Theranostics
13.3
Impact Factor
- Current Issue
- Advance Articles
- Volume 16; 2026
- Volume 15; 2025
- Volume 14; 2024
- Volume 13; 2023
- Volume 12; 2022
- Archive
- Cover Images
- Cover Suggestion
- Special Issues
Top
Introduction
Results
Discussion
Materials and Methods
Abbreviations
Acknowledgements
Supplementary Material
References
Introduction
Results
Discussion
Materials and Methods
Abbreviations
Acknowledgements
Supplementary Material
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2017; 7(11):2757-2774. doi:10.7150/thno.19551 This issue Cite
Research Paper
Cardiac Metabolic Deregulation Induced by the Tyrosine Kinase Receptor Inhibitor Sunitinib is rescued by Endothelin Receptor Antagonism
Joevin Sourdon1, Franck Lager2, Thomas Viel1, Daniel Balvay1, Rebecca Moorhouse3, Evangeline Bennana2,4, Gilles Renault2, Pierre-Louis Tharaux1, Neeraj Dhaun5, Bertrand Tavitian1, 6 ![]()
1. Paris Cardiovascular Research Center (PARCC); INSERM UMR970; Université Paris Descartes; Paris, France;
2. Institut Cochin, Université Paris Descartes, INSERM U1016, Paris 75014, France;
3. University/British Heart Foundation Centre for Cardiovascular Science, Queen's Medical Research Institute, Edinburgh, United Kingdom;
4. 3P5 proteomics facility, Université Paris Descartes, Université Sorbonne Paris Cité, Paris, France
5. University/British Heart Foundation Centre of Research Excellence, The Queen's Medical Research Institute, University of Edinburgh, United Kingdom;
6. Service de Radiologie, Hôpital Européen Georges Pompidou, Assistance Publique-Hôpitaux de Paris (AP-HP), Paris, France.
Received 2017-2-7; Accepted 2017-3-31; Published 2017-7-8
Citation:
Sourdon J, Lager F, Viel T, Balvay D, Moorhouse R, Bennana E, Renault G, Tharaux PL, Dhaun N, Tavitian B. Cardiac Metabolic Deregulation Induced by the Tyrosine Kinase Receptor Inhibitor Sunitinib is rescued by Endothelin Receptor Antagonism. Theranostics 2017; 7(11):2757-2774. doi:10.7150/thno.19551. https://www.thno.org/v07p2757.htm
Other stylesAbstract

The growing field of cardio-oncology addresses the side effects of cancer treatment on the cardiovascular system. Here, we explored the cardiotoxicity of the antiangiogenic therapy, sunitinib, in the mouse heart from a diagnostic and therapeutic perspective.
We showed that sunitinib induces an anaerobic switch of cellular metabolism within the myocardium which is associated with the development of myocardial fibrosis and reduced left ventricular ejection fraction as demonstrated by echocardiography. The capacity of positron emission tomography with [18F]fluorodeoxyglucose to detect the changes in cardiac metabolism caused by sunitinib was dependent on fasting status and duration of treatment. Pan proteomic analysis in the myocardium showed that sunitinib induced (i) an early metabolic switch with enhanced glycolysis and reduced oxidative phosphorylation, and (ii) a metabolic failure to use glucose as energy substrate, similar to the insulin resistance found in type 2 diabetes. Co-administration of the endothelin receptor antagonist, macitentan, to sunitinib-treated animals prevented both metabolic defects, restored glucose uptake and cardiac function, and prevented myocardial fibrosis.
These results support the endothelin system in mediating the cardiotoxic effects of sunitinib and endothelin receptor antagonism as a potential therapeutic approach to prevent cardiotoxicity.
Furthermore, metabolic and functional imaging can monitor the cardiotoxic effects and the benefits of endothelin antagonism in a theranostic approach.
Keywords: cardio-oncology, cardiotoxicity, positron emission tomography, echocardiography, endothelin, sunitinib, macitentan.
Introduction
The major factor limiting therapeutic administration of anticancer drugs is their toxic side effect on off-target organs. It is well known that classical anticancer drugs, e.g. anthracyclines, antimetabolites, alkylating agents, taxanes, induce serious cardiovascular toxicity (1, 2). Newer anticancer agents such as interferon and tyrosine kinase receptor (TKR) inhibitors also have cardiovascular side-effects (3) that, although often less severe than those observed with anthracyclines, are frequent and may be life-threatening (4). The clinical importance of cardiotoxicity associated with cancer therapy has led to the emergence of cardio-oncology, an interdisciplinary field that aims to better understand and limit the cardiotoxicity of cancer therapy (5).
Anti-angiogenics targeting the Vascular Endothelial Growth Factor Receptors (VEGFRs) pathway are considered as an essential asset for cancer treatment given the importance of the neovascularization process in tumor development (6,7). However, they are associated with a spectrum of cardiovascular side effects (8). The major treatment-limiting side effects of antiangiogenic TKR inhibitors include nausea, fatigue, hypertension, myocardial infarction, left ventricular cardiac and renal dysfunction, QT prolongation, neutropenia, proteinuria, thyroid impairment, thrombosis and hemorrhage (9-12).
Sunitinib (Sutent; Pfizer, USA) is an anti-angiogenic TKR inhibitor of VEGFRs, platelet-derived growth factor receptors (PDGF-Rs), and c-kit (13), approved in 2006 by the FDA for the treatment of renal cell carcinoma (14), imatinib-resistant gastrointestinal stromal tumor (15) and neuroendocrine tumors (16). In 2007, a study reported hypertension in ~50% of sunitinib-treated patients (8), decreased left ventricular ejection fraction (LVEF) in ~30% of patients, and congestive heart failure in ~10% of imatinib-resistant patients treated with sunitinib (8). Regarding sunitinib-induced hypertension, Lankhorst et al. listed as plausible mechanisms: deregulation of the nitric oxide signaling pathway, microvascular rarefaction, activation of endothelin (ET) system, salt sensitivity and oxidative stress (17). However, there are contradictory reports on the potential of sunitinib to up- or downregulate the nitric oxide pathway (17) and to cause microvascular rarefaction (18). Several lines of evidence suggest that the cardiotoxic effects of sunitinib are not only mediated by the development of hypertension but also through direct effects on the heart (8, 19). Inhibition of myocardial VEGFRs reduces response to stress, activates the pro-apoptotic pathway and maintains a high level of HIF-1α that is known to induce cardiomyopathy (20). Inhibition of PDGFRs by sunitinib induces a loss of coronary microvascular pericytes, suggesting a possible direct effect of sunitinib on myocardial blood supply (19). In rats, sunitinib leads to mitochondrial dysfunction; in mice, to increased apoptosis of cardiomyocytes (8). Ex vivo, a poor coronary flow response to bradykinin was reported in sunitinib-treated hearts, supporting microvascular dysfunction as a direct cardiac side effect of the drug (21). Finally, the fact that sunitinib inhibits AMPK (22) and induces mitochondrial damage (8) opens up the possibility that some or all of its side effects could result from a direct deregulation of cardiac metabolism.
Elevated plasma levels of endothelin-1 (ET-1) are found in animals and patients treated with sunitinib (21). An activated ET system likely contributes to vasoconstriction, hypertension, renal injury and proteinuria (23). Previous studies have shown that ET receptor blockers can reduce blood pressure (BP) and renal injury in animals and patients treated with sunitinib (21, 23-25). However, to our knowledge, the potential benefits of ET receptor antagonism in treating (or preventing) the cardiotoxic effects of sunitinib have not been studied.
Clinical imaging of the heart is a method of choice to explore the cardiac side effects of anticancer therapy (26). Echocardiography and magnetic resonance imaging (MRI) show reduced left ventricular ejection fraction (LVEF) in advanced stages of cardiotoxicity (27). In fact, most imaging studies have focused on the late stages of cardiotoxicity, either because imaging was prescribed after the clinical signs became evident, or because imaging failed to detect earlier signs. Nevertheless, a few studies have shown the capacity of nuclear imaging techniques to assess early cardiotoxicity associated with anthracyclines and trastuzumab (28). Borde et al. described a higher uptake of 2'-deoxy-2'-[18F]fluoro-D-glucose (FDG) in the myocardium of patients treated with adriamycin, highlighting the capacity of positron emission tomography (PET) to detect a deregulation of myocardial metabolism induced by a cancer treatment (29). Recently, O'Farrell et al. described an early increase of the metabolic rate of glucose in sunitinib-treated rodents (30). This is particularly interesting from a clinical perspective as PET can simultaneously stage cancer and explore cardiac metabolism.
Here, we explored cardiac metabolism after sunitinib treatment in mice using PET-FDG. We aimed to (i) better clarify the cardiac metabolic pathways deregulated during the early stages of sunitinib treatment, (ii) determine if the cardiac side effects are mediated by the endothelin pathway (iii) test the hypothesis that blockade of the endothelin system would prevent the cardiac side effects of sunitinib, and (iv) confirm that PET FDG can be useful to monitor cardiac metabolic remodeling.
Results
The study design followed a standard protocol of mouse oncology studies for monitoring of short-term response to therapy with PET in fasted, tumor-bearing, nude mice (Figure 1A). The same protocol was repeated in C57BL/6 immunocompetent mice with addition of echocardiography (Figure 1B). In another group of C57BL/6 mice, treatment duration was extended to three weeks (Figure 1C). Histology and proteome analysis were performed in all groups after treatment completion.
Sunitinib increases myocardial FDG uptake
After 5 days of sunitinib treatment, a significantly higher accumulation of FDG in the heart of fasted tumor-bearing nude mice was observed than in the vehicle group (Figure 2A). Compared to baseline, the mean standard uptake value (SUV) increased in the sunitinib group whereas it remained unchanged in the vehicle group (p <0.001, Figure 2B). Similar results were obtained in non-tumorized immunocompetent C57BL/6 mice using the same protocol: 5 days of sunitinib significantly increased FDG uptake compared to baseline (p <0.05; Figure 2B). Dynamic PET scans obtained under fasting conditions confirmed that sunitinib increased cardiac FDG metabolic flux in nude and C57BL/6 mice (Figure 2C). These findings support a sunitinib-induced shift towards glycolysis. Accordingly, hearts from sunitinib-treated mice showed a trend for higher expression of the glucose transporter protein GLUT1 and of Hexokinase II, the glucose phosphorylating enzyme, together with reduced expression of PGC1α, a key regulator of energy metabolism (Figure 3E). Moreover, 5 days of sunitinib treatment in C57BL/6 mice raised cardiac FDG uptake and lowered cardiac output (p <0.05, sunitinib vs. vehicle) (Figure 2D).
Sunitinib-induced increased myocardial FDG uptake is associated with increased cardiac fibrosis
We then explored whether the sunitinib-induced increase in FDG uptake was associated with a direct effect on the cardiac microvasculature, but found no difference in vessel density after sunitinib treatment (Figure 3A - 3B). This is in keeping with previous studies that have shown that microvascular rarefaction is not responsible for sunitinib-induced coronary dysfunction (19). By contrast, sunitinib treatment significantly increased myocardial fibrosis compared to hearts from vehicle-treated mice (p<0.001 and p<0.01 for nude and C57BL/6 mice, respectively) (Figure 3C - 3D).
Figure 1
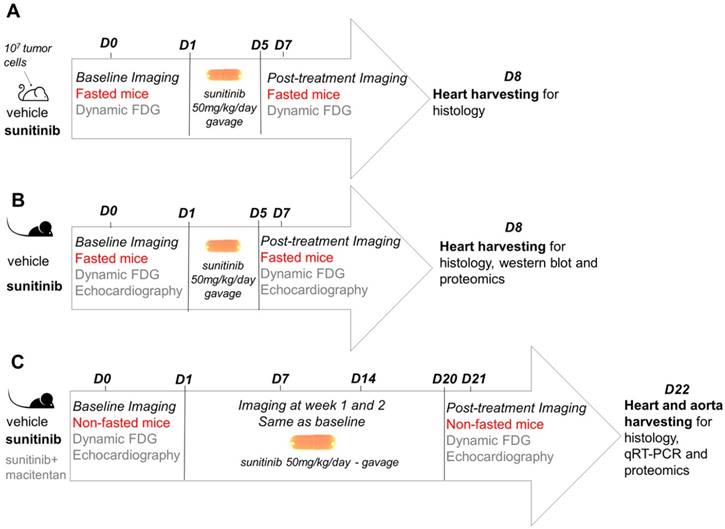
Study design: (A) represents investigation for short-term cardiotoxic effects on immunodeficient tumor-bearing mice (nude). Sunitinib-treated mice were studied at baseline and week 1 using a cancer PET protocol compared to vehicle. (B) represents investigation for short-term cardiotoxic effects on immunocompetent mice (C57BL/6). Sunitinib-treated mice were studied at baseline and week 1 using a cancer PET protocol and echocardiography compared to vehicle. (C) represents study design for long-term treatment on immunocompetent mice (C57BL/6). Sunitinib-treated mice were followed at baseline, week 1, week 2 and week 3 using a cardiac PET protocol and echocardiography compared to vehicle and sunitinib+macitentan groups. FDG: 2'-deoxy-2'-[18F]fluoro-D-glucose.
Figure 2
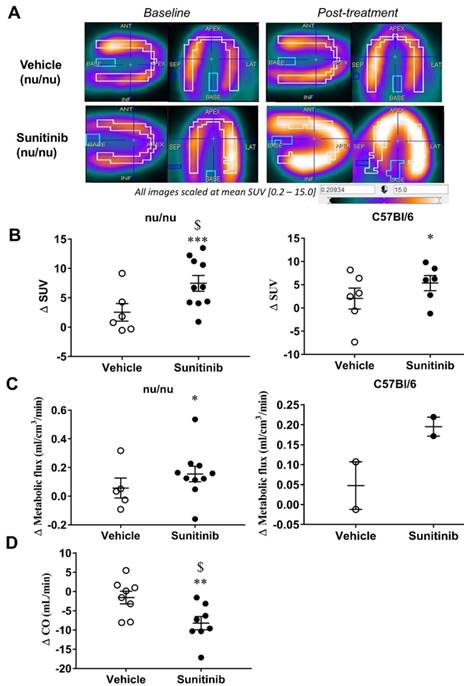
Sunitinib increases myocardial FDG uptake in fasted mice: (A) Example of PET scan images representing cardiac views of FDG-SUV at baseline (left) and post-treatment (right) in vehicle-treated mice and sunitinib-treated mice. (B) Difference post-treatment - baseline of myocardial FDG-SUV for sunitinib and vehicle groups in nude mice (n=6 for vehicle, n=10 for sunitinib) and C57BL/6 (n=6 for each). (C) Difference post-treatment - baseline of myocardial metabolic flux for sunitinib and vehicle groups in nude mice (n=5 for vehicle, n=10 for sunitinib) and C57BL/6 (n=2 for each). (D) Difference in cardiac output (CO, Heart Rate times stroke volume) for sunitinib and vehicle in C57BL/6 mice (n=8 for each groups). Data expressed as mean±SEM; *p <0.05 compared to baseline, **p <0.01 and ***p <0.001 compared to baseline, $p <0.05 compared to vehicle. CO: cardiac output; SUV: standard uptake values
Sunitinib downregulates oxidative energy metabolism pathways
Label-free pan-analysis of protein expression in mouse hearts revealed that 5 days of sunitinib treatment led to a significant downregulation of major oxygen-dependent metabolic pathways, including isocitrate dehydrogenase and succinate dehydrogenase (both enzymes of the tricarboxylic acid (TCA) cycle), phosphoenolpyruvate carboxykinase (first enzyme of gluconeogenesis), phosphoglycerate mutase and phosphorylase B kinase (key enzymes of carbohydrate metabolism) and carnitine O-palmitoyltransferase (mitochondrial transporter of fatty acids). All the proteins for which we found different levels of expression in the sunitinib and vehicle groups are listed in Table 1. Ingenuity® analysis highlighted that sunitinib treatment induced mitochondrial dysfunction and a clear switch towards anaerobic glycolytic metabolism, similar to the one seen during cardiac hypertrophy (31). In particular, proteins of the fatty acid degradation pathway such as acyl-CoA dehydrogenase (Acad8), phospholipases, and fatty acid transferases, those controlling glycogen breakdown (Pgam1, Me3) and the synthesis of the cofactor flavine adenine dinucleotide (FAD) were reduced in sunitinib-treated hearts. Taken together, these results are in agreement with a switch towards the utilization of glucose as major energy source and with the activation of glycolysis in the myocardium of sunitinib-treated mice evidenced by increased FDG uptake.
The effect of sunitinib on cardiac FDG uptake depends on glucose metabolism of the heart
Cardiac FDG uptake varies widely with plasmatic concentrations of glucose and insulinemia, hence with the post-prandial time. Oncology PET scans are performed in fasted patients and animals in order to reduce the cardiac uptake of FDG in oncology studies and better delineate the tumor uptake. Conversely, imaging of glucose metabolism in the heart is often performed under an euglycemic clamp (glucose plus insulin administration) in order to increase metabolism of glucose and FDG uptake in the heart. Since sunitinib has been shown to induce hypoglycemia in patient and animal studies (32), we tested whether the effect of the drug on cardiac FDG uptake would differ in fasted versus non-fasted mice. Interestingly, in contrast to fasted mice, the FDG SUV were similar in sunitinib and vehicle-treated non-fasted animals after 1 and 3 weeks of treatment (Figure 4A). However, the effects of sunitinib on heart function were maintained in non-fasted animals, i.e. cardiac output was reduced after 1 and 3 weeks of sunitinib with respect to pre-sunitinib baseline values (Figure 4B), with significant reductions in diastolic (Figure 4C) and stroke volumes. Sunitinib also significantly reduced blood flow as shown by the fall in aortic flow (aortic velocity tracking integral) (Figure 4D).
Figure 3
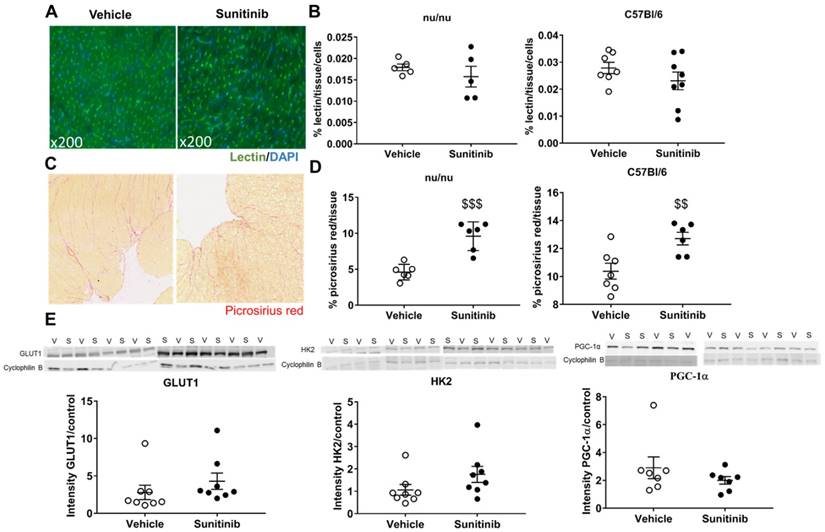
Sunitinib-induced increased FDG uptake in fasted mice is associated with increased fibrosis: (A) Representative sections of myocardium stained for blood vessels (lsolectine B4 Griffonia Simplicifolia-FITC, green) and nuclei (DAPI, blue). (B) Quantification of microvascular density normalized by cell number in nude and C57Bl/6 mice treated with vehicle (open circles) or sunitinib (black circles). (C) Representative sections of myocardium stained for fibrosis (Picrosirius red) from hearts of mice treated with vehicle (left) or sunitinib (right). (D) Quantification of fibrosis (normalized by tissue area) in nude and C57Bl/6 mice treated with vehicle (open circles) or sunitinib (filled circles). (E) Representative blots and their associated quantification for GLUT1, HK2 and PGC1α (normalized by cyclophilin B) mice treated with vehicle (open circles) or sunitinib (filled circles). Data expressed as mean ± SEM; $$p <0.01 $$$p <0.001 compared to vehicle. HK2: Hexokinase 2; GLUT1: glucose transporter 1; PGC1α; Peroxisome proliferator-activated receptor gamma coactivator 1-alpha; S: sunitinib; V: vehicle.
Endothelin receptor antagonism prevents sunitinib-induced diastolic dysfunction and myocardial flux dysfunction
It has been demonstrated that the endothelin (ET) pathway mediates the hypertension and renal complications of sunitinib (24). Macitentan, a clinically-available mixed ETA and ETB receptor antagonist, was given concomitantly with sunitinib to mice during 3 weeks. A comparison of protein expression in the sunitinib-treated group with that in the vehicle group and sunitinib plus macitentan group evidenced the protective effects of macitentan on diastolic dysfunction. This was confirmed by expression levels of several actin and myosin protein isoforms and associated fibrillar protein, for which macitentan countered the effect of sunitinib on their level of expression (Table 2). It should be noted that macitentan treatment normalized both the diastolic volume and the cardiac output, allowing the maintenance of aortic flow (Figure 4B - 4D).
Kinetics analysis of dynamic FDG-PET scans of the heart showed a trend towards a higher metabolic flux and a higher metabolic rate of glucose (MRGlu) after 1 and 2 weeks of sunitinib treatment that did not reach statistical significance (Figure 5A). In contrast, both parameters were significantly reduced after 3 weeks of sunitinib, showing that the early promotion of cardiac glycolysis by sunitinib was followed by a secondary drop in glucose utilization. Macitentan plus sunitinib co-administration maintained metabolic flux and MRGlu to their pre-treatment baseline levels after 3 weeks of treatment (Figure 5A). In line with our previous observations at 1 week of treatment, 3 weeks of sunitinib was not associated with any change in myocardial vessel density (Figure 5B - 5C). A proteomics analysis of the macitentan and sunitinib treated hearts and comparison with the sunitinib-only or vehicle-treated groups confirmed the protective effects of endothelin antagonism by macitentan on endothelial cell dysfunction (Table 2).
Table 1
Major changes in protein expression levels of the myocardium after one week of treatment
| Categories | Protein name | Gene ID | sunitinib versus vehicle | ||
|---|---|---|---|---|---|
| p-value | FC | ||||
| Glucogenolysis | Phosphoglycerate mutase 1 | Pgam1 | 0.01 | -1.28 | |
| Glucogenolysis | NADP-dependent malic enzyme 3, mitochondrial | Me3 | 0.01 | -1.17 | |
| TCA cycle | Isobutyryl-CoA dehydrogenase, mitochondrial | Acad8 | < 0.001 | -1.30 | |
| TCA cycle | Pyruvate dehydrogenase (acetyl-transferring) kinase isozyme 2, mitochondrial | Pdk2 | 0.08 | -1.21 | |
| TCA cycle | Isocitrate dehydrogenase [NAD] subunit gamma 1, mitochondrial | Idh3g | 0.03 | -1.24 | |
| TCA cycle | Isocitrate dehydrogenase [NAD] subunit alpha, mitochondrial | Idh3a | 0.05 | -1.14 | |
| TCA cycle | Succinate dehydrogenase complex flavoprotein subunit A, mitochondrial | Sdha | 0.02 | -1.12 | |
| AMPK | Phosphoenolpyruvate carboxykinase 2, mitochondrial | Pck2 | < 0.0001 | - | |
| AMPK | Carnitine O-palmitoyltransferase 1, muscle isoform | Cpt1b | 0.21 | -1.19 | |
| Hypertrophy | Carnitine O-palmitoyltransferase 1, liver isoform | Cpt1a | 0.04 | -1.31 | |
| Hypertrophy | Phospholipase C beta 3 | Plcb3 | 0.01 | - | |
| Hypertrophy | Transforming protein RhoA | RhoA | 0.02 | -1.59 | |
| Hypertrophy | Caveolin-1 | Cav1 | 0.04 | -1.30 | |
| Hypertrophy | Telethonin | Tcap | < 0.01 | -1.70 | |
| Hypertrophy | A-kinase anchor protein 1, mitochondrial | Akap1 | 0.02 | -1.45 | |
| Mitochondria | Heat shock protein beta-1 | Hspb1 | 0.02 | -1.23 | |
| Mitochondria | Heat shock 70 kDa protein 4L | Hspa4l | 0.01 | -1.44 | |
| Mitochondria | Mitochondrial import receptor subunit TOM70 | Tomm70a | 0.06 | -1.27 | |
| Mitochondria | Electron transfer flavoprotein dehydrogenase | Etfdh | 0,04 | -1.34 | |
| Mitochondria | Sulfite oxidase, mitochondrial | Suox | 0.02 | 1.22 | |
| Energy metabolism | NAD-dependent protein deacetylase sirtuin-4 | Sirt4 | - | - | |
| Energy metabolism | PGC-1 and ERR-induced regulator in muscle protein 1 | Perm1 | 0.09 | -1.35 | |
| Hypoxia | ubiquitin specific peptidase 19 | Usp19 | < 0.0001 | - | |
| Hypoxia | Hypoxia up-regulated protein 1 | Hyou1 | 0.06 | 1.26 | |
| Nitric oxide | Guanylate cyclase soluble subunit beta-1 | Gucy1b3 | 0.03 | -1.55 | |
| Oxidative stress | Glutathione reductase, mitochondrial | Gsr | 0.02 | 1.41 | |
| Glycometabolism | 1-acylglycerol-3-phosphate O-acyltransferase 2 | Agpat2 | < 0.001 | -2.13 | |
| Glycometabolism | Dolichyl-diphosphooligosaccharide-non-catalytic subunit | Ddost | 0.02 | 1.15 | |
| Glycometabolism | CDP-diacylglycerol--inositol 3-phosphatidyltransferase | Cdipt | 0.02 | 1.63 | |
| Sodium-dependent phosphate transporter 2 | Slc20a2 | 0.03 | -1.28 | ||
| Sodium/potassium-transporting ATPase subunit beta-1 | Atp1b1 | 0.03 | -1.31 | ||
| Flavin adenine dinucleotide synthetase 1 | Flad1 | 0.03 | -1.29 | ||
| Pirin | Pir | 0.04 | - | ||
Results from Label-free protein quantification showing the ratios of protein expression levels in sunitinib versus vehicle. Numbers indicate the fold change (FC) between two groups and the p-value of the two tailed Student t.test. The sign “-“ indicates undetected protein in the denominator. group. n=3 for each. AMPK: 5' AMP-activated protein kinase; cdp: cytidine diphospho; ERR: estrogen-related receptor; NAD: nicotinamide adenine dinucleotide; NADP: nicotinamide adenine dinucleotide phosphate; PGC-1: peroxisome proliferator-activated receptor gamma coactivator 1-alpha; TCA: tricarboxylic acid.
Figure 4
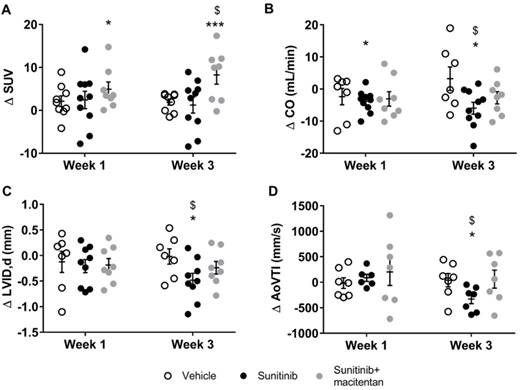
Macitentan prevents sunitinib-induced diastolic dysfunction. (A) Difference treatment - baseline of myocardial FDG-SUV for vehicle (n=9), sunitinib (n=10) and sunitinib+macitentan treated mice (n=8) at week 1 and week 3. (B) Difference treatment - baseline values of CO is represented for vehicle (n=7), sunitinib (n=10) and sunitinib+macitentan (n=8) groups at week 1 and 3. (C) Difference treatment - baseline of left ventricular internal diameter at diastole for the three groups. (D) Difference treatment - baseline of aortic velocity time integral for both groups. Data are expressed as mean ± SEM, *p <0.05 compared to baseline, ***p <0.001 compared to baseline, $p <0.05 compared to other at week 3. AoVTI: Aortic velocity tracking integral; CO: cardiac output; LVID: left ventricular internal diameter; SUV: standard uptake value.
Table 2
Major changes in protein expression levels of the myocardium after three weeks of treatments
| sunitinib versus vehicle | sunitinib versus sunitinib + macitentan | sunitinib + macitentan versus vehicle | ||||||
|---|---|---|---|---|---|---|---|---|
| Categories | Protein | ID | p-value | FC | p-value | FC | p-value | FC |
| Glucogenolysis | facilitated glucose transporter member 4 | Slc2a4 | 0,009 | 7,78 | 0,219 | 1,57 | 0,024 | 4,97 |
| Glucogenolysis | RAB10, member RAS oncogene family | Rab10 | 0,071 | -1,30 | 0,405 | 1,07 | 0,030 | -1,39 |
| Glycogen, IR | 1,4-alpha-glucan-branching enzyme 1 | Gbe1 | 0,038 | -1,22 | 0,010 | -1,23 | 0,908 | 1,01 |
| Glucogenolysis | Acyl-CoA dehydrogenase family member 9 | Acad9 | 0,010 | -1,18 | 0,114 | -1,11 | 0,172 | -1,06 |
| Glucogenolysis | phosphorylase, glycogen, muscle | Pygm | 0,046 | -1,14 | 0,213 | -1,09 | 0,424 | -1,05 |
| Glucogenolysis | Glucose-6-phosphate isomerase | Gpi | 0,003 | 1,15 | 0,276 | -1,07 | 0,003 | 1,23 |
| Glucogenolysis | Glucose-6-phosphate 1-dehydrogenase | G6pdx | 0,855 | -1,09 | 0,360 | 1,98 | 0,015 | -2,17 |
| Glucogenolysis | Phosphofructokinase, muscle | Pfkm | 0,071 | -1,20 | 0,959 | 1,00 | 0,071 | -1,20 |
| Glucogenolysis | phosphofructokinase, platelet | Pfkp | 0,024 | 1,26 | 0,013 | -1,42 | 0,001 | 1,79 |
| Glucogenolysis | Phosphoglycerate mutase 1 | Pgam1 | 0,011 | 1,48 | 0,141 | 1,18 | 0,057 | 1,26 |
| Glucogenolysis | Glyceraldehyde-3-phosphate dehydrogenase | Gapdh | 0,074 | -1,12 | 0,043 | -1,24 | 0,265 | 1,10 |
| FA metabolism, IR | CD36 molecule | Cd36 | 0,012 | 1,36 | 0,999 | 1,00 | 0,014 | 1,36 |
| FA metabolism | Acyl-CoA synthetase family member 2 | Acsf2 | 0,038 | 1,28 | 0,022 | -1,37 | 0,518 | 1,08 |
| TCA cycle, FA metabolism, IR | Malonyl-CoA decarboxylase, mitochondrial | Mlycd | 0,024 | 1,23 | 0,208 | -1,15 | 0,477 | -1,08 |
| TCA cycle | Isobutyryl-CoA dehydrogenase, mitochondrial | Acad8 | 0,026 | -1,21 | 0,002 | -1,20 | 0,932 | -1,01 |
| TCA cycle | Pyruvate dehydrogenase (E1 component) beta | Pdhb | 0,004 | -1,20 | 0,003 | -1,36 | 0,144 | 1,13 |
| TCA cycle | Pyruvate dehydrogenase (E1 component) alpha | Pdha1 | 0,041 | -1,17 | 0,009 | -1,22 | 0,491 | 1,04 |
| TCA cycle | Pyruvate dehydrogenase phosphatase regulatory subunit | Pdpr | 0,008 | -1,24 | 0,047 | -1,13 | 0,106 | -1,10 |
| TCA cycle | Dihydrolipoamide S-acetyltransferase | Dlat | 0,003 | -1,30 | 0,012 | -1,28 | 0,814 | -1,01 |
| TCA cycle | Succinate dehydrogenase; flavoprotein subunit | Sdha | 0,022 | -1,18 | 0,089 | -1,14 | 0,649 | -1,03 |
| TCA cycle | Isocitrate dehydrogenase [NAD] subunit | Idh3g | 0,017 | -1,42 | 0,001 | -1,31 | 0,496 | -1,08 |
| TCA cycle | 2-oxoglutarate dehydrogenase, mitochondrial | Ogdh | 0,074 | -1,17 | 0,041 | -1,21 | 0,446 | 1,04 |
| L-lactate dehydrogenase A | Ldha | 0,001 | 1,60 | 0,958 | -1,01 | <0,001 | 1,61 | |
| Mitochondria, FA | 3-hydroxyisobutyryl-CoA hydrolase, mitochondrial | Hibch | 0,403 | -1,06 | 0,018 | -1,18 | 0,020 | 1,12 |
| Mitochondria | 3-hydroxyisobutyrate dehydrogenase, mitochondrial | Hibadh | 0,738 | 1,04 | 0,036 | -1,26 | 0,005 | 1,30 |
| Mitochondria | Mitochondrial-processing peptidase subunit beta | Pmpcb | 0,049 | -1,17 | 0,011 | -1,36 | 0,142 | 1,16 |
| Mitochondria | Mitochondrial import receptor subunit TOM22 | Tomm22 | 0,299 | 1,16 | 0,006 | -1,46 | 0,001 | 1,70 |
| Mitochondria | Translocase of inner mitochondrial membrane 8 | Timm8a1 | 0,015 | 1,80 | 0,613 | 1,09 | 0,004 | 1,65 |
| Mitochondria | Mitochondrial Rho GTPase 1 | Rhot1 | 0,008 | 3,11 | 0,862 | 1,01 | 0,010 | 3,08 |
| Mitochondria | Translocase of outer mitochondrial membrane 70 | Tomm70a | 0,001 | -1,41 | 0,118 | -1,13 | 0,010 | -1,25 |
| Mitochondria | Mitochondrial import membrane translocase subunit | Timm50 | 0,474 | 1,08 | 0,418 | -1,08 | 0,010 | 1,17 |
| Mitochondria | Mitochondrial import receptor subunit TOM40 homolog | Tomm40 | 0,024 | 1,23 | 0,923 | 1,01 | 0,022 | 1,22 |
| Mitochondria | Translation factor Guf1, mitochondrial | Guf1 | 0,026 | -4,87 | 0,022 | -5,32 | 0,791 | 1,09 |
| Mitochondria | Elongation factor G, mitochondrial | Gfm1 | 0,011 | -1,24 | 0,021 | -1,20 | 0,422 | -1,03 |
| Mitochondria | Dynamin-like protein, mitochondrial | Opa1 | 0,099 | -1,16 | 0,069 | -1,16 | 0,994 | 1,00 |
| Mitochondria, IR | Cytochrome c oxidase subunit 6A2 | Cox6a2 | 0,021 | 3,65 | 0,369 | 1,42 | 0,113 | 2,57 |
| Mitochondria | Cytochrome c oxidase assembly factor 3 | Coa3 | 0,007 | - | 0,934 | 1,03 | 0,002 | - |
| Mitochondria | Mitochondrial transcription termination factor 2 | Mterf2 | 0,045 | -2,39 | 0,005 | -2,27 | 0,821 | -1,05 |
| Mitochondria | ATP synthase F1 complex assembly factor 2 | Atpaf2 | 0,249 | 1,33 | 0,106 | -1,21 | 0,040 | 1,62 |
| Mitochondria | ATP synthase subunit beta, mitochondrial | Atp5b | 0,145 | 1,22 | 0,277 | -1,15 | 0,006 | 1,41 |
| Mitochondria | BolA-like protein 3 | Bola3 | 0,010 | -2,77 | 0,025 | -2,26 | 0,164 | -1,22 |
| Mitochondria | A-kinase anchor protein 1, mitochondrial | Akap1 | 0,185 | -1,19 | 0,057 | -1,31 | 0,471 | 1,10 |
| Mitochondria | Aldehyde dehydrogenase, mitochondrial | Aldh2 | 0,013 | -1,20 | 0,467 | 1,05 | 0,001 | -1,26 |
| Mitochondria | 3-oxoacyl-[acyl-carrier-protein] synthase, mitochondrial | Oxsm | 0,890 | 1,01 | 0,013 | -1,20 | 0,037 | 1,22 |
| Mitochondria | Elongation factor 1-delta | Eef1d | 0,011 | 1,22 | 0,065 | 1,17 | 0,529 | 1,04 |
| Mitochondria repiratory chain | NADH dehydrogenase 1 beta subcomplex subunit 3 | Ndufb3 | 0,221 | 1,33 | 0,840 | -1,04 | 0,020 | 1,38 |
| Mitochondria repiratory chain | NADH dehydrogenase 1 alpha subcomplex subunit 13 | Ndufa13 | 0,288 | 1,22 | 0,611 | -1,08 | 0,027 | 1,32 |
| Mitochondria repiratory chain | NADH dehydrogenase 1 beta subcomplex subunit 10 | Ndufb10 | 0,171 | 1,25 | 0,915 | -1,01 | 0,006 | 1,27 |
| Mitochondria repiratory chain | NADH dehydrogenase flavoprotein 2, mitochondrial | Ndufv2 | 0,335 | 1,13 | 0,325 | -1,12 | 0,008 | 1,27 |
| Mitochondria repiratory chain | NADH dehydrogenase 1 alpha subcomplex subunit 11 | Ndufa11 | 0,010 | 1,99 | 0,704 | 1,05 | 0,009 | 1,90 |
| Mitochondria repiratory chain | NADH dehydrogenase 1 alpha subcomplex subunit 9 | Ndufa9 | 0,160 | 1,28 | 0,876 | 1,02 | 0,038 | 1,25 |
| Mitochondria repiratory chain | NADH dehydrogenase iron-sulfur protein 2 | Ndufs2 | 0,391 | -1,09 | 0,040 | -1,26 | 0,031 | 1,16 |
| Energy metabolism | Creatine kinase M-type | Ckm | 0,066 | -1,24 | 0,032 | -1,25 | 0,948 | 1,00 |
| Energy metabolism | Nucleoside diphosphate kinase 3 | Ndk3 | 0,640 | -1,38 | 0,032 | -2,65 | 0,030 | 1,92 |
| Energy metabolism | NAD-dependent protein deacetylase sirtuin-3 | Sirt3 | 0,297 | -1,16 | 0,044 | -1,47 | 0,196 | 1,27 |
| Energy metabolism | NAD-dependent protein deacylase sirtuin-5 | Sirt5 | 0,142 | -1,17 | 0,033 | -1,18 | 0,867 | 1,01 |
| Energy metabolism, | NAD-dependent protein deacetylase sirtuin-2 | Sirt2 | 0,039 | -1,63 | 0,033 | -1,72 | 0,715 | 1,06 |
| Inflammation | Macrophage migration inhibitory factor | Mif | 0,797 | 1,13 | <0,001 | -1,93 | 0,040 | 2,19 |
| Inflammation | Interferon-inducible GTPase 1 | Irga6 | 0,038 | -3,04 | 0,275 | -4,79 | 0,617 | 1,57 |
| Inflammation | Serpin family F member 2 | Serpinf2 | 0,025 | 1,58 | 0,915 | 1,02 | 0,011 | 1,55 |
| Inflammation | Fibrinogen beta chain | Fgb | 0,032 | 1,48 | 0,361 | -1,27 | 0,058 | 1,87 |
| Inflammation | Mitogen-activated protein kinase kinase 1 | Map2k1 | 0,011 | -1,32 | 0,291 | -1,14 | 0,107 | -1,16 |
| Inflammation, hypertension | Plasma kallikrein B1 | Klkb1 | 0,033 | 2,10 | 0,311 | 1,30 | 0,073 | 1,62 |
| Inflammation, hypertension | Kininogen1 | Kng1 | 0,002 | 1,72 | 0,335 | -1,13 | 0,0002 | 1,94 |
| Inflammation, atherosclerosis | Plasminogen | Plg | 0,008 | 1,60 | 0,521 | 1,09 | 0,010 | 1,47 |
| Inflammation, atherosclerosis | Ras homolog family member A | rho A | 0,010 | 1,58 | 0,117 | 1,23 | 0,086 | 1,29 |
| Inflammation, atherosclerosis | Fibronectin | Fn1 | 0,007 | 1,55 | 0,252 | 1,16 | 0,038 | 1,33 |
| Inflammation, atherosclerosis, IR | Signal transducer and activator of transcription 1 | Stat1 | 0,031 | -2,94 | 0,223 | -3,00 | 0,969 | 1,02 |
| Atherosclerosis | Intercellular adhesion molecule 1 | Icam1 | 0,037 | 2,07 | 0,273 | 1,33 | 0,270 | 1,56 |
| Atherosclerosis | Apolipoprotein A-IV | Apoa4 | 0,013 | 1,62 | 0,212 | 1,24 | 0,106 | 1,31 |
| Atherosclerosis, infarction | Glutathione S-transferase Mu 1 | Gstm1 | 0,026 | 1,25 | 1,152 | 1,15 | 0,243 | 1,08 |
| Thrombosis, | Heparin cofactor 2 | Serpind1 | 0,033 | 2,68 | 0,621 | 1,11 | 0,028 | 2,42 |
| Thrombosis, endothelial cell apoptosis | Antithrombin-III | Serpinc1 | 0,014 | 1,56 | 0,167 | 1,21 | 0,052 | 1,29 |
| Hypertension, IR | Adiponectin | Adipoq | 0,033 | 1,39 | 0,280 | 1,17 | 0,067 | 1,19 |
| Hypertension, infarction | Solute carrier family 8 member A1 | Slc8a1 | 0,004 | -1,37 | 0,034 | -1,29 | 0,550 | -1,06 |
| Nitric oxyde | Guanylate cyclase soluble subunit beta-1 | Gucy1b3 | 0,002 | -2,76 | 0,893 | 1,07 | 0,001 | -2,94 |
| Oxidative stress | Glutathione reductase, mitochondrial | Gsr | 0,005 | 1,23 | 0,066 | 1,13 | 0,153 | 1,09 |
| Endothelial cell dysfuntion, infarct | Prostacyclin synthase | Ptgis | 0,038 | 3,08 | 0,020 | 3,69 | 0,789 | -1,20 |
| Endothelial cell dysfuntion, infarct, IR | Paraoxonase 1 | Pon1 | 0,014 | - | 0,014 | - | - | - |
| Endothelial cell dysfuntion, infarct, IR | Apolipoprotein A1 | Apoa1 | 0,029 | 1,62 | 0,023 | 1,65 | 0,849 | -1,02 |
| Endothelial cell dysfuntion, infarct | Cathepsin D | Ctsd | <0,001 | 1,66 | 0,046 | 1,23 | <0,001 | 1,35 |
| Endothelial cell apoptosis | Ferritin heavy chain 1 | Fth1 | 0,014 | 1,72 | 0,122 | 1,29 | 0,035 | 1,34 |
| Endothelial cell apoptosis | Voltage-dependent anion channel 1 | Vdac1 | 0,006 | 1,25 | 0,949 | 1,00 | 0,003 | 1,25 |
| Endothelial cell apoptosis | Catenin beta-1 | Ctnnb1 | 0,017 | -1,19 | 0,156 | -1,11 | 0,351 | -1,07 |
| Endothelial cell apoptosis | Transferrin receptor protein 1 | Tfrc | 0,048 | -1,38 | 0,007 | -1,63 | 0,199 | 1,18 |
| Endothelial cell apoptosis | Xanthine dehydrogenase | Xdh | 0,011 | -1,47 | 0,635 | 1,08 | <0,001 | -1,58 |
| Cardiac function | Popeye domain-containing protein 2 | Popdc2 | 0,032 | 5,43 | 0,965 | -1,02 | 0,014 | 5,52 |
| Cellular hometostasis | ATPase Na+/K+ transporting beta 3 | Atp1b3 | 0,010 | 1,92 | 0,020 | 1,53 | 0,325 | 1,26 |
| Cellular hometostasis | ATPase Na+/K+ transporting alpha 2 | Atp1a2 | 0,017 | -1,24 | 0,667 | 1,04 | 0,012 | -1,29 |
| Cellular hometostasis | Chloride intracellular channel 4 | Clic4 | 0,017 | -1,27 | 0,886 | 1,01 | <0,001 | -1,28 |
| Cellular hometostasis | Calcium/calmodulin-dependent protein kinase type II | Camk2d | 0,001 | -1,41 | 0,341 | -1,08 | 0,001 | -1,30 |
| Cellular hometostasis | Ferritin light chain 1 & 2 | Ftl1;Ftl2 | 0,019 | 1,37 | 0,031 | 1,36 | 0,919 | 1,01 |
| Cellular hometostasis | Metalloreductase STEAP4 | Steap4 | 0,705 | -1,19 | 0,024 | 6,71 | 0,036 | -7,99 |
| Cellular morphology | Filamin-B | Flnb | 0,269 | 1,13 | 0,068 | -1,23 | 0,001 | 1,38 |
| Cellular morphology | Myosin-7 | Myh7 | 0,306 | 1,38 | 0,027 | -5,19 | 0,020 | 7,15 |
| Cellular morphology | Myosin-Ic | Myo1c | 0,461 | -1,06 | 0,066 | 1,22 | <0,001 | -1,29 |
| Cellular morphology | Capping actin protein of muscle Z-line beta | Capzb | 0,025 | -1,18 | 0,212 | 1,09 | <0,001 | -1,29 |
| Cellular morphology | Actin, alpha skeletal muscle | Acta1 | 0,107 | -1,90 | 0,029 | -3,87 | 0,128 | 2,04 |
| Cellular morphology | Myosin-XVIIIa | Myo18a | 0,014 | -1,26 | <0,001 | -1,22 | 0,620 | -1,03 |
| Cellular morphology | Titin | Ttn | 0,014 | -1,37 | 0,016 | -1,15 | 0,099 | -1,20 |
| Cellular morphology | Myocardial zonula adherens protein | Myzap | 0,001 | -1,33 | 0,004 | -1,26 | 0,286 | -1,05 |
| Cellular morphology | Myomesin-2 | Myom2 | 0,019 | -1,17 | 0,045 | -1,17 | 0,981 | -1,00 |
| Cellular morphology | Actin-related protein 2/3 complex subunit 2 | Arpc2 | 0,752 | 1,04 | 0,037 | 1,35 | 0,003 | -1,30 |
| Cellular morphology | Valine--tRNA ligase | Vars | 0,151 | -1,13 | 0,108 | 1,16 | <0,001 | -1,32 |
| Cellular morphology | GTP-binding nuclear protein Ran | Ran | 0,010 | 1,41 | 0,009 | 1,42 | 0,925 | -1,01 |
| Endothelin downstream signaling | Phospholipase C | Plcb4 | - | - | 0,009 | 2,08 | - | - |
| Endothelin downstream signaling | Mapk1; Erk2 | 0,343 | 1,25 | 0,103 | 1,53 | 0,330 | 1,22 | |
| Afamin | Afm | <0,001 | 5,15 | 0,064 | 1,40 | 0,001 | 3,67 | |
| Flavin adenine dinucleotide synthetase 1 | Flad1 | 0,010 | -1,62 | 0,005 | -1,38 | 0,249 | -1,17 | |
| Musashi RNA binding protein 2 | Msi2 | 0,022 | -1,29 | 0,011 | -1,49 | 0,298 | 1,15 | |
| Pre-mRNA-processing factor 19 | Prpf19 | 0,046 | -1,35 | 0,298 | -1,22 | 0,480 | -1,10 | |
| Adenylosuccinate lyase | Adsl | 0,031 | -1,31 | 0,017 | -1,41 | 0,584 | 1,08 | |
| Vinculin | Vcl | 0,005 | 1,15 | 0,002 | 1,13 | 0,600 | 1,02 | |
Results from Label-free protein quantification showing the ratios of protein expression levels in sunitinib versus vehicle, sunitinib versus sunitinib+macitentan and sunitinib+macitentan versus vehicle. Numbers indicate the fold change (FC) between two groups and the p-value of the two tailed Student t.test Data are expressed as Fold change (FC). The sign “-“ indicates undetected protein in the denominator group. n=6 for each. ATP: Adenosine triphosphate; CD36: cluster of differentiation 36; FA: fatty acid; GTP: Guanosine-5'-triphosphate; IR: insulin resitance; K+: potassium; Na+: sodium; NAD: nicotinamide adenine dinucleotide; RAB: Ras-related protein; RNA: Ribonucleic acid; TCA: tricarboxylic acid.
Macitentan reduces cardiac fibrosis and downregulates myocardial ETA receptors
We studied the effect of sunitinib, with or without the addition of macitentan, on cardiac fibrosis. Sunitinib treatment was associated with the development of cardiac fibrosis that was prevented by co-administration of macitentan (Figure 6B). Since the ETA receptor is known to mediate fibrosis (33) and cardiac hypertrophy (34), it can be assumed that sunitinib-induced cardiac injuries are mediated through the ETA receptor. We further explored the effects of macitentan on the ET pathway in order to define which receptor is involved in sunitinib-induced cardiac injuries. Macitentan did not affect ET-1 or ETB receptor expression, but significantly downregulated myocardial ETA receptor expression (Figure 6A).
Moreover, both ETA and ETB receptors of the aorta were increased in mice treated with sunitinib and sunitinib+macitentan (Figure S1). Since elevated ET-1 is found in patients and rodents treated with sunitinib (21) and elevated ET receptors characterizes hypertension (35), we speculated that the ET pathway was activated by sunitinib. Accordingly, the ET downstream signaling, phospholipase C, protein kinase C and MAPK1;ERK2, was high in all samples (n=6) of sunitinib-treated hearts but was undetectable in the vehicle group. In addition, these effectors were detected only in 3 samples out of 6 (50%) in the sunitinib+macitentan group and were expressed at lower level than the sunitinib-group.
Macitentan reverses the sunitinib-induced aerobic to anaerobic switch
Label-free pan-analysis of protein expression in mouse hearts collected from the three groups (vehicle, sunitinib and sunitinib+macitentan) is shown in Table 2. Data analysis using Ingenuity® and Pathway Studio® softwares showed that sunitinib induced an impairment in carbohydrate and fatty acid oxidative metabolisms and in the TCA cycle. Mitochondrial homeostasis was also deregulated, as shown by reduced expression in both the inner and outer mitochondrial membrane complexes, in mitochondrial transporters and in mitochondrial translation factors. In line with the effect of sunitinib on myocardial flux dysfunction. Several patterns of protein clusters were drastically different in the sunitinib and vehicle groups (Figure 7C). Protein clusters involved in myocardial infarction, endothelial cell dysfunction and apoptosis, atherosclerosis, thrombosis, inflammation and hypertension, were targeted by sunitinib. Comparing protein expression in the sunitinib-treated group with that in the vehicle and sunitinib plus macitentan groups highlighted once again the protective effects of macitentan, notably on the following clusters: myocardial infarction, endothelial cell dysfunction and apoptosis (Figure 7A), glycogen metabolism, TCA cycle, acetyl CoA biosynthesis. The level of expression of the pyruvate dehydrogenase (PDH) components (alpha and beta E1 components of the complex, PDH phosphatase) were maintained at control levels by macitentan in the oxidative phosphorylation deficient myocardium induced by sunitinib (Figure 7B). Interestingly, the levels of expression of some proteins involved in glycolysis were augmented by 3 weeks of sunitinib treatment (e.g. glucose-6-phosphate isomerase and phosphoglycerate mutase), while those of others were reduced, e.g. muscle-type phosphofructokinase and glyceraldehyde-3-phosphate dehydrogenase. Sunitinib-treated hearts showed higher lactate dehydrogenase (LDH) that is released during tissue hypoxia and damage. Surprisingly, sunitinib induced a dramatic (8-fold) increase in the expression of GLUT4 (SLC2a4), the insulin-regulated transporter of glucose at the plasma membrane. However, the level of Rab10, a small ras-family GTPase required for translocation of GLUT4, was significantly decreased. Together with a trend for elevated blood sugar in the sunitinib treated group (190±38 mg/dL), these results were coherent with an insulin-resistant type of diabetic metabolic profile of the sunitinib treated hearts. Addition of macitentan to sunitinib suppressed the diabetic-like clusters (Figure 7C), returned glycaemia to pre-treatment levels (171±17 mg/dL and 173±23 mg/dL in the vehicle and sunitinib+macitentan treated groups, respectively), and activated clusters for glycolysis. Macitentan only partially reverted the effects of sunitinib on the level of GLUT4 and Rab10, but significantly increased the level of expression of the pleiotropic regulatory protein sirtuin 2, in line with the role of this protein in energetic metabolism preservation.
Discussion
Sunitinib treatment is limited by its cardiovascular side effects. A recent study reported that sunitinib induces an early switch of cardiac metabolism to anaerobic glycolysis and impairs heart function (30). Here, we show that in addition to this metabolic switch, myocardial remodeling by sunitinib also induces a reduced glucose uptake resembling the one found during insulin resistance, and show that sunitinib cardiotoxicity is a combination of several complex mechanisms occurring over a sequential time course. Moreover, we show that sunitinib-induced cardiac injury and dysfunction are prevented through inhibition of endothelin signaling, strongly supporting a role for this pathway in sunitinib's cardiotoxic effects (Figure 8).
Our results confirm that FDG-PET is able to quantify the metabolic changes induced by sunitinib administration. It remains unclear why this has not been noted previously given the widespread use of FDG-PET in the staging and follow-up of cancer patients. One explanation might be that myocardial glucose metabolism is a complex, tightly regulated mechanism that varies according to nutrient and oxygen levels. Under physiological conditions, the heart preferentially uses oxidization of fatty acids as fuel and switches to glycolysis in high glucose and insulin or low oxygen conditions by activating synthesis and translocation to the plasma membrane of the glucose transporter GLUT4 (36). Therefore, myocardial FDG uptake varies according to fasting status, diet, diabetes and ischemia (37). In mouse and man, FDG uptake is higher in non-fasting than in fasting conditions (37) and in oncology studies FDG PET imaging is acquired under fasting conditions in order to minimize muscular and myocardial uptake and improve tumor detection. In contrast, cardiac PET-FDG often utilizes an euglycemic clamp (glucose load with additional insulin administration after overnight fasting) in order to maximize heart uptake (37, 38). Interestingly, in the case of vascular dysfunction, PET images of the heart look different under fasting and non-fasting conditions: ischemic territories appear as hot spots with higher FDG uptake than the intact myocardium after fasting (39) while they may not differentiate from intact tissue in non-fasted conditions after a glucose load (40-42). In the absence of sunitinib treatment, FDG-SUV and metabolic flux were lower in fasted than in non-fasted states. We show here that the increase in FDG uptake after one week of sunitinib was more pronounced in fasted than in non-fasted mice. Therefore, dietary status also influences the FDG cardiac uptake under sunitinib treatment.
Systemic administration of sunitinib rapidly induces a metabolic switch towards glycolysis with reduced expressions of key enzymes of the TCA cycle and key proteins for mitochondrial oxidative phosphorylation (OXPHOS) and for the beta-oxidation of fatty acids. We observed this switch regardless of whether the animals were fasted or not (although with a different intensity in both conditions) at one and 3 weeks after beginning of the anti-angiogenic treatment. O'Farrel et al. also showed increased FDG uptake as early 2-3 days after introduction of sunitinib in mice and 5 days in rats (30). Overall, the sunitinib-treated heart tends to switch towards an anaerobic glycolysis metabolism with accumulation of lactate, similar to the one found in hypoxic muscles and during heart failure (43). At the proteomic level, the changes in the expression of proteins involved in metabolic pathways in the hearts of non-fasted animals treated during one and three weeks were largely consistent. For instance, 3 weeks of treatment decreased the PDH complex and increased levels of LDH, confirming the glycolytic switch combining inhibition of the pathways leading to acetyl CoA biosynthesis and diversion toward anaerobic conversion of pyruvate to lactate. Sunitinib-treated hearts were also unlikely to use glycogen as energy source since they expressed very low phosphorylase B kinase, in agreement with the elevated glycogen level described by Rees et al. (32). Interestingly, the mechanism of sunitinib's cardiac toxicity is not due to a reduction in blood vessel density in the myocardium. This is different from the action on tumor vessels, whose number is drastically reduced after a short course of sunitinib (44).
Figure 5
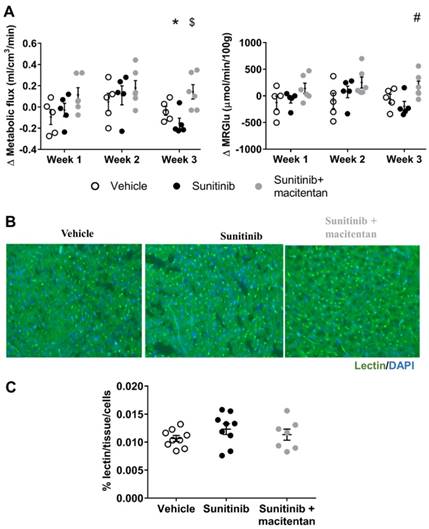
Macitentan prevents myocardial flux dysfunction induced by sunitinib. (A) Difference treatment - baseline of myocardial metabolic flux and metabolic rate of glucose (MRglu) for vehicle (n=5), sunitinib (n=5) and sunitinib+macitentan (n=6) groups. Data are expressed as mean ± SEM, *p <0.05 compared to baseline, $p <0.05 compared to other at week 3. #p <0.05 compared to sunitinib group at week 3. (B) Micrographs of cardiac sections of microvascular staining (lectin in green and nuclei in blue) at D22 in non-fasted C57Bl/6 mice. (C) Quantification of vessels area reported on number of cells for vehicle (n=9), sunitinib (n=9) and sunitinib+macitentan (n=7) groups at D22 in non-fasted C57Bl/6 mice. MRGlu: metabolic rate of glucose.
Figure 6
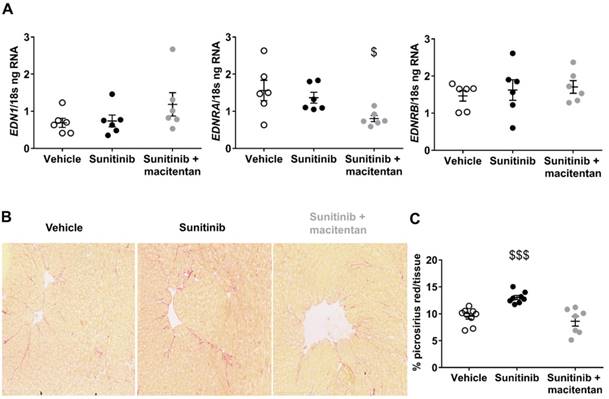
Protective effects of macitentan involve ET receptors: (A) ng of ET-1, ETA and ETB receptor mRNA reported on ng of 18s RNA in the myocardium (n=6 for each). (B) Micrographs of cardiac sections of fibrosis marker (Picrosirius red) at D22 non-fasted C57Bl/6 mice. (C) Quantification of fibrosis for vehicle (n=9), sunitinib (n=9) and sunitinib+ macitentan (n=7) groups at D22 in non-fasted C57Bl/6 mice. Data are expressed as mean ± SEM, $p <0.05; $$$p <0.001 compared to other. EDN1: Preproendothelin-1; EDNRA: endothelin receptor type A; EDNRB: endothelin receptor type B.
Figure 7
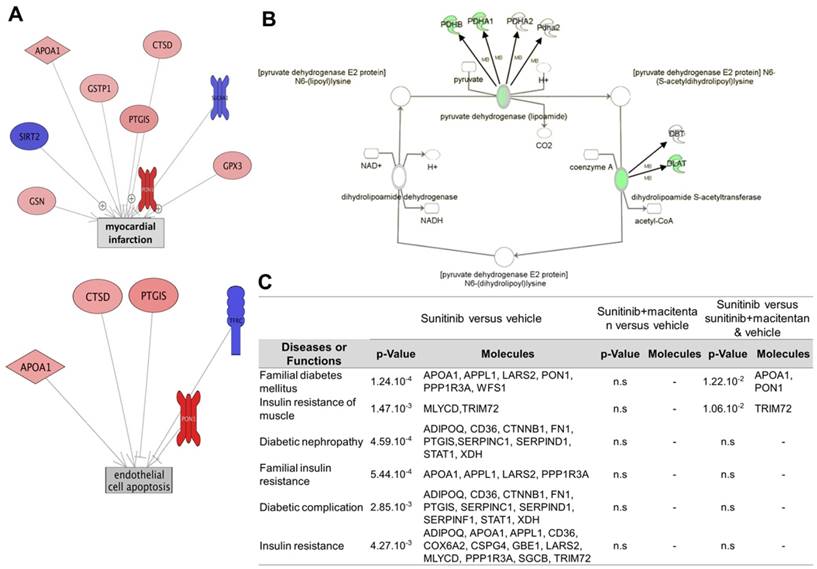
Cardioprotective effects of macitentan. (A) Pathway studio representations of proteins involved in myocardial infarction and endothelial cell apoptosis showing the changes in protein expression levels in the myocardium of the sunitinib group and the (vehicle and sunitinib+macitentan) groups. Red indicates significantly (p<0.05) upregulated proteins, blue significantly (p<0.05) down-regulated proteins. (B) Ingenuity representation of the changes in Pyruvate dehydrogenase complex expression in the myocardium of the sunitinib group in comparison to the (vehicle and sunitinib+macitentan) groups. Green shows significantly (p<0.05) downregulated proteins in the sunitinib group. (C) Table of the diabetes-related pathways most significantly perturbed by sunitinib in mouse myocardium and correction by macitentan. n=6 for each group. n.s: non-significant. Proteins described in Table 2.
Figure 8
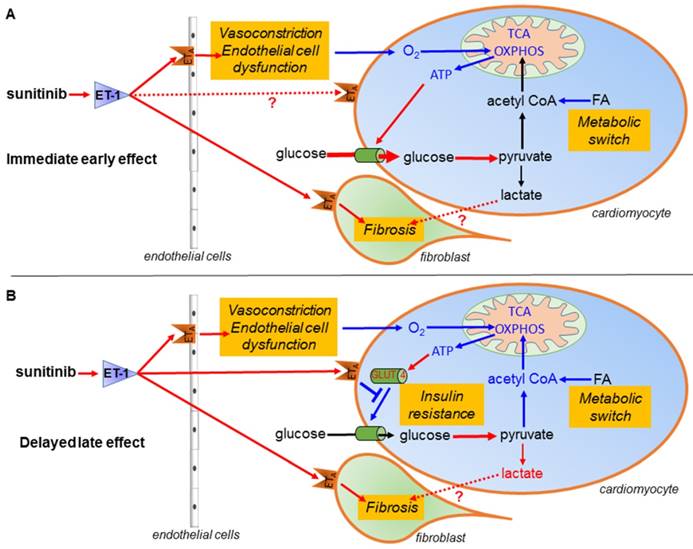
Schematic representation of the mechanism of sunitinib-induced cardiac side effects: (A) Sunitinib upregulates glycolysis and downregulates oxidative metabolism in cardiac mitochondria. (B) Sunitinib induces resistance to insulin stimulation of cardiac glucose uptake. The metabolic switch is an immediate early response to sunitinib while insulin resistance either appears later or is masked by the metabolic switch during the early stages of sunitinib treatment. Both mechanisms depend on signaling by the endothelin pathway, and lead to myocardial fibrosis and impaired cardiac function, and are reversed by the endothelin receptors antagonist macitentan. Red indicates upregulated proteins and pathways, blue indicates downregulated protein and pathways. ATP: Adenosine triphosphate; ET-1: endothelin 1; ETA: endothelin receptor type A; FA: fatty acid; GLUT: glucose transporter protein; O2: oxygen; OXPHOS: oxidative phosphorylation; TCA: tricarboxylic acid.
Here, macitentan treatment prevented the deficiency in acetyl CoA biosynthesis and impairment of the TCA cycle and suppressed the protein patterns typical of endothelial cell apoptosis and myocardial infarction. Others have shown that macitentan also prevented the hypertension associated with sunitinib (25). Our results suggest that the cardiotoxic effects of sunitinib are mediated by the ETA receptor. The current explanation for the cardiac toxicity of sunitinib is a direct toxicity on the myocardium with mitochondrial dysfunction, that is exacerbated by hypertension since cardiomyocytes use glucose as main energy source when subjected to pressure overload (45). Reduced availability of oxygen in the myocardium will also lead to rapid metabolic changes with increased FDG uptake. This is the case in the hearts of fasted mice at one day after transverse aortic constriction surgery (46). Moreover, during fasting the ischemic territories of the heart show a higher FDG uptake than healthy myocardium that prefers fatty acids as energy substrate (47). The present data support a role for the endothelin system in mediating switch towards anaerobic metabolism.
We observed a decreased metabolic flux in the hearts of mice treated with sunitinib for 3 weeks. Although they did not elaborate on this result, O'Farrell et al. observed the same trend and showed a similar metabolic rate of glucose at 3 weeks of treatment in sham and sunitinib treated animals (30). Interestingly, in their study, as in ours, the apparent reversal of the early metabolic switch induced by sunitinib was not accompanied by an improvement in the left ventricular ejection fraction. This delayed effect of sunitinib may be in line with a case report describing decreased myocardial FDG uptake in patients treated with imatinib plus sorafenib who later developed a cardiac event (48). Therefore, we explored the possible mechanisms underlying this observation. On the one hand, sunitinib-treated hearts presented anaerobic metabolism and it is known that PDH inhibition leads to a slow recovery of glucose uptake and uncoupling of glycolysis (49), and that lactate accumulation decreases glucose uptake (43). On the other hand, patterns of protein expression in the 3-week sunitinib-treated heart resembled the one seen in diabetic patients in which the FDG metabolic flux is reduced (50), as well as the pattern in diabetic rats where myocardial glucose uptake is reduced under ischemic conditions (51). Despite the lower metabolic rate of glucose in sunitinib-treated hearts, we observed a dramatic increase of GLUT4 activated in high glycaemia-high insulinemia conditions (36). However, the expression of Rab10, that controls the plasma membrane insertion of GLUT4 and whose activation is under control of the AS160 GTPase, a substrate of AMPK which is known to be inhibited by sunitinib (22), was reduced. Therefore, it is likely that although overexpressed in 3-week sunitinib treated hearts, GLUT4 was not inserted in the plasma membrane and not actively transporting glucose or FDG inside the cardiomyocytes.
Overall our results indicate that, in mice, a 3-week sunitinib course (the regimen is usually 4 weeks in patients) induces a form of cardiac insulin resistance. This is not totally surprising as sunitinib and TKI are known to affect glucose levels to the point that they have been considered as potential drugs for type 2 diabetes (52). In our study, the addition of macitentan largely improved the metabolic rate of glucose and suppressed the sunitinib-induced insulin resistance and diabetic patterns. Moreover, ETA activation is associated with impaired glucose uptake via the inhibition of the AMPK/Akt signaling pathway of the translocation of GLUT4 in skeletal muscle (53). Here, we demonstrate that the sunitinib-induced reduction in glucose uptake after prolonged treatment is mediated by the ETA receptor through the inhibition of insulin-stimulated AMPK. Other studies have shown that disruption of the endothelin pathway is associated with impaired glucose uptake in skeletal muscle. Shemyakin et al. showed decreased insulin-stimulated Akt phosphorylation by ET-1, and infusion of ET-1 reduced insulin sensitivity in humans and in animals (54,55). In addition, ET-1 blockade by ETA/ETB receptors inhibition increases glucose uptake in patients with insulin resistance (54, 55).
Our results suggest that two mechanisms are at work in the cardiotoxic effects of sunitinib: (i) in the first days after initiation of treatment, a metabolic switch towards glycolysis with increased FDG uptake and, (ii) evidenced after at least 3 weeks of treatment, a type of insulin resistance that leads to subnormal FDG uptake. Our present data show that the cardiotoxicity of sunitinib is for a good part related to a deregulation of cardiac metabolism, which in some respect is similar to the one observed in other situations. It has been demonstrated that during ischemia, the heart is not able to use its metabolic reserve (56) and is associated with lactate acidosis leading to contractile dysfunction (57). Similarly, hypertrophic hearts are not able to increase glucose uptake enough to sustain the energy demand (58), and by-products from glycolysis and glycogen utilization are required for proper myocardium relaxation (59). This metabolic dead end is concomitant with major deleterious consequences on three key components of cardiac physiology: (i) mitochondrial homeostasis; (ii); fibrosis and (iii) contractile function. Fibrosis is responsible for impaired myocardial relaxation and we show here that impaired cardiac output was due to a decreased left ventricular volume during the diastole (LVID,d), responsible for diastolic dysfunction (60, 61). This has functional consequences with decreased flow velocities, leading to cardiac dysfunction.
The therapeutic perspective is to control and, whenever possible, to prevent the cardiotoxicity of anticancer drugs. Regarding sunitinib, our results show that the administration of macitentan reverts most, if not all, the effects of sunitinib on cardiac metabolism and prevents impairment of left ventricular function and fibrosis. Macitentan is a mixed ETA/ETB antagonist approved for the treatment of pulmonary arterial hypertension and has no known direct or indirect interaction with sunitinib of other TKI (FDA). Furthermore, non-selective ET receptors antagonism prevents hypertension and renal injury (24) induced by sunitinib. In the future, it will be interesting to test if the use of a selective ETA receptor antagonism produces comparable cardioprotection since ET-1 is known to exert a pro-fibrotic action (62) and hypertrophy (34) via the ETA receptor.
We have shown here that co-administration of macitentan prevents deregulation of myocardial metabolism and cardiac fibrosis and restores the diastolic function impaired by sunitinib. Taken together, these results support the administration of a mixed ETA/ETB inhibitor such as macitentan to protect the myocardium during sunitinib treatment. Our pre-clinical results call for clinical imaging studies aiming to identify the risk of developing side effects at an early stage of sunitinib treatment, and consequently to administer preventive therapy (22). Because of its clinical importance, further exploration of the capacity of macitentan therapy to protect the heart from sunitinib-induced cardiac dysfunction is worth exploring in a theranostic approach.
Materials and Methods
Experimental design
Animal experiments were authorized by the French Ethical committee for animal experimentation under No. 15-045 and performed by certified personal following the French law of animal welfare on animal experimentation n°2013-118. Female nude (nu/nu) and C57BL/6 mice (Janvier Labs, France) 15 weeks aged were maintained in controlled temperature (24°C) and relative humidity (50%) on a 12/12-light/dark cycle and were fed ad libitum. Sunitinib malate (Sutent®, Pfizer, USA) was dissolved at 10mg/mL in DMSO/PBS (1:4). 40 mg/kg/ml is the minimum dosage that permit to demonstrate anti-tumor effect and produce comparable plasma concentrations to those found in patients (8,63,64). However, preliminary results in our institution demonstrated the variability in the results with this dosage. Based on that, we administered sunitinib daily by oral gavage in a dose of 50 mg/kg body weight to improve homogeneity and reduce number of animals. In the sunitnib+macitentan group, macitentan (20mg/mL) was added to the gavage solution, with the volume maintained at 150µL.
This study followed a standard protocol for monitoring of short-term response to therapy in fasted tumor-bearing nude mice. Mice were injected bilaterally in the left and right flanks subcutaneously with 1 x 107 Cricetulus griseus (CCL-39) tumor cells suspended in 300µl of culture medium. The PET scan was performed before and 2 days after a 5-day course of 50mg/kg sunitinib malate (treatment group) or vehicle (vehicle group) (Figure 1A). To check whether the increased SUV was independent of the presence of a tumor in immunodeficient animals, the same protocol was reproduced in C57BL/6 immunocompetent mice fasted before the PET examination. Chow diet was removed the day before the experiment for a 12 hours fasting. Cardiac echography was also performed to measure heart function (Figure 1B). Finally, to explore the pathophysiological mechanisms of sunitinib cardiotoxicity, PET-FDG and echocardiography were performed in non-fasted mice at baseline and 1 and 3 weeks of a three-week course of sunitinib or vehicle. A third group with co-administration of sunitinib and the endothelin receptor antagonist macitentan was added to test the hypothesis that the cardiac effects of sunitinib were, in part, mediated by the endothelin pathway (Figure 1C). For all the panels, animals of both groups were randomly assessed, experiment and analysis were realized blinded and animals from both groups were image the same day.
Cardiac Positron Emission Tomography (PET)
All mice from groups A and B were fasted overnight. Mice from panel C were not fasted and had free access to food and water. Mice were anesthetized (2±0.5% isoflurane in air), weighted and glycemia was measured in blood drawn from the caudal ventral artery using an Accu-Chek® Aviva Nano A (Accu-Chek, France). A catheter home-made from a 26G needle (Fischer Scientific, France) connected to a 5cm polyethylene tubing (Tygon Microbore Tubing, 0.010" x 0.030"OD; Fisher Scientific, France) was inserted in the caudal vein for radiotracer injection. Mice were then installed into the PET-CT dedicated bed and respiration and body temperature were registered. Body temperature was maintained at 37°C and anesthesia was controlled on the breathing rate throughout the entire examination. CT was acquired in a PET-CT scanner (nanoScan PET-CT; Mediso, Hungary) using the following acquisition parameters: semi-circular mode, 39kV tension, 720 projections full scan, 300ms per projection, binning 1:4. Then, PET acquisition was started and, 30 seconds later, 10MBq of 2'-deoxy-2'-[18F]fluoro-D-glucose (FDG; Advanced Applied Applications, France) in 0.2mL saline was injected via the catheter. The first scan was a dynamic acquisition of 30.5min and was followed by a gated cardiac scan of 30min duration. PET data were collected in list mode and binned using a 5ns time window, with a 400-600keV energy window and a 1:5 coincidence mode. Data were reconstructed using the Tera-Tomo reconstruction engine (3D-OSEM based manufactured customized algorithm) with expectation maximization iterations, scatter and attenuation correction. The first scan was reconstructed starting 10s before FDG injection with the following time sequence: 26 x 5s; 6 x 30s; 5 x 120s; 3 x 300s and 3 x 600s. The second scan reconstructed in a single time frame from 45 to 60min post-injection.
FDG accumulation was quantified as mean Standard Uptake Value (SUV, ratio of the radioactivity concentration in myocardium on the whole body concentration of the injected radioactivity) between 45 and 60min post-injection in 3D volumes-of-interest (VOI) delineated semi-automatically by iso-contours at 45% threshold of maximal value in the myocardium on PET/CT fusion slices using the PMOD software package (PMOD Technologies Ltd, Zürich, Switzerland). Metabolic flux was quantified using compartmental modeling tool of PMOD software using the same VOI as above and a VOI semi-automatically delineated on the vena cava for the arterial input function as previously described (65). Metabolic rate of glucose were calculated by the multiplication of the metabolic flux by [plasma glucose (mmol/l)/lumped constant (fixed at 0.69)].
Echocardiography
Conventional echocardiography was performed in anesthetized mice using a Vevo 2100 high resolution ultrasound device (Visualsonics, Toronto, Canada) with a 40MHz probe (MS-550). Mice were anesthetized with 3% isoflurane in air for induction and maintained with 1.5%. Mice were depilated in the thoracic region and then placed in the supine position on a dedicated heating platform, allowing monitoring of ECG, temperature and respiratory frequency. All acquisitions were performed within body temperature limits 36-37.5°C. Parasternal long axis views were recorded and 3 consecutive measurements in M-mode were drawn to determine Left Ventricular Internal Diameter (LVID) at diastole (d) and systole (s) and Left Ventricular Posterior Wall thickness (LVPW) in both telediastole (d) and telesystole (s). Cardiac Output (CO) and the percentage of fractional shortering (FS) were then calculated using the VevoLab Software (Visualsonics). Mitral flow deceleration time was calculated from mitral flow velocities using Pulsed Wave Doppler (PW Doppler). Ascending aorta diameter (Ao) and Left Atrium diameter (LA) were measured using M-mode in parasternal long axis. Aortic velocity tracking integral (AoVTI) was measured using PW Doppler, in suprasternal view allowing measurement of mean aortic velocity and peak aortic velocity.
Assessment of microvascular density
Cardiac microvessels were stained using Isolectine B4 Griffonia Simplicifolia-FITC (Sigma Aldrich). Nuclei were counter stained with DAPI. Microvessels and nuclei were counted in 4 fields at a magnification of x200 in 2 independent sections from each heart using Matlab® based software. Microvessel density was normalized to the number of nuclei.
Assessment of cardiac fibrosis
Frozen sections were incubated with Picrosirius red (VWR) 0.1% in picric acid (Sigma) in a Leica ST5020 automatic stainer during 30min and dehydrated in ethanol and xylene. Whole sections were observed at a magnification of x20 using a Nanozoomer HT 2.0 (Hamamatsu) and fibrosis was quantified using Matlab® based software.
Western blotting
Twenty micrograms of each heart lysate were loaded onto a 10% SDS-PAGE gels (mini-protean TGX gels, BioRad) and transferred to nitrocellulose membranes. The membranes were blocked and immunoblotted with the following primary antibodies: GLUT1 (1:1000, ab652, Abcam), PGC1α (1:1000, ab54481, Abcam) and hexokinase II (1:500, bs-3993R, Interchim). Membranes were then incubated with horseradish peroxidase (HRP)-conjugated anti-rabbit secondary antibodies (1:10000, 474-1506, KPL). Chemiluminescence detection was performed using the ECL kit (Clarity Western ECl substrate; BioRad). Quantitation of immunoblots was done on digitalized images using ImageJ software. The intensity of immunoreactive bands was normalized by the loading control (Cyclophilin B, 1:1000, ab16045, Abcam).
Proteomics
Tissue sample preparation
Frozen mouse hearts were individually ground under liquid nitrogen to yield a fine powder using a pestle and mortar. The tissue powder was weighted and solubilized in lysis buffer (4% SDS, 100mM Tris-HCl, pH 8.0). Protein extracts were clarified by centrifugation at 21,000×G, 1 hour, 4°C. Protein concentration of the supernatant was determined using bicinchoninic acid assay (BCA, Pierce). Peptides were prepared by Filter Aided Separation method (FASP) essentially as described (66). Briefly, 50µg of proteins from whole lysates were diluted to 100µL in solubilization buffer (50 mM Tris/HCl, pH8.5, SDS 2%, 20mM TCEP, 50 mM chloroacetamide) and heated for 5 min at 95°C. After cooling to room temperature, extracts were diluted with 300µL Tris Urea buffer (Urea 8M, Tris/HCl 50mM (pH 8.5) and transferred onto 30kDa centrifuged filters and prepared for digestion as described (66). Proteins were digested during 14h at 37°C with µg trypsin (Promega) and peptides were desalted on C18 StageTips (67). After drying, peptides were solubilized in 2% trifluoroacetic acid (TFA) and fractionated by strong cationic exchange (SCX) StageTips, mainly as described (68) except that fractions 1 and 2 were pooled in most experiments.
Mass spectrometry analysis
Mass spectrometry analyses were performed on a Dionex U3000 RSLC nano-LC- system coupled to a Q-Exactive mass spectrometer (Thermo Fisher Scientific). After drying, peptides from SCX StageTip fractions were solubilized in 10 µL of 0.1% TFA containing 2% acetonitrile (ACN). One µL was loaded, concentrated and washed for 3min on a C18 reverse phase precolumn (3µm particle size, 100 Å pore size, 75 µm inner diameter, 2 cm length, Dionex, Thermo Fisher Scientific). Peptides were separated on a C18 reverse phase resin (2 µm particle size, 100Å pore size, 75µm inner diameter, 25cm length from Dionex) with a 3-hour gradient starting from 99% of solvent A containing 0.1% formic acid in H2O and ending in 40% of solvent B containing 80% acetonitrile, 0.085% formic acid in H2O. The mass spectrometer acquired data throughout the elution process and operated in a data-dependent scheme with full MS scans acquired with the Orbitrap, followed by up to 10 MS/MS HCD fragmentations in the Q-Exactive (Thermo Fisher) on the most abundant ions detected. Settings were essentially as in (69) with slight modifications: the recurrent loop of the 10 most intense nLC-eluting peptides were HCD-fragmented between each full scan (data dependent mode). Resolution was set to 70,000 for full scans at AGC target 1,10e6 within 60ms MIIT. The MS scans spanned from 350 to 1500m/z. Precursor selection window was set at 2Th, and MS/MS scan resolution was set at 17,500 with AGC target 1,10e5 within 60ms MIIT. HCD Normalized Collision Energy (NCE) was set at 27%. Dynamic exclusion was set to 30s duration. Spectra were recorded in profile mode. The mass spectrometry data were analyzed using Maxquant version 1,5,2,8 (70). The database used was a concatenation of human sequences from the Uniprot-Swissprot database (Uniprot, release 2015-02) and a list of contaminant sequences from Maxquant. The enzyme specificity was trypsin. The precursor mass tolerance was set to 4.5ppm and the fragment mass tolerance to 20ppm for Q-Exactive data. Carbamidomethylation of cysteins was set as constant modification and acetylation of protein N-terminus and oxidation of methionines were set as variable modification. Second peptide search was allowed and minimal length of peptides was set at 7 amino acids. False discovery rate (FDR) was kept below 1% on both peptides and proteins. Label-free protein quantification (LFQ) was done using both unique and razor peptides. At least 2 such peptides were required for LFQ. The “match between runs” (MBR) option was allowed with a match time 0.7 min window and an alignment time window of 20min. For analysis, LFQ results from MaxQuant were imported into the Perseus software (version 1.5.1.6). Reverse and contaminant proteins were excluded from analysis. Contaminating proteins from culture medium, essentially coming from added serum, such as immunoglobulins or transferrin were also removed from the protein list. Protein copy numbers per cell were then calculated using the “Protein ruler” plugin of Perseus by standardization on total histone MS signal as described (71).
Modeling with Ingenuity® and Pathway Studio®
We used ingenuity pathway analysis (Ingenuity Systems, Redwood City, CA) and Pathway Studio (https://www.pathwaystudio.com/) to study pathways deregulated by sunitinib as already described (72). For both, data from proteomic were classified according to the fold change and p-value compared to reference group (i.e: vehicle) using a paired t.test and only significant deregulated proteins were enter in the software. Briefly, these software create hypothetical networks (including several proteins) to highlight networks that are significantly (p <0.05) different compared to a reference group (i.e: vehicle group or sunitinib + macitentan group). Those networks were assigned to a biological function(s), pathway(s) and/or disease(s) using the Ingenuity Pathways Knowledge Base, Pathway Studio database and the scientific literature.
RNA extraction and qPCR
RNA from hearts and aorta was isolated using TRI Reagent Solution (Invitrogen). Any DNA present was degraded using RQ1 RNAse-Free DNase (Promega) according to the manufacturer's instructions. cDNA was synthesized using a High Capacity cDNA Reverse Transcription kit with RNase inhibitor (Invitrogen). qRT-PCR was performed using Fast SYBR Green Master Mix (Applied Biosystems) on the ABI7900 System (Applied Biosystems). Primers for 18s, prepro-ET-1, ETA and ETB receptors were generated. The amplification reaction mixture was heated at 95°C for 20s, then subject to 40 cycles of 95°C for 1s then 60°C for 20s. Values obtained for experimental gene measurements were normalized against expression of 18s.
Statistical analysis
Statistical analysis and data comparison for proteomics data were done using either Perseus or Excel software to classify the data using the two tailed Student t.test. In modeling software, p-value cutoff was 0.05 calculated with right-tailed Fisher Exact Test (Ingenuity®) and with Mann-Whitney U-test (Pathway Studio®). Data are expressed as mean ± SEM. Analyses were performed with Graphpad Prism (7.00). Unpaired and paired t tests were used to compare two data sets, one-way ANOVA was used to compare three data sets, and two-way ANOVA was used to compare three groups followed across time.
Abbreviations
AMPK: 5' AMP-activated protein kinase; Akt: Protein kinase B; AoVTI: Aortic velocity tracking integral; CO: cardiac output; ET (EDN1): endothelin; ETA (EDNRA): endothelin receptor type A; ETB (EDNRB): endothelin receptor type B; FA: fatty acid; FC: fold change; FDG: 2'-deoxy-2'-[18F]fluoro-D-glucose; FS: fractional shortening; GLUT: glucose transporter protein; HIF: hypoxia-inducible factor; HK: hexokinase; IR: insulin resistance; LVEJ: left ventricular ejection fraction; LVID: left ventricular internal diameter; MRGlu: metabolic rate of glucose; MRI: magnetic resonance imaging; OXPHOS: oxidative phosphorylation; PDGF-Rs: platelet-derived growth factor receptors; PDH: pyruvate dehydrogenase; PET: positon emission tomography; PGC1: Peroxisome proliferator-activated receptor gamma coactivator 1-alpha; SUV: standard uptake values; TKR: tyrosine kinase receptor; TCA: tricarboxylic acid; VEGFRs: vascular endothelial growth factor; Voi: volum of interest.
Acknowledgements
In vivo imaging was performed at the Life Imaging Facility of Paris Descartes University (Plateforme Imageries du Vivant - PIV), supported by France Life Imaging (grant ANR-11-INBS-0006) and Infrastructures Biologie-Santé (IBISA). The authors are grateful to Fanny Aprahamian and Mélanie Rouil for help with histology, to Corinne Lesaffre of the PARCC Histology Core Facility, to Philippe Chafey, Morgane Le Gall and François Guillonneau from the 3P5 proteomic facility of Paris Descartes University and to Professor Elie Mousseaux for helpful discussions.
Funding
This study was supported by the Institut National de la Santé et de la Recherche Médicale, Paris Descartes University and France Life Imaging. Joevin Sourdon was supported by a scholarship of the Ministère de l'Enseignement Supérieur et de la Recherche.
Author contributions
JS conceived the study, performed all experiments and wrote the paper. FL, JS and GR performed ultrasound imaging, TV and JS performed PET imaging, RM and ND performed the PCR experiments. DB provided the software for data analysis. PLT provided macitentan. JS, ND and BT conceived the study, analyzed the results and wrote the paper.
Spectrometry proteomics data
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (73) partner repository with the dataset identifier PXD006888.
Supplementary Material
Supplementary figures.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Tolba KA, Deliargyris EN. Cardiotoxicity of cancer therapy. Cancer Invest. 1999;17(6):408-22
2. Monsuez J-J, Charniot J-C, Vignat N, Artigou J-Y. Cardiac side-effects of cancer chemotherapy. Int J Cardiol. 2010Sep24;144(1):3-15
3. Herrmann J, Yang EH, Iliescu CA, Cilingiroglu M, Charitakis K, Hakeem A. et al. Vascular Toxicities of Cancer Therapies: The Old and the New-An Evolving Avenue. Circulation. 2016Mar29;133(13):1272-89
4. Gottlieb S. Cancer drug may cause heart failure. BMJ. 2000Jul29;321(7256):259
5. Kounis NG, Koniari I, Hahalis G. Cardio-oncology, Immuno-oncology, Onco-cardiology and Onco-immunology. Int J Cardiol. 2016Nov15;223:254-7
6. Verheul HMW, Pinedo HM. Possible molecular mechanisms involved in the toxicity of angiogenesis inhibition. Nat Rev Cancer. 2007Jun;7(6):475-85
7. Kerbel R, Folkman J. Clinical translation of angiogenesis inhibitors. Nat Rev Cancer. 2002Oct;2(10):727-39
8. Chu TF, Rupnick MA, Kerkela R, Dallabrida SM, Zurakowski D, Nguyen L. et al. Cardiotoxicity associated with tyrosine kinase inhibitor sunitinib. Lancet Lond Engl. 2007Dec15;370(9604):2011-9
9. Kappers MH, van Esch JHM, Sleijfer S, Danser AHJ, van den Meiracker AH. Cardiovascular and renal toxicity during angiogenesis inhibition: clinical and mechanistic aspects. J Hypertens. 2009Dec;27(12):2297-309
10. Kappers MHW, van Esch JHM, Smedts FMM, de Krijger RR, Eechoute K, Mathijssen RHJ. et al. Sunitinib-induced hypothyroidism is due to induction of type 3 deiodinase activity and thyroidal capillary regression. J Clin Endocrinol Metab. 2011Oct;96(10):3087-94
11. Kamba T, McDonald DM. Mechanisms of adverse effects of anti-VEGF therapy for cancer. Br J Cancer. 2007Jun18;96(12):1788-95
12. Chrisoulidou A, Mandanas S, Margaritidou E, Mathiopoulou L, Boudina M, Georgopoulos K. et al. Treatment compliance and severe adverse events limit the use of tyrosine kinase inhibitors in refractory thyroid cancer. OncoTargets Ther. 2015Sep3;8:2435-42
13. Hartmann JT, Kanz L. Sunitinib and periodic hair depigmentation due to temporary c-KIT inhibition. Arch Dermatol. 2008Nov;144(11):1525-6
14. Motzer RJ, Hutson TE, Tomczak P, Michaelson MD, Bukowski RM, Rixe O. et al. Sunitinib versus interferon alfa in metastatic renal-cell carcinoma. N Engl J Med. 2007Jan11;356(2):115-24
15. Demetri GD, van Oosterom AT, Garrett CR, Blackstein ME, Shah MH, Verweij J. et al. Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib: a randomised controlled trial. Lancet Lond Engl. 2006Oct14;368(9544):1329-38
16. Raymond E, Dahan L, Raoul J-L, Bang Y-J, Borbath I, Lombard-Bohas C. et al. Sunitinib malate for the treatment of pancreatic neuroendocrine tumors. N Engl J Med. 2011Feb10;364(6):501-13
17. Lankhorst S, Kappers MHW, van Esch JHM, Danser AHJ, van den Meiracker AH. Mechanism of hypertension and proteinuria during angiogenesis inhibition: evolving role of endothelin-1. J Hypertens. 2013Mar;31(3):444-454 discussion 454
18. Meiracker AH van den, Danser AHJ, Sleijfer S, Kappers MHW. Re: Hypertension as a Biomarker of Efficacy in Patients With Metastatic Renal Cell Carcinoma Treated With Sunitinib. J Natl Cancer Inst [Internet]. 2011 Aug 23. http://jnci.oxfordjournals.org/content/early/2011/08/23/jnci.djr328
19. Chintalgattu V, Rees ML, Culver JC, Goel A, Jiffar T, Zhang J. et al. Coronary microvascular pericytes are the cellular target of sunitinib malate-induced cardiotoxicity. Sci Transl Med. 2013May29;5(187):187ra69
20. Moslehi J, Minamishima YA, Shi J, Neuberg D, Charytan DM, Padera RF. et al. Loss of hypoxia-inducible factor prolyl hydroxylase activity in cardiomyocytes phenocopies ischemic cardiomyopathy. Circulation. 2010Sep7;122(10):1004-16
21. Kappers MHW, Esch JHM van, Sluiter W, Sleijfer S, Danser AHJ, Meiracker AH van den. Hypertension Induced by the Tyrosine Kinase Inhibitor Sunitinib Is Associated With Increased Circulating Endothelin-1 Levels. Hypertension. 2010Oct1;56(4):675-81
22. Yang Y, Bu P. Progress on the cardiotoxicity of sunitinib: Prognostic significance, mechanism and protective therapies. Chem Biol Interact. 2016Sep25;257:125-31
23. Dhaun N, Webb DJ. Receptor tyrosine kinase inhibition, hypertension, and proteinuria: is endothelin the smoking gun? Hypertens Dallas Tex 1979. 2010Oct;56(4):575-7
24. Lankhorst S, Kappers MHW, van Esch JHM, Smedts FMM, Sleijfer S, Mathijssen RHJ. et al. Treatment of hypertension and renal injury induced by the angiogenesis inhibitor sunitinib: preclinical study. Hypertens Dallas Tex 1979. 2014Dec;64(6):1282-9
25. Dhaun N, MacIntyre IM, Kerr D, Melville V, Johnston NR, Haughie S. et al. Selective endothelin-A receptor antagonism reduces proteinuria, blood pressure, and arterial stiffness in chronic proteinuric kidney disease. Hypertens Dallas Tex 1979. 2011Apr;57(4):772-9
26. Kongbundansuk S, Hundley WG. Noninvasive imaging of cardiovascular injury related to the treatment of cancer. JACC Cardiovasc Imaging. 2014Aug;7(8):824-38
27. Pizzino F, Vizzari G, Qamar R, Bomzer C, Carerj S, Zito C. et al. Multimodality Imaging in Cardiooncology. J Oncol. 2015;2015:263950
28. D'Amore C, Gargiulo P, Paolillo S, Pellegrino AM, Formisano T, Mariniello A. et al. Nuclear imaging in detection and monitoring of cardiotoxicity. World J Radiol. 2014Jul28;6(7):486-92
29. Borde C, Kand P, Basu S. Enhanced myocardial fluorodeoxyglucose uptake following Adriamycin-based therapy: Evidence of early chemotherapeutic cardiotoxicity? World J Radiol. 2012May28;4(5):220-3
30. O'Farrell AC, Evans R, Silvola JMU, Miller IS, Conroy E, Hector S. et al. A Novel Positron Emission Tomography (PET) Approach to Monitor Cardiac Metabolic Pathway Remodeling in Response to Sunitinib Malate. PloS One. 2017;12(1):e0169964
31. Kolwicz SC, Tian R. Glucose metabolism and cardiac hypertrophy. Cardiovasc Res. 2011May1;90(2):194-201
32. Rees ML, Subramaniam J, Li Y, Hamilton DJ, Frazier OH, Taegtmeyer H. A PKM2 signature in the failing heart. Biochem Biophys Res Commun. 2015Apr10;459(3):430-6
33. Hafizi S, Wharton J, Chester AH, Yacoub MH. Profibrotic effects of endothelin-1 via the ETA receptor in cultured human cardiac fibroblasts. Cell Physiol Biochem Int J Exp Cell Physiol Biochem Pharmacol. 2004;14(4-6):285-92
34. Ehmke H, Faulhaber J, Münter K, Kirchengast M, Wiesner RJ. Chronic ETA receptor blockade attenuates cardiac hypertrophy independently of blood pressure effects in renovascular hypertensive rats. Hypertens Dallas Tex 1979. 1999Apr;33(4):954-60
35. Nilsson D, Wackenfors A, Gustafsson L, Edvinsson L, Paulsson P, Ingemansson R. et al. Increased ETA and ETB receptor contraction in the left internal mammary artery from patients with hypertension. J Hum Hypertens. 2007Dec13;22(3):226-9
36. Hue L, Beauloye C, Marsin A-S, Bertrand L, Horman S, Rider MH. Insulin and Ischemia Stimulate Glycolysis by Acting on the Same Targets Through Different and Opposing Signaling Pathways. J Mol Cell Cardiol. 2002Sep;34(9):1091-7
37. Kreissl MC, Stout DB, Wong K-P, Wu H-M, Caglayan E, Ladno W. et al. Influence of dietary state and insulin on myocardial, skeletal muscle and brain [ 18 F]-fluorodeoxyglucose kinetics in mice. EJNMMI Res. 2011Jul6;1(1):8
38. Vitale GD, deKemp RA, Ruddy TD, Williams K, Beanlands RS. Myocardial glucose utilization and optimization of (18)F-FDG PET imaging in patients with non-insulin-dependent diabetes mellitus, coronary artery disease, and left ventricular dysfunction. J Nucl Med Off Publ Soc Nucl Med. 2001Dec;42(12):1730-6
39. Dilsizian V. 18F-FDG Uptake as a Surrogate Marker for Antecedent Ischemia. J Nucl Med. 2008Dec1;49(12):1909-11
40. Haas F, Jennen L, Heinzmann U, Augustin N, Wottke M, Schwaiger M. et al. Ischemically compromised myocardium displays different time-courses of functional recovery: Correlation with morphological alterations? ResearchGate. 2001Sep1;20(2):290-8
41. Schinkel AFL, Poldermans D, Elhendy A, Bax JJ. Assessment of Myocardial Viability in Patients with Heart Failure. J Nucl Med. 2007Jul1;48(7):1135-46
42. Rahimtoola SH, Dilsizian V, Kramer CM, Marwick TH, Vanoverschelde J-LJ. Chronic Ischemic Left Ventricular Dysfunction. JACC Cardiovasc Imaging. 2008Jul1;1(4):536-55
43. Piper HM. Pathophysiology of Severe Ischemic Myocardial Injury. Kluwer Academic Publishers. 1990:448
44. Kumar RMR, Arlt MJ, Kuzmanov A, Born W, Fuchs B. Sunitinib malate (SU-11248) reduces tumour burden and lung metastasis in an intratibial human xenograft osteosarcoma mouse model. Am J Cancer Res. 2015Jun15;5(7):2156-68
45. Goodwin GW, Taylor CS, Taegtmeyer H. Regulation of energy metabolism of the heart during acute increase in heart work. J Biol Chem. 1998Nov6;273(45):29530-9
46. Zhong M, Alonso CE, Taegtmeyer H, Kundu BK. Quantitative PET imaging detects early metabolic remodeling in a mouse model of pressure-overload left ventricular hypertrophy in vivo. J Nucl Med Off Publ Soc Nucl Med. 2013Apr;54(4):609-15
47. Gropler RJ, Peterson LR. Imaging of Myocardial Metabolism. ResearchGate. 2010:641-56
48. Toubert M-E, Vercellino L, Faugeron I, Lussato D, Hindie E, Bousquet G. Fatal heart failure after a 26-month combination of tyrosine kinase inhibitors in a papillary thyroid cancer. Thyroid Off J Am Thyroid Assoc. 2011Apr;21(4):451-4
49. Kobayashi K, Neely JR. Effects of ischemia and reperfusion on pyruvate dehydrogenase activity in isolated rat hearts. J Mol Cell Cardiol. 1983Jun;15(6):359-67
50. Voipio-Pulkki LM, Nuutila P, Knuuti MJ, Ruotsalainen U, Haaparanta M, Teräs M. et al. Heart and skeletal muscle glucose disposal in type 2 diabetic patients as determined by positron emission tomography. ResearchGate. 1994Jan1;34(12):2064-7
51. Stanley WC, Hall JL, Hacker TA, Hernandez LA, Whitesell LF. Decreased myocardial glucose uptake during ischemia in diabetic swine. Metabolism. 1997Feb;46(2):168-72
52. Malek R, Davis SN. Tyrosine kinase inhibitors under investigation for the treatment of type II diabetes. Expert Opin Investig Drugs. 2016;25(3):287-96
53. Horinouchi T, Hoshi A, Harada T, Higa T, Karki S, Terada K. et al. Endothelin-1 suppresses insulin-stimulated Akt phosphorylation and glucose uptake via GPCR kinase 2 in skeletal muscle cells. Br J Pharmacol. 2016Mar;173(6):1018-32
54. Shemyakin A, Salehzadeh F, Böhm F, Al-Khalili L, Gonon A, Wagner H. et al. Regulation of Glucose Uptake by Endothelin-1 in Human Skeletal Muscle in Vivo and in Vitro. J Clin Endocrinol Metab. 2010May1;95(5):2359-66
55. Shemyakin A, Salehzadeh F, Duque-Guimaraes DE, Böhm F, Rullman E, Gustafsson T. et al. Endothelin-1 Reduces Glucose Uptake in Human Skeletal Muscle In Vivo and In Vitro. Diabetes. 2011Aug1;60(8):2061-7
56. Nagata D, Hirata Y. The role of AMP-activated protein kinase in the cardiovascular system. Hypertens Res. 2009Nov13;33(1):22-8
57. Orchard CH. The role of the sarcoplasmic reticulum in the response of ferret and rat heart muscle to acidosis. J Physiol. 1987Mar;384:431-49
58. Kassiotis C, Rajabi M, Taegtmeyer H. Metabolic Reserve of the Heart: The Forgotten Link Between Contraction and Coronary Flow. Prog Cardiovasc Dis. 2008;51(1):74-88
59. Kusuoka H, Marban E. Mechanism of the diastolic dysfunction induced by glycolytic inhibition. Does adenosine triphosphate derived from glycolysis play a favored role in cellular Ca2+ homeostasis in ferret myocardium? J Clin Invest. 1994Mar;93(3):1216-23
60. Burlew BS, Weber KT. Cardiac fibrosis as a cause of diastolic dysfunction. Herz. 2002Mar;27(2):92-8
61. Mandinov L, Eberli FR, Seiler C, Hess OM. Diastolic heart failure. Cardiovasc Res. 2000Mar1;45(4):813-25
62. Clozel M, Salloukh H. Role of endothelin in fibrosis and anti-fibrotic potential of bosentan. Ann Med. 2005;37(1):2-12
63. Inhibition of Metastatic Potential in Breast Carcinoma In Vivo and In Vitro through Targeting VEGFRs and FGFRs; Evid-Based Complement Altern Med ECAM; 2013. Chien M-H, Lee L-M, Hsiao M, Wei L-H, Chen C-H, Lai T-C, et al. http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3686112/
64. Mendel DB, Laird AD, Xin X, Louie SG, Christensen JG, Li G. et al. In vivo antitumor activity of SU11248, a novel tyrosine kinase inhibitor targeting vascular endothelial growth factor and platelet-derived growth factor receptors: determination of a pharmacokinetic/pharmacodynamic relationship. Clin Cancer Res Off J Am Assoc Cancer Res. 2003Jan;9(1):327-37
65. Thackeray JT, Bankstahl JP, Bengel FM. Impact of Image-Derived Input Function and Fit Time Intervals on Patlak Quantification of Myocardial Glucose Uptake in Mice. J Nucl Med Off Publ Soc Nucl Med. 2015Oct;56(10):1615-21
66. Wiśniewski JR, Zougman A, Nagaraj N, Mann M. Universal sample preparation method for proteome analysis. Nat Methods. 2009May;6(5):359-62
67. Rappsilber J, Mann M, Ishihama Y. Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat Protoc. 2007;2(8):1896-906
68. Kulak NA, Pichler G, Paron I, Nagaraj N, Mann M. Minimal, encapsulated proteomicsample processing applied to copy-number estimation in eukaryotic cells. Nat Methods. 2014;11(3):319-24
69. Wiśniewski JR, Duś K, Mann M. Proteomic workflow for analysis of archival formalin-fixed and paraffin-embedded clinical samples ot a depth o f0 0100 proteins. Proteomics Clin Appl. 2013Apr;7(3-4):225-33
70. Cox J, Matic I, Hilger M, Nagaraj N, Selbach M, Olsen JV. et al. A practical guide to the MaxQuant computational platform for SILAC-based quantitative proteomics. Nat Protoc. 2009;4(5):698-705
71. Wiśniewski JR, Hein MY, Cox J, Mann M. A “proteomic ruler” for protein copy number and concentration estimation without spike-in standards. Mol Cell Proteomics MCP. 2014Dec;13(12):3497-506
72. Prévilon M, Gall ML, Chafey P, Federeci C, Pezet M, Clary G. et al. Comparative differential proteomic profiles of nonfailing and failing hearts after in vivo thoracic aortic constriction in mice overexpressing FKBP12.6. Physiol Rep. 2013 doi: 10.1002%Fphy2.39
73. Vizcaíno JA, Csordas A, del-Toro N, Dianes JA, Griss J, Lavidas I, Mayer G, Perez- Riverol Y, Reisinger F, Ternent T, Xu QW, Wang R, Hermjakob H. 2016 update of the PRIDE database and related tools. Nucleic Acids Res. 2016;44(D1):D447-D456 (PubMed ID: 26527722)
Author contact
![]() Corresponding author: Bertrand Tavitian, Paris Cardiovascular Research Center (PARCC), INSERM UMR970, Université Paris Descartes, Paris, France bertrand.tavitianfr
Corresponding author: Bertrand Tavitian, Paris Cardiovascular Research Center (PARCC), INSERM UMR970, Université Paris Descartes, Paris, France bertrand.tavitianfr
Citation styles
APA
Sourdon, J., Lager, F., Viel, T., Balvay, D., Moorhouse, R., Bennana, E., Renault, G., Tharaux, P.L., Dhaun, N., Tavitian, B. (2017). Cardiac Metabolic Deregulation Induced by the Tyrosine Kinase Receptor Inhibitor Sunitinib is rescued by Endothelin Receptor Antagonism. Theranostics, 7(11), 2757-2774. https://doi.org/10.7150/thno.19551.
ACS
Sourdon, J.; Lager, F.; Viel, T.; Balvay, D.; Moorhouse, R.; Bennana, E.; Renault, G.; Tharaux, P.L.; Dhaun, N.; Tavitian, B. Cardiac Metabolic Deregulation Induced by the Tyrosine Kinase Receptor Inhibitor Sunitinib is rescued by Endothelin Receptor Antagonism. Theranostics 2017, 7 (11), 2757-2774. DOI: 10.7150/thno.19551.
NLM
Sourdon J, Lager F, Viel T, Balvay D, Moorhouse R, Bennana E, Renault G, Tharaux PL, Dhaun N, Tavitian B. Cardiac Metabolic Deregulation Induced by the Tyrosine Kinase Receptor Inhibitor Sunitinib is rescued by Endothelin Receptor Antagonism. Theranostics 2017; 7(11):2757-2774. doi:10.7150/thno.19551. https://www.thno.org/v07p2757.htm
CSE
Sourdon J, Lager F, Viel T, Balvay D, Moorhouse R, Bennana E, Renault G, Tharaux PL, Dhaun N, Tavitian B. 2017. Cardiac Metabolic Deregulation Induced by the Tyrosine Kinase Receptor Inhibitor Sunitinib is rescued by Endothelin Receptor Antagonism. Theranostics. 7(11):2757-2774.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.