivo therapy studies. (a) Representative bioluminescence images of mice treated by X-PDT, RT, MC540-SAO:Eu@mSiO2 and PBS on Day 1, 7, and 12. MC540-SAO:Eu@mSiO2 nanoparticles were intrathoracically injected to the mice. In the RT and X-PDT groups, a single dose X-ray radiation of 5 Gy was applied to the tumor area, with the rest of the body covered by lead. (b) Tumor growth, measured by monitoring BLI signal changes at different time points. (c) Ex vivo bioluminescence images taken immediately after tissue dissection. The organs were organized in the following order: top row (from left to right): intestine, spleen, liver and skin; bottom row (from left to right): muscle; brain; lung; heart and kidneys. (d) BLI signals from the lungs. Based on ROI analyses on (c). (e) Representative photographs of lungs taken from the X-PDT and control Groups. (f) H&E staining with tumor tissues from different treatment groups. Scale bars, 100 µm.
As a comparison, in all the treatment groups, tumor growth was efficiently slowed. On Day 16, the tumor suppression rates were 54.2% and 33.8%, for the animals bore with 1-, and 2-cm thick pork, respectively. The results suggest that tissue thickness can still affect the efficacy of X-PDT; the impact, however, was much less severe than with PDT, which lost its efficacy beyond 1 cm thickness. Such tissue impact can likely be compensated by increasing radiation doses and is less of a concern in the clinical setting, where deep-penetrating megavoltage X-rays are used [33]. The efficiency in cancer cell killing was further confirmed with H&E analysis (Figure 4b). Compared with the control animals, where cancer cells were densely packed, X-PDT dramatically reduced cell density and disrupted connective tissues (Figure 4b). Meanwhile, we observed no signs of systematic toxicities to the surrounding tissues (Figure 4c).
Next, we investigated the X-PDT efficacy by injecting nanoparticles and cell mixture to the thorax of mice and irradiated the injection sites in vivo. Due to thick soft tissues and bones, diseases at this position are not accessible by conventional PDT [34]. Specifically, we injected MC540-SAO:Eu@mSiO2 nanoparticles (4.25 mg/kg), along with 5 × 105 firefly luciferase expressing H1299 cells (H1299-Luc), into the left lateral thorax of nude mice (Figure 5a). Radiation (5 Gy) was applied to the tumor inoculation sites, with the rest of the animal body shielded by lead. Control group animals received radiation only or PBS only. The tumor growth was then monitored in vivo by bioluminescence imaging (BLI). In PBS and RT only groups, the BLI signals were detected in the lung areas on Day 7 and continued increasing afterwards (Figure 5a). In X-PDT treated animals, on the other hand, the BLI signals were significantly suppressed. By regions of interest (ROI) analysis, on Day 12, the average BLI signals were 5.6 × 105, 2.9 × 106, and 2.6 × 106 photons/sec/cm2/sr for the X-PDT, RT, and PBS groups, respectively (Figure 5b). Ex vivo imaging confirmed the efficiency of X-PDT induced tumor suppression, finding strong residual signals in the lungs of control animals but close-to-background signals from the X-PDT group (Figure 5c,d). Moreover, we found multiple large tumors in the lungs dissected from control animals, but few detectable colonies in X-PDT treated mice (Figure 5e). Such reduced tumorigenicy by X-PDT was further confirmed by H&E staining (Figure 5f).
Meanwhile, X-PDT did not cause detectable systematic toxicities. These include no signs of side effects to normal tissues from H&E staining (Figure 6a). Also, hemodiagnosis found that the serum aspartate aminotransferase (AST) and alanine aminotransferase (ALT) levels remained unchanged over the course of the treatment, again suggesting low toxicities (Figure 6b). This is in accordance with our previous observations that i.v. injected MC540-SAO:Eu@mSiO2 nanoparticles are degraded after the conclusion of therapy and efficiently cleared from the hosts [14].
Discussions
While initially developed as a PDT derivative, the current study suggests that X-PDT is essentially a PDT and RT combination. While a portion of X-ray energy was converted to activate PDT, ionizing radiation continues playing an important role in X-PDT. The PDT and RT components target different cellular components and the combination causes enhanced effect that is beyond the repairs of cells. This explains the greater cytotoxicity with X-PDT than with RT, especially for cells that are refractory to RT. In fact, previous studies have observed synergy between PDT and RT [35-38]. However, it was found that PDT and ionizing irradiation need to be given at the same time so as to override cell repairs [35, 36]. This requires photo-irradiation and X-ray irradiation to be applied simultaneously, which is difficult to achieve in the clinic, not to mention the shallow penetration of PDT. X-PDT, on the other hand, is an inherent PDT and RT combination, with both processes regulated by one deep-penetrating radiation. From this perspective, X-PDT represents a major advance for seamlessly integrating PDT and RT. In this sense, X-PDT should be viewed as not only a PDT derivative but also a RT derivative.
RT remains a major therapy option in clinical oncology. More than 50% of all cancer patients receive RT during their curative process [39]. One major limitation of RT is that not all cancer cells are well responsive to RT, and tumors that are originally responsive may develop resistance over the course of therapy [40]. Increasing radiation doses can, to a certain degree, address the issue, but will inevitably cause collateral damage to normal tissues [41]. Radiosensitizing agents of different types have been developed to sensitize cancer cells to RT; however, many radiosensitizers are cytotoxic agents [42]. As discussed above, X-PDT can be viewed as a RT derivative and as such a novel radiosensitizing technology. This was confirmed by our clonogenic assays, which observed a high DEF with X-PDT, even against cells that are refractory to RT. Unlike many conventional radiosensitizers, MC540-SAO:Eu@mSiO2 nanoparticles are not toxic in the absence of radiation. Meanwhile, they are highly biodegradable, efficiently cleared out of the hosts after treatment, and causing no side effects to normal tissues (Figure 6). These properties suggest great promise of X-PDT in clinic translation to improve RT efficacy, reduce normal tissue radiation exposure, and battle with radioresistant tumors. In the present studies, nanoparticles were directly injected into tumors. In the future studies, it is worthwhile to reduce nanoparticle size, improve photosensitizer loading, and conjugating targeting ligands to the nanoparticle surface for achieve formulations that can be systematically injected to mediate tumor selective X-PDT treatment.
Figure 6
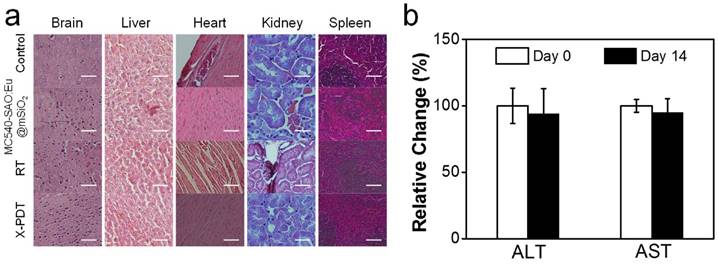
Systematic toxicities of X-PDT. (a) H&E staining with normal tissues taken from X-PDT, RT, MC540-SAO:Eu@mSiO2, and control groups. Scale bars, 100 µm. (b) Hemodiagnosis. ALT and AST level showed no significant changes before and 14 days after X-PDT treatment.
Conclusion
In summary, we investigated in this study the impacts of X-PDT on cancer cells. We found that X-PDT is not simply a PDT process; rather, it is essentially a RT and PDT combination. The combination leads to enhanced therapy effects that make the treatment much more efficient than RT alone, even when used against cells that are refractory to RT. We also show that X-PDT can be exploited to suppress tumors lain under deep tissues. The findings and advances are of great value to the developments of X-PDT as a novel treatment modality against cancer.
Materials and Methods
Nanoparticle synthesis and coating SAO:Eu was synthesized by a carbo-thermal reduction and vapor-phase deposition method that was published previously [43]. To render SAO:Eu amenable to bio-related applications, bulk SAO:Eu was ground into nanoparticles with a diameter of ~ 80 nm. The bare SAO:Eu nanoparticles were coated with a layer of solid silica by following a previously published protocol [14]. In a typical synthesis, 10 mg of SAO:Eu nanoparticles were dispersed in a mixture of 25 mL of ethanol, 2 mL of H2O, and 1 mL of ammonia (25%), and stirred for 10 min. Then, 75 µL of tetraethyl orthosilicate (TEOS) was added to solution and the mixture was stirred at room temperature for 12 hours. The resulting nanoparticles were subsequently coated with a layer of mesoporous silica. A mixture of 3-aminopropyltriethoxysilane and TEOS (5 v/v%) was used as silane precursors [24]. Solid silica coated SAO:Eu nanoparticles (10 mg) from the first step were dispersed in a mixture of 45 mL of H2O, 0.3 mL of 2 M NaOH, and 10 mg of cetyltrimethylammonium bromide (CTAB) for 30 min. After heating to 70 oC, 50 µL of TEOS and 300 µL of ethyl acetate were added, and the solution was magnetically stirred for 2 h. The raw products were collected by centrifugation and washed with ethanol for three times. The resulting nanoparticles were re-suspended in ethanol (20 mL) and mixed with NH4NO3 (100 mg) at 60 oC for 2 hours to remove CTAB. For photosensitizer loading, MC540 (Invitrogen) were dispersed in ethanol with SAO:Eu@mSiO2 nanoparticles. The mixture was incubated overnight at room temperature [14]. After centrifuging and washing several times, and the supernatant removed. The MC540-loaded nanoparticles were resuspended in PBS (Thermo Scientific) for further studies.
Characterizations of SAO:Eu and MC540-SAO:Eu@mSiO2 nanoparticles. UV-Vis absorption spectra were recorded on a Shimdzu 2450 UV-Vis spectrometer. Photoluminescence measurements were performed on a Hitachi F-7000 fluorometer. A mini-X X-ray tube (Amptek Inc.) was used as the X-ray source, and was set at 50 kV and 70 μI for all the experiments in this study. TEM samples were prepared by dripping sample solutions onto carbon-coated copper grids and evaporating the solvent. TEM images were taken on an FEI Tecnai 20 transmission electron microscope operating at 200 kV.
Cell culture H1299, a human NSCLC cell line, was used in in vitro and in vivo studies. The H1299 cells have been transfected to stably express firefly luciferase (i.e. H1299-Luc). The cells had been tested and were rodent pathogens free. H1299-Luc cells were grown in RPMI 1640 medium supplemented with 10% FBS, 100 units/mL of penicillin (ATCC) and 250 µg/mL hygromycin. The cells were maintained in a humidified, 5 % carbon dioxide (CO2) atmosphere at 37 °C.
In vitro toxicity study 104 H1299 cells were seeded in 96-well plates (Corning) and cultured to 90% confluency. For cytotoxicity studies, the cells were then incubated with 0, 5, 10, 25, 50 and 100 μg/mL MC540-SAO:Eu@mSiO2 nanoparticles for 24 h. Subsequently, they were washed with PBS for two times, and then were evaluated by MTT assay (Sigma Aldrich) by following the vendor's protocols.
In vitro viability study. 104 H1299 cells were seeded in 96-well plates (Corning) and cultured for 24 h. The cells were then divided into 5 groups: 1) PBS group, 2) MC540-SAO:Eu@mSiO2 nanoparticles only, 3) MC540 only, 4) RT only, and 5) X-PDT. For Group 2 and 5, the final concentration of MC540-SAO:Eu@mSiO2 nanoparticles was 50 μg/mL and the incubation time was 1 hour. All the cells were incubated in the dark. Cells in Group 4 and 5 were irradiated with 50 kV X-ray at 0.83, 1.67, 3.33 and 5 Gy while all the other cells received irradiation. After the treatment, all cells were washed with PBS for two times. Cell culture medium was replenished and the incubation was maintained for 24 h (5 % CO2 and 37 °C). Cell viabilities were evaluated by MTT assays (Sigma Aldrich) following the vendor's protocol.
Apoptosis/necrosis/healthy assay H1299-Luc cells treated with X-PDT (5 Gy), RT (5 Gy) and PBS were subjected to Apoptotic/Necrotic/Healthy assay (PromoCell GmbH, Heidelberg, Germany) following the vendor's protocol. Briefly, FITC-annexin V (green) binds to the apoptosis marker of phosphatidyl serine, and ethidium homodimer III (red) binds to DNA under the necrotic conditions. The cells stained with annexin V antibody alone or together with ethidium homodimer III were counted as early or late stages of apoptotic cells, respectively [34]. The cells labeled with ethidium homodimer III alone were counted as necrotic cells. In addition, membrane permeable Hoechst 33342 (blue) stains the nuclei. Healthy cells would only display blue staining. The resulted cells were then evaluated by fluorescent imaging on an Olympus IX71 fluorescent microscope (3 trials per group).
Clonogenic assay Mono-layered H1299-Luc cells were prepared in 6-well cell petri-dishes one day before the experiments. On the experimental day, the 6-well cell petri-dishes were randomly divided into the control group, the X-ray radiation group, and the X-PDT group. The two treatment groups were treated by X-ray (50 kv) for a dose of 1, 2.5, 5, 7.5 and 10 Gy while the control group received no irradiation. The irradiated and un-treated cells were then harvested using trypsin-EDTA and plated into 100 mm cell petri-dishes. Each petri-dish was seeded with 100 viable cells and was placed into a cell culture incubator (5% CO2 and 37°C) for 14 days. After incubation for 14 days, the cells were fixed by formalin and stained with 0.5% Gentian Violet. Colonies containing more than 50 cells were counted and the survival fractions were calculated by comparing to the control. Dose enhancement factor (DEF) was calculated as the ratio between RT and X-PDT radiation doses at 10% survival fractions. All the experiments were repeated 3 times.
Lipid peroxidation assay Lipid peroxidation levels were measured using Image-iT® Lipid Peroxidation Kit (Life Technologies Corporation, Carlsbad, CA) by following the vendor's protocol. Briefly, H1299-Luc cells seeded in 6-well petri-dishes were treated with RT or X-PDT under 5 Gy X-ray irradiation. Un-irradiated cells were used as control. The Image-iT reagent was incubated with cells for 30 min. The cells were washed three times with PBS and observed on an Olympus IX71 fluorescence microscope. Data were analyzed by ImageJ (NIH, Bethesta, Maryland, U.S.). The experiments were repeated 5 times.
Single cell gel electrophoresis (comet assay) Mono-layered cells were prepared in 6-well cell petri-dishes one day before the experiments. On the experimental day, the 6-well cell petri-dishes were randomly divided into the control group, the X-ray radiation group, and the X-PDT (with MC540-SAO:Eu@mSiO2) group. Then the two treatment groups were irradiated by X-ray (50 kV) for a dose of 5 Gy while the control group received 0 dose irradiation. The irradiated and un-treated cells were then harvested. 1 × 105/mL of the treated cells were immediately combined with molten LMAgarose (at 37°C) at a ratio of 1:10 (v/v). 50 μL of the solution was pipetted onto a CometSlide™ (Trevigen Inc., Gaithersburg, MD). The slide was then immersed in 4°C lysis solution for 1 hour or overnight. After that, ~850 ml 4°C 1× neutral electrophoresis buffer was added to the electrophoresis gel box and the slides were placed in a slide tray. The power supply was set at 21 volts. After 45 minutes, the slides were gently removed and immersed in DNA Precipitation Solution for 30 minutes, and then in 70% ethanol for 30 minutes at room temperature. The slides were dried and stained in SYBR® safe for 30 min in the dark. The slides were then ready for microscopic imaging.
Western blot Approximately 5 × 105 H1299-Luc cells were seeded in a 6-well plate prior to radiation therapy. Cells were harvested 1 hours and 24 hours after the treatment. The cells were lysed in a lysis buffer. Proteins were separated by pre-cast 12% Bis-Tris NuPageTM SDS-PAGE (Life SciencesTM), transferred to iBlot® polyvinylidene difluoride membrane (InvitrogenTM, Grand Island, NY, USA), and immunoblotted using primary antibodies against COX2, H2AX, and β-actin. HRP-conjugated secondary antibody (Cell Signaling Technology, Danvers, MA, USA) was applied subsequently. The protein bands were then visualized with SuperSignal™ West Pico Chemiluminescent Substrate (Thermo Fisher Scientific) using a Fluorchem HD2 chemiluminescent imaging system (Protein Simple, Santa Clara, CA).
Animal studies All the animal studies conformed to the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health, USA, and a protocol approved by the Institutional Animal Care and Use Committee (IACUC), University of Georgia. Nude mice (4-6 weeks old, Harlan) with a body weight of ~20 g were used for the animal model establishment. The animals were housed in alternating lighting conditions under a 12 h dark and 12 h light regime.
Therapy studies with subcutaneous tumor models 15 nude 5-6 week athymic nude mice were randomly divided into three groups, 1) control group, 2) 1-cm thick pork group, and 3) 2-cm thick pork group. Animal models were established by subcutaneous injection of 5 × 105 H1299-Luc tumor cells onto the hind legs of mice. When tumor sizes reached 100 nm, 100 μL PBS solution containing MC540-SAO:Eu@mSiO2 nanoparticles (4.25 mg/kg) was intratumorally injected in Group 2 and 3. The same amount of PBS was injected into the tumors of the control group. For therapy studies, irradiation (5 Gy) was applied to tumors, with the rest of the animal body covered by lead. The control group received no irradiation.
Therapy studies with cancer cells implanted into the lungs 20 nude mice were randomized to receive the following treatments group (n = 5): 1) PBS, 2) MC540-SAO:Eu@mSiO2 only, 3) RT only, and 4) X-PDT (MC540-SAO:Eu@mSiO2 plus RT). 5 × 105 H1299-Luc cells were injected into the left lateral thorax of anesthetized nude mice. For Group 1 and 3, PBS solution was premixed with the cells into a final volume of 50 µL. For group 2 and 4, nanoparticles were premixed with cells into a final volume 50 µL matrigel solutions (2.5 mg MC540-SAO:Eu@mSiO2/mL, or 1.7 mg SAO:Eu/mL). Group 1 and 2 received no X-ray irradiation. Group 3 and group 4 received X-ray irradiation (5 Gy) 5 minutes after the injection. Only one therapy dose was given to each animal. The tumor growth was monitored BLI by an IVIS Lumina scanner (PerkinElmer Inc. Waltham, Massachusetts). Tumors and major organs from the euthanized animals were harvested, weighed, and cryosectioned. The tissue sections were then subjected to standard H&E staining to assess treatment outcomes and side effects (BBC Biochemical). Hemodiagnosis assay was performed by withdrawing the blood before the treatment as well as before euthanizing the mice. The concentrations of AST and ALT were determined by using commercial kits from Thermo-Scientific.
Statistical analyses Quantitative data were expressed as mean ± s.e.m. A two-tailed Student's t-test was used for statistically comparing the treatment group and the control group. P < 0.05 was considered statistically significant.
Supplementary Material
Supplementary tables and figures.
Acknowledgements
This research was supported by a DoD CDMRP grant (CA140666, J.X.), a NSF CAREER grant (NSF1552617, J.X.), a UGA-GRU seed grant (J.X.), and a NIH P41 grant support (GM103390, L. W.).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Agostinis P, Berg K, Cengel KA, Foster TH, Girotti AW, Gollnick SO. et al. Photodynamic Therapy of Cancer: An Update. Ca-Cancer J Clin. 2011;61:250-81
2. Castano AP, Mroz P, Hamblin MR. Photodynamic therapy and anti-tumour immunity. Nat Rev Cancer. 2006;6:535-45
3. Dolmans DEJGJ, Fukumura D, Jain RK. Photodynamic therapy for cancer. Nat Rev Cancer. 2003;3:380-7
4. Juarranz A, Jaen P, Sanz-Rodriguez F, Cuevas J, Gonzalez S. Photodynamic therapy of cancer. Basic principles and applications. Clin Transl Oncol. 2008;10:148-54
5. Lucky SS, Soo KC, Zhang Y. Nanoparticles in Photodynamic Therapy. Chem Rev. 2015;115:1990-2042
6. Fink-Puches R, Hofer A, Smolle J, Kerl H, Wolf P. Primary clinical response and long-term follow-up of solar keratoses treated with topically applied 5-aminolevulinic acid and irradiation by different wave bands of light. J Photoch Photobio B. 1997;41:145-51
7. Muroya T, Suehiro Y, Umayahara K, Akiya T, Iwabuchi H, Sakunaga H. et al. [Photodynamic therapy (PDT) for early cervical cancer]. Gan to kagaku ryoho Cancer & chemotherapy. 1996;23:47-56
8. Walther MM, Delaney TF, Smith PD, Friauf WS, Thomas GF, Shawker TH. et al. Phase I trial of photodynamic therapy in the treatment of recurrent superficial transitional cell carcinoma of the bladder. Urology. 1997;50:199-206
9. Lou PJ, Jones L, Hopper C. Clinical outcomes of photodynamic therapy for head-and-neck cancer. Technology in cancer research & treatment. 2003;2:311-7
10. Simone CB, Friedberg JS, Glatstein E, Stevenson JP, Sterman DH, Hahn SM. et al. Photodynamic therapy for the treatment of non-small cell lung cancer. J Thorac Dis. 2012;4:63-75
11. Vrouenraets MB, Visser GWM, Snow GB, van Dongen GAMS. Basic principles, applications in oncology and improved selectivity of photodynamic therapy. Anticancer Res. 2003;23:505-22
12. Brandis A, Mazor O, Neumark E, Rosenbach-Belkin V, Salomon Y, Scherz A. Novel water-soluble bacteriochlorophyll derivatives for vascular-targeted photodynamic therapy: Synthesis, solubility, phototoxicity and the effect of serum proteins. Photochem Photobiol. 2005;81:983-93
13. O'Connor AE, Gallagher WM, Byrne AT. Porphyrin and Nonporphyrin Photosensitizers in Oncology: Preclinical and Clinical Advances in Photodynamic Therapy. Photochem Photobiol. 2009;85:1053-74
14. Chen H, Wang GD, Chuang YJ, Zhen Z, Chen X, Biddinger P. et al. Nanoscintillator-mediated X-ray inducible photodynamic therapy for in vivo cancer treatment. Nano Lett. 2015;15:2249-56
15. Ma L, Zou XJ, Bui B, Chen W, Song KH, Solberg T. X-ray excited ZnS:Cu,Co afterglow nanoparticles for photodynamic activation. Appl Phys Lett. 2014:105
16. Zou XJ, Yao MZ, Ma L, Hossu M, Han XM, Juzenas P. et al. X-ray-induced nanoparticle-based photodynamic therapy of cancer. Nanomedicine-Uk. 2014;9:2339-51
17. Zhang C, Zhao KL, Bu WB, Ni DL, Liu YY, Feng JW. et al. Marriage of Scintillator and Semiconductor for Synchronous Radiotherapy and Deep Photodynamic Therapy with Diminished Oxygen Dependence. Angew Chem Int Edit. 2015;54:1770-4
18. Karnkaew A, Chen F, Zhan YH, Majewski RL, Cai WB. Scintillating Nanoparticles as Energy Mediators for Enhanced Photodynamic Therapy. Acs Nano. 2016;10:3918-35
19. Chen W, Zhang J. Using nanoparticles to enable simultaneous radiation and photodynamic therapies for cancer treatment. Journal of nanoscience and nanotechnology. 2006;6:1159-66
20. Sato Y, Yamazaki T, Yasukawa K, Kaneita Y, Mochimaru J, Hanai M. et al. [Merocyanine 540-mediated photodynamic therapy inhibits P-glycoprotein (P-gp) activity in adriamycin-resistant K562 cells]. Gan to kagaku ryoho Cancer & chemotherapy. 1999;26:2195-200
21. Sung HY, Wu HG, Ahn JH, Park WY. Dcr3 inhibit p53-dependent apoptosis in gamma-irradiated lung cancer cells. Int J Radiat Biol. 2010;86:780-90
22. Kim EH, Park AK, Dong SM, Ahn JH, Park WY. Global analysis of CpG methylation reveals epigenetic control of the radiosensitivity in lung cancer cell lines. Oncogene. 2010;29:4725-31
23. Im CN, Kim BM, Moon EY, Hong DW, Park JW, Hong SH. Characterization of H460R, a Radioresistant Human Lung Cancer Cell Line, and Involvement of Syntrophin Beta 2 (SNTB2) in Radioresistance. Genomics & informatics. 2013;11:245-53
24. Hu J, Tang YA, Elmenoufy AH, Xu HB, Cheng Z, Yang XL. Nanocomposite-Based Photodynamic Therapy Strategies for Deep Tumor Treatment. Small. 2015;11:5860-87
25. Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem. 1998;273:5858-68
26. Thrall DE. Biologic basis of radiation therapy. The Veterinary clinics of North America Small animal practice. 1997;27:21-35
27. Girotti AW. Photosensitized oxidation of membrane lipids: reaction pathways, cytotoxic effects, and cytoprotective mechanisms. J Photoch Photobio B. 2001;63:103-13
28. Yasuhara S, Zhu Y, Matsui T, Tipirneni N, Yasuhara Y, Kaneki M. et al. Comparison of comet assay, electron microscopy, and flow cytometry for detection of apoptosis. J Histochem Cytochem. 2003;51:873-85
29. Paull TT, Rogakou EP, Yamazaki V, Kirchgessner CU, Gellert M, Bonner WM. A critical role for histone H2AX in recruitment of repair factors to nuclear foci after DNA damage. Curr Biol. 2000;10:886-95
30. Rappold I, Iwabuchi K, Date T, Chen J. Tumor suppressor p53 binding protein 1 (53BP1) is involved in DNA damage-signaling pathways. J Cell Biol. 2001;153:613-20
31. Ferrario A, Von Tiehl K, Wong S, Luna M, Gomer CJ. Cyclooxygenase-2 inhibitor treatment enhances photodynamic therapy-mediated tumor response. Cancer Res. 2002;62:3956-61
32. Agarwal ML, Larkin HE, Zaidi SIA, Mukhtar H, Oleinick NL. Phospholipase Activation Triggers Apoptosis in Photosensitized Mouse Lymphoma-Cells. Cancer Res. 1993;53:5897-902
33. Yoshizumi T, Brady SL, Robbins ME, Bourland JD. Specific issues in small animal dosimetry and irradiator calibration. Int J Radiat Biol. 2011;87:1001-10
34. Idris NM, Gnanasammandhan MK, Zhang J, Ho PC, Mahendran R, Zhang Y. In vivo photodynamic therapy using upconversion nanoparticles as remote-controlled nanotransducers. Nat Med. 2012;18:1580-U190
35. Montazerabadi AR, Sazgarnia A, Bahreyni-Toosi MH, Ahmadi A, Aledavood A. The effects of combined treatment with ionizing radiation and indocyanine green-mediated photodynamic therapy on breast cancer cells. Journal of photochemistry and photobiology B, Biology. 2012;109:42-9
36. Kavarnos G, Nath R, Bongiorni P. Visible-light and X irradiations of Chinese hamster lung cells treated with hematoporphyrin derivative. Radiat Res. 1994;137:196-201
37. Prinsze C, Penning LC, Dubbelman TM, VanSteveninck J. Interaction of photodynamic treatment and either hyperthermia or ionizing radiation and of ionizing radiation and hyperthermia with respect to cell killing of L929 fibroblasts, Chinese hamster ovary cells, and T24 human bladder carcinoma cells. Cancer Res. 1992;52:117-20
38. Luksiene Z, Kalvelyte A, Supino R. On the combination of photodynamic therapy with ionizing radiation. Journal of photochemistry and photobiology B, Biology. 1999;52:35-42
39. Bernier J, Hall EJ, Giaccia A. Timeline - Radiation oncology: a century of achievements. Nature Reviews Cancer. 2004;4:737-U15
40. Begg AC, Stewart FA, Vens C. GENOMIC INSTABILITY IN CANCER Strategies to improve radiotherapy with targeted drugs. Nat Rev Cancer. 2011;11:239-53
41. Prasanna PGS, Stone HB, Wong RS, Capala J, Bernhard EJ, Vikram B. et al. Normal tissue protection for improving radiotherapy: where are the Gaps? Transl Cancer Res. 2012;1:35-48
42. Wardman P. Chemical radiosensitizers for use in radiotherapy. Clin Oncol-Uk. 2007;19:397-417
43. Liu F, Budai JD, Li XF, Tischler JZ, Howe JY, Sun CJ. et al. New Ternary Europium Aluminate Luminescent Nanoribbons for Advanced Photonics. Adv Funct Mater. 2013;23:1998-2006
Author contact
![]() Corresponding author: hchenedu Or jinxieedu.
Corresponding author: hchenedu Or jinxieedu.
Citation styles
APA
Wang, G.D., Nguyen, H.T., Chen, H., Cox, P.B., Wang, L., Nagata, K., Hao, Z., Wang, A., Li, Z., Xie, J. (2016). X-Ray Induced Photodynamic Therapy: A Combination of Radiotherapy and Photodynamic Therapy. Theranostics, 6(13), 2295-2305. https://doi.org/10.7150/thno.16141.
ACS
Wang, G.D.; Nguyen, H.T.; Chen, H.; Cox, P.B.; Wang, L.; Nagata, K.; Hao, Z.; Wang, A.; Li, Z.; Xie, J. X-Ray Induced Photodynamic Therapy: A Combination of Radiotherapy and Photodynamic Therapy. Theranostics 2016, 6 (13), 2295-2305. DOI: 10.7150/thno.16141.
NLM
Wang GD, Nguyen HT, Chen H, Cox PB, Wang L, Nagata K, Hao Z, Wang A, Li Z, Xie J. X-Ray Induced Photodynamic Therapy: A Combination of Radiotherapy and Photodynamic Therapy. Theranostics 2016; 6(13):2295-2305. doi:10.7150/thno.16141. https://www.thno.org/v06p2295.htm
CSE
Wang GD, Nguyen HT, Chen H, Cox PB, Wang L, Nagata K, Hao Z, Wang A, Li Z, Xie J. 2016. X-Ray Induced Photodynamic Therapy: A Combination of Radiotherapy and Photodynamic Therapy. Theranostics. 6(13):2295-2305.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) License. See http://ivyspring.com/terms for full terms and conditions.