Impact Factor
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2017; 7(19):4805-4824. doi:10.7150/thno.20771 This issue Cite
Research Paper
Nanoparticle Delivery of miR-34a Eradicates Long-term-cultured Breast Cancer Stem Cells via Targeting C22ORF28 Directly
Xiaoti Lin1, 2*, Weiyu Chen3*, Fengqin Wei2, 4*, Binhua P. Zhou1, 5, Mien-Chie Hung6, Xiaoming Xie1 ![]()
1. Department of Breast Oncology, State Key Laboratory of Oncology in South China, Sun Yat-sen University Cancer Center, Collaborative Innovation Center for Cancer Medicine, Guangzhou 510060, China;
2. Department of Oncology, The Affiliated Xiang'an Hospital of Xiamen University, Medical College of Xiamen University, Xiamen 361003, China;
3. Department of Physiology, Zhongshan Medical School, Sun Yat-sen University, Guangzhou 510060, China;
4. Department of Emergency, Fujian Provincial 2 nd People's Hospital, Affiliated Hospital of Fujian University of Traditional Chinese Medicine, Fuzhou 350000, China;
5. Department of Molecular and Cellular Biochemistry, Markey Cancer Center, College of Medicine, University of Kentucky, Lexington, Kentucky 40506, USA;
6. Department of Molecular and Cellular Oncology, The University of Texas MD Anderson Cancer Center, Houston, Texas 77030, USA.
* These authors contributed equally to this work.
Received 2017-4-27; Accepted 2017-8-16; Published 2017-10-17
Abstract

Rationale: Cancer stem cells (CSCs) have been implicated as the seeds of therapeutic resistance and metastasis, due to their unique abilities of self-renew, wide differentiation potentials and resistance to most conventional therapies. It is a proactive strategy for cancer therapy to eradicate CSCs. Methods: Tumor tissue-derived breast CSCs (BCSC), including XM322 and XM607, were isolated by fluorescence-activated cell sorting (FACS); while cell line-derived BCSC, including MDA-MB-231.SC and MCF-7.SC, were purified by magnetic-activated cell sorting (MACS). Analyses of microRNA and mRNA expression array profiles were performed in multiple breast cell lines. The mentioned nanoparticles were constructed following the standard molecular cloning protocol. Tissue microarray analysis has been used to study 217 cases of clinical breast cancer specimens. Results: Here, we have successfully established four long-term maintenance BCSC that retain their tumor-initiating biological properties. Our analyses of microarray and qRT-PCR explored that miR-34a is the most pronounced microRNA for investigation of BCSC. We establish hTERT promoter-driven VISA delivery of miR-34a (TV-miR-34a) plasmid that can induce high throughput of miR-34a expression in BCSC. TV-miR-34a significantly inhibited the tumor-initiating properties of long-term-cultured BCSC in vitro and reduced the proliferation of BCSC in vivo by an efficient and safe way. TV-miR-34a synergizes with docetaxel, a standard therapy for invasive breast cancer, to act as a BCSC inhibitor. Further mechanistic investigation indicates that TV-miR-34a directly prevents C22ORF28 accumulation, which abrogates clonogenicity and tumor growth and correlates with low miR-34 and high C22ORF28 levels in breast cancer patients. Conclusion: Taken together, we generated four long-term maintenance BCSC derived from either clinical specimens or cell lines, which would be greatly beneficial to the research progress in breast cancer patients. We further developed the non-viral TV-miR-34a plasmid, which has a great potential to be applied as a clinical application for breast cancer therapy.
Keywords: cancer stem cells, breast cancer, targeted drug delivery, miR-34a, C22ORF28, therapeutic target.
Introduction
Cancer stem cells (CSCs), also known as tumor-initiating cells, have been implicated as the seeds of metastasis and sources of therapy resistance, due to their unique abilities of self-renew, wide differentiation potentials and resistance to most conventional therapies. It is an attractive therapy strategy for treating cancer to eradicate CSCs [1, 2]. However, long-term-cultured CSC lines are rarely generated in most investigations, and the reported treatment effects and mechanisms within CSCs have been intensively questioned. It is difficult to make a distinction between therapeutic response and self-differentiation of CSCs. With a goal of unlocking this mystery, we isolated, identified and maintained tumor tissue-derived breast CSCs (BCSC) in our recent reported investigation [3]. In sum, prolonged maintenance of CSCs could be a great benefit for enabling research progress in patients with tumors.
The tumor-initiating properties of CSCs distinguish them from the remainder of cancer cells, and play a key role in therapeutic resistance, evasion of cell death, as well as dormancy [4]. Therefore, it remains an effective approach to eradicate CSCs through regulating determinants of tumor-initiating properties. On the other hand, it is well known that microRNAs, a class of small non-coding RNAs, can post-transcriptionally mediate gene expression of neoplasm. Multiple researches indicated that microRNAs could efficiently induce pluripotent stem cells to generate cell lineage without stemness characteristics, mediate self-renewal of stem cells and even determine their cell fate [5-7]. Consequently, microRNA interference might potentially be implicated in the tumor-initiating properties and cell fate of long-term-cultured CSCs.
Since conventional therapy is not effective against CSCs, novel therapeutic strategies applying nanoparticle systems to deliver target-specific molecules have become promising approaches for potentially eliminating them [8-10]. Nanomedicine, as one of the fastest growing applications of nanotechnology, presents several outstanding advantages over conventional chemotherapeutics: enhanced permeability and retention effect, better surface modification for active tumor targeting, tunability of drug release, as well as nano-carriers with various drug molecules and imaging agents [11]. Although both viral vectors and non-viral vectors are used for systemic delivery in clinical trials, non-viral vectors hold the superior advantages of large-scale production, high level of transduction efficiencies, low immunogenicity and well-tolerated toxicity in comparison with viral vectors [8]. Currently, growing evidences have shown that lipid nanoparticle technology would be a dominant non-viral technology to enable the enormous potential of gene therapy [12].
In our previous researches, we developed VP16-GAL4-WPRE integrated systemic amplifier (VISA) as a non-viral targeting vector [13]. This versatile non-viral VISA vector was successfully shown to selectively amplify the transcriptional activity of promoters, with virtually no toxicity in pancreatic tumors [13], lung cancer [14], ovarian cancer [15], hepatocellular carcinoma [16, 17], prostate cancer [18] and breast cancer [19-22]. Furthermore, we established the hTERT-promoter-based VISA vector that drives transgene expression in treating breast cancer [18, 20, 21]. Our group also found that VISA-enabled systemic delivery of BikDD reduces the population of short-term-isolated breast cancer initiating cells [19, 23]. Collectively, our above-mentioned investigations urge us to explore the therapeutic response of long-term-cultured BCSC to hTERT-promoter-based VISA vector.
In the present study, we tried to exploit a feasible treatment for long-term-cultured BCSC based on our previous investigations. Preliminarily, we explored the most prominent microRNA for investigation in tumor tissue-derived or cell line-derived long-term-cultured BCSC. We further sought to determine the therapeutic effect of hTERT-promoter-based VISA delivery of the specific microRNA on prolonged maintained BCSC. In addition, we provided mechanistic insight into this therapeutic strategy and the tolerability of this non-viral liposomal nanoparticle.
Methods
Patients and animal study
Informed consent was obtained from all patients. Studies involved in animal experiments and human breast cancer tumor collection were approved by the research ethics committee of Sun Yat-sen University Cancer Center (SYSUCC).
Isolation and passage of long-term-cultured BCSC
Tumor tissue-derived BCSC (XM322 and XM607) were isolated by fluorescence-activated cell sorting (FACS) as previously described [3]. Cell line-derived BCSC (MDA-MB-231.SC and MCF-7.SC) were purified by magnetic-activated cell sorting (MACS). The application of culture medium was as described previously [3]. Further details are available in Supplementary methods.
Patients, tissues, tissue microarray construction (TMA) and immunohistochemistry (IHC)
A total of 134 female patients who were hospitalized in SYSUCC from 2001 to 2006 were enrolled in our study. Four fresh tissue samples of breast tumors collected during eradicative operations performed at Sun Yat-Sen University Cancer Center in July 2015 were randomly chosen for the Western blot study. See Supplementary methods for details on data processing.
Constructs and transfection
Detailed information of the VISA plasmid, hTERT promoter-driven VISA nanoparticle delivery of miR-34a (TV-miR-34a) and hTERT promoter-driven VISA nanoparticle delivery of control (TV-miR-Ctrl) were described previously [13, 22]. In brief, the miR-34a shRNA was incorporated into the Bgl II/Nhe I sites of the plasmid pGL3-T-VISA-Luc; following, the T-VISA-miR-34a fragment of pGL3-T-VISA-miR-34a was subcloned into the Not I and Sal I sites of pUK21. Transient transfections were performed as previously described [22]. Briefly, BCSC were transiently transfected with 1µg of the hTERT-VISA plasmid DNA using extruded DOTAP: cholesterol liposomes in 24-well plates (N/P ratio of 2:1). C22ORF28-Ad (addition of C22ORF28) and C22ORF28-KD (knockdown of C22ORF28) were generated as previously described [24, 25]. For site-specific mutagenesis, we mutated the regions in the C22ORF28-3'UTR and LIN28A-3'UTR complementary to the seed sequence of miR-34a using the QuikChange II Site-Directed Mutagenesis Kit. Further details are available in Supplementary methods.
Flow cytometry
Fluorescence-activated cell sorting (FACS) was performed to analyze the CD44+CD24- subpopulation, proportion of Lin and population of ALDH1 in BCSC using antibodies of CD44+, CD24-, Lin-PE and ALDH1-PE. Details are available in Supplementary methods.
RNA isolation and real-time PCR
The procedure for miR-34a expression was described previously [22]. Briefly, total RNA was extracted from BCSC using TRIzol Reagent (Invitrogen), according to the manufacturer's instructions. The amount of mRNA was determined relative to 5S ribosomal RNA. Each sample was analyzed in triplicate.
Serial passages of luciferase-labeled and green fluorescent protein (GFP)-labeled BCSC lineages
Establishment protocols were described previously [3, 13]. Further details are available in Supplementary methods.
Western blot analysis
Protein extracts of BCSC (10 µg protein) were separated using 10% SDS-PAGE, and electrophoretically transferred to polyvinylidene difluoride (PVDF) membranes. Next, antibodies were assayed. We purchased primary antibodies of hTERT, C22ORF28, LIN28A and CD44 from Sigma-Aldrich Co. St Louis, USA. GAPDH was designed as a protein loading control.
Telomerase activity assay
Quantitative Telomerase Detection Kit (Allied Biotech, Vallejo, CA) was used to evaluate the telomerase activity in 1 µg of corresponding cell lysate according to the manufacturer's instructions.
MTT assay of cell viability
MTT (Sigma) assay was used to assess cell viability according to the manufacturer's instructions. Each assay was repeated five times. Further details are available in Supplementary methods.
Clonal pair-cell analysis
In brief, BCSC (5×106 cells) were transduced with GFP-labeled C22ORF28-KD plasmid for 48 h, stained with 2 µL DAPI (10 µg/mL) for 5 min, and counterstained with anti-CD44-PE (1:200) for 30 min, respectively. Preliminarily, GFP-labeled BCSC were purified and collected. Next, images were acquired by an inverted epifluorescence microscope system and further analyzed by digital confocal microscopy.
Clonogenicity assay in soft agarose
Clonogenicity assay in soft agarose was performed to determine clonal expansion ability of BCSC as previously described [3]. Further details are available in Supplementary methods.
Tumor transplantation experiment
To determine the optimum antitumor dose of T-VISA-miR-34a plasmid in vivo, a suspension of luciferase-labeled BCSC (1×104 cells) was inoculated at the left fourth inguinal mammary gland of female BALB/c-nude mice (6-week-old; Vital River Laboratories Animal, Beijing, China). Each group of mice received 100 µL of DNA-liposome complexes that contained 5 µg TV-miR-34a, 10 µg TV-miR-34a, 20 µg TV-miR-34a, or liposomal complexes administered via tail vein injection, every other day / quaque omni die (qod) for 4 consecutive weeks. Moreover, to investigate the antitumor effect of TV-miR-34a in vivo, luciferase-labeled BCSC (1×104 cells) were injected into the left fourth inguinal mammary gland of mice. When the tumors reached ~50 mm3, the mice were noninvasively imaged using the IVIS system to confirm tumor growth and then randomly assigned to one of three following treatment groups (10 mice per group): Each group of mice received 100 µL of DNA-liposome complexes that contained TV-miR-Ctrl liposomal complexes (10 μg qod), TV-miR-34a liposomal complexes (10 μg qod) or liposomal (Ctrl) alone. The secretion levels of serum alanine transaminase (ALT), aspartate transaminase (AST), blood urea nitrogen (BUN) and creatinine (Cr) were determined with an automatic analyzer (Roche Cobas Mira Plus; Roche, Mannheim, Germany). The secretion concentrations of cytokines (TNF-α, IL-6 and IFN-γ) in mouse sera were quantified using the cytometric bead array kit for mouse inflammatory cytokines (CBA; BD Biosciences). The test was repeated more than five times. See also in Supplementary methods.
Microarray analysis
RNA was tested by capillary electrophoresis on an Agilent 2100 bioanalyzer and high quality of samples was confirmed. Acquired images and the signal intensities of microRNA spots were evaluated by GeneSpring GX (Agilent Technologies, Capital Biochip Corporation) and normalized based on global signal intensity according to the manufacturer's protocol. The results had been submitted to ArrayExpress (accession number E-MTAB-5584). The involved gene expressions of BCSC were described previously (accession number E-MTAB-5057) [3]. Further details are available in Supplementary methods.
Statistical analysis
All values are presented as the mean ± standard deviation (SD). For comparisons, Student's t test (two-tailed), Wilcoxon signed-rank test, Pearson chi-square test, Pearson correlation analysis, Log-rank test, Fisher's exact test, and nonparametric Mann-Whitney U test were performed as indicated. Survival rates were analyzed by log-rank test using SSPS 13.0 software. The significance level was set at p < 0.05. All data in our study have been recorded at Sun Yat-sen University Cancer Center for future reference (number RDDB2017000168).
Results
Identification of in vitro long-term-cultured BCSC
In our previous work, we successfully generated and characterized two BCSC lines (BCSC1/XM322, Figure 1A left panel; and BCSC2/XM607, Figure 1A right panel) derived from 2 (out of 22) clinical specimens with surgically resected breast cancer. Both BCSC lineages showed prolonged maintenance over 20 serial passages in vitro, while their tumor-initiating biological properties were retained [3]. To better investigate the innate features of BCSC, we further generated and maintained two BCSC lineages derived from breast cancer cell lines: MDA-MB-231.SC and MCF-7.SC. Here, we performed a magnetic-activated cell sorting (MACS) method to isolate cell line-derived BCSC, rather than using a fluorescence-activated cell sorting (FACS) approach for tumor tissue-derived BCSC. Both MDA-MB-231.SC and MCF-7.SC were isolated successfully and subsequently maintained on the first try. In a general sense, the success rate of cell line-derived BCSC isolated by MACS (2/2) is superior to that of tumor tissue-derived BCSC isolated by FACS (2/22) without removal of potential confounding factors (p < 0.05). Two cell line-derived BCSC sustained more than 20 passages for more than 3 years. Both of the 20th serial passages of BCSC also held the properties of mammosphere, self-renewal (MDA-MB-231.SC, Figure 1B left panel; and MCF-7.SC, Figure 1B right panel), CD44+ CD24-/low (MDA-MB-231.SC, Supplementary figure S1A left panel; and MCF-7.SC, Supplementary figure S1A right panel), Lin- (Supplementary figure S1B) ALDH1+ (Supplementary figure S1C), and clonogenicity in soft agarose (Supplementary figure S1D). Consistent to the tumor tissue-derived BCSC we described in a previous study [3], as few as 1×103 BCSC were sufficient to enable formation into tumors (Supplementary figure S1E), while as many as 1×105 breast cancer cells failed to generate tumors within two months according to the results of tumor transplantation experiment (Supplementary table S1). In sum, we successfully generated and maintained four long-term-cultured BCSC lineages.
miR-34a is the most pronounced microRNA for further research in long-term-cultured BCSC. (A) Capacities of self-renewal and sphere-formation of XM322 (left panel) and XM607 (right panel). Scale bar, 100 µm. (B) Capacities of self-renewal and sphere-formation of MDA-MB-231.SC (left panel) and MCF-7.SC (right panel). Scale bar, 100 µm. (C) Heatmap presents microRNA expressions among immortalized healthy mammary epithelial cells (HME, 184A1 and MCF-12A), breast cancer cells (BC, contained MDA-MB-231, MCF-7, MDA-MB-468 and SK-BR-3), and BCSC (contained MDA-MB-231.SC, MCF-7.SC, XM322 and XM607). (D) qRT-PCR analysis indicated that miR-34a expression of HME is significantly higher than that of BC and BCSC. miR-34a expression was normalized to the endogenous control 5S rRNA. NS: not significant. (E) qRT-PCR analyses indicated that samples within the stage I-II group had significantly lower miR-34a expression compared with those in groups of the adjacent healthy breast tissue specimens (HBT); additionally, miR-34a expression within stage III patients was lowest among the three groups. ***p<0.001.
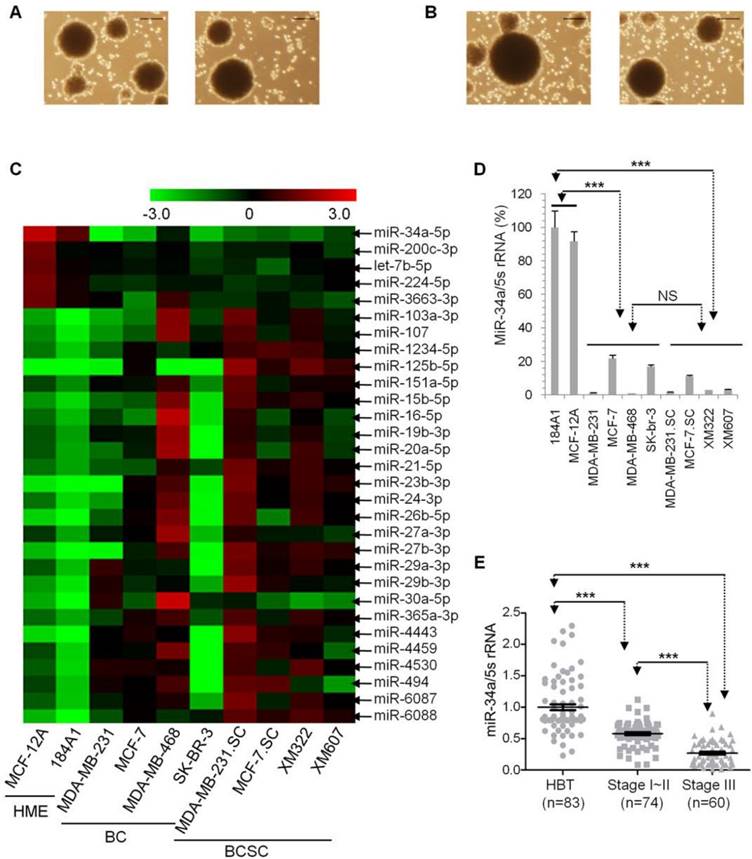
Microarray analyses of microRNA expression profiles to explore the most prominent microRNA expression in BCSC
Since the hTERT-promoter-based VISA vector drives transgene expression in multiple tumors, we aimed to develop the hTERT-promoter-based VISA delivery of specific microRNA in order to treat malignant breast cancer cells (breast cancer cells and BCSC). First of all, we sought the lowest microRNA expressions in malignant breast cancer cells in comparison with healthy mammary epithelial cells (HME). By screening a genome-wide microRNA array, we explored significant differences of microRNA expressions among breast cancer cells (BC, including MDA-MB-231, MCF-7, MDA-MB-468 and SK-BR-3), BCSC (including MDA-MB-231.SC, MCF-7.SC, XM322 and XM607), and immortal HME (184A1 and MCF-12A) (Figure 1C and Supplementary table S2). We found that miR-34a is the most pronounced microRNA in microarray analyses. We further performed quantitative RT-PCR analysis to confirm these results. Compared with these two immortalized healthy mammary epithelial cell lines, miR-34a expression is significantly down-regulated in both groups of BCs and BCSC (Figure 1D). Additionally, miR-34a expression had no significant difference between groups of BCs and BCSC (Figure 1D). To evaluate miR-34a expression in clinical specimens, we retrospectively analyzed clinicopathological data of 134 patients from previous reports (Supplementary table S3) [3]. Quantitative RT-PCR analyses indicated that the expression level of miR-34a in the stage I-II group is lower than that in their adjacent healthy breast tissue specimens (HBT), and miR-34a expression within the stage III group was lowest among the three mentioned groups (Figure 1E). So, miR-34a is downregulated in human breast cancer cells and BCSC in comparison with healthy breast cells. Therefore, miR-34a is the most prominent microRNA for further investigation in long-term-cultured BCSC.
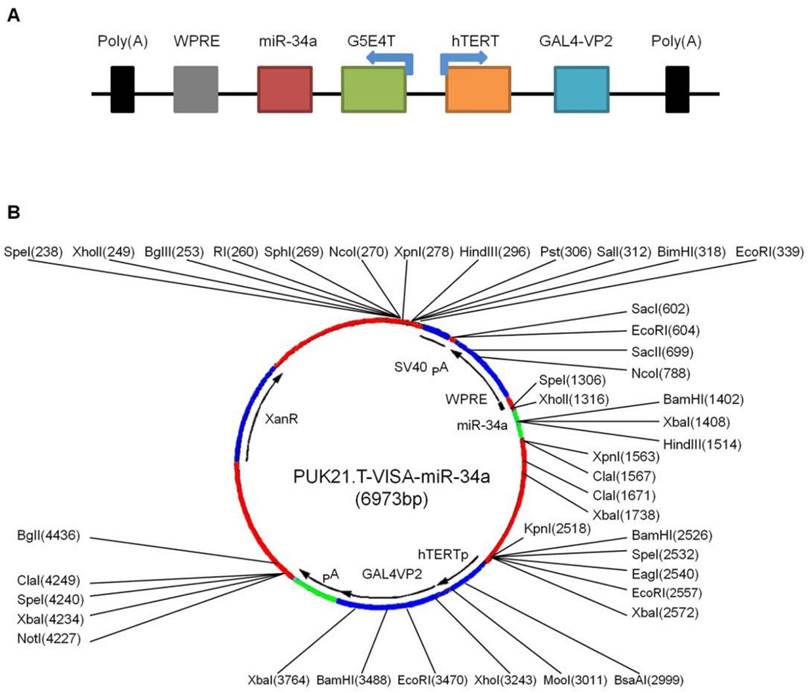
hTERT promoter-driven VISA delivery of miR-34a (TV-miR-34a) induces high throughput of miR-34a expression in BCSC
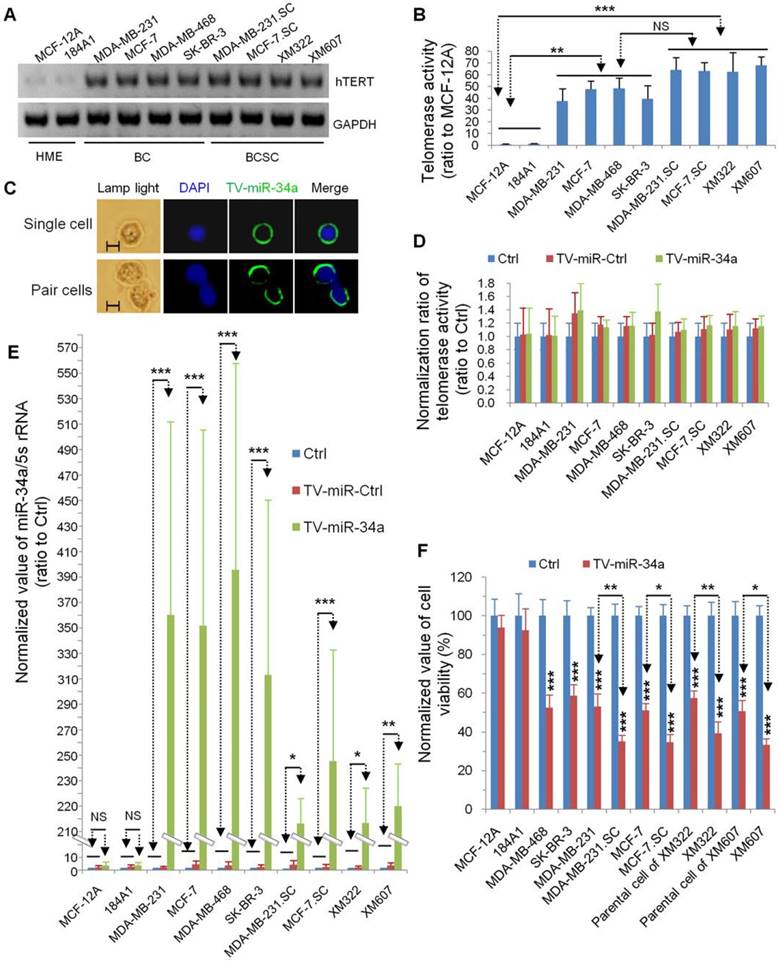
In our previous reports, our group had established TV-miR-34a plasmid (Figure 2A) and found that TV-miR-34a can lead to high throughput of miR-34a in breast cancer cells. To elucidate the correlations among gene sequences, secondary structure and function, we precisely analyzed the gene sequences (Supplementary table S4) and schematic structure (Figure 2B) of TV-miR-34a (6973bp). Here we compared the expressions of human telomerase reverse transcriptase (hTERT) among the HME, BC and BCSC. Expressions of hTERT were rarely detectable in both HME cells; nevertheless, hTERT expressions were high in all BC and BCSC (Figure 3A). Similarly, telomerase activities in HME cells were significantly lower than that in either BCs or BCSC (Figure 3B). Telomerase activity within group of BCSC seemed to be higher than that of BCs; however, this association was only on the borderline (Figure 3B). Given the nature of metabolic inertness within BCSC, the transfection efficiency of TV-miR-34a nanoparticle in BCSC remains to be fully investigated. Therefore, determination of miR-34a expression within BCSC following TV-miR-34a transduction is very crucial for this therapeutic strategy. Since transfection efficiency depends on cell proliferation, transduction rates and other complicated factors [26], we aimed to select out transfected cells for further investigation. Initially, we established plasmids of GFP-labeled Ctrl, TV-miR-Ctrl and TV-miR-34a. Then, BCSC were transfected with the above-mentioned plasmids for 48 h, respectively. Next, GFP-positive cells (both single cell and pair cells) were purified by FACS (Figure 3C, representative images of XM322). Finally, telomerase activities among the HME, BC and BCSC had not been altered significantly after the corresponding transfection with Ctrl, TV-miR-Ctrl or TV-miR-34a, respectively (Figure 3D). In addition, miR-34a expressions in purified GFP-labeled BCSC were determined by quantitative RT-PCR. The normalized value of miR-34a/5s rRNA in TV-miR-34a-infected BCSC significantly increased from 206.56 ± 27.53 (n=3) to 255.59 ± 86.97 (n=3), while the miR-34a relative value in TV-miR-34a-infected BCs enhanced significantly from 322.87 ± 137.59 (n=3) to 405.77 ± 162.17 (n=3) (Figure 3E). Here we found that there was no significant difference between BCs and BCSC following TV-miR-34a treatment (Figure 3E). In addition, cell viabilities were significantly depressed by TV-miR-34a treatment in both BCSC (MDA-MB-468, SK-BR-3, MDA-MB-231, MCF-7, parental cells of XM322 and XM607) and BCs (MDA-MB-231.SC, MCF-7.SC, XM322 and XM607) (Figure 3F). Meanwhile, this decrease was more pronounced in BCSC than in parental breast cancer cells, as depicted in Figure 3F. In sum, TV-miR-34a induces high throughput of miR-34a expression in BCSC, as well as preliminarily eradicates BCSC.
Schematic illustration of TV-miR-34a (targeted expression of micro-RNA-34a by hTERT promoter-driven VISA) plasmid. (A) Schematic diagram; (B) Schematic structure.
TV-miR-34a induces high throughput of miR-34a expression in BCSC. (A) Expression of hTERT was rarely detectable in both HME cells; while hTERT was highly expressed in all BC and BCSC. (B) Telomerase activities in HME cells were lower than BCs and BCSC. NS: not significant. (C) Representative images of isolated single cell (upper panel) and pair cells (low panel) expressing GFP-labeled TV-miR-34a in XM322. Scale bar, 5 µm (D) Telomerase activities among HME, BC and BCSC did not alter significantly following their corresponding transfection with Ctrl, TV-miR-Ctrl or TV-miR-34a, respectively. Scale bar, 5 µm.(E) qRT-PCR analysis demonstrated that TV-miR-34a plasmid significantly up-regulated miR-34a expression comparing with either TV-miR-Ctrl or Ctrl in both BCs and BCSC. (F) MTT assays of cell viabilities showed that TV-miR-34a decreased the cell viabilities in both BCSC (MDA-MB-468, SK-BR-3, MDA-MB-231, MCF-7, parental cells of XM322 and XM607) and BCs (MDA-MB-231.SC, MCF-7.SC, XM322 and XM607); whereas, cell viabilities of HME (MCF-12A and 184A1) were not found to be significantly different. The inhibitory effect of TV-miR-34a nanopartiple on cell viabilities of BCSC was stronger than their corresponding parental breast cancer cells. *p<0.05, **p<0.01, ***p<0.001
TV-miR-34a significantly inhibits tumor-initiating properties of long-term-cultured BCSC in vitro
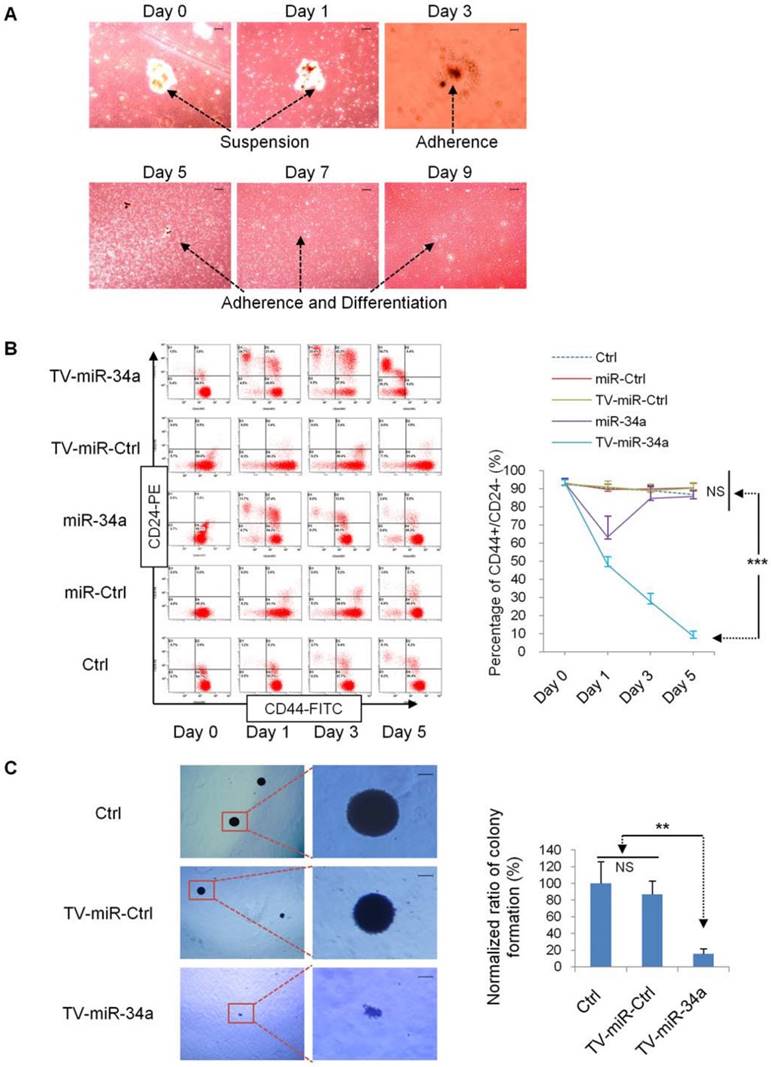
In the beginning, we explored whether TV-miR-34a mediates the biological properties of BCSC. First, we transduced BCSC mammosphere with TV-miR-34a plasmid. We found that mammosphere of XM607 initiated to adherence and differentiation on day 3 (Figure 4A). Next, we found that TV-miR-34a reduced the proportion of CD44+CD24-, and this trend was more robust and persistent in comparison with Ctrl, miR-Ctrl, TV-miR-Ctrl or miR-34a in XM607; whereas the influence of miR-34a was transient and reversible (Figure 4B). Similar results were observed in MCF-7.SC (Supplementary figure S2). These findings were exciting because they implied that TV-miR-34a leads to the inhibition of tumor-initiating properties in BCSC. Consistent with the above results, we also found that TV-miR-34a decreased the clonogenicity ability of MCF-7.SC within soft agarose (Figure 4C).
TV-miR-34a inhibits tumor-initiating properties of long-term-cultured BCSC in vitro. (A) Mammospheres of XM607 initiated adherence and differentiation following TV-miR-34a interference. Scale bar, 100 µm. (B) Representative images (left panel) and statistical results (right panel) of TV-miR-34a robustly and persistently decreasing the population of CD44+CD24- XM607; while miR-34a influence remained transient and reversible. (C) Representative images (left panel; scale bar, 100 µm) and statistical result (right panel) of clonogenesis abilities of MCF-7.SC down-regulated by TV-miR-34a. ***p<0.001.
TV-miR-34a reduces proliferation of BCSC effectively and safely in vivo
In our previous study, our group found that TV-miR-34a drastically eliminated breast cancer cells in vivo [22]. Here, we further investigated whether TV-miR-34a treatment plays a role in inhibiting BCSC proliferation. To confirm the therapeutic response of TV-miR-34a treatment in vivo, we initially established and maintained BCSC lineages expressing firefly luciferase. We found that TV-miR-34a inhibited the growth of BCSC-bearing tumors in mice by a dose-dependent manner. With the goal of achieving most efficacious antitumor activity with lowest potential toxicity, we selected an optimum dose (10 µg qod) for TV-miR-34a treatment in XM322-bearing mouse tumors (Figure 5A). Importantly, luciferase imaging revealed that treatment with TV-miR-34a led to significantly decreased bioluminescent signal from the tumors by day 10 after the first injection (Figure 5B). Additionally, TV-miR-34a prolonged mouse survival even more effectively than either Ctrl or TV-miR-Ctrl (Figure 5C).
TV-miR-34a suppresses proliferation effectively and safely in vivo. (A) TV-miR-34a inhibited the growth of luciferase-labeled XM322 bearing tumors in a dose-dependent manner. The optimum dose is 10 µg qod. (B) TV-miR-34a plasmid significantly suppressed fluorescence intensity of XM322 (upper left: representative images). (C) Kaplan-Meier survival analyses of TV-miR-34a prolonging mice survival in orthotopic xenograft models. (D) Body weights in all participant nude mice showed no significant change. (E) Side effects of TV-miR-34a treatment on serum ALT (left panel) and AST (right panel). (F) Side effects of TV-miR-34a treatment on serum BUN (left panel) and Cr (right panel). Each experiment was repeated more than three times. (G) In comparison with both groups of Ctrl and TV-miR-Ctrl, secretion levels of TNF-α (left panel), IL-6 (middle panel) and IFN-γ (right panel) in the TV-miR-34a treatment group were significantly increased from day 0 to day 10. Each experiment was repeated more than three times. *p<0.05, **p<0.01, ***p<0.001.
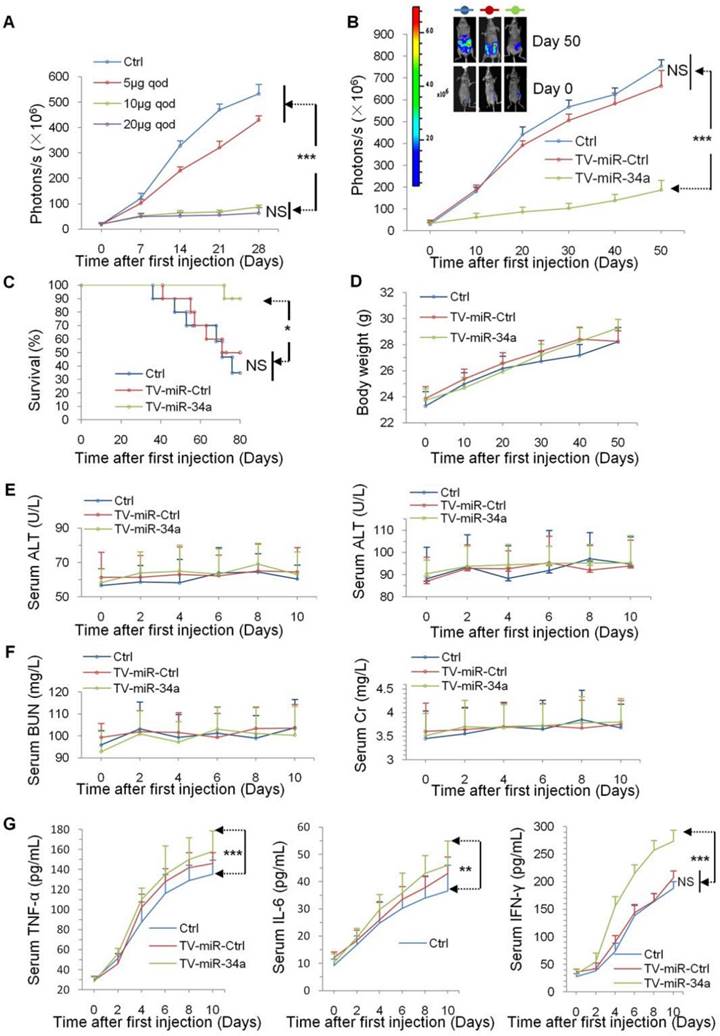
Besides the efficacy of TV-miR-34a treatment, we also paid attention on its safety. Inconsistent to our previous work, [3] we found that body weights for all participating MDA-MB-231.SC-bearing nude mice did not change significantly (Figure 5D); suggesting that the dosage of 10 µg qod is better tolerated than 15 µg biw for TV-miR-34a, based on the treatment's influence on body metabolism. In addition, liver injury in BALB/c-nude mice was assessed by measuring the levels of key liver enzymes in the serum. TV-miR-34a treatment did not affect serum alanine aminotransferase (ALT) (Figure 5E, left panel) and aspartate aminotransferase (AST) (Figure 5E, right panel) in MDA-MB-231.SC-bearing mice. Similarly, the effect of TV-miR-34a treatment on renal function of mice was further evaluated in MDA-MB-231.SC-bearing mice. We found no difference in levels of both blood urea nitrogen (BUN) (Figure 5F, left panel) or creatinine (Cr) (Figure 5F, right panel) among Ctrl, TV-miR-Ctrl and TV-miR-34a. All these parameters confirmed BCSC-bearing mice with TV-miR-34a treatment were physiologically healthy.
The potential immunotoxicities of nanomaterials have received substantial attention. It is generally agreed that secretion amounts of proinflammatory cytokines can be useful tools in evaluating nanomaterial immunotoxicity [27]. In this study, we measured the serum proinflammatory cytokines (TNF-α, IL-6 and IFN-γ) to evaluate immunotoxicity produced by TV-imR-34a treatment in vivo. The differences in cytokine concentrations (TNF-α, IL-6 and IFN-γ) between the groups of Ctrl-treated and TV-miR-Ctrl-treated mice were not statistically significant (Figure 5G). In comparison with groups of Ctrl and TV-miR-Ctrl, concentrations of TNF-α, IL-6 and IFN-γ of mice in the study group of TV-miR-34a treatment were significantly increased from day 0 to day 10 (Figure 5G). Remarkably, 10 days after injection of empty vector (liposome 100 µL) in mice, TNF-α was increased by 3.5-fold, relative to day 0 (p < 0.001). The level of IL-6 was increased by ~3.0-fold relative to day 0 (p < 0.001). Similarly, the secretion amount of IFN-γ was increased by 5.7-fold as compared with day 0 (p < 0.001). Liposome injection increased immunomodulatory activities in mice in vivo, suggesting that there remains much space to develop the versatile empty vector. Although the mouse immune system was found to release significantly higher levels of proinflammatory cytokines (TNF-α, IL-6 and IFN-γ), we did not observe lethal inflammatory responses in mice throughout these tests. Therefore, treatment of TV-miR-34a for BCSC is well-tolerated in vivo.
C22ORF28 (chromosome 22 open reading frame 28) acts as a direct and functional target of TV-miR-34a treatment in long-term-cultured BCSC
Although multiple molecules have been reported to be direct targets of miR-34a in stem cells [5, 28], the precise molecular mechanisms of long-term-cultured BCSC repressed by TV-miR-34a remain to be established. In our recent research, we found that POMC, AKT2 and GSK3β could regulate the tumor-initiating properties of long-term-cultured BCSC [3]. Next, we speculated whether these molecules and their related signaling pathways are involved in the treatment mechanism of TV-miR-34a on BCSC. First, we used miRWalk 2.0 (http://zmf.umm.uniheidelberg.de/apps/zmf/mirwalk2) to predict RNA-binding proteins related to genes of AKT2, GSK3β and POMC (Supplementary table S5). C22ORF28-3'-UTR (Figure 6A) and LIN28A3'-UTR (Supplementary figure S3A) were determined to contain miR-34a-conserved binding sites. Second, we constructed a luciferase reporter vector comprising the 3'-UTR or 3'-UTR with mutation sites of C22ORF28 (Figure 6A) and LIN28A (Supplementary figure S3A). Encouragingly, TV-miR-34a significantly repressed luciferase expressions of Luc-C22ORF28-3'UTR rather than Luc-LIN28A-3'UTR; whereas the mutation of five nucleotides at the miR-34a-binding site of C22ORF28-3'UTR led to complete abrogation of the suppressive effect (Figure 6B and Supplementary figure S3B). In order to determine which molecule is acting as the functional target of TV-miR-34a in long-term-cultured BCSC, we performed Western blotting to measure expressions of C22ORF28, LIN28A and CD44. LIN28A expression was rarely detectable in all four BCSC. We further found that TV-miR-34a interference significantly inhibited expressions of C22ORF28 and CD44 rather than LIN28A (Figure 6C). Taken together, we conclude that C22ORF28 was the direct and functional target of TV-miR-34a in long-term-cultured BCSC.
TV-miR-34a plays an inhibitory effect on tumor-initiating properties of long-term-cultured BCSC via directly targeting C22ORF28. (A) Schematic diagram of Luc-C22ORF28-3'UTR and mutation of Luc-C22ORF28-3'UTR (Luc-C22ORF28-3'UTR-mut) for presence of miR-34a conversed binding sites. (B) Mutating the predicted miR-34a binding sites within the Luc-C22ORF28-3'UTR luciferase reporter significantly abolished TV-miR-34a-dependent repression. (C) LIN28A is rarely detected in BCSC. Both C22ORF28 and CD44, rather than LIN28A, were the inhibitory targets of TV-miR-34a treatment. (D) Constructions of C22ORF28-Ad and C22ORF28-KD can effectively up-regulate and down-regulate expressions of C22ORF28 and CD44 in BCSC, respectively. (E) C22ORF28-Ad increased percentage of CD44+CD24- subpopulation cells in BCSC, whereas C22ORF28-KD decreased these subpopulation cells. (F) Representative images (left panel) and statistical results (right panel) of C22ORF28-Ad promoting sphere-formation ability within soft agarose of XM322, but C22ORF28-KD eliminating this ability. Scale bar, 100 µm. (G) Representative images (left panel) and statistical result (right panel) of TV-miR-34a suppressing CD44 expression measured by clonal pair-cell analyses in XM607. Scale bar, 10 µm; C.E.: co-expressed; M.E.: mutually exclusive. Each experiment was repeated at least three times. *p<0.05, **p<0.01, ***p<0.001.
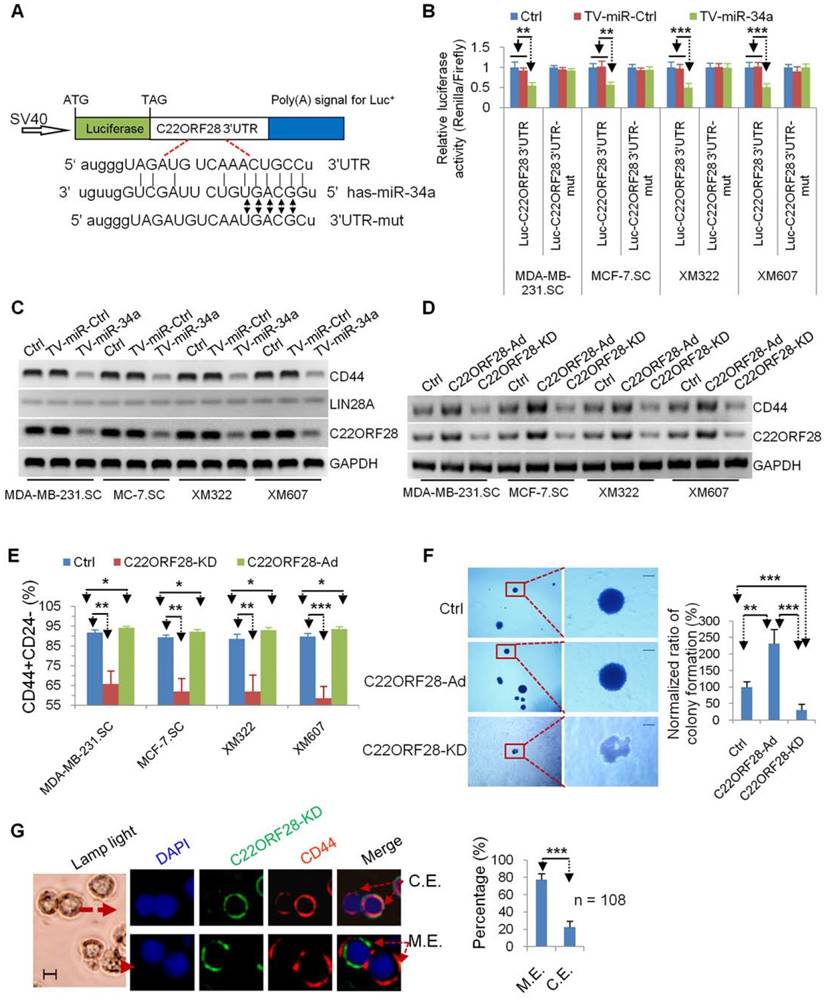
C22ORF28 mediates the inhibition effect of TV-miR-34a on tumor-initiating properties in long-term-cultured BCSC
We further explored the role of C22ORF28 in the effect of TV-miR-34a on BCSC. Regulators of C22ORF28 were preliminarily established. We found that addition of exogenous C22ORF28 (C22ORF28-Ad) up-regulated expressions of C22ORF28 and CD44; conversely, knockdown of C22ORF28 (C22ORF28-KD) down-regulated these expressions in BCSC (Figure 6D). Interestingly, C22ORF28-Ad increased the proportion of CD44+CD24- in BCSC; while C22ORF28-KD decreased their population (Figure 6E). Similarly, C22ORF28-KD inhibited the clonogenicity ability of XM322 within soft agarose; while C22ORF28-Ad promoted this ability (Figure 6F). Furthermore, clonal pair-cell assay was performed to evaluate the suppressive effect of C22ORF28-KD on XM607. Encouragingly, we indicated that C22ORF28-KD and CD44 were mutually exclusive of each other more often than they were co-expressed (Figure 6G). Collectively, these processes in vitro demonstrate that knockout of C22ORF28 could dramatically eliminate the tumor-initiating properties of long-term-cultured BCSC.
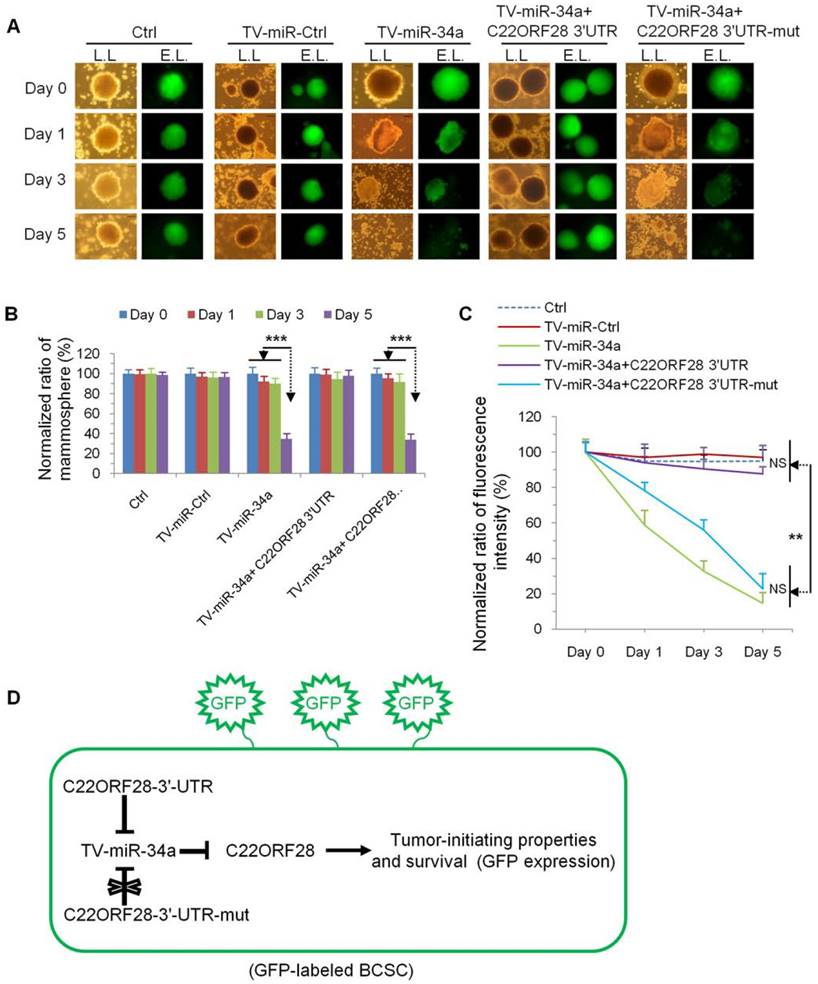
With a goal of evaluating the effect of C22ORF28 on the tumor-initiating biological properties of long-term-cultured BCSC, we generated GFP-labeled MDA-MB-231.SC (Supplementary figure S4). In comparison with the effect on day 0, day 1 and day 3, TV-miR-34a treatment significantly inhibited mammosphere formation of MDA-MB-231.SC on day 5; while this effect was reversed by co-transfection of C22ORF28-3'-UTR rather than C22ORF28-3'-UTR-mut (Figure 7A and Figure 7B). These differences were further reflected by differences in GFP intensity. Compared with treatment of both Ctrl and TV-miR-Ctrl, TV-miR-34a significantly reduced the fluorescence intensity of BCSC to a similar extent in a time-dependent manner. Moreover, co-transfection of C22ORF28-3'-UTR could counteract the growth suppression mediated by TV-miR-34a in BCSC, whereas a mutation in the C22ORF28-3'-UTR binding sites of miR-34a did not have this effect (Figure 7A and Figure 7C). As presented in Figure 7D, C22ORF28-3'-UTR plasmid inhibited the eradication effect of TV-miR-34a on GFP-labeled BCSC; hence, C22ORF28-3'-UTR-mut plasmid conserved GFP expression during TV-miR-34a intervention. This result lent further credence to the notion that TV-miR-34a targeted C22ORF28 directly and functionally in long-term-cultured BCSC. In a word, by targeting to C22ORF28 directly, TV-miR-34a attenuated the tumor-initiating properties of long-term-cultured BCSC.
Evaluation of the inhibitory effect of C22ORF28 intervene on long-term-cultured BCSC during TV-miR-34a treatment. (A) Representative images of mammosphere formation and fluorescence expression of GFP-labeled BCSC following transduction with Ctrl, TV-miR-Ctrl, TV-miR-34a, co-transfection of TV-miR-34a plus C22ORF28-3'-UTR, as well as co-transfection of TV-miR-34a plus C22ORF28-3'-UTR-mut, respectively (L.L.: lamp light; E.L.: exciting light). Scale bar, 100 µm. (B) Statistical results of differences in mammosphere formation. (C) Statistical results of fluorescence intensity. Each experiment was repeated at least in triplicate. (D) Schematic diagram of C22ORF28 mediating TV-miR-34a treatment in GFP-labeled long-term-cultured BCSC. **p<0.01, ***p<0.001.
TV-miR-34a synergizes with docetaxel for inhibition of BCSC
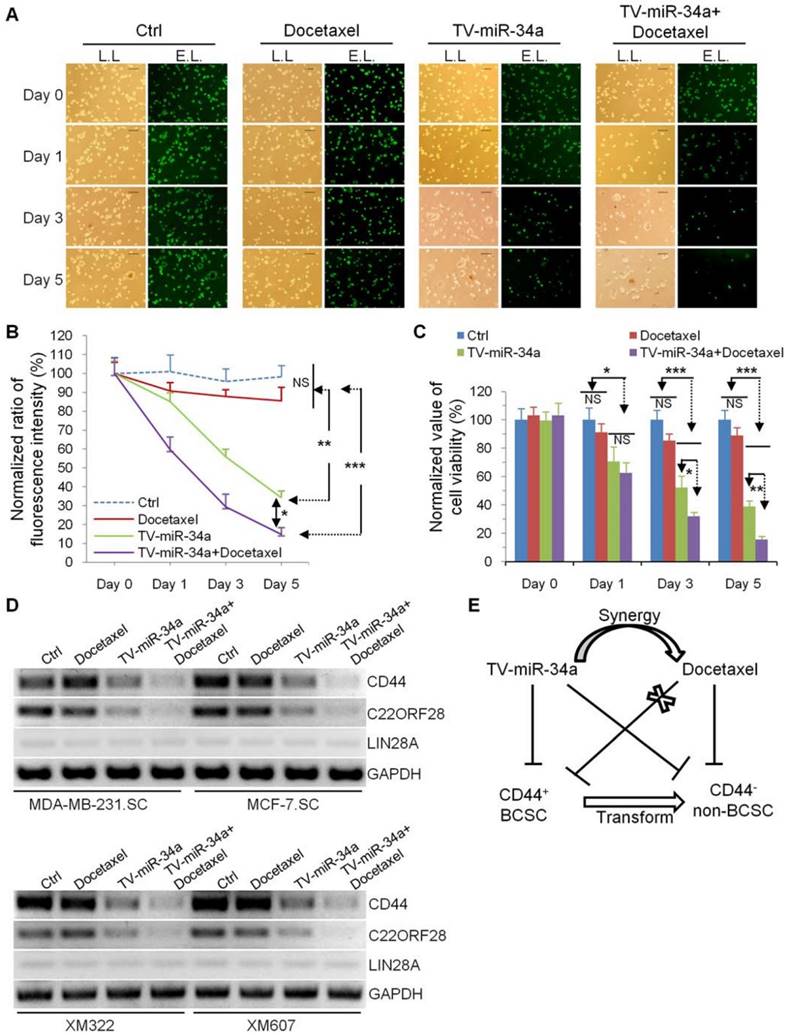
In current clinical practice, the chemotherapeutic taxanes, represented by docetaxel and paclitaxel, are standard therapy for patients with invasive breast cancer. In this study, we examined whether the combination of TV-miR-34a and docetaxel may enhance the therapeutic effect on BCSC. Limitedly, docetaxel alone exerted no effect against MDA-MB-231.SC (Figure 8A and Figure 8B). However, we found that TV-miR-34a effectively sensitized GFP-labeled MDA-MB-231.SC to docetaxel. Docetaxel played a rare role in eradicating MDA-MB-231.SC: the inhibition effect of TV-miR-34a plus docetaxel on MDA-MB-231.SC was superior than that of TV-miR-34a alone (Figure 8A and Figure 8B). Similarly, docetaxel alone did not significantly affect cell viability of XM322; while XM322 with TV-miR-34a treatment showed a significant decrease in cell viability. In particular, combination treatment of TV-miR-34a plus docetaxel significantly decreased cell viability in XM322 more than single treatment alone using TV-miR-34a (Figure 8C). In line with these above-mentioned results, we found that TV-miR-34a alone significantly inhibited expressions of CD44 and C22ORF28 compared with either the vector or docetaxel treatment alone (Figure 8D). Further, the combined usage of TV-miR-34a plus docetaxel demonstrated even better inhibitory efficacy than single-agent application of TV-miR-34a plasmid (Figure 8D). Here we also found that LIN28A expression was rarely detectable in all four BCSC lines (Figure 8D). From the results of our previous investigations, TV-miR-34a plasmid significantly depressed the tumor-initiating properties of BCSC, thereby transforming their stem-like properties to non-stemness, along with inhibiting cell proliferation of BCSC (Figure 8E). These results suggested that the enhanced eradication effect induced by synergy of TV-miR-34a and docetaxel may contribute to their synergistic effects on the cell fate of BCSC, owing to the transformation of CD44 positive BCSC to non-stem-like tumor cells (Figure 8E).
TV-miR-34a has synergistic effects with docetaxel in eradication of BCSC. (A) MDA-MB-231.SC was observed with lamp light (L.L., left panel) or exciting light (E.L., right panel) to detect the depression effects. Scale bar, 100 µm. (B) Statistical results of differences in fluorescence expressions among groups of Ctrl, TV-miR-34a, and presence or absence of docetaxel 5 days following TV-miR-34a transduction. (C) MTT assay. (D) Docetaxel alone did not significantly alter expressions of CD44 and C22ORF28 in BCSC, compared to untreated cells. Combined application of TV-miR-34a plus docetaxel on BCSC for 48 h significantly decreased expressions of CD44 and C22ORF28 compared with TV-miR-34a alone. Cell line-derived (upper panel); tumor tissue-derived BCSC (low panel). (E) Schematic Diagram. All data corresponds to the mean ± SD of three independent experiments.. *p<0.05, **p<0.01, ***p<0.001.
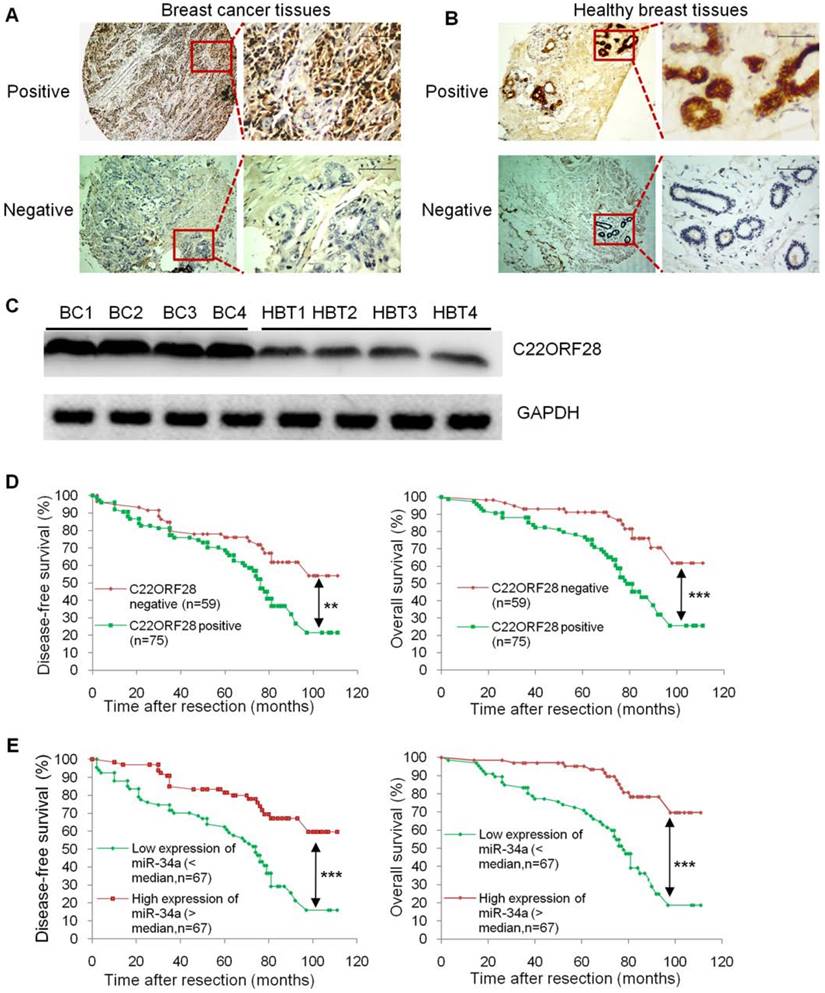
C22ORF28 is frequently up-regulated in breast tumor tissues, and positive expression is correlated with an unfavorable prognosis
Given the striking effect of C22ORF28 on TV-miR-34a treatment in long-term-cultured BCSC, we then questioned whether there was an association between C22ORF28 expression and clinical prognosis in patients with breast cancer. Here we evaluated C22ORF28 expression and other clinicopathological data from a previously reported study [3]. Representative slides of breast cancer tissues and corresponding adjacent healthy breast tissues are shown in Figure 9A and 9B. Expressions of C22ORF28 were assessed by immunohistochemistry. We found that expression of C22ORF28 was conversely related to miR-34a level in breast cancer tissues (Supplementary table S6). These results were consistent with the response of miR-34a repressing C22ORF28 in BCSC. In addition, C22ORF28 positive probability in HBT was lower than that in patients with stage I-II, which was less than that in stage III (Supplementary table S6). To confirm these results of TMA and IHC, we further evaluated four cases of breast cancer tissues. Compared with HBT by Western blotting, the tumor tissues had a significantly higher expression of C22ORF28 (Figure 9C). Further Kaplan-Meier survival analyses showed that breast cancer patients with C22ORF28 positive expression had shorter disease-free survival (DFS) and overall survival (OS) than those patients with negative expression (Figure 9D). Moreover, we found a more favorable DFS and OS in the group that had high expression of miR-34a than in the group with low expression of miR-34a (Figure 9E). Therefore, up-regulation of C22ORF28 was common in patients with breast cancer, and C22ORF28 positive expression was associated with a more unfavorable patient prognosis.
C22ORF28 is frequently up-regulated in breast tumor tissues, and C22ORF28 positive expression is correlated with a poor prognosis. (A) Representative cases of breast cancer tissues immunostained with positive C22ORF28 (upper panel) and negative C22ORF28 (low panel). Scale bar, 100 µm. (B) Representative cases of adjacent healthy breast tissues immunostained with positive C22ORF28 (upper panel) and negative C22ORF28 (low panel). Scale bar, 100 µm. (C) Tumor tissues had a significantly higher expression of C22ORF28 than their adjacent healthy breast tissues (HBT). (D) Kaplan-Meier survival curves of disease-free survival (left panel) and overall survival (right panel) for C22ORF28 status in surgically resected breast cancer. (E) Kaplan-Meier survival curves of disease-free survival (left panel) and overall survival (right panel) for miR-34a status in surgically resected breast cancer. **p<0.01, ***p<0.001.
Discussion
In this study, we have successfully generated four long-term-cultured BCSC that presented prolonged maintenance over 20 serial passages, while retaining their tumor-initiating properties. Analyses of microarray and qRT-PCR showed that miR-34a is the most pronounced microRNA for further BCSC investigation. We established hTERT promoter-driven VISA delivery of miR-34a plasmid, which can induce high throughput of miR-34a expression in BCSC. TV-miR-34a significantly inhibited tumor-initiating properties of the long-term-cultured BCSC in vitro, along with suppressing the proliferation of BCSC in vivo in an efficient and well-tolerated manner. Further mechanistic studies revealed that TV-miR-34a attenuates the tumor-initiating properties of long-term-cultured BCSC via targeting to C22ORF28 directly. Furthermore, TV-miR-34a plasmid synergizes with docetaxel for eradicating BCSC. In addition, investigation of clinical implication showed that miR-34 and C22ORF28 have a negative correlation in breast tumor tissues. Also, positive expression of C22ORF28 is associated with an unfavorable prognosis.
In term of BCSC generation, we found that the success rate for isolation of cell line-derived BCSC purified by MACS is higher than that for isolation of tumor tissue-derived BCSC purified by FACS. However, this result did not denote that MACS is always better than FACS in isolation of CSCs. Separating the cells based on their own cellular physical or biochemical properties, FACS and MACS have been developed into the most widely used techniques for cell sorting [29]. FACS method vibrates and analyzes each cell individually [30], therefore it may not be fit for maintenance of CSCs. However, the tumor tissue-derived CSCs were derived from the primary cells of human cancer tissues. In this case, FACS is efficient enough to exclude the extracellular matrix and the cells without stem-like properties. Alternatively, the MACS approach is beneficial due to its simplicity and robustness, not requiring sophisticated fluid handling [29]. So, we speculate that development of primary cell lines from tumor tissues will bring about a great advance in isolation of tumor tissue-derived CSCs. Multiple CSCs have been isolated and identified in recent decades [31]. However, long-term-cultured CSCs are rarely claimed. Here, we report four BCSC lineages that have been cultured for more than 20 serial passages and more than 3 years. We have to emphasize that application of proper culture medium, avoiding fungi infection and reducing unnecessary intense shock are crucial for long-term maintenance of BCSC, whether derived from tumor tissues or cell lines [3]. So, the feature of inertia within CSCs may reveal that they prefer a “quiet microenvironment” rather than dynamic conditions.
The hypometabolism status of CSCs demands a highly specific and efficient amplification system to deliver anti-tumor molecules to eradicate them. VISA system is a non-viral system, which contains two basic elements: the two-step transcriptional amplification system (GAL4-VP2) and the WPRE sequence [13, 22]. WPRE plays a role in stabilizing 3'UTR of microRNA and enhancing the half-life of RNA transcripts. In addition, GAL4-VP2 combines and activates a therapeutic molecule. It is stated that the active human telomerase enzyme is mainly composed of hTERT, human telomerase RNA (hTR), and dyskerin. In this study, we found that both hTERT expression and telomerase activity in HME cells was significantly inferior to either BC or BCSC. We further found that hTERT promoter within TV-miR-34a can direct preferential transgene expression in cancer cells and BCSC rather than HME. Our results were consistent with the findings of Cao et al. for HME cells [32]. They interpreted that mRNA and protein expression of hTERT could be up-regulated in expressing exogenous hTERT amplified HME, but hTR limits telomerase activity and telomere maintenance in the context of hTERT amplification. This explanation indicated that TV-miR-34a drives the expression of miR-34a selectively in BCSC and CS, but does not drive its expression in HME.
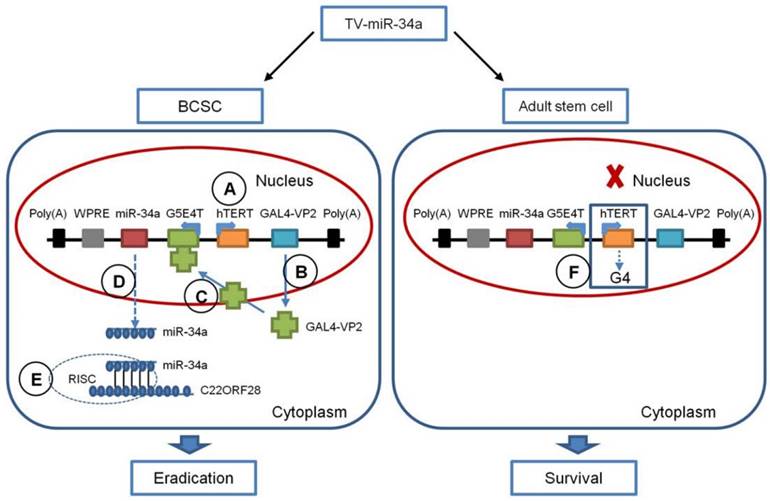
As illustrated in Figure 10, TV-miR-34a selectively controls the expression of miR-34a in BCSC in five steps: (1) The hTERT promoter drives expression of the GAL4-VP2 (two copies of VP16) fusion protein specifically in BCSC, rather than in adult stem cells; (2) the activator GAL4-VP2 is expressed; (3) GAL4-VP2 discerns the promoter G5E4T, due to G5E4T containing five GAL4 sites positioned upstream of the adenovirus E4 TATA box; (4) G5E4T induces the expression of the miR-34a shRNA transcripts, and then generates miR-34a; (5) the mature miR-34a are incorporated into the RNA-induced silencing complex (RISC), which induces downregulation of C22ORF28 leading to eradication of BCSC. In untransformed human tissues, most somatic cells lack telomerase activity, while somatic stem and progenitor cells express low levels of telomerase [33]. Of note, telomeres can form G-quadruplex structures (G4) in adult stem cells. Structures of G4 are highly stable and are difficult to resolve during replication, which can cause replication fork stalling (Figure 10) [33]. Additionally, chromosomal instability coming from replication stress can be ruled out by efficient checkpoints, which ensures the elimination of damaged stem cells and prevents stem cell-derived tumorigenesis [33]. It is well known that miR-34a has been identified as a tumor suppressor by several researches. It has been elucidated that p53 checkpoints and the p53-miR-34 network can perform the second line of defense for avoiding abnormal proliferation in adult stem cells [33]. Therefore, the hTERT promoter has much less influence on adult stem cells, resulting in very limited target gene expression and therefore cell survival. Similarly, recent studies in human embryonic stem cells showed that increased TERT expression is not functionally associated with an increase in active telomerase, as hTR, but not TERT, is limiting [34]. Collectively, TV-miR-34a plasmid has the appropriate construction to eradicate BCSC via an efficient and safe approach. In the present study, in order to provide guidance for this novel therapeutic avenue, we have specifically analyzed the gene sequences and schematic structure of TV-miR-34a in detail.
Schematic Diagram of the TV-miR-34a System. (A) The hTERT promoter drives expression of the GAL4-VP2 fusion protein selectively in BCSC, but not in adult stem cells (B) GAL4-VP2 is expressed; (C) GAL4-VP2 discerns the promoter G5E4T; (D) G5E4T promotes the expression of the miR-34a protein; (E) the mature miR-34a are incorporated into RISC, which inhibits C22ORF28 expression leading to eradication of BCSC. (F) Telomeres (mainly composed by hTERT) form G-quadruplex structures (G4), which along with G4 can cause replication fork stalling.
CSCs hold the tumor-initiating properties of self-renewal, proliferation, specific marker and differentiation [4, 35]. Interferences with adherence and differentiation are the critical directions for elimination of these progenitor cells. The therapeutic approach with TV-miR-34a plasmid shows us the “dual steps” to eradicate BCSC. On the first step, findings regarding CD44+ CD24-/low differences, clonal pair-cell analyses and synergistic effects of docetaxel confirm that CD44+ CD24-/low BCSC are asymmetrically divided into CD44 negative heterogeneous lineages of breast cancer cells [3]. TV-miR-34a might restrict the acquisition of expanded cell fate potential in pluripotent stem cells through overexpression of miR-34a [5]. On the second step, as our previous study described, TV-miR-34a suppresses proliferation and invasion of breast cancer cells [22]. Therefore, miR-34a overexpressed by hTERT promoter-driven VISA plasmid can effectively eradicate BCSC.
In the current research, we indicated that the non-viral TV-miR-34a nanoparticle showed great therapeutic efficacy in orthotopic xenograft models with a high degree of safety. TV-miR-34a treatment had no significant side effects on functions of liver and kidney in the participant mice. No lethal inflammatory responses were observed throughout the study. Here, we review plausible explanations for the increase of proinflammatory cytokines. First of all, this is essential for the nanomaterial to play a potential antitumor role in tumor-bearing mice [27]. Of note, the immune response is also influenced by the animal model. Kim and colleagues found that various proinflammatory cytokine levels are much higher in nude mice than wild-type mice after treatment [36]. So, the involved nude mice are more likely to produce a cytokine surge than wild-type mice. In sum, TV-miR-34a treatment for tumor-bearing mice is well-tolerated.
There is increasing evidence that hypoxic microenvironment promotes renewal, differentiation, angiogenesis and accumulation of CSCs in tumor tissues. The hypoxia-inducible factors (HIFs) family, including HIF-1α, HIF-2α and HIF-3α, orchestrate signaling events regulating cell survival, proliferation and differentiation, angiogenesis, metabolism, invasion and metastasis of cancer cells, and is mediated prominently by HIF-1α and HIF-2α [37]. More recently, Li and colleagues found that miR-34a is directly repressed by HIF-1α under hypoxic conditions, and ectopic miR-34a prevents hypoxia-induced invasion and migration in hypoxic cancer cells [38]. Therefore, it is conceivable that miR-34a restored by TV-miR-34a may have therapeutic potential for hypoxia-driven formation of invasion and metastasis in tumor tissues.
C22ORF28 is highly homologous to HSPC117 (hematopoietic stem/progenitor cell 117) in humans, also previously known as focal adhesion associated protein (FAAP) in mice, and RTCB (RNA 2', 3'-cyclic phosphate and 5'-OH ligase) in prokaryotes. The high degree of conservation of C22ORF28/RTCB proteins from E. coli to human is suggestive of shared important roles for this protein family in organisms [39]. FAAP facilitates vinculin-paxillin association, decreases interaction of paxillin-focal adhesion kinase and reduces the phosphorylation of extracellular signal-regulated kinase in murine [40]. In the research of moving cells, Carmen et al. found that HSPC117 might be involved in initiation of cell spreading by virtue of cell adhesion [41]. Recent investigations indicated that HSPC117 mediates tRNA [39, 42] and mRNA [25, 43] splicing. Herein, for the first time to our knowledge, we found that C22ORF28 maintains properties of self-renewal, proliferation, tumor formation, CD44+CD24- subpopulation and CD44 expression in BCSC. In summary, we indicated that TV-miR-34a promotes adherence and differentiation in long-term maintenance of BCSC via targeting to C22ORF28.
In conclusion, we generated long-term maintenance of BCSC, either from clinical samples or cell lines. We established the non-viral TV-miR-34a plasmid, which could eradicate long-term-cultured BCSC in a safe and efficient approach both in vitro and in vivo. The combination of TV-miR-34a and docetaxel enhanced the therapeutic effect on BCSC. Mechanistic studies further indicated that TV-miR-34a suppresses tumor-initiating properties, promotes adherence and boosts differentiation in long-term maintenance of BCSC through targeting C22ORF28 directly. Thus, we developed the non-viral TV-miR-34a plasmid, which has great potential for clinical application in breast cancer therapies targeting to BCSC.
Abbreviations
ALT: alanine transaminase; AST: aspartate transaminase; BUN: blood urea nitrogen; BC: breast cancer cells; BCSC: breast cancer stem cells; C22ORF28-Ad: addition of C22ORF28; C22ORF28-KD: knockdown of C22ORF28; CSCs: cancer stem cells; Cr: creatinine; DFS: disease-free survival; FACS: fluorescence-activated cell sorting; FAAP: focal adhesion associated protein; G4: G-quadruplex structures; GFP: green fluorescent protein; HBT: healthy breast tissue specimens; HME: healthy mammary epithelial cells; HSPC117: hematopoietic stem/progenitor cell 117; hTR: human telomerase RNA; HIFs: hypoxia-inducible factors; IHC: immunohistochemistry; MACS: magnetic-activated cell sorting; OS: overall survival; PVDF: polyvinylidene difluoride; qod: quaque omni die; RISC: RNA-induced silencing complex; SD: standard deviation; TMA: tissue microarray construction; TV-miR-34a: hTERT promoter-driven VISA delivery of miR-34a; TV-miR-Ctrl: hTERT promoter-driven VISA nanoparticle delivery of control; VISA: VP16-GAL4-WPRE integrated systemic amplifier.
Supplementary Material
Supplementary methods, tables (S1-S6) and figures (S1-S4).
Acknowledgements
The authors would like to thank all the patients that provided samples for the study. This work was supported by funds from the Key Program of the National Natural Science Foundation of China (31030061), the National Natural Science Foundation of China (81272514 and 81472575), the China Postdoctoral Science Foundation (2017M610570), Natural Science Foundation of Guangdong (2013B060300009), Science and Technology Planning Project of Guangzhou (2014J4100169), Guiding Project of Science and Technology Department of Fujian Province (2016Y0020), Training Project Funding Plan of Young and Middle-aged Talent of Health System in Fujian Province (2016-ZQN-18), Training Project Funding Plan of Youth Innovative Talents of Xiamen City (2017), and Sisters Hospital Network Fund (SINF) between University of Texas MD Anderson Cancer Center and Sun Yat-sen University Cancer Center.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Crea F, Nur Saidy NR, Collins CC, Wang Y. The epigenetic/noncoding origin of tumor dormancy. Trends Mol Med. 2015;21:206-11
2. Willyard C. Stem cells: bad seeds. Nature. 2013;498:S12-3
3. Lin X, Chen W, Wei F, Zhou BP, Hung MC, Xie X. POMC maintains tumor-initiating properties of tumor tissue-derived long-term-cultured breast cancer stem cells. Int J Cancer. 2017;140:2517-25
4. Kreso A, Dick JE. Evolution of the cancer stem cell model. Cell Stem Cell. 2014;14:275-91
5. Choi YJ, Lin CP, Risso D, Chen S, Kim TA, Tan MH. et al. Deficiency of microRNA miR-34a expands cell fate potential in pluripotent stem cells. Science. 2017;355:1927-47
6. Miki K, Endo K, Takahashi S, Funakoshi S, Takei I, Katayama S. et al. Efficient Detection and Purification of Cell Populations Using Synthetic MicroRNA Switches. Cell Stem Cell. 2015;16:699-711
7. Lu X, Li X, He Q, Gao J, Gao Y, Liu B. et al. miR-142-3p regulates the formation and differentiation of hematopoietic stem cells in vertebrates. Cell Res. 2013;23:1356-68
8. Mitra AK, Agrahari V, Mandal A, Cholkar K, Natarajan C, Shah S. et al. Novel delivery approaches for cancer therapeutics. J Control Release. 2015;219:248-68
9. Meacham CE, Morrison SJ. Tumour heterogeneity and cancer cell plasticity. Nature. 2013;501:328-37
10. Magee JA, Piskounova E, Morrison SJ. Cancer stem cells: impact, heterogeneity, and uncertainty. Cancer Cell. 2012;21:283-96
11. He Q, Guo S, Qian Z, Chen X. Development of individualized anti-metastasis strategies by engineering nanomedicines. Chem Soc Rev. 2015;44:6258-86
12. Cullis PR, Hope MJ. Lipid Nanoparticle Systems for Enabling Gene Therapies. Mol Ther. 2017;25:1467-75
13. Xie X, Xia W, Li Z, Kuo HP, Liu Y, Li Z. et al. Targeted expression of BikDD eradicates pancreatic tumors in noninvasive imaging models. Cancer Cell. 2007;12:52-65
14. Sher YP, Tzeng TF, Kan SF, Hsu J, Xie X, Han Z. et al. Cancer targeted gene therapy of BikDD inhibits orthotopic lung cancer growth and improves long-term survival. Oncogene. 2009;28:3286-95
15. Xie X, Hsu JL, Choi MG, Xia W, Yamaguchi H, Chen CT. et al. A novel hTERT promoter-driven E1A therapeutic for ovarian cancer. Mol Cancer Ther. 2009;8:2375-82
16. Li LY, Dai HY, Yeh FL, Kan SF, Lang J, Hsu JL. et al. Targeted hepatocellular carcinoma proapoptotic BikDD gene therapy. Oncogene. 2011;30:1773-83
17. Dai HY, Chen HY, Lai WC, Hung MC, Li LY. Targeted expression of BikDD combined with metronomic doxorubicin induces synergistic antitumor effect through Bax activation in hepatocellular carcinoma. Oncotarget. 2015;6:23807-19
18. Xie X, Kong Y, Tang H, Yang L, Hsu JL, Hung MC. Targeted BikDD expression kills androgen-dependent and castration-resistant prostate cancer cells. Mol Cancer Ther. 2014;13:1813-25
19. Lang JY, Hsu JL, Meric-Bernstam F, Chang CJ, Wang Q, Bao Y. et al. BikDD eliminates breast cancer initiating cells and synergizes with lapatinib for breast cancer treatment. Cancer Cell. 2011;20:341-56
20. Xie X, Tang H, Liu P, Kong Y, Wu M, Xiao X. et al. Development of PEA-15 using a potent non-viral vector for therapeutic application in breast cancer. Cancer Lett. 2015;356:374-81
21. Xie X, Li L, Xiao X, Guo J, Kong Y, Wu M. et al. Targeted expression of BikDD eliminates breast cancer with virtually no toxicity in noninvasive imaging models. Mol Cancer Ther. 2012;11:1915-24
22. Li L, Xie X, Luo J, Liu M, Xi S, Guo J. et al. Targeted expression of miR-34a using the T-VISA system suppresses breast cancer cell growth and invasion. Mol Ther. 2012;20:2326-34
23. Rahal OM, Nie L, Chan LC, Li CW, Hsu YH, Hsu J. et al. Selective expression of constitutively active pro-apoptotic protein BikDD gene in primary mammary tumors inhibits tumor growth and reduces tumor initiating cells. Am J Cancer Res. 2015;5:3624-34
24. Heath RJ, Leong JM, Visegrady B, Machesky LM, Xavier RJ. Bacterial and host determinants of MAL activation upon EPEC infection: the roles of Tir, ABRA, and FLRT3. PLoS Pathog. 2011;7:e1001332-46
25. Ray A, Zhang S, Rentas C, Caldwell KA, Caldwell GA. RTCB-1 mediates neuroprotection via XBP-1 mRNA splicing in the unfolded protein response pathway. J Neurosci. 2014;34:16076-85
26. Schlaeger TM, Daheron L, Brickler TR, Entwisle S, Chan K, Cianci A. et al. A comparison of non-integrating reprogramming methods. Nat Biotechnol. 2015;33:58-63
27. Elsabahy M, Wooley KL. Cytokines as biomarkers of nanoparticle immunotoxicity. Chem Soc Rev. 2013;42:5552-76
28. Rokavec M, Li H, Jiang L, Hermeking H. The p53/miR-34 axis in development and disease. J Mol Cell Biol. 2014;6:214-30
29. Murray C, Pao E, Tseng P, Aftab S, Kulkarni R, Rettig M. et al. Quantitative Magnetic Separation of Particles and Cells Using Gradient Magnetic Ratcheting. Small. 2016;12:1891-9
30. Greve B, Kelsch R, Spaniol K, Eich HT, Gotte M. Flow cytometry in cancer stem cell analysis and separation. Cytometry A. 2012;81:284-93
31. Qureshi-Baig K, Ullmann P, Haan S, Letellier E. Tumor-Initiating Cells: a criTICal review of isolation approaches and new challenges in targeting strategies. Mol Cancer. 2017;16:40-55
32. Cao Y, Huschtscha LI, Nouwens AS, Pickett HA, Neumann AA, Chang AC. et al. Amplification of telomerase reverse transcriptase gene in human mammary epithelial cells with limiting telomerase RNA expression levels. Cancer Res. 2008;68:3115-23
33. Günes C, Rudolph KL. The role of telomeres in stem cells and cancer. Cell. 2013;152:390-3
34. Chiba K, Johnson JZ, Vogan JM, Wagner T, Boyle JM. Cancer-associated TERT promoter mutations abrogate telomerase silencing. Elife. 2015;4:e07918-37
35. Clarke MF, Dick JE, Dirks PB, Eaves CJ, Jamieson CH, Jones DL. et al. Cancer stem cells-perspectives on current status and future directions: AACR Workshop on cancer stem cells. Cancer Res. 2006;66:9339-44
36. Kim KD, Zhao J, Auh S, Yang X, Du P, Tang H. et al. Adaptive immune cells temper initial innate responses. Nat Med. 2007;13:1248-52
37. Sotiropoulou PA, Christodoulou MS, Silvani A, Herold-Mende C, Passarella D. Chemical approaches to targeting drug resistance in cancer stem cells. Drug Discov Today. 2014;19:1547-62
38. Li H, Rokavec M, Jiang L, Horst D, Hermeking H. Antagonistic Effects of p53 and HIF1A on microRNA-34a Regulation of PPP1R11 and STAT3 and Hypoxia-induced Epithelial to Mesenchymal Transition in Colorectal Cancer Cells. Gastroenterology. 2017;153:505-20
39. Popow J, Englert M, Weitzer S, Schleiffer A, Mierzwa B, Mechtler K. et al. HSPC117 is the essential subunit of a human tRNA splicing ligase complex. Science. 2011;331:760-4
40. Hu J, Teng J, Ding N, He M, Sun Y, Yu AC. et al. FAAP, a novel murine protein, is involved in cell adhesion through regulating vinculin-paxillin association. Front Biosci. 2008;13:7123-31
41. de Hoog CL, Foster LJ, Mann M. RNA and RNA binding proteins participate in early stages of cell spreading through spreading initiation centers. Cell. 2004;117:649-62
42. Popow J, Jurkin J, Schleiffer A, Martinez J. Analysis of orthologous groups reveals archease and DDX1 as tRNA splicing factors. Nature. 2014;511:104-7
43. Tanaka N, Meineke B, Shuman S. RtcB, a novel RNA ligase, can catalyze tRNA splicing and HAC1 mRNA splicing in vivo. J Biol Chem. 2011;286:30253-7
Author contact
![]() Corresponding author: Xiaoming Xie, MD, PhD, Department of Breast Oncology, Sun Yat-sen University Cancer Center, Guangzhou 510060, China, Tel.: 86-20-87343805; Fax: 86-20-38320368; E-mail: xiexiaoming001com
Corresponding author: Xiaoming Xie, MD, PhD, Department of Breast Oncology, Sun Yat-sen University Cancer Center, Guangzhou 510060, China, Tel.: 86-20-87343805; Fax: 86-20-38320368; E-mail: xiexiaoming001com