Theranostics
13.3
Impact Factor
- Current Issue
- Advance Articles
- Volume 16; 2026
- Volume 15; 2025
- Volume 14; 2024
- Volume 13; 2023
- Volume 12; 2022
- Archive
- Cover Images
- Cover Suggestion
- Special Issues
Top
Introduction
Materials and Methods
Results
Discussion
Conclusions
Supplementary Material
Acknowledgements
References
Introduction
Materials and Methods
Results
Discussion
Conclusions
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2017; 7(9):2495-2508. doi:10.7150/thno.19863 This issue Cite
Research Paper
High-Yield Synthesis of Monomeric LMWP(CPP)-siRNA Covalent Conjugate for Effective Cytosolic Delivery of siRNA
Junxiao Ye1, 2, Ergang Liu1, 2, Junbo Gong2, Jianxin Wang3, Yongzhuo Huang4, Huining He1 ![]() , Victor C. Yang1, 5
, Victor C. Yang1, 5 ![]()
1. Tianjin Key Laboratory on Technologies Enabling Development of Clinical Therapeutics and Diagnostics, School of Pharmacy, Tianjin Medical University, Tianjin 300070, China;
2. Collaborative Innovation Center of Chemical Science and Chemical Engineering and Technology, Tianjin University, Tianjin 300072, China;
3. Department of Pharmaceutics, School of Pharmacy, Fudan University
Key Laboratory of Smart Drug Delivery, Ministry of Education & PLA, Shanghai 201201, China
4. Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai 201203, China;
5. Department of Pharmaceutical Sciences, College of Pharmacy, University of Michigan, Ann Arbor, Michigan 48109-1065, USA.
Received 2017-2-28; Accepted 2017-4-17; Published 2017-6-25
Citation:
Ye J, Liu E, Gong J, Wang J, Huang Y, He H, Yang VC. High-Yield Synthesis of Monomeric LMWP(CPP)-siRNA Covalent Conjugate for Effective Cytosolic Delivery of siRNA. Theranostics 2017; 7(9):2495-2508. doi:10.7150/thno.19863. https://www.thno.org/v07p2495.htm
Other stylesAbstract

Because of the unparalleled efficiency and universal utility in treating a variety of disease types, siRNA agents have evolved as the future drug-of-choice. Yet, the inability of the polyanionic siRNA macromolecules to cross the cell membrane remains as the bottleneck of possible clinical applications. With the cell penetrating peptides (CPP) being discovered lately, the most effective tactic to achieve the highest intracellular siRNA delivery deems to be by covalently conjugating the drug to a CPP; for instance, the arginine-rich Tat or low molecular weight protamine (LMWP) peptides. However, construction of such a chemical conjugate has been referred by scientists in this field as the “Holy Grail” challenge due to self-assembly of the cationic CPP and anionic siRNA into insoluble aggregates that are deprived of the biological functions of both compounds. Based on the dynamic motion of PEG, we present herein a concise coupling strategy that is capable of permitting a high-yield synthesis of the cell-permeable, cytosol-dissociable LMWP-siRNA covalent conjugates. Cell culture assessment demonstrates that this chemical conjugate yields by far the most effective intracellular siRNA delivery and its corresponded gene-silencing activities. This work may offer a breakthrough advance towards realizing the clinical potential of all siRNA therapeutics and, presumably, most anionic macromolecular drugs such as anti-sense oligonucleotides, gene compounds, DNA vectors and proteins where conjugation with the CPP encounters with problems of aggregation and precipitation. To this end, the impact of this coupling technique is significant, far-reaching and wide-spread.
Keywords: cell penetrating peptide, siRNA delivery, covalent CPP-siRNA chemical conjugate, mass preparation.
Introduction
Since the Nobel-prize observation by Fire and Mello of double-stranded RNA (dsRNA)-induced RNA interference (RNAi) in 1998 [1, 2], such double-stranded short interfering RNA duplexes, termed siRNA, that can lead to gene-specific silencing have evolved as a new paradigm of powerful therapeutics. In principle, RNAi offers the potential to modulate any and all disease-related mRNAs, allowing personalized medical treatment of diseases of almost all types. In addition, because of the unparalleled prowess in regulating gene expression and capability in carrying out countless rounds of mRNA cleavage, siRNA has been recognized as the future choice of therapeutics in the treatment of diseases of genetic disorders, viral infections [3-5], cancer [6-11], etc. The IC50 values (i.e. the dose that causes 50% cell death) for siRNA are normally in the pica-molar range [12], comparing to the micro-molar range for conventional small anti-tumor agents such as doxorubicin [13], thus allowing these drugs to eradicate a tumor at significantly lower, by 6-8 orders of magnitudes, bioavailable concentrations at the target tissues. However, siRNAs are highly negatively charged (about -40 charges) large molecules (~13,300 Da), they cannot successfully diffuse across cell membrane to enter the cytosolic compartment where the RNA-induced silencing complex (RISC) machineries locate; because cell membranes are permeable to only small (<700 Da) and hydrophobic compounds [14-16]. Hence, for siRNA-induced RNAi to realize even a fraction of its therapeutic potential, an intracellular delivery system for the siRNA drugs must be established.
Recent discovery of the class of cell-penetrating peptides (CPPs), also termed peptide transduction domains (PTDs), offers hope of resolving this critical membrane barrier [16-18]. Via covalent linkage, CPPs were shown to be capable of delivering a wide variety of cargos into primary cells and most tissues in preclinical models [17, 19]. Ample evidence also demonstrated that even through electrostatic condensation of the polyanionic DNA or siRNA with the polycationic CPP was also capable of inducing reasonable intracellular uptake of these drugs via the mechanism of macropinocytosis [18-20]; albeit that CPP-mediated cell translocation of such non-covalently linked complexes were far less effective than the covalent conjugates [21]. Hence, the most ideal means in realizing the therapeutic potential of the siRNA drugs is by formulating a soluble, monomeric CPP-siRNA conjugate through a covalent yet cytosol-cleavable linkage, so that CPP can mediate a potent intracellular delivery of siRNA via its unique endocytosis- independent pathway, but is then detached from the drug to allow siRNA remain in the cytosol, executing its gene-silencing therapeutic functions by means of the RISC system.
Yet, direct covalent conjugation of the cationic CPP to anionic siRNA faces a host of daunting challenges, as these two substrates would self-assemble into insoluble ionic aggregates, resulting in charge neutralization and inactivation of both siRNA and CPP [22-28]. It has been recognized that the dense cationic arginine motifs on CPP are crucial for inducing cellular uptake, by engaging with the anionic components on the cell surface, and charge neutralization has been proven to inhibit CPP-mediated cell internalization [27]. Even if covalently linked monomeric CPP-siRNA chimera could be successfully synthesized, this conjugate might still lack cell transduction activity, due to the likelihood of forming intra-molecular hairpin structure between CPP and siRNA, causing their charge neutralization. Moreover, most CPPs are known to possess nuclear-localization properties [16, 29, 30], and would thus ferry the attached cargo into nucleus as opposed to the desired destination of cytosol where machineries of the RISC system locate. Indeed, an authoritative review by Dowdy and co-workers has referred the task of realizing a covalently linked yet cytosol-dissociable monomeric CPP-siRNA chemical conjugate as the “Holy Grail” challenge; apparently reflecting the intractable technical difficulties in avoiding the charge-induced substrate aggregation and in achieving a highly effective intracellular and, specifically, cytosolic delivery of the siRNA drug. [27].
As known the PEG polymer chain, due to its dynamic spatial movement, could shield its attached cargo from recognition and interaction by the host immune system, we developed an innovative strategy to formulate an 1:1 monomeric LMWP (note: a well-established CPP [31])-siRNA covalent conjugate via a cytosol-cleavable disulfide linkage (Fig, 1A), without confronting the charge-induced neutralization and aggregation by the two oppositely charged substrates. Presented herein were cell culture results that offered a proof-of-concept demonstration of the plausibility of this chemical conjugation method. Comparing with the charge-complexed CPP-siRNA aggregated product, this chemical conjugate displayed far superior efficacy in delivering fully functional siRNA into the cytosolic compartment in executing presumably the most operational gene-silencing event (Fig. 1B).
Materials and Methods
Materials
Anti EGFP siRNA-cysteine (Sequence: 5′-GGCUACGUCCAGGAGCGCACC-3′, 3′-UUCCGAUGCAGGUCCUCGCGU-5′) and negative control siRNA (Sequence: 5′-AGCAUGCAUGAGUACCCAGCC-3′, 3′-UUUCGUACGUACUCAUGGGUC-5′) were both synthesized by Guangzhou Ribobio Co., Ltd. A single strand oligonucleotide 2′-O-MOE (AS-ODN, sequence: 5′-GGCCAAACCTCGGCTTACCTGAAAT-3′) was synthesized by Shanghai GenePharma Co., Ltd. LMWP (VSRRRRGGRRRRRR) was synthesized according to our previously developed protocol [32, 33]. Heterobifunctional PEG derivatives (Maleimide PEG NHS Ester, MW 3500) were obtained from Jenkem technology Co., Ltd. N-succinimidyl-3-(2-pyridyldithiol) propionate (SPDP) and Dithiothreitol (DTT) were purchased from Thermo Fisher Scientific Inc. Fluorescein isothiocyanate isomer I (FITC), 4',6-diamidino-2-phenylindole (DAPI) and paraformaldehyde were from Sigma-Aldrich. Human breast cancer cell line MDA-MB-231 and EGFP stable transfected cell line MDA-MB-231-EGFP cells were kindly donated by Prof. Xiaoyue Tan (School of Medicine, Nankai University), where the former was used in cell uptake studies while the latter was applied for gene silencing efficacy study. All of the cell culture reagents were purchased from Invitrogen. The transfection agent Lipofector as well as the Diethy pyrocarbonate (DEPC) was both supplied by Beyotime Institute of Biotechnology. Rabbit monoclonal antibody to β-actin and EGFP antibodies, goat anti-rabbit IgG-HRP were all from Santa Cruz. Water was distilled and deionized. Unless otherwise stated, chemicals were obtained from Sinopharm chemical reagent Beijing Co., Ltd.
Synthesis of the Conjugates
As displayed in Fig. S1, LMWP was first linked with a short (~3,500 Da) PEG chain containing hetero-bifunctional activated ends of N-hydroxysuccinimide ester (NHS ester) and maleimide (Mal). Since LMWP possesses no -SH group and only one reactive -NH2 group at its N-terminal, it could be linked only to the NHS-end of the PEG polymer. Cysteine was then added to the LMWP-PEG-Mal conjugate thus synthesized to convert the maleimide group to a -NH2 end, via the formation of a thioester bond between the -SH group on cysteine and maleimide group on LMWP-PEG. The LMWP-PEG-NH2 conjugate thus prepared was then activated with SPDP and thiolated with DTT to yield a reactive LMWP-PEG-SH compound (Step A; Fig. S1). For coupling, the siRNA drug, pre- synthesized to contain an extra cysteine residue at the 5'-end of the sense strand, was added to the afore-activated LMWP-PEG-SH to yield the LMWP-PEG-S-S-siRNA final conjugate through the formation of a cytosol-cleavable disulfide (S-S) linkage (Step B; Fig. S1). As noted, PEG polymers possess low polydispersity in molecular weight and unique solubility in both aqueous and organic solvents [34, 35]. To synthesize the LMWP-linked single-strand RNA product, 2′-O-MOE, a randomly selected single-strand anti-sense oligodeoxynulceotide (AS-ODN) that was pre-synthesized to contain a cysteine residue at the 5'-end, was reacted with the LMWP-PEG-SH according to the same procedure described above.
Figure 1
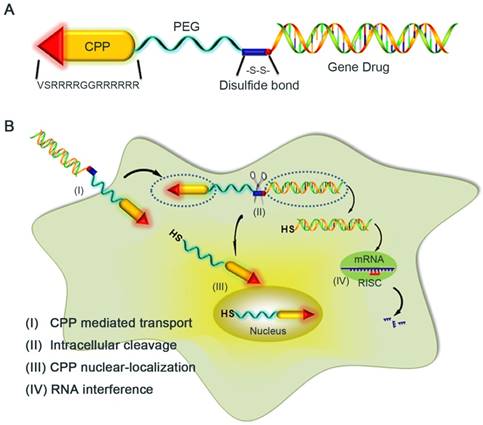
Schematic illustration of the reaction mechanism of the CPP-siRNA conjugates. A) The siRNA duplex with the 3'-end of the sense strand being modified with a sulfhydryl group was coupled, via a disulfide linkage, to a PEG-protected cell-penetrating peptide (LMWP), thereby avoiding charge-induced complexing/aggregation between siRNA and LMWP. B) Intracellular uptake of the LMWP-PEG-S-S-siRNA conjugate includes four separate events: (I) CPP-mediated cell transduction; (II) Cleavage of the disulfide bond resulting in the detachment of LMWP from siRNA; (III) Nucleus localization of LMWP; and (IV) Specific gene silencing by the cytosol-released siRNA via the RISC system.
Alternatively, the non-reducible LMWP-PEG-mal-S-siRNA conjugate was prepared by directly reacting LMWP-PEG-Mal compound with the above thiol-modified anti-EGFP siRNA.
Purification and Characterization of the Conjugates
Purification of the desired LMWP-PEG-S-S-siRNA conjugates was achieved via an anion exchange HiTrapTM DEAE FF (1ml, GE Healthcare, Sweden) chromatography using 20 mM sodium phosphate solution containning 10 mM EDTA at pH 7.2 (solution A) as the equilibrium buffer and the same solution containing 1M NaCl (solution B) as the elution buffer. The optimum elution conditions were as follows: Flow rate: 1 ml/min; Linear Gradient: 10-70% of solution B; Detection Wavelength: 260 nm. For comparison, the retention times on the DEAE column of LMWP-PEG-SH and siRNA were examined under identical conditions, with detection wavelength being set at 215nm for LMWP-PEG-SH.
Characterization of the purified conjugates was carried out using the matrix-assisted laser desorption/ionization time of fight (MALDI-TOF) mass spectrometry, according to a well-established procedure [36-38].
Preparation of the Lipofecter/siRNA Complex and the LMWP/siRNA Physical Mixtures
The Lipofecter/siRNA complex was produced by blending 5μl Lipofecter with 2μg siRNA according to the protocol provided by the supplier. The physical mixtures of LMWP-PEG/siRNA and LMWP/siRNA were prepared by adding equal moles of either LMWP-PEG or unmodified LMWP to siRNA (100 nM) prepared in the DEPC buffer containing 0.15 M NaCl and 10 mM EDTA at pH 7.2. The reaction mixtures were incubated for 5 minutes and then centrifuges at 1200 rpm to collect the final products.
Dynamic light scattering measurement
The particle sizes of the physical mixtures of LMWP/siRNA and LMWP-PEG/siRNA, as well as the LMWP-PEG-S-S-siRNA covalent conjugate were measured using Brookhaven Goniometer Light Scattering system, with deionized water as reference. Samples were prepared in light-scattering vials at the same concentration of siRNA. Dynamic light scattering was performed in high resistivity water as indicated below. All data points for dynamic light scattering were the average of three measurements performed on the same sample, and error bars represented the standard deviation.
Cell Uptake and Intracellular Localization Studies
Human breast cancer cell line MDA-MB-231 cells were utilized for these studies. Briefly, cells were grown at 37 °C in humidified atmosphere in L15 medium supplemented with 1% antibiotics, 1% NEAA and 10 % FBS, according to a previously established protocol [39]. Formulations containing FITC-labeled siRNA (including: PBS, native siRNA, Lipofecter/siRNA complex, LMWP-PEG/siRNA physical mixture, LMWP/siRNA physical mixture, the LMWP-PEG-S-S-siRNA (Conj-1) and LMWP-PEG-mal-S-siRNA (Conj-2) were then incubated the MDA-MB-231 cells with at 37 °C for 2 h. Cells were washed twice with PBS, fixed with 4% paraformaldehyde for 30 min, treated with the DAPI (2μg/ml) nucleic acid staining, and then subjected to confocal laser scanning microscopy analysis using an Olympus FV-300 laser scanning microscope operated with FLUOVIEW software (Olympus, Tokyo, Japan).
Intracellular Distribution of the Conjugates Using Raman Imaging
The MDA-MB-231 cells were utilized for the Raman imaging studies. In brief, cells seeded on glass slides were incubated overnight and then treated with either the bio-reducible Conj-1 or the non-reducible Conj-2 at an equal concentration (100 nM) at 37 °C for 2 h. Cells were then washed twice with PBS and fixed with 4% paraformaldehyde solution for 30 min at room temperature. Cell localization of these conjugates was analyzed by the intensity of the Raman laser light using the DXR Raman Spectrophotometer (Thermal Scientific, USA) in the presence of water. The excitation laser wavelength used was 532 nm, where the laser intensity was set at 2 mW. Laser light filtered by a plasma filter was focused onto the samples with a 50x objective (numerical aperture, N.A. = 0.9). A line map scan was performed along a single cell using a step size of 100 nm. Statistical averages were obtained from at least 5 images, with the exposure time being set at 10 second for each image. Spectra were acquired in the range from 700 to 1800 cm-1, and images were presented pertaining to the FITC band at 1172 cm-1.
Transfection and In vitro Inhibition on EGFP Expression by the siRNA Formulations
The EGFP stable transfected cell line MDA-MB-231-EGFP cells were used for these studies. Briefly, the cells were grown at 37 °C in humidified atmosphere in L15 medium supplemented with 1% antibiotics, 1% NEAA and 10 % FBS. In vitro inhibition on EGFP expression was evaluated by treating the cells with formulations containing PBS, negative control siRNA, naked siRNA, lipofecter/siRNA complex, LMWP/siRNA physical mixture, the bio-reducible Conj-1, or the non-reducible Conj-2). The concentration of siRNA was maintained at 50 nM in all of these formulations. The treated cells were incubated for 12h, followed by the replacement with fresh culture media and further incubation for up to 3 days at 37 °C. The siRNA gene silencing down effect were assessed by the inhibition on EGFP expression, using different methodologies including confocal microscopy, FACS, fluorescence intensity and Western-blot analysis. Experimental details were described in each of the following sections.
In vitro Confocal Laser Scanning Microscopy
MDA-MB-231-EGFP cells were seeded at 2×104 cells per well on a glass cover slip for 24 h. After transfection using the aforementioned procedure, cells were rinsed with PBS, fixed with 4% paraformaldehyde solution for 30min at room temperature, and then washed three times with PBS. The media was replaced with 2 mM DAPI (Sigma, USA) Hanks balanced salt solution to stain the nuclei for 10 min, and the cover slip was then placed on a sticky microscopy slide in the presence of the antifade mounting medium (Genemed, USA). The cells were imaged immediately using the Olympus FV-300 laser scanning confocal microscope which was equipped with the FLUOVIEW software (Olympus, Tokyo, Japan).
Fluorescence Intensity Studies
After transfection by using the aforementioned procedure, cells were washed twice with PBS and then lysed on ice with 200μl cell lysis buffer containing 20mM Tris, 150mM NaCl,1% Triton X-100, and 1mM PMSF at pH=7.5. The cell lysate was then centrifuged at 10,000g for 10 min at 4℃ and the supernatant was collected. All of the samples were adjusted to the same protein concentration using the BCA protein detection kit to avoid heterogeneity in cell density among the treated groups. The EGFP fluorescence intensity in the supernatant was recorded using a microplate reader (SpectraMax M3, Molecular Devices, California USA), with the excitation and emission wavelength being set at 488 nm and at 507 nm, respectively.
Fluorescence-Activated Cell Sorter Analysis of EGFP Expression
The flow cytometry (BD Biosciences, Bradford, MA) method was used to quantify the EGFP expression in the MDA-MB-231-EGFP cells treated with test formulations. The treated cells were washed twice with PBS and then fixed with 4% paraformaldehyde solution. Expression of EGFP in the MDA-MB-231 cells was analyzed with using the BD FACS Canto II flow cytometry equipped with the FlowJo Software.
Western-Blotting Analysis
Transfected cells were lysed (n=5) and quantified for the EGFP expression using the western-blot method and β-actin as the internal reference, according to a well-established procedure [40, 41]. The gene down-regulate efficiency was quantified by densitometry (Image J 1.48u, National Institutes of Health, USA).
Statistical Analysis
Data were collected from three separate experiments and expressed as the mean ± standard deviation (SD). Differences between groups were analyzed using unpaired two-tailed Student's t-test. A P-value of <0.05 was considered to be statistically significant.
Results
The double stranded anti-EGFP siRNA, (5′-GGCUACGUCCAGGAGCGCACC-3′, 3′-UUCCGAUGCAGGUCCUCGCGU-5′), was selected as the primary representative siRNA drug. To validate the utility of the conjugation method towards single stranded RNA, however, a randomly selected anti-sense oligodeoxynucleotide (AS-ODN), 2′-O-MOE (5′-GGCCAAACCTCGGCTTACCTGAAAT-3′), was also examined. Alternatively, the LMWP peptide (VSRRRRRRGGRRRR) developed in our laboratory, which was proven to possess potent cell-penetrating ability [32, 33, 42], was selected as the model CPP based on several unmatched advantages. First, among all of the existing CPPs, LMWP was the only natural product derived directly from native protamine by enzymatic digestion [43], and was also suitable for mass production/manufacturing. Secondly, the toxicological profiles of LMWP were already established [7]. In vivo results showed that LMWP possessed significantly reduced antigenicity, mutagenicity and complement activating activity, when comparing with native protamine; a FDA-approved drug currently in clinical use [44-46].
Synthesis, Purification and Characterization of the LMWP-PEG-S-S-siRNA Conjugate
The proposed conjugation method was illustrated in the Supplementary Section (Fig. S1). In brief, the strategy was based on the unique and unparalleled dynamic movement of the PEG polymer and its yielded shielding effect on the charged molecules to override all of the aforementioned obstacles [34, 35]. As noted, the hydrated state of the PEG chain was demonstrated to possess an unparalleled spatial dynamic mobility thereby enhancing the solubility of its linked compound and diminishing it from aggregation and detection by the host immune system [34]. In agreement with these reported observations, our results showed that while strong charge-induced aggregation occurred when unmodified LMWP was mixed with siRNA (the turbid solution; Fig. 2A.1). In sharp contrast, the reaction mixture remained transparent when either the pegylated LMWP was physically mixed with siRNA (Fig. 2A.2) or following a successful synthesis of the final LMWP-PEG-S-S-siRNA chemical conjugates (Fig. 2A.3). These findings provide sound evidence to confirm our hypothesis: i.e. the PEG chain on LMWP-PEG would shield LMWP from charge-induced complexation with siRNA during the coupling process (Fig. 2A.2), and the extraordinarily dynamic spatial movement of PEG would also prevent the intra- or inter-molecular self-assembly event in the final LMWP-PEG-siRNA conjugates (Fig. 2A.3). Since both reactants involved in the coupling process contain only a single reactive -SH group, formation of 1:1 (siRNA: LMWP) covalently linked conjugates is therefore warranted. Analysis of the particle size of the reaction mixtures also offered strong support to the above findings. As shown in Fig. 2B.2, the average particle size of the mixture of native LMWP and siRNA was in the range between 300 - 400 nm, explicitly suggesting the formation of aggregated particles due to charge complexation. In sharp contrast, particle sizes of the physical mixture of LMWP-PEG and siRNA (Fig. 2B.3) as well as the final LMWP-PEG-S-S-siRNA conjugates (Fig. 2B.4) were almost non-detectable and similar to that of the ultra-pure water (Fig. 2B.1), reflecting masking of the electrostatic interaction between the two oppositely charged LMWP and siRNA by PEG. These results further confirmed that the final LMWP-PEG-S-S-siRNA conjugates existed in a hydrated and soluble state.
DEAE anion exchange chromatography was employed to purify the LMWP-PEG-S-S-siRNA conjugate, where the siRNA-containing products would bind to the DEAE column and subsequently be eluted using a gradient NaCl solutions. As seen in Fig. 2C, while LMWP-PEG-SH (detected at 215nm; Fig. 2C.1) and anti-EGFP siRNA (detected at 260nm; Fig. 2C.2) were eluted from the DEAE column at NaCl concentrations of 0 M and 0.4 M, due to the absence or presence of strong binding affinity towards DEAE, respectively, the LMWP-PEG-S-S-siRNA conjugate was eluted in-between of these two salt concentrations (0.2 M NaCl; the middle peak in Fig. 2C.3). However, the by-product of the reaction, siRNA-S-S-siRNA, was also eluted in this step at 0.5 M NaCl (the trailing peak in Fig. 2C.3), as it had a stronger polyanionic nature than native siRNA, thereby resulting stronger binding to DEAE.
The MALDI-TOF analysis of these products yielded a molecular weight of 5897.2 Da, 13158.8 Da and 18299.8 Da for LMWP-PEG-SH, siRNA and LMWP-PEG-S-S-siRNA, respectively. These results were in excellent agreement with the calculated molecular weights for these products, confirming the success of synthesizing the monomeric LMWP-PEG-S-S-siRNA conjugate. The recovery yield of the final chemical conjugate was about 30%. However, with an further optimized protocol and a bench purification process, our preliminary findings indicated that a final yield as high as 55% can be readily attained; indicating the suitability of this conjugation method for scaling-up purpose in mass manufacturing of the final LMWP-PEG-S-S-siRNA covalent conjugates.
In a separate sequence of experiments, we confirmed the utility of this conjugation method towards single-stranded RNA. The elution profiles and MALDI-TOF data for the LMWP-PEG-MOE (an AS-ODN) product were presented in the Supplementary Fig. S2 and Fig. S3, respectively. Consistent with findings observed above for the double-stranded anti-EGFP siRNA, the elution peaks of LMWP-PEG, MOE and LMWP-PEG-S-S-MOE occurred at 0 M, 0.4 M and 0.2 M NaCl, whereas their molecular weights, determined by MALDI-TOF, for these three compounds were 5897.2 Da, 6601.1 Da and 12355.5 Da, respectively. Compounding these findings suggested the universal applicability of this conjugation strategy towards all types of nucleic acid-based therapeutic agents.
Intracellular Uptake and Localization of the Test Conjugates
Cell uptake studies were carried out on MDA-MB-231 cells using confocal microscope. Six samples including: (1) PBS, (2) anti-EGFP siRNA alone, (3) Lipofecter and anti-EGFP siRNA complex, (4) physical mixture (molar ratio: 1:1) of LMWP-PEG and anti-EGFP siRNA (5) physical mixture (molar ratio: 1:1) of LMWP and anti-EGFP siRNA, and (6) the LMWP-PEG-S-S-anti-EGFP siRNA conjugate were prepared and subsequently examined. As expected, confocal images (Fig. 3A) showed insignificant cell uptake of the FITC-labeled anti-EGFP siRNA (Sample #2), as the fluorescence intensity (FI) (Fig. 2B) inside the cells (FI = 23.3) was almost identical to the background intensity (FI = 4.7) shown by the PBS buffer; primarily due to the lack of cell internalizing ability by the anti-EGFP siRNA alone. On the other hand, both the Lipofecter and anti-EGFP siRNA complex (Sample #3) as well as the physical mixture of LMWP-PEG and anti-EGFP siRNA (Sample #4) displayed slightly improved (over Sample #2) yet still fairly weak cellular FITC intensities (FI = 90.7 & 78.0, respectively; Fig. 3B), suggesting the capable yet ineffective cell internalization by these two samples. Obviously, the lack of cell uptake of Sample #4 was presumably due to the interference of the PEG moiety on LWMP-PEG that prohibited the formation of the cell-permeable LMWP/siRNA charge complexes. In agreement with our previous findings [31] and also literature reports by many other investigators [31, 35], non-covalent complexing of the anionic anti-EGFP siRNA with cationic LMWP (Sample #5) resulted in moderately successful CPP-mediated cell entry (FI = 509); albeit this FITC intensity was only 43% of that (FI = 1181) mediated by the LMWP-PEG-S-S-siRNA covalent conjugate (Sample #6). Overall, cellular uptake of the LMWP-PEG-S-S-siRNA conjugates was 50.6-fold higher than that displayed by native siRNA without LMWP modification, and 2.3-fold higher than that by charge complexing of siRNA with CPP - the most efficient method to date in delivering siRNA drugs into cells. These results confirmed two very important aspects of utilizing our conjugation strategy in cellular delivery of siRNA. First, under the same CPP concentration and an equal molar ratio of CPP vs siRNA, the covalent and monomeric (1:1) chemical conjugate (sample #6) would yield a far more effective, by over 2 folds higher, intracellular siRNA uptake than that by the charge-associated aggregates produced via the conventional method of physically mixing CPP with siRNA (sample #5).
Figure 2
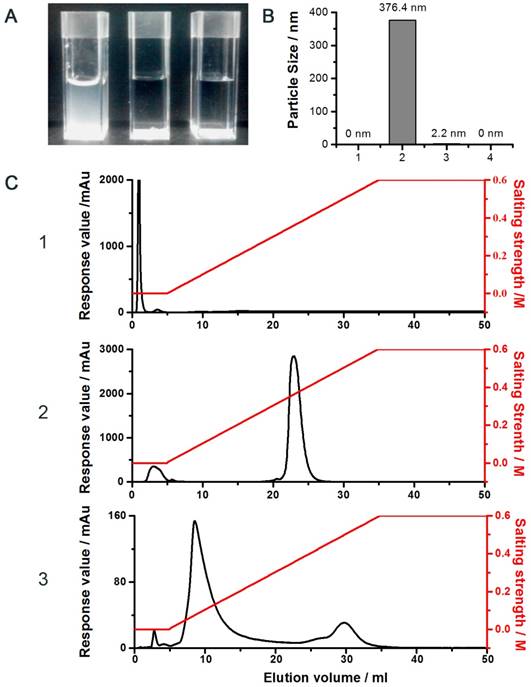
Characterization of the conjugate. A) Photographs of the reaction mixtures of: (1) unmodified siRNA and LMWP, (2) siRNA and LMWP-PEG, and (3) the LMWP-PEG-siRNA conjugate; B) Particle size analysis of: (1) pure water, (2) physical mixture of unmodified LMWP and siRNA, (3) physical mixture of LMWP-PEG and siRNA, and (4) the LMWP-PEG-S-S-siRNA conjugate; C) DEAE chromatograms of: (1) LMWP-PEG-SH, (2) native siRNA, and (3) the LMWP-PEG-S-S-siRNA conjugates.
Figure 3
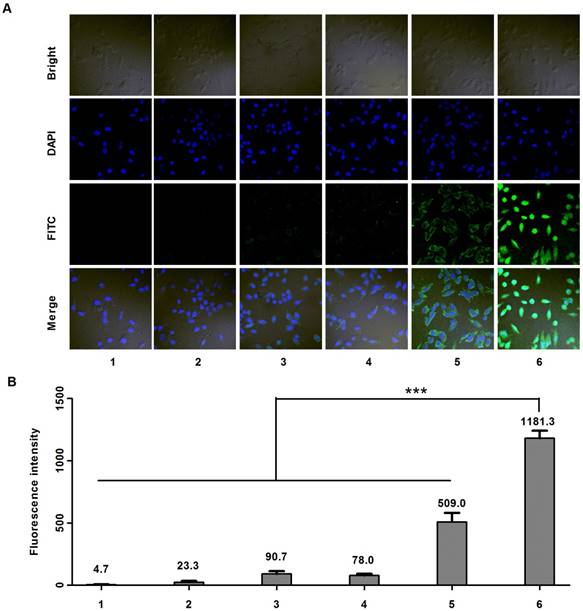
Cell uptake studies carried out on MDA-MB-231 cells using FITC-labeled anti-EGFP siRNA. A) Confocal microscopy; and B) Fluorescence intensity results of samples containing: (1) PBS, (2) anti-EGFP siRNA alone, (3) Lipofecter and anti-EGFP siRNA complex, (4) physical mixture (molar ratio: 1:1) of LMWP-PEG and anti-EGFP siRNA, (5) physical mixture (molar ratio: 1:1) of LMWP and anti-EGFP siRNA, and (6) the LMWP-PEG-S-S-anti-EGFP siRNA conjugate. For comparison, LMWP concentration and molar ratio of LMWP vs. anti-EFGP siRNA employed in all of these studies were maintained identical. (n=5, p<0.001)
Secondly, our conjugation strategy apparently eliminated the formation of the charge-induced intra-molecular hairpin structure and inter-molecular aggregation between CPP and siRNA or, otherwise the LMWP-siRNA chemical conjugate would not be able to internalize cells due to charge neutralization of the cell-penetrating arginine motifs on LMWP. Consistently, the covalent conjugate containing the anti-sense drug (i.e. LMWP-PEG-S-S-MOE) displayed similar intracellular uptake efficiency (Fig. S4) when compared with the conjugate containing the anti-EGFP siRNA (i.e. LMWP-PEG-S-S-siRNA).
It was known that many CPPs including TAT and LMWP displayed nucleus localization preference [47-49]. Yet, the RISC machineries required for siRNA to carry out the specific gene silencing functions resided in the cytosol. To magnify the partitioning of siRNA in the cytosol as opposed to the nucleus, we purposely connected siRNA and LMWP via a disulfide linkage, thereby once internalizing the cells via CPP-mediated trans-membrane function, this -S-S- bond would be broken automatically due to the elevated levels of reductase and glutathion activities in the cytosol. To verify this aspect, a comparison cell localization study was carried out by using LMWP-siRNA conjugates linked with a cytosol-cleavable disulfide bond (LMWP-PEG-S-S-siRNA; termed Conj-1) or a non-cleavable maleimide-sulfide bond (LMWP-PEG-Mal-S-siRNA; termed Conj-2). Chemical structures of these two conjugates were illustrated in Fig. 4A. It was expected that the LMWP-PEG-S-S-siRNA conjugate (Conj-1) would be reduced in the cytosol into two components, LMWP-PEG and free siRNA; of which the former would further internalize the nucleus whereas the latter would retain in the cytosol. In contrast, the LMWP-PEG-Mal-S-siRNA conjugate (Conj-2) would stay intact in the cytosol and eventually be partitioning, as a whole, into the nucleus phase, due to the preferable nucleus localization behaviour of LMWP. Confocal images in Fig. 4B revealed that the fluorescent intensities of these two conjugates in the cells were almost identical, suggesting that both conjugates possessed a similar cell internalization ability and were thus taken by the MDA-MB-231 cells in an equally effective manner. Nevertheless, the exact locations of these two conjugates could not be clearly identified, as the cells appeared to be too bright due to the high power (60μW) of the laser beam employed in this study. To address this issue, the laser power was reduced to 20μW to help visually examine cell distribution of these two conjugates more precisely. As shown by the enlarged confocal images in Fig. 4C, after 2h of incubation Conj-1 displayed a higher partitioning ratio in the cytosol over the nucleus, owing to dissociation of the disulfide bond by the cytoplasmic reducing environment. In contrast, the non-reducible Conj-2 accumulated more profoundly in the nucleus, due to the nucleus localization preference of the linked LMWP.
Raman images based on lateral Raman scans were generated to further differentiate cell localization of these two conjugates. Fig. 4D presented the Raman spectral images pertaining to FITC (1172 cm-1 band) [50-52], where the colors reflected the fluorescent intensities (blue: low intensity, red: high intensity) corresponding to the concentrations of FITC-labeled siRNA at each spot. As noted, the plot for Conj-1 in Fig. 4D revealed that high FITC intensities resided predominantly in cytoplasmic region with relatively low intensities being found in the nucleus. This finding confirmed our hypothesis that once entered the cells the disulfide bond in Conj-1 was rapidly hewn in the cytosol by chemical reduction, rendering the detached siRNA to stay in the cytosolic compartment. In contrast, for the non-reducible Conj-2 in Fig. 4D, FITC intensities were seen to spread over the entire cell, with high intensities being specifically identified in the nucleus, suggesting that the fluorescent-labeled siRNA not only entered the cells but, giving sufficient time, also destined preferably towards the nucleus compartment, apparently attributing to the nucleus localization function by the undetached LMWP. The corresponding simulated intensity 3D maps displayed in Fig. 4E, where X, Y axes represented the two-dimensional location in the cell and Z-axis denoted the fluorescent intensity, further revealed a distinctive difference in the cell distribution patterns of these two conjugates. As reflected by the locations of the intensity maxima of these two conjugates, deposition of Conj-1 occurred primarily in close vicinity of the membrane areas but was distance away from the nucleus, owing to a rapid detachment of siRNA from LMWP by the action of the cytoplasmic reducing agents (e.g. glutathione, reductase, etc.) immediately following its cellular uptake. It should be noted that a certain fraction of Conj-1 was able to escape cytosolic reduction and, with the undeleted LMWP unit, was able to translocate, as a whole conjugate body, into the nucleus; as evidenced by impermeable FITC-labeled siRNA in the nucleus. Distinctively, because of the presence of the non-reducible bond between siRNA and LMWP, most of Conj-2 remained stable in the cytosolic compartment. While a small portion of the conjugates was retained in the cytosol, the majority of Conj-2 continued to migrate into the nucleus via the nucleus-localization function of LMWP; as reflected by the distribution of FITC-labeled siRNA in both the cytosolic (minor) and nucleus (major) compartments.
Figure 4
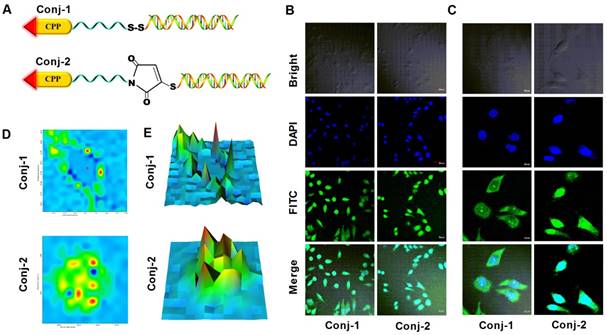
Comparative cell-localization studies on MDA-MB-231 cells of the bio-reducible (LMWP-PEG-S-S-siRNA; Conj-1) and non-reducible (LMWP-PEG-S-mal-siRNA; Conj-2) conjugates using FITC-labeled anti-EGFP siRNA. A) Chemical structures of the linkages employed in Conj-1 and Conj-2; B) Confocal images under a laser power of 60μW; C) Enlarged confocal images under a reduced laser power of 20μW; D) Confocal Raman images at the band of 1172 cm-1; and E) The corresponding simulated intensity 3D maps.
In Vitro Gene Silencing Efficacy on EGFP Expression
The gene silencing down-regulating effect by anti-EGFP siRNA was assessed using EGFP over-expressed MDA-MB-231-EGFP cells. In practice, the MDA-MB-231 cells transfected with EGFP genes evinced a green fluorescent color, and thus a diminution in green color intensity would symbolize the down regulation of EGFP expression and hence the gene-silencing efficacy of the tested anti-EGFP siRNA formulations (for details, see legend in Fig. 5).
Figure 5
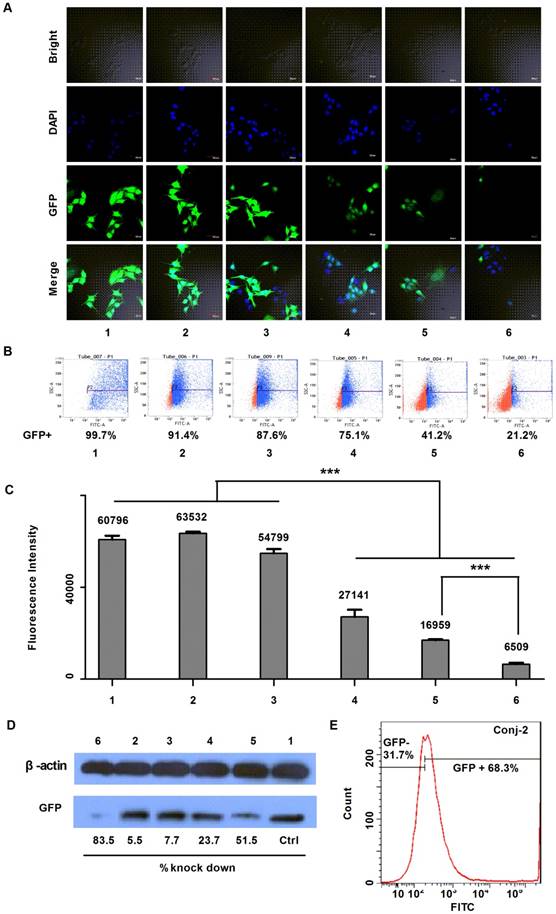
The gene silencing down effect by anti-EGFP siRNA on EGFP over-expressed steady transfection MDA-MB-231-EGFP cells. A) Confocal fluorescent images; B) FACS quantification results; C) Fluorescence intensity results of samples being excited at a wavelength of 488 nm and emitted at 507 nm (n=5, p<0.001); and D) Western blot analysis of EGFP levels after gene knockdown, determined 72 h after activation. The test samples in A to D are: (1) PBS, (2) siRNA negative control, (3) anti-EGFP siRNA alone, (4) Lipofecter and anti-EGFP siRNA complex, (5) Physical mixture (molar ratio: 1:1) of LMWP and anti-EGFP siRNA, and (6) the LMWP-PEG-S-S-anti-EGFP siRNA conjugate; and E) Gene-silencing activity of LMWP-PEG-S-Mal-siRNA conjugate. For comparison, LMWP concentration and molar ratio of LMWP vs anti-EFGP siRNA employed in all of these studies were maintained identical.
As displayed by the laser scanning microscopy images in Fig. 5A, both the siRNA negative control (Sample #2) and anti-EGFP siRNA alone (Sample #3) yielded negligible inhibition on EGFP expression, as the green color intensities in the MDA-MB-231-EGFP cells of these two samples remained similar to the background intensity of cells treated with PBS (Sample #1). This was apparently due to the lack of uptake of the cell-impermeable siRNA compounds. On the other hand, while both the Lipofecter/siRNA complex (Sample #4) and LMWP/siRNA physical mixture (Sample #5) displayed obvious inhibition on EGFP expression in the MDA-MB-231-EGFP cells, the most profound EGFP gene-silencing effect was observed in cells treated with the LMWP-PEG-S-S-siRNA conjugates (Sample #6); as judged by the degree of reduction in the fluorescent intensity in the treated cells. To further validate this event, the down-regulating effect on EGFP expression in MDA-MB-231-EGFP cells was assessed using flow cytometry. In agreement with confocal image results, flow cytometry measurements presented in Fig. 5B revealed that the levels of EGFP expression (Blue Dot: EGFP over-expressed cells; Red Dot: Cells with EGFP at low concentration or without EGFP) in the MDA-MB-231-EGFP cells treated with siRNA negative control (Sample #2) and naked anti-EGFP siRNA (Sample #3) were 91.4% and 87.6%, respectively, of the background FACS number shown by cells treated with PBS (Sample #1). Similarly, while both the Lipofecter/siRNA complex (Sample #4) and LMWP/siRNA physical mixture (Sample #5) unveiled noteworthy suppression on EGFP gene expression to 75.1% and 41.2% of the background level, respectively, the LMWP-PEG-S-S-siRNA conjugate (Sample #6) again yielded the most substantial down-regulation on EGFP expression, by reducing the FACS quantity to the least extent of 21.2%. The EGFP fluorescence intensity on the lysed MDA-MB-231-EGFP cells treated with different groups also offered consistent findings (Fig. 5C). While cells treated with PBS, siRNA negative control and naked siRNA (Samples #1, #2 & #3, respectively) exhibited virtually no significant difference in fluorescence intensity (FI), cells treated with the lipofecter/siRNA complex (Sample #4), the LMWP/siRNA physical mixture (Sample #5) and the LMWP-PEG-S-S-siRNA conjugates (Sample #6) all displayed statistically significant reduction on the FI values (27,141, 16,959 and 6,509, respectively); demonstrating an inhibition on EGFP expression by 55.4%, 72.1% and 89.3%, respectively; when comparing their FI numbers with the background total of 60,796 shown by Sample #1. It is important to note that the gene-silencing potency, judged by the remaining EGFP levels in the cells, was 2.6-fold more overwhelming in Sample #6 than in Sample #5 (10.7% vs 27.9%, respectively, as calculated from the FI data), consistent with the revealed results in Figure 3 that cell uptake of the LMWP-PEG-S-S-siRNA conjugates was 2.3-fold higher than that of the siRNA and CPP mixture made by charge complexation. Such results indicated that the difference on the amount of cytosol-delivered free siRNA molecules by these two different formulations, which were prepared under an identical amount of CPP and siRNA, were likely to be also around a ratio of ~2.5:1.0; suggesting that both preparations yielded similar degree of cytosol localization, and the better gene silencing effect of the LMWP-PEG-S-S-siRNA conjugates was primary due to their enhanced cell internalization ability over the charge complexes. In other words, the gene-silencing ability depends primarily on the amount of free siRNA molecules in the cytosol and not necessarily upon the stability of these two formulations in the cells. It should be specifically pointed out that up-till-now formulating the CPP/siRNA charge aggregates has been considered as the most competent strategy over all other existing methods in delivering siRNA (or DNA) compounds into cells.
The above findings were further validated by examining the EGFP expression in homogenized cell lysates supplied with protease inhibitors using the western-blot method. Among the aforementioned six test samples, the LMWP-PEG-S-S-siRNA conjugate once again yielded the strongest gene silencing effect on MDA-MB-231-EGFP cells (marked as Sample #6 in Fig. 5D), with the EGFP down-regulating ratio being about 83.5%; consistent with the fluorescent intensity and FACS analysis data. However, the charge-associated LMWP/siRNA aggregates (Sample #5; Fig. 5D) and cationic lipofecter/siRNA complex (Sample #4; Fig. 5D) also displayed comparatively low gene down-regulating efficiency, at a level of 51.5% and 23.7%, respectively. For comparison, flow cytometry examination of the non-reducible LMWP-PEG-S-Mal-siRNA (anti-EGFP siRNA) conjugate (Conj-2) was also carried out. As depicted in Fig. 5E, Conj-2 yielded a much weaker down-regulation on EGFP expression, reducing its level to 68.3% of the baseline total; as compared to that (21.2%) by the cytosol-reducible LMWP-PEG-S-S-siRNA (anti-EGFP siRNA) conjugate (Sample #6 in Fig. 5B). This finding demonstrated the importance of retaining the cell-delivered siRNA in the cytosolic compartment, should one want to achieve the most potent gene-silencing efficacy.
Discussion
The unmatched selectivity, efficiency, and potentially the universal utility in treating diseases of all types render the siRNA agents to evolve as the new generation of drug-of-choice. Nevertheless, owing to the inability of naked siRNA to cross the cell membrane barrier and induce RNA interfering response, developing methods that can lead to successful intracellular delivery of these agents becomes a formidable necessity. Recent discovery of the so-called cell-penetrating peptides (CPP) shed light of solving this dilemma. Via physical mixing or covalent conjugation of CPP with siRNA, both the charge-associated aggregates and covalent conjugates were shown to be capable of delivering the siRNA cargos into cells; albeit that the covalent conjugates was demonstrated to be far superior than the charge-linked aggregates in transducing siRNA into cells as well as in inducing potent gene-silencing activity. Despite continuous efforts and pursuit, however, formulation of the 1:1 CPP: siRNA chemical conjugate was still being considered by most experts as an insurmountable and, indeed, a “Holy Grail” task; owing to the self-assembly process that occurred concurrently between the highly cationic LMWP and anionic siRNA. As noted, formation of intra-molecular crosslinks or inter-molecular charge aggregates would instigate significant impairment on the biological functions of both CPP and siRNA.
Since the report of CPP-mediated cell transduction of macro-molecular cargos in 1999, till to-date there were only very few reports claiming the success in obtaining the desirable CPP-siRNA chemical conjugates [16, 23, 27, 53, 54]. Yet, these works all failed to demonstrate that the charge-induced self-aggregation event in their process was completely eliminated, or the yield of the final chemical product was actually meaningful. More specifically, none of these methods deemed capable of producing sufficient quantities of the final covalent conjugates that were essential for carrying out in vivo or clinical trials. Hence, one of the primary impacts of the presented conjugation strategy was that it might offer, for the first time, a “breakthrough” advance on mass synthesis of the functional CPP-siRNA covalent conjugates, thereby paving the road for future clinical translation of the siRNA therapeutics. The presence of a transparent fluid state during the conjugation process (Fig. 2A) and the particle size measurements (Fig. 2B) all provided indisputable evidence of completely avoiding the charge-induced aggregation during chemical synthesis as well as attaining the desirable monomeric LMWP-PEG-S-S-siRNA conjugates. Importantly, even under our current laboratory settings we were already able to attain a recovery yield of approximately 30% of the pure conjugate (results from Fig. 2).
Considering that the chromatographic purification method was limited for scale-up purpose, we actually took the advantages of the so-called “batch purification” strategy which was suitable for industry-scale product production. Results showed a convenient increase of the final yield of this chemical conjugate to over 50% from the initial reactants, making this method suitable for mass production, thus fulfilling the need of chemicals in conducting animal studies and clinical efficacy trials. Additionally, improving the efficiency of the coupling reaction and reducing the formation of the by-product siRNA-S-S-siRNA would also increase the final yield, which we intended to realize by optimizing the conjugation method.
Comparing with all of the CPP-aided siRNA delivery systems to date, data presented in this paper proved that formulating the covalent conjugate via our method deemed to be the most efficient means in delivering siRNA into cells. While cell uptake of both the bio-reducible and non-reducible conjugates (Conj-1 & Conj-2 in Fig. 4A) remained similar, as reflected by the presence of almost identical cell fluorescence intensities for both compounds (see Fig. 4B), these two chemical conjugates nevertheless yielded a 2.3-fold enhancement on intracellular siRNA delivery over that by the charge-linked LMWP/siRNA aggregates prepared via the conventional mixing method (i.e. by comparing the FI values in Sample #6 vs. Sample #5; see Fig. 3B). Yet, results related to Figure 5E also demonstrated that the gene-silencing potency did not rely solely on the level of cell uptake, but was also dependent on the localization of these aggregates/conjugates within the cells. As shown from the flow cytometry results, the presence of a disulfide linkage thereby permitting cytosolic detachment of siRNA from LMWP enabled the LMWP-PEG-S-S-siRNA conjugate (Conj-1) to be far more potent than the non-reducible LMWP-PEG-S-Mal-siRNA conjugate (Conj-2) in inducing RNAi response (the remaining EFGP levels in cells were 21.2% for Sample #6 in Fig. 5B versus 68.3% in Fig. 5E, respectively); although cell uptake of both conjugates observed in Fig. 4B appeared to be nearly identical. On the other hand, despite the fact that Conj-2 exhibited a significantly higher cell uptake than the LMWP/siRNA charge aggregates (refer to the FI values of Sample #6 versus Sample #5 in Fig. 3B), flow cytometry results nevertheless revealed a much weaker gene-silencing potency by Conj-2 than that by the LMWP/siRNA charge aggregates (68.3% versus 41.2%, according to the FACS results shown in Fig. 5E & 5B, respectively). This phenomenon could be accounted for in terms of the presence a higher nucleus partitioning ratio of the covalent conjugates than the charge-associated LMWP/siRNA aggregates, due to the nucleus localization preference caused by of the non-dissociable LMWP moiety on Conj-2.
Conclusions
In conclusion, in vitro findings confirmed that the proposed novel conjugation strategy would not only yield the most successful cellular uptake of the siRNA agents but also produce the highest gene-silencing therapeutic efficacy. Since this method was proven to possess the utility towards both single- or double-stranded RNA compounds, it could serve as a platform technology to realize clinical translation of virtually all nucleic acid-based drugs including therapeutic genes. Furthermore, because of the simplicity and robotic nature of the synthesis protocol of this conjugation method, it is envisioned that the technology could be readily transformed into a kit made of two cartridges, one comprises of a chemical-stabilized, pre-made LMWP-PEG-SH compound capable for cell-penetration, whereas the other consists of a selected siRNA (or DNA) drug pre-synthesized to contain a cysteine residue (and thus an active -SH functional group) at the 3' end of the sense strand and also stabilized with the addition of certain specific chemicals [55]. For synthesis, one could simply mingle these two cartridges to attain the final LMWP-PEG-S-S-siRNA conjugate, which in turns would produce the most effective cytosol-specific delivery of siRNA and subsequently the most potent RNA interference (RNAi) response. Lastly, it should be reiterated that, albeit the current study was performed only in cell culture, the capability of CPP-mediated intracellular delivery of large or charged cargos such as proteins, genes and drug carriers has already been thoroughly confirmed in animal studies [16-18]. Yet, the real bottleneck to achieve successful intracellular siRNA delivery in vivo lies in the inability to synthesize sufficient quantity of the CPP-siRNA covalent conjugates in vitro [27]. To this regard, the current study offers a breakthrough stride towards realizing clinical translation of the siRNA therapeutics. However, effective in vivo delivery of these conjugates requires a carefully designed delivery system that can protect the conjugates from proteolytic degradation thereby maintaining their in vivo stability, as well as enhance the tumor targeting efficiency and reduce the systemic toxic effects over normal tissues. To this regard, integration of these covalent conjugates with our ongoing research of designing a magnetic iron oxide nano-carrier (MION)-based delivery system is essential to achieve the milestone goal of realizing a MRI-guided, cocktail-type siRNA brain tumor therapy.
Supplementary Material
Supplementary figures.
Acknowledgements
This work was supported in part by the National Natural Science Foundation of China (NSFC) on Grants 81402856 and 81361140344 (A3 project). This research was also partially sponsored by National Key Research and Development Plan (2016YFE0119200) and Tianjin Municipal Science and Technology Commission (Grant 15JCYBJC28700) and the National Key Basic Research Program of China (Grant 2013CB932502).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Fire A, Xu S, Montgomery MK. et al. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature. 1998;391:806-11
2. Yang S, Chen Y, Ahmadie R. et al. Advancements in the field of intravaginal siRNA delivery. J Control Release. 2013;167:29-39
3. Thi EP, Mire CE, Lee AC. et al. Lipid nanoparticle siRNA treatment of Ebola-virus-Makona-infected nonhuman primates. Nature. 2015;521:362-5
4. Draz MS, Fang BA, Zhang P. et al. Nanoparticle-mediated systemic delivery of siRNA for treatment of cancers and viral infections. Theranostics. 2014;4:872-92
5. Van Rij RP, Andino R. The silent treatment: RNAi as a defense against virus infection in mammals. Trends Biotechnol. 2006;24:186-93
6. Shen J, Kim HC, Su H. et al. Cyclodextrin and polyethylenimine functionalized mesoporous silica nanoparticles for delivery of siRNA cancer therapeutics. Theranostics. 2014;4:487-97
7. Choi YS, Lee JY, Suh JS. et al. The systemic delivery of siRNAs by a cell penetrating peptide, low molecular weight protamine. Biomaterials. 2010;31:1429-43
8. Guo P, Yang J, Jia D. et al. ICAM-1-Targeted, Lcn2 siRNA-Encapsulating Liposomes are Potent Anti-angiogenic Agents for Triple Negative Breast Cancer. Theranostics. 2016;6:1-13
9. Lee J, Saw PE, Gujrati V. et al. Mono-arginine Cholesterol-based Small Lipid Nanoparticles as a Systemic siRNA Delivery Platform for Effective Cancer Therapy. Theranostics. 2016;6:192-203
10. Gavrilov K, Saltzman WM. Therapeutic siRNA: principles, challenges, and strategies. Yale J Biol Med. 2012;85:187-200
11. Fan B, Kang L, Chen L. et al. Systemic siRNA Delivery with a Dual pH-Responsive and Tumor-targeted Nanovector for Inhibiting Tumor Growth and Spontaneous Metastasis in Orthotopic Murine Model of Breast Carcinoma. Theranostics. 2017;7:357-76
12. Sakurai K, Amarzguioui M, Kim DH. et al. A role for human Dicer in pre-RISC loading of siRNAs. Nucleic Acids Res. 2011;39:1510-25
13. Vrudhula VM, Svensson HP, Senter PD. Cephalosporin derivatives of doxorubicin as prodrugs for activation by monoclonal antibody-beta-lactamase conjugates. J Med Chem. 1995;38:1380-5
14. Meade BR, Dowdy SF. The road to therapeutic RNA interference (RNAi): Tackling the 800 pound siRNA delivery gorilla. Discov Med. 2009;8:253-6
15. Meade BR, Gogoi K, Hamil AS. et al. Efficient delivery of RNAi prodrugs containing reversible charge-neutralizing phosphotriester backbone modifications. Nat Biotechnol. 2014;32:1256-61
16. Lonn P, Dowdy SF. Cationic PTD/CPP-mediated macromolecular delivery: charging into the cell. Expert Opin Drug Deliv. 2015:1-10
17. Fonseca SB, Pereira MP, Kelley SO. Recent advances in the use of cell-penetrating peptides for medical and biological applications. Adv Drug Deliv Rev. 2009;61:953-64
18. Shi NQ, Qi XR, Xiang B. et al. A survey on "Trojan Horse" peptides: opportunities, issues and controlled entry to "Troy". J Control Release. 2014;194:53-70
19. Sawant R, Torchilin V. Intracellular transduction using cell-penetrating peptides. Mol Biosyst. 2010;6:628-40
20. Freire JM, Rego de Figueiredo I, Valle J. et al. siRNA-cell-penetrating peptides complexes as a combinatorial therapy against chronic myeloid leukemia using BV173 cell line as model. J Control Release. 2017;245:127-36
21. Reischl D, Zimmer A. Drug delivery of siRNA therapeutics: potentials and limits of nanosystems. Nanomedicine. 2009;5:8-20
22. Endoh T, Ohtsuki T. Cellular siRNA delivery using cell-penetrating peptides modified for endosomal escape. Adv Drug Deliv Rev. 2009;61:704-9
23. Presente A, Dowdy SF. PTD/CPP peptide-mediated delivery of siRNAs. Curr Pharm Des. 2013;19:2943-7
24. Crombez L, Divita G. A non-covalent peptide-based strategy for siRNA delivery. Methods Mol Biol. 2011;683:349-60
25. Crombez L, Morris MC, Heitz F. et al. A non-covalent peptide-based strategy for ex vivo and in vivo oligonucleotide delivery. Methods Mol Biol. 2011;764:59-73
26. Gooding M, Browne LP, Quinteiro FM. et al. siRNA delivery: from lipids to cell-penetrating peptides and their mimics. Chem Biol Drug Des. 2012;80:787-809
27. Meade BR, Dowdy SF. Exogenous siRNA delivery using peptide transduction domains/cell penetrating peptides. Adv Drug Deliv Rev. 2007;59:134-40
28. Deshayes S, Morris M, Heitz F. et al. Delivery of proteins and nucleic acids using a non-covalent peptide-based strategy. Adv Drug Deliv Rev. 2008;60:537-47
29. Zorko M, Langel U. Cell-penetrating peptides: mechanism and kinetics of cargo delivery. Adv Drug Deliv Rev. 2005;57:529-45
30. Uusna J, Langel K, Langel U. Toxicity, Immunogenicity, Uptake, and Kinetics Methods for CPPs. Methods Mol Biol. 2015;1324:133-48
31. He H, Sheng J, David AE. et al. The use of low molecular weight protamine chemical chimera to enhance monomeric insulin intestinal absorption. Biomaterials. 2013;34:7733-43
32. Park YJ, Liang JF, Ko KS. et al. Low molecular weight protam ine as an efficient and nontoxic gene carrier: in vitro study. J Gene Med. 2003;5:700-11
33. Park YJ, Chang LC, Liang JF. et al. Nontoxic membrane translocation peptide from protamine, low molecular weight protamine (LMWP), for enhanced intracellular protein delivery: in vitro and in vivo study. FASEB J. 2005;19:1555-7
34. Kolate A, Baradia D, Patil S. et al. PEG - a versatile conjugating ligand for drugs and drug delivery systems. J Control Release. 2014;192:67-81
35. Jung S, Lee SH, Mok H. et al. Gene silencing efficiency of siRNA-PEG conjugates: effect of PEGylation site and PEG molecular weight. J Control Release. 2010;144:306-13
36. Kirpekar F, Berkenkamp S, Hillenkamp F. Detection of double-stranded DNA by IR- and UV-MALDI mass spectrometry. Anal Chem. 1999;71:2334-9
37. Beverly MB. Applications of mass spectrometry to the study of siRNA. Mass Spectrom Rev. 2011;30:979-98
38. Shimizu H, Jinno F, Morohashi A. et al. Application of high-resolution ESI and MALDI mass spectrometry to metabolite profiling of small interfering RNA duplex. J Mass Spectrom. 2012;47:1015-22
39. Seo YH, Carroll KS. Profiling protein thiol oxidation in tumor cells using sulfenic acid-specific antibodies. Proc Natl Acad Sci USA. 2009;106:16163-8
40. Seeley-Fallen MK, Liu LJ, Shapiro MR. et al. Actin-binding protein 1 links B-cell antigen receptors to negative signaling pathways. Proc Natl Acad Sci USA. 2014;111:9881-6
41. Song W, Liu C, Seeley-Fallen MK. et al. Actin-mediated feedback loops in B-cell receptor signaling. Immunol Rev. 2013;256:177-89
42. Suh JS, Lee JY, Choi YS. et al. Peptide-mediated intracellular delivery of miRNA-29b for osteogenic stem cell differentiation. Biomaterials. 2013;34:4347-59
43. Chang LC, Lee HF, Yang Z. et al. Low molecular weight protamine (LMWP) as nontoxic heparin/low molecular weight heparin antidote (I): Preparation and characterization. AAPS PharmSci. 2001;3:7-14
44. Thu MS, Bryant LH, Coppola T. et al. Self-assembling nanocomplexes by combining ferumoxytol, heparin and protamine for cell tracking by magnetic resonance imaging. Nat Med. 2012;18:463-7
45. Liang JF, Li YT, Connell ME. et al. Synthesis and characterization of positively charged tPA as a prodrug using heparin/protamine-based drug delivery system. AAPS PharmSci. 2000;2:E7
46. Arbab AS, Yocum GT, Rad AM. et al. Labeling of cells with ferumoxides-protamine sulfate complexes does not inhibit function or differentiation capacity of hematopoietic or mesenchymal stem cells. NMR Biomed. 2005;18:553-9
47. Torchilin VP. Tat peptide-mediated intracellular delivery of pharmaceutical nanocarriers. Adv Drug Deliv Rev. 2008;60:548-58
48. Choi YS, Lee JY, Suh JS. et al. Cell penetrating peptides for tumor targeting. Curr Pharm Biotechnol. 2011;12:1166-82
49. Lee TY, Park YS, Garcia GA. et al. Cell permeable cocaine esterases constructed by chemical conjugation and genetic recombination. Mol Pharm. 2012;9:1361-73
50. Franzen L, Windbergs M. Applications of Raman spectroscopy in skin research-From skin physiology and diagnosis up to risk assessment and dermal drug delivery. Adv Drug Deliv Rev. 2015;89:91-104
51. Mikoliunaite L, Rodriguez RD, Sheremet E. et al. The substrate matters in the Raman spectroscopy analysis of cells. Sci Rep. 2015;5:13150
52. Yano TA, Ichimura T, Kuwahara S. et al. Tip-enhanced nano-Raman analytical imaging of locally induced strain distribution in carbon nanotubes. Nat Commun. 2013;4:2592
53. Eguchi A, Dowdy SF. siRNA delivery using peptide transduction domains. Trends Pharmacol Sci. 2009;30:341-5
54. Beloor J, Zeller S, Choi CS. et al. Cationic cell-penetrating peptides as vehicles for siRNA delivery. Ther Deliv. 2015;6:491-507
55. Yang VC, He HN, Ye JX. et al. A novel covalently-linked, monomertic and bioreducible CPP-siRNA conjugates and its application. Chinese National Invention Patent. 2016 10083905.9.China2016
Author contact
![]() Corresponding authors: Email: hehuiningedu.cn (H. He) and E-mail: vcyangumich.edu (V. C. Yang)
Corresponding authors: Email: hehuiningedu.cn (H. He) and E-mail: vcyangumich.edu (V. C. Yang)
Citation styles
APA
Ye, J., Liu, E., Gong, J., Wang, J., Huang, Y., He, H., Yang, V.C. (2017). High-Yield Synthesis of Monomeric LMWP(CPP)-siRNA Covalent Conjugate for Effective Cytosolic Delivery of siRNA. Theranostics, 7(9), 2495-2508. https://doi.org/10.7150/thno.19863.
ACS
Ye, J.; Liu, E.; Gong, J.; Wang, J.; Huang, Y.; He, H.; Yang, V.C. High-Yield Synthesis of Monomeric LMWP(CPP)-siRNA Covalent Conjugate for Effective Cytosolic Delivery of siRNA. Theranostics 2017, 7 (9), 2495-2508. DOI: 10.7150/thno.19863.
NLM
Ye J, Liu E, Gong J, Wang J, Huang Y, He H, Yang VC. High-Yield Synthesis of Monomeric LMWP(CPP)-siRNA Covalent Conjugate for Effective Cytosolic Delivery of siRNA. Theranostics 2017; 7(9):2495-2508. doi:10.7150/thno.19863. https://www.thno.org/v07p2495.htm
CSE
Ye J, Liu E, Gong J, Wang J, Huang Y, He H, Yang VC. 2017. High-Yield Synthesis of Monomeric LMWP(CPP)-siRNA Covalent Conjugate for Effective Cytosolic Delivery of siRNA. Theranostics. 7(9):2495-2508.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.