Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Experimental section
Results and Discussion
Conclusions
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2017; 7(8):2305-2313. doi:10.7150/thno.19554 This issue Cite
Research Paper
A Fluorogenic Probe for Ultrafast and Reversible Detection of Formaldehyde in Neurovascular Tissues
Xing-Guang Liang1, 3*, Bo Chen2*, Ling-Xiao Shao1, Juan Cheng1, Ming-Zhu Huang3, Yu Chen3, Yong-Zhou Hu1, Yi-Feng Han2, Feng Han1 ![]() , Xin Li1
, Xin Li1 ![]()
1. College of Pharmaceutical Sciences, Zhejiang University, Hangzhou 310058, China;
2. Department of Chemistry, Zhejiang Sci-Tech University, Hangzhou 310018, China;
3. Central Laboratory, First Affiliated Hospital, School of Medicine, Zhejiang University, Hangzhou 310003, China.
* These authors contributed equally to this work.
Received 2017-2-7; Accepted 2017-3-16; Published 2017-6-1
Abstract

Formaldehyde (FA) is endogenously produced in live systems and has been implicated in a diverse array of pathophysiological processes. To disentangle the detailed molecular mechanisms of FA biology, a reliable method for monitoring FA changes in live cells would be indispensable. Although there have been several fluorescent probes reported to detect FA, most are limited by the slow detection kinetics and the intrinsic disadvantage of detecting FA in an irreversible manner which may disturb endogenous FA homeostasis. Herein we developed a coumarin-hydrazonate based fluorogenic probe (PFM) based on a finely-tailored stereoelectronic effect. PFM could respond to FA swiftly and reversibly. This, together with its desirable specificity and sensitivity, endows us to track endogenous FA in live neurovascular cells with excellent temporal and spatial resolution. Further study in the brain tissue imaging showed the first direct observation of aberrant FA accumulation in cortex and hippocampus of Alzheimer's mouse model, indicating the potential of PFM as a diagnostic tool.
Keywords: fluorescent probe, formaldehyde, neurodegenerative disorder, neurovascular cell.
Introduction
Formaldehyde (FA) has traditionally been viewed as a notorious environmental toxin for its carcinogenic effects in mammals. However, it is now evident that FA is also endogenously produced by enzymatic systems mediated by semicarbazide-sensitive amine oxidase (SSAO) [1, 2], carboxymethyl transferase [3], lysine-specific demethylase 1 [4, 5], et al. The presence of these three kinds of syntheses in healthy brain suggests the continuous production of endogenous formaldehyde [6-9]. It has been proposed that endogenous FA participates in spatial memory formation through DNA methylation [10]. However, aberrant FA has been implicated in the progression of cognitive impairment [11-14]. To better understand the Janus biological properties of FA, it is essential to have practical methods and tools to detect dynamic changes of FA in vivo. Traditional methods for FA analysis have relied on in vitro techniques, including gas chromatography [15, 16], high-performance liquid chromatography [17, 18], selected ion flow tube mass spectrometry [19, 20], and radiometry [21]. Although these methods have facilitated FA biology study to some extent, they nevertheless suffer from tedious biological sample preparation procedures and the intrinsic inability to detect FA in complex live biological systems.
Fluorescence imaging using small-molecule probes is attracting increasingly more interest. Due to the good biocompatibility of probes, and the straightforward dosing and observation, their scope of applications is extending [22-29]. Recently, there have emerged several elegant fluorescent probes for imaging FA in live cells. However, most of the probes were designed based on an FA-triggered 2-aza-Cope rearrangement and are therefore intrinsically limited by the sluggish detection kinetics and by being irreversible [30-34]. Although the application of the amine-FA condensation reaction has resulted in the development of phenylhydrazine-based probes with improved detection rates [35-37], still improvements could be envisioned including developing faster and reversible probes. Furthermore, previously reported probes are irreversible ones which perform detection by consuming FA permanently and may disturb native FA homeostasis. Moreover, these irreversible probes are incapable of tracking the dynamic homeostasis of endogenous FA. Therefore, fast and reversible probes capable of tracking native FA with high spatiotemporal resolution remains an unmet challenge, which is imperative to understand the pathophysiological roles of FA more accurately.
Herein we present a novel FA fluorescent probe, PFM, which was designed by incorporating a stereoelectronic effect to increase the rate of detection and render the probe reversible. Strikingly, PFM could detect FA with ultrafast kinetics, and complete response is observed for PFM (10 μM) towards FA (200 μM) in less than 1 minute. More impressively, reversibility was demonstrated by first observing the fluorescent response of PFM and then scavenging FA led to decreased fluorescence. Re-addition of FA restored the response, and the reversibility tolerates at least three cycles. Moreover, the applicability of PFM to image endogenous FA has been confirmed by experiments on neurovascular cells. Further work with APP/PS1 transgenic mice highlighted the potential of PFM for the diagnosis of aberrant FA levels during pathological development.
Experimental section
Materials
Anhydrous toluene was distilled from Na prior to use. Dry CH2Cl2 was distilled from CaH2. Other chemicals and reagents were from commercial supplies and used without further purification. Reactions were monitored by thin layer chromatography using TLC Silica gel 60 F254 supplied by Qingdao Puke Separation Material Corporation, Qingdao, P. R. China, and UV light was used as the visualizing agent. Flash column chromatography was performed using 200-300 mesh silica gel and was supplied by Qingdao Marine Chemical Factory, Qingdao, P. R. China.
Instruments
1H and 13C NMR spectra were recorded on a Bruker Fourier transform 500 NMR spectrometer at ambient temperature. CDCl3 was used as solvent except otherwise indicated and the spectra were calibrated referencing residual undeuterated solvent as an internal reference (1H NMR = 7.26, 13C NMR = 77.16). Chemical shifts were given in ppm and coupling constants (J) in Hz. Following abbreviations were used to designate multiplicities: s = singlet, d = doublet, m = multiplet. High-resolution mass data were obtained on an Agilent 6224 TOF LC/MS spectrometer using ESI-TOF (electrospray ionization-time of flight). IR spectra were obtained on a Bruker VECTOR FTIR. UV-Vis spectra were taken on a HITACHI U-3010 Spectrophotometer. Fluorescence measurements were performed on an Agilent Cary Eclipse Fluorescence Spectrophotometer with slit widths to be 10 and 10 nm for excitation and emission respectively except otherwise indicated, and the photomultiplier (PMT) detector voltage was set at medium.
Synthesis of PFM
A mixture of 7-diethylamino-coumarin (0.50 g, 2.3 mmol) and Lawesson reagent (1.9 g, 4.6 mmol) in dry benzene (15 mL) was heated under reflux with an inert atmosphere of nitrogen for 6 h. After being cooled to ambient temperature, the solid precipitate was removed by filtration. The filtrate was concentrated by rotary evaporation, diluted with ethyl acetate (100 mL) and washed subsequently with H2O (1×50 mL) and brine (1×50 mL). After being dried over anhydrous Na2SO4, the solution was concentrated under reduced pressure to give the crude product which was pure enough for further reaction. Part of this crude product (100 mg, ca. 0.43 mmol) was then dissolved in ethanol (5 mL), to which under an inert atmosphere of nitrogen was added hydrazine hydrate (80%, 0.16 mL, 1.6 mmol). The mixture was heated to reflux and kept under reflux for 3 h, then was cooled to ambient temperature. After removal of ethanol by rotary evaporation, the mixture was diluted by the addition of H2O (10 mL). The mixture was extracted with CH2Cl2 (3×20 mL). The combined organic phases were washed with brine (1×20 mL) and dried over anhydrous Na2SO4. Concentration under reduced pressure gave the crude product which was purified by column chromatography on silica gel eluted with CH2Cl2 to give PFM as a reddish brown solid (64 mg, 66% in two steps). M.p.: 100.5 - 102.5 oC; IR (cm-1): 3332, 3177, 2965, 2925, 1605, 1522, 1421, 1354, 1200, 1137, 1078; 1H NMR (500 MHz, CDCl3): δ 6.98 (d, J = 8.8 Hz, 1H), 6.65 (d, J = 9.6 Hz, 1H), 6.37 (m, 2H), 5.98 (d, J = 9.7 Hz, 1H), 3.37 (q, J = 7.1 Hz, 4H), 1.19 (t, J = 7.1 Hz, 6H); 13C NMR (125 MHz, CDCl3): δ 154.60, 148.99, 146.10, 128.51, 127.84, 113.43, 108.88, 106.82, 97.90, 44.64, 12.66; ESI-HRMS (m/z): [M+H]+ calc'd. for C13H18N3O: 232.1450, found 232.1458.
Fluorometric analysis
All the photophysical characterization experiments were carried out at ambient temperature. Deionized water was used to prepare all aqueous solutions. Phosphate buffer saline (PBS, 10 mM) was purged with N2 for 5 min before use. PFM was dissolved in DMSO to make a 10 mM stock solution. Stock solutions of FA and other bio-relevant species were prepared by dissolving commercial chemicals in deionized water or DMSO. To test the fluorescent response of PFM towards FA or other species, aliquots of probe stock solution were diluted with PBS and treated with analytes to make sure both probes and analytes were kept at desired final concentrations. After quick and vigorous shaking, the mixture was allowed standing in the dark for desired time and then the fluorescence spectra were taken with λex 451 nm. All fluorometric experiments were performed in triplicate, and data shown were the average.
Cell culture
Human brain microvascular endothelial cell (HBMEC) and human brain vascular pericyte (HBVP) were purchased from Sciencell Research Laboratories (Carlsbad, CA, USA). The HBMECs were maintained in 1640 medium (Invitrogen) and HBVPs were cultured in Dulbecco's modified Eagle medium (DMEM, Invitrogen) supplemented with 10% heat-inactivated FBS (Invitrogen), penicillin (100 U/mL) (Invitrogen), and streptomycin (100 U/mL) (Invitrogen). The cultures were maintained at 37 oC in a 95% humidified atmosphere with 5% CO2.
Cytotoxicity assay for PFM
HBMECs and HBVPs were seeded in 96-well plates at a density of 5×103 cells/well at 37 oC in a 95% humidified atmosphere with 5% CO2 for 24 h. After washing with PBS twice, PFM with concentration of 1 μM, 5 μM, 10 μM, 25 μM, 50 μM, and 100 μM were added to the cells, which were allowed an incubation period of 3 h, 6 h, 12 h, 24 h, 48 h. After introducing 10 μL of CCK8 solution for 1 h, the absorption at 450 nm was measured by Microplate Spectrophotometer (MD I3X). Each experiment was repeated three times, and the average values were taken in analyses.
Flow cytometric analysis
Samples of HBMECs and HBVPs for flow cytometry were prepared by passaging and seeding in 25T Flask (Corning) before experiments. Cells of 70% confluency were trypsinized, pelleted via centrifugation, resuspended in medium. For analysis of exogenous FA in HBMECs and HBVPs, cells were fist stained with PFM (10 μM) for 15 min at 37 oC, and then incubated with 0.2 mM, 0.5 mM, 2 mM FA, or 5 mM FA at 37 oC for 15 min. For analysis of endogenous FA in living HBMECs and HBVPs, cells were incubated with 10 μM PFM with or without NaHSO3 pre-treatment. Excitation was provided by the 488 nm HeNe laser. Each plot represented 10,000 viable cells, non-viable cells were excluded from FACS analysis by appropriate gating. All data analyses were carried out using Cell Quest software (Becton Dickinson).
Brain slices preparation
APPswe/PS1 (B6.Cg-Tg (APPswe, PSEN1dE9) 85Dbo/Mmjax mice (Stock No. 34832, Jackson Laboratory)) and wild-type C57BL/6 mice were housed under a 12 h light/dark schedule and had access to food and water ad libitum. All experiments and protocols were approved by the Zhejiang University Animal Experimentation Committee and were in complete compliance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. APP/PS1 transgenic mice or C57BL/6 mice were rapidly decapitated, and then the brain was quickly removed and submerged in a 4 oC modified artificial cerebrospinal fluid (ACSF) containing (in mM): 75 sucrose, 87 NaCl, 2.5 KCl, 15 NaH2PO4, 7 MgCl2, 0.5 CaCl2, 25 NaHCO3 and 25 glucose. The brain was then extracted and glued on the platform of a semiautomatic vibrating blade microtome (VT1000; Leica). The platform was then placed in the slicing chamber containing modified ACSF at 4 oC. Coronal sections of 300 µm containing the hippocampus were collected in a holding chamber filled with ACSF saturated with 95% O2 and 5% CO2, containing (in mM): 124 NaCl, 3 KCl, 1.25 NaH2PO4, 1.0 MgSO4, 2 CaCl2, 26 NaHCO3, 10 glucose for a minimum of 30 min at 34ºC, then the temperature was maintained at room temperature prior to use.
Confocal fluorescence imaging
For confocal fluorescent imaging experiments to detect exogenous FA in living HBMECs and HBVPs, cells were treated with 10 μM PFM for 15 min, washed with PBS (pH 7.4), followed without or with the incubation of 200 μM FA for 15 min. For the fluorescence imaging experiments of endogenous FA in living brain cells, HBMECs and HBVPs were incubated without or with 200 μM NaHSO3 for 30 min, and then washed with PBS (pH 7.4), followed by 10 μM PFM incubation for 15 min. The residual probe was washed three times before imaging. Digital images were captured using the FV10-ASW 3.0 viewer software (Olympus). Cell counts were performed using a 4×, 20×, 40×, or 60× objective in at least five fields of view randomly selected from each coverslip. At least 3 independent experiments were counted. For real-time visualization of endogenous and exogenous FA in HBMECs, cells were incubated with 10 μM PFM after being treated with exogenous FA (500 μM), thapsigargin (5 μM), or NaHSO3 (200 μM) for 30 min, and fluorescence were obtained with a confocal laser scanning microscope (Olympus, FV1000). For imaging endogenous FA in living brain tissue slides, tissue slides were first incubated without or with 200 μM NaHSO3 for 30 min, followed by incubation with 20 μM PFM for another 30 min. The fluorescence density was analyzed using Image J software (NIH, Bethesda, MD, USA).
Results and Discussion
Due to its good fluorescent properties and the nature as a privileged structure in drug discovery which might endow the probe with physicochemical properties desirable for in vivo application [38], the coumarin scaffold was chosen as the prime building block for probe development and PFM was devised by introducing a hydrazonate group to C-2 position of 7-(diethylamino) coumarin. We envisioned that due to the hyperconjugation of the lone pair electrons on the oxygen atom to the antibonding C=N (Figure 1), the hydrazonate in PFM should be of potent nucleophilicity to readily react with FA to form an imine bond which, in turn, may change the fluorescence profile of PFM due to the altered degree of electronic polarization in the coumarin electronic system. To test this hypothesis on the stereoelectronic effect, calculations on the electron density of the terminal nitrogen were conducted with 7-(diethylamino)-2-hydrazononaphthalen as a control (Figure S1). It turned out that the hydrazone terminal nitrogen of the control compound has less electron density than that of PFM, justifying the rationality of the hydrazonate group as a FA-targeting moiety. Furthermore, it is also hypothesized that because of the neighboring electron-withdrawing C=N bond, the newly formed imine bond might be hydrolyzed easily in aqueous solution to fulfill the reversible detection of FA. PFM were facilely prepared as detailed above.
Structure and design philosophy of PFM.
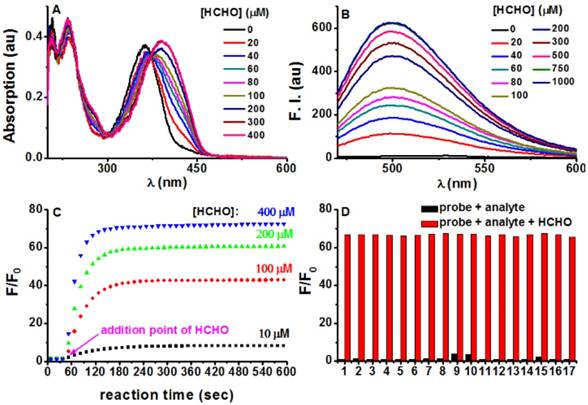
With PFM in hand, its spectroscopic responses towards FA in aqueous solution were first characterized. As shown in Figure 2A, PFM in phosphate buffer solution (PBS, 10 mM, pH 7.4) showed its maximum absorption at 364 nm (ε 18 710 M-1 cm-1, Ф 0.003), whereas the treatment of FA caused a dose-dependent bathochromic-shift of its absorption and reached the plateau at 393 nm (ε 19 325 M-1 cm-1, Ф 0.029). In contrast to the highly emissive 7-(diethylamino) coumarin, PFM was almost non-fluorescent in PBS, presumably due to the substitution of the less electron-withdrawing hydrazonate group which decreased the electronic push-pull effect. However, FA treatment induced a dose-dependent increase of the fluorescence centered at 500 nm (Figure 2B), suggesting the conversion of the hydrazonate group to Nˈ-methylenehydrazonate which made the probe a more electron-donor-acceptor system. Coinciding with the spectroscopic responses, NMR titration experiments provided direct evidence in support of the formation of Nˈ-methylenehydrazonate group (Figure S2). These results taken together, implied an “intramolecular charge transfer (ICT)” mechanism for PFM fluorescence quenching [39]. Furthermore, plot of the fluorescent intensity (F) versus the concentrations of FA gave an exponential dependence (Figure S3), with concentrations of FA higher than 500 μM bringing the response to its maximum (Fmax). Interestingly, the Napierian logarithm of Fmax minus F correlated linearly with the corresponding FA concentrations ranging from 0 to 200 μM (Figure S4), which happens to fall into the reported biologically relevant range of FA [40], implying the great potential of the probe to quantify endogenous FA. The detection limit of PFM was found to be as low as 0.4 μM (Figure S5). To further confirm the rapid detection kinetics of PFM, we recorded its intensity at emission maximum (λem 500 nm) as time lapsed upon the treatment of various concentrations of FA, and it turned out that the reaction ran fast enough to complete in 1 min, even with FA concentration as low as 10 μM (Figure 2C). This ultrafast detection kinetics and the low detection limit highlight PFM as the most sensitive fluorescent probe for FA ever. Notably, the fluorogenic response of PFM was found to be selective for FA over other potentially competing biological carbonyl species such as glucose, glyoxal, methylglyoxal, acetaldehyde, etc (Figure 2D, Figure S6). In fact, treating PFM (10 μM) with FA (300 μM) caused a 68-fold fluorescence enhancement. While among the other analytes, only methylglyoxal, glyoxal and HClO caused intensity enhancement of 3.8, 3.5, and 2.2-fold respectively (Figure S7), suggesting the high specificity of PFM towards FA. It is noteworthy that PFM retains its sensitivity for FA even in the presence of the above-mentioned analytes (Figure 2D, Figure S8), indicating the potential of PFM to detect FA in more complicated biological contexts. We also studied the influence of surrounding pH changes to the detection reaction, and it turned out that although a gradual fluorescence increase occurred to PFM alone as pH values decreased from pH 7.4 to pH 4.5 as a result of protonation of the amino group (Figure S9), this effect causes little detriment to the detection sensitivity of PFM towards FA in the pH range of 7.4-6.0 (Figure S10). Photostability of both PFM and the reaction product was evaluated by keeping the samples under continuous excitation. The results showed that PFM retained its sensitivity for FA even after a long irradiation time, and that the fluorescence intensity of PFM-FA remained unchanged after continuous excitation as long as 50 min (Figure S11).
Photophysical responses of PFM to FA. (A) PFM (20 μM) was treated with various amounts of FA for 5 min, and then the UV-vis spectra were recorded. (B) Fluorescent spectra of PFM (10 μM) after the treatment of various concentrations of FA for 5 min. (C) Reaction time course of PFM (10 μM) with FA (10, 100, 200, 400 μM). (D) Fluorescent responses of PFM (10 μM) toward various analytes (300 μM) after a reaction time of 30 min without (black) or with (red) the presence of FA (300 μM). The analytes tested were PFM blank (1), acetaldehyde (2), malonaldehyde (3), ascorbic acid (4), glucose (5), glucosone (6), oxalic acid (7), pyruvate (8), methylglyoxal (9), glyoxal (10), p-methoxybenzaldehyde (11), trichloroacetaldehyde (12), p-nitrobenzaldehyde (13), acetone (14), HClO (15), H2O2 (16), GSH (17). For C and D, F represents the fluorescent intensity of PFM at 500 nm after the treatment of FA for various time or after the treatment of various analytes, and F0 represents the intensity of blank PFM solution at 500 nm. All data were collected in PBS (pH 7.4, 10 mM) at ambient temperature with λex 451 nm.
Finally, to test if PFM could detect FA in a reversible way, NaHSO3 (200 μM) was added as a FA scavenger to the probe (10 μM)-FA (200 μM) solution. The emission intensity was decreased to almost the original level of the probe blank solution. However, re-addition of another portion of FA (200 μM) restored the fluorescent intensity to just about the level before NaHSO3 was added (Figure S12). This cycle could be repeated for at least twice, indicting the reversibility of the detection. Furthermore, based on the dose-response plot of PFM to FA (Figure S13), we were able to calculate the dissociation constant of PFM toward FA (Kd, FA) to be around 100 μM employing a literature method [41].
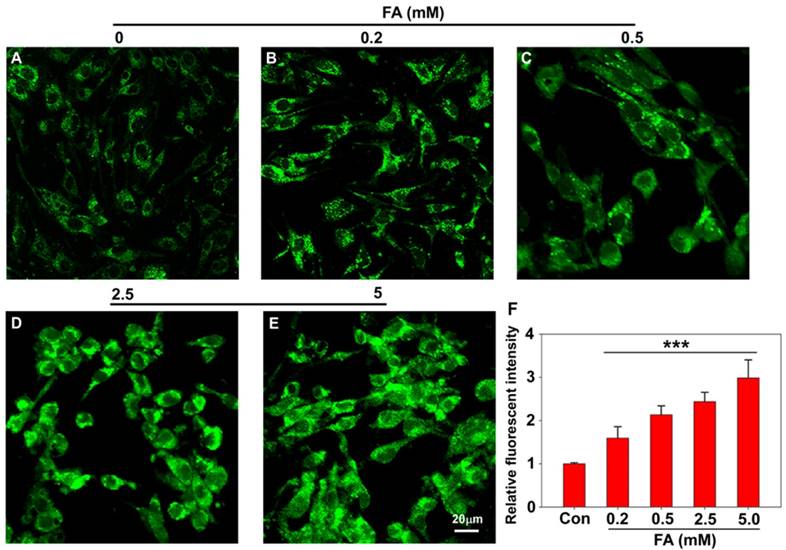
Having established PFM as a robust probe to detect FA in aqueous solution, we next moved to investigate its applicability to image FA in live cells, and the cytotoxicity of PFM was first assayed for this purpose. CCK8 assay carried out in Human brain microvessel endothelial cells (HBMECs) and human brain vascular pericytes (HBVPs) showed that PFM remained weakly cytotoxic to neurovascular cells at concentrations up to 100 μM with prolonged treatment time of 48 h (Figure S14). Confocal microscopy and flow cytometry were then used to investigate the capability of PFM for real-time bioimaging of exogenous FA in live neurovascular cells. HMBEC or HBVP cells were treated with PFM (10 μM) for 15 min followed by washing to remove excess probe. The fluorescence intensities of PFM were monitored following various concentrations of FA treatment (0, 0.2, 0.5, 2.5 and 5 mM), and a significant increase of intracellular probe fluorescence was observed which correlated with FA concentrations (Figure 3 and S15). We also quantified the fluorescence intensities by flow cytometry analysis, and found that significant intracellular PFM fluorescence increased in a dose-dependent manner following FA treatment (Figure S16C, S17C, S18), demonstrating the feasibility of PFM as a bioimaging probe for detecting FA in neurovascular cells.
Confocal imaging of PFM for detecting exogenous FA in living HBMECs (A, B, C, D, E). Cells are treated with 10 μM PFM for 15 min at 37 oC, then washed with PBS (pH 7.4), and followed by FA incubation for 15 min. control (A); 0.2 mM FA (B); 0.5 mM FA (C); 2.5 mM FA (D); 5 mM FA (E). (F) Quantification of image data in (A-E). Data are expressed as mean±S.E.M. A minimum of 3 images for each condition were quantified and averaged. ***P<0.01 versus control. The PFM fluorescence was monitored at 515-545 nm (λex=488 nm). Scale bar=20 μm.
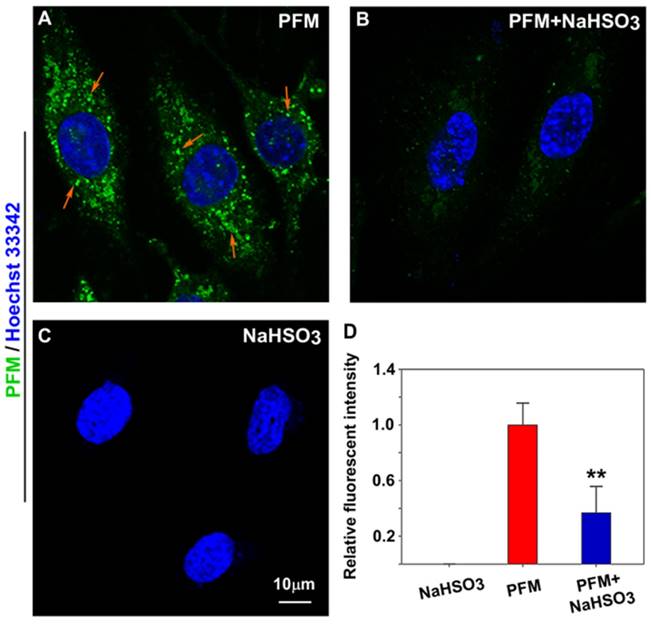
As endogenous FA has been reported to be expressed in neurovascular cells [11, 14], we were therefore keen to know if PFM could be sensitive enough to image native FA in intact cells. As shown in Figure 4A and Figure S19B, probe-loaded HBMECs and HBVPs exhibited significant intracellular fluorescence even without exogenous FA. However, pre-incubation of the cells with NaHSO3 (a FA scavenger) blunted intracellular fluorescence (Figure 4B and Figure S19C) [35], indicating the presence of endogenous FA. We thus conclude that PFM is a cell permeable, non-toxic small molecule probe capable of imaging native FA in live neurovascular cells, which should be the prerequisite for dynamically tracking FA fluxes in live cells.
Imaging endogenous FA in live HBMECs. (A) HBMECs treated with PFM (10 μM). (B) HBMECs were pre-treated with NaHSO3 (200 μM) for 30 min, then washed with PBS (pH 7.4) and followed by PFM (10 μM) incubation for 15 min at 37 oC. (C) HBMECs treated with NaHSO3 (200 μM). Nuclei were stained with Hoechst 33342. (D) Quantification of image data. Data are expressed as mean±S.E.M. A minimum of 3 images for each condition were quantified and averaged. **P<0.01 versus PFM alone. The PFM fluorescence was monitored at 515-545 nm (λex=488 nm).
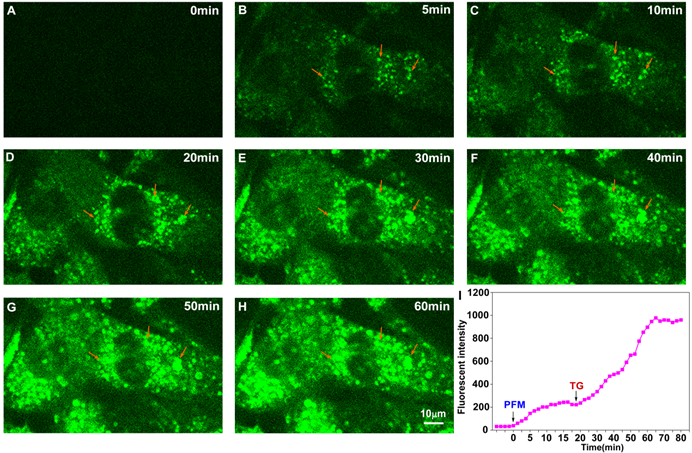
We next addressed whether PFM retained its ultrafast response towards FA in neurovascular cells. As shown in Figure S20 and video S1, significant intracellular fluorescence could be observed even 1 min after PFM incubation. Fluorescence intensity of PFM peaked after 20 min of incubation. Moreover, further increase of PFM fluorescence were observed when cells were exposed to exogenous FA (video S2 and Figure S21), whereas NaHSO3 (200 μM) pre-treatment partially reduced the PFM response to exogenous FA (video S3 and Figure S22). These results thus show time-dependent and reversible character of probe PFM. Since excess formaldehyde has been reported to cause hyper-modification and misfolding of proteins, leading to endoplasmic reticulum stress [42], we therefore employed an ER stress model to stimulate the burst of native FA in live cells. As shown in Figure S23, ER stress caused by thapsigargin (TG) or tunicamycin (TM) treatment resulted in significantly increased intracellular PFM fluorescence, while time lapse live cell imaging of HBMECs revealed a 5-fold increase in the fluorescence intensity of PFM probe after being subjected to thapsigargin (TG, 5 μM) stimulation within one-hour time frame (Figure 5, video S4). Our present results implicated that PFM is suitable to monitor the dynamic changes of endogenous FA in neurovascular cells, which could be useful to address the mechanisms involved in the pathological process of neurovascular disorders
Real-time visualization of endogenous FA in living HBMECs upon ER stress. The time-series images are individual frames from a continuous time-lapse movie and show dynamic fluorescence elevation with PFM (10 μM) treatment (A, B, C) and thapsigargin (TG, 5 μM) stimulation (D, E, F, G, H). (I) Mean values of fluorescence intensity from time-lapse movie (video S4) were measured to quantify the endogenous FA production upon TG treatment. The arrowheads indicate the same position of the target cell. The PFM fluorescence was monitored at 515-545 nm (λex=488 nm).
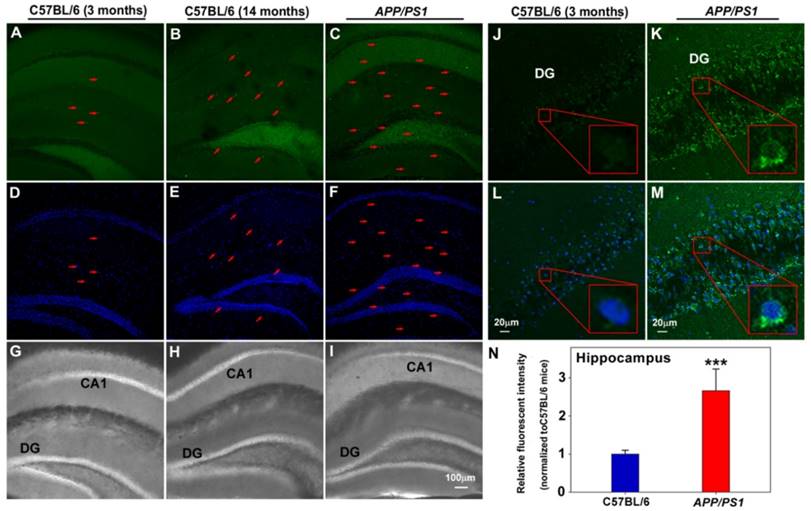
Finally, the capability of PFM to image endogenous FA in mice brain tissues was tested. To this end, PFM was first examined for its sensitivity and specificity to detect exogenous FA in brain tissues from 3-month old mice, with and without NaHSO3 pretreatment. In good agreement with the above cell-based results, our data demonstrated significantly higher PFM fluorescence in cerebral cortex (Figure S24) pretreated with FA compared with those pretreated with NaHSO3. Since excessive elevation of endogenous FA has been implicated in the pathological progression of neurodegenerative pathology [11-14], we therefore assessed whether PFM could be used to monitor aberrant FA levels in brain tissues of aging or APP/PS1 transgenic mice. This translation to ex vivo pathological model imaging showed that significant PFM fluorescence elevation was observed in hippocampus (Figure 6) and brain cortex (Figure S25 and S26) of 14-month-old C57BL/6 mice and APP/PS1 transgenic mice compared with 3-month-old C57BL/6 mice, indicating the correlation between FA levels and neurodegenerative process.
Application of PFM for monitoring endogenous FA in living brain slices of C57BL/6 and APP/PS1 transgenic mice. Fluorescence images of hippocampus of C57BL/6 and APP/PS1 transgenic mice incubated with PFM (20 μM) for 30 min, (A, D, G) 3 month old C57BL/6 mice, (B, E, H) 14 month old C57BL/6 mice, (C, F, I) APP/PS1 transgenic mice. The arrow indicated the cells with high fluorescence. Scale bar=100 μm. Fluorescence images of hippocapus of C57BL/6 (J, L) and APP/PS1 transgenic mice (K, M), Scale bar=20 μm. (N) Quantification of image data represented by D1 and D2. Data are expressed as mean±S.E.M., a minimum of 3 images for each condition were quantified and averaged. ***P<0.001 versus C57BL/6 mice. The PFM fluorescence was monitored at 515-545 nm (λex=488 nm). Nuclei were stained with Hoechst 33342 (blue).
Conclusions
We have developed a coumarine-hydrazonate based fluorogenic probe for the ultrafast detection of FA in intact biological samples. Apart from its desired biocompatibility, this probe has the advantage of detecting FA with fast kinetics and in a reversible manner. In combination with a pharmacological approach, we have confirmed that PFM can detect endogenous FA in neurovascular cells swiftly and specifically with good spatiotemporal sensitivity. Moreover, PFM-assisted fluorescence detection of FA in pathological samples obtained from APP/PS1 transgenic mice further highlights the diagnostic potentials of PFM for neurodegenerative diseases. PFM may be used in future to unlock the molecular basis behind the correlation between FA levels and progression of neurodegenerative diseases.
Supplementary Material
Additional File 1Figure S1-S26.
Video S1.
Video S2.
Video S3.
Video S4.
Acknowledgements
This work was supported in part by The National Key Research and Development Program of China (2016YFE0125400), Projects of National Natural Science Foundations of China (81573411, 81300991, 81503048, 21642007), Zhejiang Provincial Natural Science Foundation (Z16H310003, LY15H300003).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Yu PH, Wright S, Fan EH, Lun ZR, Gubisne-Harberle D. Physiological and pathological implications of semicarbazide-sensitive amine oxidase. Biochim Biophys Acta. 2003;1647:193-9
2. O'Sullivan J, Unzeta M, Healy J, O'Sullivan MI, Davey G, Tipton KF. Semicarbazide-sensitive amine oxidases: enzymes with quite a lot to do. Neurotoxicology. 2004;25:303-15
3. Lee ES, Chen H, Hardman C, Simm A, Charlton C. Excessive S-adenosyl-L-methionine-dependent methylation increases levels of methanol, formaldehyde and formic acid in rat brain striatal homogenates: possible role in S-adenosyl-L-methionine-induced Parkinson's disease-like disorders. Life Sci. 2008;83:821-7
4. Shi Y, Lan F, Matson C, Mulligan P, Whetstine JR, Cole PA, Casero RA, Shi Y. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell. 2004;119:941-53
5. Wang Y, Zhang H, Chen Y, Sun Y, Yang F, Yu W, Liang J, Sun L, Yang X, Shi L, Li R, Li Y, Zhang Y, Li Q, Yi X, Shang Y. LSD1 is a subunit of the NuRD complex and targets the metastasis programs in breast cancer. Cell. 2009;138:660-72
6. Diliberto EJ Jr, Axelrod J. Regional and subcellular distribution of protein carboxymethylase in brain and other tissues. J Neurochem. 1976;26:1159-65
7. Obata T. Semicarbazide-sensitive amine oxidase (SSAO) in the brain. Neurochem Res. 2002;27:263-8
8. Zhang YZ, Zhang QH, Ye H, Zhang Y, Luo YM, Ji XM, Su YY. Distribution of lysine-specific demethylase 1 in the brain of rat and its response in transient global cerebral ischemia. Neurosci Res. 2010;68:66-72
9. Zuo DM, Yu PH. Semicarbazide-sensitive amine oxidase and monoamine oxidase in rat brain microvessels, meninges, retina and eye sclera. Brain Res Bull. 1994;33:307-11
10. Tong Z, Han C, Luo W, Li H, Luo H, Qiang M, Su T, Wu B, Liu Y, Yang X, Wan Y, Cui D, He R. Aging-associated excess formaldehyde leads to spatial memory deficits. Sci Rep. 2013;3:1807
11. Lu J, Miao J, Su T, Liu Y, He R. Formaldehyde induces hyperphosphorylation and polymerization of Tau protein both in vitro and in vivo. Biochim Biophys Acta. 2013;1830:4102-16
12. Unzeta M, Solé M, Boada M, Hernández M. Semicarbazide-sensitive amine oxidase (SSAO) and its possible contribution to vascular damage in Alzheimer's disease. J Neural Transm (Vienna). 2007;114:857-62
13. Tong Z, Zhang J, Luo W, Wang W, Li F, Li H, Luo H, Lu J, Zhou J, Wan Y, He R. Urine formaldehyde level is inversely correlated to mini mental state examination scores in senile dementia. Neurobiol Aging. 2011;32:31-41
14. Ferrer I, Lizcano JM, Hernández M, Unzeta M. Overexpression of semicarbazide sensitive amine oxidase in the cerebral blood vessels in patients with Alzheimer's disease and cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy. Neurosci Lett. 2002;321:21-4
15. Takeuchi A, Takigawa T, Abe M, Kawai T, Endo Y, Yasugi T, Endo G, Ogino K. Determination of formaldehyde in urine by headspace gas chromatography. Bull Environ Contam Toxicol. 2007;79:1-4
16. Ebeler SE, Clifford AJ, Shibamoto T. Quantitative analysis by gas chromatography of volatile carbonyl compounds in expired air from mice and human. J Chromatogr B Biomed Sci Appl. 1997;702:211-5
17. Yu PH, Cauglin C, Wempe KL, Gubisne-Haberle D. A novel sensitive high-performance liquid chromatography/electrochemical procedure for measuring formaldehyde produced from oxidative deamination of methylamine and in biological samples. Anal Biochem. 2003;318:285-90
18. Luo W, Li H, Zhang Y, Ang CY. Determination of formaldehyde in blood plasma by high-performance liquid chromatography with fluorescence detection. J Chromatogr B Biomed Sci Appl. 2001;753:253-7
19. Kato S, Burke PJ, Koch TH, Bierbaum VM. Formaldehyde in human cancer cells: detection by preconcentration-chemical ionization mass spectrometry. Anal Chem. 2001;73:2992-7
20. Spanel P, Smith D, Holland TA, Al Singary W, Elder JB. Analysis of formaldehyde in the headspace of urine from bladder and prostate cancer patients using selected ion flow tube mass spectrometry. Rapid Commun Mass Spectrom. 1999;13:1354-9
21. Szarvas T, Szatlóczky E, Volford J, Trézl L, Tyihák E, Rusznák I. Determination of endogenous formaldehyde level in human blood and urine by dimedone-14C radiometric method. J Radioanal Nucl Chem. 1986;106:357-367
22. Avraham R, Haseley N, Brown D, Penaranda C, Jijon HB, Trombetta JJ, Satija R, Shalek AK, Xavier RJ, Regev A, Hung DT. Pathogen Cell-to-Cell Variability Drives Heterogeneity in Host Immune Responses. Cell. 2015;162:1309-21
23. Shachar S, Voss TC, Pegoraro G, Sciascia N, Misteli T. Identification of Gene Positioning Factors Using High-Throughput Imaging Mapping. Cell. 2015;162:911-23
24. Dean KM, Palmer AE. Advances in fluorescence labeling strategies for dynamic cellular imaging. Nat Chem Biol. 2014;10:512-23
25. Kocaoglu O, Carlson EE. Progress and prospects for small-molecule probes of bacterial imaging. Nat Chem Biol. 2016;12:472-8
26. Headley MB, Bins A, Nip A, Roberts EW, Looney MR, Gerard A, Krummel MF. Visualization of immediate immune responses to pioneer metastatic cells in the lung. Nature. 2016;531:513-7
27. O'Herron P, Chhatbar PY, Levy M, Shen Z, Schramm AE, Lu Z, Kara P. Neural correlates of single-vessel haemodynamic responses in vivo. Nature. 2016;534:378-82
28. Chen KH, Boettiger AN, Moffitt JR, Wang S, Zhuang X. RNA imaging. Spatially resolved, highly multiplexed RNA profiling in single cells. Science. 2015;348(6233):aaa6090
29. Morisaki T, Lyon K, DeLuca KF, DeLuca JG, English BP, Zhang Z, Lavis LD, Grimm JB, Viswanathan S, Looger LL, Lionnet T, Stasevich TJ. Real-time quantification of single RNA translation dynamics in living cells. Science. 2016;352:1425-9
30. Brewer TF, Chang CJ. An Aza-Cope Reactivity-Based Fluorescent Probe for Imaging Formaldehyde in Living Cells. J Am Chem Soc. 2015;137:10886-9
31. Roth A, Li H, Anorma C, Chan J. A Reaction-Based Fluorescent Probe for Imaging of Formaldehyde in Living Cells. J Am Chem Soc. 2015;137:10890-3
32. Li JB, Wang QQ, Yuan L, Wu YX, Hu XX, Zhang XB, Tan W. A two-photon fluorescent probe for bio-imaging of formaldehyde in living cells and tissues. Analyst. 2016;141:3395-402
33. He L, Yang X, Liu Y, Kong X, Lin W. A ratiometric fluorescent formaldehyde probe for bioimaging applications. Chem Commun (Camb). 2016;52:4029-32
34. He L, Yang X, Ren M, Kong X, Liu Y, Lin W. An ultra-fast illuminating fluorescent probe for monitoring formaldehyde in living cells, shiitake mushrooms, and indoors. Chem Commun (Camb). 2016;52:9582-5
35. Tang Y, Kong X, Xu A, Dong B, Lin W. Development of a Two-Photon Fluorescent Probe for Imaging of Endogenous Formaldehyde in Living Tissues. Angew Chem Int Ed Engl. 2016;55:3356-9
36. Tang Y, Kong X, Liu Z. R, Xu A, Lin W. Lysosome-Targeted Turn-On Fluorescent Probe for Endogenous Formaldehyde in Living Cells. Anal Chem. 2016;88:9359-9363
37. Lee YH, Tang Y, Verwilst P, Lin W, Kim JS. A biotin-guided formaldehyde sensor selectively detecting endogenous concentrations in cancerous cells and tissues. Chem Commun (Camb). 2016;52:11247-11250
38. Torres FC, Brucker N, Andrade SF, Kawano DF, Garcia SC, Poser GL, Eifler-Lima VL. New insights into the chemistry and antioxidant activity of coumarins. Curr Top Med Chem. 2014;14:2600-23
39. Demchenko AP. Introduction to Fluorescence Sensing. New York, USA: Springer Science+Business media. 2009
40. Tong Z, Han C, Luo W, Wang X, Li H, Luo H, Zhou J, Qi J, He R. Accumulated hippocampal formaldehyde induces age-dependent memory decline. Age (Dordr). 2013;35:583-96
41. Umezawa K, Yoshida M, Kamiya M, Yamasoba T, Urano Y. Rational design of reversible fluorescent probes for live-cell imaging and quantification of fast glutathione dynamics. Nat Chem. 2017;9:279-286
42. Li X, Zhang KY, Zhang P, Chen LX, Wang L, Xie M, Wang CY, Tang XQ. Hydrogen sulfide inhibits formaldehyde-induced endoplasmic reticulum stress in PC12 cells by upregulation of SIRT-1. PLoS One. 2014;9:e89856
Author contact
![]() Corresponding authors: Prof F. Han & Prof X. Li, Zhejiang University, 866 Yuhangtang Road, Hangzhou 310058, China. Email: changhuahanedu.cn; lixin81edu.cn
Corresponding authors: Prof F. Han & Prof X. Li, Zhejiang University, 866 Yuhangtang Road, Hangzhou 310058, China. Email: changhuahanedu.cn; lixin81edu.cn