Theranostics
13.3
Impact Factor
- Current Issue
- Advance Articles
- Volume 16; 2026
- Volume 15; 2025
- Volume 14; 2024
- Volume 13; 2023
- Volume 12; 2022
- Archive
- Cover Images
- Cover Suggestion
- Special Issues
Top
Introduction
Materials and Methods
Results and Discussion
Conclusion
Abbreviations
Acknowledgements
Supplementary Material
References
Introduction
Materials and Methods
Results and Discussion
Conclusion
Abbreviations
Acknowledgements
Supplementary Material
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2017; 7(15):3638-3652. doi:10.7150/thno.20028 This issue Cite
Research Paper
Cancer Nanomedicines Stabilized by π-π Stacking between Heterodimeric Prodrugs Enable Exceptionally High Drug Loading Capacity and Safer Delivery of Drug Combinations
Hangxiang Wang1 ![]() , Jianmei Chen1, Chang Xu2, Linlin Shi3, Munire Tayier2, Jiahui Zhou2, Jun Zhang1, Jiaping Wu1, Zhijian Ye4, Tao Fang4, Weidong Han3
, Jianmei Chen1, Chang Xu2, Linlin Shi3, Munire Tayier2, Jiahui Zhou2, Jun Zhang1, Jiaping Wu1, Zhijian Ye4, Tao Fang4, Weidong Han3 ![]()
1. The First Affiliated Hospital; Collaborative Innovation Center for Diagnosis and Treatment of Infectious Diseases; Key Laboratory of Combined Multi-Organ Transplantation, Ministry of Public Health, School of Medicine, Zhejiang University, Hangzhou, 310003, PR China;
2. School of Medicine, Zhejiang University, Hangzhou, 310027, PR China;
3. Department of Medical Oncology; Sir Run Run Shaw Hospital; School of Medicine, Zhejiang University, Hangzhou, 310003, PR China.
4. Jinhua People's Hospital, Jinhua, Zhejiang Province, 321000, PR China.
Received 2017-3-11; Accepted 2017-7-18; Published 2017-8-23
Citation:
Wang H, Chen J, Xu C, Shi L, Tayier M, Zhou J, Zhang J, Wu J, Ye Z, Fang T, Han W. Cancer Nanomedicines Stabilized by π-π Stacking between Heterodimeric Prodrugs Enable Exceptionally High Drug Loading Capacity and Safer Delivery of Drug Combinations. Theranostics 2017; 7(15):3638-3652. doi:10.7150/thno.20028. https://www.thno.org/v07p3638.htm
Other stylesAbstract

Combination therapy using distinct mode-of-action drugs has sparked a rapidly growing interest because this paradigm holds promise for improving the therapeutic efficacy of anticancer therapy. However, the current drug combination therapy refers to administering individual drugs together, which is far from a perfect regimen for cancer patients. The aim of this work was to demonstrate that synergistic delivery of two chemotherapeutic drugs in a single nanoparticle reservoir could be achieved through the rational chemical ligation of the drugs followed by supramolecular nano-assembly via blending of the drugs with a minimal amount of matrix. Choosing 7-ethyl-10-hydroxycamptothecin and taxanes, which are rich in aromatic structures, as model compounds, we show that the heterodimeric conjugates of the two agents are miscible with lipids to form systemically injectable nanomedicines. The compatibility between the prodrug conjugates and lipid carriers is substantially augmented by the intermolecular π-π stacking and alleviated polarity, thus enabling an exceptionally high drug loading (DL) capacity (~92%) and a gratifyingly long drug retention time within the micellar core. We further observed superior therapeutic outcomes in a mouse tumor model without detecting accompanying systemic toxicity. This structure-based, self-assembled cancer nanomedicine increased the potency and drug tolerability in animals and thus offers a robust strategy for simultaneously formulating two or more drugs in single nanovehicles.
Keywords: cancer nanomedicine, heterodimeric prodrugs, drug combination, SN38, taxane.
Introduction
Cancer remains one of the world's most life-threatening diseases. Advanced cancers acquire complex phenotypes and genetic diversity during their multistep development [1]. Numerous studies have confirmed that cancer therapies relying on a single “magic bullet” approach may be a rare commodity for the management of these disastrous diseases due to factors including network robustness, crosstalk, compensatory actions, and counter-target activities [2]. Consequently, it is rationally envisioned that drug combinations that rewire distinct cellular targets could be a promising therapeutic paradigm for cancers [3-6]. Unlike therapy using single agents, combinatorial chemotherapy has the potential to target distinct pathways in one population of cancer cells, thereby maximizing the therapeutic index or overcoming drug resistance [7, 8]. Indeed, many regimens have achieved certain successes, and some have been clinically approved for the treatment of cancers in the clinic [9, 10]. However, the current combination therapy, which refers simply to administering individual drugs together, is far from a perfect regimen for cancer patients. Consequently, unifying the pharmacokinetics, biodistribution, and cellular uptake of individual and chemically dissimilar drugs in vivo remains extremely challenging.
To address these therapeutic challenges, packaging multiple drugs or drug candidates into single nanocarriers with synthetic polymers is of particular interest for achieving synergistic activi-ties [3, 11-21]. However, this approach possesses several disadvantages. For example, when drugs are physically encapsulated in polymeric matrices, weak noncovalent associations between the drugs and carriers could be destabilized by the presence of abundant serum proteins after systemic injection. This mixing effect could result in suboptimal drug release kinetics in the blood and fail to properly exploit the enhanced permeability and retention (EPR) effect [22,23]. To enhance the stability and retention of drug payloads in carrier matrices, various elegant strategies have been attempted [24-33]. For instance, a platinum (IV)-prodrug tether with two axial ligands (e.g., hexanoic acid) was prepared, which tailored the hydrophobicity and miscibility of the parent cisplatin toward a polymer matrix [28, 34]. More interestingly, by exploiting the unique π-π stacking interaction between drugs and polymeric micelles, Hennink et al. demonstrated that paclitaxel (PTX) could be stabilized within a π-rich micellar structure, thereby augmenting drug delivery to tumors [29]. However, in these approaches, large quantities of adjuvant materials are required to encapsulate the drug payloads, which inevitably results in low drug loadings (typically less than 10%). This limitation may also raise concern with regard to excipient-associated side effects as a consequence of poor metabolism and elimination of carrier materials. Additionally, integrating heterogeneous drug components into single vehicles is a critical challenge, which suffers from broad diversity in the release kinetics of multiple payloads into the blood circulation. Therefore, in spite of these advances, alternative sophisticated strategies are necessary to abrogate these limitations without enhancing the complexity of the delivery systems.
We hereby propose a heterodimeric prodrug approach through ligation of chemotherapeutics that are lipophilic and rich in aromatic structures to facilitate intermolecular π-π stacking when assembled in polymeric nanoparticles (NPs). To validate this concept, we chose 7-ethyl-10-hydroxycamptothecin (SN38) and taxanes as model agents for constructing heterodimeric conjugates. SN38, a potent inhibitor of DNA topoisomerase I, being a planar, π-rich structure, has been actively re-explored for better therapeutic outcomes [35-38]. Esterification of this agent has further improved the drug-carrier association, as evidenced by our studies [27, 39]. On the other hand, taxanes (e.g., paclitaxel [PTX], docetaxel [DTX], cabazitaxel [CTX]) are widely used cytotoxic agents in oncology, which primarily target microtubules, resulting in the inhibition of mitosis and eventual cell death [40]. Unfortunately, all of these agents are poorly water-soluble. To solubilize SN38, a water-soluble prodrug, CPT-11, was developed and clinically approved for the treatment of cancers [41]. However, the enzymatic conversion of CPT-11 to active SN38 is quite low (usually 2-8%) in the body. Taxanes are also water insoluble and require surfactants (i.e., Cremophor EL or polysorbate 80) and ethanol as additives for drug solubilization; however, this approach has led to concerns with regard to in vivo excipient-associated toxicity. Moreover, previous studies have suggested that the combination of taxane with SN38 (or CPT-11) yields superior anticancer outcomes compared to individual agents in various cancer cells or in clinical trials, strongly supporting our rationale [42-45].
In the current study, we used a clinically available amphiphilic lipid, 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy-(polyethylene glycol) 2000] (DSPE-PEG2000), to formulate three types of synthetic taxane-SN38 conjugates (i.e., compounds 1-3 in Fig. 1B). DSPE-PEG2000 can form micelles in aqueous media and exhibits excellent miscibility with versatile hydrophobic drugs [46, 47]. Upon assembling with DSPE-PEG2000, we obtained systemically injectable and PEGylated nanomedicines encapsulating heterodimeric conjugates without the use of exogenous organic solvents. Due to the π-π stacked structures and alleviated polarity, these prodrug-assembled nanomedicines exhibited high drug loading capacity and strong drug retention within the inner core of NPs. We carefully examined the combinatorial effects using the drug pair of DTX and SN38 in cancer cells, and the results showed a synergistic effect. Furthermore, in vivo near-infrared fluorescence (NIR)-based tracking and imaging of the nanomedicines demonstrated that the DTX-SN38 conjugate 2-assembled NP exhibited highly tumor-specific accumulation. This approach ultimately improved the therapeutic efficacy and alleviated the systemic toxicity relative to the individual drugs in a BALB/c mouse model bearing human colon HCT-116 xenografts.
Materials and Methods
A full description of the methods is provided in the Supplementary Information.
Materials
7-Ethyl-10-hydroxycamptothecin (SN38), paclitaxel (PTX), docetaxel (DTX), and cabazitaxel (CTX) were purchased from Knowshine Pharmachemicals Inc. (Shanghai, China). 1,2-Distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy (polyethylene glycol) 2000] (DSPE-PEG2000) was purchased from Advanced Vehicle Technology (Shanghai, China). Cyanine5.5 (Cy5.5) NHS ester was purchased from Lumiprobe (Florida, USA). Click-iT® EdU Alexa Fluor® 488 Assay Kit was purchased from Invitrogen (Shanghai, China). Cell culture reagents were purchased from Life Technologies (Shanghai, China). All other compounds and solvents were purchased from J&K Chemical (Shanghai, China). Deionized (DI) Milli-Q water (Millipore) was used throughout the experiments.
Synthesis of taxane-SN38 conjugates 1, 2, and 3
To synthesize conjugates 1-3, two-step reactions were performed (Fig. 1C): i) carboxyl-terminated taxanes were prepared by reaction with succinic anhydride, and ii) activated taxanes were condensed with SN38. For the first reaction, anhydrous pyridine containing taxanes (PTX, DTX or CTX) was mixed with succinic anhydride and 4-dimethylaminopyridine (DMAP) [48] at 25°C, followed by stirring for 3 h. After removing the solvent, dichloromethane (DCM) was added, and the organic solvent was washed with 0.1 N HCl and brine. The organic layer was dried over anhydrous Na2SO4, filtered, and evaporated under vacuum. The residue was purified by flash column chromatography with silica gel to afford carboxyl-terminated derivatives 4, 5, and 6.
Activated taxane derivatives 4, 5, and 6 were further used to react with SN38 (molar ratio at 1:1) in anhydrous dimethylformamide (DMF) in the presence of condensation agents. The reactants were stirred at 25°C overnight. After removing the solvent, DCM was added, and the organic solvent was washed with 5% citric acid, saturated NaHCO3 and brine. The organic layer was dried over anhydrous Na2SO4. The residue was purified by flash column chromatography with silica gel to afford the final conjugates 1, 2, and 3. The data from 1H NMR, 13C NMR (Bruker 400 spectrometer) and high-resolution mass spectra (recorded on AB TripleTOF 5600+System, SCIEX, Framingham, USA), as well as the synthetic details, are available in the supplementary information.
Co-assembly of prodrug conjugates with DSPE-PEG2000
Prodrug conjugate 1, 2, or 3 was first blended with DSPE-PEG2000 in DMSO at pre-determined various weight ratios, while keeping the drug concentration at 40 mg/mL. Thereafter, the mixture solution (100 μL) was rapidly injected into DI water (1 mL) under ultrasonication. Prior to further use, the solutions were dialyzed against DI water to remove the organic solvent, and the drug concentration was determined by reverse-phase high-performance liquid chromatography (RP-HPLC) analysis using a C18 reverse-phase column (5 μm, 250 mm × 4.6 mm, YMC Co., Ltd., Kyoto, Japan). A gradient of 20-80% acetonitrile in water was adopted as the mobile phase at a flow rate of 1 mL/min together with UV detection at a wavelength of 220 nm.
Cell culture
The human colon carcinoma cell lines HCT-116 and LoVo were grown in RPMI 1640 medium. All of the culture media were supplemented with 10% (v/v) fetal bovine serum (FBS), penicillin (100 units/mL), and streptomycin (100 μg/mL). The cells were cultured at 37°C in a humidified atmosphere containing 5% CO2.
Animals
BALB/c nude mice (5 weeks old) were used in the animal studies and were purchased from the Shanghai Experimental Animal Center, Chinese Academy of Science. All studies on the mice were conducted in accordance with the National Institute of Health Guide for the Care and Use of Laboratory Animals.
In vivo antitumor efficacy of 2-NP
HCT-116 cells were grown to 80% confluence in 90 mm tissue culture dishes. After the cells were harvested, they were suspended in phosphate buffer saline (PBS) at 4°C to a final concentration of 2.5 × 107 cells/mL. The nude mice were subcutaneously injected in the right flank with 200 μL of a cell suspension containing 5×106 cells in a 1 mL disposable syringe. For each mouse, one tumor was implanted.
Mice bearing HCT-116 tumor xenografts (~60 mm3) were randomly separated into four experimental groups (n = 10 in each group). The mice were injected intravenously (IV) with solutions containing 2-NP (at SN38- and DTX-equivalent doses of 10 and 20.6 mg/kg, respectively; weight ratio of DSPE-PEG2000/2 = 1:10) three times on days 0, 3, and 6. CPT-11 (15 mg/kg) in saline, free DTX formulated in Tween 80/ethanol (20.6 mg/kg), and saline were administered following the same schedule. Each injection contained 200 μL of solution. The tumor volumes and body weights were monitored and recorded at predetermined time intervals. The tumor volume was calculated according to the following formula: V = (length × width2)/2, where the long axis is the length, and the short axis is the width.
Figure 1
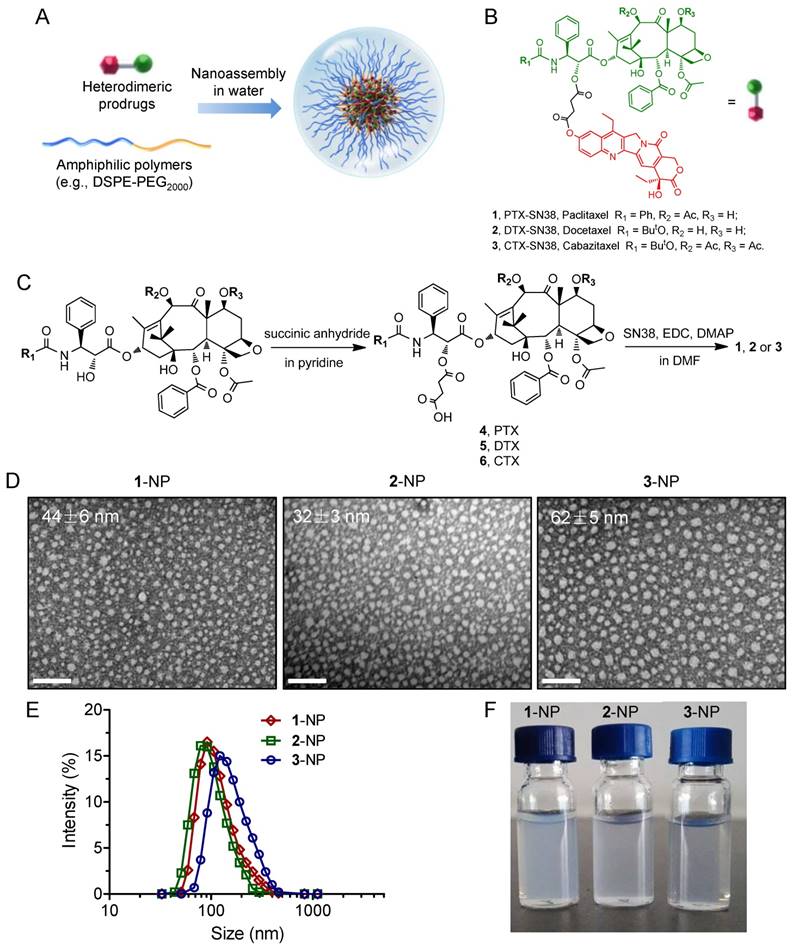
(A) Schematic illustration of the nano-assembly of heterodimeric prodrugs with amphiphilic matrix, DSPE-PEG2000, to form colloidal stable nanocomplexes for systemic administration. (B) Molecular structures of prodrug conjugates 1-3 used in this study. (C) Synthetic scheme for prodrug conjugates 1-3. TEM images (D) and hydrodynamic diameters (DH) (E) of 1-3-assembled NPs (the weight ratio of drug to DSPE-PEG2000 was set at 1:5). Scale bars, 200 nm. (F) Photographs of drug-formulated NPs in DI water.
Statistical analysis
All quantitative data are presented as the mean ± standard deviation of three independent experiments. The statistical significance was evaluated using the two-tailed unpaired Student's t-test. The threshold for significance was p<0.05.
Results and Discussion
Synthesis of heterodimeric prodrug conjugates and their assembly to form PEGylated NPs
To generate taxane-SN38 heterodimeric prodrugs, we employed a simple two-step esterification process to first ligate PTX and SN38 (Fig. 1). The 2'-hydroxyl group of PTX is fairly reactive compared with the other two hydroxyl groups, thus providing a simple approach to selectively esterify 2'-OH and convert it to other functional groups. Accordingly, a succinate derivative, 4, was constructed from the parent PTX by stirring PTX and succinic anhydride in pyridine for 3 h (Fig. 1C). This carboxyl-terminated derivative was further ligated with other chemotherapeutics. Therefore, we condensed compound 4 with the SN38 agent, which bears a phenolate moiety, to afford the final adduct, namely, PTX-SN38 1. The resulting conjugate 1 was unambiguously characterized by 1H and 13C NMR and high-resolution mass spectrometry (see ESI, Fig. S1-S6). This synthetic strategy produced a unique heterodimeric prodrug, in which the therapeutically active components could be released by hydrolysis and/or intracellular esterases.
Subsequently, we exploited a reprecipitation method to validate whether conjugate 1 could be miscible with amphiphilic matrices such as DSPE-PEG2000. Rapid injection of a mixture of 1 and DSPE-PEG2000 at a weight ratio of 1:5 (dissolved in dimethyl sulfoxide [DMSO]) into an antisolvent (e.g., water) resulted in a homogeneous solution without visible precipitations. Further dialysis against DI water to remove DMSO did not yield large precipitates, suggesting that solubilization of extremely hydrophobic conjugate 1 is independent of organic solvents. This procedure could PEGylate the surface of particles, rendering them with the stability in aqueous solutions and improved pharmacokinetics in vivo [49, 50].
Prompted by this finding, we next explored the application of this strategy to the nanoformulation of other taxane-based chemotherapeutics, such as DTX and CTX. Accordingly, the prodrugs DTX-SN38 (compound 2, Fig. S7-S12) and CTX-SN38 (compound 3, Fig. S13-S18) were constructed using a similar synthetic route by sharing the same linker with the conjugate 1. Analogous to the above procedure, injection of 2 or 3 into DI water in the presence of DSPE-PEG2000 gave transparent solutions, indicating that the heterodimeric prodrugs could be assembled with this matrix in aqueous solution. We designated these prodrugs 1, 2, and 3-assembled NPs as 1-NP, 2-NP, and 3-NP, respectively. To depict the necessity of our dimeric strategy, we further evaluated the ability of the parent taxanes and SN38 agents to assemble with the DSPE-PEG2000 lipid separately. Injection of a DMSO solution containing each parent drug with DSPE-PEG2000 into DI water instantaneously resulted in precipitates, presumably due to their incompatibility with this matrix. These results clearly imply that the ligation of two chemotherapeutics using a simple linker (i.e., succinic acid) and chemical derivatization (i.e., ester bonds) can adjust the miscibility of the parent drugs with conventionally used materials. Moreover, because both of anticancer drugs are rich in aromatic structures, strong intermolecular π-π stacking interactions can be envisaged. Hence, our results provide a rational paradigm for reformulating double structurally dissimilar drugs in an ideal delivery vehicle.
Characterization of prodrug-assembled NPs
Having obtained these prodrug/DSPE-PEG2000 coassemblies, we next subjected them to morphology studies using transmission electron microscopy (TEM) and dynamic light scattering (DLS) analysis. All the prodrug-formulated assemblies exhibited uniform monodispersities and spherical nanostructures with small diameters ranging from 32 to 62 nm in the TEM images (Fig. 1D). The peaks derived from DLS analysis revealed that the average hydrodynamic diameters (DH) for 1-, 2- and 3-NP were 122, 106 and 162 nm, respectively (Fig. 1E). The smaller sizes from the TEM images compared to the DLS measurements can be ascribed to the shrinkage of the particles when the samples were subjected to the dry conditions of the TEM grids. All of the dual prodrug-loaded NPs exhibited monomodal distributions, as evidenced by relatively low polydispersity indices (PDIs), e.g., the PDIs of 1-, 2-, and 3-NP were 0.122, 0.124, and 0.106 nm, respectively. Nevertheless, these NPs are larger in size than the renal clearance threshold (ca. 6-8 nm) but smaller than the leakage size of tumor vasculature (ca. 200 nm); thus, these NPs should be applicable for preclinical studies and passive tumor targeting by exploiting the EPR effect. Notably, among three types of NPs, 2-assembled NP displayed smaller, more stable particles than the other two. Therefore, 2-NP was used for subsequent in vitro characterization and optimization.
Detailed characterization of prodrug 2-formulated nanoassemblies
To validate our dimeric design strategy that features exceptionally high drug loading (DL), we carefully characterized the physicochemical properties of blended 2/DSPE-PEG2000. Gradually decreasing the weight ratios of the DSPE-PEG2000 lipid from 5:1 to 1:10 (DSPE-PEG2000/2, w/w) still enabled the formation of stable nanocomplexes. No substantial change was observed in the heterogeneity of the formulations. Upon blending with the lipid, the 2-NP suspension showed a typical Tyndall effect when irradiated with a green laser beam, suggesting the presence of colloidal nanoparticles rather than bulky agglomerations. We additionally found that the constructed 2-NPs were monomodal in size, yielding DH values of 102, 76, 87, and 74 nm for DSPE-PEG2000/2 weight ratios of 5:1, 1:1, 1:5, and 1:10, respectively (Fig. 1B and Table 1, entries 5-8). The encapsulation efficiency (EE) and DL values are summarized in Table 1, and these nanoformulations exhibited high EE values that exceeded 99%. Notably, when the weight ratio of DSPE-PEG2000/2 was fixed at 1:10, the DL was extrapolated to be as exceptionally high as 92.3%. This value is greater than that of most currently existing nanoformulations, and the improved DL could be beneficial for alleviating excipient-associated side effects in patients. Most importantly, these NPs exhibited remarkable colloidal stability in DI water, PBS, and 20% rat serum over the long term (up to at least 14 days) when stored at room temperature (Fig. 2C and Fig. S23). DLS analysis further confirmed that only negligible variations in DH were observed for 2-NPs (Fig. 2D and Fig. S23). In sharp contrast, the parent SN38 or DTX was not miscible with DSPE-PEG2000 lipid (w/w at 1:10); we observed a cloudy solution or micrometer-sized aggregates, respectively (Fig. 2A and Table 1, entries 1-4). Furthermore, precipitates were obvious in the samples of entry 1-4 after 24 h incubation at room temperature. These results demonstrate that the miscibility of drug payloads with polymeric matrices can be tailored by the rational heterodimeric conjugation of distinct mode-of-action drugs, thereby augmenting the drug-carrier compatibility to be suitable for clinical translation.
Improved compatibility between drug and matrix by dimeric prodrug strategy
Undesirable and premature drug release from NPs during systemic circulation in the blood is a critical challenge, which might impair tumor-specific accumulation of drug payloads via the EPR effect. Therefore, to augment delivery into target tumors, sustained drug release is favored. We thus quantified the release kinetics using conjugate 2 embedded in DSPE-PEG2000 as a model compound. Intriguingly, as shown in Fig. 2E, the respective NPs exhibited a fairly slow and sustained SN38 release rate, which was extended over several days, thereby completely eliminating the burst release effect. We further assessed the release kinetics of DTX molecule by HPLC analysis (Fig. S24). Both drug release patterns were similar but DTX plateaued at a lower release percentage than the SN38 molecule, probably due to the poor solubility of DTX in the releasing media. Moreover, the drug release kinetics varied depending on the feed ratios of lipid to drug conjugate; that is, decreasing the amount of DSPE-PEG2000 lipid delayed the drug release rate. For instance, the cumulative release of total drugs at pH 7.4 was approximately 59.9% and 36.7% after 13 days when the weight ratios of lipid/2 were 5:1 and 1:10, respectively. Therefore, we suggest that a small amount of amphiphilic lipid layer (~10%) could serve as molecular barrier for delaying the hydrolysis of prodrug conjugates and hence the release kinetics. Additionally, PEG cloaking could reduce the uptake by the reticuloendothelial system (RES) and yield nanodrugs with long-term circulation upon systemic injection. Prior studies indicate that π-π stacking interactions are important driving forces for enhancing the robustness of nanoassemblies [29, 33, 51]. In those cases, aromatic groups tethered to polymer chains were exploited to stabilize individual anticancer drugs but they suffered from low drug loading capacity. However, our nanosystems constructed from small-molecule prodrug entities and a minimal amount of matrix featured exceptionally high drug loading and long-term drug retention within NPs.
Table 1
Characterization of nanoformulations of parent SN38, DTX, and conjugate 2 with DSPE-PEG2000.[a]
| Entry | Drug | DSPE-PEG2000 /Drug (w/w) | Particle size (nm) | PDI [b] | EE (%) [c] | DL (%) [d] |
|---|---|---|---|---|---|---|
| 1 | SN38 | 10:1 | 619±70 | - | - | - |
| 2 | DTX | 1:1 | 5250±351 | - | - | - |
| 3 | DTX | 5:1 | N.D. [e] | N.D. | - | - |
| 4 | DTX | 10:1 | N.D. | N.D. | - | - |
| 5 | 2 | 5:1 | 102±5 | 0.124 | 99.0 | 19.3 |
| 6 | 2 | 1:1 | 76±3 | 0.138 | 99.3 | 54.6 |
| 7 | 2 | 1:5 | 87±2 | 0.208 | 99.6 | 85.6 |
| 8 | 2 | 1:10 | 74±8 | 0.237 | 99.4 | 92.3 |
[a] A mixture of DSPE-PEG2000/drug in DMSO (at a drug-equivalent concentration of 10 mg/mL) was rapidly injected into DI water at 1 mg/mL. Prior to measurement, the drug-equivalent concentration was adjusted to 0.1 mg/mL.
[b] PDI: Polydispersity index.
[c] EE: Drug encapsulation efficiency.
[d] DL: Drug loading of authentic content.
[e] N.D: Not detectable in particle sizes.
Figure 2
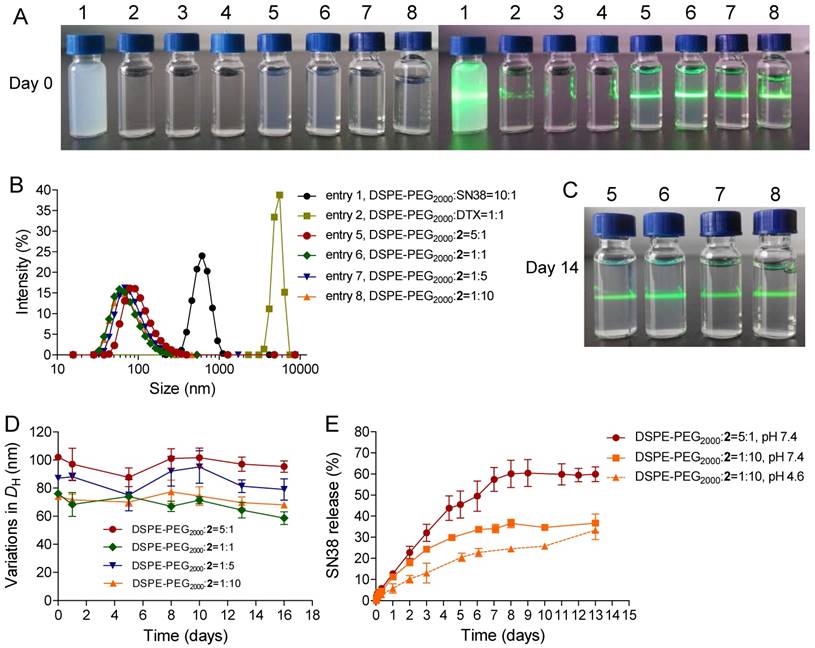
(A) Photographs of parent SN38 (0.1 mg/mL), DTX (0.1 mg/mL), and 2 (0.1 mg/mL) that were assembled with DSPE-PEG2000 at varying weight ratios. The 2-formulated solution (entry 5-8) exhibited Tyndall effect when the suspension was irradiated with a green laser, suggesting no visible particle aggregation in solution, whereas strong laser scattering was observed in SN38 or DTX due to the presence of extensive aggregation upon blending with DSPE-PEG2000. (B) Distribution of the hydrodynamic diameters (DH) of the NPs when conjugate 2 and DSPE-PEG2000 were assembled at different weight ratios. (C) Photographs of the NP solutions corresponding to entries 5-8 after storage at room temperature for 14 days. (D) Variation in the particle sizes of the NPs. (E) Drug release profiles (i.e., SN38) from the assembled NPs against PBS buffer.
In vitro cytotoxicity
To examine whether the use of DTX could synergize with the SN38 effect in cancer cells, we initially conducted in vitro cytotoxicity assays in which a fixed dose of DTX was added to various concentrations of SN38 agent. Two human colon cancer cell lines (i.e., HCT-116 and LoVo cells) were used to validate this synergy. The cells were treated with varying concentrations of free SN38, whereas the concentrations of free DTX were fixed (i.e., 0, 5, and 20 nM). Cell viability, which was analyzed by MTS assay, is indicated in Fig. 3A and B. The combination of DTX and SN38 exhibited a robust increase in cytotoxicity in both cell lines. For example, the viabilities of HCT-116 cells treated with SN38 (160 nM) and co-incubated with DTX at 20 and 40 nM were 61% and 48%, respectively. These values were 1.1 to 1.4-fold lower than that of free SN38 alone (65%), showing the enhanced potency of the DTX plus SN38 combination. We further determined the combination index (CI) to assess whether synergy could be observed when a free DTX/SN38 mixture was used in vitro. The results are presented as a synergy “heat map” (Table S1 and S2 for HCT-11 and LoVo cells, respectively), where CI values reflecting synergy, additivity, and antagonism are displayed in green, yellow, and red, respectively. Interestingly, co-administration of DTX/SN38 in a 1:1 molar ratio rendered the DTX/SN38 mixture combination synergistic: the CI values were observed to be below 1 across various concentrations in both cancer cells. These results validated the rationale of our design for drug ligation in a 1:1molar ratio.
Figure 3
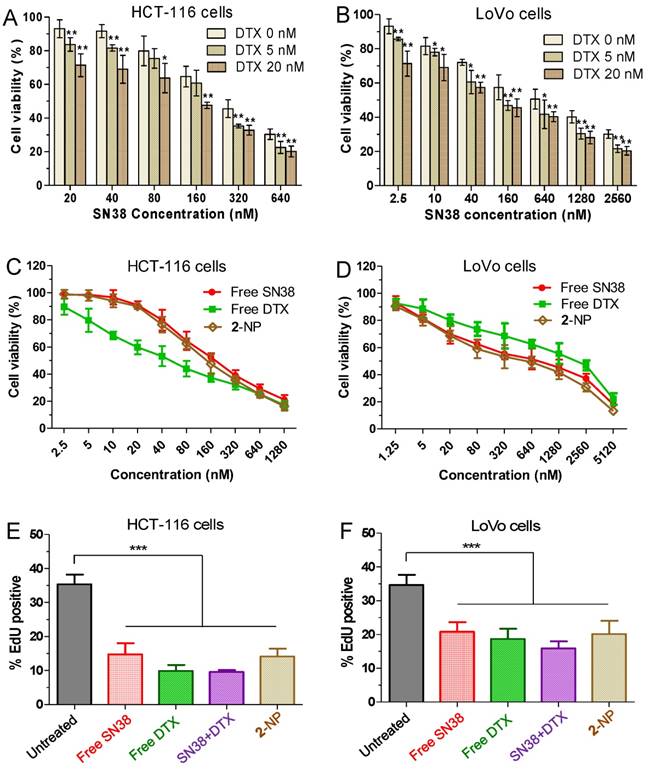
(A and B) In vitro cytotoxicity of SN38 alone (light yellow) or in combination with 5 nM DTX (medium yellow) or 20 nM DTX (yellow). The cell viability was assessed by the MTS assay after 24 h of drug incubation. The addition of DTX increased the dose-dependent inhibition by SN38 at all of the tested concentrations. Higher doses of DTX increased the efficacy of the combinations. The cell viability for free SN38, DTX and conjugate 2-assembled NPs in HCT116 (C) and LoVo (D) cells measured by MTS assay. (E-F) Effects of free SN38, free DTX, the [SN38+DTX] combination and 2-NP on the proliferation of cancerous cells. Cells were incubated with various drugs (at 50 nM). The cell proliferation ratio was expressed as the ratio of EdU-positive cells to total Hoechst-positive cells. The data are presented as the means ± SD. *p< 0.05, **p< 0.01, ***p<0.001 vs. the control.
After achieving this synergistic cytotoxicity, we evaluated the potency of the DTX-SN38 conjugate 2-assembled NP by measuring the half maximal inhibitory concentration (IC50). The viability was quantified after treatment of cells with SN38, DTX or 2-NP (formulated with a weight ratio of lipid to 2 of 1:10) for 24 h. Impressively, in both cancer cells, 2-NP exhibited comparable or slightly lower cytotoxicity compared to SN38 or the DTX agent (Fig. 3C and D). We further assessed the effect of 2-NP on the proliferation of cancerous cells using the EdU incorporation assay. As indicated in Fig. 3E-F and Fig. S25, exposure to 2-NP at a 50 nM concentration significantly reduced the EdU-positive cells (in green), leading to 2.5-fold and 1.7-fold reductions compared to untreated HCT-116 and LoVo cells, respectively. In general, nanoformulation of covalently ligated drugs could substantially compromise the in vitro anti-proliferative activity. However, we did not observe reduced activity, presumably due to the effective intracellular uptake of NPs and the subsequent effective liberation of both therapeutically active compounds.
Cell cycle distribution and apoptosis induced by synergistic drug combination
To determine whether the growth inhibition induced by SN38, DTX or 2-NP was a result of cell cycle arrest, we analyzed the effect of the drugs on cell cycle progression in HCT-116 cells. After the cells were treated for 24 h, free SN38 induced S and G2M cell cycle arrest, whereas free DTX led to significant G2M arrest (Fig. 4A). Interestingly, 2-NP mainly arrested cells at the S and G2M stages, which was similar to the SN38 agent alone.
Figure 4
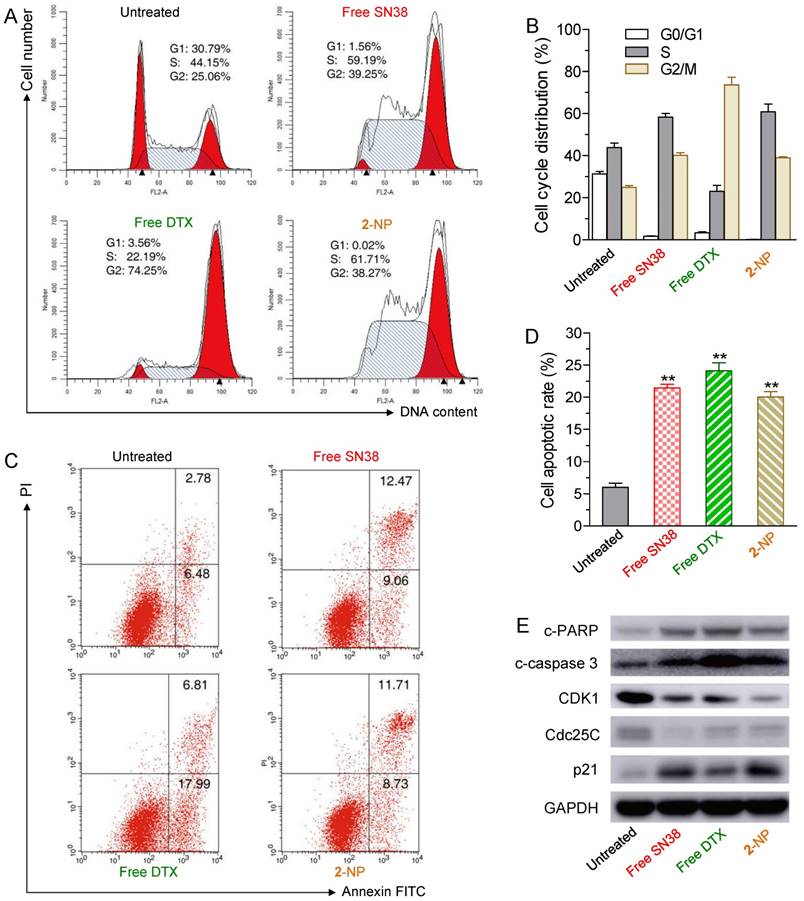
The effects of free SN38 (50 nM), free DTX (50 nM) and conjugate 2-assembled NP (at 50 nM) on HCT-116 cell cycle and apoptosis based on flow cytometry analysis. The cells were treated with various drugs for 24 h. (A) Effect of 2-NP on cell cycle of HCT-116 cells. (B) The histogram shows the cell cycle distribution. (C) Apoptosis analysis of HCT-116 cells determined by FACS (Alexa Fluor 488 Annexin V/PI assay). Viable cells are indicated in the lower left quadrant; early and late apoptotic cells are indicated in the lower right and upper right quadrants, respectively; necrotic or dead cells are indicated in the upper left quadrant. (D) The percentage of apoptotic cells in each group is shown in the histogram. (E) Western blot detection of related cell cycle and apoptosis markers in HCT-116 cells treated for 24 h with free SN38, free DTX, or 2-NP.
We further examined whether the cell death was a consequence of apoptosis induced by 2-NP. HCT-116 cells were exposed to various drugs (i.e., the concentrations of free SN38, free DTX, and 2 were 50 nM) for 24 h. The drug-induced apoptotic capabilities were assessed using the Alexa Fluor 488 Annexin V/PI assay. Fluorescently labeled Annexin V can specifically detect the presence of phosphatidylserine, which is expressed on the outer leaflet of the cell membrane during apoptosis [52]. We observed ~20.4% of the cell populations at early and late apoptotic stages when exposed to 2-NP (Fig. 4C and D). This activity was comparable to that of free SN38 and DTX (i.e., 21.5% and 24.8% of SN38- and DTX-treated cells, respectively) and was in good agreement with the MTS data.
Moreover, to elucidate the mechanism of cell cycle arrest and apoptosis caused by 2-NP in HCT116 cells, we performed western blot analysis of several key protein markers, including cleaved poly (ADP-ribose) polymerase (c-PARP), cleaved caspase 3 (c-caspase 3), cyclin-dependent kinase 1 (CDK1), Cdc25C, and p21. Among these proteins, PARP and caspase 3 are crucial mediators of cell apoptosis that, can be proteolytically activated by various death stimuli to form cleaved products [53, 54]. On the other hand, the expression of CDK1 and Cdc25c is pivotal for the G2/M transition, and both of these proteins decrease during the G2/M phases of the cell cycle [55]. CDK1 is negatively regulated by p21, a CDK1 inhibitor [56]. The results derived from western blots clearly showed that 2-NP produced an increase in c-PARP, c-caspase 3 and p21, whereas decreased expression levels of CDK1 and Cdc25c were observed, suggesting that cell death can be attributed to apoptotic events (Fig. 4E). Moreover, the decreased level of CDK1 and Cdc25c indicated that HCT-116 cells were arrested at the G2M stage, which corresponds to the cell cycle analysis illustrated in Fig.4A and 4B. However, compared with free SN38 and DTX treatments, 2-NP did not exhibit an obviously superior effect in HCT-116 cells. These data imply that the sustained drug release from 2-NP could slightly impair the cytotoxic effects.
In vivo nanodrug tumor targeting evaluated by near-infrared fluorescence imaging
To validate the ability of 2-assembled NPs to preferentially target tumors, we performed whole-body NIR imaging to study the retention and biodistribution of nanodrugs in vivo. To better mimic the heterodimeric prodrug, a NIR probe Cy5.5 was covalently linked to DTX (Fig. S19-22), and the corresponding Cy5.5-DTX conjugate 7 was further co-assembled with DSPE-PEG2000 to construct NPs simultaneously containing conjugates 2 and 7 (designated as 2/7-NP, Fig. 5A and Fig. S26). Cy5.5-integrated 2-NP was also prepared using the same protocol. Both types of NPs exhibited spherical nanostructures and monomodal distributions via TEM and DLS measurements, which is similar to unlabeled 2-NP (Fig. S26). More interestingly, the fluorescence of Cy5.5 was not quenched upon co-assembly with 2-NP compared with free Cy5.5 in DMSO (Fig. S27). Therefore, the resulting 2/7-NP is expected to be well suited for tracking the biodistribution of the nanodrug in animals. BALB/c nude mice bearing subcutaneous HCT-116 colon tumor xenografts were intravenously administered with 2/7-NP (i.e., at a Cy5.5 equivalent dose of 20 μg per mouse) via the tail vein. For comparison, solutions of free Cy5.5 and free Cy5.5-loaded NPs were also injected. Real-time whole-body imaging at 0, 6, and 24 h post-administration was conducted using an in vivo imaging system and the results are shown in Fig. 5B-D. As expected, the NIR signals derived from free Cy5.5 were observed to be rapidly eliminated upon systemic injection with negligible signals observed within the tumor tissues. In sharp contrast, the tumor contours with intense NIR signals in mice receiving Cy5.5-loaded NP and 2/7-NP were maintained for up to at least 24 h, indicative of the long retention ability of the nanodrugs in the blood.
We further performed ex vivo fluorescence imaging to elucidate the in vivo biodistribution of 2-NP after harvesting the major organs and tumor tissues from mice that were sacrificed at 24 h post-injection. As shown in the ex vivo images of the organs (Fig. 5B-D) and the quantitative data (Fig. 5E), the mice that received 2/7-NP exhibited stronger fluorescence signals in the tumors relative to the mice that received free Cy5.5 and free Cy5.5-loaded NP. Interestingly, low accumulation of the nanoparticles in liver was observed, suggesting the successful avoidance of uptake by the RES in this organ. These results strongly support the notion that dimeric prodrugs can be constrained within NPs in the systemic circulation and preferentially reach tumors via the EPR effect. Moreover, compared with parent Cy5.5, the ligation of the DTX agent augmented the retention of Cy5.5 in NPs, thereby enhancing the drug accumulation in tumors.
Pharmacokinetic study of prodrug-assembled NPs
Long-term circulation of nanoparticles after systemic administration will enhance the accumulation of drugs in tumors via the EPR effect, which ultimately improves the therapeutic efficacy. Therefore, we investigated the blood clearance of 2-NP in comparison with free DTX and CPT-11 after a single IV administration of the drugs in Sprague Dawley (SD) rats (~250 g). Both drugs (i.e., DTX and SN38) in the plasma were analyzed by HPLC and the results are provided in Fig. S28 and Table S3. Compared to the relatively rapid clearance of free DTX and CPT-11 from the bloodstream, 2-NP clearly exhibited prolonged retention in the blood. For example, both drugs were maintained for up to 24 h when 2-NP was used, producing a significantly higher area under the blood concentration curve (AUC0-24h) compared to CPT-11 (1258.8 vs. 230.1 μg·h/mL, Table S3) and free DTX (2606.1 vs. 613.5 μg·h/mL). Taken together, these results indicate that this dimeric prodrug strategy significantly prolong the presence of the drug in the blood and that the structural stability of the nanoparticles contributes favorably to in vivo pharmacokinetics. These data also imply that intermolecular π-π stacking interactions between the drug entities within the nanoassemblies can be reasonably exploited to create stable systems that ultimately could contribute to the prolonged circulation of nanodrugs in vivo.
Figure 5
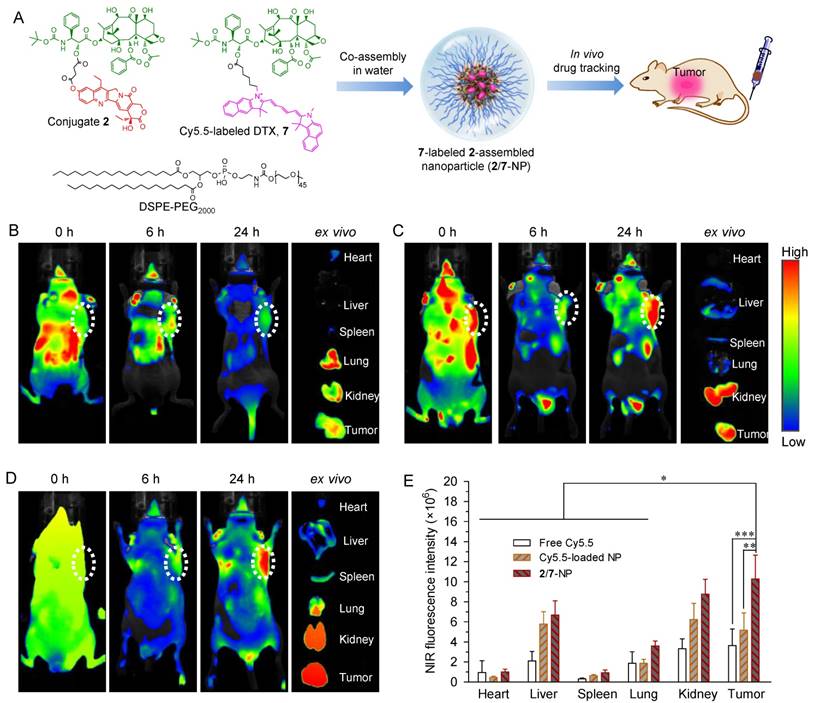
(A) Schematic illustration of the co-assembly of Cy5.5-labeled DTX 7 into NPs for in vivo nanodrug tracking. (B-D) In vivo fluorescence imaging of HCT-116-bearing nude mice and the dissected organs after IV injection of free Cy5.5 (B), free Cy5.5-loaded 2-NP (C), and 2/7-co-assembled NP (2/7-NP) via the tail wein (D). (E) Quantitative evaluation of the fluorescence intensity in the excised organs and tumors. White dotted circles indicate tumor regions. The data are presented as the mean ± SD (n = 5). *p < 0.05, **p < 0.01, ***p < 0.001.
In vivo antitumor activity in a colorectal xenograft mouse model
Encouraged by the synergistic cytotoxicity and in vivo tumor-targeting capacity of the dual drug-loaded nanodrugs, we evaluated their therapeutic potential in a BALB/c nude mouse model bearing HCT-116 colorectal xenografts. Mice (n = 6 in each group) were IV injected with 2-NP (at SN38- and DTX-equivalent doses of 5 and 10.3 mg/kg, respectively). Several control formulations via IV injection were included via IV injection in this experiment (Fig. S29). 2-NP outperformed the other formulations but showed comparable tumor inhibition compared with the [SN38+DTX] combination in terms of therapeutic efficacy in this mouse model. Impressively, the mice receiving 2-NP at a total drug dose of 15.3 mg/kg did not show body weight loss, whereas the mice receiving the [SN38+DTX] combination or DTX suffered from significant weight loss throughout the course of the studies and two out of six mice died at day 15, indicating a significantly improved toxicity profile for our NP approach.
We further intensified the dosage to 10 and 20.6 mg/kg for the SN38- and DTX-equivalence, respectively. Unfortunately, three injection of DTX at a dose of 20.6 mg/kg led to significant animal death. We thus terminated the therapeutic procedure using free DTX. The therapeutic efficacy of the other groups is illustrated in Figure 6A. After three successive injections of 2-NP, we observed a significant delay in tumor growth in the mice (n = 10 in each group), which was more pronounced than in those that received CPT-11 (p<0.01 versus CPT-11-treated groups). For example, the tumor volume in the mice receiving 2-NP was 2.8-fold smaller than in those that received CPT-11. By sharp contrast, the tumor volume in the saline-treated mice exhibited a rapid increase, exceeding 10% of the animal's body weight at day 18.
Figure 6
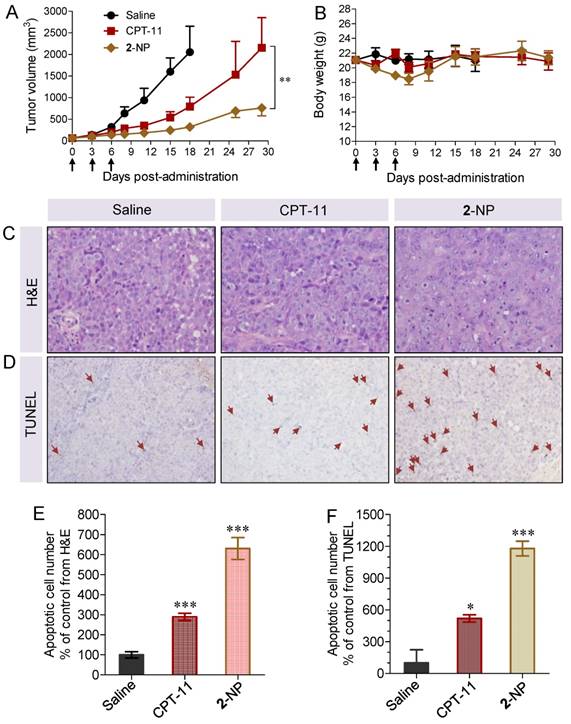
In vivo therapeutic effects of 2-assembled NP (2-NP) in comparison with controls, i.e., free CPT-11 (15 mg/kg), free DTX (20.6 mg/kg), and saline. HCT-116 tumor xenograft-bearing BALB/c nude mice were administered with drugs via IV injection at days 0, 3 and 6. (A) Tumor growth curve of different groups. 2-NP delayed tumor growth significantly after day 8 compared to CPT-11 (**p<0.01). (B) Body weight changes in mice receiving various treatments. (C) Representative histological features of tumors. Intense intratumoral apoptosis was observed in the 2-NP-treated tumor tissues. The image magnification is 400×. (D) TUNEL-stained sections from tumors in each group. Quantitative analysis of apoptotic cells from H&E staining (E) and TUNEL assay (F). An increased number of TUNEL-positive apoptotic cells were observed in the 2-NP-treated mouse tumor tissues relative to the saline or CPT-11-treated mouse tumor tissues.
The intratumoral effectiveness was further assessed by histological analysis of H&E stained tissue sections. As shown in Figure 6C, the injection of 2-NP evoked intensive apoptosis of the tumor cells. Compared with the saline and CPT-11 treatments, the tumor tissues in the mice receiving 2-NP lacked the normal architecture of tumor cells while exhibiting vacuolization and typical apoptotic characteristics. We further performed TUNEL staining to analyze the apoptotic cells. The results were similar to those of H&E staining, indicating that prodrug 2-formulated NP caused widespread apoptosis in the tumor tissues. These data also suggest that our dimeric prodrug strategy can constrain drugs within the particles, thereby contributing to sustained drug release (Fig. 2E) and preferential accumulation in tumors possibly via the EPR effect (Fig. 6). Ultimately, 2-NP that resides in tumor tissues could undergo gradual hydrolysis to liberate the two parent drugs at therapeutically relevant concentrations and possibly serve as long-term therapeutic-generating depots.
Finally, we assessed the toxicity of the nanodrugs because of the high dosage used in this study. Although the mice receiving 2-NP at a total drug dosage of 30.6 mg/kg showed a loss of body weight upon successive injections, a gradual restoration of weight occurred after stopping the injections (Fig. 6B). Impressively, the toxicity caused by 2-NP was substantially lower than that caused by free DTX formulations at both dosages. We further examined the major organs by histological analysis. Microscopic changes in the major organs (i.e., heart, liver, spleen, lung, kidney and ileum) were characterized by H&E staining at the end point of the study. As indicated in Fig. 7, the pathological images of the organs from 2-NP-treated mice were similar to those from the saline-treated mice. However, we observed destruction of the glandular structures and epithelial layer in the ileum portion of small intestine in mice receiving CPT-11. Collectively, these results indicate that at a high dosage (i.e., 30.6 mg/kg), 2-NP produced negligible damage to organs and systemic toxicity.
Conclusion
In summary, we have successfully constructed novel heterodimeric prodrugs bearing two distinct mode-of-action chemotherapeutics (i.e., SN38 and taxanes) via simple two-step esterification. Both parent therapeutics are immiscible with the clinically approved lipid DSPE-PEG2000; however, this can be simply resolved by the chemical ligation of the two drugs through an appropriate linker, enabling the formation of systemically injectable nanoassemblies upon blending with the lipid as well as synergistic drug combination for cancer therapy. In the DTX-SN38 conjugate 2-assembled nanostructures, strong intermolecular π-π stacking interactions between prodrug entities can be reasonably envisaged. These nanoassemblies showed impressively high DL (e.g., exceeding 92% for compound 2) that were comparable to previously reported high values [57-59]. Additionally, burst drug release profiles from assembled NPs were completely eliminated, thereby extending drug retention in particle systems during systemic circulation. Most significantly, the tolerability of the nanomedicine in animals was improved compared to the clinical formulation of free DTX. Collectively, our studies provide an alternative strategy for co-delivering two structurally diverse chemotherapeutics using clinically available matrices, thereby permitting drug combination therapy and the avoiding the use of toxic excipients.
Figure 7
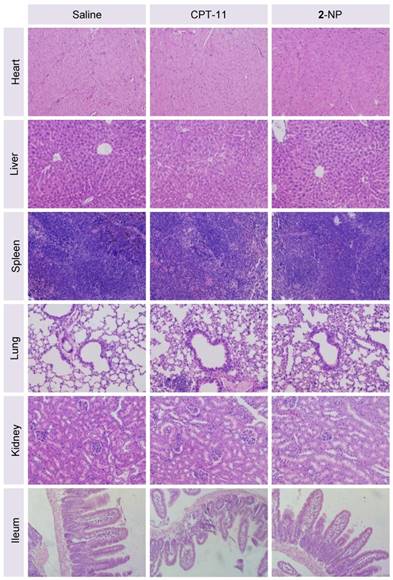
Representative H&E staining of major organs from mice sacrificed at day 8 following administration of saline, CPT-11, or 2-NP. There was no significant damage to the heart, liver, spleen, lung, kidney, or ileum in the nanodrug-treated mice compared to the saline-treated mice, indicating the low systemic toxicity of this dimeric prodrug approach (n = 3 in each group; the image magnification is 200×).
Abbreviations
DL, drug loading; EPR, enhanced permeability and retention; PTX, paclitaxel; DTX, docetaxel; CTX, cabazitaxel; NPs, nanoparticles; SN38, 7-ethyl-10-hydroxycamptothecin; DSPE-PEG2000, 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy (polyethylene glycol) 2000]; NIR, near-infrared fluorescence; Cy5.5, Cyanine5.5; DI, Deionized; DMAP, 4-dimethylaminopyridine; DCM, dichloromethane; DMF, dimethylformamide; NMR, nuclear magnetic resonance; RT-HPLC, reverse-phase high-performance liquid chromatography; FBS, fetal bovine serum; PBS, phosphate buffer saline; DMSO, dimethyl sulfoxide; TEM, transmission electron microscopy; DLS, dynamic light scattering; DH, hydrodynamic diameters; PDIs, low polydispersity indices; EE, encapsulation efficiency; RES, the reticuloendothelial system; CI, combination index; c-PARP, cleaved poly (ADP-ribose) polymerase; c-caspase 3, cleaved caspase 3; CDK1, cyclin-dependent kinase 1; FACS, fluorescence-activated cell sorting; SD, Sprague Dawley.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Nos. 81571799, 81572361, 81372621, 81773193 and 21202147), the Zhejiang Province Preeminence Youth Fund (No. LR16H160001), the Science and Technology Research Program of Jinhua City (No. 2017-3-020), and the Project of Public Technology and Application Research of Zhejiang Province (No. 2017C37139).
Supplementary Material
Supplementary figures and tables.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646-74
2. Jia J, Zhu F, Ma X. et al. Mechanisms of drug combinations: interaction and network perspectives. Nat Rev Drug Discov. 2009;8:111-28
3. Zhang L, Xiao H, Li J. et al. Co-delivery of doxorubicin and arsenite with reduction and pH dual-sensitive vesicle for synergistic cancer therapy. Nanoscale. 2016;8:12608-17
4. Sui J, Cui Y, Cai H. et al. Synergistic chemotherapeutic effect of sorafenib-loaded pullulan-Dox conjugate nanoparticles against murine breast carcinoma. Nanoscale. 2017;9:2755-67
5. Wang K, Hu QD, Zhu W. et al. Structure-invertible nanopaticles for triggered co-delivery of nucleic acids and hydrophobic drugs for combination cancer therapy. Adv Funct Mater. 2015;25:3380-92
6. Su JG, Sun HP, Meng QS. et al. Enhanced blood suspensibility and laser-activated tumor-specific drug release of theranostic mesoporous silica nanoparticles by functionalizing with erythrocyte membranes. Theranostics. 2017;7:523-37
7. Kolishetti N, Dhar S, Valencia PM. et al. Engineering of self-assembled nanoparticle platform for precisely controlled combination drug therapy. Proc Natl Acad Sci U S A. 2010;107:17939-44
8. Ma L, Kohli M, Smith A. Nanoparticles for combination drug therapy. ACS Nano. 2013;7:9518-25
9. Woodcock J, Griffin JP, Behrman RE. Development of novel combination therapies. N Engl J Med. 2011;364:985-7
10. Humphrey RW, Brockway-Lunardi LM, Bonk DT. et al. Opportunities and challenges in the development of experimental drug combinations for cancer. J Natl Cancer Inst. 2011;103:1222-6
11. Sun R, Liu Y, Li SY. et al. Co-delivery of all-trans-retinoic acid and doxorubicin for cancer therapy with synergistic inhibition of cancer stem cells. Biomaterials. 2015;37:405-14
12. Yang T, Zhao PX, Rong Z. et al. Anti-tumor efficiency of lipid-coated cisplatin nanoparticles co-loaded with microRNA-375. Theranostics. 2016;6:142-54
13. Zhang XD, Yang Y, Liang X. et al. Enhancing therapeutic effects of docetaxel-loaded dendritic copolymer nanoparticles by co-treatment with autophagy inhibitor on breast cancer. Theranostics. 2014;4:1085-95
14. Hu XL, Hu JM, Tian J. et al. Polyprodrug amphiphiles: hierarchical assemblies for shape-regulated cellular internalization, trafficking, and drug delivery. J Am Chem Soc. 2013;135:17617-29
15. Hu XL, Liu GH, Li Y. et al. Cell-penetrating hyperbranched polyprodrug amphiphiles for synergistic reductive milieu-triggered drug release and enhanced magnetic resonance signals. J Am Chem Soc. 2015;137:362-8
16. Yan LS, Yang LX, He HY. et al. Photo-cross-linked mPEG-poly(gamma-cinnamyl-L-glutamate) micelles as stable drug carriers. Polym Chem. 2012;3:1300-7
17. Yan LS, Wu WB, Zhao W. et al. Reduction-sensitive core-cross-linked mPEG-poly(ester-carbonate) micelles for glutathione-triggered intracellular drug release. Polym Chem. 2012;3:2403-12
18. Hao TN, Chen DW, Liu KX. et al. Micelles of d-alpha-tocopheryl polyethylene glycol 2000 succinate (TPGS 2K) for doxorubicin delivery with reversal of multidrug resistance. ACS Appl Mater Interfaces. 2015;7:18064-75
19. Jiang CJ, Wang HX, Zhang XM. et al. Deoxycholic acid-modified chitooligosaccharide/mPEG-PDLLA mixed micelles loaded with paclitaxel for enhanced antitumor efficacy. Int J Pharm. 2014;475:60-8
20. Wang JG, Wang HX, Li J. et al. iRGD-decorated polymeric nanoparticles for the efficient delivery of vandetanib to hepatocellular carcinoma: preparation and in vitro and in vivo evaluation. ACS Appl Mater Interfaces. 2016;8:19228-37
21. Miao L, Guo S, Zhang J. et al. Nanoparticles with precise ratiometric co-loading and co-delivery of gemcitabine monophosphate and cisplatin for treatment of bladder cancer. Adv Funct Mater. 2014;24:6601-11
22. Matsumoto Y, Nichols JW, Toh K. et al. Vascular bursts enhance permeability of tumour blood vessels and improve nanoparticle delivery. Nat Nanotechnol. 2016;11:533-8
23. Fang J, Nakamura H, Maeda H. The EPR effect: Unique features of tumor blood vessels for drug delivery, factors involved, and limitations and augmentation of the effect. Adv Drug Deliv Rev. 2011;63:136-51
24. Han X, Chen J, Jiang M. et al. Paclitaxel-paclitaxel prodrug nanoassembly as a versatile nanoplatform for combinational cancer therapy. ACS Appl Mater Interfaces. 2016;8:33506-13
25. Lin W, Sun T, Xie Z. et al. A dual-responsive nanocapsule via disulfide-induced self-assembly for therapeutic agent delivery. Chem Sci. 2016;7:1846-52
26. Wang Y, Liu D, Zheng Q. et al. Disulfide bond bridge insertion turns hydrophobic anticancer prodrugs into self-assembled nanomedicines. Nano Lett. 2014;14:5577-83
27. Wang H, Xie H, Wu J. et al. Structure-based rational design of prodrugs to enable their combination with polymeric nanoparticle delivery platforms for enhanced antitumor efficacy. Angew Chem Int Ed Engl. 2014;53:11532-7
28. Dhar S, Kolishetti N, Lippard SJ. et al. Targeted delivery of a cisplatin prodrug for safer and more effective prostate cancer therapy in vivo. Proc Natl Acad Sci U S A. 2011;108:1850-5
29. Shi Y, Van dMR, Theek B. et al. Complete regression of xenograft tumors upon targeted delivery of paclitaxel via pi-pi stacking stabilized polymeric micelles. ACS Nano. 2015;9:3740-52
30. Alferiev IS, Iyer R, Croucher JL. et al. Nanoparticle-mediated delivery of a rapidly activatable prodrug of SN-38 for neuroblastoma therapy. Biomaterials. 2015;51:22-9
31. Zhao YM, Fay F, Hak S. et al. Augmenting drug-carrier compatibility improves tumour nanotherapy efficacy. Nat Commun. 2016;7:11221-31
32. Sengupta P, Basu S, Soni S. et al. Cholesterol-tethered platinum II-based supramolecular nanoparticle increases antitumor efficacy and reduces nephrotoxicity. Proc Natl Acad Sci U S A. 2012;109:11294-9
33. Wei X, Wang Y, Xiong X. et al. Codelivery of a pi-pi stacked dual anticancer drug combination with nanocarriers for overcoming multidrug resistance and tumor metastasis. Adv Funct Mater. 2016;26:8266-80
34. Dhar S, Gu FX, Langer R. et al. Targeted delivery of cisplatin to prostate cancer cells by aptamer functionalized Pt(IV) prodrug-PLGA-PEG nanoparticles. Proc Natl Acad Sci U S A. 2008;105:17356-61
35. Bala V, Rao SS, Boyd BJ. et al. Prodrug and nanomedicine approaches for the delivery of the camptothecin analogue SN38. J Control Release. 2013;172:48-61
36. Fang T, Dong YH, Zhang XM. et al. Integrating a novel SN38 prodrug into the PEGylated liposomal system as a robust platform for efficient cancer therapy in solid tumors. Int J Pharm. 2016;512:39-48
37. Zhang HF, Wang JQ, Mao WW. et al. Novel SN38 conjugate-forming nanoparticles as anticancer prodrug: In vitro and in vivo studies. J Control Release. 2013;166:147-58
38. Xie HY, Xu X, Chen JM. et al. Rational design of multifunctional small-molecule prodrugs for simultaneous suppression of cancer cell growth and metastasis in vitro and in vivo. Chem Commun (Camb). 2016;52:5601-4
39. Wang HX, Xie HY, Wang JG. et al. Self-assembling prodrugs by precise programming of molecular structures that contribute distinct stability, pharmacokinetics, and antitumor efficacy. Adv Funct Mater. 2015;25:4956-65
40. Jordan MA, Wilson L. Microtubules as a target for anticancer drugs. Nat Rev Cancer. 2004;4:253-65
41. Venditto VJ, Simanek EE. Cancer therapies utilizing the camptothecins: a review of the in vivo literature. Mol Pharm. 2010;7:307-49
42. Roy A, Cunningham D, Hawkins R. et al. Docetaxel combined with irinotecan or 5-fluorouracil in patients with advanced oesophago-gastric cancer: a randomised phase II study. Br J Cancer. 2012;107:435-41
43. Teramoto M, Suzuki T, Satohisa S. et al. Low-dose SN-38 with paclitaxel induces lethality in human uterine cervical adenocarcinoma cells by increasing caspase activity. Med Mol Morphol. 2014;47:31-7
44. Burtness B, Gibson M, Egleston B. et al. Phase II trial of docetaxel-irinotecan combination in advanced esophageal cancer. Ann Oncol. 2009;20:1242-8
45. Hawkes E, Okines AFC, Papamichael D. et al. Docetaxel and irinotecan as second-line therapy for advanced oesophagogastric cancer. Eur J Cancer. 2011;47:1146-51
46. Kastantin M, Ananthanarayanan B, Karmali P. et al. Effect of the lipid chain melting transition on the stability of DSPE-PEG (2000) micelles. Langmuir. 2009;25:7279-86
47. Wei T, Liu J, Ma HL. et al. Functionalized nanoscale micelles improve drug delivery for cancer therapy in vitro and in vivo. Nano Lett. 2013;13:2528-34
48. Wang H, Koshi Y, Minato D. et al. Chemical cell-surface receptor engineering using affinity-guided, multivalent organocatalysts. J Am Chem Soc. 2011;133:12220-8
49. Cabral H, Kataoka K. Progress of drug-loaded polymeric micelles into clinical studies. J Control Release. 2014;190:465-76
50. Ishi T, Miyata K, Anraku Y. et al. Enhanced target recognition of nanoparticle by cocktail PEGylation with chains of varying lengths. Chem Commun. 2016;52:1517-9
51. Liang Y, Deng X, Zhang L. et al. Terminal modification of polymeric micelles with π-conjugated moieties for efficient anticancer drug delivery. Biomaterials. 2015;71:1-10
52. Logue SE, Elgendy M, Martin SJ. Expression, purification and use of recombinant annexin V for the detection of apoptotic cells. Nat Protoc. 2009;4:1383-95
53. Porter AG, Janicke RU. Emerging roles of caspase-3 in apoptosis. Cell Death Differ. 1999;6:99-104
54. Huang Y, Yang X, Xu T. et al. Overcoming resistance to TRAIL-induced apoptosis in solid tumor cells by simultaneously targeting death receptors, c-FLIP and IAPs. Int J Oncol. 2016;49:153-63
55. Sherr CJ. Cancer cell cycles. Science. 1996;274:1672-7
56. Serrano M, Hannon GJ, Beach D. A new regulatory motif in cell-cycle control causing specific inhibition of cyclin D/CDK4. Nature. 1993;366:704-7
57. Cai K, He X, Song Z. et al. Dimeric drug polymeric nanoparticles with exceptionally high drug loading and quantitative loading efficiency. J Am Chem Soc. 2015;137:3458-61
58. Kasai H, Murakami T, Ikuta Y. et al. Creation of pure nanodrugs and their antitumor properties. Angew Chem Int Ed Engl. 2012;51:10315-8
59. Ikuta Y, Koseki Y, Onodera T. et al. The effect of molecular structure on the anticancer drug release rate from prodrug nanoparticles. Chem Commun. 2015;51:12835-8
Author contact
![]() Corresponding authors: Weidong Han, E-mail: hanwdedu.cn; Hangxiang Wang, E-mail: wanghxedu.cn
Corresponding authors: Weidong Han, E-mail: hanwdedu.cn; Hangxiang Wang, E-mail: wanghxedu.cn
Citation styles
APA
Wang, H., Chen, J., Xu, C., Shi, L., Tayier, M., Zhou, J., Zhang, J., Wu, J., Ye, Z., Fang, T., Han, W. (2017). Cancer Nanomedicines Stabilized by π-π Stacking between Heterodimeric Prodrugs Enable Exceptionally High Drug Loading Capacity and Safer Delivery of Drug Combinations. Theranostics, 7(15), 3638-3652. https://doi.org/10.7150/thno.20028.
ACS
Wang, H.; Chen, J.; Xu, C.; Shi, L.; Tayier, M.; Zhou, J.; Zhang, J.; Wu, J.; Ye, Z.; Fang, T.; Han, W. Cancer Nanomedicines Stabilized by π-π Stacking between Heterodimeric Prodrugs Enable Exceptionally High Drug Loading Capacity and Safer Delivery of Drug Combinations. Theranostics 2017, 7 (15), 3638-3652. DOI: 10.7150/thno.20028.
NLM
Wang H, Chen J, Xu C, Shi L, Tayier M, Zhou J, Zhang J, Wu J, Ye Z, Fang T, Han W. Cancer Nanomedicines Stabilized by π-π Stacking between Heterodimeric Prodrugs Enable Exceptionally High Drug Loading Capacity and Safer Delivery of Drug Combinations. Theranostics 2017; 7(15):3638-3652. doi:10.7150/thno.20028. https://www.thno.org/v07p3638.htm
CSE
Wang H, Chen J, Xu C, Shi L, Tayier M, Zhou J, Zhang J, Wu J, Ye Z, Fang T, Han W. 2017. Cancer Nanomedicines Stabilized by π-π Stacking between Heterodimeric Prodrugs Enable Exceptionally High Drug Loading Capacity and Safer Delivery of Drug Combinations. Theranostics. 7(15):3638-3652.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.