Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Results
Discussion
Materials and Methods
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2017; 7(8):2325-2338. doi:10.7150/thno.18225 This issue Cite
Research Paper
Atg9b Deficiency Suppresses Autophagy and Potentiates Endoplasmic Reticulum Stress-Associated Hepatocyte Apoptosis in Hepatocarcinogenesis
Ning Wang, Hor-Yue Tan, Sha Li, Yibin Feng ![]()
School of Chinese Medicine, The University of Hong Kong, Hong Kong S.A.R, PR of China
Received 2016-11-6; Accepted 2017-3-8; Published 2017-6-2
Abstract

The aim of this study was to investigate the mechanism underlying autophagy deficiency during hepatic carcinogenesis. For this purpose, we used choline-deficient, amino acid-defined (CDAA) hepatocarcinogenesis model in mice. miRNA microarrays combined with computational target predictions and GO analysis were used to identify molecular processes involved in carcinogenesis. PCR profiler array was employed to detect the dysregulated autophagy-related genes during carcinogenesis. We observed induction of hepatic tumours with increased inflammation, DNA damage, and cell death. These cellular processes were particularly detected upon oncogenic transformation of hepatocytes in which ER stress was excessively induced. Microarray combined with GO analysis showed that transformation of hepatocytes resulted in dysregulated events associated with cytoplasmic vesicle formation, which, in turn, was related to ER stress-induced autophagy. Defects of autophagy were observed in livers harbouring tumours and suffered a loss of expression of autophagy-related protein 9b (Atg9b). Hepatocytes lacking Atg9b were vulnerable to cell death induced by ER stress stimulus mainly caused by accumulation of ubiquitinated proteins. Loss of Atg9b also blocked recruitment of p62-associated ubiquitinated protein for autophagosome-lysosome degradation as Atg9b-driven phagophores may facilitate docking of both LC3 and p62 to initiate autophagy-associated degradation. miR-3091-3p from tumour-derived exosomes, which were internalised by hepatocytes, could suppress Atg9b expression. Observations from this study advance our knowledge about the regulation of autophagy during hepatocarcinogenesis.
Keywords: Hepatic carcinogenesis, Autophagy, Atg9b, ER stress, Tumour-derived exosome, microRNA.
Introduction
Several lines of evidence have suggested the protective role of autophagy during carcinogenesis of the liver. Autophagy may prevent inflammation, fibrosis, and carcinogenesis in the liver of alpha-1-antitrypsin (AT)-deficient rodents by mediating the degradation of this mutant protein [1, 2]. Using autophagy-enhancing chemical successfully decreased the hepatic load of protein aggregates and prevented liver from injury and fibrogenesis [2]. The essential role of autophagy was further proven by the observation that lysosome inhibitors may accelerate hepatocarcinogenesis in genotoxin-treated rodents [3]. Blocking initiation of autophagy by chronic activation of mTOR signalling, an upstream negative regulator of autophagy, is able to initialise endoplasmic reticulum (ER) stress in hepatocytes, which contributes to hepatic damage and inflammation-associated carcinogenesis [4]. Liver-specific knockout of autophagy-associated gene 5 (Atg5), which impairs autophagy in the liver, acquires hepatocarcinogenesis in mice at 10-months of age [5]. Defects of autophagy render hepatocytes vulnerable to a variety of metabolic stresses resulting in cell death and hepatic inflammation [6] while inducing hepatic autophagy in mice under carcinogen treatment can suppress oxidative stress and genome instability [7]. Although these observations provide evidence that autophagy is a promising target for cancer prevention, how autophagy is regulated during hepatocarcinogenesis remains unclear.
Autophagy is a highly conserved biological process that maintains intracellular homeostasis by recycling damaged cell components and scavenging harmful substrates such as damaged organelles and protein aggregates [8]. Initiation and regulation of cellular autophagy are under strict control and involve regulation of a series of autophagy-associated proteins [9]. Ablation or mutation of these genes, which commonly occurs in cancer patients, abrogates autophagy initiation and may be of pathological and prognostic significance. Autophagy was found down-regulated in hepatocellular carcinoma (HCC) specimens collected from patients with or without detectable hepatitis viruses and low expression of Atg5 was found correlated with increased virus burden [10]. Interestingly, p62 accumulation caused by autophagy deficiency could be observed in HCC specimens regardless of the virus status [10, 11]. Down-regulation of autophagy-related genes has been shown to be correlated with poor prognosis in HCC patients [12]. Similarly, a significant decrease in expression of Beclin-1 was found in HCC tissues, which was correlated with diseases-free survival and overall survival in patients with Bcl-xL expression [13]. Beclin-1 expression in HCC patients was reported to have a negative correlation with cirrhosis as well as vascular invasion [14]. These observations point to the clinical importance of the expression of autophagy-related genes for HCC prognosis.
In this report, we used choline deficient, amino acid-defined (CDAA) hepatocarcinogenesis model in mice to study the expression of autophagy-associated genes during early hepatocarcinogenesis. Feeding mice with CDAA diet leads to the development of non-alcoholic steatohepatitis/non-alcoholic fatty liver disease (NASH/NAFLD) and subsequent hepatocellular adenomas and carcinoma, which imitate hepatocarcinogenesis in humans [15]. We hypothesized that the link between autophagy deficiency and hepatocarcinogenesis can be elaborated with profiling of microRNAs (miRNAs) in combination with computational analysis. With PCR profiler array of autophagy-related genes, we identified the role of autophagy-related protein 9b (Atg9b), the deletion of which causes early embryonic lethality [16], in mediating autophagy deficiency during hepatocarcinogenesis. Furthermore, we elucidated the detailed mechanism underlying the role of Atg9b in initiating autophagy in response to ER stress.
Results
NAFLD mice accelerate hepatic inflammation and hepatocyte death during hepatocarcinogenesis
HCC commonly arises in patients with chronic liver diseases, especially NAFLD and is therefore considered a typical inflammation-related cancer. The cellular and molecular process of cancerous switch of NAFLD has just begun to be understood. To observe the pathological changes during hepatocarcinogenesis, we collected specimens of CDAA diet-fed mice at week 32, 56 and 72. While there was no hepatic tumour in mice after 32weeks, carcinogenesis in the liver was evident after 56 weeks (Fig.1a). Five out of ten mice presented hepatic tumours after 56-week treatment and all CDAA diet-fed mice had liver tumors after 72 weeks. No hepatic tumour was observed in mice fed with CSAA diet of standard chow. Descriptive analysis on the specimen is shown in Supplemental Table 1. Biochemical analysis showed significantly elevated activity of the serum ALT in CDAA diet-fed mice compared with the standard chow-fed group (Supplemental Figure 1). CDAA diet-fed mice presented serious NASH accompanied with significant accumulation of lipid droplets in the liver (Supplemental Figure 2). Histological analysis of liver tumours in mice with CDAA feeding exhibited various patterns predominantly with fatty changes and solid growth. Some cases developed cytoplasmic inclusions and trabecular growth (Fig.1b and Supplemental Figure 3). The microscopic analysis of the liver from individual CDAA diet-fed mice is shown in Fig.1c. Mice fed with CDAA diet showed increased infiltration of macrophages into the liver (Fig.1d). Specifically, CDAA diet-fed mice with hepatic tumours (T) exhibited a significantly higher level of liver inflammation (Fig.1e) as well as inflammatory monocytes in the peripheral blood (Supplemental Figure 4) than those without tumour (NT). This observation indicated that hepatocarcinogenesis may be accompanied by increased chronic inflammation that facilitates progression of liver tumour. This was further supported by the evidence that expression of inflammation markers IL6, IL1β and TNFα were increased particularly in the livers of CDAA diet-fed mice with hepatic tumours (Supplemental Figure 5). Increased apoptosis of hepatocytes in the livers of mice with hepatic tumours was observed (Fig.1f), while mice without tumours showed decreased cell death in hepatocytes (Fig.1g)
Hepatocarcinogenesis is accompanied by excessive inflammation and cell death of hepatocytes in CDAA diet-fed mice a) macromorphology of livers. Tumours could be observed after mice received CDAA diet for 56 and 72 weeks; tumor size increased with feeding time. CSAA diet did not cause formation of visible liver tumors in mice; b) histological analysis of livers. Mice developed liver tumors with various growth patterns. c) growth patterns in CDAA diet-fed mice. Columns depict individual mice; d) representative pictures of macrophage infiltration in the livers from CSAA diet-fed mice (CSAA), CDAA diet-fed mice with tumor (T) or without hepatic tumor (NT) after 56 weeks. E) analysis of grade of hepatic inflammation as represented by macrophage infiltration. Each dot depicts one individual mouse; f) increase in cell death of hepatocytes in CDAA diet-fed mice at 56 weeks; g) analysis of grade of cell death. Each dot depicts one individual mouse.
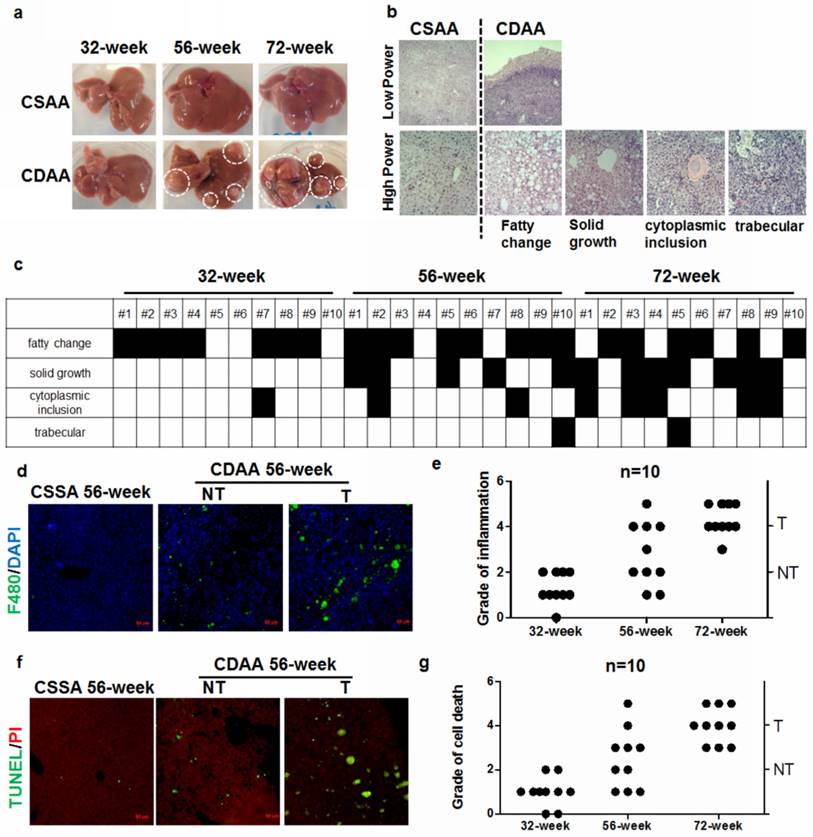
Peritumor hepatocytes present increased ER stress during hepatocarcinogenesis
Progressive ER stress was previously implicated in hepatocyte apoptosis in Acyl-Coenzye A oxidase 1-deficient mice during hepatocarcinogenesis [17]. CDAA diet progressively increased ER stress in the liver of mice compared with CSAA diet (CS). However, there was a remarkable increase in ER stress in mice with tumors (T) when compared with those without tumors (NT) (Fig. 2a). Analysis of expression of related genes revealed that livers with tumors possessed accelerated ER stress (Fig.2b), though non-tumor livers from CDAA diet-fed mice also had a moderate level of ER stress. In particular, when we isolated hepatocytes from livers with or without tumors, hepatocytes from livers with tumors had a significantly higher level of ER stress and apoptosis than those from non-tumor livers (Fig. 2c & d). These observations suggested that there were some molecular changes during the cancerous transformation of hepatocytes in CDAA diet-induced NAFLD mice. To identify the molecular events during hepatocarcinogenesis, microarray study was conducted to analyse the differential expression of hepatic murine microRNAs (miRNAs) that may regulate broad biological processes in hepatocytes. Differential expression profiles of miRNAs in hepatocytes from non-tumour or tumoral livers of CDAA diet-fed mice were collected (Fig. 2e). Subsets of miRNAs that were up-regulated or down-regulated in non-tumour livers compared with miRNAs expressed in livers with tumour were identified (Supplemental Table 2). 8 miRNAs exhibited a significant up-regulation in liver with tumours compared with non-tumour liver while 16 were significantly down-regulated (Fig. 2e). To understand the possible biological process involved, we computationally predicted potential targets of miRNAs with a significant difference between groups by target mining at miRDB (http://mirdb.org/miRDB/). The predicted genes with most abundance equivalence were then followed with GO analysis on DAVID (https://david.ncifcrf.gov/). The possible cellular components regulated by genes with potential change in expression were annotated with count threshold of three genes. The analysis revealed that both up- and down-regulated genes were mainly involved in the cellular processes associated with cytoplasmic vesicles and membrane delivery (Fig. 2f).
Hepatocarcinogenesis accelerates ER stress in the livers of mice a) mice with hepatic tumors exhibit increased expression of the ER stress markers GRP78 and CHOP at 56 weeks; b) expression of ER stress-related genes in hepatocytes from CSAA diet-fed mice (CSAA), CDAA diet-fed mice with tumors (T) or without hepatic tumors (NT) at 56 weeks; c) quantification of ER stress-related proteins in CDAA diet-fed mice with tumors (T) or without hepatic tumors (NT); d) quantification of DNA damage markers in CDAA diet-fed mice with (T) or without hepatic tumour (NT); e) miRNAs with significant differences in expression across different samples; f) functional annotation of predicted target genes of miRNAs by GO analysis. Target prediction was conducted by miRDB and functional annotation for cellular component of predicted genes was performed with GO analysis.
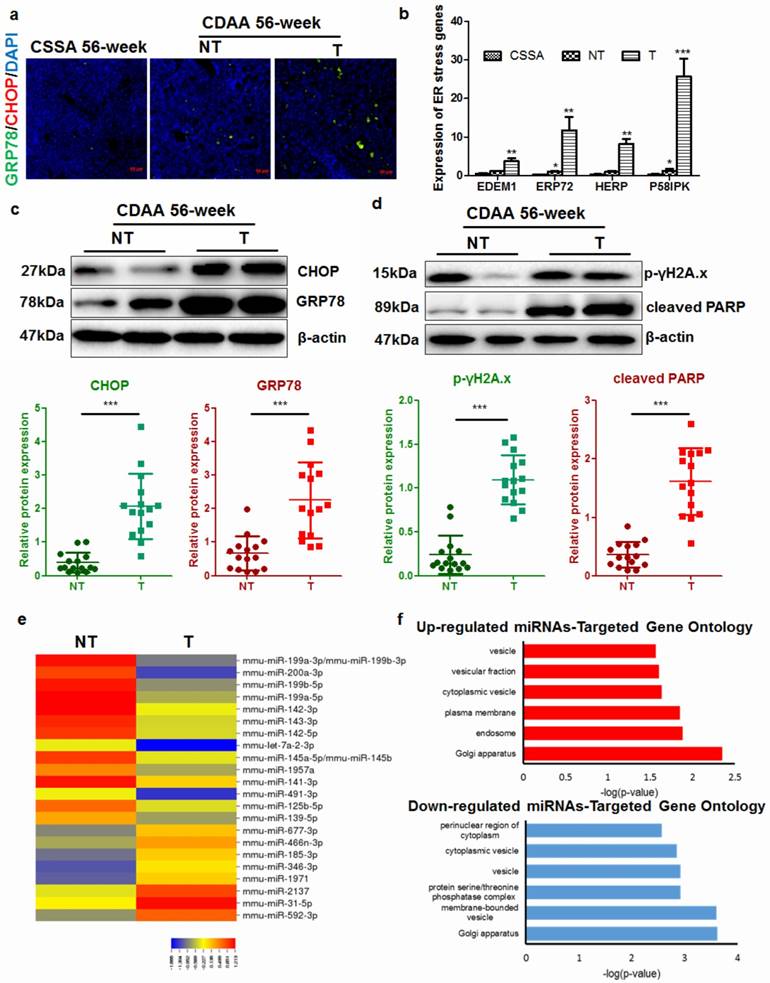
Autophagy is defective in hepatocytes during hepatic carcinogenesis
It has previously been shown that during ER stress, an important cellular process associated with cytoplasmic vesicles is the autophagy [18]. ER stress can trigger autophagy as a critical step in relieving cellular stress and prevent cell death and genomic instability [19]. Activation of unfolded protein response (UPR) could initiate autophagy to deposit misfolded proteins, an adaptive mechanism to prevent cell death in response to excessive intracellular ER stress [20]. We therefore assessed if autophagy was induced upon UPR in the liver of CDAA diet-fed mice. Intensive autophagy was observed in the liver of mice without hepatic tumor, however, autophagic punctuation was surprisingly blocked in those with tumor (Fig. 3a). Immunoblotting showed that hepatocytes from mice with tumors had defective autophagy, as evidenced by defective LC3 conversion as well as accumulation of p62 adaptor protein (Fig. 3b). This collectively showed that in hepatocytes from livers with tumors, the level of autophagy was comparatively lower than that in hepatocytes from livers without tumors, suggesting that carcinogenic process was accompanied with deficient autophagy. To further clarify the issue, we also checked the mRNA expression of LC3B in human samples and murine tissues. By analysing data from the Oncomine database, we found that expression of LC3B was not significantly changed in HCC tissues compared with adjacent non-tumor hepatic tissues (Supplemental Figure 6). Using quantitative real-time PCR (qRT-PCR), we confirmed that mRNA expression of LC3B was not significantly suppressed in hepatocytes from livers with tumors (Supplemental Figure 6). Previous study has shown that although transcription of LC3B may be induced to replenish the cytoplasmic level of LC3B protein that was turned over during excessive autophagy, transcription initiation of LC3B is not required for autophagy induction [21]. p62 is an adaptive protein involved in the autophagic deposition of misfolded proteins during ER stress [22]. Accumlation of p62, a gene that positively regulates the autophagy process, has been observed during diethylnitrosamine (DEN)-induced hepatocarcinogenesis in MAP1S-deficient mice [7]. Co-localization of p62 with ubiquitinated proteins facilitates autophagic degradation of these protein aggregates. We detected a high concentration of co-localized p62 and ubiquitin-tagged proteins in the hepatocytes from livers that harboured tumors (Fig. 3c). Interestingly, deposition of p62-ubuiqinated protein complex was specifically observed in the peritumor hepatocytes indicating its potential role in the cancerous transformation of the cells. Activation of autophagy was observed in the liver of CSAA diet-fed mice compared with mice fed with standard chow, possibly due to the moderate ER stress. Surprisingly, induction of autophagy was less observed in the hepatocytes of livers with tumors in CDAA diet-fed mice.
Autophagy was inhibited in hepatocytes during carcinogenesis a) reduced LC3 punctuation in the tumour-bearing livers of CDAA diet-fed mice at 56-weeks; b) defective activation of LC3 in CDAA diet-fed mice with liver tumors at 56-weeks leading to accumulation of p62; c)p62 was specifically accumulated in the peritumor areas and co-localized with ubiquitin-tagged proteins in CDAA diet-fed mice with hepatic tumors (T) compared with those without tumour (NT); d) scatter plots of expression of autophagy-related genes between different groups. Expression of autophagy-related genes had no significant changes in non-tumour livers compared with livers of CSAA diet-fed mice, but was significantly altered in the presence of tumors; e) changes in gene expression; expression of Atg9b was significantly reduced.
To better understand the defect of autophagy in hepatocarcinogenesis of CDAA diet-fed mice, we performed PCR profiler array to analyze the global regulation of autophagy-related genes in hepatocytes from livers with or without tumors (Fig. 3d). Subsets of down-regulated or up-regulated autophagy-related genes were identified. Only minor changes in expression of autophagy-related genes were observed in hepatocytes from CSAA diet-fed mice and CDAA diet-fed mice without tumors. However, significant changes in the expression of autophagy-related genes were detected in hepatocytes from mice bearing hepatic tumors. These expression changes included up-regulation of lysosome-related genes (Hsp90aa1, Lamp1, Hspa8) in response to accumulated ubiquitinated proteins and down-regulation of genes that encode proteins involved in autophagy regulation (Fig. 3e).
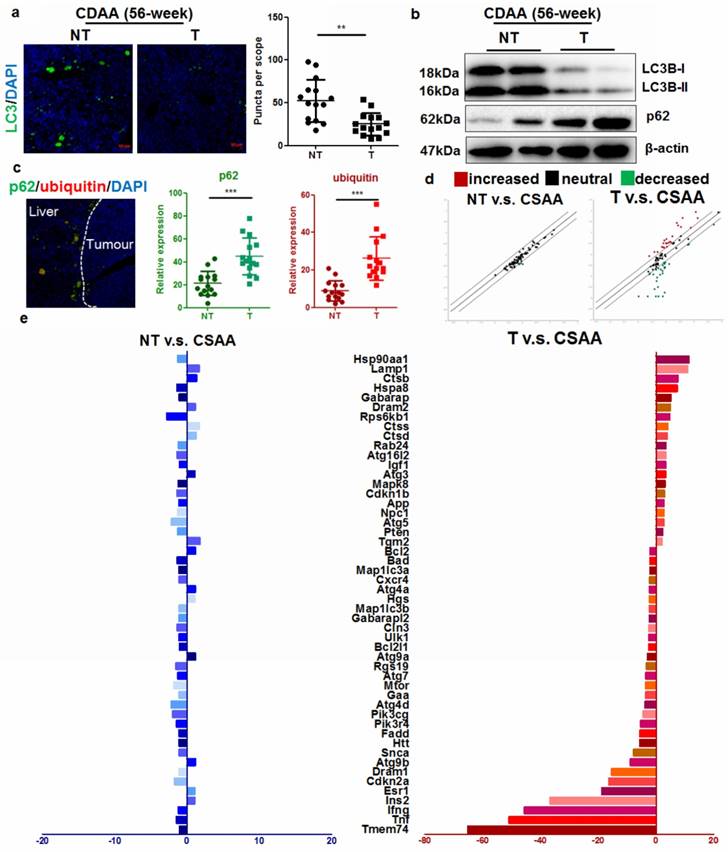
Loss of Atg9b is sufficient to increase ER stress and cell death in hepatocytes
Among the down-regulated genes, Atg9b was specifically involved in vacuole formation during autophagy initiation. Interestingly, expression of the other Atg9 homolog Atg9a was minimally changed. Compared with its expression in the livers of mice fed with CSAA diet, hepatic Atg9b was gradually repressed in the livers of CDAA diet-fed mice (Fig. 4a), but its expression was not significantly reduced (Fig.4a). The protein level of Atg9b was particularly reduced in hepatocytes from livers with tumors compared with those without tumors (Fig. 4b). We further analyzed the expression of Atg9b in human clinical specimens by qRT-PCR, The results revealed that expression of Atg9b was significantly higher in the normal liver than in the liver with HCC (Fig.4c). We then knock-downed Atg9b expression in the hepatocyte cell line AML12 using RNA interference. Immunoblotting analysis showed that RNA interference against Atg9b could successfully reduce Atg9b expression without affecting Atg9a (Fig.4d). Reduced expression of Atg9b caused significantly increased cell death induced by ER stress stimulus including 1μM thapsigargin (TG), 0.5μM calcium ionophore A23187, and 5μM tunicamycin (TM) (Fig. 4d). These results indicated that Atg9b may have a protective role in hepatocytes under ER stress stimulus. Although ER stress did not alter the expression of Atg9b in hepatocytes (Supplemental Figure 7), Atg9b silencing further induced an increasing expression of ER stress markers in hepatocytes exposed to the stimulus (Fig.4e). This may possibly be due to the accumulation of ubiquitinated proteins upon loss of Atg9b in hepatocytes as an ectopically expressed ubiquitinated protein CL1 was increased in Atg9b-deficient hepatocytes (Fig. 4f). These findings suggest that loss of Atg9b in hepatocytes during hepatic carcinogenesis is sufficient to cause decrease in autophagy that results in aggregation of ubiquitinated prone proteins and subsequent ER stress-associated cell death.
Atg9b deficiency causes accumulation of ubiquitinated protein aggregates and increases cell death in hepatocytes a) expression of Atg9b but not Atg9a was significantly suppressed in the livers of mice fed with CDAA diet; b) expression of Atg9b was particularly suppressed in CDAA diet-fed mice with hepatic tumors (T) but not those without tumors (NT); c) expression of Atg9b was reduced in human HCC samples compared with non-tumour tissues; d) suppression of Atg9b resulted in increased cell death of hepatocytes in the presence of ER stress inducers 1μM TG, 5μM TM or 0.5μM A23187 for 6 hours; e) suppression of Atg9b resulted in increased ER stress in hepatocytes; f)suppression of Atg9b resulted in accumulation of ubiquitinated CL1 proteins.
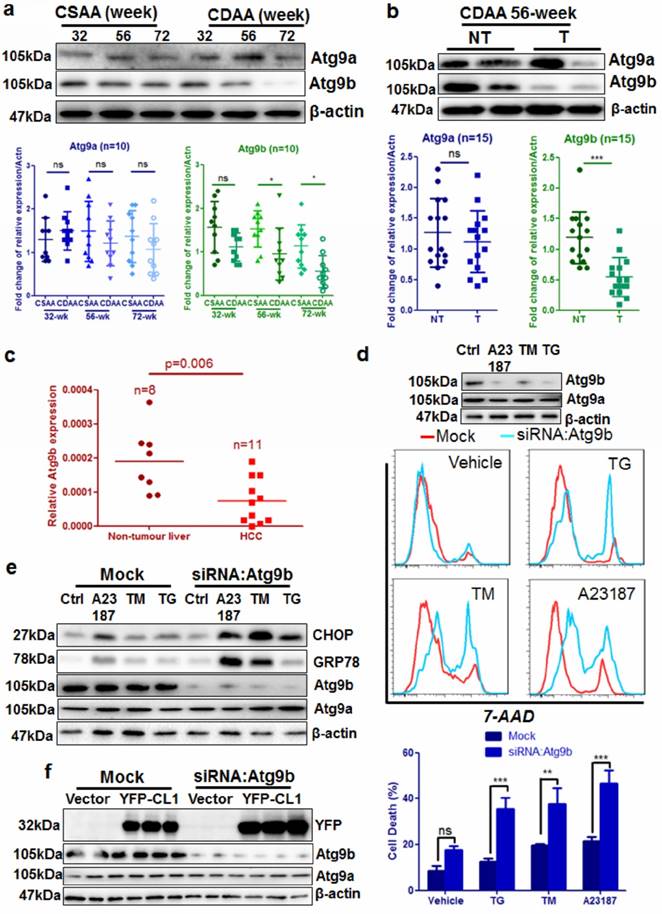
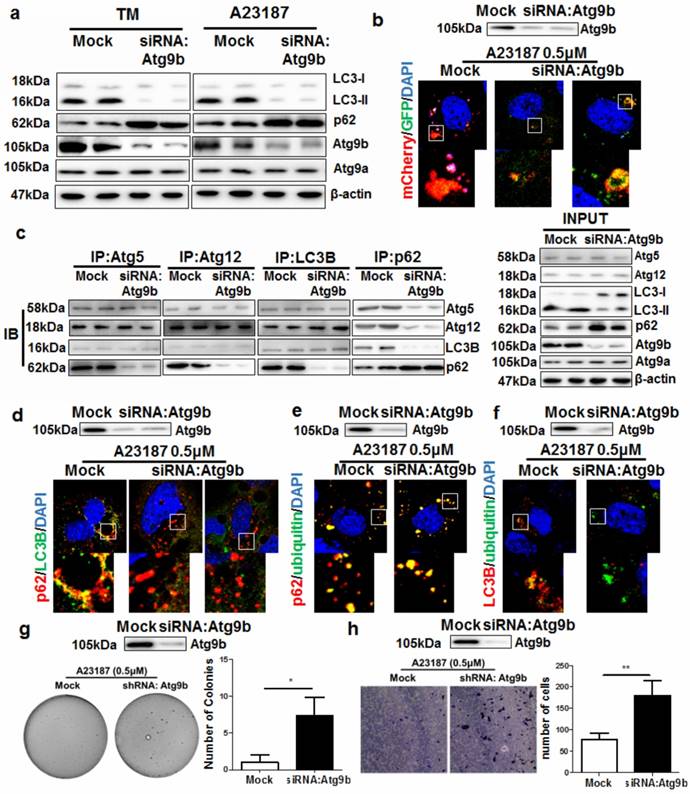
Atg9b loss leads to defects in autophagic clearance of misfolded proteins in hepatocytes
Autophagy is normally initiated by fusing the autophagosomes with lysosomes during ER stress to degrade damaged or misfolded proteins [23]. By treating AML12 cells with different ER stress stimuli including calcium ionophore A23187 and tunicamycin (TM), we observed a potent ER stress (Supplemental Figure 7) and autophagy initiation. Silencing Atg9b reduced the conversion of LC3 proteins leading to accumulation of p62 (Fig. 5a). To further elucidate the role of Atg9b in mediating induction of autophagy, we expressed LC3 proteins tagged with EGFP-mCherry in AML12 cells. Presentation of red but not green signals in A23187-treated AML12 cells revealed that cells underwent a complete process of autophagy; while expression of both green and red signal in cells with reduced Atg9b expression indicated blockade of autophagy (Fig.5b). EGFP signal was quenched in the acidic environment when LC3-formed autophagosomes were fused with lysosomes for degradation while the mCherry signal was pH insensitive. This result indicated that loss of Atg9b was sufficient to block the lysosomal degradation of the autophagic compartment. Suppression on Atg9b expression had minimal effect on the expression or cellular localization of LAMP-2, the docking protein expressing on lysosome which is necessary for the lysosomal degradation of the autophagic compartment (Supplemental Figure 8). To further understand which process of autophagy was blocked in Atg9b-deficient cells, we conducted endogenous co-immunoprecipitation assays. Pull down of Atg5, Atg12, LC3 or p62-containing protein complex was performed with corresponding antibodies, and protein-protein interactions were measured by immunoblotting. Interestingly, interactions of Atg5-Atg12, Atg5-LC3 and Atg12-LC3 remained normal when we blocked Atg9b expression in AML12 cells. Loss of Atg9b was sufficient to cause reduced recruitment of p62 to Atg5-Atg12-LC3 compartments (Fig.5c). This may indicate that p62 accumulation in Atg9b-deficient hepatic cells was partially, if not entirely, due to the inability of p62 to attach to phagophore membrane. This was further confirmed by the observation that p62 and LC3 were not co-localized in Atg9b-deficient cells (Fig. 5d). To understand that p62 may function as a cargo between ubiquitinated protein aggregates and LC3 during autophagic degradation of misfolded proteins [24], we examined if decreased Atg9b expression could block the associations of the protein complex. It was evident that p62 could be recruited to ubiquitinated proteins during ER stress regardless of the expression status of Atg9b (Fig. 5e). However, co-localization of LC3 and ubiquitinated proteins could not be observed when Atg9b was repressed (Fig. 5f) resulting in further accumulation of ubiquitinated proteins in cells undergoing ER stress. To investigate if knockdown of Atg9b would lead to cancerous transformation-like changes in hepatocytes, we examined the proliferation and migration of Atg9b-deficient hepatocytes using colony formation in soft agar and migration assays. We observed that suppression of Atg9b could increase formation of cell colonies in soft agar in the presence of sustained treatment with ER stress inducer A23187 (Fig. 5g). Increased migration away from the ER stress inducer A23187 was also detected in Atg9b-deficient cells (Fig. 5h). Thus, it appears plausible that repression of Atg9b can shift the non-cancerous cells to a more cancerous phenotype. These findings may further explain the essential role of Atg9b in autophagic clearance of ER stress-produced ubiquitinated proteins during hepatocarcinogenesis.
Suppression of Atg9b leads to failure in autophagic degradation of ubiquitinated protein aggregates a) suppression of Atg9b caused deficiency in autophagy initiation. Inhibiting Atg9b expression in hepatocytes led to failure in LC3 conversion and accumulation of p62; b) Inhibition of Atg9b reduced fusion of lysosomes with autophagosomes; c) Inhibition of Atg9b reduced association of p62 with proteins forming autophagic vacuoles; d) Atg9b suppression reduced co-localisation of p62 with LC3B; e) inhibition of Atg9b did not reduce co-localization of p62 with ubiquitinated-prone proteins; f) suppression of Atg9b led to failure of co-localization of LC3B with ubiquitinated protein aggregates. g) suppression of Atg9b led to anchorage-independent growth of A23187-treated AML12 cells on soft agar; h) suppression of Atg9b increased migratory ability of AML12 cells away from the stress inducer A23187.
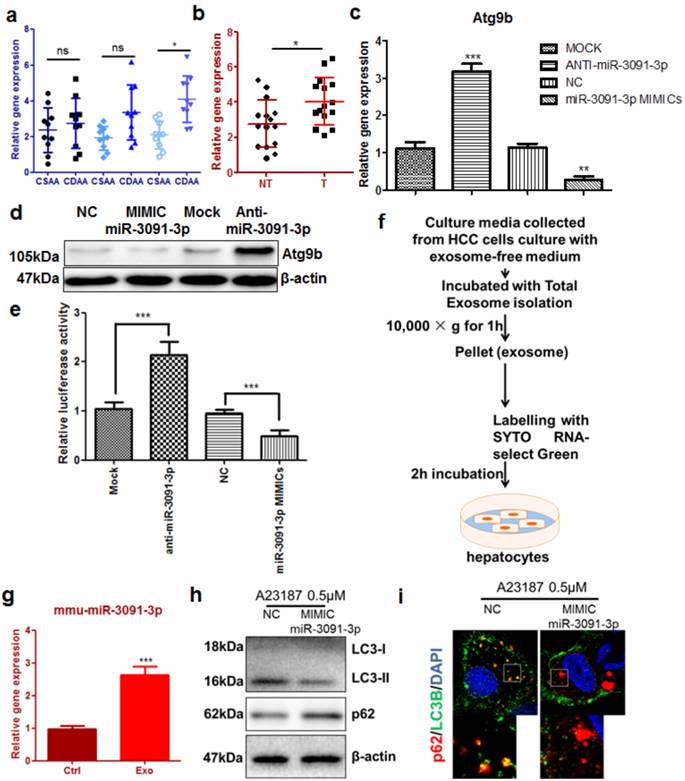
microRNA-3091-3p from tumor-derived exosomes can suppress Atg9b expression in hepatocytes
To better understand how Atg9b was aberrantly regulated in hepatocytes during hepatic carcinogenesis, we examined the expression of microRNAs that were predicted to target 3'UTRs of murine Atg9b. Interestingly, in CDAA diet-fed mice, hepatic mmu-miR-3091-3p was considerably elevated (Fig. 6a). In particular, this miRNA was significantly increased in hepatocytes from livers with tumors compared with those without tumors (Fig. 6b). Furthermore, expression of miR-3091-3p was higher in murine tumour cell lines (Supplemental Figure 9). Overexpression of mmu-miR-3091-3p in AML12 cells could suppress Atg9b while knockdown of the miRNA restored Atg9b at both mRNA and protein levels (Fig.6c&d). To prove that mmu-miR-3091-3p can target 3'UTR of murine Atg9b, we expressed luciferase-tagged 3'UTR of Atg9b in AML12 hepatocytes. Co-expression of the inhibitor against mmu-miR-3091-3p restored luciferase signal intensity while miRNA mimics further reduced 3'UTR transcription (Fig.6e). It was previously reported that hepatic cancer cells can secrete exosomes to shuttle molecules that facilitate the oncogenic transformation of normal hepatocytes [25]. We harvested exosomes from the conditioned medium of Hepa1-6 cells to determine their involvement in the regulation of Atg9b in hepatocytes (Fig.6f). Incubation of SYTO RNA-select Green-stained exosomes with AML12 cells reveals that normal hepatocytes can absorb tumour-derived exosomes (Supplemental Figure 10). Expression of miR-3091-3p was up-regulated in hepatocytes internalising tumour-derived exosomes (Fig.6g). Initiation of autophagy by ER stress inducer was attenuated by transfection with miR-3091-3p mimics. Furthermore, miR-3091-3p can reduce the formation of autophagic punctas, leading to accumulation of p62 (Fig. 6h&i). Taken together, these observations indicated that miR-3091-3p in tumour cell-derived exosomes may be able to suppress Atg9b expression during the cancerous transformation of hepatocytes.
Tumour cell-derived exosomes lead to Atg9b repression in hepatocytes a) expression of miR-3091-3p was induced in liver of mice with CDAA diet; b) expression of miR-3091-3p was significantly elevated in CDAA diet-fed mice with hepatic tumors (T) compared with those without tumors (NT); c) miR-3091-3p reduced mRNA expression of Atg9b in murine hepatocytes which were transfected with either mimics or inhibitor of miR-3091-3p; d) mimics of miR-3091-3p reduced Atg9b protein expression while inhibitor of miR-3091-3p restored Atg9b protein; e) miR-3091-3p bound 3'UTR of Atg9b mRNA; f) protocol of isolation and staining of tumor cells-derived exosomes; g) addition of exosomes induced expression of miR-3091-3p in hepatocytes; h) miR-3091-3p mimics could suppress LC3 conversion and accumulation of p62 in the presence of A23187; i) miR-3091-3p mimics could reduce LC3 punctuation leading to increased intracellular p62 level.
Discussion
In this study, we observed that excessive ER stress was a distinctive feature of hepatocytes from livers with tumours, which showed an elevated expression of ER stress-related proteins such as GRP78. This observation could reflect the clinical characteristics of the early stage of HCCs, as expression of GRP78 mRNA had a positive correlation with staging of HCC as well as ER stress response in hepatocytes [26]. Accelerated ER stress accompanies autophagy deficiency during carcinogenesis, leading to increased death of hepatocytes. We identified that an Atg9 homolog Atg9b was down-regulated in hepatocytes during carcinogenesis of the liver. Atg9 proteins were identified as the integral proteins that are required for the formation of autophagosomes and initiation of autophagy in mammalian cells [27]. Knockdown of Atg9b reduced LC3 lipidation as well as autophagic punctuations which appear to reflect the essential roles of Atg9 proteins in autophagy [28]. Interestingly, in our study, we did not observe any increase in the expression of Atg9b or its assembly into the PAS membrane during the ER stress. Although the expression of Atg9 proteins could be regulated via a transcriptional mechanism [29], the function of Atg9 proteins is dependent on a transient and dynamic interaction with autophagosomes [30]. This activity of Atg9 proteins requires a series of post-transcriptional modifications [31], but a reduction in Atg9 expression leads to failure of autophagosome formation and autolysosome compartmentalisation [32]. Atg9 protein is essential in autophagy induced by oxidative stress because of its ability to function as a source for autophagosome assembly [33]. We found that cells with reduced expression of Atg9b underwent exacerbated ER stress and increased UPR. Autophagy for the quality control of ER was independent of UPR. On the contrary, Atg9 proteins were necessary for ER stress-related protein deposition via autophagy-lysosome pathway [34]. As a progressive ER stress was shown to largely contribute to hepatocarcinogenesis, our findings together with previous studies define the important role of Atg9b during liver carcinogenesis. Interestingly, consistent with our current data, a recent study showed that decreased expression of Atg9b but not its ortholog Atg9a could influence breast cancer and directly accelerated tumour progression [35]. This was the only published study showing difference of clinical significance of Atg9 homologs. The study also showed that expression of Atg9a was related to the risk of cancer metastasis while decreased Atg9b was related to the ability to protect the normal epithelial tissues [35], indicating that the molecular functions of Atg9a and Atg9b may not be exactly complementary and biologically equivalent.
We observed accumulation of p62 in hepatocytes in response to autophagy deficiency which was consistent with a previous study reporting the same in tumour cells [36]. The role of p62 in hepatocarcinogenesis has been extensively studied and was comprehensively reviewed in some recent publications [37, 38]. An Atg7/p62 DKO mice showed decreased liver tumoriogenesis supporting the notion that p62 accumulation can promote hepatocarcinogenesis [39]. Multiple pathways have been reported to be involved in this process. Accumulation of p62 can persistently activate Nrf2 and its downstream target genes, which in turn promote sustained anchorage-independent growth of HCC cells during hepatoma development [40]. Accumulation of p62 can also impair DNA damage response by binding to nuclear RNF168 and inhibiting histone H2A ubiquitination. Consequently, the DNA repair proteins such as BRCA1, RAP80, and Rad51 cannot be recruited to DNA double-strand breaks sites leading to genome instability and carcinogenesis [41].By activating NRF2, and mTORC1 and inducing c-Myc, p62 sustains the stressed tumour-initiating cells in HCC. p62 is necessary and sufficient for HCC induction in mice and is indicative of high recurrence of tumours after curative resection [42]. The elevated level of p62 in hepatocytes from livers with tumors compared to those without tumors may be due to a deficiency of autophagic degradation. It could also be due to a positive feedback loop of transcriptional activation of p62 in response to deposition of protein aggregates, asp62 has been shown to mediate their removal via autophagy to ensure genome stability [7, 22].
Our study indicated that p62 accumulation was probably due to the loss of assembly of protein aggregates in autophagic vacuoles in Atg9b-deficient cells. Since we did not observe dissociation of Atg5-Atg12-LC3 complex, accumulation of p62 could have been due to the loss of essential phagophores that mediate p62 docking on autophagic membranes. Whether lipidated LC3 has a direct association with p62 during autophagic protein degradation is so far controversial [43, 44]. Data from our study support the concept that Atg9b-driven phagophores may enable docking of both LC3 and p62 during autophagic protein degradation. Furthermore, accumulation of p62 may activate inflammation-related NF-κB signalling and induce production of ROS, which, in turn, leads to double-strained DNA break (DSB)-associated genome instability [37]. Transgenic mice overexpressing p62 were prone to tumorigenesis when challenged by carcinogens [45]. These oncogenic consequences of p62 accumulation in hepatocytes further suggest deficiency of autophagy by Atg9b loss may play a fundamental role in mediating hepatic carcinogenesis.
Tumour-derived exosomes selectivity target peritumour hepatocytes, , although the mechanism of this selectivity remains obscure. A recent study showed that the protein Vps4A, which is critical for exosome biogenesis and uptake, was down-regulated in human HCC tissue reducing the exosome uptake by HCC cells [46]. Another study reported the possibility that tumour cells, which have a distinct miRNA profile, could selectively transfer specific miRNAs, that do not favor their survival and proliferation, to exosomes [47]. This selectivity may allow tumour cells to maintain partial ability to induce autophagy, which is still important for their survival and tolerance to multiple environmental stresses. Another possible scenario is that, tumour-derived exosomes bring epigenetic factors that can alter the miRNA profile of recipient cells, which, in turn, regulate the cellular biological processes. The influence of exosomes on miRNA profile on recipient cells could be global and non-specific with the overall consequence of tumorigenesis [48].
In conclusion, we elucidated the important role of Atg9b in hepatocarcinogenesis. Atg9b was down-regulated in human HCC specimens and was particularly suppressed in hepatocytes during carcinogenesis. Down-regulation of Atg9b resulted in deficient autophagy, accumulation of p62 and increased ER stress-related inflammation, genome instability, and cell death of hepatocytes. In vitro silencing of Atg9b was sufficient to suppress autophagy in response to ER stress. Hepatocytes without cytoprotective autophagy were more susceptible to exacerbated ER stress caused by accumulation of ubiquitinated proteins and were vulnerable to cell death. Loss of Atg9b blocked recruitment of p62-associated ubiquitinated proteins towards autophagosome-lysosome degradation. Atg9b-driven phagophores likely facilitated docking of both LC3 and p62 to initiate autophagy-associated degradation. miR-3091-3p from tumour-derived exosomes, which were internalized by hepatocytes, could repress Atg9b expression. Thus, observations of this study extend our knowledge about regulation of autophagy during hepatocarcinogenesis.
Materials and Methods
Choline deficient, amino acid-defined (CDAA) hepatocarcinogenesis model in mice
Protocol of animal study was approved by the Committee on the Use of Live Animals in Teaching and Research (CULATR) of The University of Hong Kong. Mice were fed with CDAA diet for 32, 56, and 72 weeks, or with CSAA diets as controls which were identical to CDAA diets in every respect except that choline was not omitted from the formulation [15]. Animals were monthly weighted. At the end of each study, mice were sacrificed and subjected to biomedical analysis.
Plasmids, siRNAs, cells, and cell culture
pBABE-puro Plasmids expressing mCherry-EGFP-LC3B were obtained from Addgene (USA, item #22418). All siRNAs, miRNA mimics, and miRNA inhibitors were obtained from Qiagen (Germany). Murine hepatocyte cell line AML12 and murine HCC cell line Hepa1-6 were obtained from ATCC (USA). AML12 cells were maintained in Dulbecco's Modified Eagle's Medium (DMEM) and Ham's F12 Nutrient Mixture (F12) supplemented with 10% Fetal Bovine Serum (FBS), 1% antibiotics, 0.005 mg/ml insulin, 0.005 mg/ml transferrin, 5 ng/ml selenium, and 40 ng/ml dexamethasone. Hepa1-6 cells were cultured in DMEM medium supplemented with 10% FBS and 1% antibiotics. All cells were cultured under humidified condition at 37℃ with 5%CO2.
PCR array and data analysis
Murine Autophagy PCR array assay was conducted according to the manufacturer's instructions (Qiagen, Germany). Results were normalized by expression of B2m, Actb and Gusb genes in average arithmetically. Scatter plot of differential expression between two groups was generated based on normalized expression of genes.
On-Chip microarray
RNA samples were labelled with miRCURY LNA™ microRNA Hi-Power Labelling Kit (Exiqon, Denmark), Hy3™/Hy5™ and hybridized on the miRCURY LNA™ microRNA Array (7th Gen, Exiqon, Denmark) following a dual-color experimental design. Data analysis was conducted following normalization of the quantified signals (background corrected) using the global Lowess regression algorithm of Agilent.
Quantitative Real-time PCR
Total RNA was isolated using Trizol and cDNA was synthesized with robust kit (Takara, Japan) or miRNA-specific kit (Exiqon, Denmark). Gene expression was determined by quantitative real-time PCR (LightCycler 480, Roche, USA) with SYBR Green Probe (Takara, Japan) or LNA Probe (Exiqon, Demark) with gene-specific primers.
Co-immunoprecipitation
Cells were lysed with NF-40 cell lysis buffer (Life Technologies, USA) supplemented with cocktail proteinase inhibitor (Roche, USA). Cell lysate was then incubated with Protein G magnetic beads (Millipore, USA) pre-conjugated with specific antibody to pull down the protein complex.
Immunoblotting
Total proteins were separated by electrophoresis on an SDS-PAGE and then transferred to PVDF membrane (Bio-rad, USA). The membrane was then blocked with 5%BSA and incubated with specific primary antibodies overnight at 4℃, followed by incubation with corresponding secondary antibodies. Target blots were read under chemiluminescence (Biorad, USA) with ECL select as substrate (GE healthcare, UK).
Exosome isolation and labelling
Tumour cell-derived exosomes were collected from culture supernatants of Hepa1-6 cells. Briefly, cells were cultured in DMEM supplemented with exosomes-free FBS. Culture medium was collected and filtered, and exosomes were enriched with total exosome isolation reagent following the manufacturer's instructions (Life Technologies, USA). For exosome labelling, SYTO RNA select (Life Technologies, USA) was used. Labelled exosomes were added to the culture medium of AML12 cells and incubated 1.5 hr to observe exosome uptake by hepatocytes.
Soft Agar Assay
1,000 AML12 cells was seeded in 0.7% top agarose in DMEM/F12 medium supplemented with 0.5μM A23187 on top of 0.5% Agar base. Medium was replenished every 3 days for 21 days. At the end of study, cells were stained with 0.005% crystal violet in methanol for 2 hours and washed cell colonies were counted.
Migration assay
50,000 AML12 cells in serum free-DMEM/F12 medium were seeded onto the transwell chamber (8μm, Corning, USA). Receiving chamber was supplemented with DEMEM/F12 medium with 10% FBS. ER stress inducer A23187 was added to the transwell chamber with final concentration of 0.5μM. 6 hours after cell seeding, cells at the bottom of transwell chamber were fixed in 4% PFA and stained with 2% crystal violet. Number of cells were counted under light microscope.
Statistical Analysis
Results were presented in mean±standard deviation (SD). Statistical analysis was conducted with ONE-WAY ANOVA and p value less than 0.05 was considered significant statistically.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
The study was financially supported by grants from the research council of the University of Hong Kong (Project Codes: 104003422, 201511159159), the Research Grants Committee (RGC) of Hong Kong, HKSAR (Project Codes: 766211 and 17152116), Wong's Donation on Modern Oncology of Chinese Medicine (Project code: 200006276), Gala Family Trust (Project Code: 200007008), Government-Matching Grant Scheme (Project Code: 207060411), and National Natural Science Foundation of China (Project code 81302808). The authors would like to express thanks to Mr. Keith Wong, Ms. Cindy Lee, Mr. Alex Shek, and Faculty Core Facility for their technical supports.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Perlmutter DH. Autophagic disposal of the aggregation-prone protein that causes liver inflammation and carcinogenesis in alpha-1-antitrypsin deficiency. Cell death and differentiation. 2009;16:39-45
2. Hidvegi T, Ewing M, Hale P, Dippold C, Beckett C, Kemp C. et al. An autophagy-enhancing drug promotes degradation of mutant alpha1-antitrypsin Z and reduces hepatic fibrosis. Science. 2010;329:229-32
3. Sun K, Guo XL, Zhao QD, Jing YY, Kou XR, Xie XQ. et al. Paradoxical role of autophagy in the dysplastic and tumor-forming stages of hepatocarcinoma development in rats. Cell Death Dis. 2013;4:e501
4. Menon S, Yecies JL, Zhang HH, Howell JJ, Nicholatos J, Harputlugil E. et al. Chronic activation of mTOR complex 1 is sufficient to cause hepatocellular carcinoma in mice. Sci Signal. 2012;5:ra24
5. Tian Y, Kuo CF, Sir D, Wang L, Govindarajan S, Petrovic LM. et al. Autophagy inhibits oxidative stress and tumor suppressors to exert its dual effect on hepatocarcinogenesis. Cell Death Differ. 2015;22:1025-34
6. Ni HM, Woolbright BL, Williams J, Copple B, Cui W, Luyendyk JP. et al. Nrf2 promotes the development of fibrosis and tumorigenesis in mice with defective hepatic autophagy. J Hepatol. 2014;61:617-25
7. Xie R, Wang F, McKeehan WL, Liu L. Autophagy enhanced by microtubule- and mitochondrion-associated MAP1S suppresses genome instability and hepatocarcinogenesis. Cancer Res. 2011;71:7537-46
8. Wang N, Feng Y. Elaborating the role of natural products-induced autophagy in cancer treatment: achievements and artifacts in the state of the art. Biomed Res Int. 2015;2015:934207
9. Lin H, Yan J, Wang Z, Hua F, Yu J, Sun W. et al. Loss of immunity-supported senescence enhances susceptibility to hepatocellular carcinogenesis and progression in Toll-like receptor 2-deficient mice. Hepatology. 2013;57:171-82
10. Lan SH, Wu SY, Zuchini R, Lin XZ, Su IJ, Tsai TF. et al. Autophagy suppresses tumorigenesis of hepatitis B virus-associated hepatocellular carcinoma through degradation of microRNA-224. Hepatology. 2014;59:505-17
11. Bao L, Chandra PK, Moroz K, Zhang X, Thung SN, Wu T. et al. Impaired autophagy response in human hepatocellular carcinoma. Exp Mol Pathol. 2014;96:149-54
12. Thomas HE, Mercer CA, Carnevalli LS, Park J, Andersen JB, Conner EA. et al. mTOR inhibitors synergize on regression, reversal of gene expression, and autophagy in hepatocellular carcinoma. Sci Transl Med. 2012;4:139ra84
13. Ding ZB, Shi YH, Zhou J, Qiu SJ, Xu Y, Dai Z. et al. Association of autophagy defect with a malignant phenotype and poor prognosis of hepatocellular carcinoma. Cancer Res. 2008;68:9167-75
14. Qiu DM, Wang GL, Chen L, Xu YY, He S, Cao XL. et al. The expression of beclin-1, an autophagic gene, in hepatocellular carcinoma associated with clinical pathological and prognostic significance. BMC Cancer. 2014;14:327
15. Wang B, Majumder S, Nuovo G, Kutay H, Volinia S, Patel T. et al. Role of microRNA-155 at early stages of hepatocarcinogenesis induced by choline-deficient and amino acid-defined diet in C57BL/6 mice. Hepatology. 2009;50:1152-61
16. Saitoh T, Fujita N, Hayashi T, Takahara K, Satoh T, Lee H. et al. Atg9a controls dsDNA-driven dynamic translocation of STING and the innate immune response. Proc Natl Acad Sci U S A. 2009;106:20842-6
17. Huang J, Viswakarma N, Yu S, Jia Y, Bai L, Vluggens A. et al. Progressive endoplasmic reticulum stress contributes to hepatocarcinogenesis in fatty acyl-CoA oxidase 1-deficient mice. The American journal of pathology. 2011;179:703-13
18. Ciechomska IA, Gabrusiewicz K, Szczepankiewicz AA, Kaminska B. Endoplasmic reticulum stress triggers autophagy in malignant glioma cells undergoing cyclosporine a-induced cell death. Oncogene. 2013;32:1518-29
19. Grigaravicius P, Kaminska E, Hubner CA, McKinnon PJ, von Deimling A, Frappart PO. Rint1 inactivation triggers genomic instability, ER stress and autophagy inhibition in the brain. Cell death and differentiation. 2016;23:454-68
20. Hoyer-Hansen M, Jaattela M. Connecting endoplasmic reticulum stress to autophagy by unfolded protein response and calcium. Cell death and differentiation. 2007;14:1576-82
21. Rouschop KM, van den Beucken T, Dubois L, Niessen H, Bussink J, Savelkouls K. et al. The unfolded protein response protects human tumor cells during hypoxia through regulation of the autophagy genes MAP1LC3B and ATG5. J Clin Invest. 2010;120:127-41
22. Johansen T, Lamark T. Selective autophagy mediated by autophagic adapter proteins. Autophagy. 2011;7:279-96
23. Rao RV, Bredesen DE. Misfolded proteins, endoplasmic reticulum stress and neurodegeneration. Curr Opin Cell Biol. 2004;16:653-62
24. Lee J, Giordano S, Zhang J. Autophagy, mitochondria and oxidative stress: cross-talk and redox signalling. Biochem J. 2012;441:523-40
25. He M, Qin H, Poon TC, Sze SC, Ding X, Co NN. et al. Hepatocellular carcinoma-derived exosomes promote motility of immortalized hepatocyte through transfer of oncogenic proteins and RNAs. Carcinogenesis. 2015;36:1008-18
26. Jiang X, Kanda T, Nakamoto S, Miyamura T, Wu S, Yokosuka O. Involvement of androgen receptor and glucose-regulated protein 78 kDa in human hepatocarcinogenesis. Exp Cell Res. 2014;323:326-36
27. He C, Baba M, Cao Y, Klionsky DJ. Self-interaction is critical for Atg9 transport and function at the phagophore assembly site during autophagy. Mol Biol Cell. 2008;19:5506-16
28. Chen D, Chen X, Li M, Zhang H, Ding WX, Yin XM. CCCP-Induced LC3 lipidation depends on Atg9 whereas FIP200/Atg13 and Beclin 1/Atg14 are dispensable. Biochem Biophys Res Commun. 2013;432:226-30
29. Jin M, Klionsky DJ. Transcriptional regulation of ATG9 by the Pho23-Rpd3 complex modulates the frequency of autophagosome formation. Autophagy. 2014;10:1681-2
30. Orsi A, Razi M, Dooley HC, Robinson D, Weston AE, Collinson LM. et al. Dynamic and transient interactions of Atg9 with autophagosomes, but not membrane integration, are required for autophagy. Mol Biol Cell. 2012;23:1860-73
31. Mack HI, Zheng B, Asara JM, Thomas SM. AMPK-dependent phosphorylation of ULK1 regulates ATG9 localization. Autophagy. 2012;8:1197-214
32. Bader CA, Shandala T, Ng YS, Johnson IR, Brooks DA. Atg9 is required for intraluminal vesicles in amphisomes and autolysosomes. Biol Open. 2015;4:1345-55
33. Tang HW, Liao HM, Peng WH, Lin HR, Chen CH, Chen GC. Atg9 interacts with dTRAF2/TRAF6 to regulate oxidative stress-induced JNK activation and autophagy induction. Dev Cell. 2013;27:489-503
34. Lipatova Z, Segev N. A Role for Macro-ER-Phagy in ER Quality Control. PLoS Genet. 2015;11:e1005390
35. Zhang X, Li C, Wang D, Chen Q, Li CL, Li HJ. Aberrant methylation of ATG2B, ATG4D, ATG9A and ATG9B CpG island promoter is associated with decreased mRNA expression in sporadic breast carcinoma. Gene. 2016;590:285-92
36. Mathew R, Karp CM, Beaudoin B, Vuong N, Chen G, Chen HY. et al. Autophagy suppresses tumorigenesis through elimination of p62. Cell. 2009;137:1062-75
37. Manley S, Williams JA, Ding WX. Role of p62/SQSTM1 in liver physiology and pathogenesis. Experimental biology and medicine. 2013;238:525-38
38. Komatsu M. Potential role of p62 in tumor development. Autophagy. 2011;7:1088-90
39. Takamura A, Komatsu M, Hara T, Sakamoto A, Kishi C, Waguri S. et al. Autophagy-deficient mice develop multiple liver tumors. Genes & development. 2011;25:795-800
40. Inami Y, Waguri S, Sakamoto A, Kouno T, Nakada K, Hino O. et al. Persistent activation of Nrf2 through p62 in hepatocellular carcinoma cells. The Journal of cell biology. 2011;193:275-84
41. Wang Y, Zhang N, Zhang L, Li R, Fu W, Ma K. et al. Autophagy Regulates Chromatin Ubiquitination in DNA Damage Response through Elimination of SQSTM1/p62. Molecular cell. 2016;63:34-48
42. Umemura A, He F, Taniguchi K, Nakagawa H, Yamachika S, Font-Burgada J. et al. p62, Upregulated during Preneoplasia, Induces Hepatocellular Carcinogenesis by Maintaining Survival of Stressed HCC-Initiating Cells. Cancer cell. 2016;29:935-48
43. Birgisdottir AB, Lamark T, Johansen T. The LIR motif - crucial for selective autophagy. J Cell Sci. 2013;126:3237-47
44. Itakura E, Mizushima N. p62 Targeting to the autophagosome formation site requires self-oligomerization but not LC3 binding. J Cell Biol. 2011;192:17-27
45. Kessler SM, Laggai S, Barghash A, Schultheiss CS, Lederer E, Artl M. et al. IMP2/p62 induces genomic instability and an aggressive hepatocellular carcinoma phenotype. Cell Death Dis. 2015;6:e1894
46. Wei JX, Lv LH, Wan YL, Cao Y, Li GL, Lin HM. et al. Vps4A functions as a tumor suppressor by regulating the secretion and uptake of exosomal microRNAs in human hepatoma cells. Hepatology. 2015;61:1284-94
47. Kogure T, Lin WL, Yan IK, Braconi C, Patel T. Intercellular nanovesicle-mediated microRNA transfer: a mechanism of environmental modulation of hepatocellular cancer cell growth. Hepatology. 2011;54:1237-48
48. Whiteside TL. Exosomes and tumor-mediated immune suppression. J Clin Invest. 2016;126:1216-23
Author contact
![]() Corresponding author: Feng Yibin. School of Chinese Medicine, The University of Hong Kong,10 Sassoon Road, Pokfulam, Hong Kong S.A.R, PR of China. Tel.: +852 25890482; Fax: +852 28725476; E-mail address: yfenghk.
Corresponding author: Feng Yibin. School of Chinese Medicine, The University of Hong Kong,10 Sassoon Road, Pokfulam, Hong Kong S.A.R, PR of China. Tel.: +852 25890482; Fax: +852 28725476; E-mail address: yfenghk.