Theranostics
13.3
Impact Factor
- Current Issue
- Advance Articles
- Volume 16; 2026
- Volume 15; 2025
- Volume 14; 2024
- Volume 13; 2023
- Volume 12; 2022
- Archive
- Cover Images
- Cover Suggestion
- Special Issues
Top
Introduction
Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References
Introduction
Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2017; 7(19):4753-4762. doi:10.7150/thno.21687 This issue Cite
Research Paper
Mutational Landscape of cfDNA Identifies Distinct Molecular Features Associated With Therapeutic Response to First-Line Platinum-Based Doublet Chemotherapy in Patients with Advanced NSCLC
Tao Jiang1*, Xuefei Li2*, Jianfei Wang3*, Chunxia Su1, Wenbo Han3, Chao Zhao2, Fengying Wu1, Guanghui Gao1, Wei Li1, Xiaoxia Chen1, Jiayu Li1, Fei Zhou1, Jing Zhao1, Weijing Cai1, Henghui Zhang3, Bo Du3, Jun Zhang4 ![]() , Shengxiang Ren1
, Shengxiang Ren1 ![]() , Caicun Zhou1
, Caicun Zhou1 ![]() , Hui Yu5, Fred R. Hirsch5
, Hui Yu5, Fred R. Hirsch5
1. Department of Medical Oncology, Shanghai Pulmonary Hospital, Thoracic Cancer Institute, Tongji University School of Medicine, Shanghai, P.R. China;
2. Department of Lung Cancer and Immunology, Shanghai Pulmonary Hospital, Tongji University School of Medicine, Shanghai, P.R. China;
3. Beijing Genecast Biotechnology Co., Beijing, P.R. China;
4. Division of Hematology, Oncology and Blood & Marrow Transplantation, Department of Internal Medicine, Holden Comprehensive Cancer Center, University of Iowa Carver College of Medicine, Iowa City, IA, USA;
5. Department of Medicine, Division of Medical Oncology, University of Colorado Cancer Center, Anschutz Medical Campus, Aurora, CO, USA.
* These authors make the same contributions to this paper.
Received 2017-6-29; Accepted 2017-9-13; Published 2017-10-17
Citation:
Jiang T, Li X, Wang J, Su C, Han W, Zhao C, Wu F, Gao G, Li W, Chen X, Li J, Zhou F, Zhao J, Cai W, Zhang H, Du B, Zhang J, Ren S, Zhou C, Yu H, Hirsch FR. Mutational Landscape of cfDNA Identifies Distinct Molecular Features Associated With Therapeutic Response to First-Line Platinum-Based Doublet Chemotherapy in Patients with Advanced NSCLC. Theranostics 2017; 7(19):4753-4762. doi:10.7150/thno.21687. https://www.thno.org/v07p4753.htm
Other stylesAbstract

Rationale To investigate whether the mutational landscape of circulating cell-free DNA (cfDNA) could predict and dynamically monitor the response to first-line platinum-based chemotherapy in patients with advanced non-small-cell lung cancer (NSCLC).
Methods Eligible patients were included and blood samples were collected from a phase III trial. Both cfDNA fragments and fragmented genomic DNA were extracted for enrichment in a 1.15M size panel covering exon regions of 1,086 genes. Molecular mutational burden (MMB) was calculated to investigate the relationship between molecular features of cfDNA and response to chemotherapy.
Results In total, 52 eligible cases were enrolled and their blood samples were prospectively collected at baseline, every cycle of chemotherapy and time of disease progression. At baseline, alterations of 17 genes were found. Patients with partial response (PR) had significantly lower baseline MMB of these genes than those patients with either stable disease (SD) (P = 0.0006) or progression disease (PD) (P = 0.0074). Further analysis revealed that the mutational landscape of cfDNA from pretreatment blood samples were distinctly different among patients with PR vs. SD/PD. For patients with baseline TP53 mutation, those with PR experienced a significant reduction in MMB whereas patients with SD or PD experienced an increase after two, three or four cycles of chemotherapy. Furthermore, patients with low MMB had superior response rate and significantly longer progression-free survival than those with high MMB.
Conclusion This study indicated that the mutational landscape of cfDNA has potential clinical value to predict the therapeutic response to first-line platinum-based doublet chemotherapy in NSCLC patients. At the single gene level, dynamic change of molecular mutational burden of TP53 is valuable to monitor efficacy (and, therefore, might aid in early recognition of resistance and relapse) in patients harboring this mutation at baseline.
Keywords: Non-small-cell lung cancer, circulating cell-free DNA, chemotherapy, molecular mutational burden
Introduction
Although molecular targeted therapy and checkpoint immunotherapy have shaped therapeutic strategies and significantly improved prognosis for subpopulations of patients with advanced non-small-cell lung cancer (NSCLC) [1-4], lung cancer still remains the leading cause of cancer-related death worldwide [5, 6]. More than 50% of NSCLC do not harbor targetable driver mutations, and acquired resistance is inevitable for those suitable for targeted therapy [7-9]. As for immunotherapy, only ~20% of unselected patients with advanced NSCLC respond to immune checkpoint inhibitors [10-12]. Therefore, the classical platinum-based doublet chemotherapy still remains the backbone for the majority of patients with advanced NSCLC [13]. Despite its effectiveness, platinum-based chemotherapy in NSCLC is unfortunately still used in a historical “one-size fits all” approach, which has been left behind in the era of precision medicine. Although much effort has been exerted to explore predictive biomarkers or molecular features to predict response to chemotherapy, to date, none of them had been successfully implemented in routine daily clinical practice [13].
Circulating cell-free DNA (cfDNA), released by tumor or normal cells, is a potential surrogate for the genomic information of the whole tumor [14]. Previously, we and other researchers have reviewed the clinical application of cfDNA in NSCLC, and proposed cfDNA as a promising predictive factor for various treatments including targeted therapy and chemotherapy [14-16]. Indeed, it was shown to be effective for real-time monitoring of tumor burden in response to therapy. For example, a proof-of-concept study indicated that circulating tumor DNA could provide the earliest assessment of chemotherapeutic response and prediction of impending relapse when compared with traditional circulating markers [17]. A recent study suggested that detection and dynamic change of epidermal growth factor receptor (EGFR) mutation from cfDNA could be a predictor of treatment effect and survival outcomes in patients with NSCLC treated with first-line intercalated erlotinib and chemotherapy [18]. Wu and colleagues also demonstrated that EGFR mutation detection in cfDNA was significantly associated with more advanced disease and poorer prognosis [19]. In spite of the recent progress, there is little data on molecular analysis to link specific genetic variations with chemotherapeutic response in NSCLC patients, and there is a paucity of studies utilizing cfDNA as an alternative to tumor tissue to investigate the predictive value of the mutational landscape in NSCLC patients treated with platinum-based chemotherapy.
With the aim to address the predictive significance of molecular features of cfDNA for first-line platinum-based doublet chemotherapy in patients with advanced NSCLC, we performed a biomarker exploratory analysis in patients who participated in a randomized phase III trial [20]. To further explore the relationship between molecular features of cfDNA and response to chemotherapy, we developed a new concept, molecular mutational burden (MMB, details are listed in the Methods section: Definition and Algorithm of MMB). We found that total MMB of frequently altered genes was significantly associated with chemotherapeutic response. There was a distinct copy number variations (CNVs) pattern for cfDNA from pretreatment blood samples among patients with partial response (PR) vs. stable disease (SD)/progression disease (PD). For patients who had baseline mutation of tumor protein p53 (TP53), its dynamic change in MMB revealed in the cfDNA could be used to monitor the objective response to chemotherapy in NSCLC patients. Moreover, we found that patients with low MMB had significantly longer progression-free survival (PFS) than those with high MMB.
Methods
Patient recruitment and sample collection
Eligible patients were consecutively enrolled from a randomized phase III trial. Details of the study design and patient eligibility criteria were previously described [20]. Briefly, eligible patients should meet the following criteria: pathologically confirmed and previously untreated stage IIIB and stage IV NSCLC without activated EGFR mutation, age ranging from 18 to 70 years old, Eastern Cooperative Oncology Group (ECOG) performance status of 0 or 1, no need for palliative radiotherapy, expected survival time ≥ 3 months and adequate organ function.
Enrolled patients were randomly assigned to receive mecapegfilgrastim (recombinant human granulocyte-colony stimulating factor) 100 mg/kg, a 6 mg fixed dose, or placebo (control arm) on day 3 in cycle 1. All patients were scheduled to receive up to 4 cycles of platinum-based chemotherapy (docetaxel 75 mg/m2 combined with cisplatin at a dose of 75 mg/m2, or carboplatin with an area under the curve of 5, on day 1, every 21 days). Blood samples were collected at the baseline, every cycle of chemotherapy and the time of disease progression. The protocol was approved by the institutional ethics committee of each participating medical center. All patients signed informed consent forms before the initiation of any study-related procedure.
DNA extraction and sequencing
Peripheral blood lymphocytes and plasma were separated by centrifugation at 1600 × g for 10 min. Supernatant plasma was then transferred to a new 2 mL centrifuge tube and centrifuged at 16,000 × g for 10 min. MagMAXTM Cell-Free DNA isolation kit (Life Technologies, California, USA) was used to extract cfDNA in the plasma according to its manufacturer's instruction. Tiangen whole blood DNA kit (Tiangen, Beijing, PRC) was used to extract DNA from peripheral blood lymphocytes according to the manufacturer's instructions. DNA concentration was measured using Qubit dsDNA HS Assay kit or Qubit dsDNA BR Assay kit (Life Technologies, California, USA).
Genomic DNA was sheared into 150-200 bp fragments with Covaris M220 Focused-ultrasonicatorTM Instrument (Covaris, Massachusetts, USA). Fragmented DNA and cfDNA libraries were constructed by KAPA HTP Library Preparation Kit (Illumina platforms) (KAPA Biosystems, Massachusetts, USA) following manufacturer's instruction. DNA libraries were captured with a designed Genescope panel of 1086 genes (Genecast, Beijing, China) that included major tumor related genes. The captured samples were subjected to Illumina HiSeq X-Ten for paired end sequencing.
Bioinformatics pipeline
Paired-end reads generated from Hiseq X-Ten platform were mapped to the hg19 reference genome with BWA 0.7.12 (default parameters), then sorted, filtered and indexed with SAM tools. In order to identify somatic SNP and indel mutations, the obtained BAM files from both blood cell and plasma samples for each patient were processed for pairwise variant calling using VarScan (v2.4.2) [21] using the following parameters: i) minimum coverage for calling somatic variants in blood cell samples is 8, and 6 for calling in plasma samples. P-value threshold to call a somatic site was 0.05; ii) variants with ≤ 90% strand bias were kept for further study. The generated candidate mutations were annotated use Annovar software tool [22], the dbNSFP and Exome Aggregation Consortum (ExAC) database were used to filter out either the benign mutations with pp2_hdiv score ≤ 0.452 or the population polymorphic sites. Finally, the resulted nonsynonymous mutations at the exonic regions were kept. The average variant allele frequency was calculated for each gene during the obtained somatic mutations and was treated as mutation burden for each patient.
We used all of the blood cell samples from patients to construct copy number baseline as negative control and used CNV kit to call copy number variation from the plasma samples for each patient. Target region sequencing would induce some poor coverage homogeneity between different enrichment regions due to biases related to the efficiency of target capture and library preparation, which is a limitation for precise copy number detection. We used a software package CNV kit that applies both the sequencing target region reads and nonspecifically captured off-target reads to help reduce that bias and, therefore, improved somatic CNV detection resolution. During the software working procedure, three main sources of bias that induce the extraneous variability of sequencing read depth, which included GC content, target footprint size and spacing, and repetitive sequences, were also evaluated and corrected.
Definition and Algorithm of MMB
For each patient, Mpileup files generated from Samtools were used as input for the mutation detection algorithm (VarScan2). VarScan2 algorithm performs pair-wise comparisons of base calls and normalizes sequence depth at each position for both tumor and normal samples simultaneously. During variant detection, the genotype for normal and tumor samples were determined independently based on adjustable minimum thresholds for coverage, base quality, strand bias, variant allele frequency and statistical significance. The value was computed by Fisher's exact test of the read counts supporting each allele (both reference and variant) compared to the expected distribution based on sequencing error alone. At each position where one or both samples has a variant, VarScan2 performs a direct comparison between tumor and normal genotypes and supporting read counts to identify the somatic mutations. The mutation frequency value for each somatic mutation was calculated as its variants supporting reads divided by reference supporting reads. Then a mean frequency was calculated among obtained mutations for each gene as its MMB.
Statistical analysis
Both Wilcoxon signed rank test and t test were applied for comparison of copy number variation and mutation frequency between defined patients' groups, and P < 0.05 was considered statistically significant. Circos-0.69-4 was used to generate circos plots for SNP and indel distributions. A python package python 3.6-seaborn was used for copy number clustering and heat-map presentation.
Results
Patient collection and baseline characteristics
In total, 52 eligible cases were enrolled and their blood samples were prospectively collected at baseline, every cycle of chemotherapy and time of disease progression. Baseline features of included patients are listed in Table 1. Briefly, 35 (67.31%) of them were male and 34 (65.4%) of them had ECOG PS=1. The vast majority of patients had histology-confirmed adenocarcinoma and squamous cell carcinoma (76.92%). Most of them received docetaxel plus cisplatin (67.31%). There were 11, 29 and 12 patients who experienced PR, SD or PD to first line chemotherapy respectively.
Detection of cfDNA at baseline
We collected 52 blood samples at baseline. Among them, 48 samples had measurable concentrations of cfDNA, but 4 of them (PR, n = 1; SD, n = 2; PD, n = 1) were excluded due to negligible levels. Although the median levels of cfDNA were numerically lower in patients with SD and PD than those in patients with PR, there was no significant difference of baseline cfDNA concentrations among patients with PR, SD and PD (P > 0.05) (Figure S1).
Relationship between MMB level and chemotherapeutic response
To assess the association between mutational landscape of cfDNA and treatment response, both cfDNA fragment and genomic DNA were subjected to enrichment for a 1.15M size panel covering exon regions from 1,086 genes (Table S1). We firstly focused on the known driver mutations and/or the genes that are known to correlate with the effect of chemotherapy. The results showed that alterations of 17 genes including ALK, BCL2, BRAF, CD74, CDKN2A, EML4, GSTP1, KIF5B, KRAS, MLH1, MTHFR, NRAS, RRM1, PIK3CA, SLC34A2, XPC and XRCC1 were frequently noticed in the 48 blood samples at baseline (Figure 1). Since no patients with EGFR mutation were enrolled in this cohort, EGFR mutation was not identified. By summing up the total MMB of these genes, we found that patients with PR had significantly lower values of MMB than those with SD (P = 0.0006) and PD (P = 0.0074). Although patients with SD appeared to have the lower value of MMB than patients with PD, there was no statistically significant difference (P = 0.1516) (Figure 1).
To further explore whether the level of MMB could stratify patients with distinct response and PFS, we selected the median level of MMB at baseline among all cases as the cutoff value to divide the patients into two groups (high vs. low MMB group). As shown in Table 1, there were no significant association between clinicopathological features (age, gender, PS, histology and regimen) and level of MMB. The results showed that the objective response rate (ORR) and disease control rate (DCR) in the low MMB group was higher than in the high MMB group (ORR: 33.3% vs. 8.3%, P = 0.076; DCR: 95.8% vs. 58.3%, P = 0.006) (Figure 2A). Moreover, patients with low MMB had markedly longer PFS than those with high MMB (median PFS: 7.0 months vs. 3.6 months, HR = 0.31, 95% CI 0.09-0.35, P < 0.0001) (Figure 2B).
Table 1
Baseline characteristics of included patients (n = 52).
| Total | % | Low MMB | % | High MMB | % | P value | ||
|---|---|---|---|---|---|---|---|---|
| Age | < 65 | 32 | 61.5% | 19 | 63.3% | 11 | 36.7% | 0.100 |
| ≥ 65 | 20 | 38.5% | 7 | 38.9% | 11 | 61.1% | ||
| Gender | Male | 35 | 67.3% | 15 | 46.9% | 17 | 53.1% | 0.838 |
| Female | 17 | 32.7% | 7 | 43.8% | 9 | 56.3% | ||
| ECOG PS | 0 | 18 | 34.6% | 9 | 52.9% | 8 | 47.1% | 0.464 |
| 1 | 34 | 65.4% | 13 | 41.9% | 18 | 58.1% | ||
| Histology | Adenocarcinoma | 26 | 50.0% | 13 | 56.5% | 10 | 43.5% | 0.252* |
| Squamous | 14 | 26.9% | 6 | 42.9% | 8 | 57.1% | ||
| Adenosquamous | 2 | 3.8% | 1 | 50.0% | 1 | 50.0% | ||
| Large cell carcinoma | 1 | 1.9% | 0 | 0.0% | 1 | 100.0% | ||
| NOS | 9 | 17.3% | 3 | 37.5% | 5 | 62.5% | ||
| Regimen | Docetaxel + cisplatin | 35 | 67.3% | 18 | 54.5% | 15 | 45.5% | 0.938 |
| Docetaxel + carboplatin | 17 | 32.7% | 8 | 53.3% | 7 | 46.7% | ||
| Mecapegfilgrastim | Yes | 33 | 63.5% | 20 | 76.9% | 18 | 81.8% | 0.953 |
| No | 19 | 36.5% | 6 | 23.1% | 4 | 18.2% | ||
| Response rate | CR | 0 | 0.0% | 0 | 0.0% | 0 | 0.0% | |
| PR | 11 | 21.2% | 8 | 80.0% | 2 | 20.0% | 0.249 | |
| SD | 29 | 55.8% | 15 | 55.6% | 12 | 44.4% | ||
| PD | 12 | 23.1% | 1 | 9.1% | 10 | 90.9% |
ECOG PS: Eastern Cooperative Oncology Group performance status; NOS: not otherwise specified; CR: complete response; PR: partial response; SD: stable disease; PD: progression disease; MMB, molecular mutational burden.
*, p value refers to adenocarcinoma vs. non-adenocarcinoma.
**, 52 blood samples were collected at baseline and 48 samples had measurable concentration (4 of them were excluded due to negligible level).
Figure 1
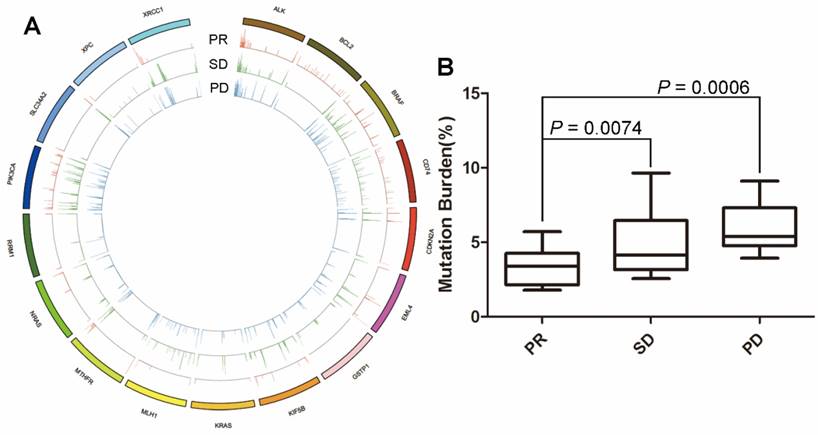
The association between molecular mutational burden and treatment response. (A) alterations of 17 genes including ALK, BCL2, BRAF, CD74, CDKN2A, EML4, GSTP1, KIF5B, KRAS, MLH1, MTHFR, NRAS, RRM1, PIK3CA, SLC34A2, XPC and XRCC1 were frequently observed in 48 blood samples at baseline; (B) patients with PR had significantly lower molecular mutational burden of these genes than patients with SD. Although patients with SD appeared to have lower molecular mutational burden than those with PD, there was no statistically significant difference.
Figure 2
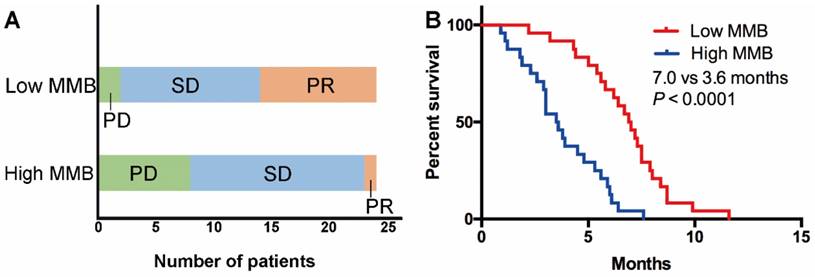
Low molecular mutational burden was associated with superior response rate and longer PFS. (A) The objective response rate and disease control rate in the low molecular mutational burden group was higher than in the high molecular mutational burden group; (B) patients with low molecular mutational burden had markedly longer PFS than those with high molecular mutational burden.
Among the 17 frequently found gene alterations at baseline, 8 of them (i.e., BCL2, CD74, GSTP1, MTHFR, MLH1, RRM1, XPC and XRCC1) were associated with chemotherapeutic effect in previous reports in NSCLC. Thus, we performed a further subgroup analysis about the relationship between these 8 genes' MMB level and chemotherapeutic response. We found that patients with PR had significantly lower values of MMB than those with PD (P = 0.0166), but not those with SD (P =0.1460). Although patients with SD appeared to have the lower value of MMB than patients with PD, there was no statistically significant difference (P = 0.2152) (Figure S4). As for PFS prediction, we found that the 8 genes' MMB showed a similar trend to the MMB of 17 genes. The median PFS was 5.6 months in patients with low MMB, which was numerically longer than the 4.8 months in patients with high MMB (P = 0.9193) (Figure S5).
Figure 3
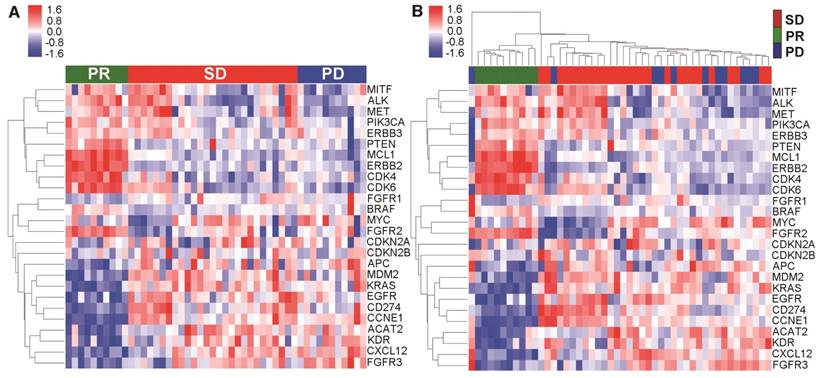
Copy number variation (CNV) # from cfDNA was associated with the effect of first-line chemotherapy. (A) CNV profiles from cfDNA was significantly different between patients with PR vs. SD/PD; (B) Clear segregation according to objective response was observed via hierarchical clustering of cfDNA. # To reduce the noise signal, the average CNV value of all patients with PR was compared to determine the relative CNV value of each patient.
Copy number variation clearly segregated patients with distinct therapeutic response
To further explore the relationship between mutational landscape of cfDNA and effect of first-line chemotherapy, we surveyed the CNVs across the genome for all the blood samples at baseline. We used the genomic DNA extracted from white blood cells and cfDNA in parallel to identify meaningful CNV patterns. Based on previously reported NSCLC-related CNVs in the literature and online database (TCGA), we further looked into each individual gene and identified 26 genes whose CNV pattern could clearly segregate the CNV pattern of patients with PR from those with either SD or PD (Figure 3A). Interestingly, when we performed hierarchical clustering based on these genes, only in cfDNA (Figure 3B) but not genomic DNA (Figure S2), did we see clear segregation associated with treatment response to the first-line platinum-based chemotherapy. These results suggested that CNV profiles of cfDNA might have a clinically predictive value.
Changes of TP53 MMB dynamically monitored the efficacy of chemotherapy
Several studies demonstrated that cfDNA dynamics could be associated with treatment response. However, no study has ever investigated the predictive value of such dynamic change of tumor-specific gene mutational burden in cfDNA in NSCLC patients treated with chemotherapy. Here, we chose TP53, one of the most common altered genes in various types of cancer, to explore whether dynamic changes in TP53 MMB (defined as the sum of alterations in TP53 including SNV and indels) in cfDNA could predict the objective response in NSCLC patients treated with platinum-based first-line chemotherapy. 28 cases (58.3%) had TP53 mutations at baseline. There was no significant difference of baseline MMB of TP53 among patients with PR, SD and PD (P > 0.05) (Figure S3). In 11 patients with PR, 6 patients had subsequent cfDNA specimens available to compare with their baseline TP53 mutation burden. When we grouped samples drawn after the same chemotherapy cycle together, we found that there was a significant reduction in MMB of TP53 after two (patients P11 and P12, TP53: 1.5→0.9), three (patient P6, TP53: 0.6→0.4; patients P8 and P9, TP53: 1.6→1.2→0.7) or four (patient P1, TP53: 2.6→1.9→0.8→0.3) cycles of chemotherapy (Figure 4). In 27 patients with SD, 7 of them had cfDNA available for comparison, which showed a slight increase in MMB of TP53 after two (patients P13, P14, P15, P16 and P29, TP53: 1.8→2.1) or three (patients P21 and P23, TP53: 1.1→1.6→1.9) cycles of chemotherapy (Figure 4). For patients with PD, 5 patients were available for comparison. Interestingly, they all experienced a remarkable increase in TP53 MMB after either two (patients P10, P28 and P30, TP53: 1.7→2.7) or four (patients P18 and P31, TP53: 1.5→8.3) cycles of chemotherapy (Figure 4). These results indicated that dynamic changes in TP53 MMB in cfDNA could well monitor the response to first-line platinum-based chemotherapy in patients with advanced NSCLC.
Figure 4
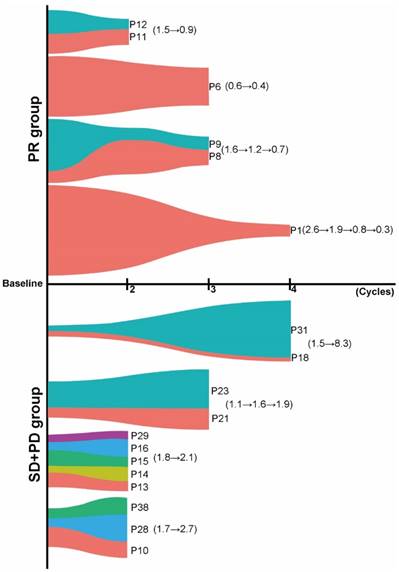
Dynamic changes in TP53 mutational burden in cfDNA could predict the objective response. Patients with PR experienced a significant reduction in TP53 mutational burden, while patients with SD or PD experienced increased TP53 mutational burden after several cycles of chemotherapy (different colors stand for different patients).
Discussion
To our best knowledge, the present study is the first one to reveal that the mutational landscape of cfDNA is significantly associated with the effect of first-line platinum-based doublet chemotherapy in patients with advanced NSCLC. We found that patients with low MMB had superior response rates and significantly longer PFS than those with high MMB. We also observed that patients with distinct therapeutic response presented with quite different mutational features of cfDNA from pretreatment blood samples. Furthermore, in patients with baseline TP53 mutation, we observed a significant reduction in TP53 MMB in patients with PR but increased burden in patients with SD and PD after two, three or four cycles of chemotherapy, indicating that changes in TP53 MMB might dynamically monitor the therapeutic response.
Although the biology of cfDNA remains debatable, the clinical application of cfDNA has been extensively studied [23]. Several proof-of-principle studies have demonstrated the promising role of cfDNA for cancer diagnosis, prognosis, evaluation of recurrence risk, selection of optimal treatment, and monitoring of tumor burden and therapeutic response [14, 16, 17, 24-29]. Using cfDNA as a predictor for therapeutic response in NSCLC has been investigated in many previous studies [30-33]. However, most focused on cfDNA levels and the results are inconsistent. In 2004, Gautschi et al. reported that tumor progression was significantly associated with increasing cfDNA level and patients who responded to chemotherapy had lower serum cfDNA concentration [30]. In line with this result, another two studies also found that cfDNA levels could predict chemotherapeutic response in patients with advanced NSCLC [31, 32]. However, a recent large prospective study evaluated the predictive value of total plasma cfDNA concentration for systemic therapy response in NSCLC, and found that the baseline cfDNA level did not correlated with PFS (HR = 1.06, P = 0.41) and OS (HR = 1.04, P = 0.51), and dynamic changes in total plasma cfDNA did not correlate with radiologic response[33], which suggested that total cfDNA concentration is not a suitable predictive biomarker for outcome of systemic therapy and recommended that future investigations in cfDNA should focus on other biomarkers such as tumor-specific genomic aberrations of cfDNA via next-generation sequencing. Consistent with their finding, this current prospective study reiterated that the median baseline cfDNA level was similar between patients with PR vs. SD/PD.
To further investigate the relationship between the mutational landscape of cfDNA and chemotherapeutic response, we developed a new concept in this study, named MMB. MMB was distinguished from tumor mutation burden. Tumor mutation burden measures the overall number of somatic protein coding mutations per area of sequence in the tumor specimen [34] and is a discrete variable. The exploratory analysis of CheckMate 026 showed that, in the chemotherapy group, patients with low/medium tumor mutation burden had similar ORR as patients with high tumor mutation burden (33% vs. 28%; P = 0.544) [35]. In contrast, MMB was defined as the sum of mean mutation frequency value for each somatic mutation and is a continuous variable. Thus, its continuous and dynamic change could be applied to real-time and dynamic monitoring of treatment response. In the current study, alterations of 17 genes were frequently found at baseline. Nine of them were known drivers in carcinogenesis (ALK, BRAF, CDKN2A, EML4, KIF5B, KRAS, NRAS, PIK3CA, and SLC34A2) and 8 of them were associated with chemotherapeutic effect in previous reports (BCL2, CD74, GSTP1, MTHFR, MLH1, RRM1, XPC and XRCC1) in NSCLC. Although the association of each individual gene with chemotherapeutic effect remains controversial, a robust correlation between the total MMB of these genes in cfDNA and chemotherapeutic response was observed. Moreover, patients in the low MMB group also had significantly longer PFS than those with high MMB. This result indicated that the total MMB of this 17-genes panel could be valuable to predict chemotherapeutic response and PFS of first-line platinum-based doublet chemotherapy in patients with advanced NSCLC.
CNV is a type of genetic alteration characterized by chromosomal amplification and deletion that results in considerable change in the genome [36]. It is considered one of the major types of genome aberrations that contribute to tumor initiation, maintenance and progression. Previous studies have demonstrated that CNV pattern in cfDNA could mirror the primary tumors of prostate and breast cancer [37, 38], suggesting CNV in cfDNA might act as a surrogate of primary tumor. Clinical application of cfDNA CNV has been extensively explored and most of the studies focused on the relationship between CNV of specific genes with clinicopathological features or prognosis in NSCLC [39, 40]. Recently, Louise and colleagues examined the CNV in circulating tumor cells (CTC) from pretreatment blood samples of small cell lung cancer (SCLC) patients and reported that CNV-based classification could distinguish chemosensitive from chemorefractory cases with an accuracy of 83.3% [41]. In line with this result, we found that cfDNA CNV pattern was significantly different between patients of PR with SD or PD to chemotherapy, and clear segregation of response matching CNV pattern was observed by using hierarchical clustering. Taken together, these results indicated that CNV might also potentially be used as a first step toward identification of a proper biomarker that may predict response to chemotherapy in NSCLC patients.
The tumor-specific genomic aberration from cfDNA also showed promising results for real-time monitoring of the response to systemic therapy. Mok T et al. performed a prospective analysis of EGFR mutation detected from cfDNA in the FASTACT-2 trial and found that patients with negative detection of EGFR mutation at cycle 3 predicted longer PFS and OS, indicating that dynamic change of blood-based EGFR status could be applied to predict long-term benefit of EGFR tyrosine kinase inhibitor (TKI) [18]. Zhou et al. further found there were 2 different dynamic changes of EGFR L858R mutation at the time of disease progression, which have different prognosis after EGFR TKI treatment [42]. However, to date, no biomarkers have been developed to predict or monitor the efficacy of chemotherapy in NSCLC. Previous studies reported that TP53 specific mutation status was associated with cisplatin sensitivity [43]. Therefore, we further investigated the dynamic changes of TP53 mutational burden in this study, and we observed that patients with PR experienced a significant reduction in TP53 mutational burden, whereas contrary results were observed for patients with SD or PD after several cycles of chemotherapy. These findings suggested that the dynamic changes of TP53 mutational burden might have monitoring value for the efficacy of first-line platinum-based doublet chemotherapy in patients with advanced NSCLC, and further large-scale investigation is warranted to validate these results.
In conclusion, we found that the mutational landscape of cfDNA is associated with therapeutic response to first-line platinum-based doublet chemotherapy in patients with advanced NSCLC. Both the MMB and CNVs have value in predicting therapeutic effects, and tumor-specific TP53 mutation burden has value in dynamically monitoring therapeutic efficacy and is worth further investigation. Collectively, our study shed a light on the use of cfDNA to predict and monitor the efficacy of first line chemotherapy in patients with advanced NSCLC; therefore, future valuable biomarkers should be developed using this non-invasive approach.
Abbreviations
cfDNA: cell-free DNA; CNVs: copy number variations; CTC: circulating tumor cells; DCR: disease control rate; ECOG: Eastern Cooperative Oncology Group; EGFR: epidermal growth factor receptor; MMB: molecular mutational burden; NSCLC: non-small-cell lung cancer; ORR: objective response rate; PD: progression disease; PR: partial response; SCLC: small cell lung cancer; SD: stable disease; TKI: tyrosine kinase inhibitor; TP53: tumor protein p53.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
This study was sponsored by the Beijing Genecast Biotechnology Co., Beijing, China, and supported in part by grants from the National Natural Science Foundation of China (No. 81672286, 81401890 and 81772467), an IASLC Fellowship award (S. Ren), “Shuguang Program” supported by Shanghai Education Development Foundation and Shanghai Municipal Education Commission (No. 16SG18), the Chronic Diseases Program of Shanghai Shen Kang Pharmaceutical Development Co. Ltd (No. SHDC 12015314), Shanghai Committee of Science and Technology, China (No. 134119b1001) and the Outstanding Young Doctor Program of the Shanghai Municipal Commission of Health and Family Planning (No. XYQ2013097).
Authors' contributions
Tao Jiang, Xuefei Li, Chunxia Su, Shengxaing Ren, Bo Du, Jun Zhang and Caicun Zhou designed this study. Tao Jiang, Xuefei Li, Chao Zhao, Fengying Wu, Guanghui Gao, Wei Li, Jie Zhang, Xiaoxia Chen, Weijing Cai, Fei Zhou, Jing Zhao and Anwen Xiong collected the blood samples and clinical data. Tao Jiang, Wenbo Han, Jianfei Wang, Henghui Zhang, Jun Zhang and Bo Du performed the statistical analyses. Tao Jiang, Chunxia Su, Shengxaing Ren, and Caicun Zhou drafted the manuscript. Caicun Zhou, Jun Zhang, Hui Yu and Fred R. Hirsch provided critical comments, suggestions and revised the manuscript. All authors read and approved the final version of the manuscript.
Competing Interests
Wenbo Han, Jianfei Wang, Henghui Zhang and Bo Du are employees of Beijing Genecast Biotechnology Co., Beijing, China. The other authors declare no potential conflict of interest.
References
1. Zhou C, Wu YL, Chen G, Feng J, Liu XQ, Wang C. et al. Erlotinib versus chemotherapy as first-line treatment for patients with advanced EGFR mutation-positive non-small-cell lung cancer (OPTIMAL, CTONG-0802): a multicentre, open-label, randomised, phase 3 study. Lancet Oncol. 2011;12:735-42
2. Reck M, Rodriguez-Abreu D, Robinson AG, Hui R, Csoszi T, Fulop A. et al. Pembrolizumab versus Chemotherapy for PD-L1-Positive Non-Small-Cell Lung Cancer. N Engl J Med. 2016;375:1823-1833
3. Solomon BJ, Mok T, Kim DW, Wu YL, Nakagawa K, Mekhail T. et al. First-line crizotinib versus chemotherapy in ALK-positive lung cancer. N Engl J Med. 2014;371:2167-77
4. Tan WL, Jain A, Takano A, Newell EW, Iyer NG, Lim WT. et al. Novel therapeutic targets on the horizon for lung cancer. Lancet Oncol. 2016;17:e347-62
5. Chen W, Zheng R, Baade PD, Zhang S, Zeng H, Bray F. et al. Cancer statistics in China, 2015. CA Cancer J Clin. 2016
6. Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65:87-108
7. Hirsch FR, Scagliotti GV, Mulshine JL, Kwon R, Curran WJ Jr, Wu YL. et al. Lung cancer: current therapies and new targeted treatments. Lancet. 2017;389:299-311
8. Hirsch FR, Suda K, Wiens J, Bunn PA Jr. New and emerging targeted treatments in advanced non-small-cell lung cancer. Lancet. 2016;388:1012-24
9. Tan CS, Gilligan D, Pacey S. Treatment approaches for EGFR-inhibitor-resistant patients with non-small-cell lung cancer. Lancet Oncol. 2015;16:e447-59
10. Chen DS, Mellman I. Elements of cancer immunity and the cancer-immune set point. Nature. 2017;541:321-330
11. Gibney GT, Weiner LM, Atkins MB. Predictive biomarkers for checkpoint inhibitor-based immunotherapy. Lancet Oncol. 2016;17:e542-e551
12. Topalian SL, Taube JM, Anders RA, Pardoll DM. Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat Rev Cancer. 2016;16:275-87
13. Olaussen KA, Postel-Vinay S. Predictors of chemotherapy efficacy in non-small-cell lung cancer: a challenging landscape. Ann Oncol. 2016;27:2004-2016
14. Wan JC, Massie C, Garcia-Corbacho J, Mouliere F, Brenton JD, Caldas C. et al. Liquid biopsies come of age: towards implementation of circulating tumour DNA. Nat Rev Cancer. 2017
15. Jiang T, Ren S, Zhou C. Role of circulating-tumor DNA analysis in non-small cell lung cancer. Lung Cancer. 2015;90:128-34
16. Siravegna G, Marsoni S, Siena S, Bardelli A. Integrating liquid biopsies into the management of cancer. Nat Rev Clin Oncol. 2017
17. Dawson SJ, Tsui DW, Murtaza M, Biggs H, Rueda OM, Chin SF. et al. Analysis of circulating tumor DNA to monitor metastatic breast cancer. N Engl J Med. 2013;368:1199-209
18. Mok T, Wu YL, Lee JS, Yu CJ, Sriuranpong V, Sandoval-Tan J. et al. Detection and Dynamic Changes of EGFR Mutations from Circulating Tumor DNA as a Predictor of Survival Outcomes in NSCLC Patients Treated with First-line Intercalated Erlotinib and Chemotherapy. Clin Cancer Res. 2015;21:3196-203
19. Wu YL, Sequist LV, Hu CP, Feng J, Lu S, Huang Y. et al. EGFR mutation detection in circulating cell-free DNA of lung adenocarcinoma patients: analysis of LUX-Lung 3 and 6. Br J Cancer. 2017;116:175-185
20. Zhou C, Huang Y, Wang D, An C, Zhou F, Li Y. et al. A Randomized Multicenter Phase III Study of Single Administration of Mecapegfilgrastim (HHPG-19K), a Pegfilgrastim Biosimilar, for Prophylaxis of Chemotherapy-Induced Neutropenia in Patients With Advanced Non-Small-Cell Lung Cancer (NSCLC). Clin Lung Cancer. 2016;17:119-27
21. Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L. et al. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012;22:568-76
22. Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164
23. Bennett CW, Berchem G, Kim YJ, El-Khoury V. Cell-free DNA and next-generation sequencing in the service of personalized medicine for lung cancer. Oncotarget. 2016;7:71013-71035
24. Jiang T, Zhai C, Su C, Ren S, Zhou C. The diagnostic value of circulating cell free DNA quantification in non-small cell lung cancer: A systematic review with meta-analysis. Lung Cancer. 2016;100:63-70
25. Diehl F, Schmidt K, Choti MA, Romans K, Goodman S, Li M. et al. Circulating mutant DNA to assess tumor dynamics. Nat Med. 2008;14:985-90
26. Newman AM, Bratman SV, To J, Wynne JF, Eclov NC, Modlin LA. et al. An ultrasensitive method for quantitating circulating tumor DNA with broad patient coverage. Nat Med. 2014;20:548-54
27. Tie J, Wang Y, Tomasetti C, Li L, Springer S, Kinde I. et al. Circulating tumor DNA analysis detects minimal residual disease and predicts recurrence in patients with stage II colon cancer. Sci Transl Med. 2016;8:346ra92
28. Garcia-Murillas I, Schiavon G, Weigelt B, Ng C, Hrebien S, Cutts RJ. et al. Mutation tracking in circulating tumor DNA predicts relapse in early breast cancer. Sci Transl Med. 2015;7:302ra133
29. Reinert T, Scholer LV, Thomsen R, Tobiasen H, Vang S, Nordentoft I. et al. Analysis of circulating tumour DNA to monitor disease burden following colorectal cancer surgery. Gut. 2016;65:625-34
30. Gautschi O, Bigosch C, Huegli B, Jermann M, Marx A, Chasse E. et al. Circulating deoxyribonucleic Acid as prognostic marker in non-small-cell lung cancer patients undergoing chemotherapy. J Clin Oncol. 2004;22:4157-64
31. Kumar S, Guleria R, Singh V, Bharti AC, Mohan A, Das BC. Plasma DNA level in predicting therapeutic efficacy in advanced nonsmall cell lung cancer. Eur Respir J. 2010;36:885-92
32. Pan S, Xia W, Ding Q, Shu Y, Xu T, Geng Y. et al. Can plasma DNA monitoring be employed in personalized chemotherapy for patients with advanced lung cancer? Biomed Pharmacother. 2012;66:131-7
33. Li BT, Drilon A, Johnson ML, Hsu M, Sima CS, McGinn C. et al. A prospective study of total plasma cell-free DNA as a predictive biomarker for response to systemic therapy in patients with advanced non-small-cell lung cancers. Ann Oncol. 2016;27:154-9
34. Schrock AB, Li SD, Frampton GM, Suh J, Braun E, Mehra R. et al. Pulmonary sarcomatoid carcinomas commonly harbor either potentially targetable genomic alterations or high tumor mutational burden as observed by comprehensive genomic profiling. J Thorac Oncol. 2017
35. Carbone DP, Reck M, Paz-Ares L, Creelan B, Horn L, Steins M. et al. First-Line Nivolumab in Stage IV or Recurrent Non-Small-Cell Lung Cancer. N Engl J Med. 2017;376:2415-2426
36. Sharp AJ, Locke DP, McGrath SD, Cheng Z, Bailey JA, Vallente RU. et al. Segmental duplications and copy-number variation in the human genome. Am J Hum Genet. 2005;77:78-88
37. Heitzer E, Ulz P, Belic J, Gutschi S, Quehenberger F, Fischereder K. et al. Tumor-associated copy number changes in the circulation of patients with prostate cancer identified through whole-genome sequencing. Genome Med. 2013;5:30
38. Shaw JA, Page K, Blighe K, Hava N, Guttery D, Ward B. et al. Genomic analysis of circulating cell-free DNA infers breast cancer dormancy. Genome Res. 2012;22:220-31
39. Seo AN, Yang JM, Kim H, Jheon S, Kim K, Lee CT. et al. Clinicopathologic and prognostic significance of c-MYC copy number gain in lung adenocarcinomas. Br J Cancer. 2014;110:2688-99
40. Choi S, Kim HR, Sung CO, Kim J, Kim S, Ahn SM. et al. Genomic Alterations in the RB Pathway Indicate Prognostic Outcomes of Early-Stage Lung Adenocarcinoma. Clin Cancer Res. 2015;21:2613-23
41. Carter L, Rothwell DG, Mesquita B, Smowton C, Leong HS, Fernandez-Gutierrez F. et al. Molecular analysis of circulating tumor cells identifies distinct copy-number profiles in patients with chemosensitive and chemorefractory small-cell lung cancer. Nat Med. 2017;23:114-119
42. Zhou Q, Yang JJ, Chen ZH, Zhang XC, Yan HH, Xu CR. et al. Serial cfDNA assessment of response and resistance to EGFR-TKI for patients with EGFR-L858R mutant lung cancer from a prospective clinical trial. J Hematol Oncol. 2016;9:86
43. Karekla E, Liao WJ, Sharp B, Pugh J, Reid H, Le Quesne JP. et al. Ex vivo explant cultures of non-small cell lung carcinoma enable evaluation of primary tumor responses to anticancer therapy. Cancer Res. 2017;77(8):2029-2039
Author contact
![]() Corresponding authors: Prof C. Zhou, Department of Medical Oncology, Shanghai Pulmonary Hospital & Thoracic Cancer Institute, Tongji University School of Medicine, No. 507, Zheng Min Road, Shanghai, 200433, P.R. China. Tel: +86-21-65115006; Fax +86-21-65111298; E-mail: tonyjiangdredu,cn; Prof S. Ren, Department of Medical Oncology, Shanghai Pulmonary Hospital & Thoracic Cancer Institute, Tongji University School of Medicine, No. 507, Zheng Min Road, Shanghai, 200433, P.R. China. Tel: +86-21-65115006; Fax +86-21-65111298; E-mail: harry_rencom; Prof J.Zhang, Division of Hematology, Oncology and Blood & Marrow Transplantation, Department of Internal Medicine, Holden Comprehensive Cancer Center, University of Iowa Carver College of Medicine, Iowa City, IA, USA. Tel: 01004152444731, E-mail: jun-zhang-1edu
Corresponding authors: Prof C. Zhou, Department of Medical Oncology, Shanghai Pulmonary Hospital & Thoracic Cancer Institute, Tongji University School of Medicine, No. 507, Zheng Min Road, Shanghai, 200433, P.R. China. Tel: +86-21-65115006; Fax +86-21-65111298; E-mail: tonyjiangdredu,cn; Prof S. Ren, Department of Medical Oncology, Shanghai Pulmonary Hospital & Thoracic Cancer Institute, Tongji University School of Medicine, No. 507, Zheng Min Road, Shanghai, 200433, P.R. China. Tel: +86-21-65115006; Fax +86-21-65111298; E-mail: harry_rencom; Prof J.Zhang, Division of Hematology, Oncology and Blood & Marrow Transplantation, Department of Internal Medicine, Holden Comprehensive Cancer Center, University of Iowa Carver College of Medicine, Iowa City, IA, USA. Tel: 01004152444731, E-mail: jun-zhang-1edu
Citation styles
APA
Jiang, T., Li, X., Wang, J., Su, C., Han, W., Zhao, C., Wu, F., Gao, G., Li, W., Chen, X., Li, J., Zhou, F., Zhao, J., Cai, W., Zhang, H., Du, B., Zhang, J., Ren, S., Zhou, C., Yu, H., Hirsch, F.R. (2017). Mutational Landscape of cfDNA Identifies Distinct Molecular Features Associated With Therapeutic Response to First-Line Platinum-Based Doublet Chemotherapy in Patients with Advanced NSCLC. Theranostics, 7(19), 4753-4762. https://doi.org/10.7150/thno.21687.
ACS
Jiang, T.; Li, X.; Wang, J.; Su, C.; Han, W.; Zhao, C.; Wu, F.; Gao, G.; Li, W.; Chen, X.; Li, J.; Zhou, F.; Zhao, J.; Cai, W.; Zhang, H.; Du, B.; Zhang, J.; Ren, S.; Zhou, C.; Yu, H.; Hirsch, F.R. Mutational Landscape of cfDNA Identifies Distinct Molecular Features Associated With Therapeutic Response to First-Line Platinum-Based Doublet Chemotherapy in Patients with Advanced NSCLC. Theranostics 2017, 7 (19), 4753-4762. DOI: 10.7150/thno.21687.
NLM
Jiang T, Li X, Wang J, Su C, Han W, Zhao C, Wu F, Gao G, Li W, Chen X, Li J, Zhou F, Zhao J, Cai W, Zhang H, Du B, Zhang J, Ren S, Zhou C, Yu H, Hirsch FR. Mutational Landscape of cfDNA Identifies Distinct Molecular Features Associated With Therapeutic Response to First-Line Platinum-Based Doublet Chemotherapy in Patients with Advanced NSCLC. Theranostics 2017; 7(19):4753-4762. doi:10.7150/thno.21687. https://www.thno.org/v07p4753.htm
CSE
Jiang T, Li X, Wang J, Su C, Han W, Zhao C, Wu F, Gao G, Li W, Chen X, Li J, Zhou F, Zhao J, Cai W, Zhang H, Du B, Zhang J, Ren S, Zhou C, Yu H, Hirsch FR. 2017. Mutational Landscape of cfDNA Identifies Distinct Molecular Features Associated With Therapeutic Response to First-Line Platinum-Based Doublet Chemotherapy in Patients with Advanced NSCLC. Theranostics. 7(19):4753-4762.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.