Impact Factor
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2017; 7(3):717-732. doi:10.7150/thno.17154 This issue Cite
Research Paper
HER Family Receptors are Important Theranostic Biomarkers for Cervical Cancer: Blocking Glucose Metabolism Enhances the Therapeutic Effect of HER Inhibitors
Olga Martinho1,2,3 ![]() , Renato Silva-Oliveira3, Fernanda P. Cury3, Ana Martins Barbosa1,2, Sara Granja1,2, Adriane Feijó Evangelista3, Fábio Marques4, Vera Miranda-Gonçalves1,2, Diana Cardoso-Carneiro1,2, Flávia E. de Paula3, Maicon Zanon3, Cristovam Scapulatempo-Neto3, Marise A.R. Moreira4, Fátima Baltazar1,2, Adhemar Longatto-Filho1,2,3,5, Rui Manuel Reis1,2,3
, Renato Silva-Oliveira3, Fernanda P. Cury3, Ana Martins Barbosa1,2, Sara Granja1,2, Adriane Feijó Evangelista3, Fábio Marques4, Vera Miranda-Gonçalves1,2, Diana Cardoso-Carneiro1,2, Flávia E. de Paula3, Maicon Zanon3, Cristovam Scapulatempo-Neto3, Marise A.R. Moreira4, Fátima Baltazar1,2, Adhemar Longatto-Filho1,2,3,5, Rui Manuel Reis1,2,3 ![]()
1. Life and Health Sciences Research Institute (ICVS), School of Medicine, University of Minho, Braga, Portugal;
2. ICVS/3B's - PT Government Associate Laboratory, Braga/Guimarães, Portugal;
3. Molecular Oncology Research Center (CPOM), Barretos Cancer Hospital, Barretos, São Paulo, Brazil;
4. Department of Pathology of the School of Medicine of the Federal University of Goiás, Brazil;
5. Laboratory of Medical Investigation (LIM) 14, Faculty of Medicine, São Paulo State University, Brazil.
Received 2016-8-10; Accepted 2016-11-21; Published 2017-1-15
Abstract

Persistent HPV infection alone is not sufficient for cervical cancer development, which requires additional molecular alterations for tumor progression and metastasis ultimately leading to a lethal disease. In this study, we performed a comprehensive analysis of HER family receptor alterations in cervical adenocarcinoma. We detected overexpression of HER protein, mainly HER2, which was an independent prognostic marker for these patients. By using in vitro and in vivo approaches, we provided evidence that HER inhibitors, allitinib and lapatinib, were effective in reducing cervical cancer aggressiveness. Furthermore, combination of these drugs with glucose uptake blockers could overcome the putative HIF1-α-mediated resistance to HER-targeted therapies. Thus, we propose that the use of HER inhibitors in association with glycolysis blockers can be a potentially effective treatment option for HER-positive cervical cancer patients.
Keywords: ErbB, EGFR, HER2, cervical cancer, prognosis, targeted therapy.
Introduction
Cervical cancer is a major malignant disease among women worldwide and is estimated to account for 8% of the total cancer related mortality. Around 500,000 new cases of cervical cancer and 250,000 cervical cancer-related deaths occur worldwide every year [1]. Screenings with cytology and high-risk human papillomavirus (HPV) DNA testing have reduced the incidence and mortality of this disease. However, women who do not have access to health care and those living in underserved areas remain at high risk of death from this disease [2]. Although early-stage disease can be cured by radical surgery and locally advanced disease by chemo-radiotherapy, women with metastatic and non-operable recurrent disease have few treatment options [2]. Platinum-based chemotherapy in this setting is palliative and is associated with median overall survival of 8 to 12 months [2-4]. Thus, a deeper molecular understanding and effective therapeutic strategies are desperately needed for the treatment of recurrent/metastatic cervical cancer.
Receptor tyrosine kinases (RTKs) are membrane-spanning proteins that possess a ligand-controlled intracellular kinase activity [5]. Deregulation of RTKs due to overexpression, mutation, amplification or autocrine stimulation has been causally linked to cancer development and progression [5]. We had been studying the molecular pathology of cervical cancer, with emphasis on RTK alterations and reported important alterations in KIT, PDGFRA, VEGFR2 and EGFR family of receptors [3, 6-8]. RTKs and intracellular signaling pathways constitute promising targets for cervical cancer therapy, since several oncogenic alterations were already identified that can be potentially predictive of response to anti-RTKs and anti-signaling protein drugs [9, 10].
Inhibitors of HER family receptors, or ErBb, have been among the most successful examples of targeted cancer therapies to date, including antibodies (e.g., trastuzumab and cetuximab) and small-molecule tyrosine kinase inhibitors (e.g., erlotinib, gefitinib, lapatinib and allitinib) [11-13]. A large number of novel HER2-targeted agents are also in clinical trials [14].
HER receptors belong to family I of RTKs which includes EGFR, HER2, HER3 and HER4. These RTKs recognize 13 different but structurally related growth factors like EGF. However, for HER2, no soluble ligand has yet been identified [15]. Amplification, overexpression, and co-expression of the HER family receptors have been implicated in the genesis or progression of human mammary, ovarian, gastric and brain cancers [14, 15]. Anti-HER2 therapy is currently approved for breast, gastric, and gastroesophageal cancers that overexpress HER2 protein or carry amplified HER2 gene. HER2 aberrations have also been reported in other diverse malignancies. Indeed, about 1-37% of tumors, such as bladder, colon, endometrium, germ cell, glioblastoma, head and neck, liver, lung, ovarian, pancreas, and salivary duct, harbor HER2 aberrations [14].
In cervical cancer, HER receptor expression has been reported to be upregulated, but its prognostic and therapeutic value remains unclear [11, 16-22]. Some clinical trials are using RTK inhibitors in the treatment of cervical cancer patients, but the expected benefits have not been demonstrated, mainly because patients were not pre-selected for the molecular alterations in the targets of interest [23]. Hence, besides bevacizumab, no other targeted therapies, including those designed to target RTKs, approval for cervical cancer treatment [3, 24, 25].
Thus, in this work we aimed to identify RTK targets, mainly HER receptors, for personalized therapeutic options for cervical cancer and to elucidate the putative molecular mechanisms underlying the response to the tested inhibitors.
Materials and Methods
Tissue samples
In the present study 229 cervical cancer tissues, which included 194 adenocarcinomas and 35 adenosquamous carcinomas, were analyzed. The paraffin samples included in tissue microarrays (TMA) were retrieved from the files from School of Medicine of the Federal University of Goiás (Goiânia, Goias State, Brazil). All histopathological diagnoses were reviewed by the authors and categorized according to the WHO classification [26]. All patients with cervical cancer were of Brazilian origin, with a mean age of 47 years (range 21-84 years). Follow-up data was available for 137 patients, and collected through direct interview with patients or their relatives and by review of in-hospital patient files. The median follow-up time was 96.19±11.23 months (range, 1-780 months).
Cell lines and Cell culture
Four cervical cancer cell lines, HeLa, SiHa, C-33A and Caski, were used in the present study. HeLa cell line was a gift by Drª Elsa Logarinho (IBMC, Portugal), SiHa, C-33A and Caski cell lines were kindly provided by Drª Luisa Villa (INCT-HPV, Brazil) [27]. All cell lines were grown and maintained at 37ºC and 5% CO2 in Dulbecco's Modified Eagle's Medium (DMEM 1x, High Glucose; Gibco, Invitrogen) supplemented with 10% Fetal Bovine Serum (FBS; Gibco, Invitrogen) and 1% penicillin/streptomycin solution (Gibco, Invitrogen).
Downregulation of HIF1-α was performed with Silencer Select Validated siRNAs from Ambion (siRNA ID #42840) as well as with a non-targeting control siRNA (Silencer Select Negative Control No.1 siRNA, 4390843), using Lipofectamine RNAiMAX (13778-075, Invitrogen) as a transfection reagent, according to the manufacturer's instructions, and as described [28].
Authentication of cell lines was performed in our lab by short tandem repeat (STR) DNA typing according to the International Reference Standard for Authentication of Human Cell Lines using a panel of 8 (D5S818, D13S317, D7S820, D16S539, vWA, TH01, TPOX and CSF1P0) STR loci plus gender determination (AMEL) [29]. The genotyping confirmed the identity of all cell lines.
Drugs
Cediranib (CD), sunitinib (SU), imatinib (IM), erlotinib (ER), lapatinib (LA), allitinib (AST1306 - AST), MK2206 (MK) and selumetinib (SE) were obtained from Selleck Chemicals (Houston, USA) and 2-Deoxy-D-glucose (2-DG) was purchased from Sigma-Aldrich (Sintra, Portugal). All the drugs were prepared as stock solutions in dimethyl sulfoxide (DMSO) and stored at -20ºC, as described [30]. The 2-DG was always freshly prepared and diluted in water to a 10 mM stock solution. In all experimental conditions the drugs were diluted in 0.5% FBS culture medium. The vehicle control was also used in all experiments.
Real-time quantification of HER2/neu gene amplification
For HER2 gene amplification detection, DNA from paraffin tumors was isolated as previously described [6]. The copy numbers of the gene were detected using the LightMix® Kit HER2/neu (Cat. number 40-0333-16) from Roche Molecular Biochemicals, following the manufacture recommendations. The calculation of the relative amounts of HER2 DNA compared to the reference gene DNA was performed, and the final results were expressed as a ratio of HER2:reference gene copies in the sample, normalized with the ratio of HER2:reference gene copies in the calibrator DNA. The ratio HER2: reference gene copies in the calibrator DNA was set to one. A ratio of < 2.0 was considered to be negative for HER2 over-amplification, a ratio of ≥ 2.0 was assumed to be positive for HER2 amplification.
Immunohistochemistry analysis (IHC)
Histological slides with 4 μm-thick tissue sections were subjected to immunohistochemistry analysis according to the streptavidin-biotin peroxidase complex system (UltraVision Large Volume Detection System Anti-Polyvalent, HRP; LabVision Corporation), as previously described [27]. Briefly, deparaffinized and rehydrated slides were submitted heat-induced antigen retrieval for 20min at 98ºC with 10mM citrate buffer (pH 6.0) for GLUT1, HIF-1α and Ki-67, or in microwave at 600W in 1 mM EDTA buffer (pH 7.8) for HER4, and treated with pronase (1mM) for 30 min at 37°C for HER2, HER3 and EGFR. The sources of and their dilutions were as follows: EGFR (Zymed, dilution 1:100); HER2 (Cell Signaling, dilution 1:100); HER3 (Cell Signaling, dilution 1:100); HER4 (Cell Signaling, dilution of 1:50); GLUT1 (Abcam, dilution 1:500); HIF-1α (BD Biosciences, dilution 1:100); and Ki-67 (Gennova, dilution 1:100). All primary antibodies were incubated ON at 4ºC. The secondary biotinylated goat anti-polyvalent antibody was applied for 10 minutes followed by incubation with the streptavidin-peroxidase complex. The immune reaction was visualized by 3, 3'-Diamonobenzidine (DAB) as a chromogen. All sections were counterstained with Gill-2 haematoxylin. For negative controls, primary antibodies were omitted and replaced by a universal negative control antibody (CEA, rabbit anti-human, DAKO Corporation).
Breast carcinoma tissues were used as positive controls. Stained slides were evaluated and then photographed using a bright field microscope Olympus BX61.
The immunostaining of the tissues was semi-quantitatively evaluated by an experienced professional considering extension and intensity of staining for EGFR, HER2, HER3, HER4 and GLUT1, as previously described [31]. The score for immunoreactive extension was as follows: score 0, 0% of immunoreactive cells; score 1, <5% of immuno-reactive cells; score 2, 5%-50% immunoreactive cells; and score 3, >50% of immunoreactive cells. For intensity, the score was as follows: 0, negative; 1, weak; 2, intermediate; and 3, strong. The final score was defined as the sum of these 2 semiquantitative scores, and for statistical analysis, final score >3 was considered to be positive. Cellular localization of staining (cytoplasm/membrane/nucleus) of the studied markers was also evaluated. For the nuclear markers Ki-67 and HIF-1α, the sections were scored semi-quantitatively as follows [6, 27]: (-), 0% of immunoreactive cells; (+), <5% of immunoreactive cells; (++), 5-50% of immunoreactive cells; and (+++), >50% of immunoreactive cells. Samples with scores (-) and (+) were considered negative, and those with scores (++) and (+ + +) were considered positive.
Immunofluorescence
The cells were plated until ~60% of confluency on glass slides and allowed to adhere overnight. Afterwards, the cells were fixed in methanol 100% at -20ºC and blocked in a solution of PBS containing 10% FBS for 30 min. The primary antibodies used were the same as for the IHC, and incubated overnight at 4ºC. The FITC Alexa Fluor- conjugated secondary antibodies (Molecular Probes, Invitrogen) were used at a dilution of 1:500. For negative controls, primary antibodies were omitted and replaced by a universal negative control antibody (CEA, rabbit anti-human, DAKO Corporation).
The nuclei were stained with DAPI and slides were covered using VECTSHIELD mounting solution. The slides were examined and images were obtained by fluorescence microscopy (BX16; Olympus).
Western blot and human RTK arrays
For in vitro assays, the cells were plate in a 6 well plate at a density of 8x105 cells per well and allowed to adhere for at least 24 hours, then serum starved for 2 hours and incubated with the drugs for 2 hours, as previous described [30, 32]. When necessary, the cells were also stimulated with 10 ng/ml of EGF for 15 minutes before the end of the time point. To assess apoptosis, cells were incubated with only one concentration of each drug and incubated for 24 hours. At the indicated time points, the cells were washed and scraped in cold PBS and lysed in buffer, as previously described [30]. For the in vivo assays, the treated tumors at the desired concentration of the drugs for four days, were divided in half washed in PBS and lysed in the same buffer as for cell lines. Western blotting was done using standard 10% SDS-PAGE, loading 40 µg of protein per lane. All antibodies were used as recommended by the manufacturers, and were the same as described for IHC and IF. All other antibodies (apoptosis and intracellular pathways assessment) were used as previously described [30]. Anti-GAPDH (Santa Cruz Biotechnology, dilution 1:2000) was used as a loading control.
For the human RTK arrays, 500 μg of fresh protein lysates, before and after treatment with the drugs, were incubated overnight at 4ºC with nitrocellulose membranes dotted with duplicated spots for 42 anti-RTK and control antibodies. Bound phospho-RTKs were incubated with a pan anti-phosphotyrosine-HRP antibody for 2 h at room temperature [30].
Blots detection was done by chemiluminescence (Thermo Scientific Pierce ECL Western Blotting) using the ChemiDoc™ XRS+ System (Bio-Rad).
Cell viability assay
To determine the concentration at which 50% of the cell growth is inhibited by drug treatment (IC50 concentration), cells were plated in 96-well plates at a density of 4x103 cells per well and allowed to adhere overnight in DMEM containing 10% FBS. Subsequently, the cells were treated with increasing concentrations of the drugs or with DMSO alone, both diluted in 0.5% FBS culture medium to a final concentration of 1% DMSO. When necessary the cells were incubated simultaneously with 2.5 µM of selumetinib or MK2206, and 1.5 mM 2-DG. After 72 hours, cell viability was quantified using the Cell Titer96 Aqueous cell proliferation assay (Promega). The results were expressed as the mean percentage ± SD of viable cells relative to the DMSO alone (considered as 100% viability). The IC50 concentration was calculated by nonlinear regression analysis using GraphPad Prism software.
To assess the effect of a fixed concentration of the drug in cellular viability over time, the cells were plated onto 96-well plates at a density of 2x103 cells per well and allowed to adhere overnight in complete DMEM medium. Next, the viable cells were quantified using the Cell Titer96 Aqueous cell proliferation assay (Promega), and used for time point 0. Then, the cells were incubated with fixed concentrations of the drugs or with DMSO alone, both diluted in 0.5% FBS culture medium to a final concentration of 1% DMSO, over 24, 48 and 72 hours. At the end of each time point, cell viability was again assessed using the Cell Titer96 Aqueous cell proliferation assay. The results were calibrated to the starting viability (time 0h, considered as 100% viability) and expressed as the mean ± SD. Both assays were performed in triplicate at least three times [30].
Wound healing migration assay
The cells were seeded in 6-well plates and cultured to at least 95% confluency, as described [27, 30]. Monolayer cells were washed with PBS and scraped with a plastic 200 µL pipette tip and then incubated with fresh DMEM medium without serum. The ''wounded'' areas were photographed by phase contrast microscopy at 24, 48 and 72 hours' time points. The relative migration distance was calculated by the following formula: percentage of wound closure (%) = 100 (A-B)/A, where A was the width of cell wounds before incubation, and B was the width of cell wounds after incubation. Results were expressed as the mean ± SD. The assay was done in triplicate at least three times.
Clonogenicity assay
The cells (1x103) were seeded in 12-well plates and incubated for 24h to adhere. Medium was replaced with DMEM-0.5 with or without drugs and the cells incubated for 10-15 days, with medium renewal after every 3 days. The colonies were stained with 5% Giemsa for 45min and manually counted. Results were expressed as the mean colonies ± SD. The assay was done in triplicate at least three times.
Extracellular lactate measurements
Cells were plated in 48-well plates at a density of 3x104 cells per well and allowed to adhere overnight. Lactate content was analyzed in cell culture medium after 24h of drugs treatment, using a commercial kit (Spinreact), as described [33]. The total protein (expressed as total biomass) was assessed using the sulforhodamine B assay (SRB, Sigma-Aldrich). Results were expressed as mean mg of lactate/ total biomass ± SD and adjusted for the control (considered as 1).
In vivo Chick chorioallantoic membrane (CAM) assay
To assess in vivo tumor proliferation we used the CAM assay as previously described [27, 30], with some modifications. On day 9 of development, the cells (2x106 cells) were suspended in matrigel and incubated on the CAM to allow the formation of a 3D tumor, for the next 4 days. On day 13 of development the tumors were treated with the desired drugs for 4 more days. On day 17 of development, the tumor formed was photographed in ovo and ex ovo using a stereomicroscope (Olympus S2x16), and the perimeter of the tumor was measured using Cell B software (Olympus). The results were expressed as the mean perimeter ± SD. The vasculature area was calculated using AngioTool software (National Cancer Institute), and the results expressed as vessels area mean ± SD (pixel). The tumors were then lysed or embedded in paraffin and processed for Western blot and histological analysis, respectively, as described above.
In Silico analysis
The cBioPortal for Cancer Genomics (http://www.cbioportal.org) is a repository of cancer genomics datasets [34, 35] that was used to investigate the molecular profiles of HER2, HER3, HER4, and EGFR in cervical cancer. In the present study it was used for mutations, putative copy number alterations (CNA), transcriptomic, and survival data from the provisional TCGA cervical squamous cell carcinoma and endocervical adenocarcinoma (CESC) project with 307 samples currently available. The repository had samples containing both sequencing and CNA data, corresponding to 191 patients by August 2015. According to the TCGA guidelines (http://cancergenome.nih.gov/publications/publicationguidelines). This dataset has no limitations or restrictions.
The frequency of alterations of the genes across the set of samples was summarized in the format of OncoPrint plots. The mutation mapper tool of cBioPortal was used to plot the distribution of mutations. Significant CNA was obtained by GISTIC method. The dysregulation of gene expression was determined by a Z-score threshold of ± 2.0 when compared with normal samples. Finally, the genes analyzed alone and in combinations were considered for associations with overall survival using the Kaplan-Meier method with log rank testing with p < 0.05.
Statistical analysis
Correlations between proteins expression and clinical data from patients were performed using the chi-square test (χ2-test). Cumulative survival probabilities were calculated using the Kaplan-Meier method. Differences between survival rates were tested with the log-rank test. Multivariate analysis was done using the Cox proportional hazards model. The statistical analysis was performed using SPSS software for Windows, version 17.0. For in vitro and in vivo assays, single comparisons between the different conditions studied were done using Student's t test, and differences between groups were tested using two-way analysis of variance (ANOVA). Statistical analysis was done using Graph Pad Prism version 5. The level of significance in all the statistical analysis was set at p<0.05.
Results
Overexpression of HER receptors in cervical cancer tissues as an independent prognostic marker
Immunohistochemistry analysis was carried out to determine the frequency of HER protein receptor expression and distribution in a series of cervical cancer. As shown in Figure 1A, EGFR positivity was found in the cytoplasm, membrane, and stroma of tumor cells, in 18.8% (33/176) of all tumors. A positive HER2 expression was observed in the membrane and cytoplasm of tumor cells in 53.5% (91/170) of the cases, whereas HER3-specific staining was present in the membrane, cytoplasm, stroma and in a few nuclei of the tumor cells in 74.7% (124/166) of patients. Finally, only 23.4% (37/158) of cases exhibited lower positivity (all with the score<4) for HER4 in the cytoplasm, nuclei, and stroma of the tumor cells. With the exception of HER2 and HER3 that were co-expressed in 61.7% (71/115) of the cases (p=0.007), no significant correlations were found between the co-expression of various receptors.
Molecular characterization of HER receptors in human cervical cancer specimens. (A) Immunohistochemistry analysis of HER family receptors in representative cervical cancer tissues. All pictures were taken at 400x magnification. (B) Overall survival according to HER2 expression in cervical cancer. Cases with HER2 overexpression (p= 0.014) have a poor prognosis. (C) OncoPrint plot of TCGA CESC data generated by cBioPortal showing CNA, mutation and mRNA frequency of alterations of HER2, HER3, HER4 and EGFR in a total of 190 patients analyzed (only 66 cases are represented). (D) Boxplot of TCGA CESC data from cBioPortal showing protein upregulation relative to amplification of ERBB2. (E) Survival analysis of ERBB2 mRNA of TCGA CESC patients with Kaplan Meier plot generated using cBioPortal (p<0.05).
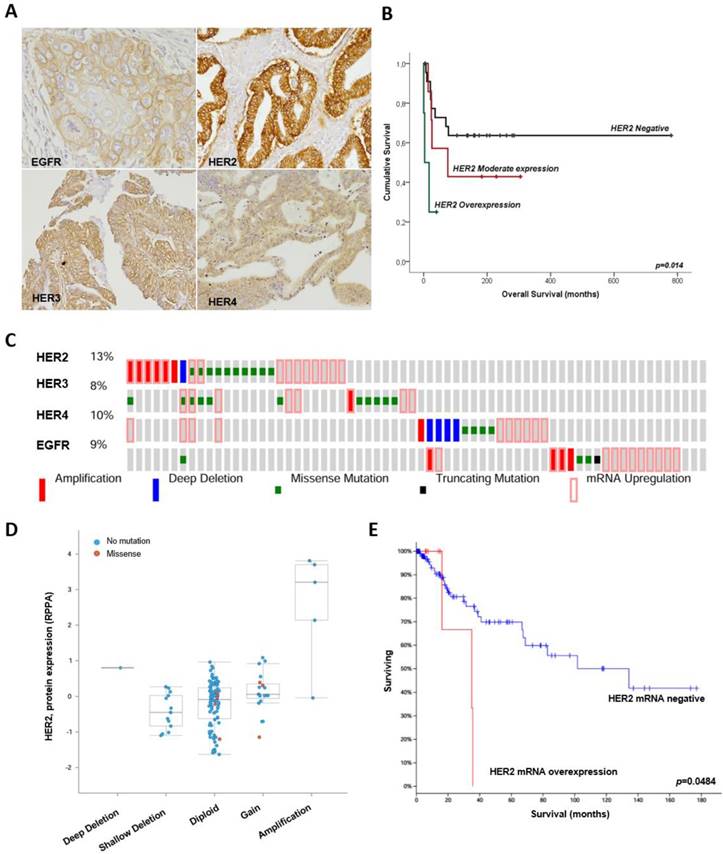
The clinical impact of the expression of HER family of receptors in cervical cancer patients was evaluated in relation with the available clinical features (Supplementary Table 1). EGFR was differentially expressed (p<0.05) between the two histological types in study, being associated with the adenosquamous subtype (44.4%). HER3, was significantly more expressed in older patients (>47 years old), and it was also associated with the presence of metastasis (p<0.05) (Supplementary Table 1).
Using the Kaplan-Meier analysis (Table 1), we detected a tendency (p=0.07) for an association between HER2 positivity and poor prognosis by univariate analysis. Importantly, stratifying the levels of HER2 expression by negative (score 0 and 2), moderate positivity (score 3 and 4) and overexpression (score 5 and 6), we observed a significant association (p=0.014) between the overexpression of HER2 and poor survival (Figure 1B). This association was also observed by multivariate analysis where metastasis and HER2 overexpression constituted independent prognostic markers for cervical cancer patients (Table 1).
Univariate and multivariate analyses between the clinicopathological features and overall survival of the patients.
| Parameter | N | Univariate Analysis | Multivariate Analysis | |||
|---|---|---|---|---|---|---|
| (months ± SD) | p | HR (95% CI) | p | |||
| Metastasis | ||||||
| Absent | 24 | 586.2 ± 68.2 | 1 | |||
| Present | 4 | 17.5 ± 3.8 | 0.001 | 0.097 (0.02-0.42) | 0.002 | |
| HER2 expression | ||||||
| Negative | 24 | 508.5 ± 76.7 | 1 | |||
| Positive | 11 | 120.3 ± 41.8 | 0.071 | 3.4 (0.9-12.2) | 0.059 | |
HER2 overexpression is strongly associated with HER2 gene amplification in breast cancer patients [36]. We used quantitative PCR in a subset of cases (n=17) to screen for the presence of HER2 amplification in cervical cancer. Eight of the 17 samples evaluated had HER2 overexpression by IHC and HER2 amplification was detected in only 1 case (12.5% - 1/8); all other cases showed absence of HER2 gene amplification.
In silico validation of the role of HER receptors in cervical cancer
We extended our analysis by corroborating with the microarray profiling datasets of cervical cancer tissues available from the cBioPortal for Cancer Genomics and from the TCGA database. As is seen in Figure 1C, HER receptors are altered with respect to mRNA expression, gene copy numbers and mutations with the most frequent alteration (13% - 25/190) detected in HER2. EGFR and HER2 presented mutations in more than one tyrosine kinase phosphorylation site (Supplementary figure S1), highlighting their potential use as molecular targets for tyrosine kinase inhibitors. Furthermore, HER2 gene amplification present in 3% of the cases (6/190) appeared to be exclusively present in the cases with HER2 mRNA upregulation and protein overexpression (Figure 1C and D).
Importantly, we were able to confirm in a different cohort of patients that HER2 overexpression, even at mRNA level, was significantly associated with poor survival in cervical cancer patients (Figure 1E).
HER receptors are targetable proteins on cervical cancer cell lines
To determine whether RTKs, especially the HER family of receptors, could be targetable proteins for cervical cancer therapy, we initially characterized the in vitro models for the expression and activation of these proteins. According to the Cancer Cell Lines Project Database (COSMIC - http://cancer.sanger.ac.uk/cell_lines), there are no genetic mutations of RTKs on HeLa, SiHa, Caski and C-33A cervical cancer cell lines. We used a phospho-RTK array to identify the basal levels of RTK activation and interestingly found that only the HER family of receptors was activated (Figure 2A). These results were also validated by Western blotting showing the basal activation levels of EGFR on SiHa, HeLa and Caski cell lines, and of HER4 and HER2 on C-33A and Caski cell lines, respectively (Figure 2B). Upon EGF stimulation, phosphorylated EGFR was observed in all cell lines, and phosphorylated HER2 was detectable in SiHa, HeLa and Caski cell lines (Figure 2B). Since plasma membrane localization is important for predicting the therapeutic efficacy, we confirmed the surface localization of HER receptors by immunofluorescence (Figure 2C).
HER inhibitors are targeted drugs for cervical cancer cell lines. (A) Phospho-RTK arrays were done at basal conditions for all cell lines, and after 2 hours treatment with 1.5 µM of AST1306 (AST) and lapatinib (LA) for Caski and C-33A cell lines. Each RTK is represented in duplicat in the arrays (two spots side-by-side), and three pairs of phospho-tyrosine positive controls are located in the corners of each array. (B-C) Characterization of cervical cancer cell lines for HER receptors expression at basal conditions before and after stimulation with 10ng/ml EGF by 15 minutes (B) by western blot and (C), by immunofluorescence. D) Western blot analysis of HER receptors and ERK/AKT intracellular pathways for all cell lines treated with 2.5 µM of all drugs for a period of 2 hours with 10ng/ml EGF for 15 minutes.
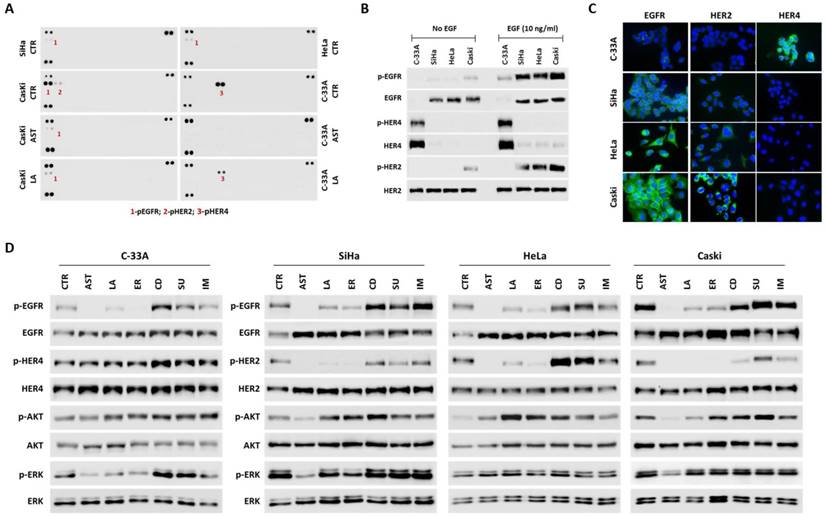
To evaluate whether the HER family of receptors can be specifically and efficiently targeted on cervical cancer, we screened the sensitivity of several cell lines to different pan-RTKs inhibitors (Table 2 and supplementary Figure 2A). As summarized in Table 2, all cell lines were very responsive to the two pan-HER inhibitors used, allitinib (AST1306 - an irreversible inhibitor) and lapatinib (a reversible inhibitor). Compared to the other inhibitors, which were designed to target only EGFR (erlotinib) or different RTKs such as KIT, PDGFRA and VEGFR2 (cediranib, sunitinib and imatinib), the IC50 values for AST1306 and lapatinib were significantly lower. Next, to gain a better insight into the mechanism of cytotoxicity of the inhibitors, we studied the effect of each drug in HER signaling pathways inhibition (Figure 2A and 2D). It appeared that the highest cytotoxic effect of AST1306 and lapatinib could be mainly due to its specificity to concomitantly inhibit the phosphorylation of HER2, HER4, EGFR and intracellular signaling pathways such as ERK and AKT (summarized on Table 2). Interestingly, at higher doses, cediranib was a potent inhibitor on our panel of cell lines, especially for HeLa cells (Table 2 and supplementary Figure 2A). Western blotting revealed that cediranib was able to inhibit HER2 in Caski cells (Figure 2D) and ERK in all cell lines without EGF-stimulation (supplementary Figure 2B), partly explaining its potency. We have previously reported that cediranib is a very potent inhibitor of ERK and can also block other receptors, such as EGFR, AXL and EphA [30]. It is of note that erlotinib, which efficiently inhibited the activation of both EGFR and HER2 under EGF-stimulated conditions (Figure 2D), was non-toxic for HeLa and SiHa cell lines (Table 2). These two cell lines appeared to be less dependent on EGF for intracellular activation, mainly via AKT pathway, than the other cell lines (Supplementary Figure 2C). In fact, potential secondary bypass mechanisms of intracellular signaling activation have been identified as inducing cellular resistance to specific inhibitors, which might be the case for HeLa and SiHa cells. To clarify whether the concomitant inhibition of RTKs and intracellular pathways are crucial for an effective response to these inhibitors, we combined lapatinib with AKT (MK2206) and MAPK (selumetinib) pathway inhibitors in C-33A and SiHa cell lines with different responses and dependencies on these pathways. Both drugs were able to lower the IC50 value of lapatinib, but the effect was more pronounced in the less responsive and EGF dependent cell line, SiHa. Furthermore, the combined inhibition of EGFR (with lapatinb) and AKT pathway (with MK2206) was more pronounced increasing almost twice the response of SiHa cells to lapatinib (Supplementary Figure 2D). Thus, we can conclude that HER receptors present on cervical cancer cell lines are good theranostic markers for HER specific inhibitors (such as AST1306 and lapatinib) and the cellular response to these drugs can be improved by concomitantly inhibiting the receptors and their intracellular signaling pathways.
Inhibitory effect of anti-RTK drugs on cervical cancer cell lines.
| Caski | C-33A | SiHa | HeLa | |||||
|---|---|---|---|---|---|---|---|---|
| IC50 (µM ± SD) | Signalling Inhibition* | IC50 (µM ± SD) | Signalling inhibition* | IC50 (µM ± SD) | Signalling inhibition | IC50 (µM ± SD) | Signalling inhibition | |
| Cediranib (CD) | LR | HER2 | LR | n.i | LR | n.i | S | n.i |
| (2.73 ± 0.48) | (4.26 ± 0.08) | (7.22 ± 1.23) | (0.53 ± 0.49) | |||||
| Sunitinib (SU) | LR | n.i | LR | n.i | LR | n.i | LR | n.i |
| (3.23 ± 0.91) | (9.96 ± 5.48) | (10.21 ± 3.24) | (4.77 ± 1.35) | |||||
| Imatinib (IM) | LR | n.i | LR | n.i | LR | n.i | LR | n.i |
| (>20) | (>20) | (>20) | (>20) | |||||
| Erlotinib (ER) | S | EGFR/HER2 | S | EGFR/ERK | LR | EGFR/HER2 | LR | EGFR/HER2 |
| (2.41 ± 1.19) | (2.30 ± 0.89) | (>20) | (>20) | |||||
| Lapatinib (LA) | S | EGFR/HER2/AKT | S | HER4/EGFR/ERK | S | EGFR/HER2/AKT | S | EGFR/HER2 |
| (0.54 ± 0.023) | (1.78 ± 0.43) | (2.36 ± 0.63) | (2.19 ± 0.98) | |||||
| AST1306 (AST) | S | EGFR/HER2/AKT/ERK | S | HER4/EGFR/ERK | S | EGFR/HER2/AKT/ERK | S | EGFR/HER2 |
| (0.024 ± 0.023) | (0.10 ± 0.12) | (2.31 ± 0.28) | (2.34 ± 0.41) | |||||
*Data from Figure 2A and 2D; IC50 ≤ 2.5µM - Sensitive (S); IC50 > 2.5µM - Lower Responsive (LR); n.i - No Inhibition.
Inhibition of HER receptors can reverse aggressiveness of cervical cancer cells in vitro and in vivo
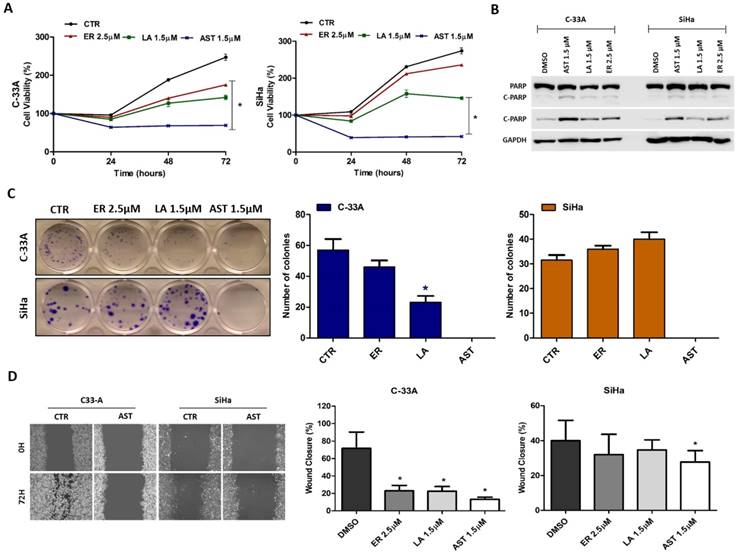
To determine the biological impact of HER pan-inhibitors on viability, migration and proliferation of cells both in vitro and in vivo, we choose a highly sensitive cell line, C-33A and a relatively less sensitive, SiHa cell line. The viability assay overtime showed that lapatinib and especially AST1306, even at lower doses, were more efficient than erlotinib due to their strong cytotoxic effect on both cell lines (Figure 3A). The PARP cleavage measurement revealed the efficacy of AST1306 was also due to its enhanced capability of apoptosis induction (Figure 3B). Furthermore, using clonogenicity and migration assays, which measure important features of cancer cell aggressiveness, we have observed a significant inhibitory effect of AST1306 in both cell lines (Figure 3C and 3D), while erlotinib and lapatinib were effective only in the more sensitive C-33A cell line (Figure 3C and 3D).
Biological effect of HER inhibitors in vitro (C-33A and SiHa). (A) Cell viability was measured at 24, 48, and 72 hours by MTS after treatment with erlotinib (ER), AST1306 (AST) and lapatinib (LA). (B) Apoptosis was measured by Western blotting for PARP cleavage (C-PARP) after 24 hours treatment with AST, LA and ER. (C) Colony formation was measured by a clonogenicity assay. At the left are representative pictures of the assays done in 12-well plates (D) Migration was measured by wound healing migration assay after 72 hours of treatment with drugs. At the left are representative pictures of wounds (40x magnification). The results are expressed in relation to the DMSO control as the mean ± SD.
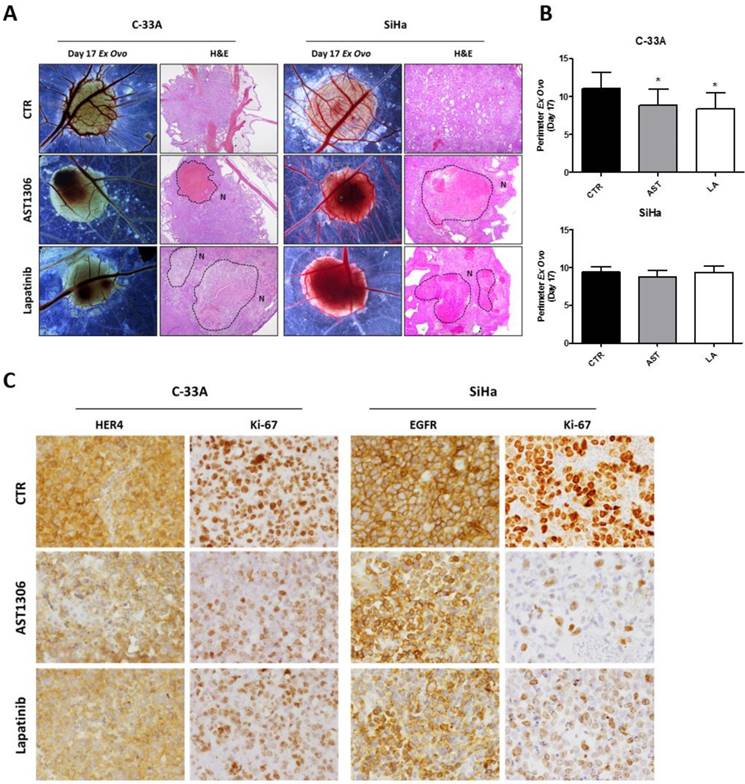
We further performed an in vivo CAM assay by inducing the formation of 3D tumors that were highly aggressive and exhibited histological features that were similar to those observed in human tumors with high rates of angiogenesis and collapsed blood vessels (Figure 4A and Supplementary Figure 3). In agreement with the in vitro assays, we found that AST1306 and lapatinib treatment reduced the size of the tumors formed by injecting C-33A cells but not the SiHa cells (Figure 4B), although both inhibitors were anti-angiogenic for SiHa-induced tumors (Supplementary Figure 3B). Histological analysis revealed that both drugs were biological active in suppressing tumorigenesis in both models. There was a lack of cellular viability and integrity in the treated tumors with formation of large necrotic areas (Figure 4A) and a decrease in the expression of the respective HER targets and in the staining of number/intensity of Ki-67 positive cells (Figure 4C and Supplementary Figure 3C). Therefore, even when the tumor mass was not reduced, probably because the experimental window in the CAM assay was too small, induction of cell death and inhibition of proliferation was observed in vivo, highlighting the potential usage of pan-HER inhibitors for cervical tumors.
In vivo effect of HER inhibitors on cervical cancer. (A) After tumor formation at day 13 of development, the treatment with AST and LA (1.5 µM) was performed for 4 days. Representative pictures (16X magnification) of CAM assay ex ovo at day 17 with the respective H&E pictures (40X magnification), where the necrosis areas (N) are marked. (B) Tumor growth was measured as described in materials and methods in an average of 15 tumors per condition. The results are expressed as the mean ± SD. (C) Representative pictures of the immunohistochemistry (IHC) analysis for HER4, EGFR and ki-67 of the tumors (200X magnification).
Combination of lapatinib with metabolic blockers can prevent the putative in vivo resistance of cervical tumors to HER inhibitors
The resistance to the RTK inhibitors is a significant clinical problem, since even patients who initially respond to the therapy have a high probability to develop acquired resistance to the drugs with time [37]. A mechanism that has been recently associated with resistance to conventional chemotherapy or oncogene-targeted therapies is the deregulated glucose metabolism, in which HIF-1α has a crucial role. The clinical potential of targeted therapies can be enhanced by combining them with glycolysis inhibitors [38]. There is compelling evidence linking HER inhibitors' response to the regulation of energy metabolism in HER-dependent tumors [38-40]. To address whether glucose metabolism could be putatively involved in the response of cervical cancer cells to HER inhibitors, we focused on SiHa cells which have been well characterized previously and showed relatively low responsive to HER inhibitors.
We first interrogated the expression level of HIF-1α upon treatment with HER inhibitors. Using IHC on SiHa-tumor sections, we observed that HIF-1α expression varies with the inhibitors. There was a tendency of decreased expression in the tumors treated with AST1306, whereas lapatinib-treated tumors maintained high levels of HIF-1α (Figure 5A and 5B). These experiments were also performed using the C-33A cell line-derived tumors where the increase in HIF-1α expression was even more pronounced, probably because its basal expression level was very low (Supplementary Figure 4A and 4B).
Effective combinations to improve lapatinib therapy response in vitro. (A) Representative pictures of the immunohistochemistry (IHC) analysis for HIF1-α in SiHa-induced tumors (200X magnification). (B) Graphic representation of the scores obtained in IHC analysis of in vivo tumors for HIF1-α. The horizontal lines represent the mean score obtained in the samples for each condition. (C) Quantification of extracellular lactate levels in vitro, in SiHa cell line after 24 hours of treatment with 1.5 µM of AST or LA, 1.5 mM of 2-DG or LA in combination with 2-DG. (D) At the left is the Western blot analysis of HIF1-α in SiHa cells transfected with siRNA for HIF1-α or with the scramble control. At the right is the cell viability measured in vitro by MTS after 72 hours of treatment to determine the half maximal inhibitory concentrations (IC50) for lapatinib before and after siRNA for HIF1-α transfection or in combination with 2-DG. All experiments were done at least three times in triplicate and the graphs are presented as the mean ± SD relative to DMSO alone (100% viability).
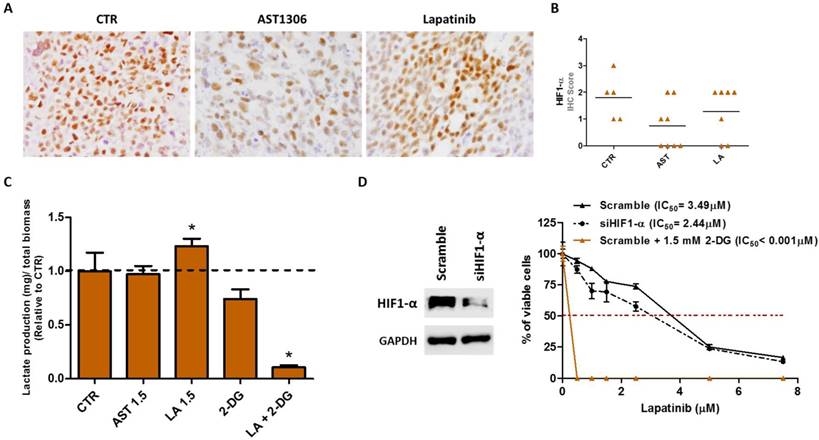
Then, we explored whether changes occur in the cellular metabolism, by measuring the levels of extracellular lactate, upon treatment of cells with the drugs. We observed a significant increase in the production of lactate only upon lapatinib treatment (Figure 5C), a phenomenon not observed in the sensitive cell line C-33A (Supplementary Figure 4C). It was not clear whether the greater efficacy of AST1306 and lapatinib in C-33A cells compared to that in SiHa cells could be related to their basal expression levels of HIF-1α. To explore this, we re-assessed the sensitivity of SiHa cells to lapatinib upon HIF-1α downregulation and found that in fact the cells became more responsive in the absence of HIF-1α (Figure 5D). However, we also observed that besides maintaining or increasing the expression levels of HIF-1α in both cell lines, lapitinib increases the glycolytic metabolism only in the less responsive SiHa cells. It has been reported that alterations in the glucose metabolism ultimately dictate the resistance phenotype of the cells to the RTK targeted therapies [38]. We therefore tested whether the inhibition of the lactate production in lapatinib-treated tumors can revert the sensitivity of cells to this drug. We used combinations of lapatinib with a non-metabolizable glucose analogue, 2-Deoxy-D-glucose (2-DG) (Figure 5D), at a dose that alone does not significantly affect the cellular viability (Supplementary Figure 4D). The combination resulted in a significant reduction of the lactate level (Figure 5C and Supplementary Figure 4C) and was much more efficient in sensitizing cells to lapatinib treatment than the inhibition of HIF-1α expression alone (Figure 5D). The same synergetic effect was observed for C-33A cells (Supplementary Figure 4E). However, since the cells were already highly sensitive to lapitinib, it is plausible that the combination of the drug with 2-DG could be indicated for tumors less sensitive to lapatinib and for which metabolic changes can be expected upon treatment, as was the case with SiHa cells. The combinations of 2-DG with AST1306 were also effective (Supplementary Figure 4F).
To validate our results in vivo, we induced SiHa tumors in CAM and treated with HER inhibitors alone or in combination with 2-DG (Figure 6). The tumors were divided in half for simultaneous histological and Western blot analysis. With this approach, we confirmed in vivo that both drugs (lapatinib and AST1306) were effective in the reduction of EGFR phosphorylation and activation of ERK/AKT pathways (Figure 6A and 6B). Once more, AST1306 proved to be more cytotoxic, since the majority of cells exhibited an apoptotic phenotype, with no proliferating cells (Figure 6A), total cleavage of PARP, and absence of HIF-1α positive cells (Figure 6B). In contrast, upon lapatinib treatment, the tumors retained some activation levels of EGFR and signaling pathways as well as a considerable number of Ki-67 positive cells (Figure 6A and 6B). Importantly, while expression of GLUT1 was maintained at high levels under all conditions, expression of HIF-1α varied considerably (Figure 6A) with overexpression in the lapatinib-treated tumors. The increased expression of HIF-1α was abolished after the combined treatment with 2-DG (Figure 6A and B) resulting in a significant reduction in tumor size and vasculature, while 2-DG alone has no major impact on tumor growth despite being a potent anti-angiogenic drug (Figure 6C and 6D).
In vivo efficacy of lapatinib combination with the glucose analogue 2-DG. After tumor formation, at day 13 of development, treatment with AST and LA (1.5 µM) or 2-DG (1.5 mM) alone and in combination with LA was done for 4 days. The tumors were divided in half for simultaneous histological and Western blot analyses. (A) Representative pictures of the immunohistochemistry (IHC) analysis for EGFR, Ki-67, GLUT1 and HIF-1α, with the respective H&E for the in vivo tumors: CTR (tumor 7), AST (tumor 3), LA (tumor 4), LA+2-DG (tumor 14) and 2-DG (tumor 8) (200X magnification). (B) Western blot analysis of the same tumors shown in Figure 6A. (C) Quantification of tumors perimeter and (D) vessels area as described in materials and methods. The results are expressed as the mean ± SD.
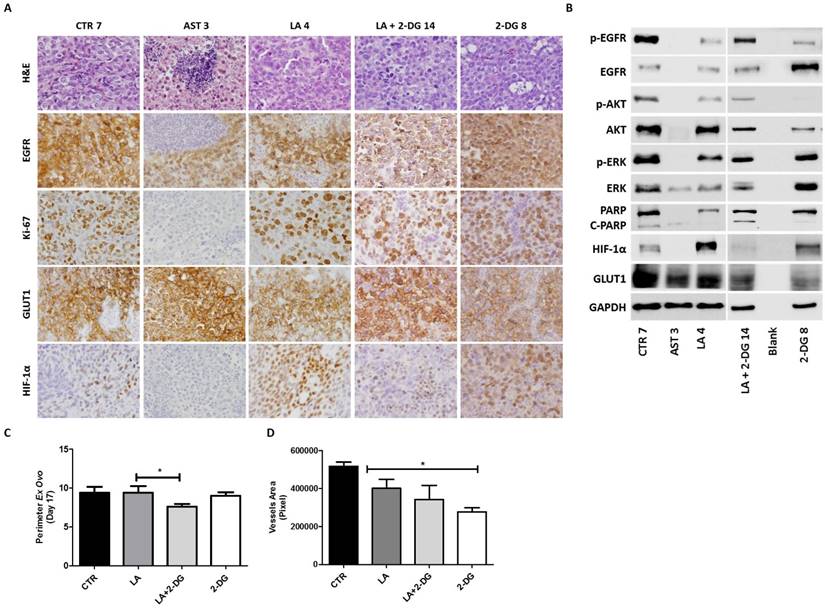
Thus, in conjunction with glucose inhibitors the responsiveness of cervical cancer cells to lapatinib increased both in vitro and in vivo. This appears to be a good strategy to prevent the putative HIF1-α-induced resistance due to long term exposure to lapatinib of cervical cancer cells.
Discussion
Cervical cancer is a major women health problem worldwide [2]. The addition of platin-based chemotherapy to primary radiotherapy has increased the 5-year survival of advanced-stage cervical cancer patients, which is, however, still only 66% [2]. In view of this, there is a critical need to identify molecularly targeted therapies that prolong life but do not create additional toxicities further compromising the patients' quality of life [37, 41]. However, no targeted drugs have been developed specifically for cervical cancer, mainly because data on targetable genetic alterations in these tumors are far behind the knowledge for other tumor types. Recent whole-exome sequencing studies have provided evidence for the presence of targetable oncogenic mutations that warrant further investigation on the feasibility of specific targeted therapies for these tumors [9, 42]. In the present work, we aimed to study the frequency of alterations in the ErbB family of receptors and to explore their potential for specific targeting in cervical cancer.
Initially, we analyzed a large series of cervical cancer samples by immunohistochemistry. Our results revealed that HER2 was frequently expressed (53.5%), whereas EGFR was least frequently expressed (23.4%) in this set of cervical tumors. This could be explained by the fact that EGFR is preferentially expressed in squamous tumors, as also reported by other studies [7, 19, 43, 44] and the majority of our cases were adenocarcinomas. Importantly, we observed that HER2 overexpression constituted an independent prognostic marker for cervical cancer patients. Since squamous carcinomas are more common, the majority of the studies were conducted on this histologic type [45, 46], making a comparison with our work difficult. ErbB receptor overexpression is generally considered in the literature as a rare event in cervical cancer with highly discrepant frequencies among studies. HER2 expression varies between 16-77% [44, 47, 48] and is associated with poor prognosis in some studies, while others failed to find such clinical association [49-52]. EGFR expression in squamous cervical cancer varies from 6% to 90% [53] and its prognostic role remains controversial [16, 17]. Among other HER receptors, HER3 was overexpressed in 74.4% (58/78) of patients and was correlated with poor prognosis [54], whereas HER4 was overexpressed in 79.5% of squamous tumors with no association with clinical parameters [54]. In our study, HER3 overexpression was detected in 75% of the tumors and was associated with poor prognosis markers such as age and metastasis. In contrast, only 23% of the tumors were positive for HER4 in our study.
Our immunohistochemistry findings were further corroborated by in silico analysis. HER receptors were differentially altered in terms of mRNA expression, gene copy numbers, and mutations with HER2 being the most frequently altered (13% - 25/190) gene. EGFR and HER2 harbored mutations in more than one tyrosine kinase phosphorylation site. These results were supported by comprehensive genetic landscape analysis studies, which described HER2 amplification in a small percentage of cervical cancer patients and evidence of HER2 activation by somatic mutation, amplification, and human papilloma virus (HPV) integration in cervical cancer [9, 42]. Recent work by Xiang et al. also reported the presence of HER2 (4.2%) and EGFR (0.4%) oncogenic mutations [42].
We further characterized four commercial cell lines (Caski, SiHa, HeLa and C-33A) and found that only HER family of receptors was activated in these cell lines, an observation that was mostly validated using other methods. A substantially higher proportion of cervical cancer cell lines, either established from fresh tumors or from commercial sources, were reported to express HER2 when evaluated by flow cytometry [55]. It was also reported that HER2-positive cells may have a selective advantage over HER2-negative cells both in vitro and in vivo [46], and that HER2-negative cervical cancers can be transformed into HER2-expressing ones when they recur or metastasize [53, 55, 56].
We screened the sensitivity of cervical cancer cell lines to different pan-RTKs inhibitors and observed that the drugs that target one or more HER receptors (such as erlotinib, AST1306 and lapatinib), were more potent than the ones designed to target different RTKs such as KIT, PDGFRA and VEGFR2 (cediranib, sunitinib and imatinib). Importantly, we found that all cell lines were very responsive to the two pan-HER inhibitors used, AST1306 (allitinib - an irreversible agent) and lapatinib, probably due to their ability to inhibit the phosphorylation of HER2, HER4, and EGFR and its downstream signaling pathways. Hence, we showed that pan-HER inhibitors are effective anti-tumorigenic drugs for HER positive cervical tumors, both in vitro and in vivo.
None of the inhibitors designed to target HER receptor received approval for cervical cancer treatment. Pre-clinical studies showed that the growth of cervical cancer cell lines expressing HER2 was inhibited when incubated with trastuzumab [55]. Importantly, it was found that afatinib - also an irreversible agent as allitinib- was highly effective against HER2-mutated cervical cancer in vitro [57]. Another study reported that combination therapy with trastuzumab and lapatinib induced significant in vivo anti-tumor activity and suggested that HER2-amplified cervical cancer patient-derived xenograft models may accurately predict the outcomes and treatment response to these inhibitors [58]. In one case report, a HER2-negative cervical cancer exhibited HER2 positivity after peritoneal metastasis and the patient had a dramatic treatment response after combined trastuzumab and lapatinib treatment [14]. The RTK inhibitors have not shown the expected benefits in clinical trials mainly because the patients were not pre-screened or pre-selected for the molecular alterations in the targets of interest [23]. Consistent with our in vitro results that showed the absence of KIT, PDGFRA and VEGFR2 on cervical cell lines, no evidence of response or stabilization of disease was observed after imatinib or sunitinib treatment in the clinical trials [59, 60]. In contrast, cediranib had a significant efficacy when added to carboplatin and paclitaxel in the treatment of metastatic or recurrent cervical cancer though it was accompanied by an increased toxic effect [61]. Also, the results of targeted therapy based on EGFR blockage in cervical cancer have not been as promising as those obtained for other tumors with tyrosine kinase inhibitors such as gefitinib and erlotinib [3, 62, 63]. A Phase II, open-label study of specific inhibitors of HER2 protein, pazopanib or lapatinib, in patients with advanced and recurrent cervical cancer showed that pazopanib was well tolerated and resulted in improved progression free survival. The response rates were 9% and 5% for pazopanib and lapatinib, respectively [64]. Although this study was not powered for overall survival, an updated publication of this trial reported a median OS of 44.1 weeks for lapatinib and of 49.7 weeks for patients who received pazopanib [65]. However, no phase III trials are yet registered with these agents. Taken together, these data indicate that cell growth regulation by the HER pathways could be a dominant oncogenic driver in cervical cancer.
One of the major challenges in oncology research is to overcome the inherent resistance to molecular targeted therapies following prolonged treatment periods. Secondary activation of intracellular signaling pathways upon inhibition of RTKs is considered a potential bypass mechanism underlying cellular resistance RTK inhibitors. Another mechanism that was recently associated with resistance to classical chemotherapy or oncogene-targeted therapies is the deregulated glucose metabolism [38]. It has been shown that resistance to known oncogene targeted therapies can be driven by enhanced glycolytic metabolism by the resistant cells in contrast to the sensitive cells [40, 66-70]. The HIF-1α, as well as glucose transporters are considered as the possible predictive factors regulating this “reprogramming” in the glycolytic metabolism [40, 66-69]. For cervical cancer in particular, overexpression of HIF-1α has also been identified as an independent negative prognostic marker [71]. However, the relevance of tumor metabolism in the modulation of cervical tumors response to targeted therapies remains to be elucidated.
Herein, we found by immunohistochemistry that HIF-1α expression was maintained or increased upon lapatinib treatment in vivo, and that HIF-1α downregulation sensitized the cells to lapatinib. Also, in the low responsive cells to this drug we detected an increased lactate production upon treatment, indicating reprogramming of glycolytic metabolism. This phenomenon was also reported for breast cancer [40]; Komurov and colleagues found that lapatinib (EGFR and HER2 inhibitor) induction of toxicity in HER2-positive breast cancer cells was associated with glucose deprivation, and prolonged lapatinib treatment led to acquired resistance that was characterized by increased expression of networks involved in glucose deprivation or hypoglycemic response [40]. Furthermore, it has been shown that cetuximab (monoclonal antibody for EGFR) treatment downregulated glycolysis through HIF-1α inhibition, which was dependent on effective inhibition of PI3K/AKT pathway in non-small-cell lung carcinoma [67, 68]. In contrast, colorectal cancer models with acquired resistance to cetuximab had a significantly higher production of lactate [72], and resistant cells of head and neck squamous cell carcinoma models overexpressed HIF-1α and were highly glycolytic [73]. Moreover, overexpression of HIF-1α conferred cellular resistance to cetuximab-induced apoptosis in sensitive lung adenocarcinoma cells [67, 68].
In our in vivo experiments, the increased expression of HIF-1α upon lapatinib treatment was abolished after the combined treatment with 2-DG, which resulted in a significant reduction in the tumor vasculature and size. There was no major impact on tumor growth of the treatment with 2-DG alone which was potent anti-angiogenic drug. Interestingly, in lung adenocarcinoma, targeting glycolysis with 2-DG was an effective therapeutic option to overcome the limited efficacy of afatinib in patients with EGFR resistant mutations [39].
In conclusion, in this study, we performed the most comprehensive analysis of HER family receptors alterations in cervical adenocarcinoma. We observed the presence of HER protein overexpression, mainly HER2, which was an independent prognostic marker for these patients. By using HER specific inhibitors in both in vitro and in vivo assays, we provided evidence for an important biological and oncogenic role of these proteins in cervical cancer cells. More significantly, we demonstrated that the combination of lapatinib with 2-DG increased the in vitro and in vivo responsiveness of cervical cancer cells to lapatinib. This may likely be a good strategy to prevent a putative HIF1-α-induced resistance due to long term exposure to lapatinib. Thus, we propose the usage of HER family inhibitors in association with glucose metabolism blockers as potentially effective anti-cancer agents for the treatment of cervical cancer patients.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
This study was partially supported by FINEP (MCTI/FINEP/MS/SCTIE/DECIT-BIOPLAT (1302/13), Brazil and co-funded by the project “ON.2 SR&TD Integrated Program (NORTE-07-0124-FEDER-000017)” co-funded by Programa Operacional Regional do Norte (ON.2- O Novo Norte), Quadro de Referência Estratégico Nacional (QREN), through Fundo Europeu de Desenvolvimento Regional (FEDER). OM is recipient of a post-doc fellowship (SFRH/BPD/108351/2015) from Fundação para a Ciência e Tecnologia (FCT), Portugal. FC is recipient of a master fellowship (2014/03684-0) from Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP). VMG is recipient from a PhD fellowship (SFRH/BD/51997/2012) from Fundação para a Ciência e Tecnologia (FCT), Portugal.
Conflict of Interest
The authors have no conflict of interest to declare.
References
1. Jemal A, Center MM, DeSantis C, Ward EM. Global patterns of cancer incidence and mortality rates and trends. Cancer Epidemiol Biomarkers Prev. 2010;19:1893-907
2. Quinn MA, Benedet JL, Odicino F, Maisonneuve P, Beller U, Creasman WT. et al. Carcinoma of the Cervix Uteri. Int J Gynaecol Obstet. 2006;95:S43-S103
3. Duenas-Gonzalez A, Serrano-Olvera A, Cetina L, Coronel J. New molecular targets against cervical cancer. Int J Womens Health. 2014;6:1023-31
4. Seol HJ, Ulak R, Ki KD, Lee JM. Cytotoxic and targeted systemic therapy in advanced and recurrent cervical cancer: experience from clinical trials. Tohoku J Exp Med. 2014;232:269-76
5. Lemmon MA, Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2010;141:1117-34
6. Martinho O, Goncalves A, Moreira MA, Ribeiro LF, Queiroz GS, Schmitt FC. et al. KIT activation in uterine cervix adenosquamous carcinomas by KIT/SCF autocrine/paracrine stimulation loops. Gynecol Oncol. 2008;111:350-5
7. Longatto-Filho A, Pinheiro C, Martinho O, Moreira MA, Ribeiro LF, Queiroz GS. et al. Molecular characterization of EGFR, PDGFRA and VEGFR2 in cervical adenosquamous carcinoma. BMC cancer. 2009;9:212
8. Gonzalez Martin A. Molecular biology of cervical cancer. Clin Transl Oncol. 2007;9:347-54
9. Ojesina AI, Lichtenstein L, Freeman SS, Pedamallu CS, Imaz-Rosshandler I, Pugh TJ. et al. Landscape of genomic alterations in cervical carcinomas. Nature. 2014;506:371-5
10. Wright AA, Howitt BE, Myers AP, Dahlberg SE, Palescandolo E, Van Hummelen P. et al. Oncogenic mutations in cervical cancer: genomic differences between adenocarcinomas and squamous cell carcinomas of the cervix. Cancer. 2013;119:3776-83
11. Arteaga CL. ErbB-targeted therapeutic approaches in human cancer. Exp Cell Res. 2003;284:122-30
12. Reichert JM, Valge-Archer VE. Development trends for monoclonal antibody cancer therapeutics. Nat Rev Drug Discov. 2007;6:349-56
13. Shawver LK, Slamon D, Ullrich A. Smart drugs: tyrosine kinase inhibitors in cancer therapy. Cancer cell. 2002;1:117-23
14. Yan M, Parker BA, Schwab R, Kurzrock R. HER2 aberrations in cancer: implications for therapy. Cancer Treat Rev. 2014;40:770-80
15. Lemmon MA, Schlessinger J, Ferguson KM. The EGFR family: not so prototypical receptor tyrosine kinases. Cold Spring Harb Perspect Biol. 2014;6:a020768
16. Noordhuis MG, Eijsink JJ, Roossink F, de Graeff P, Pras E, Schuuring E. et al. Prognostic cell biological markers in cervical cancer patients primarily treated with (chemo)radiation: a systematic review. Int J Radiat Oncol Biol Phys. 2011;79:325-34
17. Soonthornthum T, Arias-Pulido H, Joste N, Lomo L, Muller C, Rutledge T. et al. Epidermal growth factor receptor as a biomarker for cervical cancer. Ann Oncol. 2011;22:2166-78
18. Reyes HD, Thiel KW, Carlson MJ, Meng X, Yang S, Stephan JM. et al. Comprehensive profiling of EGFR/HER receptors for personalized treatment of gynecologic cancers. Mol Diagn Ther. 2014;18:137-51
19. Lee CM, Shrieve DC, Zempolich KA, Lee RJ, Hammond E, Handrahan DL. et al. Correlation between human epidermal growth factor receptor family (EGFR, HER2, HER3, HER4), phosphorylated Akt (P-Akt), and clinical outcomes after radiation therapy in carcinoma of the cervix. Gynecol Oncol. 2005;99:415-21
20. Ocana A, Vera-Badillo F, Seruga B, Templeton A, Pandiella A, Amir E. HER3 overexpression and survival in solid tumors: a meta-analysis. J Natl Cancer Inst. 2013;105:266-73
21. Campiglio M, Ali S, Knyazev PG, Ullrich A. Characteristics of EGFR family-mediated HRG signals in human ovarian cancer. J Cell Biochem. 1999;73:522-32
22. Feng SM, Sartor CI, Hunter D, Zhou H, Yang X, Caskey LS. et al. The HER4 cytoplasmic domain, but not its C terminus, inhibits mammary cell proliferation. Mol Endocrinol. 2007;21:1861-76
23. Tomao F, Di Tucci C, Imperiale L, Boccia SM, Marchetti C, Palaia I. et al. Cervical cancer: are there potential new targets? An update on preclinical and clinical results. Curr Drug Targets. 2014;15:1107-20
24. Tewari KS, Sill MW, Long HJ 3rd, Penson RT, Huang H, Ramondetta LM. et al. Improved survival with bevacizumab in advanced cervical cancer. N Engl J Med. 2014;370:734-43
25. Tomao F, Papa A, Rossi L, Zaccarelli E, Caruso D, Zoratto F. et al. Angiogenesis and antiangiogenic agents in cervical cancer. Onco Targets Ther. 2014;7:2237-48
26. Pecorelli S, Zigliani L, Odicino F. Revised FIGO staging for carcinoma of the cervix. Int J Gynaecol Obstet. 2009;105:107-8
27. Martinho O, Pinto F, Granja S, Miranda-Goncalves V, Moreira MA, Ribeiro LF. et al. RKIP inhibition in cervical cancer is associated with higher tumor aggressive behavior and resistance to cisplatin therapy. PloS one. 2013;8:e59104
28. Miranda-Goncalves V, Granja S, Martinho O, Honavar M, Pojo M, Costa BM. et al. Hypoxia-mediated upregulation of MCT1 expression supports the glycolytic phenotype of glioblastomas. Oncotarget. 2016 [Epub ahead of print]
29. Silva-Oliveira RJ, Silva VA, Martinho O, Cruvinel-Carloni A, Melendez ME, Rosa MN. et al. Cytotoxicity of allitinib, an irreversible anti-EGFR agent, in a large panel of human cancer-derived cell lines: KRAS mutation status as a predictive biomarker. Cell Oncol (Dordr). 2016;39(3):253-63
30. Martinho O, Silva-Oliveira R, Miranda-Goncalves V, Clara C, Almeida JR, Carvalho AL. et al. In Vitro and In Vivo Analysis of RTK Inhibitor Efficacy and Identification of Its Novel Targets in Glioblastomas. Transl Oncol. 2013;6:187-96
31. Martinho O, Granja S, Jaraquemada T, Caeiro C, Miranda-Goncalves V, Honavar M. et al. Downregulation of RKIP is associated with poor outcome and malignant progression in gliomas. PloS one. 2012;7:e30769
32. Martinho O, Zucca LE, Reis RM. AXL as a modulator of sunitinib response in glioblastoma cell lines. Exp Cell Res. 2015;332:1-10
33. Miranda-Goncalves V, Honavar M, Pinheiro C, Martinho O, Pires MM, Pinheiro C. et al. Monocarboxylate transporters (MCTs) in gliomas: expression and exploitation as therapeutic targets. Neuro Oncol. 2013;15:172-88
34. Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO. et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6:pl1
35. Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA. et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401-4
36. Saez R, Molina MA, Ramsey EE, Rojo F, Keenan EJ, Albanell J. et al. p95HER-2 predicts worse outcome in patients with HER-2-positive breast cancer. Clin Cancer Res. 2006;12:424-31
37. Jurgensmeier JM, Eder JP, Herbst RS. New strategies in personalized medicine for solid tumors: molecular markers and clinical trial designs. Clin Cancer Res. 2014;20:4425-35
38. Granja S, Pinheiro C, Reis RM, Martinho O, Baltazar F. Glucose Addiction in Cancer Therapy: Advances and Drawbacks. Curr Drug Metab. 2015;16:221-42
39. Kim SM, Yun MR, Hong YK, Solca F, Kim JH, Kim HJ. et al. Glycolysis inhibition sensitizes non-small cell lung cancer with T790M mutation to irreversible EGFR inhibitors via translational suppression of Mcl-1 by AMPK activation. Mol Cancer Ther. 2013;12:2145-56
40. Komurov K, Tseng JT, Muller M, Seviour EG, Moss TJ, Yang L. et al. The glucose-deprivation network counteracts lapatinib-induced toxicity in resistant ErbB2-positive breast cancer cells. Mol Syst Biol. 2012;8:596
41. Blume-Jensen P, Hunter T. Oncogenic kinase signalling. Nature. 2001;411:355-65
42. Xiang L, Li J, Jiang W, Shen X, Yang W, Wu X. et al. Comprehensive analysis of targetable oncogenic mutations in chinese cervical cancers. Oncotarget. 2015;6:4968-75
43. Iida K, Nakayama K, Rahman MT, Rahman M, Ishikawa M, Katagiri A. et al. EGFR gene amplification is related to adverse clinical outcomes in cervical squamous cell carcinoma, making the EGFR pathway a novel therapeutic target. Br J Cancer. 2011;105:420-7
44. Leung TW, Cheung AN, Cheng DK, Wong LC, Ngan HY. Expressions of c-erbB-2, epidermal growth factor receptor and pan-ras proto-oncogenes in adenocarcinoma of the cervix: correlation with clinical prognosis. Oncol Rep. 2001;8:1159-64
45. Davey DD. Cervical cytology classification and the Bethesda System. Cancer J. 2003;9:327-34
46. English DP, Roque DM, Santin AD. HER2 expression beyond breast cancer: therapeutic implications for gynecologic malignancies. Mol Diagn Ther. 2013;17:85-99
47. Fukazawa EM, Baiocchi G, Soares FA, Kumagai LY, Faloppa CC, Badiglian-Filho L. et al. Cox-2, EGFR, and ERBB-2 expression in cervical intraepithelial neoplasia and cervical cancer using an automated imaging system. Int J Gynecol Pathol. 2014;33:225-34
48. Ndubisi B, Sanz S, Lu L, Podczaski E, Benrubi G, Masood S. The prognostic value of HER-2/neu oncogene in cervical cancer. Ann Clin Lab Sci. 1997;27:396-401
49. Kihana T, Tsuda H, Teshima S, Nomoto K, Tsugane S, Sonoda T. et al. Prognostic significance of the overexpression of c-erbB-2 protein in adenocarcinoma of the uterine cervix. Cancer. 1994;73:148-53
50. Ngan HY, Cheung AN, Liu SS, Cheng DK, Ng TY, Wong LC. Abnormal expression of epidermal growth factor receptor and c-erbB2 in squamous cell carcinoma of the cervix: correlation with human papillomavirus and prognosis. Tumour Biol. 2001;22:176-83
51. Perez-Regadera J, Sanchez-Munoz A, De-la-Cruz J, Ballestin C, Lora D, Garcia-Martin R. et al. Negative prognostic impact of the coexpression of epidermal growth factor receptor and c-erbB-2 in locally advanced cervical cancer. Oncology. 2009;76:133-41
52. Zagouri F, Sergentanis TN, Chrysikos D, Filipits M, Bartsch R. Molecularly targeted therapies in cervical cancer. A systematic review. Gynecol Oncol. 2012;126:291-303
53. Berchuck A, Rodriguez G, Kamel A, Soper JT, Clarke-Pearson DL, Bast RC Jr. Expression of epidermal growth factor receptor and HER-2/neu in normal and neoplastic cervix, vulva, and vagina. Obstet Gynecol. 1990;76:381-7
54. Fuchs I, Vorsteher N, Buhler H, Evers K, Sehouli J, Schaller G. et al. The prognostic significance of human epidermal growth factor receptor correlations in squamous cell cervical carcinoma. Anticancer Res. 2007;27:959-63
55. Bellone S, Palmieri M, Gokden M, Joshua J, Roman JJ, Pecorelli S. et al. Selection of HER-2/neu-positive tumor cells in early stage cervical cancer: implications for Herceptin-mediated therapy. Gynecol Oncol. 2003;91:231-40
56. Chavez-Blanco A, Perez-Sanchez V, Gonzalez-Fierro A, Vela-Chavez T, Candelaria M, Cetina L. et al. HER2 expression in cervical cancer as a potential therapeutic target. BMC cancer. 2004;4:59
57. Lopez S, Cocco E, Stefania B, Bortolomai I, Bonazzoli E, Nicoletti R. et al. Abstract 4498: Afatinib, an irreversible ErbB family inhibitor, demonstrates activity against HER2 mutated cervical cancer in vitro. Cancer Res. 2014;74:4498
58. Oh DY, Kim S, Choi YL, Cho YJ, Oh E, Choi JJ. et al. HER2 as a novel therapeutic target for cervical cancer. Oncotarget. 2015;6:36219-30
59. Candelaria M, Arias-Bonfill D, Chavez-Blanco A, Chanona J, Cantu D, Perez C. et al. Lack in efficacy for imatinib mesylate as second-line treatment of recurrent or metastatic cervical cancer expressing platelet-derived growth factor receptor alpha. Int J Gynecol Cancer. 2009;19:1632-7
60. Mackay HJ, Tinker A, Winquist E, Thomas G, Swenerton K, Oza A. et al. A phase II study of sunitinib in patients with locally advanced or metastatic cervical carcinoma: NCIC CTG Trial IND.184. Gynecol Onco. 2010;116:163-7
61. Symonds RP, Gourley C, Davidson S, Carty K, McCartney E, Rai D. et al. Cediranib combined with carboplatin and paclitaxel in patients with metastatic or recurrent cervical cancer (CIRCCa): a randomised, double-blind, placebo-controlled phase 2 trial. Lancet Oncol. 2015;16:1515-24
62. Nogueira-Rodrigues A, Moralez G, Grazziotin R, Carmo CC, Small IA, Alves FV. et al. Phase 2 trial of erlotinib combined with cisplatin and radiotherapy in patients with locally advanced cervical cancer. Cancer. 2014;120:1187-93
63. Santin AD, Sill MW, McMeekin DS, Leitao MM Jr, Brown J, Sutton GP. et al. Phase II trial of cetuximab in the treatment of persistent or recurrent squamous or non-squamous cell carcinoma of the cervix: a Gynecologic Oncology Group study. Gynecol Onco. 2011;122:495-500
64. Monk BJ, Mas Lopez L, Zarba JJ, Oaknin A, Tarpin C, Termrungruanglert W. et al. Phase II, open-label study of pazopanib or lapatinib monotherapy compared with pazopanib plus lapatinib combination therapy in patients with advanced and recurrent cervical cancer. J Clin Oncol. 2010;28:3562-9
65. Monk BJ, Pandite LN. Survival data from a phase II, open-label study of pazopanib or lapatinib monotherapy in patients with advanced and recurrent cervical cancer. J Clin Oncol. 2011;29:4845
66. Nilsson MB, Zage PE, Zeng L, Xu L, Cascone T, Wu HK. et al. Multiple receptor tyrosine kinases regulate HIF-1alpha and HIF-2alpha in normoxia and hypoxia in neuroblastoma: implications for antiangiogenic mechanisms of multikinase inhibitors. Oncogene. 2010;29:2938-49
67. Lu Y, Liang K, Li X, Fan Z. Responses of cancer cells with wild-type or tyrosine kinase domain-mutated epidermal growth factor receptor (EGFR) to EGFR-targeted therapy are linked to downregulation of hypoxia-inducible factor-1alpha. Mol Cancer. 2007;6:63
68. Li X, Lu Y, Liang K, Pan T, Mendelsohn J, Fan Z. Requirement of hypoxia-inducible factor-1alpha down-regulation in mediating the antitumor activity of the anti-epidermal growth factor receptor monoclonal antibody cetuximab. Mol Cancer Ther. 2008;7:1207-17
69. Kominsky DJ, Klawitter J, Brown JL, Boros LG, Melo JV, Eckhardt SG. et al. Abnormalities in glucose uptake and metabolism in imatinib-resistant human BCR-ABL-positive cells. Clin Cancer Res. 2009;15:3442-50
70. Strohecker AM, White E. Targeting mitochondrial metabolism by inhibiting autophagy in BRAF-driven cancers. Cancer Discov. 2014;4:766-72
71. Huang M, Chen Q, Xiao J, Yao T, Bian L, Liu C. et al. Overexpression of hypoxia-inducible factor-1alpha is a predictor of poor prognosis in cervical cancer: a clinicopathologic study and a meta-analysis. Int J Gynecol Cancer. 2014;24:1054-64
72. Monteleone F, Rosa R, Vitale M, D'Ambrosio C, Succoio M, Formisano L. et al. Increased anaerobic metabolism is a distinctive signature in a colorectal cancer cellular model of resistance to antiepidermal growth factor receptor antibody. Proteomics. 2013;13:866-77
73. Lu H, Li X, Luo Z, Liu J, Fan Z. Cetuximab reverses the Warburg effect by inhibiting HIF-1-regulated LDH-A. Mol Cancer Ther. 2013;12:2187-99
Author contact
![]() Corresponding authors: Olga Martinho, PhD. Life and Health Sciences Research Institute (ICVS), School of Medicine, University of Minho, Campus de Gualtar, 4710-057 Braga, Portugal. Phone: + 351 253 604868 E-mail: olgamartinhouminho.pt AND Rui Manuel Reis, PhD. Molecular Oncology Research Center, Barretos Cancer Hospital, Rua Antenor Duarte Villela, 1331, CEP 14784-400, Barretos, S. Paulo, Brazil. E-mail: ruireis.hcbcom.
Corresponding authors: Olga Martinho, PhD. Life and Health Sciences Research Institute (ICVS), School of Medicine, University of Minho, Campus de Gualtar, 4710-057 Braga, Portugal. Phone: + 351 253 604868 E-mail: olgamartinhouminho.pt AND Rui Manuel Reis, PhD. Molecular Oncology Research Center, Barretos Cancer Hospital, Rua Antenor Duarte Villela, 1331, CEP 14784-400, Barretos, S. Paulo, Brazil. E-mail: ruireis.hcbcom.