Impact Factor
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2017; 7(16):3972-3988. doi:10.7150/thno.18990 This issue Cite
Research Paper
MiR-205/YAP1 in Activated Fibroblasts of Breast Tumor Promotes VEGF-independent Angiogenesis through STAT3 Signaling
Yan-e Du1, Gang Tu2, Guanglun Yang2, Genyou Li3, Dan Yang1, Lei Lang1, Lei Xi1, Kexin Sun1, Yanlin Chen1, Kunxian Shu5, Huadong Liao5, Manran Liu1 ![]() , Yixuan Hou1, 4
, Yixuan Hou1, 4 ![]()
1. Key Laboratory of Laboratory Medical Diagnostics, Chinese Ministry of Education, Chongqing Medical University, Chongqing 400016, China;
2. Department of Endocrine and Breast Surgery, the First Affiliated Hospital of Chongqing Medical University, Chongqing 400016, China;
3. Department of Laboratory Medicine, Chongqing Hechuan Blood Centre, Chongqing 401520, China;
4. Experimental Teaching Center of Basic Medicine Science, Chongqing Medical University, Chongqing 400016, China;
5. Institute of Bioinformatics, Chongqing University of Posts and Telecommunications, Chongqing 400065, China.
Received 2016-12-30; Accepted 2017-8-14; Published 2017-9-15
Abstract

Tumor microenvironment contributes to tumor angiogenesis. However, the role of the activated cancer associated-fibroblasts (CAFs) in angiogenesis is still unclear. Here we report that miR-205/YAP1 signaling in the activated stromal fibroblasts plays a critical role in VEGF-independent angiogenesis in breast tumor. Methods: miR-205 expression was assessed by quantitative real-time polymerase chain reaction (qRT-PCR); YAP1 expression by qRT-PCR, western blotting and immunohistochemistry; IL11 and IL15 expression by qRT-PCR, western blotting and ELISA. Tube formation and three-dimensioned sprouting assays in vitro, and orthotopic Xenografts in vivo were conducted as angiogenesis experiments. The mechanism of miR-205/YAP1-mediated tumor angiogenesis was analyzed via overexpression and shRNA, siRNA, or antibody neutralization experiments in combination with anti-VEGF antibody or Axitinib. Results: miR-205/YAP1 signaling axis activates breast normal fibroblasts (NFs) into CAFs, promotes tubule formation and sprouting of Human Umbilical Vein Endothelial Cells (HUVECs). Rescue of miR-205 in CAFs blunts angiogenesis processes. YAP1, a target of miR-205, does not regulate VEGF expression but specifically enhances IL11 and IL15 expressions, maintaining tumor angiogenesis even in the presence of Axitinib or after exhaustion of VEGF by neutralizing VEGF antibody. IL11 and IL15 released from CAFs activate STAT3 signaling in HUVECs. Blockage of IL11 and IL15 expression in CAFs results in the inactivation of STAT3-signaling in HUVECs and repression of the CAF-induced angiogenesis. The blunt angiogenesis halts the invasion and metastasis of breast cancer cells in vivo. Conclusions: These results provide a novel insight into breast CAF-induced tumor angiogenesis in a VEGF-independent manner.
Keywords: activated fibroblasts, miR-205, YAP1, VEGF-independent angiogenesis.
Introduction
Tumor angiogenesis is critical to tumor growth, progression, and metastasis [1]. In the absence of blood vessels, tumor growth stays at dormancy within the diameter of 1-2 mm. Tumor cells produce chemical compounds to trigger tumor angiogenesis, in which endothelial cells from the existing blood vessels degrade basement membrane, invade through extracellular matrix, and migrate toward the tumors, then proliferate and sprout to form new blood vessels [2].
Angiogenesis is regulated by a balance between proangiogenic and antiangiogenic molecules in the tumor. One of these molecules, vascular endothelial growth factor A (VEGF) activates a crucial signaling pathway in tumor angiogenesis by binding to its cognate receptor (VEGFR) [3, 4]. The Food and Drug Administration (FDA) has already approved antiangiogenic drugs including Bevacizumab, Axitinib, Sorafenib and Sunitinib that target VEGF or VEGFR, which have shown clinical efficacies in some patients with colon cancer, non-small-cell lung cancer, and renal carcinoma [5]. However, blocking VEGF alone has no meaningful benefit for some cancer patients [6, 7]. For example, anti-VEGF therapy failed to establish overall survival effects for patients with metastatic breast cancer; only 12% of patients experienced prolonged significant overall survival in phase III randomized clinical trials [8]. This suggests that additional factors or pathways independent of VEGF signaling may be implicated in tumor angiogenesis. Indeed, multiple mechanisms have been revealed to induce resistance to VEGF-targeted therapy, including angiogenic factors bFGF, PIGF and PDGF [9], and pathways of Notch-DLL4 and NRP1-ABL1 [10]. These VEGF-independent mechanisms represent potential new therapeutics valuable for improving outcomes for cancer patients.
Recently the tumor angiogenic roles of stromal cells in the tumor microenvironment have been investigated extensively [11]. Tumor-associated macrophages (TAMs), neutrophils and mast cells directly stimulate the migration and proliferation of endothelial cells via secreting proangiogenic factors VEGF, IL-8, and TGF-α [12]. Bone marrow (BM)-derived precursor cells mobilize to tumor tissue and contribute to the newly formed vessels and growth [13]. Extracellular matrix (ECM) components such as fibronectin, collagen, and vitronectin transduce angiogenic signals by binding to integrins on the endothelial cells, inducing their proliferation and migration [14, 15].
Cancer-associated fibroblasts (CAFs), activated fibroblasts secreting higher levels of cytokines and growth factors, are the major stromal residents in tumor stroma and favor tumor growth and metastasis partly through promoting tumor angiogenesis [16, 17]. The mouse embryonic fibroblasts are recognized to promote angiogenesis and tumorigenesis via VEGF [18]. Furthermore, several studies have demonstrated that CAF also contributes to angiogenesis by expressing angiopoietin-1 and angiopoietin-2 in ovarian carcinoma [19] or by depositing ECM proteins involved in angiogenesis [20]. Fibroblasts that have been “educated” by skin carcinoma cells have been shown to mediate innate immune cell recruitment and promote tumor growth by stimulating angiogenesis [21]. Interestingly, CAFs from resistant EL4 tumors can also mediate resistance to antiangiogenic therapy via VEGF-independent PDGF-C signaling [22]. However, the complicated mechanism underlying how the CAFs in breast cancers promote VEGF-independent angiogenesis is not well understood.
Recent studies have shown that microRNAs (miRNAs) may be an important regulator of tumor angiogenesis. miRNAs have been shown to signal in tumor cells by regulating angiogenic factors such as VEGF [23]. Specifically, miRNAs may be involved in the control of endothelial cell recruitment and function [24]. miRNA regulates crosstalk between tumor and microenvironment, but miRNAs in breast CAFs that drive the angiogenesis have not been identified. Our previous work identified a set of dysregulated miRNAs in breast CAFs, and miR-205 was found to be significantly downregulated in CAFs [25], suggesting their essential role in CAF function. It has been revealed that reduced miR-205 expression in breast cancer cells promotes tumor growth, migration and invasion [26]. However, whether the stromal miR-205 in breast CAFs could promote tumor growth and metastasis by orchestrating activation of fibroblasts and stimulating tumor angiogenesis needs to be determined.
Yes-associated protein (YAP1) is a core downstream effector of Hippo pathway, regulating organ development, cell proliferation and differentiation [27]. As a transcriptional coactivator, YAP1 interacts with transcription factors (e.g., TEAD family, SMAD), regulating its target genes' functions in many biological processes [28, 29]. Enhanced YAP1 expression has been found in some carcinomas of the lung, prostate, colon, and breast, triggering tumor initiation, reprogramming of cancer cells into cancer stem cells (CSCs), or promoting tumor growth, proliferation and metastasis [27]. Recently, it has been reported that overexpressed YAP1 in cholangiocarcinoma promotes angiogenesis by regulating pro-angiogenic MFAP5 expression [30]. Whether stromal YAP1 in breast CAFs regulates VEGF-independent angiogenesis mechanisms remains to be determined.
In this study, we demonstrate that breast CAFs are closely associated with tumor angiogenesis in a VEGF-independent manner. Reduced miR-205 expression in breast fibroblasts activates NFs into CAFs by targeting YAP1. Particularly, miR-205 and YAP1-mediated CAF activation promotes angiogenesis even in the absence of VEGF signaling. Except for VEGF, IL11 and IL15 are regulated by YAP1 in CAFs and stimulate angiogenesis resistance to anti-VEGF therapy and activate STAT3 signaling in endothelial cells. Rescue of miR-205 or loss of YAP1 in CAFs suppresses tumor growth and metastasis by reducing tumor angiogenesis in vivo. Therefore, our study discloses a previously unknown mechanism by which breast stromal CAF contributes to cancer growth and metastasis by tumor angiogenesis via a VEGF-independent manner, suggesting potential novel therapeutic strategies for anti-angiogenic therapy.
Materials and Methods
Clinical samples
Breast tumor tissues and their adjacent normal tissues used in this study were obtained from patients with breast cancer at the First Affiliated Hospital of Chongqing Medical University. None of the patients had previously received radiotherapy or neoadjuvant chemotherapy treatment. The investigation was approved by the ethics committee of Chongqing Medical University.
Stromal fibroblasts isolation, immortalization, and cell culture
Cancer-associated fibroblasts (CAFs) and normal fibroblasts (NFs) were isolated from breast tumor tissues or their paired normal tissues as described previously [31]. Briefly, fresh tumor tissue and the corresponding normal tissue were washed with sterile PBS containing antibiotics (100 U/mL penicillin, 100 μg/mL streptomycin and 50 μg/mL gentamycin), minced into small pieces and digested for 8 h at 37 °C in DMEM containing 10% FBS and 0.5% mg/mL collagenase type I (Sigma, USA). The digested tissues were carefully pipetted using culture DMEM (Invitrogen, USA), and the mixtures were centrifuged to remove the fat and tissue debris. The mammary tissues were cultured in DMEM containing 10% FBS (GIBCO-BRL, USA) until the fibroblasts adhered to the dish, and other cellular types in the suspension were removed. Cell purity of stromal fibroblasts was identified by the test of fibroblast biomarker fibronectin (FN), CAFs-specific markers α-smooth muscle actin (α-SMA) and fibroblast activation protein (FAP), epithelial cell markers cytokeratin 8 and cytokeratin 18 (CK8+18) and endothelial cell marker CD31. NFs and CAFs were immortalized by human telomerase reverse transcriptase gene (hTERT) as described previously [32]. NFs and CAFs were culture in DMEM with 10% FBS, and HUVECs and MDA-MB-231 were cultured in RPMI 1640 medium (Invitrogen) containing 10% FBS at 37 °C in humidified atmosphere containing 5% CO2.
Plasmid constructs and reagents
LV3-puro-miR-205 and LV3-puro-anti-miR-205 (the shRNA specifically targeting miR-205) were purchased from GenePharma (Shanghai, China). The pSUPER-vector and pSUPER-miR-205 vector were kindly provided by Dr Jörg Hackermüller (RNomics Group, Germany). The pBABE-YAP1 vector was obtained from Addgene (Boston, USA). Full-length IL11 and IL15 cDNA were amplified by PCR and cloned into the pBABE-puro vector at BamHI and EcoRI sites. The shRNAs specifically against YAP1 were cloned into the lentiviral vector pLVX-shRNA1 (Clontech, USA) at BamHI and EcoRI sites. The siRNA sequences specifically against miR-205 and the genes are listed in Table S1. To generate WT-YAP1 3'UTR-Luc, Mut-YAP1 3'UTR-Luc, IL11 promoter-Luc, and IL15 promoter-Luc reporters, the synthetic oligonucleotides (Invitrogen) that correspond to the wild-type or the mutated binding sites of miR-205 in the 3'-UTR of YAP1, and the IL11 or IL15 promoter were separately cloned into the PMIR-Reporter vector (Ambion, USA) at Spe I and Hind III sites.
The reagents used in this study in vitro are as follows: Axitinib (Selleck, USA), 5 nM; S3I-201 (Selleck, USA), 20 μM; MTT (Beyotime, China), 5 mg/mL; for preparation of conditioned medium, the neutralizing antibody against VEGF (Bevacizumab; Roche/Genentech), 0.2 mg/mL; Neutralizing antibody against IL11 (ab89887, Abcam), 5 μg/mL; IL15 (MAB2471, R&D Systems), 5 μg/mL; control antibody (MAB002, MAB003, R&D Systems), 5 μg/mL.
Luciferase assay
For 3'-UTR luciferase reporter assay, cells were seeded at a density of 1 × 105 cells in 24-well plates and co-transfected with pSUPER-miR-205 and pMIR-YAP1 3'-UTR (wild-type or mutant) and the control plasmid pRL-TK (Promega, USA) by using lipofectamine 2000 (Invitrogen). For promoter reporter assay, cells were co-transfected with shRNA (Control or YAP1), pMIR-promoter (IL11 or IL15) and pRL-TK. Luciferase activities were performed by using a Dual-Luciferase Reporter System (Promega, USA) after 48 h, and normalized by using pRL-TK reporters as an internal control.
Immunohistochemistry (IHC) and immunofluorescence (IF)
The deparaffinized tissue sections at 4 μm thick were heated for antigen retrieval at 95 °C in citric acid buffer (pH 6.0). After treating with 3% H2O2, the sections were blocked with 5% goat normal serum, and incubated with primary antibody against α-SMA (1:100, ab5694, Abcam), CD31 (1:50, ab9498, Abcam, UK) and YAP1 (1:200, sc-398182, Santa Cruz, USA), separately. Microvessel density (MVD) was assessed by CD31 staining as described previously [33, 34]. α-SMA+ CAFs and CD31+ endothelial cells were scored by mean optical density (density/area) using Image-pro plus 6.0 software. YAP1 staining in stromal tissues was scored into 5 intensities: 0, no staining; 1+, 1%-25%; 2+, 26%-50%; 3+, 51%-75%; 4+, 76%-100%. For immunofluorescence, cells were grown on pre-prepared coverslips for 24 h. After being fixed with 4% paraformaldehyde, treated by 0.1% triton-100, and incubated with 5% goat serum, the cells were separately stained with antibody specifically against FN (1:150, ab32419, Abcam), α-SMA (1:150), FAP (1:150, ab53066, Abcam), cytokeratin (CK8+CK18) (1:200, ab53280, Abcam), and CD31 (1:80) at 4 °C for overnight, then labelled with FITC-labeled secondary antibody (ZSBIO, China). The nuclei were stained with DAPI and the images were captured by a Nikon Eclipse 80i microscope.
MTT and Flow cytometric analysis
Stromal fibroblast cells (3 × 103 per well) were seeded into a 96-well plate, and cell growth was measured every day by MTT bromide assay at a wavelength of 492 nm on an ultraviolet spectrophotometric reader. Cells in the S-phase of the cell cycle (using standard propidium iodide staining), apoptotic cells (using standard Annexin V-FITC and Propidium Iodide Kit (Beyotime, China)), and stromal fibroblasts were analyzed by flow cytometry with an FCdevice (Beckman, USA). Experiments were repeated in triplicate.
RNA extraction and qRT-PCR
Total RNA was extracted using Trizol (Invitrogen). The purified RNA was reverse-transcribed to form cDNA using a reverse transcriptase kit (Takara, China), quantitative real-time PCR was performed with SYBR Premix Ex TaqTM II (Takara, Dalian, China). The primers used in qRT-PCR are listed in the Table S2.
Invasion assays
Transwell invasion assays were conducted using a 8 μm pore chamber coated with Matrigel (Corning BioCoat, USA) as described previously [32]. Cells were allowed to invade toward medium in the lower chamber, and the invaded cells on the opposite side of the filter were stained with hematoxylin in methanol and counted after 8 h for fibroblasts and 20 h for HUVECs.
Preparation of conditioned medium (CM) and antibody neutralization test
The stromal fibroblasts (1 × 106) were seeded into a 6 well plate in growth medium for 6 h. After removing the growth medium, FBS-free medium (1 mL) was added to further culture for 30 h, and the supernate was collected as conditioned medium (CM). To exclude the potential side effects of cell proliferation and apoptosis of CAFs and NFs (e.g. the engineered CAFs and NFs , and their control cells) on CM, total cell numbers, cell proliferation, cell apoptosis, and protein concentration (using Bradford assay kit, Beyotime, China) of CM were carefully determined at CM collection time point. For neutralization experiments, the neutralizing antibody against VEGF, IL11, or IL15 was preincubated at 4 °C with 1 mL supernatant for 6 h, then used for HUVECs co-culture in tubulogenesis or sprouting experiments.
Tubule formation and three-dimensional sprouting assays
HUVECs (1 × 104) were cultured with 150 μL conditioned medium in 96-well plates precoated with 50 μL of growth factor-deprived Matrigel. After incubating at 37 °C for 6 h, tubule formations were analyzed by using Image J software. For endothelial cell sprouting assays, HUVECs (1 × 103) were cultured with medium containing 0.24% carboxymethylcellulose in non-adherent round-bottom 96-well plates overnight. Spheroids were collected by centrifugation at 400 xg and mixed with 2 mg/mL fibrinogen (Corning) and FBS-free medium. The three-dimensional fibrin gel was then covered with CM and incubated at 37 °C for at least 24 h. Endothelial sprouting was analyzed with at least 10 spheroids per group and quantified by recording the branch number under a microscope.
Western blotting and ELISA assays
Cell lysates were separated by 10% SDS-PAGE. The specific primary antibodies used in Western blotting analysis are as follows: FAP (1:1000, ab53066, Abcam, UK), α-SMA (1:1000, ab5694, Abcam), IL11 (1:1000, ab89887, Abcam), IL15 (1:800, ab7213, Abcam); VEGFA (1:1000, wl00009b, Wanleibio, China); p-VEGFR2 (1:800, D155165, Sangon, China), T-VEGFR2 (1:1000, D151118, Sangon), p-KIT (1:800, D151515, Sangon), p-PDGFRβ (1:800, D151409, Sangon); FXR1 (1:1000, BS70701, bioworld, China); YAP1 (1:1000, sc-398182, Santa Cruz, USA); T-STAT3 (1:1000, #4904, CST, USA), p-STAT3 (1:1000, #9131, CST), T-AKT (1:1000, #9272, CST), p-AKT (1:1000, #2965, CST), T-ERK (1:1000, #9102, CST), p-ERK (1:1000, #4348, CST), T-P38 (1:1000, #9212, CST), p-P38 (1:1000, #4631, CST) and β-Actin (1:1500, ZSBIO, China). For ELISA assay, conditioned medium was collected from 1 × 106 NFs or CAFs in a 6-well plate, and the concentrations of IL11 and IL15 were measured using a standard ELISA Kit (Raybiotech, USA) according to the manufacturer's instructions.
Orthotopic Xenografts
The orthotopic tumor xenografts models were as described previously [32]. Animal experiments were approved by the animal care ethics committees at Chongqing Medical University. MDA-MB-231 cells (1 × 106) were mixed with equal number of CAF/Ctrl (CAF/miR-Ctrl, CAF/sh-Ctrl) or engineered CAF cells (CAF/miR-205, CAF/sh-YAP1) in 200 μL of PBS:Matrigel at a 1:1 ratio and subcutaneously injected into 4-week-old female nude mice. Tumor size was assessed by caliper measurements every three days; volume was calculated ((L × w2) × 0.5). When the tumor volume was around 100 mm3, the mice implanted with mixtures of MDA-MB-231 and CAF/sh-Ctrl or CAF/sh-YAP1 were separately fed oral Axitinib twice a day at low- (30 mg/kg, 1 group) or high-concentrations (40 mg/kg, 1 group), or intraperitoneally administered with anti-VEGF antibody (10 mg/kg, three times a week; 1 group) (Bevacizumab; Roche/Genentech). Control mice received 0.5% Carboxymethylcellulose (CMC; 1 group) or appropriate sterile saline (1 group). At the end of animal experiments, tumors or mice lungs were serially sectioned into 4 μm sections and stained with hematoxylin and eosin (H&E) for subsequent blinded evaluation of MVD and metastases in the lungs.
TCGA Bioinformatic Analysis
Bioinformatic analysis was performed as described [35]. We obtained the clinically annotated data from The Cancer Genome Atlas (TCGA) focused on expressions of miR-205, YAP1 and CD31 in breast cancer patients. To determine the progression free survival of patients, approximately 2/3 of the samples were used as a training cohort to obtain the thresholds yielding the most significant log rank test p-values (one-tailed test), which can divide the data into good and poor prognosis groups. The thresholds were then used to analyze the data in the remaining 1/3 of the samples, which functioned as validation cohort, and p-values were computed for those cohorts. All datasets were applied to assess survival differences between the low-expression and high-expression groups by the Kaplan-Meier curves.
Statistical analysis
Statistical significance was determined using SPSS 17.0 software. The results are shown as means ± SD. Multiple groups were analyzed using ANOVA followed by the Student-Newman-Keuls multiple comparison test, and single comparison between two groups was analyzed using Student's t-test. A p-value less than 0.05 was considered to be statistically significant.
Results
Breast stromal CAFs closely correlate with tumor angiogenesis in VEGF-independent signaling
We observed abundant vessel-like structures in breast tumor stroma compared with its non-tumor stromal area by HE staining (Figure 1A) and CD31 staining (a specific biomarker of the vascular endothelial cell) (Figure 1B). Activated CAFs account for nearly 60-80% of breast cancer stroma cells [16], suggesting that the activated stromal fibroblasts may be involved in tumor angiogenesis. We examined α-SMA and CD31 protein levels by immunohistochemical staining in tumor tissues and their para-cancer (non-tumor) tissues from 93 breast cancer patients, including 21 with ductal carcinoma in situ (DCIS), 42 with invasive ductal carcinoma (IDC), and 30 with metastatic carcinoma. α-SMA was negative in stromal fibroblasts in the non-tumor tissues (Figure S1A), while gradually stronger α-SMA expressions in CAFs and more plentiful microvessel density (MVD) were observed in the advanced breast tumor tissues (Figure 1C, Figure S1B). A positive correlation between the enhanced α-SMA and MVD was further disclosed by the Pearson's correlation analysis (Figure 1D). These results suggest that CAFs are the potential promoter of breast tumor angiogenesis.
Breast CAFs are closely correlated with tumor angiogenesis. (A) HE staining to show vessel-like structures in the stroma of para-cancer and cancer tissues. Scale bar, 200 μm. (B) CD31 staining to show the different distribution of MVD in breast cancer stroma and the cancer nest area. Scale bar, 50 μm. (C) Expressions of CD31 and α-SMA (CAFs marker) in ductal carcinoma in situ (DCIS), invasive ductal carcinoma (IDC) and metastatic carcinoma. Scale bar, 200 μm. (D) A positive correlation is shown between α-SMA protein levels and MVD in different clinical breast cancer tissues. (Pearson's correlation test, P<0.01). (E) Heatmap to illustrate the hierarchical clustering of altered angiogenesis-associated genes in CAFs and their paired NFs. (F-I) The cell proliferation (F), invasion (G), and tubule formation (H-I) (Scale bar, 300 μm) abilities of human endothelial cells (HUVECs) were tested under co-culture with the conditioned medium (CM) from CAFs or the paired NFs with Axitinib (H), or the CM neutralized with the anti-VEGF antibody (I). (n=3; *P<0.05; **P<0.01).
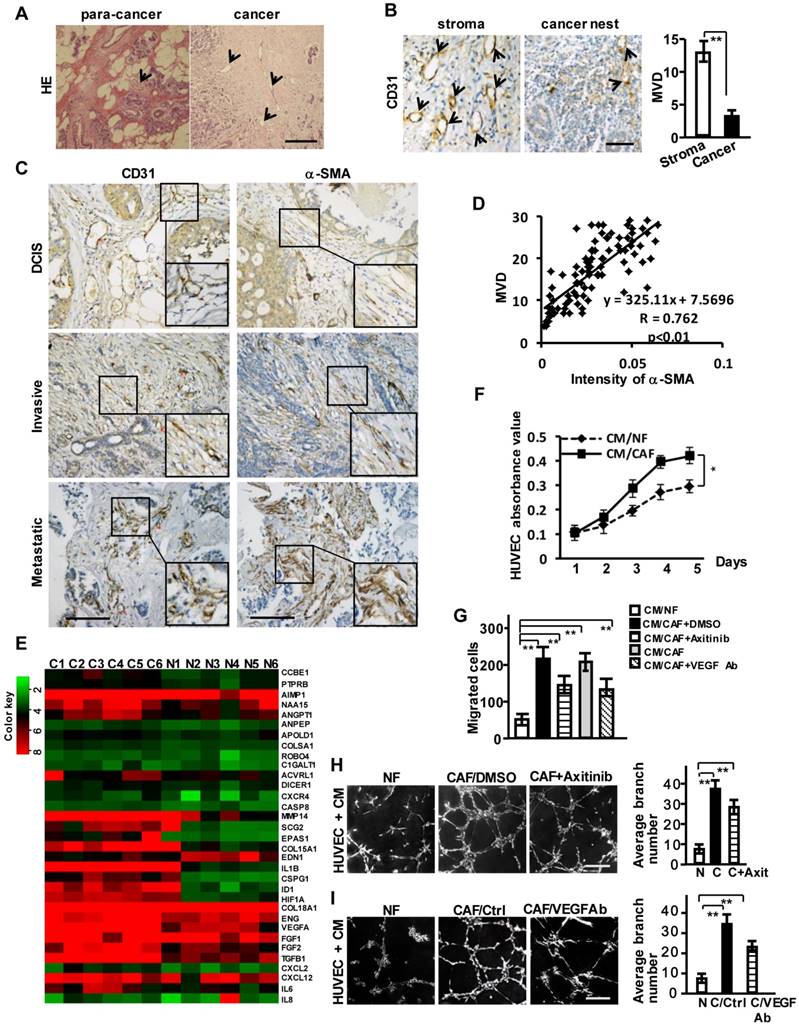
Furthermore, the angiogenesis-associated genes and factors classified by gene ontology analysis from mRNA microarray were significantly up-regulated in the primary CAFs (Figure 1E), whose purities were identified by fibroblast biomarker FN, the activated fibroblasts biomarkers, epithelial markers, and endothelium marker (Figure S1C-S1D). To assess whether CAFs affect the angiogenic behavior of vascular endothelial cells, a co-culture system including human umbilical vein endothelial cells (HUVECs) and conditioned medium (CM) derived from CAFs or NFs was employed. The proliferation (Figure 1F), invasion (Figure 1G) and tubule formation abilities (Figure 1H-1I) of HUVECs were dramatically enhanced by CM from CAFs. The addition of Axitinib (an effective inhibitor for activated VEGFR of vascular endothelial cells) (Figure S1E) in CM derived from CAFs or the CM treated with anti-VEGF antibody (Figure S1F) could not completely impede the CAF-induced invasion and tubule formation (Figure 1G-1I). These data suggest that the breast cancer stromal CAFs may contribute to VEGF-independent angiogenesis.
Decreased miR-205 in CAFs contributes to breast tumor angiogenesis
Our previous studies identified a set of dysregulated miRNAs in CAFs [25]. MiR-205 was down-regulated in CAFs (Figure S2A-S2B), the MDA-MB-231 cell-educated fibroblasts (Figure S2C), and a set of primary CAFs freshly isolated from 21 patients with breast cancer (Figure 2A). Decreased miR-205 expression was found to be associated with higher CD31 levels and worse overall survival of breast cancer patients (Figure 2B, Figure S2D) after analysis of the Cancer Genome Atlas (TCGA) database. Similarly, the higher level of CD31 and the abundance of MVD were oppositively corresponding to lower levels of stromal miR-205 in the detected breast tumor tissues (Figure 2C). Bioinformatics analysis revealed that the predicted targets of miR-205 were involved in angiogenesis and cell differentiation (Figure S2E). The several known angiogenic factors including VEGFA, TGFB1, IL6 were increased in NF/anti-miR-205 (loss of miR-205 by miR-205 inhibitor in NFs), decreased in CAF/miR-205 (over-expression of miR-205 in CAFs) in comparison to their control cells (Figure S2F). These data indicate that miR-205 in stromal fibroblasts potentially affects angiogenesis in breast tumors.
Downregulated miR-205 in CAFs is a core player in breast tumor angiogenesis. (A) The expressions of miR-205 were evaluated by qRT-PCR in 21 paired NFs and CAFs freshly isolated from breast carcinoma tissues. (B) Pearson's correlations are shown between miR-205 and CD31 expressions analyzed using the Cancer Genome Atlas (TCGA) database. (C) Representative images of MVD (Left) in breast tissues with high miR-205 expression of NFs or low miR-205 expression of CAFs. Scale bar, 200 μm. Quantity of MVD between breast tissues with high or low miR-205 expression (Middle panel). Pearson's correlations of CD31 expression in breast tissues and miR-205 expression in fibroblasts (Right). (D-G) HUVECs were co-cultured on Matrigel in the presence of CM from NFs/anti-miR205 or its control cells with Axitinib, or the CM neutralized with the anti-VEGF antibody. The representative images of tubule formation (D, F; Scale bar, 300 μm) and sprouting (E, G; Scale bar, 150 μm) are shown. (α-miR205: anti-miR205, inhibitor of miR-205; n=3; **P<0.01).
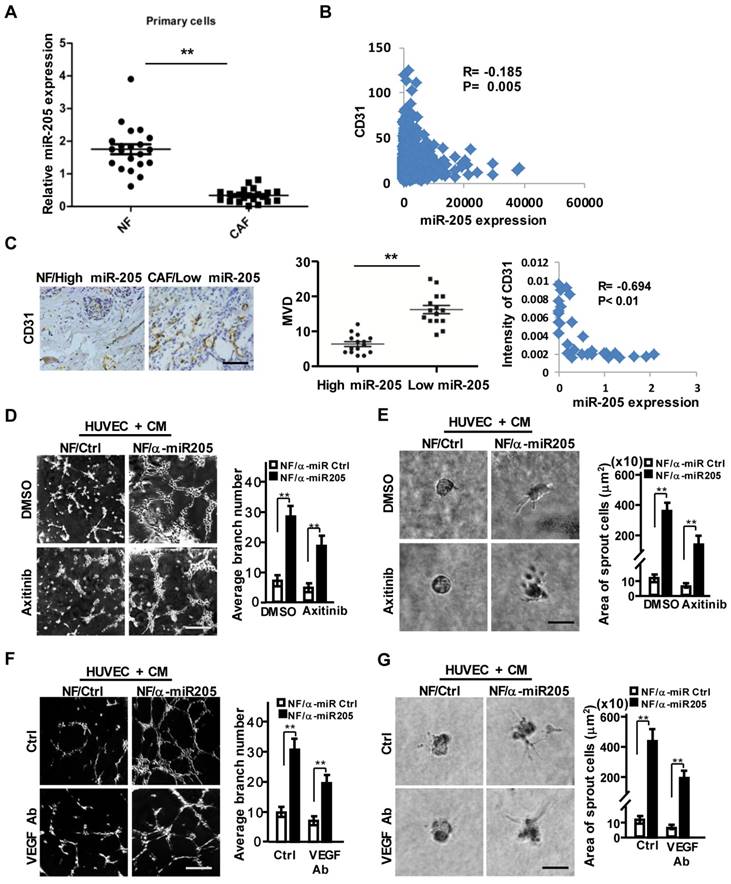
Indeed, miR-205 in stromal fibroblasts is closely related to the pro-angiogenic phenotypes of HUVECs. Impeded miR-205 expression in NFs (NFs/anti-miR-205) was observed using its antisense sequences (anti-miR-205) (Figure S3A), and CM derived from the NFs/anti-miR-205 cells significantly promoted the migration, invasion (Figure S3B-S3C, upper), tubule formation and sprouting of HUVECs (Figure 2D-2G, upper). Furthermore, after rescuing miR-205 expression in CAFs (CAFs/miR-205) (Figure S3A), CM from these CAFs (miR-205 overexpressing) disrupted the angiogenic potentials of HUVECs (Figure S3B-S3C, down; and Figure S3D-S3G, upper). These findings show that down-regulation of miR-205 in CAFs indeed promotes angiogenesis.
VEGF, a major driving factor for tumor angiogenesis, was elevated in miR-205-silenced NFs or repressed in miR-205-overexpressing CAFs (Figure S3H). Interestingly, CM derived from CAFs still kept the ability to promote tubule formation and sprouting of HUVECs in the presence of Axitinib (Figure 2D-2E and Figure S3D-S3E, down panel) or anti-VEGF antibody (Figure 2F-2G and Figure S3F-S3G, down panel). These data suggest a VEGF-independent proangiogenic function of miR-205 in CAFs.
YAP1, a downstream target of miR-205, is associated with tumor angiogenesis
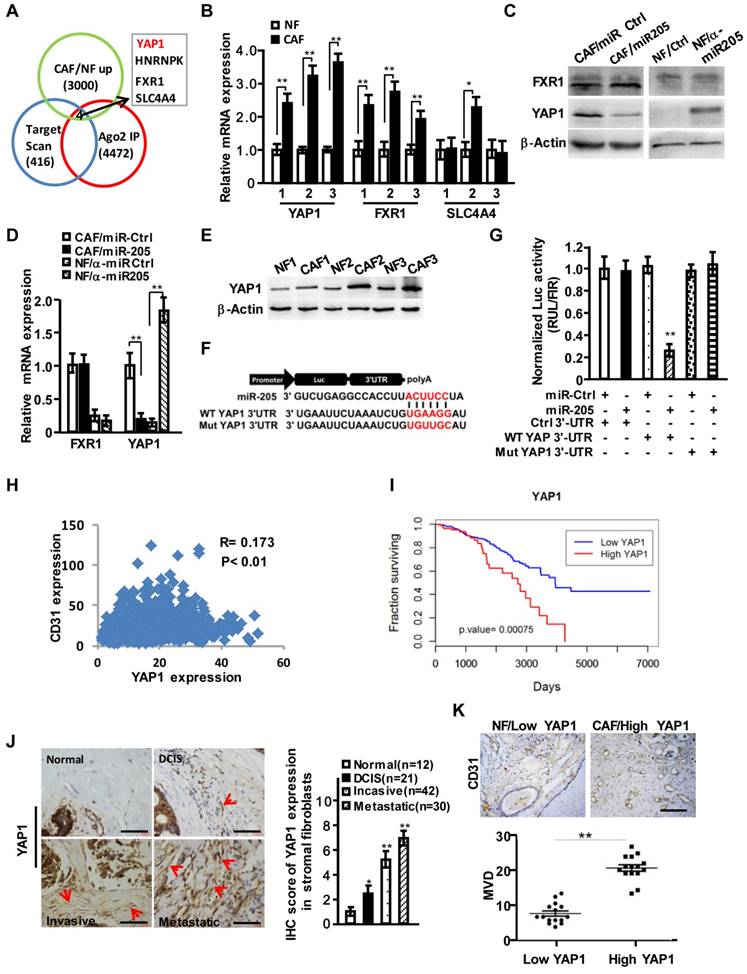
To further explore the functions of stromal miR-205 in regulating angiogenesis, we identified target genes of miR-205 by merging results (GSE39735) acquired from GEO database (http://www.ncbi.nlm.nih.gov/geo/) with miR-205 targets predicted by Target Scan and the up-regulated genes (fold change >2.0) in our previous mRNA expression profiles. We obtained 4 candidate genes including YAP1, HNRNPK, FXR1 and SLC4A4 (Figure 3A). Further analysis confirmed that only YAP1 was significantly changed at mRNA and protein levels in CAFs (Figure 3B-3D). The enhanced expression of YAP1 was observed in 3 immortalized CAFs (Figure 3E). In addition, the potential binding sites of miR-205 were identified in the YAP1 3'-UTR (Figure 3F), which led to a dramatic transcriptional suppression of YAP1 under ectopic expression of miR-205 (Figure 3G). Mutation of the binding sites in the YAP1 3'-UTR abolished the repression of miR-205 to YAP1 (Figure 3G), suggesting that YAP1 is a direct target of miR-205 in CAFs. Using the Cancer Genome Atlas (TCGA) database, high level of YAP1 was found to be positively related to CD31 expression in breast cancer (Figure 3H) and worse overall survival of breast cancer patients (Figure 3I). The high levels of YAP1 proteins in CAFs were further confirmed in the developed breast tumor tissues (Figure 3J), revealing a close correlation between YAP1 in stromal fibroblasts and MVD in these tissues (Figure 3K).
YAP1 is directly targeted by miR-205 in CAFs and is positively related to CD31 expression in breast cancer. (A) Venn diagram to show the putative miR-205 targets significantly dysregulated in CAFs. (B) YAP1, FXR1 and SLC4A4 expressions were determined by qRT-PCR in randomly selected 3 paired NFs and CAFs. (C, D) Protein (C) or mRNA levels (D) of YAP1 and FXR1 in CAFs/miR-205 or in NFs /anti-miR-205 were determined by Western blotting and qRT-PCR. (E) Protein levels of the endogenous YAP1 in 3 paired NFs and CAFs were determined by Western blot analysis. (F) Schematic diagram of luciferase reporters with the wild-type (WT) YAP1 3'-UTR or the mutant (Mut) YAP1 3'-UTR binding sites for miR-205. (G) The effect of miR-Ctrl or miR-205 on luciferase activities in CAFs co-transfected with either the wild-type YAP1 3'-UTR reporter or the mutant miR-205 binding sites reporter. (H) Pearson's correlations are shown between YAP1 and CD31 expression levels in breast cancer. (I) Kaplan-Meier survival analysis of breast cancer patients with high- or low-expression of YAP1. (J) Representative images of YAP1 expressions by IHC staining in normal and different stages of cancerous mammary tissues. The red arrows indicate the YAP1 high-expressing fibroblasts in the representative tumor tissues. Scale bar, 200 μm. The chart shows pathology scoring in breast tumor stroma. (K) Representative images of MVD by CD31 staining in breast para-cancer tissues with YAP1 low-expressed NFs or carcinoma tissues with YAP1 high-expressed CAFs. Scale bar, 200 μm. (n=3; *P<0.05; **P<0.01).
YAP1 activating NFs into CAFs promotes angiogenesis via VEGF independent manner
To investigate the pro-angiopoietic roles of YAP1 in CAFs, we transfected a retrovirus-mediated ectopic YAP1 construct into NFs, and suppressed YAP1 expression in CAFs using shRNA (Figure 4A, Figure S4A). YAP1-overexpression in NFs (NF/YAP1) or loss of endogenous YAP1 in CAF (CAF/sh-YAP1) resulted in activated NF or inactivated CAF, identified by α-SMA levels, cell proliferation and migration abilities (Figure 4A, Figure S4B-S4C), indicating that YAP1, as an effector of miR-205, is essential for CAF activation.
YAP1 is critical for miR-205-induced tumor angiogenesis via VEGF-independent signaling. (A-H) NFs were transfected with pBABE-flag-YAP1 or control vector, and CAFs were transfected with sh-YAP1 or control shRNA. (A) Western blot analysis of α-SMA, FAP, VEGF and YAP1 expression in the indicated NFs or CAFs cells. β-Actin is a loading control. (B) Cell invasion of HUVECs toward CM from the indicated fibroblasts were tested by Transwell assay. (C-F) HUVECs mixed with CM from CAF/sh-YAP1 or the control cells were cultured on Matrigel or fibrin gel and treated with Axitinib (C-D) or CM neutralized with anti-VEGF antibody (E-F). The representative images of tubule formation (C, E) or sprouting (D, F) were taken. Tubule formation (G) and sprouting (H) potential of HUVECs were assessed following incubation with CM from CAF/miR-Ctrl and CAF/miR-205 cells treated with control, YAP1 or YAP1+Axitinib. Scale bar, 300 μm for tubule formation; 150 μm for sprouting. (n=3; **P<0.01).
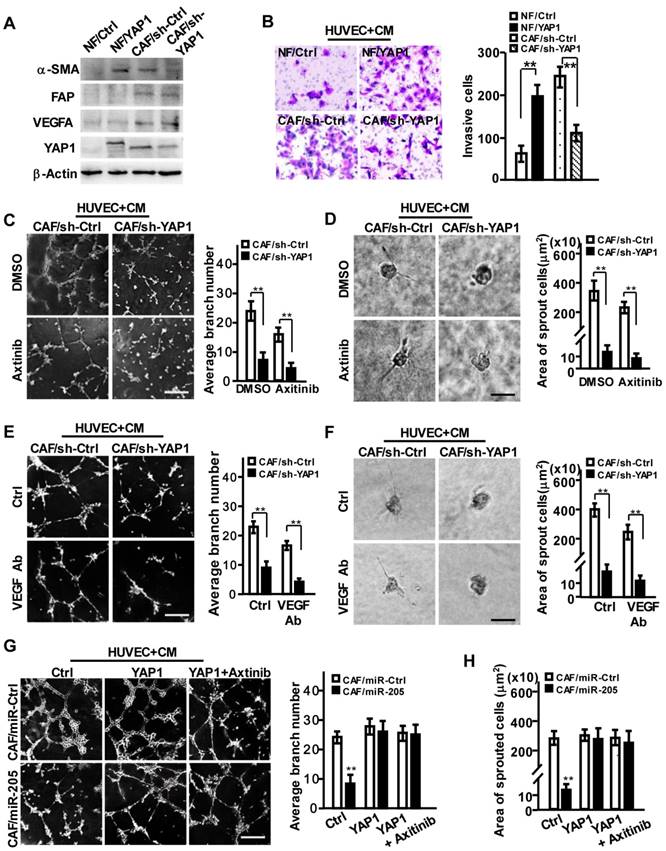
Furthermore, the acquirement of ectopic YAP1 in NFs or loss of endogenous YAP1 in CAFs had no effect on VEGF expression (Figure 4A, Figure S4D). However, the CM from YAP1-overexpressing NFs promoted cell migration and invasion of HUVECs, and the CM from YAP1 knocked-down CAFs impeded these behaviors in HUVECs (Figure 4B). Correspondingly, CM from YAP1-overexpressing NF cells obviously facilitated the tubule formation and sprouting of HUVECs (Figure S4E-S4H, upper), and CM from YAP1 knocked-down CAFs decreased HUVECs' tubule formation and sprouting abilities (Figure 4C-4F, upper). Axitinib or anti-VEGF antibody could augment these observed effects of YAP1-silenced CAFs (Figure 4C-4F, down). However, overexpression of YAP1 in NFs attenuated the suppressive functions of Axitinib or anti-VEGF antibody on tubule formation and sprouting of HUVECs (Figure S4E-S4H, down). These findings were further confirmed by YAP1 rescue in miR-205 overexpressing CAFs (CAFs/miR-205), which notably abrogated the anti-angiogenic effects of miR-205 (Figure 4G-4H). These data suggest that YAP1 promotes angiogenesis of vascular endothelial cells.
IL11 and IL15, the YAP1 targets, display a concordant effect on VEGF-independent angiogenesis
Based on the above findings, we proposed that some secreting factors other than VEGF in CAF's CM play a critical role in pro-angiogenesis. However, the known pro-angiogenic factors including SDF-1, VEGFA and FGF2 were not influenced by YAP1 in CAFs and YAP1-overexpressing NFs (NFs/YAP1) (Figure S4D). After careful analysis of the gene profiles from GEO database (GSE7700), which may be directly regulated by YAP1 and our previous mRNA profiles of CAFs by bioinformatics, HMGB1, CXCL14, IL6, IL11 and IL15 were identified as the YAP1-regulated candidate cytokines (Figure 5A). Actually, the mRNA expressions of IL6, IL11 and IL15 were down-regulated in YAP1 knock-down CAFs, and IL11 and IL15 were significantly affected by YAP1 in CAFs (Figure 5B). Overexpression of YAP1 in the miR-205-rescued CAFs (CAF/miR-205/YAP1) dramatically abrogated the suppressive effects of miR-205 on IL11 and IL15 (Figure 5C-5E). Luciferase assay showed that YAP1 could directly regulate IL11 and IL15 transcript expressions (Figure 5F).
IL11 and IL15 are required for YAP1-induced VEGF-independent angiogenesis in breast CAFs. (A) Venn diagram showing identification of cytokines as YAP1 putative targets significantly dysregulated in CAFs. (B) VEGFA, HMGB1, CXCL14, IL11 and IL15 expressions were determined by qRT-PCR in CAF/sh-Ctrl and CAF/sh-YAP1. (C) mRNA expressions of IL11 and IL15 were determined by qRT-PCR in CAF/miR-Ctrl and CAF/miR-205 transfected with YAP1 vector. (D, E) Protein levels of IL11 (D) and IL15 (E) in supernatant from the indicated engineered fibroblasts were determined by ELISA. (F) A luciferase reporter driven by IL11 or IL15 promoter in CAFs was co-transfected with sh-Ctrl or sh-YAP1 vector. (G, H) The representative images of tubule formation of HUVECs cultured with CM from the siRNA transfected-CAFs under treatment with or without Axitinib (G), or from CAF neutralized by control antibody, or IL11 and IL15 antibodies (H). Scale bar, 300 μm. (n=3; *P<0.05; **P<0.01).
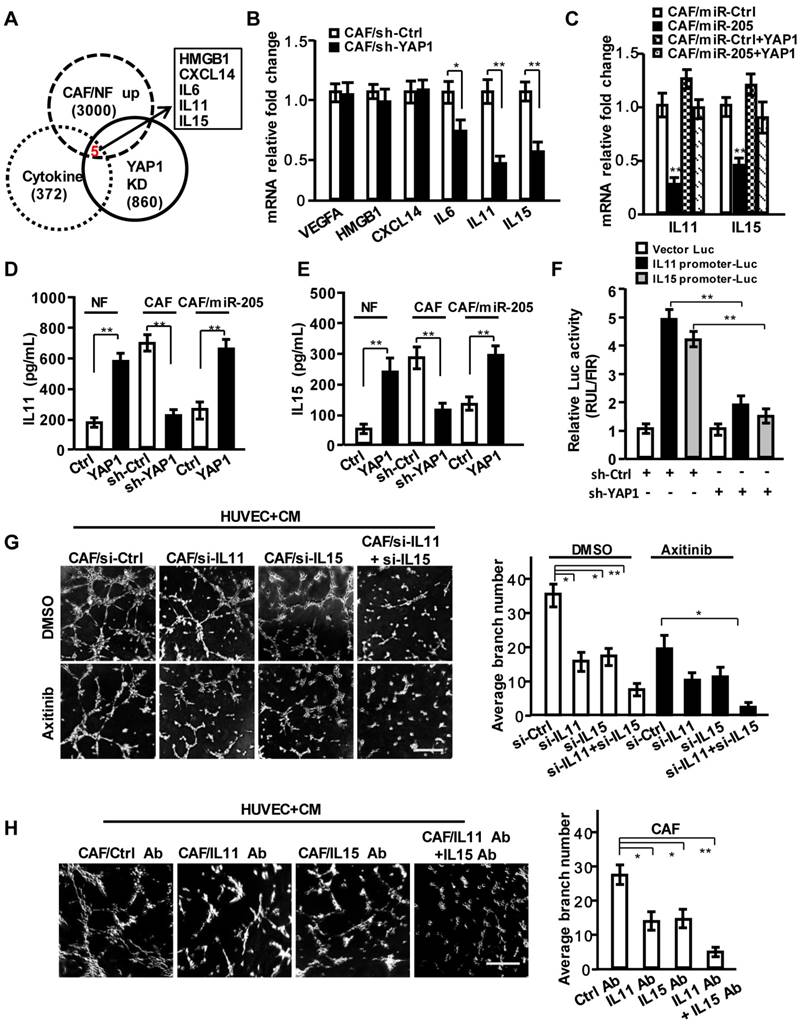
Following efficient knock-down of IL11 and IL15 in CAFs using their specific siRNA (Figure S5A-S5E), the corresponding CM decreased cell invasion (Figure S5F) and tubule formation of HUVECs (Figure 5G, upper; Figure S5G). Axitinib or anti-VEGF antibody treatment further augmented the suppression of tubule formation of HUVECs (Figure 5G, down; Figure S5G). Similarly, CM from CAFs (Figure 5H) or YAP1-overexpressing NFs (NF/YAP1) (Figure S5H) neutralized with anti-IL11 and/or anti-IL15 antibodies reduced the tubule formation of HUVECs. These data suggest that IL11 and IL15, the secreting proteins of CAFs, are essential to breast tumor angiogenesis.
IL11 and IL15 derived from CAFs activate STAT3 signaling in endothelial cells
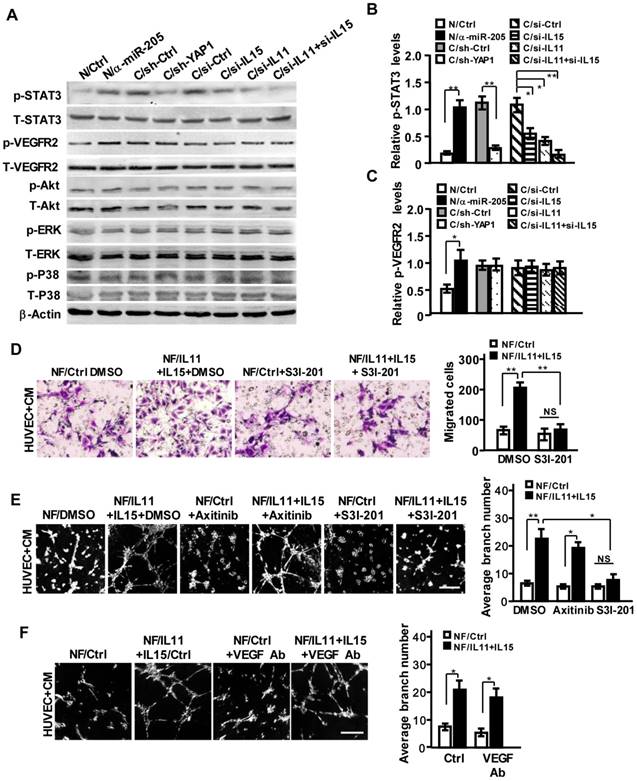
To understand the functional impact of IL11 and IL15 secreted by CAFs on pro-angiogenesis, the potential downstream signaling of interleukin family (e.g., JAK/STAT, ERK, PI3K/AKT) in HUVECs was examined. As shown in Figure 6A, a high level of phosphorylated STAT3 was detected in HUVECs that were co-cultured with CM derived from NFs/anti-miR-205 and CAFs. Knockdown of YAP1, or IL11, and IL15 in CAFs mitigated the activation of STAT3; simultaneous loss of IL11 and IL15 in CAFs could augment the inactivation of STAT3 in HUVECs (Figure 6A, 6B). However, the activation of VEGFR2 was not affected by these factors, except miR-205 (Figure 6A, 6C). Blockage of STAT3 signaling by inhibitor S3I-201 (Figure S6A) reduced cell invasion and tubule formation abilities of HUVECs (Figure S6B-S6C). Furthermore, transfection of ectopic IL11 and IL15 into NFs significantly increased IL11 and IL15 secretion in the supernatant (Figure S6D-S6F), thus leading to the activation of STAT3 signaling (Figure S6G) and increased invasion and tubule formation abilities of HUVECs (Figure 6D-6E), which could be remarkably abolished by S3I-201 but not Axitinib or anti-VEGF antibody (Figure 6E-6F).
IL11 and IL15 derived from CAFs synergistically activate STAT3 signaling in endothelial cells. (A-C) The total and phosphorylated proteins of STAT3, VEGFR2, PI3K/AKT, ERK and P38/MAPK in HUVECs cells were determined by Western blotting. β-Actin is the loading control. Levels of p-STAT3 (B) and p-VEGFR2 (C) were quantified as relative pixel intensity to β-Actin. (D-F) Cell invasion (D) and tubule formation (E,F) abilities of HUVECs were tested in the presence of CM from IL11 and IL15 overexpressing NFs with Axitinib, S3I-201 (E) or neutralizing anti-VEGF antibody (F); Scale bar, 300 μm. (n=3; NS, not significant; *P<0.05; **P<0.01).
In order to adequately assess the pro-angiogenic functions of IL11/IL15 in CM derived from the CAFs, we first evaluated the different types of vectors on some of the molecular biological characteristics of NFs or CAFs. There were no significant differences in cell proliferation (Figure S7A), miR-205 expression (Figure S7B), protein levels of YAP1/IL11/IL15 (Figure S7C), and pro-angiogenic functions (Figure S7D-S7E) between NF/anti-miR-Ctrl and NF/pBABE-Ctrl; or among CAF/miR-Ctrl, CAF/sh-Ctrl and CAF/si-Ctrl (Figure S7A-7E), suggesting negligible side effects of vectors on the inherent biological characteristics of NFs or CAFs. Next, total cell numbers, cell proliferation, cell apoptosis of the used stromal fibroblasts and protein concentrations in the CM from all used stromal fibroblasts were determined at the terminal time point of CM collection. Despite overexpression of miR-205, or knockdown of YAP1, IL11 and IL15 in CAFs decreased cell proliferation, while knockdown of miR-205, or overexpression of YAP1, IL11, and IL15 in NFs increased cell proliferation (Figure S4B, S7F-S7H). In growth medium, the total cell numbers of parent CAFs and NFs, and the engineered CAFs and NFs were almost the same (Figure S8A), and cell proliferation of these cells also had no detectable changes at the terminal time point of CM (Figure S8B). However, the percentage of apoptotic and viable cells were found to be a little bit different among these cell populations (around 2%, P>0.05) (Figure S8C), thus leading to a tiny difference in protein concentrations in their CM (Figure S8D). Protein concentrations in FBS-containing growth medium from these CAFs and NFs also had no significant difference, which may be obscured by serum (Figure S8E). To understand whether IL11 and IL15 in CM are affected by cell apoptosis, the protein concentration of CM derived from activated fibroblasts was normalized using the protein concentration deviation value between activated fibroblasts and the control fibroblasts, and adjusted to the corresponding protein concentration of control cells; then IL11 and IL15 were detected by ELISA assay. IL11 and IL15 protein levels in CM derived from the activated fibroblasts were obviously higher than that in CM derived from the control fibroblasts (Figure S8F-S8G), which is similar to the above results (Figure 5D-5E, S5D-S5E and S6E-S6F). These data demonstrate that CM, especially angiogenic factors IL11 and IL15 derived from the activated stromal fibroblasts, are not affected by cell proliferation and cell apoptosis, but are closely governed by miR-205/YAP1 signaling and play essential roles in breast tumor angiogenesis.
CAFs promote tumor angiogenesis in a VEGF-independent manner to fuel breast tumor growth and metastasis in vivo
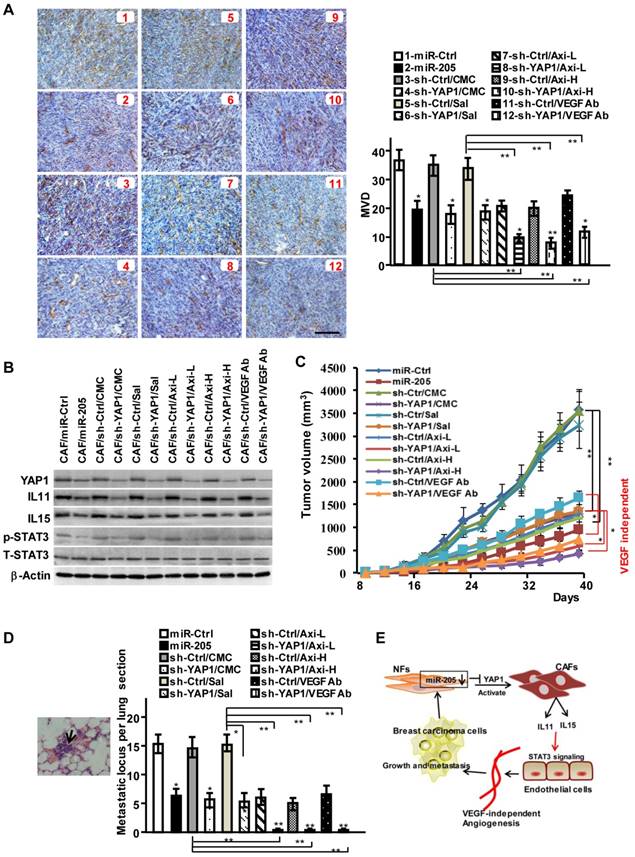
To investigate the function of miR-205/YAP1 signaling axis in pro-angiogenesis, MDA-MB-231 cells mixed with CAFs (labeled CAF/miR-Ctrl), CAFs/miR-205 or CAFs/sh-YAP1 were subcutaneously injected into nude mice. Compared with the tumor burden mice injected with MDA-MB-231 cells and CAFs, the tumor burden mice injected with MDA-MB-231 cells mixed with engineered CAFs (CAF/miR-205, CAF/sh-YAP1 (labeled CAF/sh-YAP1/CMC or Sal in Fig 7A)) had fewer blood vessels on the tumor surface, and less MVD and CD31 staining in tumor tissues (Figure 7A; Figure S9A-S9B). In contrast to blockage of VEGF signaling alone by Axitinib or by anti-VEGF antibody, YAP1-knock-down in CAFs combined with VEGF signaling inhibitors could augment the suppressive efficiencies to tumor angiogenesis (Figure 7A; Figure S9A-S9B), indicating that YAP1 acts as an obviously pro-angiogenic effector in the VEGF-independent pathway. Consistently, the levels of IL11 and IL15, the critical pro-angiogenic factors, which are regulated by miR-205 and YAP1 signaling axis, were reduced in the tumors with CAF/miR-205 or CAF/sh-YAP1 compared to their control tumors (Figure 7B). Moreover, phosphorylated STAT3 in tumors with CAF/miR-205 or CAF/sh-YAP1 significantly decreased (Figure 7B). Correspondingly, blockage of VEGF-independent tumor angiogenesis led to decreased tumor growth (Figure 7C, Figure S9C-S9D) and metastases in mice lung tissues (Figure 7D). Collectively, these data support the notion that the decreased miR-205 in CAFs increases the expression of YAP1, IL11 and IL15, and promotes tumor angiogenesis to fuel tumor cell growth, invasion and metastasis (Figure 7E).
MiR-205 and shRNA against YAP1 block VEGF independent angiogenesis in vivo. (A-D) Breast cancer cell MDA-MB-231 were mixed with CAFs/miR-205, CAFs/sh-YAP1 or their control cells, and subcutaneously injected into nude mice. The mice were treated with low (30mg/kg) or high concentration (40mg/kg) of Axitinib, or VEGF antibody as described in materials, carboxymethylcellulose (CMC) or saline as control. (A) Representative images of IHC staining of CD31 are shown; Scale bar, 200 μm; (B) Protein levels of YAP1, IL11 and IL15, phosphorylated and total STAT3 in tumors were determined by Western blotting. (C) The tumor growth curves in mice. (D) Representative images of pulmonary metastases examined by H&E-stained in the lung section; the arrows indicate metastases; Graph shows the metastatic locus per lung section. (n=5 per group; *P<0.05;**P<0.01). (E) Schematic representation depicts the role of miR-205/YAP1 in CAFs-mediated VEGF independent angiogenesis via IL11, IL15 and STAT3 signaling.
Discussion
Tumor angiogenesis plays a critical role in tumor development. However, how the activated fibroblasts in human breast tumor microenvironment regulate tumor VEGF-independent angiogenesis remains unknown. In this study, we found that the stromal CAFs are closely involved in tumor angiogenesis in breast tumor, in which the activated fibroblasts can recruit and stimulate vascular endothelial cells to form vascular tubes and sprouts, thus enhancing breast tumor cell growth and metastasis in vivo. The downregulated miR-205 and its target YAP1 activate NFs into CAFs and enhance CAF proliferation and migration activities. Furthermore, these activated CAFs induced by YAP1 cannot promote VEGF, but their IL11 and IL15 expressions stimulate angiogenesis, even under the blockage of VEGF signaling by Axitinib and anti-VEGF antibody, suggesting that CAFs can modulate tumor angiogenesis resistance to anti-VEGF therapy via IL11 and IL15. Collectively, our results provide a novel insight into breast CAFs pro-angiogenesis role in a VEGF-independent manner.
In this study, we found that the downregulated stromal miR-205 plays a key role in the activation of stromal fibroblasts, thus enhancing angiogenesis. Our findings support that the dysregulated miRNAs are involved in multiple CAF-driven tumor biological processes. For example, downregulation of miR-31 and miR-21 in ovarian CAFs [36], and miR-15 and miR-16 in prostate CAFs [37] have been shown to promote tumor migration and invasion through reprogrammed CAFs. Furthermore, the downregulated miR-320 in mammary fibroblasts induces oncogenic secretome, which in turn promotes tumor angiogenesis and tumor invasion [38].
Our studies disclose that YAP1, a target of miR-205 in breast CAFs, functions as a critical regulator in activated CAF-mediated tumor angiogenesis. YAP1, the nuclear co-transcriptional factor of the Hippo pathway, has been reported to govern organ size and stem cell differentiation in embryonic development [39]. YAP1 is also recognized as a functional mediator in lung fibroblast differentiation and fibrogenesis [29, 40]. In addition, the oncogenic YAP1 in ovarian cancer promotes tumor cell growth and metastasis [41], resulting in a poor survival for tumor patients [42]. Here, we found that miR-205 targets YAP1 in CAFs to promote VEGF-independent angiogenesis in breast tumor tissues. Although a high level of VEGF was detected in breast CAFs, YAP1 does not regulate VEGF expression. Furthermore, inhibition of VEGF signaling can't block YAP1-mediated pro-angiogenesis. Similarly, high level of YAP1 has been found in human cholangiocarcinoma to promote angiogenesis by regulating NFAP5 expression [30]. These findings support that YAP1 induces a VEGF-independent pro-angiogenic signature in tumor angiogenesis.
Our studies prove that inflammatory factor IL11 and IL15, aside from previously reported FGF2 and SDF1 [43], are novel pro-angiogenic factors regulated by YAP1 in breast stromal CAFs. Previous studies proved that IL11 and IL15 are the cytokines with biological activities towards T-cells [44]. IL15 secreted from uterine nature killer cells induce angiogenesis in endometrium [45]. IL11 and IL15 were then found to have metastatic potential in colorectal cancer cells [46] and act as a chemoresistant factor in lung adenocarcinoma [47]. IL11 or IL15 also have the capacity to promote proliferation of cancer cells [48]. Here, we confirmed that IL11 and IL15 in stromal CAFs, directly regulated by YAP1, act as powerful mediators between CAFs and breast cancer cells to facilitate tumor angiogenesis. The aberrant YAP1 induced by miR-205 in stromal CAFs enhances IL11 and IL15 expressions, but not the known pro-angiogenic factors such as VEGF, SDF, FGFs, or PDGF, to stimulate the STAT3 activation and promote tubule formation and sprouting of HUVECs. Application of Axitinib or anti-VEGF antibody alone in either the co-culture system of HUVECs and CM derived from CAF, or in tumor-burdened mice in vivo, resulted in vascular endothelial cells retaining their angiogenic properties. Axitinib or anti-VEGF antibody combination with siRNA against IL11 and IL15 in CAFs severely blunted tubule formation and sprouting of HUVECs. Moreover, IL11 and IL15 from breast CAFs can activate STAT3 in HUVECs. This is in line with the previous findings by Tao [47] and Angiolillo [49]. These findings suggest a complicated VEGF-dependent and VEGF-independent angiogenesis in breast stromal CAFs.
Our findings support that VEGF-independent angiogenesis is pivotal to clinical tumor therapy. Tumor resistance to anti-VEGF therapy and sustained tumor angiogenesis might occur in a VEGF-independent manner [50]. Excluding tumor cells themselves, various stromal cells contribute to resistance to anti-VEGF signaling, ultimately promoting tumor progression [51]. For example, CD11b+Gr1+ cells, initiated by G-CSF, mediate tumor refractoriness to anti-VEGF therapy [52]. CAFs have been confirmed to be involved in resistance to anti-VEGF treatment. For example, TAFs isolated from invasive breast cancer secrete SDF-1 to recruit hematopoietic cells [53], or anti-VEGFA refractory subcutaneous tumor produces abundant PDGF-C [22] to maintain tumor angiogenesis. Here, we prove that IL11 and IL15 are novel pro-angiogenic factors in breast CAF. Particularly, the angiogenic factors IL11 and IL15 derived from CAFs are regulated by miR-205/YAP1, which is not affected by the different types of control vectors, cell proliferation and cell apoptosis of fibroblasts during preparation of CM. Thus, miR-205/YAP1/IL11/IL15 signaling axis, a VEGF-independent signaling in breast stromal CAFs, plays a specific role to tumor angiogenesis. However, the combined targeting YAP1 and VEGF did not completely block angiogenesis and tumor growth in vivo, suggesting additional mechanisms involved in VEGF-independent angiogenesis.
In conclusion, our study demonstrates that activated CAFs promote VEGF-independent proangiogenic processes in the breast tumor microenvironment. Secretion of IL11 and IL15 governed by miR-205/YAP1 signaling in CAFs specifically stimulates STAT3 signaling in vascular endothelial cells, contributing to tumor angiogenesis and resistance to anti-VEGF, thus triggering tumor growth and metastasis in breast cancer.
Abbreviations
CAF: cancer associated-fibroblast; miR-205: micoRNA-205; YAP1: Yes-associated protein 1; VEGF: vascular endothelial growth factor A; IL11: interleukin 11; IL15: interleukin 15; NF: normal fibroblast; HUVECs: Human Umbilical Vein Endothelial Cells; STAT3: signal transducer and activator of transcription 3; VEGFR: vascular endothelial growth factor receptor; FDA: Food and Drug Administration; DLL4: delta like canonical Notch ligand 4; NRP1: neuropilin 1; ABL1: ABL proto-oncogene 1, non-receptor tyrosine kinase; FGF: fibroblast growth factor; PDGF: platelet derived growth factor; TAMs: Tumor-associated macrophages; TGF: transforming growth factor; SDF1: stromal cell-derived factor 1; G-CSF: colony stimulating factor 3; ECM: Extracellular matrix; CSCs: cancer stem cells; MFAP5: microfibril associated protein 5; H: hour; α-SMA: α-smooth muscle actin; FAP: fibroblast activation protein; CK8+18: cytokeratin 8 and cytokeratin 18; CD31: Platelet endothelial cell adhesion molecule (PECAM); LV: lentivirus; shRNA: short hairpin RNA; siRNA: small interfering RNA; MVD: microvessel density; IHC: immunohistochemistry; IF: immunofluorescence; FN: fibronectin; FITC: fluorescein isothiocyanate; qRT-PCR: quantitative real-time polymerase chain reaction; CM: conditioned medium; ELISA: enzyme-linked immuno sorbent assay; TCGA: The Cancer Genome Atlas; DCIS: carcinoma in situ; IDC: invasive ductal carcinoma; HNRNPK: heterogeneous nuclear ribonucleoprotein K; SLC4A4: solute carrier family 4 member 4; Ctrl: control; CMC: carboxymethylcellulose; Sal: saline.
Supplementary Material
Supplementary Figure S1-S9; Supplementary Table S1-S2.
Acknowledgements
This work was supported in part by National Natural Science Foundation of China (NSFC 81402180, NSFC 81773078) for Yixuan Hou; and National Natural Science Foundation of China (NSFC 81472476, NSFC 31171336, NSFC 81072147) for Manran Liu; And also supported in part by the outstanding Doctor Fund of Chongqing Medical University (2015) and Chongqing education committee (CYB15090) for Yane Du; We also thank Prof. Jörg Hackermüller for his kind donation of pSUPER-vector and pSUPER-miR-205.
Author contributions
Y.D. carried out most of the experiments, G.T. and G.Y. provided primary fibroblasts and contributed to the experimental design. G.L., D.Y., L.L. and K.S. performed cell culture and IHC staining. L.X. and Y.C. contributed to the data analysis. K.S. and H.L. performed the bioinformatic analysis. M.L. and Y.H. conceived and designed the experiments and wrote the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Naumov GN, Akslen LA, Folkman J. Role of angiogenesis in human tumor dormancy: animal models of the angiogenic switch. Cell Cycle. 2006;5:1779-1787
2. Ausprunk DH, Folkman J. Migration and proliferation of endothelial cells in preformed and newly formed blood vessels during tumor angiogenesis. Microvasc Res. 1977;14:53-65
3. Ferrara N. VEGF and the quest for tumour angiogenesis factors. Nat Rev Cancer. 2002;2:795-803
4. Backer MV, Backer JM. Imaging key biomarkers of tumor angiogenesis. Theranostics. 2012;2:502-515
5. Claesson-Welsh L, Welsh M. VEGFA and tumour angiogenesis. J Intern Med. 2013;273:114-127
6. Welti J, Loges S, Dimmeler S, Carmeliet P. Recent molecular discoveries in angiogenesis and antiangiogenic therapies in cancer. J Clin Invest. 2013;123:3190-3200
7. Torok S, Rezeli M, Kelemen O, Vegvari A, Watanabe K, Sugihara Y. et al. Limited Tumor Tissue Drug Penetration Contributes to Primary Resistance against Angiogenesis Inhibitors. Theranostics. 2017;7:400-412
8. Verma S, McLeod D, Batist G, Robidoux A, Martins IR, Mackey JR. In the end what matters most? A review of clinical endpoints in advanced breast cancer. Oncologist. 2011;16:25-35
9. Kerbel RS. Tumor angiogenesis. N Engl J Med. 2008;358:2039-2049
10. Li JL, Sainson RC, Oon CE, Turley H, Leek R, Sheldon H. et al. DLL4-Notch signaling mediates tumor resistance to anti-VEGF therapy in vivo. Cancer Res. 2011;71:6073-6083
11. Jung YD, Ahmad SA, Liu W, Reinmuth N, Parikh A, Stoeltzing O. et al. The role of the microenvironment and intercellular cross-talk in tumor angiogenesis. Semin Cancer Biol. 2002;12:105-112
12. Yu JL, Rak JW. Host microenvironment in breast cancer development: inflammatory and immune cells in tumour angiogenesis and arteriogenesis. Breast Cancer Res. 2003;5:83-88
13. Lyden D, Hattori K, Dias S, Costa C, Blaikie P, Butros L. et al. Impaired recruitment of bone-marrow-derived endothelial and hematopoietic precursor cells blocks tumor angiogenesis and growth. Nat Med. 2001;7:1194-1201
14. Rundhaug JE. Matrix metalloproteinases and angiogenesis. J Cell Mol Med. 2005;9:267-285
15. Pan D, Schmieder AH, Wang K, Yang X, Senpan A, Cui G. et al. Anti-angiogenesis therapy in the Vx2 rabbit cancer model with a lipase-cleavable Sn 2 taxane phospholipid prodrug using alpha(v)beta(3)-targeted theranostic nanoparticles. Theranostics. 2014;4:565-578
16. Bhowmick NA, Neilson EG, Moses HL. Stromal fibroblasts in cancer initiation and progression. Nature. 2004;432:332-337
17. Kalluri R, Zeisberg M. Fibroblasts in cancer. Nat Rev Cancer. 2006;6:392-401
18. Dong J, Grunstein J, Tejada M, Peale F, Frantz G, Liang WC. et al. VEGF-null cells require PDGFR alpha signaling-mediated stromal fibroblast recruitment for tumorigenesis. EMBO J. 2004;23:2800-2810
19. Granot D, Addadi Y, Kalchenko V, Harmelin A, Kunz-Schughart LA, Neeman M. In vivo imaging of the systemic recruitment of fibroblasts to the angiogenic rim of ovarian carcinoma tumors. Cancer Res. 2007;67:9180-9189
20. Weis SM, Cheresh DA. Tumor angiogenesis: molecular pathways and therapeutic targets. Nat Med. 2011;17:1359-1370
21. Erez N, Truitt M, Olson P, Arron ST, Hanahan D. Cancer-Associated Fibroblasts Are Activated in Incipient Neoplasia to Orchestrate Tumor-Promoting Inflammation in an NF-kappaB-Dependent Manner. Cancer Cell. 2010;17:135-147
22. Crawford Y, Kasman I, Yu L, Zhong C, Wu X, Modrusan Z. et al. PDGF-C mediates the angiogenic and tumorigenic properties of fibroblasts associated with tumors refractory to anti-VEGF treatment. Cancer Cell. 2009;15:21-34
23. Zhou B, Ma R, Si W, Li S, Xu Y, Tu X. et al. MicroRNA-503 targets FGF2 and VEGFA and inhibits tumor angiogenesis and growth. Cancer Lett. 2013;333:159-169
24. Png KJ, Halberg N, Yoshida M, Tavazoie SF. A microRNA regulon that mediates endothelial recruitment and metastasis by cancer cells. Nature. 2011;481:190-194
25. Zhao L, Sun Y, Hou Y, Peng Q, Wang L, Luo H. et al. MiRNA expression analysis of cancer-associated fibroblasts and normal fibroblasts in breast cancer. Int J Biochem Cell Biol. 2012;44:2051-2059
26. Wu H, Zhu S, Mo YY. Suppression of cell growth and invasion by miR-205 in breast cancer. Cell Res. 2009;19:439-448
27. Piccolo S, Dupont S, Cordenonsi M. The biology of YAP/TAZ: hippo signaling and beyond. Physiological reviews. 2014;94:1287-1312
28. Lin Z, Zhou P, von Gise A, Gu F, Ma Q, Chen J. et al. Pi3kcb links Hippo-YAP and PI3K-AKT signaling pathways to promote cardiomyocyte proliferation and survival. Circulation research. 2015;116:35-45
29. Guo SC, Tao SC, Yin WJ, Qi X, Yuan T, Zhang CQ. Exosomes derived from platelet-rich plasma promote the re-epithelization of chronic cutaneous wounds via activation of YAP in a diabetic rat model. Theranostics. 2017;7:81-96
30. Marti P, Stein C, Blumer T, Abraham Y, Dill MT, Pikiolek M. et al. YAP promotes proliferation, chemoresistance, and angiogenesis in human cholangiocarcinoma through TEAD transcription factors. Hepatology. 2015;62:1497-1510
31. Peng Q, Zhao L, Hou Y, Sun Y, Wang L, Luo H. et al. Biological characteristics and genetic heterogeneity between carcinoma-associated fibroblasts and their paired normal fibroblasts in human breast cancer. PLoS One. 2013;8:e60321
32. Tang X, Hou Y, Yang G, Wang X, Tang S, Du YE. et al. Stromal miR-200s contribute to breast cancer cell invasion through CAF activation and ECM remodeling. Cell Death Differ. 2016;23:132-145
33. Weidner N. Current pathologic methods for measuring intratumoral microvessel density within breast carcinoma and other solid tumors. Breast Cancer Res Treat. 1995;36:169-180
34. Bai YY, Gao X, Wang YC, Peng XG, Chang D, Zheng S. et al. Image-guided pro-angiogenic therapy in diabetic stroke mouse models using a multi-modal nanoprobe. Theranostics. 2014;4:787-797
35. Pecot CV, Rupaimoole R, Yang D, Akbani R, Ivan C, Lu C. et al. Tumour angiogenesis regulation by the miR-200 family. Nat Commun. 2013;4:2427
36. Mitra AK, Zillhardt M, Hua Y, Tiwari P, Murmann AE, Peter ME. et al. MicroRNAs reprogram normal fibroblasts into cancer-associated fibroblasts in ovarian cancer. Cancer Discov. 2012;2:1100-1108
37. Musumeci M, Coppola V, Addario A, Patrizii M, Maugeri-Sacca M, Memeo L. et al. Control of tumor and microenvironment cross-talk by miR-15a and miR-16 in prostate cancer. Oncogene. 2011;30:4231-4242
38. Bronisz A, Godlewski J, Wallace JA, Merchant AS, Nowicki MO, Mathsyaraja H. et al. Reprogramming of the tumour microenvironment by stromal PTEN-regulated miR-320. Nat Cell Biol. 2011;14:159-167
39. Camargo FD, Gokhale S, Johnnidis JB, Fu D, Bell GW, Jaenisch R. et al. YAP1 increases organ size and expands undifferentiated progenitor cells. Curr Biol. 2007;17:2054-2060
40. Liu F, Lagares D, Choi KM, Stopfer L, Marinkovic A, Vrbanac V. et al. Mechanosignaling through YAP and TAZ drives fibroblast activation and fibrosis. Am J Physiol Lung Cell Mol Physiol. 2015;308:L344-357
41. Lamar JM, Stern P, Liu H, Schindler JW, Jiang ZG, Hynes RO. The Hippo pathway target, YAP, promotes metastasis through its TEAD-interaction domain. Proc Natl Acad Sci U S A. 2012;109:E2441-2450
42. Hall CA, Wang R, Miao J, Oliva E, Shen X, Wheeler T. et al. Hippo pathway effector Yap is an ovarian cancer oncogene. Cancer Res. 2010;70:8517-8525
43. Nyberg P, Salo T, Kalluri R. Tumor microenvironment and angiogenesis. Front Biosci. 2008;13:6537-6553
44. Curti A, Ratta M, Corinti S, Girolomoni G, Ricci F, Tazzari P. et al. Interleukin-11 induces Th2 polarization of human CD4(+) T cells. Blood. 2001;97:2758-2763
45. Li XF, Charnock-Jones DS, Zhang E, Hiby S, Malik S, Day K. et al. Angiogenic growth factor messenger ribonucleic acids in uterine natural killer cells. The Journal of clinical endocrinology and metabolism. 2001;86:1823-1834
46. Calon A, Espinet E, Palomo-Ponce S, Tauriello DV, Iglesias M, Cespedes MV. et al. Dependency of colorectal cancer on a TGF-beta-driven program in stromal cells for metastasis initiation. Cancer Cell. 2012;22:571-584
47. Tao L, Huang G, Wang R, Pan Y, He Z, Chu X. et al. Cancer-associated fibroblasts treated with cisplatin facilitates chemoresistance of lung adenocarcinoma through IL-11/IL-11R/STAT3 signaling pathway. Sci Rep. 2016;6:38408
48. Yoshizaki A, Nakayama T, Yamazumi K, Yakata Y, Taba M, Sekine I. Expression of interleukin (IL)-11 and IL-11 receptor in human colorectal adenocarcinoma: IL-11 up-regulation of the invasive and proliferative activity of human colorectal carcinoma cells. Int J Oncol. 2006;29:869-876
49. Angiolillo AL, Kanegane H, Sgadari C, Reaman GH, Tosato G. Interleukin-15 promotes angiogenesis in vivo. Biochem Biophys Res Commun. 1997;233:231-237
50. Sennino B, McDonald DM. Controlling escape from angiogenesis inhibitors. Nat Rev Cancer. 2012;12:699-709
51. Ferrara N. Pathways mediating VEGF-independent tumor angiogenesis. Cytokine Growth Factor Rev. 2010;21:21-26
52. Shojaei F, Wu X, Qu X, Kowanetz M, Yu L, Tan M. et al. G-CSF-initiated myeloid cell mobilization and angiogenesis mediate tumor refractoriness to anti-VEGF therapy in mouse models. Proc Natl Acad Sci U S A. 2009;106:6742-6747
53. Orimo A, Gupta PB, Sgroi DC, Arenzana-Seisdedos F, Delaunay T, Naeem R. et al. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell. 2005;121:335-348
Author contact
![]() Corresponding authors: Yixuan Hou and Manran Liu, Key Laboratory of Laboratory Medical Diagnostics, Chinese Ministry of Education, Chongqing Medical University, Chongqing 400016, China. #1 You-Yi Rd., Yu-zhong District, Chongqing 400016, Chongqing, China Tel: +86 23 68485938; Fax: +86 23 68485239; E-mail: Yixuan_Houcom.cn and mliu-hncqcom
Corresponding authors: Yixuan Hou and Manran Liu, Key Laboratory of Laboratory Medical Diagnostics, Chinese Ministry of Education, Chongqing Medical University, Chongqing 400016, China. #1 You-Yi Rd., Yu-zhong District, Chongqing 400016, Chongqing, China Tel: +86 23 68485938; Fax: +86 23 68485239; E-mail: Yixuan_Houcom.cn and mliu-hncqcom