Theranostics
13.3
Impact Factor
- Current Issue
- Advance Articles
- Volume 16; 2026
- Volume 15; 2025
- Volume 14; 2024
- Volume 13; 2023
- Volume 12; 2022
- Archive
- Cover Images
- Cover Suggestion
- Special Issues
Top
Introduction
Materials and Methods
Results
Discussion
Supplementary Material
Acknowledgements
References
Introduction
Materials and Methods
Results
Discussion
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2017; 7(1):40-50. doi:10.7150/thno.16676 This issue Cite
Review
Preclinical Applications of 3'-Deoxy-3'-[18F]Fluorothymidine in Oncology - A Systematic Review
Sonja Schelhaas1, Kathrin Heinzmann2, Vikram R. Bollineni3, Gerbrand M. Kramer4, Yan Liu3, John C. Waterton5, Eric O. Aboagye2, Anthony F. Shields6, Dmitry Soloviev7,* ![]() , Andreas H. Jacobs1,8,*
, Andreas H. Jacobs1,8,* ![]()
1. European Institute for Molecular Imaging (EIMI), Westfälische Wilhelms-Universität (WWU) Münster, Münster, Germany.
2. Comprehensive Cancer Imaging Centre, Imperial College London, UK.
3. European Organization for Research and Treatment of Cancer Headquarters, Brussels, Belgium.
4. Department of Radiology and Nuclear Medicine, VU University Medical Center, Amsterdam, The Netherlands.
5. Imaging Sciences, University of Manchester, Manchester, UK.
6. Department of Oncology, Karmanos Cancer Institute, Wayne State University, Detroit, Michigan, USA.
7. Cancer Research UK Cambridge Institute, University of Cambridge, UK.
8. Department of Geriatric Medicine, Johanniter Hospital, Bonn, Germany.
* Equal contribution.
Received 2016-7-1; Accepted 2016-9-16; Published 2017-1-1
Citation:
Schelhaas S, Heinzmann K, Bollineni VR, Kramer GM, Liu Y, Waterton JC, Aboagye EO, Shields AF, Soloviev D, Jacobs AH. Preclinical Applications of 3'-Deoxy-3'-[18F]Fluorothymidine in Oncology - A Systematic Review. Theranostics 2017; 7(1):40-50. doi:10.7150/thno.16676. https://www.thno.org/v07p0040.htm
Other stylesAbstract

The positron emission tomography (PET) tracer 3'-deoxy-3'-[18F]fluorothymidine ([18F]FLT) has been proposed to measure cell proliferation non-invasively in vivo. Hence, it should provide valuable information for response assessment to tumor therapies. To date, [18F]FLT uptake has found limited use as a response biomarker in clinical trials in part because a better understanding is needed of the determinants of [18F]FLT uptake and therapy-induced changes of its retention in the tumor. In this systematic review of preclinical [18F]FLT studies, comprising 174 reports, we identify the factors governing [18F]FLT uptake in tumors, among which thymidine kinase 1 plays a primary role. The majority of publications (83 %) report that decreased [18F]FLT uptake reflects the effects of anticancer therapies. 144 times [18F]FLT uptake was related to changes in proliferation as determined by ex vivo analyses. Of these approaches, 77 % describe a positive relation, implying a good concordance of tracer accumulation and tumor biology. These preclinical data indicate that [18F]FLT uptake holds promise as an imaging biomarker for response assessment in clinical studies. Understanding of the parameters which influence cellular [18F]FLT uptake and retention as well as the mechanism of changes induced by therapy is essential for successful implementation of this PET tracer. Hence, our systematic review provides the background for the use of [18F]FLT in future clinical studies.
Keywords: positron emission tomography, FLT, Oncology.
Introduction
Non-invasive molecular imaging with positron emission tomography (PET) is used in cancer research to identify and stage tumors and assess tumor response to anti-cancer treatments [1]. The most established tracer for PET applications is 2-[18F]-fluoro-2-deoxy-D-glucose ([18F]FDG), whose uptake is regulated by glucose metabolism [2]. Due to the Warburg effect, uptake of [18F]FDG in tumors is generally high [3], making it a good candidate for visualization of neoplastic lesions. On the other hand, metabolically active organs like heart and brain, and glycolytic cells in inflammatory lesions accumulate this tracer as well. To overcome the drawbacks of [18F]FDG, Shields et al. proposed 3'-deoxy-3'-[18F]fluorothymidine ([18F]FLT) as a radiotracer for imaging actively proliferating cells [4]. [18F]FLT is taken up by cells by the same mechanisms as the nucleoside thymidine. This transport step is facilitated by nucleoside transporters, especially by the human equilibrative nucleoside transporter 1 (hENT1) [5]. Once within the cell, [18F]FLT is phosphorylated by the enzyme thymidine kinase 1 (TK1), which results in the intracellular accumulation of the tracer. Accumulation of [18F]FLT reflects the thymidine salvage pathway, specifically activity of the cytoplasmic form of TK1, which in turn is considered to be tightly linked to the S-phase of the cell proliferation cycle [6] (Fig. 1). The other important thymidine-to-DNA pathway is the de novo DNA synthesis pathway [7]. The balance of de novo and salvage pathways is of crucial importance for the accumulation rate of [18F]FLT [8,9].
[18F]FLT PET has previously been rarely used for tumor therapy follow-up in clinical trials. This is in part due to limited knowledge of the factors determining [18F]FLT uptake and therapy-induced changes of its retention. The Innovative Medicine Initiative Joint Undertaking funded project QuIC-ConCePT aims to qualify several imaging biomarkers for assessing the pharmacodynamic response of tumors to anti-cancer drugs [10]. [18F]FLT uptake is one of these imaging biomarkers. To better understand how uptake of this radiotracer reflects cellular proliferation, we herein summarize the current literature on its preclinical and in vitro applications in oncology in a systematic review. We focus on the uptake of this tracer in untreated cells and tumors and evaluate the use of [18F]FLT in monitoring therapy response to anti-cancer treatments. A detailed descriptive analysis and discussion of the articles can be found in the Supplementary Results. In this manuscript we present the overall summary of the preclinical studies, discuss the confounding factors of [18F]FLT uptake and review its utility to monitor tumor response to therapy. These data are complemented by an overview of the selected study designs and recommendations and implications for future preclinical and clinical studies.
Fig 1
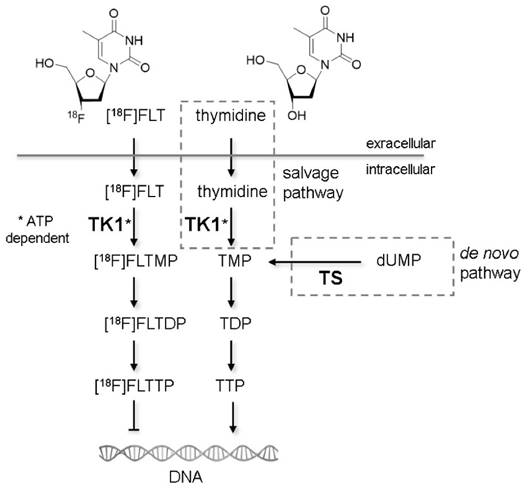
Mechanisms of cellular [18F]FLT retention. Similar to thymidine in the salvage pathway, [18F]FLT is taken up from the extracellular milieu by specialized nucleoside transporters or via passive diffusion. Within the cell [18F]FLT is phosphorylated by thymidine kinase 1 (TK1), the enzyme also responsible for phosphorylation of thymidine. TK1 activity is dependent on adenosine triphosphate (ATP). The phosphorylated form of thymidine (TMP) is further phosphorylated to thymidine diphosphate (TDP) and thymidine triphosphate (TTP), which is subsequently incorporated into the DNA. The phosphorylated form of [18F]FLT cannot be incorporated into DNA but is trapped within the cell [60]. Techniques like PET or gamma counter measurements are capable of quantifying the rate of accumulation of [18F]FLT within cells. An alternative thymidine metabolism pathway is the de novo synthesis. The key enzyme of this pathway is thymidylate synthase (TS), which methylates deoxyuridine monophosphate (dUMP) to TMP. The two pathways merge at the level of TMP. Studies describing the importance of the different factors for [18F]FLT uptake are described in detail in the Supplementary Results.
Materials and Methods
We performed a systematic search to address the question whether [18F]FLT accumulation reflects cell biology in untreated and treated tumors in preclinical model systems. Thereby, we adhered to the Preferred Reporting Items for Systematic Reviews and Meta-analyses (PRISMA) statement [11] (see PRISMA checklist in Supplementary Table S1). The search was performed in the bibliographic database Embase.com, which also includes the Medline database. The search was conducted with a combination of terms related to [18F]FLT and neoplasm (see Supplementary Methods) either to be included in the title or abstract and the study type filter “preclinical” was applied. Selection criteria were (i) published in English as a full paper in a peer-reviewed journal (no conference abstracts), (ii) published between January 1998 and January 2016 and (iii) preclinical and / or in vitro data. A single person screened the manuscripts to confirm that inclusion criteria were met. Further details of the search strategy can be found in the Supplementary Methods.
Results
Study identification and selection
The search strategy identified a total of n = 388 publications describing the use of [18F]FLT in the field of oncology. These were manually screened to confirm that the inclusion criteria were met. In addition, n = 9 papers were found through non-automated literature search, that were missed by the search strategy. These were also included, resulting in a total of n = 174 publications being eligible for inclusion in this review. Fig. 2 describes the process of study identification according to the PRISMA guideline.
Design of experimental studies
The studies varied considerably in design, which is described in detail in the Supplementary Results (chapter 3.8). Overall, colorectal cancer is the most commonly used cancer type addressed. [18F]FLT PET images were most frequently acquired 60 min after tracer injection for a duration of 10 min. However, the optimal time window might depend on the tumor model chosen. In contrast, therapy protocols were less harmonized. For instance, drug applications varied from daily to weekly intervals, hampering a direct comparison and meta-analysis of the data. These variances appear to recapitulate the variances in the clinical situation [12]. More harmonized protocols are needed to reliably evaluate the role of [18F]FLT PET in predicting therapy response in the clinic.
[18F]FLT uptake in untreated tumors
n = 45 publications characterize [18F]FLT uptake in untreated tumors. A detailed descriptive analysis of the confounding factors identified in these studies can be found in the Supplementary Results (chapter 3.2). In summary, it appears that uptake of the tracer is primarily determined by TK1 activity and the balance between salvage and de novo DNA synthesis pathways. But also other factors such as the presence of hENT1 or thymidine are of importance (see Table 1 and Fig. 1). Furthermore, [18F]FLT appears to be more tumor specific than [18F]FDG with regards to inflammatory lesions, even though [18F]FLT is also detected in some inflammatory tissues involving proliferating immune cells (see also Supplementary Results, chapter 3.2.2).
Fig 2
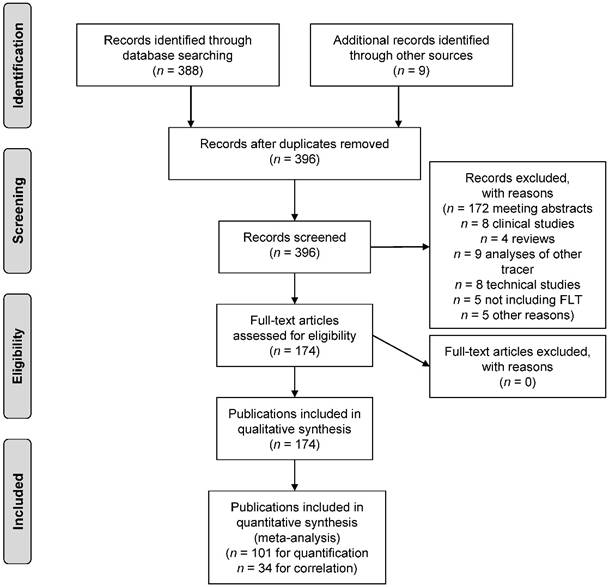
PRISMA flow diagram.
Table 1
Summary of the factors important for cellular [18F]FLT uptake.
| Baseline [18F]FLT uptake in tumors and its change in response to therapy varies greatly as a result of an overall balance of some or all of the factors below: |
| - TK1 expression levels |
| - TK1 activity |
| - ATP levels, as a cofactor for TK1 activity |
| - TS levels and activity (de novo DNA pathway), competing with the salvage DNA synthesis |
| - TP expression and activity, influencing endogenous tumor thymidine levels |
| - Thymidine levels in tumor, reflecting the nucleotide turnover rates |
| - Expression of nucleoside transporters, which are essential for the transport of [18F]FLT (notably hENT1) |
| - Delivery of the tracer |
| - Blood-brain barrier hampers [18F]FLT uptake in brain tumors |
| - Thymidine levels in blood plasma, which are competing with [18F]FLT |
| - Animal body temperature |
| - Anesthetics used |
| - Oxygen breathing |
| - Tumor vascularity, and the changes thereof by antiangiogenic treatments |
Changes of [18F]FLT uptake in treated tumors
n = 147 studies use [18F]FLT to monitor treatment response. Of these, next to radiotherapy, n = 21 different chemotherapeutic agents and n = 46 targeted agents were employed. A comprehensive description and discussion of these publications is presented in the Supplementary Results (chapter 3.3-3.7).
In summary, n = 122 studies describe a decline of [18F]FLT after tumor treatment in responsive tumor models (83 % of the 147 studies evaluating treatment response, Fig. 3A) which is in line with reduced proliferation of the tumor. Several of these studies report unchanged [18F]FLT when proliferation is not changed, for instance at very early time points after therapy initiation. Also in resistant models, [18F]FLT uptake is not altered upon treatment, underlining that changes in [18F]FLT accumulation are specific for changes in tumor proliferation, as demonstrated in n = 14 studies [13-25].
n = 14 of the 122 reports with reduced [18F]FLT accumulation after therapy, demonstrate that at the end of a treatment cycle, [18F]FLT levels might not be distinguishable from baseline, reflecting recovery of tumor proliferation as shown by respective immunohistochemistry [9,15,26-37]. Hence, for successful treatment monitoring, the imaging time point is of crucial importance. However, since therapy protocols differ substantially in timing (single administration, daily administration, therapy on two consecutive days, weekly therapy, etc.) no common conclusion can be drawn on a general time window that is best suitable for assessment of treatment response in preclinical studies. Furthermore, these findings also suggest that an early decline in [18F]FLT uptake does not rule out the possibility that at later time points treatment efficacy is not maintained, which can partly be related to a limited half-life of drugs within the body. Consequently, early imaging might help to answer the question whether the drug reaches the target and affects DNA synthesis pathways. However, it cannot predict treatment outcomes without respective growth or survival studies.
n = 12 studies (8.2 %) employ agents targeting TS and describe an increased [18F]FLT uptake early after treatment initiation. Most of these studies report a transient upregulation of TK1, which explains the increase in tracer uptake. This transient increase in [18F]FLT uptake can be referred to as flare phenomenon and can be attributed to the cellular mechanism of the therapeutic agent. Nevertheless, reduced cellular proliferation can be observed by [18F]FLT PET at later time points. In the following, these agents are grouped together as TS inhibitors, comprising agents of the group of antimetabolites, but also kinase inhibitors [38] or combination therapies [39].
Fig 3
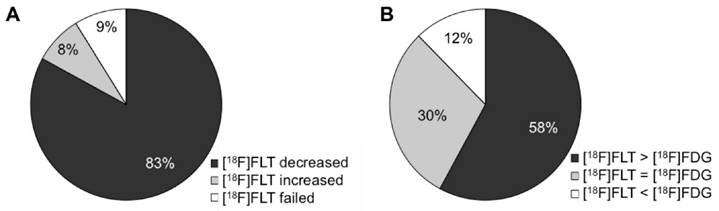
Graphical demonstration of the performance of [18F]FLT after therapeutic treatment of preclinical in vivo tumor models. (A) Of the n = 147 therapy follow up studies included in this review, [18F]FLT is decreased in n = 122 cases and hence readily parallels therapy response. n = 12 studies employ agents that inhibit TS, inducing an upregulation of the thymidine salvage pathway and hence [18F]FLT uptake. Therefore, increased [18F]FLT in early time-frames after drug administration reflects the mechanism of action of the drug. (B) n = 57 studies employed [18F]FLT and [18F]FDG for monitoring of treatment response. In n = 33 of these studies [18F]FLT appears to be superior to [18F]FDG.
In n = 13 therapy approaches (8.8 %) [18F]FLT failed to predict treatment response (i.e. unchanged tracer uptake despite effective therapy as assessed by tumor growth inhibition, immunohistochemistry of proliferation or similar). These studies cannot be attributed to a specific drug class. In most cases a reason for the failure is hypothesized, such as suboptimal timing of imaging in the case of antibody therapy [40], or low uptake of [18F]FLT in the tumors [9,41,42]. The latter might be related to preferential use of the thymidine de novo pathway by the tumor, which further complicates interpretation of changes in [18F]FLT uptake after treatment [43]. Two studies describe that unchanged [18F]FLT uptake is in line with unaltered Ki67 staining [24,44]. Therefore, [18F]FLT accurately reflects the proliferative activity of the tumor (true negative). Other studies describe that the pharmacological properties of the respective drug (like mild upregulation of TK1 or genetic alterations of the tumor, like p53 knockout) might not allow [18F]FLT to be used for therapy follow up [45-49]. This leaves only one study (0.7 %) describing an inexplicable failure of [18F]FLT in predicting therapy response [13].
Of note, all of the n = 15 studies employing radiotherapy show that [18F]FLT reliably predicts treatment response. For all other therapy approaches, failures could be noted. Consequently, [18F]FLT uptake appears to be an excellent imaging biomarker to monitor response to radiotherapy.
Whenever studies also employed [18F]FDG PET to monitor therapy response, we reported this as well in the detailed description in the Supplementary Results. Of these n = 57 studies, [18F]FLT is superior to [18F]FDG (i.e. changes are more pronounced or occur at an earlier time point) in n = 33 cases (58 %), and it is comparable in n = 17 studies (30 %) (Fig. 3B). However, these two tracers provide different information as they measure different pharmacodynamic events.
Some in vivo studies provided numeric quantitative imaging data, either from PET images or from gamma counter measurements. This allowed us to estimate the changes of [18F]FLT uptake relative to baseline or to a respective control, as presented in Fig. 4 (see also Supplementary Table S2). Each data point represents one result of a study. Most studies provide more than one data point since they report on different time points, different model systems, etc. (see Supplementary Table S2 for details). When respective data were available, standard deviations were displayed.
Of the overall n = 299 data points displayed in Fig. 4, n = 190 are statistically significant. The other data points originate from resistant tumors (blue) or tumors that recovered (green). In other cases, [18F]FLT uptake might have been determined under circumstances that also do not result in a change of tracer accumulation, for instance if the therapy dose employed was too low or the time point was very early after start of treatment. Furthermore, for some data points, statistical information was not provided.
43 % of the results displayed, relate the change to a control group, whereas the majority are comparisons with baseline uptake. We were not able to see any major differences between these analyses. In the clinical setting changes in tracer uptake are generally compared to a baseline scan before treatment initiation. However, under certain circumstances, comparison to a control group might be preferable. For instance, [18F]FLT uptake might be unchanged upon treatment, while the signal from an untreated control is significantly increased. This is possible with cytostatic treatments, which do affect tumor growth, whereas no change of [18F]FLT PET measurement before and after treatment can be noted (see e.g. [50]). This is supported by the observation that individual cell uptake of [18F]FLT is not always affected by cytostatic treatments [51].
A maximum [18F]FLT reduction of 97 % relative to baseline was reported [52]. No differences between the different therapeutic classes were noted. However, a definite conclusion is hampered by the great variability of the experimental setups and low sample size of some treatment groups. This impedes proper statistical evaluation of the data.
We also sorted the data according to tumor type investigated, quantification mode employed, or day of image acquisition relative to treatment initiation (Supplementary Fig. S9). However, we were not able to detect any specific groups that have a very pronounced reduction of [18F]FLT. The only remarkably different group is the one comprising the TS inhibitors, inducing an increase in tracer uptake. We displayed the results in a separate graph due to the variances in scaling. The increase of [18F]FLT uptake in this group is up to a factor of five with high variabilities.
Fig 4
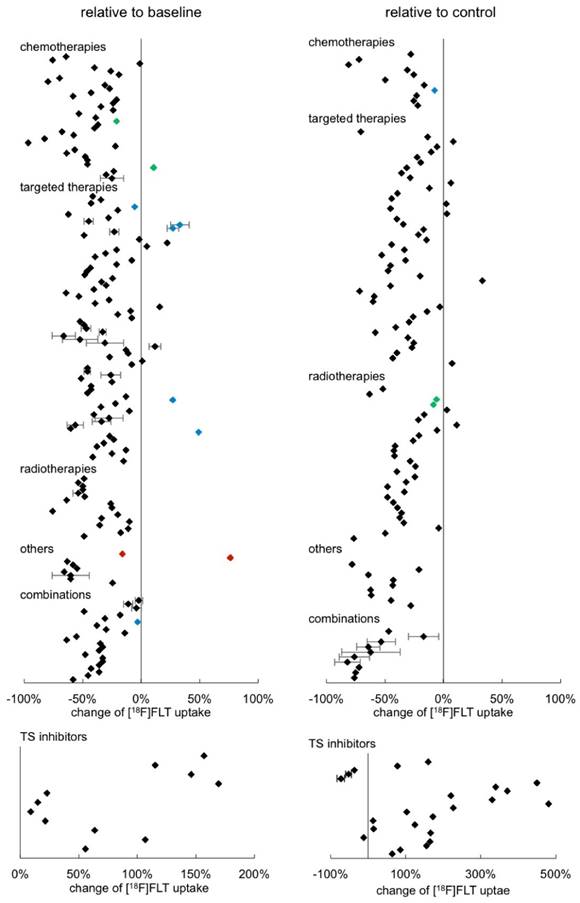
Overview of the relative change of [18F]FLT after tumor therapy in preclinical in vivo models sorted according to therapy approach. Wherever possible, quantitative data were extracted from the publications and calculated as percent change either relative to baseline (left column) or to a respective control (right column). Each data point represents a single result of a study. The standard deviation could not be extracted from all publications. Rectangles represent results of a single analysis. Green rectangles represent data points of tumors that have regained their proliferative capacity at the end of a treatment cycle (as proven by immunohistochemistry). Blue rectangles represent data points of resistant tumors. Red rectangles represent results of tumors with very low baseline [18F]FLT uptake. Detailed results are displayed in Supplementary Table S2.
Relation of [18F]FLT uptake to proliferation
Several studies employ ex vivo tumor analyses to validate in vivo imaging findings. In doing so, many studies pursued more than one approach of ex vivo validation. n = 32 publications calculated correlations of [18F]FLT with proliferation as assessed by tumor growth or histological markers of proliferation, as summarized in Supplementary Table S3. A multitude of studies describe that changes in [18F]FLT are linked to changes in tumor volume. Of these, n = 14 reports calculated a correlation. n = 102 approaches related [18F]FLT uptake to histological proliferation markers (mostly Ki67, but also PCNA or BrdU). Of these, only n = 21 studies did not detect any association of [18F]FLT accumulation and proliferation. A link of [18F]FLT with TK1 expression or activity was assessed in n = 42 studies; of these only n = 12 were not able to relate these two parameters. Association of [18F]FLT accumulation with either of the proliferation markers TK1 and Ki67 appears to be comparable. Overall, 20 % of the approaches calculated a correlation (Fig. 5). 57 % report that changes in [18F]FLT can be attributed to changes in proliferation markers. However, no correlation was calculated, which does not necessarily imply that there is no correlation. In the remaining 23 % the proliferation markers were not related to tracer uptake. This number reflects the multitude of factors that can affect uptake and retention of [18F]FLT (see Table 1), confounding the link between [18F]FLT uptake and cellular proliferation. Nevertheless, 83 % of the studies successfully employed [18F]FLT to monitor treatment response in tumors (see Fig. 3A). It is likely that the number of false negative studies is higher than reflected in this review because negative results are not published on a regular basis. Also from the experience within the QuIC-ConCePT consortium we know that changes in [18F]FLT uptake might be disconnected from the tumor response to treatment despite of the use of a suitable model (unpublished observations).
Fig 5
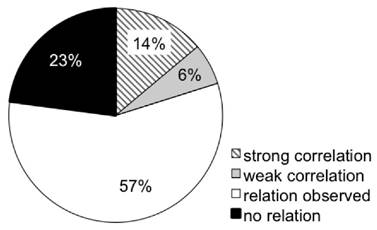
Overview of relations of [18F]FLT with parameters of cellular proliferation. This pie chart depicts the relations observed in the studies investigated in this review. Cellular proliferation comprises immunohistochemistry of Ki67, PCNA, BrdU and TK1, as well as TK1 activity or expression analyses. Supplementary Fig. S10 depicts the respective data sorted more specifically for TK1 and other markers of proliferation. “Relation observed” relates to non-quantitative observational studies. Correlations with correlation coefficients > 0.7 were considered strong.
To assess the preferential publication of positive results (i.e. publication bias), statistical approaches like a funnel plot would be needed. However, due to heterogeneities in study design and the generally low sample sizes usually employed in preclinical studies, this is not feasible here. In addition, many studies are not fully compliant with the ARRIVE guidelines for reporting preclinical studies [53], so it is sometimes impossible to verify that valid experimental designs and analyses were employed.
Discussion
Here, we systematically review the literature of preclinical [18F]FLT applications in oncology. Despite a range of confounding factors affecting [18F]FLT uptake, especially TK1 expression and activity (see also Table 1), numerous studies demonstrate that [18F]FLT can be used to monitor treatment response in appropriate tumor models. However, when interpreting [18F]FLT PET findings to delineate proliferating tumors or assess therapy response, certain considerations and pre-requisites have to be taken into account, like the mechanism of action of the therapy.
Complementary to a recently published review focusing on [18F]FDG and [18F]FLT PET imaging in preclinical studies for monitoring anti-cancer treatments [54], we attempted to systematically cover experimental applications of [18F]FLT in the field of oncology to answer the question whether changes in [18F]FLT accumulation are indeed related to tumor biology.
A biomarker, such as an imaging biomarker, is “a physical sign or laboratory measurement that occurs in association with a pathological process and that has putative diagnostic or prognostic utility” [55]. In oncology the gold standard for prognostic utility is overall survival (or at least progression-free survival). This is seldom measured in preclinical studies. In animals usually tumor size is measured which is the preclinical equivalent of objective response in humans using response evaluation criteria in solid tumors (RECIST) [1]. Only rarely is objective response acceptable as a surrogate for overall survival. The ability of changes in [18F]FLT uptake as a biomarker to predict tumor shrinkage (which is just another biomarker) is of some interest. More important here, however, is association of [18F]FLT with underlying pathological processes (biological validation). Key questions include [56]: (i) Do drug-induced changes in [18F]FLT uptake faithfully reflect drug-induced changes in histopathological proliferation? (ii) Does the temporal evolution of drug-induced changes in [18F]FLT uptake faithfully reflect the temporal evolution of drug-induced changes in histopathological proliferation? (iii) Do increasing doses of drug induce graded changes in the [18F]FLT uptake which reflect graded changes in histopathological proliferation? (iv) For which drug classes do these relationships hold? (v) Where do these correlations break down and why? (vi) Are findings reproducible between laboratories? Preclinical studies play an essential role in imaging biomarker validation, as it is difficult to address these questions in man, and the studies reviewed here help to address every single one of these questions.
Following more than 15 years of research using [18F]FLT, recent reports note that tumor proliferation imaging with this tracer may not be straightforward [57]. From clinical experiences [18F]FLT PET is not always correlated with the Ki67 proliferation index, considered as the gold standard of proliferation, and even a reverse correlation is observed in some tumors. Therefore, in a systematic review Chalkidou et al. point out that apart from technical difficulties inherent to PET imaging, there might be biochemical reasons underlying tumor biology explaining the variability of results [58]. Moreover, results from Vanderbilt University show that the magnitude of [18F]FLT uptake should not be considered as a surrogate of proliferative index [8]. Zhang et al. concluded that “[18F]FLT PET imaging does not always 'light up' proliferating tumors” [31].
Various factors can affect [18F]FLT uptake (summarized in Table 1), specifically TK1, TS and hENT1 expression and activity, as well as endogenous thymidine levels. It appears that different parameters play different roles in various experimental and therapeutic scenarios. Even though the balance of the thymidine de novo and salvage pathway is of crucial importance, there is not a single factor responsible for [18F]FLT uptake in general. The interplay between the different determinants is complex. This should be considered when designing and interpreting [18F]FLT PET studies.
Even though [18F]FLT is not a marker of cellular proliferation per se, it is capable of reflecting therapy-induced changes in cell proliferation of many tumor types. 83 % of the studies investigating alterations in [18F]FLT uptake after tumor treatment report that tracer accumulation is reduced, reflecting reduced tumor proliferation. n = 111 approaches underpin these findings by respective ex vivo analyses of cellular proliferation. The specificity of the tracer to reflect changes in tumor proliferation is emphasized by n = 14 studies showing that [18F]FLT uptake is not altered in resistant models. These studies imply that [18F]FLT PET can be employed for monitoring of response to anti-cancer treatments in the clinical situation. However, it is likely that the results described in this review might have literature bias as negative results are not always published. Potentially, the inclusion of conference abstracts, which are more likely to describe negative results, would reduce this bias.
It should be kept in mind, that in general the study design of preclinical research is less standardized than clinical approaches. Parameters that vary include tumor type, PET imaging protocol, mode of PET quantification, number of subjects and groups, number of imaging time points, and type and number of interventions. The impact of these factors on study outcome is difficult to estimate. Therefore, the number of directly comparable studies is very limited. This hampers statistical evaluation as usually done in meta-analyses. Analyses such as forest plots, funnel plots or ROC analysis could not be pursued. Even though we summed up some factors, conclusions are rather based on a qualitative approach. This is one of the major limitations of this systematic review.
Even though our review supports the implementation of [18F]FLT PET in clinical research, it needs to be determined in future larger scale clinical trials the extent to which our findings can be translated. The number of clinical studies up to now is limited. The QuIC-ConCePT consortium recently published a systematic review on [18F]FLT PET uptake as a measure of treatment response in cancer patients, following an analogous search strategy. This review revealed that [18F]FLT PET is indeed a good predictor of early response to systemic-, radio- and concurrent chemotherapy [59]. Furthermore, this article points out that the time point of imaging is of importance to assess treatment response accurately. This finding is well in line with the observations made in preclinical studies, as presented here. The promising nature of [18F]FLT PET as a tool to monitor tumor therapy response has also been highlighted by other clinical [18F]FLT reviews [12,60-62]. However, it should be kept in mind that [18F]FLT uptake might not be a reliable biomarker of proliferation in all instances, as pointed out in both, clinical [58,62] and preclinical [54, this review] systematic reviews.
Sanghera et al. noted a vast diversity in study protocols [12], which is in line with our personal observations. Variances in uptake time, acquisition method, and time per bed position could influence quantitative measurements and analysis and thereby decrease comparability of results. As the optimal time window for imaging might depend on the model employed, we recommend that this time frame should be determined and used in preclinical studies. Future clinical trials should follow harmonized protocols in analogy to those proposed for [18F]FDG as outlined e.g. by the PERCIST criteria [1] or respective recommendations by the European Association of Nuclear Medicine [63]. Some further suggestions for harmonizing clinical [18F]FLT study protocols have been proposed by Peck et al. [64]. The missing standardization of protocols probably represents one of the major challenges for reliable implementation of [18F]FLT PET in clinical trials.
Only when clinical acquisition protocols have been standardized, the full clinical validation of [18F]FLT will be possible, i.e. demonstration that acute changes in [18F]FLT accumulation help forecast overall survival or progression free survival. Biological validation of [18F]FLT uptake is feasible via histological examination of tumor specimens, allowing relation of changes in tracer uptake to tumor biology. This can sometimes be done in clinical trials, where it is possible to recover biopsy specimens or tumors excised by surgery. However, this is not always possible in patients. To comprehensively understand the tumor biology underlying [18F]FLT uptake, preclinical studies of imaging pathology correlation are of great value. The aim of this review on preclinical [18F]FLT applications was to substantially improve the understanding of these factors.
Whether or not preclinical studies employing [18F]FLT PET for monitoring cancer therapy response are predictive for cancer patients is the subject of another systematic review by the QuIC-ConCePT consortium, which is currently in preparation. Frequently, preclinical studies have employed subcutaneous xenografts, arising from cells cultured under laboratory conditions, as they are often reproducible, straightforward to implement, and risks to welfare are easily managed. However, patient derived xenografts [65] or genetic cancer models [66] appear to more closely reflect clinical cancers, and may be more predictive. Moreover, the use of combination therapies, as also employed in the clinic, might help in gaining more clinically relevant information.
Based on the first promising clinical results and the detailed understanding of the biology underlying [18F]FLT accumulation in tumors, gained especially from preclinical studies, as described in our review, we are positive that [18F]FLT PET holds promise as a therapy response assessment tool in cancer patients. Moreover, it reports on completely different biology than does the more clinically familiar tracer [18F]FDG, which accumulates in glycolytic inflammatory cells and cancer cells. Hence, [18F]FLT accumulation and its changes should be further explored as an imaging biomarker in treatment response studies in the clinical situation. The following issues should be taken into account, when applying [18F]FLT measurements in the preclinical setting:
- Low [18F]FLT uptake at baseline (SUVmax < 1; T/B < 1) is likely an indication that the investigated tumor primarily depends on de novo thymidine synthesis or there may be high levels of thymidine in the rodent strain being studied. Consequently, [18F]FLT is likely not to be a reliable tracer in these models.
- The optimal time window for PET image acquisition should be carefully chosen and should be determined for each model in dynamic imaging studies.
- All imaging parameters and protocol details should be thoroughly documented and included in future publications to allow readers to assess the quality of the study performed and to relate findings to other publications. These parameters include PET acquisition protocol (time of image acquisition, duration of scan), time point of image acquisition (relative to treatment initiation and / or tumor implantation), PET image analysis details (reconstruction algorithms, quantitation parameters), therapy protocol (drug dose, route of administration, application schedule), numbers of animals studied, and numerical results, including deviations. The ARRIVE guidelines should be followed [53] and negative results should be published.
- Drugs impacting TK1 (like TS antagonists or antifolates) may induce an [18F]FLT flare effect. This might mask possible reduction of [18F]FLT uptake due to decreased proliferation early after drug administration. Up to now it is not known, how this flare effect might be related to treatment outcome. Hence, early [18F]FLT imaging of treatment response to drugs affecting TK1 cannot be recommended if the primary objective of the study is prediction of tumor growth arrest rather than target modulation.
- Whenever possible, ex vivo correlation analyses with proliferation markers should be performed to validate in vivo PET results.
- Tumors are heterogeneous dynamic tissues which might demonstrate heterogeneous [18F]FLT uptake and therapy response uptake patterns. Care should be taken to use comparable parameters and corresponding tumor areas in correlation analyses to avoid sampling errors.
Supplementary Material
Supplementary tables and figures.
Acknowledgements
We thank E.P. Jansma (Medical Library, VU University Medical Center, Amsterdam, The Netherlands) for assistance with the search strategy and S. Collette (European Organization for Research and Treatment of Cancer Headquarters, Brussels, Belgium) for assistance in writing the manuscript.
Funding
The research leading to these results has received support from the Innovative Medicines Initiative Joint Undertaking (www.imi.europa.eu) under grant agreement number 115151, resources of which are composed of financial contribution from the European Union's Seventh Framework Programme (FP7/2007-2013) and European Federation of Pharmaceutical Industries and Associations (EFPIA) companies' in kind contribution. Please refer to the Supplementary Information for a list of all consortium members.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Wahl RL, Jacene H, Kasamon Y, Lodge MA. From RECIST to PERCIST: Evolving Considerations for PET response criteria in solid tumors. J Nucl Med. 2009May;50(Suppl 1):122S-50S
2. Herholz K, Rudolf J, Heiss WD. FDG transport and phosphorylation in human gliomas measured with dynamic PET. J Neurooncol. 1992Feb;12(2):159-65
3. Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009May22;324(5930):1029-33
4. Shields AF, Grierson JR, Dohmen BM, Machulla HJ, Stayanoff JC, Lawhorn-Crews JM. et al. Imaging proliferation in vivo with [F-18]FLT and positron emission tomography. Nat Med. 1998Nov;4(11):1334-6
5. Paproski RJ, Ng AML, Yao SYM, Graham K, Young JD, Cass CE. The role of human nucleoside transporters in uptake of 3'-deoxy-3'-fluorothymidine. Mol Pharmacol. 2008Nov;74(5):1372-80
6. Aufderklamm S, Todenhöfer T, Gakis G, Kruck S, Hennenlotter J, Stenzl A. et al. Thymidine kinase and cancer monitoring. Cancer Lett. 2012Mar;316(1):6-10
7. Rahman L, Voeller D, Rahman M, Lipkowitz S, Allegra C, Barrett JC. et al. Thymidylate synthase as an oncogene: a novel role for an essential DNA synthesis enzyme. Cancer Cell. 2004Apr;5(4):341-51
8. McKinley ET, Ayers GD, Smith RA, Saleh SA, Zhao P, Washington MK. et al. Limits of [18F]-FLT PET as a biomarker of proliferation in oncology. PLoS One. 2013;8(3):e58938
9. Moroz MA, Kochetkov T, Cai S, Wu J, Shamis M, Nair J. et al. Imaging colon cancer response following treatment with AZD1152: a preclinical analysis of [18F]fluoro-2-deoxyglucose and 3'-deoxy-3'-[18F]fluorothymidine imaging. Clin Cancer Res. 2011Mar;17(5):1099-110
10. Waterton JC, Pylkkanen L. Qualification of imaging biomarkers for oncology drug development. Eur J Cancer. 2012Mar;48(4):409-15
11. Moher D, Liberati A, Tetzlaff J, Altman DG. Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. PLoS Med. 2009Jul21;6(7):e1000097
12. Sanghera B, Wong WL, Sonoda LI, Beynon G, Makris A, Woolf D. et al. FLT PET-CT in evaluation of treatment response. Indian J Nucl Med. India. 2014Apr;29(2):65-73
13. Kawai H, Toyohara J, Kado H, Nakagawa T, Takamatsu S, Furukawa T. et al. Acquisition of resistance to antitumor alkylating agent ACNU: A possible target of positron emission tomography monitoring. Nucl Med Biol. 2006;33(1):29-35
14. Perumal M, Stronach EA, Gabra H, Aboagye EO. Evaluation of 2-deoxy-2-[18F]fluoro-D-glucose- and 3'-deoxy-3'-[18F]fluorothymidine-positron emission tomography as biomarkers of therapy response in platinum-resistant ovarian cancer. Mol imaging Biol. 2012Dec;14(6):753-61
15. Munk Jensen M, Erichsen KD, Bjorkling F, Madsen J, Jensen PB, Sehested M. et al. [18F]FLT PET for Non-Invasive Assessment of Tumor Sensitivity to Chemotherapy: Studies with Experimental Chemotherapy TP202377 in Human Cancer Xenografts in Mice. PLoS One. 2012;7(11):e50618
16. Iommelli F, De Rosa V, Gargiulo S, Panico M, Monti M, Greco A. et al. Monitoring Reversal of MET-Mediated Resistance to EGFR Tyrosine Kinase Inhibitors in Non-Small Cell Lung Cancer Using 3'-Deoxy-3'-[18F]-Fluorothymidine Positron Emission Tomography. Clin Cancer Res. 2014Jul;20(18):4806-15
17. Ullrich RT, Zander T, Neumaier B, Koker M, Shimamura T, Waerzeggers Y. et al. Early detection of erlotinib treatment response in NSCLC by 3'-deoxy-3'-[F]-fluoro-L-thymidine ([F]FLT) positron emission tomography (PET). PLoS One. 2008;3(12):e3908
18. Wei LH, Su H, Hildebrandt IJ, Phelps ME, Czernin J, Weber WA. Changes in tumor metabolism as readout for Mammalian target of rapamycin kinase inhibition by rapamycin in glioblastoma. Clin Cancer Res. 2008Jun;14(11):3416-26
19. Solit DB, Santos E, Pratilas CA, Lobo J, Moroz M, Cai S. et al. 3'-Deoxy-3'-[18F]fluorothymidine positron emission tomography is a sensitive method for imaging the response of BRAF-dependent tumors to MEK inhibition. Cancer Res. 2007;67(23):11463-9
20. Fuereder T, Wanek T, Pflegerl P, Jaeger-Lansky A, Hoeflmayer D, Strommer S. et al. Gastric cancer growth control by BEZ235 in vivo does not correlate with PI3K/mTOR target inhibition but with [18F]FLT uptake. Clin Cancer Res. 2011Aug;17(16):5322-32
21. Graf N, Li Z, Herrmann K, Weh D, Aichler M, Slawska J. et al. Positron emission tomographic monitoring of dual phosphatidylinositol-3-kinase and mTOR inhibition in anaplastic large cell lymphoma. Onco Targets Ther. 2014;7:789-98
22. Li Z, Graf N, Herrmann K, Junger A, Aichler M, Feuchtinger A. et al. FLT-PET is superior to FDG-PET for very early response prediction in NPM-ALK-positive lymphoma treated with targeted therapy. Cancer Res. 2012Oct;72(19):5014-24
23. Zheng Y, Yang Z, Zhang Y, Shi Q, Bao X, Zhang J. et al. The preliminary study of 18F-FLT micro-PET/CT in predicting radiosensitivity of human nasopharyngeal carcinoma xenografts. Ann Nucl Med. 2015Jan;29(1):29-36
24. Manning HC, Merchant NB, Foutch AC, Virostko JM, Wyatt SK, Shah C. et al. Molecular imaging of therapeutic response to epidermal growth factor receptor blockade in colorectal cancer. Clin Cancer Res. 2008Nov;14(22):7413-22
25. Cawthorne C, Burrows N, Gieling RG, Morrow CJ, Forster D, Gregory J. et al. [18F]-FLT positron emission tomography can be used to image the response of sensitive tumors to PI3-kinase inhibition with the novel agent GDC-0941. Mol Cancer Ther. 2013May;12(5):819-28
26. Mudd SR, Holich KD, Voorbach MJ, Cole TB, Reuter DR, Tapang P. et al. Pharmacodynamic evaluation of irinotecan therapy by FDG and FLT PET/CT imaging in a colorectal cancer xenograft model. Mol Imaging Biol. 2012Oct;14(5):617-24
27. Yang M, Gao H, Yan Y, Sun X, Chen K, Quan Q. et al. PET imaging of early response to the tyrosine kinase inhibitor ZD4190. Eur J Nucl Med Mol Imaging. 2011Jul;38(7):1237-47
28. Jensen MM, Erichsen KD, Bjorkling F, Madsen J, Jensen PB, Hojgaard L. et al. Early detection of response to experimental chemotherapeutic Top216 with [18F]FLT and [18F]FDG PET in human ovary cancer xenografts in mice. PLoS One. 2010;5(9):e12965
29. Brepoels L, Stroobants S, Verhoef G, De Groot T, Mortelmans L, De Wolf-Peeters C. (18)F-FDG and (18)F-FLT uptake early after cyclophosphamide and mTOR inhibition in an experimental lymphoma model. J Nucl Med. 2009Jul;50(7):1102-9
30. Cullinane C, Waldeck KL, Binns D, Bogatyreva E, Bradley DP, de Jong R. et al. Preclinical FLT-PET and FDG-PET imaging of tumor response to the multi-targeted Aurora B kinase inhibitor, TAK-901. Nucl Med Biol. 2014Feb;41(2):148-54
31. Zhang CC, Yan Z, Li W, Kuszpit K, Painter CL, Zhang Q. et al. [(18)F]FLT-PET imaging does not always “light up” proliferating tumor cells. Clin Cancer Res. 2012Mar;18(5):1303-12
32. Bao X, Wang M-W, Zhang Y-P, Zhang Y-J. Early monitoring antiangiogenesis treatment response of Sunitinib in U87MG Tumor Xenograft by (18)F-FLT MicroPET/CT imaging. Biomed Res Int. 2014;2014:218578
33. Yang M, Gao H, Sun X, Yan Y, Quan Q, Zhang W. et al. Multiplexed PET probes for imaging breast cancer early response to VEGF(1)(2)(1)/rGel treatment. Mol Pharm. 2011Apr;8(2):621-8
34. Yue J-B, Yang J, Liu J, Lee J, Cabrera AR, Sun X-D. et al. Histopathologic validation of 3'-deoxy-3'-(18)F-fluorothymidine PET for detecting tumor repopulation during fractionated radiotherapy of human FaDu squamous cell carcinoma in nude mice(18)F-FLT PET repopulation. Radiother Oncol. 2014Jun;111(3):475-81
35. Murayama C, Harada N, Kakiuchi T, Fukumoto D, Kamijo A, Kawaguchi AT. et al. Evaluation of D-18F-FMT, 18F-FDG, L- 11C-MET, and 18F-FLT for monitoring the response of tumors to radiotherapy in mice. J Nucl Med. 2009;50(2):290-5
36. Jensen MM, Erichsen KD, Bjorkling F, Madsen J, Jensen PB, Sehested M. et al. Imaging of treatment response to the combination of carboplatin and paclitaxel in human ovarian cancer xenograft tumors in mice using FDG and FLT PET. PLoS One. 2013;8(12):e85126
37. Armeanu-Ebinger S, Griessinger CM, Herrmann D, Fuchs J, Kneilling M, Pichler BJ. et al. PET/MR imaging and optical imaging of metastatic rhabdomyosarcoma in mice. J Nucl Med. 2014Sep;55(9):1545-51
38. Murakami M, Zhao S, Zhao Y, Yu W, Fatema CN, Nishijima K-I. et al. Increased intratumoral fluorothymidine uptake levels following multikinase inhibitor sorafenib treatment in a human renal cell carcinoma xenograft model. Oncol Lett. 2013Sep;6(3):667-72
39. Lee HJ, Oh SJ, Lee EJ, Chung JH, Kim Y, Ryu J-S. et al. Positron emission tomography imaging of human colon cancer xenografts in mice with [18F]fluorothymidine after TAS-102 treatment. Cancer Chemother Pharmacol. 2015May;75(5):1005-13
40. Buck AK, Kratochwil C, Glatting G, Juweid M, Bommer M, Tepsic D. et al. Early assessment of therapy response in malignant lymphoma with the thymidine analogue [18F]FLT. Eur J Nucl Med Mol Imaging. 2007Nov;34(11):1775-82
41. Kukuk D, Reischl G, Raguin O, Wiehr S, Judenhofer MS, Calaminus C. et al. Assessment of PET tracer uptake in hormone-independent and hormone-dependent xenograft prostate cancer mouse models. J Nucl Med. 2011Oct;52(10):1654-63
42. Shah C, Miller TW, Wyatt SK, McKinley ET, Olivares MG, Sanchez V. et al. Imaging biomarkers predict response to Anti-HER2 (ErbB2) therapy in preclinical models of breast cancer. Clin Cancer Res. 2009Jul;15(14):4712-21
43. Honndorf VS, Schmidt H, Wehrl HF, Wiehr S, Ehrlichmann W, Quintanilla-Martinez L. et al. Quantitative correlation at the molecular level of tumor response to docetaxel by multimodal diffusion-weighted magnetic resonance imaging and [18F]FDG/[18F]FLT positron emission tomography. Mol Imaging. 2014 Jan;13
44. Cao Q, Li Z-B, Chen K, Wu Z, He L, Neamati N. et al. Evaluation of biodistribution and anti-tumor effect of a dimeric RGD peptide-paclitaxel conjugate in mice with breast cancer. Eur J Nucl Med Mol Imaging. 2008;35(8):1489-98
45. Honer M, Ebenhan T, Allegrini PR, Ametamey SM, Becquet M, Cannet C. et al. Anti-angiogenic/vascular effects of the mTOR inhibitor everolimus are not detectable by FDG/FLT-PET. Transl Oncol. 2010;3(4):264-75
46. Stelter L, Fuchs S, Jungbluth AA, Ritter G, Longo VA, Zanzonico P. et al. Evaluation of arginine deiminase treatment in melanoma xenografts using 18F-FLT PET. Mol Imaging Biol. 2013;15(6):768-75
47. Honndorf VS, Schmidt H, Wiehr S, Wehrl HF, Quintanilla-Martinez L, Stahlschmidt A. et al. The Synergistic Effect of Selumetinib/Docetaxel Combination Therapy Monitored by [(18) F]FDG/[ (18) F]FLT PET and Diffusion-Weighted Magnetic Resonance Imaging in a Colorectal Tumor Xenograft Model. Mol imaging Biol. 2016Apr;18(2):249-57
48. Katz SI, Zhou L, Ferrara TA, Wang W, Mayes PA, Smith CD. et al. FLT-PET may not be a reliable indicator of therapeutic response in p53-null malignancy. Int J Oncol. 2011Jul;39(1):91-100
49. Keen HG, Ricketts S-A, Maynard J, Logie A, Odedra R, Shannon AM. et al. Examining changes in [18 F]FDG and [18 F]FLT uptake in U87-MG glioma xenografts as early response biomarkers to treatment with the dual mTOR1/2 inhibitor AZD8055. Mol imaging Biol. 2014Jun;16(3):421-30
50. Viel T, Schelhaas S, Wagner S, Wachsmuth L, Schwegmann K, Kuhlmann M. et al. Early assessment of the efficacy of temozolomide chemotherapy in experimental glioblastoma using [18F]FLT-PET imaging. PLoS One. 2013;8(7):e67911
51. Dittmann H, Dohmen BM, Kehlbach R, Bartusek G, Pritzkow M, Sarbia M. et al. Early changes in [18F]FLT uptake after chemotherapy: an experimental study. Eur J Nucl Med Mol Imaging. 2002Nov;29(11):1462-9
52. Fushiki H, Miyoshi S, Noda A, Murakami Y, Sasaki H, Jitsuoka M. et al. Pre-clinical validation of orthotopically-implanted pulmonary tumor by imaging with18F-fluorothymidine-positron emission tomography/computed tomography. Anticancer Res. 2013Nov;33(11):4741-50
53. Kilkenny C, Browne WJ, Cuthill IC, Emerson M, Altman DG. Improving bioscience research reporting: the ARRIVE guidelines for reporting animal research. PLoS Biol. 2010;8(6):e1000412
54. Jensen MM, Kjaer A. Monitoring of anti-cancer treatment with (18)F-FDG and (18)F-FLT PET: a comprehensive review of pre-clinical studies. Am J Nucl Med Mol Imaging. 2015;5(5):431-56
55. Lesko LJ, Atkinson AJ. Use of biomarkers and surrogate endpoints in drug development and regulatory decision making: criteria, validation, strategies. Annu Rev Pharmacol Toxicol. 2001;41:347-66
56. O'Connor JPB, Aboagye EO, Adams JE, Aerts HJWL, Barrington, Sally F, Beer AJ. et al. Imaging Biomarker Roadmap for Cancer Studies. Nat Rev Clin Oncol. 2016 Oct 11
57. Shields AF. PET imaging of tumor growth: not as easy as it looks. Clin Cancer Res. United States. 2012Mar;18(5):1189-91
58. Chalkidou A, Landau DB, Odell EW, Cornelius VR, O'Doherty MJ, Marsden PK. Correlation between Ki-67 immunohistochemistry and 18F-fluorothymidine uptake in patients with cancer: A systematic review and meta-analysis. Eur J Cancer. 2012Dec;48(18):3499-513
59. Bollineni VR, Kramer GM, Jansma EP, Liu Y, Oyen WJG. A systematic review on [18F]FLT-PET uptake as a measure of treatment response in cancer patients. Eur J Cancer. 2016;55:81-97
60. Been LB, Suurmeijer AJH, Cobben DCP, Jager PL, Hoekstra HJ, Elsinga PH. [18F]FLT-PET in oncology: current status and opportunities. Eur J Nucl Med Mol Imaging. 2004Dec;31(12):1659-72
61. Soloviev D, Lewis D, Honess D, Aboagye E. [(18)F]FLT: an imaging biomarker of tumour proliferation for assessment of tumour response to treatment. Eur J Cancer. England. 2012Mar;48(4):416-24
62. Salskov A, Tammisetti VS, Grierson J, Vesselle H. FLT: measuring tumor cell proliferation in vivo with positron emission tomography and 3'-deoxy-3'-[18F]fluorothymidine. Semin Nucl Med. United States. 2007Nov;37(6):429-39
63. Boellaard R, Delgado-Bolton R, Oyen WJG, Giammarile F, Tatsch K, Eschner W. et al. FDG PET/CT: EANM procedure guidelines for tumour imaging: version 2.0. Eur J Nucl Med Mol Imaging. 2015Feb;42(2):328-54
64. Peck M, Pollack HA, Friesen A, Muzi M, Shoner SC, Shankland EG. et al. Applications of PET imaging with the proliferation marker [(18)F]-FLT. Q J Nucl Med Mol Imaging. 2015Mar4;59(1):95-104
65. Gao H, Korn JM, Ferretti S, Monahan JE, Wang Y, Singh M. et al. High-throughput screening using patient-derived tumor xenografts to predict clinical trial drug response. Nat Med. Nature Research. 2015Oct19;21(11):1318-25
66. Tuveson D, Hanahan D. Translational medicine: Cancer lessons from mice to humans. Nature. Nature Research. 2011Mar17;471(7338):316-7
Author contact
![]() Corresponding authors: Andreas H. Jacobs, phone: +49 251 8349300, fax: +49 251 8349313, email: ahjacobsde; Dmitry Soloviev: phone: +44 1223 769704, fax: +44 1223 766002, email: dmitry.solovievcam.ac.uk
Corresponding authors: Andreas H. Jacobs, phone: +49 251 8349300, fax: +49 251 8349313, email: ahjacobsde; Dmitry Soloviev: phone: +44 1223 769704, fax: +44 1223 766002, email: dmitry.solovievcam.ac.uk
Citation styles
APA
Schelhaas, S., Heinzmann, K., Bollineni, V.R., Kramer, G.M., Liu, Y., Waterton, J.C., Aboagye, E.O., Shields, A.F., Soloviev, D., Jacobs, A.H. (2017). Preclinical Applications of 3'-Deoxy-3'-[18F]Fluorothymidine in Oncology - A Systematic Review. Theranostics, 7(1), 40-50. https://doi.org/10.7150/thno.16676.
ACS
Schelhaas, S.; Heinzmann, K.; Bollineni, V.R.; Kramer, G.M.; Liu, Y.; Waterton, J.C.; Aboagye, E.O.; Shields, A.F.; Soloviev, D.; Jacobs, A.H. Preclinical Applications of 3'-Deoxy-3'-[18F]Fluorothymidine in Oncology - A Systematic Review. Theranostics 2017, 7 (1), 40-50. DOI: 10.7150/thno.16676.
NLM
Schelhaas S, Heinzmann K, Bollineni VR, Kramer GM, Liu Y, Waterton JC, Aboagye EO, Shields AF, Soloviev D, Jacobs AH. Preclinical Applications of 3'-Deoxy-3'-[18F]Fluorothymidine in Oncology - A Systematic Review. Theranostics 2017; 7(1):40-50. doi:10.7150/thno.16676. https://www.thno.org/v07p0040.htm
CSE
Schelhaas S, Heinzmann K, Bollineni VR, Kramer GM, Liu Y, Waterton JC, Aboagye EO, Shields AF, Soloviev D, Jacobs AH. 2017. Preclinical Applications of 3'-Deoxy-3'-[18F]Fluorothymidine in Oncology - A Systematic Review. Theranostics. 7(1):40-50.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) License. See http://ivyspring.com/terms for full terms and conditions.