Theranostics
13.3
Impact Factor
- Current Issue
- Advance Articles
- Volume 16; 2026
- Volume 15; 2025
- Volume 14; 2024
- Volume 13; 2023
- Volume 12; 2022
- Archive
- Cover Images
- Cover Suggestion
- Special Issues
Top
Introduction
Results
Discussion
Materials and Methods
Abbreviations
Acknowledgements
Supplementary Material
References
Introduction
Results
Discussion
Materials and Methods
Abbreviations
Acknowledgements
Supplementary Material
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2018; 8(3):676-692. doi:10.7150/thno.21463 This issue Cite
Research Paper
AKR1C1 Activates STAT3 to Promote the Metastasis of Non-Small Cell Lung Cancer
Hong ZHU*, Lin-Lin CHANG*, Fang-Jie YAN, Yan HU, Chen-Ming ZENG, Tian-Yi ZHOU, Tao YUAN, Mei-Dan YING, Ji CAO, Qiao-Jun HE ![]() , Bo YANG
, Bo YANG ![]()
Zhejiang Province Key Laboratory of Anti-Cancer Drug Research, College of Pharmaceutical Sciences, Zhejiang University, Hangzhou China
* These authors contributed equally to this study.
Received 2017-6-14; Accepted 2017-10-23; Published 2018-1-1
Citation:
ZHU H, CHANG LL, YAN FJ, HU Y, ZENG CM, ZHOU TY, YUAN T, YING MD, CAO J, HE QJ, YANG B. AKR1C1 Activates STAT3 to Promote the Metastasis of Non-Small Cell Lung Cancer. Theranostics 2018; 8(3):676-692. doi:10.7150/thno.21463. https://www.thno.org/v08p0676.htm
Other stylesAbstract

Metastasis is the leading cause of mortality for human non-small cell lung cancer (NSCLC). However, it is difficult to target tumor metastasis because the molecular mechanisms underlying NSCLC invasion and migration remain unclear. Methods: GEO data analyses and IHC analyses were performed to identify that the expression level of AKR1C1, a member of human aldo-keto reductase family, was highly elevated in patients with metastasis or metastatic foci of NSCLC patients. Functional analyses (in vitro and in vivo) and quantitative genomic analyses were preformed to confirm the pro-metastatic effects of AKR1C1 and the underlying mechanisms. The correlation of AKR1C1 with the prognosis of NSCLC patients was evaluated using Kaplan-Meier analyses. Results: in NSCLC patients, AKR1C1 expression was closely correlated with the metastatic potential of tumors. AKR1C1 overexpression in nonmetastatic cancer cells significantly promoted metastasis both in vitro and in vivo, whereas depletion of AKR1C1 in highly metastatic tumors potently alleviated these effects. Quantitative genomic and functional analyses revealed that AKR1C1 directly interacted with STAT3 and facilitated its phosphorylation—thus reinforcing the binding of STAT3 to the promoter regions of target genes—and then transactivated these genes, which ultimately promoted tumor metastasis. Further studies showed that AKR1C1 might facilitate the interaction of STAT3 with its upstream kinase JAK2. Intriguingly, AKR1C1 exerted these pro-metastatic effects in a catalytic-independent manner. In addition, a significant correlation between AKR1C1 and STAT3 pathway was observed in the metastatic foci of NSCLC patients, and the AKR1C1-STAT3 levels were highly correlated with a poor prognosis in NSCLC patients. Conclusions: taken together, we show that AKR1C1 is a potent inducer of NSCLC metastasis. Our study uncovers the active function of AKR1C1 as a key component of the STAT3 pathway, which promotes lung cancer metastasis, and highlights a candidate therapeutic target to potentially improve the survival of NSCLC patients with metastatic disease.
Keywords: AKR1C1, Metastasis, Non-Small Cell Lung Cancer, STAT3.
Introduction
Non-small cell lung cancer (NSCLC) is the leading cause of cancer death worldwide [1], accounting for ~80% of all lung cancer cases [2]. Despite remarkable advances in targeted therapy to treat NSCLC patients, survival rates have not improved. >79% of all lung cancer patients develop metastases, and the 5-year survival rate of patients with distant metastases is <5% [3], which means that metastases account for fatal outcomes and high mortality rates in NSCLC [4, 5]. Thus, there is an urgent need to improve our understanding of the molecular underpinnings that drive NSCLC metastasis and to leverage that understanding toward better therapeutic options.
AKR1C1 (aldo-keto reductase 1, family member, C1) is a member of the human aldo-keto reductase family that is composed of four enzymes, AKR1C1-AKR1C4, which catalyze NADPH-dependent reductions [6, 7]. Mounting evidence reveals that AKR1C1 is upregulated in a variety of cancers, including lung cancer, gastric cancer, and cervical cancer [8-13], and the ectopic expression of AKR1C1 could lead to malignant transformations in NIH-3T3 cells [14]. The overexpression of AKR1C1 has been found to be associated with cancer progression [14-16]. For instance, AKR1C1 overexpression may result in drug resistance in different cancer types, including leukemia, NSCLC and bladder cancers, due to its critical role in biosynthesis, intermediary metabolism and detoxification [15]. In the bronchial epithelial cells of tobacco smokers, AKR1C1 was found to be overexpressed [17]. Since AKR1C1 and its family are responsible for the metabolic activation of polycyclic aromatic hydrocarbons, which are lung carcinogens, the induction of AKR1C1 in tobacco-exposed lung tissues might contribute to the pathogenesis of smoking-related lung cancer [18]. However, the mechanism by which AKR1C1 promotes cancer metastasis remains to be fully understood.
As the best known function of AKR1C1 is to convert progesterone into its inactive form, 20-alpha-dihydroxyprogesterone [19, 20], AKR1C1 was found to be aberrantly expressed in endometrial cancers [16, 20, 21] and contributed to the inhibition of progestin receptor production in cells [16]. The loss of progestin receptor was often found to be inversely correlated to an invasive phenotype in endometrial cancers [22]. These studies suggest a critical role for the canonical function of AKR1C1 in the aggression of hormone-related cancers. However, AKR1C1 is extensively overexpressed in non-hormone-related cancer, particularly in NSCLC, thus suggesting a non-enzymatic role for AKR1C1 in NSCLC progression.
Recent evidence highlights the importance of non-canonical functions of reductase because the inhibition of enzymatic activities of AKR1Cs fail to kill cancer cells efficiently [15]; and, some of the family members, such as AKR1C3, exhibit a catalytic-independent ability to bind and stabilize E3 ligase Siah2 by blocking its self-ubiquitination and degradation [23], which may ultimately promote tumor progression [24, 25]. In this context, it is intriguing to ask whether and how AKR1C1 functions as a pro-metastatic factor in NSCLC cells in a non-canonical manner.
Here, we show that AKR1C1 promotes the metastasis of NSCLC cells in vitro and in vivo in a catalytic-independent manner because the loss of reductase activity fails to impair the pro-metastatic activity of AKR1C1. Quantitative genomic and functional analyses indicate that AKR1C1 acts as a key component of the STAT3 pathway, which promotes NSCLC metastasis, by facilitating STAT3 phosphorylation and reinforcing the transactivation of STAT3 target genes.
Results
High AKR1C1 expression is associated with metastasis and poor prognosis in NSCLC patients
According to the GEO datasets (GDS2865 and GDS3091) [26, 27], which display the expression profiles of primary tumors vs. metastatic tumors, the AKR1C1 mRNA levels were markedly upregulated in metastatic tumor samples (Fig. S1A). This finding was supported by our previous study, which showed that AKR1C1 was significantly higher in a metastatic subline established from a step-wise enrichment of cancer cells with a higher capacity to migrate (unpublished data). Thus, we were encouraged to speculate that AKR1C1 might play critical roles in the metastasis and progression of NSCLC.
To determine the clinical relevance of AKR1C1 expression in patients with NSCLC, we first analyzed the expression of AKR1C1 in cancer patient tumor samples [28]. It was found that most cancer samples harbored a higher level of AKR1C1 than corresponding normal tissues. Notably, among all cancer types, NSCLC samples displayed the greatest upregulation of AKR1C1 (Fig. 1A and Fig. S1B) [28]. In particular, as shown in the Yamagata Lung Dataset [29] (Fig. 1A, B), the AKR1C1 mRNA levels were increased in cancer compared to normal tissues by 24.36-fold in squamous cell carcinoma (P = 7.7×10-7), 6.59-fold in large cell carcinoma (P = 0.018) and 4.95-fold in adenocarcinoma (P = 0.004). Similarly, we found that the protein level of AKR1C1 was distinctly high in 4 of 6 tested NSCLC cell lines (PC-9, A549, SK-MES-1, NCI-H460), but that it was barely detectable in BEAS-2B, a human bronchial epithelial cell line (Fig. S2A), which suggests that AKR1C1 is highly expressed in NSCLC.
NSCLC cells harboring higher levels of AKR1C1 traversed more efficiently into the lower chamber than did those with lower AKR1C1 levels (Fig. S2B). To further identify the relationship between AKR1C1 and NSCLC metastasis, we evaluated the expression level of AKR1C1 in 40 primary NSCLC tumors with corresponding lymph node metastatic tumors from the same patients (6 small cell undifferentiated carcinoma, 17 squamous cell carcinoma and 17 adenocarcinoma). Interestingly, AKR1C1 expression in metastatic tumors was significantly elevated compared to the corresponding primary tumors (Fig. 1C), suggesting that high AKR1C1 expression is associated with NSCLC metastasis.
Because there is a high degree of sequence similarity shared between AKR1C1-3, we next compared the pro-metastatic activity of AKR1C1 with its family members AKR1C2 and AKR1C3. Using a Kaplan-Meier plotter analysis [30], we found that the expression level of AKR1C1 was inversely correlated with reduced survival time in NSCLC patients (P < 0.01). In contrast, the expression levels of AKR1C2 and AKR1C3 were not related to survival time (P > 0.05) (Fig. 2A), indicating that the function of AKR1C1 in metastasis is specific. In line with these findings, AKR1C1 stood out among the three members of the AKR1C family for its potent ability to promote the invasion of NCI-H1299 cells (Fig. 2B, C). Collectively, these findings suggest that compared to the other 2 members of the AKR1C family, AKR1C1 is closely correlated with a poor prognosis in NSCLC patients and may play important roles in the metastasis and progression of NSCLC.
Figure 1
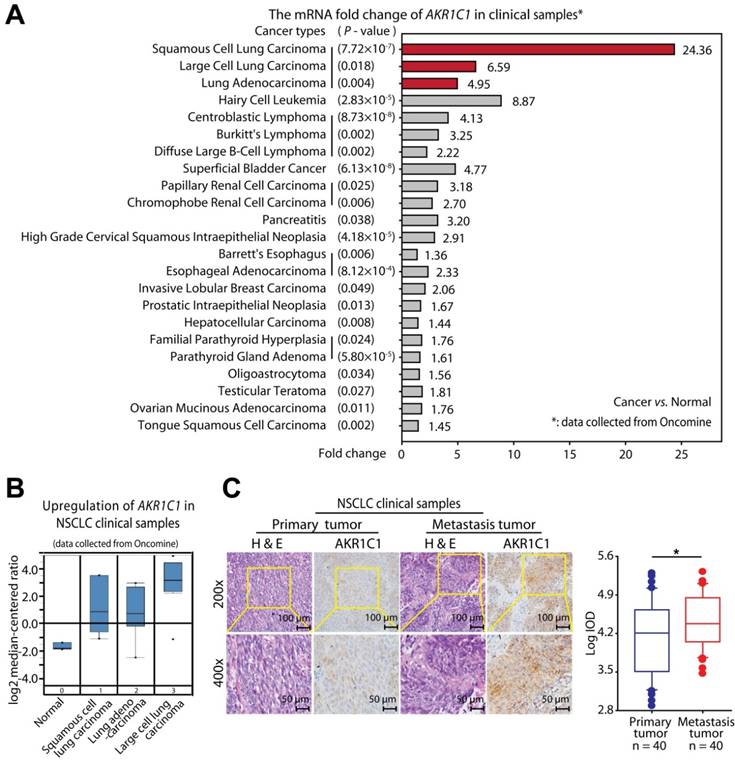
High expression levels of AKR1C1 were observed in NSCLC clinical samples, particularly in metastatic tumors. (A) NSCLC patient tumor samples displayed the highest upregulation of AKR1C1 among different cancer types [28]. Gene: AKR1C1; Analysis Type: Cancer vs. Normal Analysis; Data Type: mRNA; Sample Type: Clinical Specimen. (B) Box plot analysis of the AKR1C1 mRNA levels in NSCLC tissue samples [29]. Gene: AKR1C1; Analysis Type: Cancer vs. Normal Analysis; Data Type: mRNA; Sample Type: Clinical Specimen; Yamagata Lung. (C) Immunohistochemical staining showed that the expression levels of AKR1C1 were significantly upregulated in metastatic tumors compared with corresponding primary tumors obtained from the same NSCLC patient (n = 40, P < 0.05). The expression scores of AKR1C1 in tissue microarrays were analyzed by Image-Pro Plus. (Primary tumor tissue: n = 40; matched lymph node metastatic tumor tissue: n = 40). Statistical significance was determined by Student's test. *, P < 0.05.
Figure 2
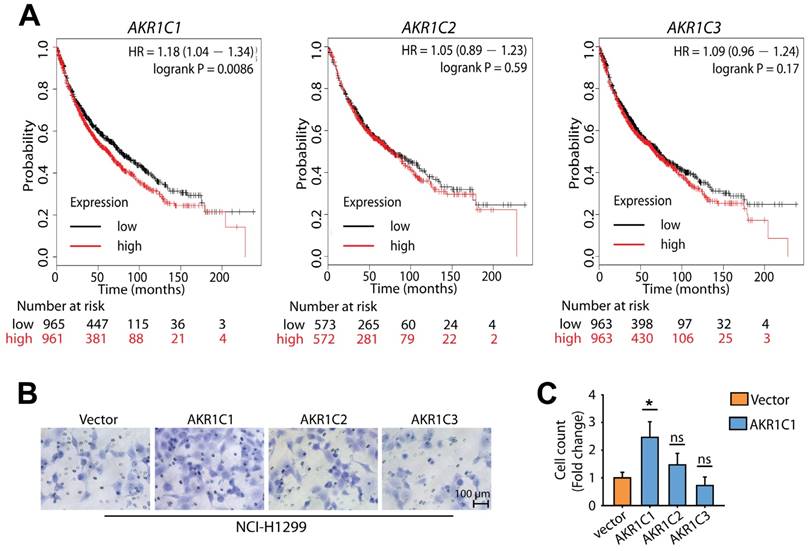
AKR1C1 possesses more potential to promote NSCLC malignancy, compared with AKR1C2 and AKR1C3. (A) The effects of AKR1C1, AKR1C2 and AKR1C3 on the overall survival of NSCLC patients [30]. (B) A transwell assay was conducted to evaluate the motility of NCI-H1299 overexpressing AKR1C1, AKR1C2 or AKR1C3, respectively. AKR1C1 stood out for its superior potential to promote cell migration. (C) Statistical significance of cell migration in (B) was determined by a two-tailed, Student's test. *, P < 0.05.
AKR1C1 drives NSCLC cell metastatic potential both in vitro and in vivo
To further address the effect of AKR1C1 on NSCLC cell metastasis, we evaluated the effects of manipulating the AKR1C1 expression levels. The exogenous introduction of wide-type (wt) AKR1C1 into those non-metastatic cells harboring low AKR1C1 expression levels (Fig. S2A) conferred NCI-H1650 and NCI-H1299 cells with significantly enhanced metastatic potential as indicated by their enhanced invasive ability (Fig. 3A). In contrast, the depletion of AKR1C1 using specific siRNA without silencing effect on AKR1C2 or AKR1C3 (Fig. S3) greatly reduced the invasive abilities of metastatic PC-9 and NCI-H460 cells (Fig. 3A) harboring higher AKR1C1 expression (Fig. S2A). Similar results were also achieved in A549 cells, which are other NSCLC cells with high AKR1C1 expression (Fig. S2C, D). The results obtained from migration assay and wound-healing assay further supported the potent metastatic driving function of AKR1C1 (Fig. S2E, F). Therefore, AKR1C1 may promote the metastatic ability of NSCLC cells in vitro.
Since transcriptional regulation is an essential component of tumor metastasis, the mRNA levels of metastasis-related genes, including Fibronectin and N-cadherin, were monitored. As expected, the exogenous introduction of AKR1C1 in NCI-H1650 cells increased the mRNA levels of Fibronectin and N-cadherin, whereas these genes in PC-9 cells were downregulated by AKR1C1 depletion (Fig. 3B). These data suggest that AKR1C1 might reprogram the transcription profiles of metastasis-related genes in NSCLC cells.
To extend our in vitro observations concerning AKR1C1's role in promoting NSCLC metastasis, we evaluated whether AKR1C1 could regulate metastasis in vivo. First, we stably overexpressed AKR1C1 in NCI-H1299 cells (cells harboring low AKR1C1 expression), which conferred NCI-H1299 cells with an invasive phenotype (Fig. S4A). Subsequently, these cells were intravenously injected into nude mice. Compared with the parental NCI-H1299 cells, AKR1C1 transfectants produced significantly increased metastatic foci in the livers of nude mice, as detected by PET-CT (Fig. 3C, D). Intriguingly, on lung tissues, minimal difference was observed in the foci numbers between the vector- and AKR1C1-groups (Fig. S5A), whereas the total foci numbers from both lung and liver were greatly increased by AKR1C1 transfectants (Fig. S5B). Taken together, these data suggested that AKR1C1 overexpression promotes metastasis of NSCLC in vivo, while imposing little effect on the lung homing.
Figure 3
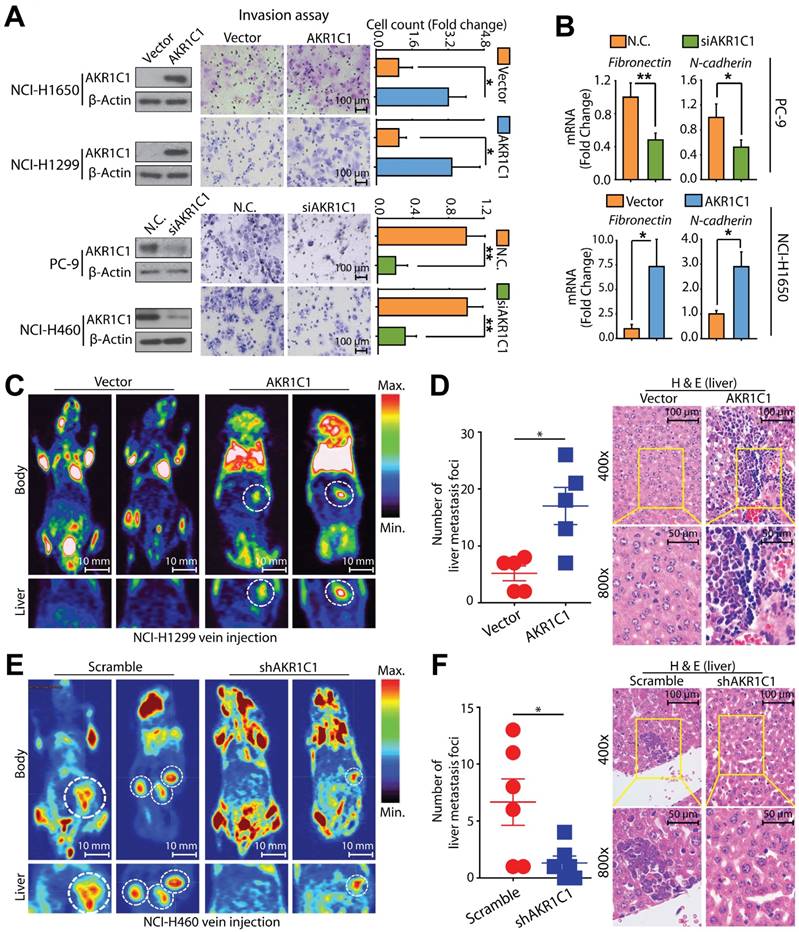
AKR1C1 promotes NSCLC invasion both in vitro and in vivo. (A) The invasion assay showed different cell motilities in AKR1C1 ectopically expressed or silenced NSCLC cells. The ectopic expression of AKR1C1 promoted the invasion of NCI-1650 and NCI-H1299 cells. Conversely, the depletion of AKR1C1 clearly inhibited the invasion of PC-9 and NCI-H460 cells. The relative numbers of invasive cells were counted and presented as the mean ± SD from three independent experiments. (B) The mRNA expression of metastasis-related genes with AKR1C1 ectopic expression or depletion was evaluated by qRT-PCR. (C-D) Stably over-expressed AKR1C1 in NCI-H1299 cells promoted liver metastasis in nude mice models. (E-F) Stable silencing of AKR1C1 reduced the capacity of NCI-H460 cells to form liver metastases. Micro PET-CT was utilized to assess metastatic potential (C&E). Representative micrographs with metastatic nodules were shown by hematoxylin and eosin staining, and the number of metastatic nodules was counted under a microscope (D&F). Statistical significance was determined by Student's t-test. *, P < 0.05; **, P < 0.01, ***, P < 0.001.
We further assessed the metastatic potential of AKR1C1-depleted NCI-H460 cells (cells harboring higher AKR1C1 expression) in nude mice. As expected, compared to the scramble group, there was a significant reduction in liver metastasis detected in the AKR1C1-depleted cell group in nude mice (Fig. 3E, F). Similarly, the cell mobility of NCI-H460 cells was also impaired by depleting AKR1C1 using shRNA (Fig. S4B) in vitro. In addition, neither the overexpression of AKR1C1 in NCI-H1299 cells nor the depletion of AKR1C1 in NCI-H460 cells imposed any effects on cell proliferation under our experimental conditions (Fig. S4A, B). These data indicate that AKR1C1 is important for the aggressive, metastatic phenotype of NSCLC cells in vivo. Taken together, the AKR1C1 overexpression or depletion results obtained from in vitro and in vivo studies and the findings from NSCLC patient biopsies demonstrate the sufficient and necessary roles of AKR1C1 in driving the metastatic potential of NSCLC cells; further, the transcription profile of metastasis-related genes was reprogrammed by AKR1C1 manipulation.
AKR1C1 increases the transcriptional activity of STAT3 by reinforcing its phosphorylation levels
The data reveal that AKR1C1 may modulate the mRNA levels of metastasis-related genes (Fig. 3B); therefore, it is interesting to know how AKR1C1 reprograms the transcription profile of these genes. A microarray analysis was conducted to gain specific insight into the AKR1C1-modulated signaling pathway(s), which led to the shift in the transcriptional profiles and facilitated NSCLC cells to metastasize. Annotation cluster analyses showed that when AKR1C1 was depleted by siRNAs, those genes associated with nucleus function, transcription regulation and DNA binding were significantly downregulated with an enrichment score of 4.63 (Fig. S6A); simultaneously, phosphoprotein was highlighted by functional annotation analyses with a p-value of 3.8×10-44 (Fig. S6B). More interestingly, Signal Transducer and Activator of Transcription 3 (STAT3) was among the 35 most significantly downregulated transcription factors and one of the top decreased genes belonging to phosphoproteins following overlay analysis (Fig. S6C). As a transcriptional factor, STAT3 controls the invasive and aggressive phenotype of cancer cells [31-33], and we consistently found that a large number of metastasis-related genes that were downregulated by AKR1C1 siRNA were tightly regulated by STAT3 (Fig. 4A). qRT-PCR analyses were further utilized to confirm the reduction of the mRNA levels of a variety of STAT3 target genes, including ZEB1, CCND1 and MCL1 [34], in AKR1C1-depleted cells (Fig. 4B).
To further identify the effects of AKR1C1 on STAT3 target genes, particularly those that are closely related to tumor metastasis [34], we monitored the mRNA levels of several metastasis-related STAT3 target genes, such as MMP2, TWIST, and SOX2. AKR1C1 knockdown greatly impeded the mRNA expression of these genes in metastatic PC-9 and NCI-H460 (Fig. 4C). In contrast, the ectopic expression of AKR1C1 could markedly upregulate MMP2, TWIST, and SOX2 in non-metastatic NCI-H1650 and NCI-H1299 cells (Fig. 4D), suggesting that AKR1C1 increased the transcriptional activity of STAT3.
We next asked whether AKR1C1 could affect STAT3 phosphorylation levels because phosphorylation modification is indispensable for the transcriptional activity of this factor [35]. Fig. 4E shows that constitutive p-STAT3 was markedly impaired by AKR1C1 knockdown in a variety of NSCLC cells harboring higher levels of AKR1C1, including in the NCI-H460, A549, PC-9 and SK-MES-1 cell lines. Moreover, the inducible phosphorylation of STAT3 by IL-6 stimulation was attenuated by AKR1C1 depletion (Fig. 4F). Inversely, an enhanced induction of p-STAT3 by IL-6 was also achieved in NCI-H1299 cells with stably overexpressed AKR1C1 (Fig. S4C). These findings indicate AKR1C1 plays an indispensable role in the hyperactivation of the STAT3 pathway and is frequently observed in aggressive NSCLC cells.
AKR1C1 enhances the association of STAT3 to its target genes by directly interacting with STAT3
As a transcription factor, the nuclear accumulation of phosphorylated STAT3 is critical for its activity. Consequently, we analyzed the p-STAT3 levels in nuclear and cytoplasmic sub-fractions, and found that ectopic AKR1C1 in NCI-H1299 cells greatly reinforces the nuclear p-STAT3 levels upon IL-6 treatment (Fig. 5A and Fig. S7); but that depletion of AKR1C1 in NCI-H460 cells markedly impaired nuclear STAT3 phosphorylation, as evidenced by both cell fraction (Fig. 5B) and immunofluorescence analyses (Fig. 5C).
We considered potential scenarios for AKR1C1-activated STAT3 signaling, including whether AKR1C1 could enhanced the association of STAT3 at the promoter regions of its target genes. To answer this question, we performed ChIP assay, and amplified the DNA fragments pulled down by STAT3 using ChIP-PCR primers for MMP2, TWIST and SOX2, respectively [36-38]. Consistent with previous studies, STAT3 binding to the promoter regions of these tested genes (MMP2, TWIST, SOX2) was increased by IL-6 stimulation (Fig. 5D, E). Importantly, the association of STAT3 with its consensus binding elements locating in the promoter regions of these target genes was greatly enhanced by AKR1C1 overexpression. These findings further unravel the mechanism by which AKR1C1 modulates the function of STAT3, that is, AKR1C1 may induce strong binding of STAT3 to its consensus binding elements and lead to high expression of target genes leading to NSCLC metastasis.
We next speculated that the observed activation of STAT3 caused by AKR1C1 was probably due to a potential interaction between these proteins. To test this hypothesis, immunoprecipitation analyses were introduced. As shown in Fig. 5F, ectopically expressed AKR1C1 and STAT3 were reciprocally precipitated from NCI-H1299 cells overexpressing both proteins, suggesting that AKR1C1 directly interacts with STAT3.
Figure 4
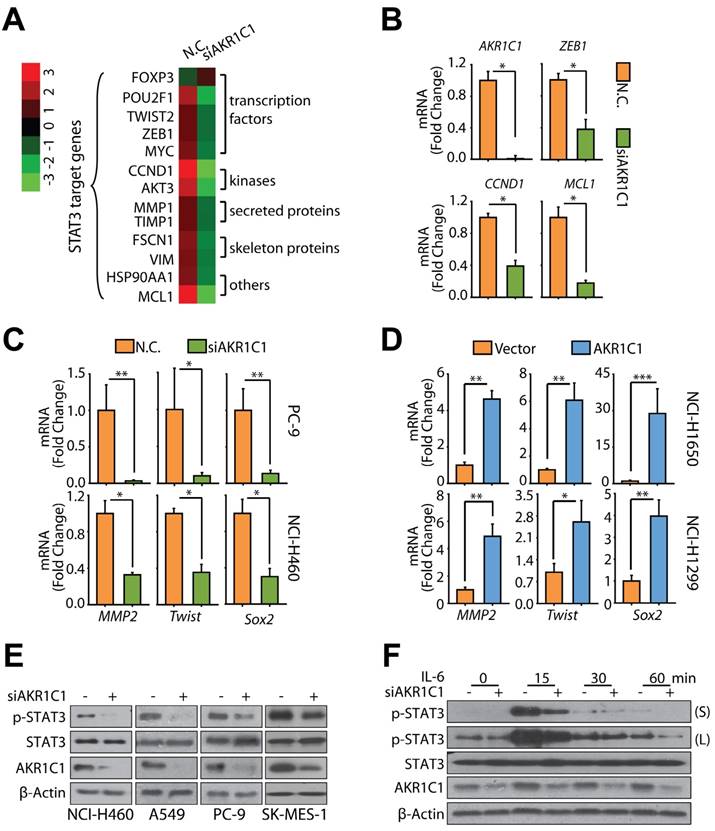
Depletion of AKR1C1 impairs the transcription activity of STAT3. (A) Heat map of expression levels of STAT3 target genes (siAKR1C1 vs. NC). (B) qRT-PCR was used to verify the expression profile of several STAT3 target genes, including HIF1A, ZEB1, CCND1, and MCL1. (C) AKR1C1 depletion downregulated the metastasis-related STAT3 target genes MMP2, Twist and Sox2. (D) Ectopic expression of AKR1C1 induced mRNA expression of MMP2, Twist and Sox2. (E) Knockdown of AKR1C1 decreased the level of p-STAT3. (F) AKR1C1 silence abrogated the inducible level of p-STAT3 treated with IL-6 (20 ng/mL). Statistical significance was determined by Student's t-test. *, P < 0.05; **, P < 0.01, ***, P < 0.001.
Figure 5
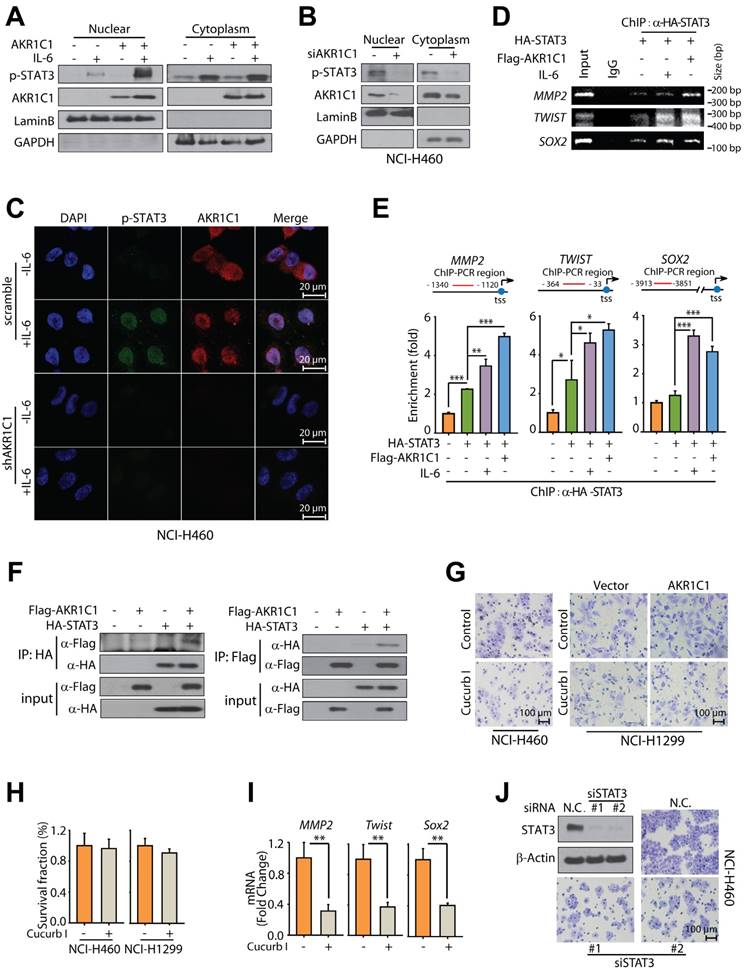
AKR1C1 interacts with STAT3 to promote metastasis. (A) AKR1C1 increases p-STAT3 nuclear translocation. NCI-H1299 cells stably expressing AKR1C1 were treated with IL-6 for 30 min after starving overnight. Cytoplasmic and nuclear fractions from NCI-H1299 cells were separated and subjected to western blot as indicated. The nuclear marker Lamin B and the cytoplasmic marker GAPDH were used to demonstrate the purity of the fractions. (B) AKR1C1 knockdown decreased endogenous p-STAT3 nuclear translocation. NCI-H460 cells were starved overnight with AKR1C1 interference. Cytoplasmic and nuclear proteins from NCI-H460 cells were collected and subjected to western blot as indicated. (C) NCI-H460-scramble and NCI-H460-shAKR1C1 cells were treated with IL-6 (20 ng/mL) for 30 min after overnight starvation, and then the expression and localization of p-STAT3 and AKR1C1 was detected using immunofluorescence staining with antibodies against p-STAT3 and AKR1C1. (D-E) Twenty-four hours post-transfection, ChIP assay was carried out using anti-HA antibody, and irrelevant anti-IgG antibody was introduced as negative control. The protein-chromatin immunoprecipitates were subjected to PCR (D) and qPCR (E) analyses. (D) Input lane, total genomic DNA used as control for the PCR reaction; IgG lane, negative control. (E) Upper: map of the primers used for amplifying STAT3-binding regions of MMP2, TWIST and SOX2; tss: transcription starting site. Bottom: ChIP-qPCR analysis of 293FT cells transfected with STAT3 or/and AKR1C1, using anti-HA antibody and PCR primers. IgG was used as a negative control. Enrichment of HA-STAT3 on the promoter regions of the target genes was calculated. Data are mean ± SD. (F) Immunoblotting analysis of lysates after immunoprecipitation from NCI-H1299 cells transfected with Flag-AKR1C1 and HA-STAT3. (G) Cucurbitacin I potently suppress the migration promoted by AKR1C1. (H) STAT3 inhibitor, Cucurbitacin I imposed little impact on the viability of NCI-H1299 (low AKR1C1 expression) and NCI-H460 (high AKR1C1 expression). (I) qRT-PCR analysis was used to assess the function of STAT3 upon exposure to Cucurbitacin I. (J) STAT3 knockdown suppressed the motility of NCI-H460 (high AKR1C1 expression). Statistical significance was determined by Student's t-test. *, P < 0.05; **, P < 0.01, ***, P < 0.001.
Given the observed interaction of AKR1C1 and STAT3, we sought to test whether the activation of STAT3 is largely responsible for AKR1C1-promoted metastasis. To this end, a specific inhibitor of STAT3, Cucurbitacin I, was employed [39-41]. Fig. 5H shows that minimal loss of cell viability was observed in the presence of Cucurbitacin I. However, under these experimental conditions, Cucurbitacin I exerted robust suppressing effects on the invasion caused by exogenous AKR1C1 in NCI-H1299 cells as well as endogenous AKR1C1 in NCI-H460 cells (Fig. 5G) and reduced the mRNA levels of STAT3 target genes such as MMP2, Twist and Sox2 (Fig. 5I). Furthermore, the employment of 2 different siRNA sequences targeting STAT3 could significantly abolish the invasive capacity of metastatic NCI-H460 cells (Fig. 5J) with high AKR1C1 expression (Fig. S2A). These findings suggest that AKR1C1 interacts with STAT3 to facilitate its phosphorylation and that STAT3 activation plays an indispensable role in AKR1C1-promoted metastasis of NSCLC cells.
AKR1C1 modulates JAK2-STAT3 interaction to drive the metastasis in NSCLC
To explore the mechanisms through which AKR1C1 induced STAT3 phosphorylation, we firstly employed an in vitro kinase assay to test whether AKR1C1 was a protein kinase and recognized STAT3 as a substrate. Human recombinant AKR1C1 was purified and incubated with recombinant STAT3 in a cell-free kinase-reaction system; human recombinant JAK2, the canonical kinase of STAT3, was introduced as a positive control. Figure 6A showed that JAK2 could phosphorylate STAT3 in the cell-free system; in contrast, AKR1C1 (and the other 2 family members) failed to directly phosphorylate recombinant STAT3, excluding the possibility that AKR1C1 functioned as an upstream protein kinase of STAT3.
Figure 6
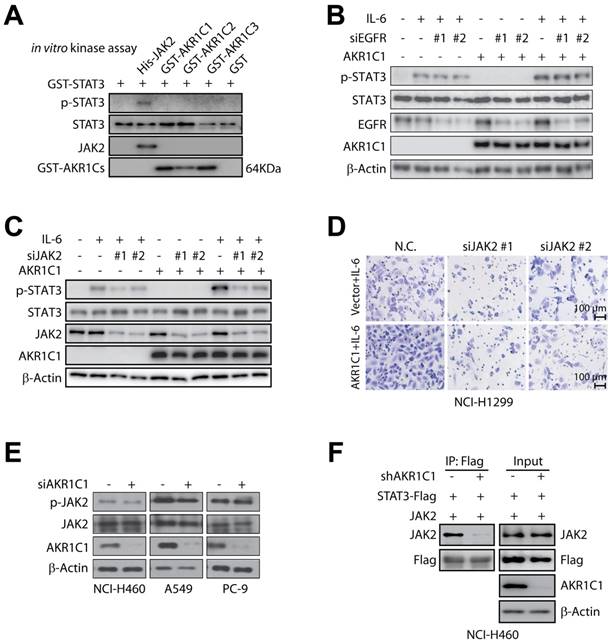
AKR1C1 facilitates the interaction of STAT3 and JAK2 in NSCLC. (A) AKR1C1/2/3 possessed no kinase activity. Purified proteins including His-JAK2, GST-AKR1C1, GST-AKR1C2, GST-AKR1C3 with GST-STAT3 were subjected to in vitro kinase assay, and the phosphorylation level of STAT3 was examined. (B) EGFR deletion had no effect on the p-STAT3 level induced by IL-6 and AKR1C1 in NCI-H1299 cells. (C) JAK2 was required for p-STAT3 induction by AKR1C1 in NCI-H1299 cells. (D) JAK2 depletion suppressed the invasive ability of AKR1C1-overexpressed NCI-H1299 cells. (E) Western blot analysis of the p-JAK2 and JAK2 expression in NSCLC cells with AKR1C1 depletion. (F) AKR1C1 silence abolished the interaction of STAT3 and JAK2 proteins in NCI-H460 cells. NCI-H460-scramble and NCI-H460-shAKR1C1 cells were transfected with STAT3-Flag and JAK2, then harvested for immunoprecipitation using anti-Flag beads, the precipitated JAK2 was detected to indicate the JAK2-STAT3 interaction.
In this context, we then focused on EGFR and JAK2, two of the most important upstream kinases of STAT3 [42, 43], by asking whether AKR1C1-enhanced p-STAT3 would be affected by the suppression of these two kinases. It appeared that EGFR depletion imposed minimal impact on the inducible STAT3 phosphorylation by IL-6 and AKR1C1 (Fig. 6B), whereas the deletion of JAK2 could significantly impair the ability of AKR1C1 to trigger STAT3 phosphorylation (Fig. 6C). In line with this, the invasive ability of AKR1C1-overexpressed NCI-H1299 cells was greatly suppressed by JAK2 depletion (Fig. 6D). These findings indicate that JAK2, but not EGFR, is required for AKR1C1-promoted STAT3 activation and cell migration.
Furthermore, we examined the phosphorylation levels of JAK2, and found that AKR1C1 showed minimal influence on p-JAK2 levels (Fig. 6E). Since JAK2 phosphorylates STAT3 by direct interaction [44, 45], we were thus inspired to ask whether AKR1C1 could affect this interaction. As shown in Fig. 6F, JAK2 could be precipitated by STAT3 in AKR1C1-intact cells, while this JAK2-STAT3 interaction was greatly abrogated by AKR1C1 depletion, implying that AKR1C1 was critical for the interaction of JAK2 and STAT3. Taken together, these results underline the crucial role of JAK2 in AKR1C1-provoked STAT3 activation and metastasis, and suggest that AKR1C1 may facilitate the activation of STAT3 by JAK2 via modulating their interaction.
AKR1C1 promotes NSCLC metastasis in a canonical reductase activity-independent manner
Since AKR1C1 is best known for its aldo-keto reductase activity, we were interested in determining whether the ability to promote metastasis is dependent on its canonical reductase activity. Therefore, we introduced 3-bromo-5-phenylsalicylic acid (5-BPSA), the most potent inhibitor of AKR1C1 enzyme activity [15], and 3 AKR1C1 mutants, E127D, H222I and R304L, with a significant loss of reductase activity [46]. Consistent with a previous study [15], 5-BPSA imposed a potent inhibitory effect against the reductase activity of recombinant AKR1C1 (Fig. 7A). However, the invasion in ectopic AKR1C1-overexpressing NCI-H1299 cells or NCI-H460 cells with high endogenous AKR1C1 was minimally suppressed by 5-BPSA (Fig. 7B, C). Similar to wild-type AKR1C1, the loss of enzymatic activities by AKR1C1 mutants (Fig. 7A) still processed prominent pro-metastatic effects on NSCLC cells (Fig. 7D), suggesting that the enzymatic activity of AKR1C1 is dispensable for its pro-metastatic capability.
In line with the abovementioned findings, AKR1C1 mutants showed a comparable ability to induce STAT3 target genes with that of wild-type AKR1C1 (Fig. 7E). STAT3 transactivates downstream target genes by directly binding the APRE sequence in the promoter region. Therefore, we introduced an APRE-responsive luciferase reporter system to compare the effects on STAT3 transcriptional activities by using AKR1C1 depletion or enzymatic activity inhibition. As displayed in Fig. 7F, AKR1C1 knock-down greatly reduced the luciferase activity induced by STAT3, which remained undisturbed in the 5-BPSA-exposed groups (Fig. 7F). Similar observations were achieved in the STAT3 phosphorylation levels, and 5-BPSA could not reduce the p-STAT3 protein levels as the siAKR1C1 did (Fig. 7G). Thus, these results indicate that the inhibitor of AKR1C1's enzymatic activity did not interfere with STAT3's transcriptional activities.
To further confirm the dispensable roles of the canonical reductase activity in an AKR1C1-driven metastasis, we next examined the interaction of AKR1C1 mutants with STAT3. As shown in Fig. 7H, the ectopically expressed mutants of AKR1C1 (E127D, H222I and R304L) could be precipitated by STAT3 from NCI-H1299 cells, which implies that despite the loss of their enzymatic activities, these mutants could interact with STAT3 and exert similar pro-metastatic effects as the wt AKR1C1. The catalytic-independent roles in STAT3 signaling by AKR1C1 were also supported by the following evidences: 1) the other two family members, AKR1C2 and AKR1C3, share similar catalytic activities [47], but show minimal binding affinity with STAT3 (Fig. 7I); 2) the depletion of AKR1C2 and AKR1C3 impose little effect on STAT3 phosphorylation (Fig. 7J). These results were in line with the above-mentioned data that AKR1C2 and AKR1C3 could not induce the invasive phenotype of NSCLC (Fig. 2), and also support the notion that AKR1C1-primed metastasis is not dependent on canonical enzymatic activity.
AKR1C1-STAT3 signaling pathway is associated with progression and poor prognosis in patients with NSCLC
To determine whether AKR1C1 and STAT3 coordinate to promote NSCLC metastasis in patients, we analyzed the potential correlation between the expression levels of AKR1C1 and the risk factors of NSCLC patients (GSE30219) [48, 49]. The mRNA levels of AKR1C1 and STAT3 target genes (Sox2, MMP9, MMP2 and Twist1), were highly correlated with high risk NSCLC patients (Fig. 8A, B). Those NSCLC patients with hyperactivation of AKR1C1-STAT3 signaling had a significantly shorter survival time (P < 0.01) (Fig. 8C). In addition, we performed an immunohistochemical analysis of the AKR1C1 and p-STAT3 levels on the metastatic tumor tissue samples collected from 40 NSCLC patients. The results in Fig. 8D show that p-STAT3 positively correlated with AKR1C1 expression in metastatic NSCLC tumor tissues (R = 0.52, P < 0.001). These data suggest that an activated AKR1C1-STAT3 signaling pathway correlates with a poor prognosis in NSCLC patients.
Figure 7
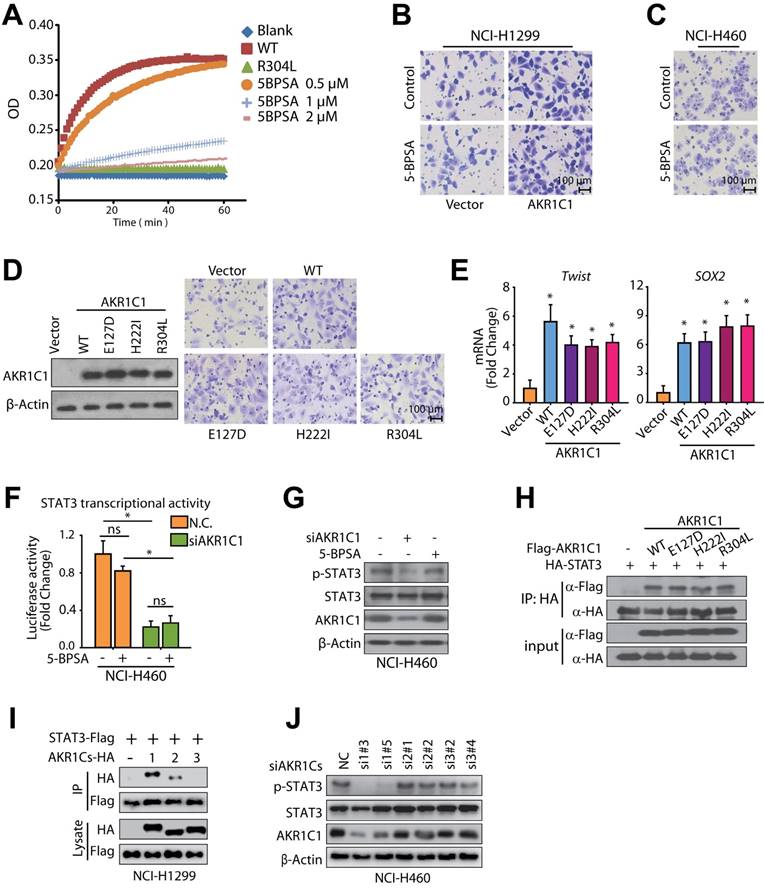
AKR1C1 primes NSCLC metastasis in a catalytic-independent manner. (A) 5-BPSA (the inhibitor of AKR1C1 enzymatic activity) and the mutant R304L both decrease the enzymatic activity of AKR1C1. The reaction mixture consisted of 0.1 M potassium phosphate (pH 6.7), 1 mM S-tetralol, 0.1 mM NADPH and enzyme. (B-C) Cell invasion assay showed that 5-BPSA (20 μM) imposed little impact on the pro-metastasis ability of AKR1C1 in NCI-H1299 (B) and NCI-H460 cells (C). (D) AKR1C1 mutants (E127D, H222I, and R304L) with a loss of enzyme activities possessed comparable pro-metastatic effects as wt AKR1C1, as indicated by invasion assay. (E) qRT-PCR was used to verify the expression profile of STAT3 target genes by AKR1C1 transfection (wide type and enzymatic mutations E127D, H222I, R304L). (F) NCI-H460 cells were transfected with APRE-luciferase gene reporter plasmid, and the impact of AKR1C1 siRNA and/or 5-BPSA (20 μM) on the luciferase activity was calculated. (G) Western blot analysis displayed that 5-BPSA failed to affect the phosphorylation of STAT3, whereas AKR1C1 depletion reduced the level of p-STAT3. (H) The interaction between AKR1C1 and STAT3 was not impaired by the loss of catalytic activity of AKR1C1. WT and mutant AKR1C1 plasmids were cotransfected with HA-STAT3 into NCI-H1299 cells as indicated, and immunoprecipitation assay were carried out. (I) STAT3-AKR1C1/2/3 interactions were also assessed by co-immunoprecipitation in NCI-H1299 cells. (J) NCI-H460 cells were interfered through two different specific targeting sequences for AKR1C1/2/3. p-STAT3 levels were evaluated by Western blot. Statistical significance was determined by Student's t-test. *, P < 0.05.
Figure 8
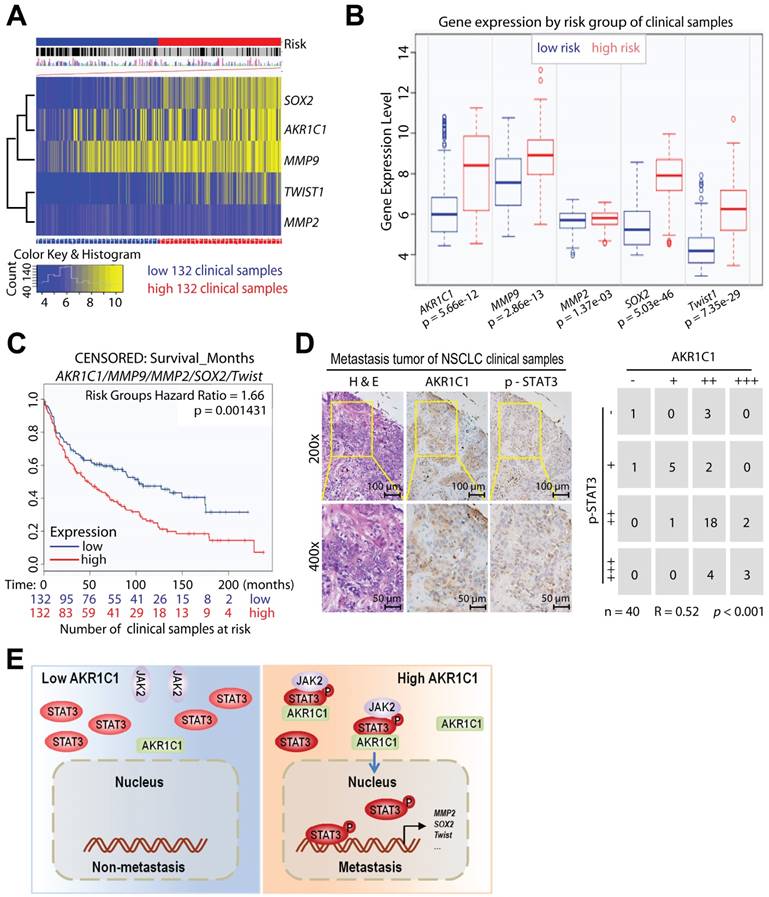
AKR1C1-STAT3 signaling pathway correlates with NSCLC patients' survival and prognosis. The data from (A-C) were obtained from the bioinformatica database (GSE30219) [49]. (A) Heat map of AKR1C1-STAT3 mRNA expression in lung cancer patient tumor samples along with the risk factor (high- and low-risk groups were split into the same size, depending on the Prognostic Index (risk score) estimated by beta coefficients multiplied by gene expression values.) (B) Gene expression levels of AKR1C1-STAT3 in the low- (n = 132) and high- (n = 132) risk groups of lung cancer patients. (C) The relapse-free survival curve of lung cancer clinical patients with low (n = 132) and high (n = 132) levels of AKR1C1-STAT3 expression. (D) The correlation analysis of AKR1C1 and p-STAT3 expression in metastatic tumors (n = 40). The expression scores of AKR1C1 and p-STAT3 in the tissue microarrays that were analyzed by Image-Pro Plus. (E) The working model for the pro-metastatic effects by AKR1C1 through activating STAT3 in NSCLC cells.
Discussion
Interference from metastases may be a promising alternative to conventional chemotherapy for NSCLC, as most patients diagnosed with distant metastasis succumb to a severely decreased survival rate. Increasing evidence has shown that AKR1C1 is frequently expressed in many cancer types including cervical cancer [10], gastric cancer [11], and endometrial cancer [50]; NSCLC harbors the most overexpression of AKR1C1 among all cancer types (Fig. 1A). Liu found that exposure to PM2.5 extract on bronchial epithelial cells resulted in the aberrant upregulation of AKR1C1 and may lead to malignant transformation [51]. In addition, AKR1C1/1C2 in NSCLC cells induced chemoresistance through the protein-kinase-C pathway [52]. However, a comprehensive mechanism for AKR1C1-mediated NSCLC progression has remained largely elusive; in particular, whether AKR1C1 can promote NSCLC metastasis has yet to be explored. In the present study, we found that NSCLC cells that harbor higher levels of AKR1C1 are conferred with an enhanced invasive ability in vitro, resulting in significantly increased metastatic foci in vivo. Conversely, the depletion of AKR1C1 by siRNA/shRNA impeded in vitro invasion and ameliorated in vivo metastasis in NSCLC cells (Fig. 3). In addition, the expression of AKR1C1 in NSCLC tumors was positively correlated with increased metastasis and reduced survival time in NSCLC patients (Fig. 1, 2 and 8). Thus, our findings establish, for the first time, a causal link between AKR1C1 and NSCLC metastasis, which may provide a potential target that interferes with malignant metastases.
We were particularly interested in exploring the potential pro-metastatic roles of AKR1C1 because this gene was highly overexpressed in our previously enriched metastatic subline. In addition, according to the GEO dataset displaying the expression profiling of primary tumors vs. metastatic tumors, AKR1C1 was significantly upregulated in metastatic samples (Fig. S1A). When we were preparing the current manuscript, Matsumoto reported a similar phenomenon that AKR1C1 is enhanced in three metastatic bladder tumor sublines compared with a primary UM-UC-3-bladder cell line [53]. And Tian Hu et al. found that high expression of AKR1C1 is associated with proliferation and migration of small-cell lung cancer cells, another type of lung cancer that makes up 15-20% of all lung cancer cases [54]. Thus, our finding that AKR1C1 may play indispensable roles in NSCLC metastasis was also supported by these two studies. Nevertheless, our present study provides mechanistic insights that AKR1C1 mediates metastasis specifically and rules out the involvement of catalytic activities, and importantly, NSCLC not only represents 80-85% of lung cancer cases, but also accounts for the first leading cause of cancer-related deaths worldwide.
Previous studies linking AKR1C1 with cancer have mostly implicated its catalytic role as a reductase. Although several lines of evidence have revealed the employment of AKR1C1 inhibitors to interfere with malignant disease, the efficacy of these inhibitors against cancer cells have been limited and even 5-BPSA, one of the most potent inhibitors of AKR1C1's enzymatic activity, fails to suppress or kill cancer cells effectively [15]. Therefore, the strong link between the overexpression of AKR1C1 and the poor prognosis of cancer patients indicate a non-catalytic biological function exerted by AKR1C1 in malignant progression. It has been revealed that the other family members, AKR1C2 and AKR1C3, possess catalytic-independent roles in tumor progression. AKR1C3 can function as an androgen receptor-selective coactivator [55]. AKR1C2 is involved in apoptosis induced by Panax ginseng polysaccharide [56]. In line with these reports, our present study found that AKR1C1 promoted metastasis of NSCLC in a catalytic-independent manner. The loss of AKR1C1 catalytic activities, either by treatment with 5-BPSA [15] or the introduction of AKR1C1 mutants [46], imposed little effect on invasion (Fig. 7). Thus, our study reveals a novel biological role of AKR1C1 in cancer progression and highlights the demand for exploring interactive pathways to develop potent strategies to interfere with AKR1C1-induced metastasis.
A large number of studies have provided strong evidence that STAT3 is critical for metastasis in different cancer models, including NSCLC, by participating in multiple steps of metastasis [57]. STAT3 is tightly regulated by phosphorylation and transduces signals from a variety of signaling pathways, such as by transactivating the target genes mediating metastasis and proliferation [34]. According to our microarray data, STAT3 was among the most significantly downregulated 35 transcription factors and among the top decreased genes belonging to phosphoproteins following overlay analysis on AKR1C1 siRNA vs. N.C. siRNA samples (Fig. S6). Importantly, we found that the mRNA levels of most STAT3 target genes involved in metastasis were distinctly reduced by AKR1C1 depletion (Fig. 4A-C). In addition, the prohibition of STAT3 greatly attenuated the invasion caused by exogenous AKR1C1 in NCI-H1299 cells, as well as endogenous AKR1C1 in NCI-H460 cells (Fig. 5G-J). In addition, we found that AKR1C1 may facilitate the interaction of STAT3 with its upstream kinase JAK2, thereby activating STAT3 pathway and inducing cell migration (Fig. 6). These data explained the mechanism underlying AKR1C1-promoted metastasis, by which the STAT3 pathway was regulated by AKR1C1 and played an indispensable role in this process.
In summary, our present study identified AKR1C1 as a new pro-metastatic factor in NSCLC. AKR1C1 interacts with STAT3 to facilitate its phosphorylation, leading to the activation of target genes and the enhancement of cell motility (Fig. 8E). A significant correlation between AKR1C1 and p-STAT3 was observed in metastatic NSCLC tissues; the co-expression of AKR1C1 and STAT3 target genes in NSCLC patients correlated with reduced survival time and poor prognosis. With these newly acquired insights, the present study not only provided this newly identified AKR1C1-STAT3 pathway as promising predictions and therapeutic targets for NSCLC treatment, but it also directed us toward the future exploration of AKR1C1-STAT3 inhibitors, thus shedding light on the design of new anti-metastatic therapies against NSCLC.
Materials and Methods
Cell culture
The NCI-H1650, NCI-H1299, PC-9 and NCI-H460 cell lines were maintained in RPMI 1640 medium. A549 was maintained in F12 medium. SK-MES-1 was maintained in MEM medium. BEAS-2B was maintained in DMEM. All the culture medium was supplemented with 10% (v/v) fetal bovine serum (FBS, Gibco, Grand Island, NY, USA). Cells were maintained at 37℃ in a humidified 5% CO2 incubator. All the cell lines were purchased from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China).
Transwell assay
A Boyden chamber system (Costar Corp., Cambridge, MA, USA) was used for transwell migration assays. A total of 2×104 NSCLC cells (serum-starved overnight) were seeded into each insert (with Matrigel for invasion assay and without Matrigel for migration assay) in serum-free media, while 600 μL media supplemented with 10% FBS was placed in the wells below. Matrigel (BD, 356234) was diluted at 1:10. After incubation for 24 h, cells that had migrated onto the lower surface of the porous membrane were photographed and counted with an inverted microscope (Leica DMI 4000 B, Wetzlar, Germany) after crystal violet staining.
Microarray analysis
Total RNA was extracted from NCI-H460 with AKR1C1 siRNA and N.C. siRNA samples. Microarray analysis was performed using SurePrint G3 Human Gene Expression 8 × 60K v2 (Agilent Array®). Gene expression was analyzed by Cluster 3.0 and TreeView software.
Transfection
Cells were transfected with the indicated plasmids or short hairpin RNA using jetPRIME® (Polyplus, NY, USA), according to the manufacturer's instructions. Human AKR1C1 and STAT3 siRNA and control siRNA were obtained from GenePharma Co. Ltd (Shanghai, China). For shRNA experiments, NCI-H460 cells were transfected with pLKO.1 vector expressing shRNA for AKR1C1 knockdown (pLKO.1-shAKR1C1). Cells expressing AKR1C1 were generated by transfection of pLenti-CMV-AKR1C1-HA-P2A-EGFP-T2A-Puro into NCI-H1299 cells. Stable cells were selected by 4 mg/mL puromycin after infection. Positive clones were then selected and amplified for further analyses. Human short RNA target sequences are provided in Supplementary Material (Table S1).
Luciferase assay
Relative luciferase activity was determined in NCI-H460 cells. An APRE luciferase reporter was co-transfected with indicated vectors in the presence or absence of siAKR1C1. Following treatment, cells were harvested and lysates were assayed for luciferase activity using the Luciferase assay kit (Promega, Madison, WI, USA) in accordance with the manufacturer's instructions.
Western blotting
Protein samples were size-fractionated by SDS-PAGE and transferred to PVDF membranes (Millipore, Bedford, UK). Blots were blocked for 1 h in 5% milk/0.1% Tween 20 in phosphate buffered saline (PBS-T) and then incubated with primary antibodies (1: 1000) at 4°C overnight. Blots were then washed three times for 25 min in PBS-T, followed by incubation with secondary antibody (according to different primary antibodies, HRP-conjugated goat anti-mouse, anti-rabbit, and rabbit anti-goat IgG were used (1: 5000, Santa Cruz, Dallas, TX) in 5% milk/PBS-T for 1 h, and then washed three times for 15 min in PBS-T. The membranes were briefly incubated with ECL detection reagent (Amersham Biosciences, Castle Hill, Australia) to visualize the proteins and were then exposed on X-ray film. Primary antibodies used were as follows: p-STAT3 (Cell Signaling Technology, 9145S), STAT3 (Cell Signaling Technology, 9139S), p-JAK2 (Cell Signaling Technology, 3776S), JAK2 (Cell Signaling Technology, 3230S), p-EGFR (Cell Signaling Technology, 2236), EGFR (Cell Signaling Technology, 4267), GAPDH (Santa Cruz, sc-25778), LaminB (Santa Cruz, sc-6216), β-Actin (Santa Cruz, sc-1615), HA (Santa Cruz, sc-805), AKR1C1 (GeneTex, GTX105620), flag (Sigma, 7425), GST (Santa Cruz, sc-138).
Immunofluorescence
Cells were plated at a confluence of 30%. After starving overnight, cells were treated with IL-6 (20 ng/mL, Novus, NBP2-34901) for 30 min and then fixed with paraformaldehyde. The antibodies used were as follows: p-STAT3 (1:50, Signaling Technology, 4113), AKR1C1 (1:400, GeneTex, GTX105620), Donkey anti-Mouse IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 488 (Invitrogen, A21202), Donkey anti-Rabbit IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 568 (Invitrogen, A10042). Nuclei were stained with DAPI (DOJINDO, Kumamoto, Japan).
Immunoprecipitation
Cells were lysed in lysis buffer (25 mM Tris (pH 8.0), 150 mM NaCl, 10% Glycerol, 1% NP40) supplemented with protease inhibitor cocktail. Whole-cell lysates were incubated with the indicated tag-beads for 2 h. The beads were washed using wash buffer (25 mM Tris (pH 8.0), 150 mM NaCl, 0.2% NP40) at least five times, and then boiled in SDS loading buffer. Immunoprecipitated protein complexes were detected using Western blotting. Anti-HA magnetic beads were bought from Biotool (B26202) and anti-FLAG Tag Agarose Resin were from Genscript (L00425).
RNA extraction and qRT-PCR
Total RNA was prepared using the EasyPure RNA Kit (Cat# H30828; TransGen Biotech, Beijing, China). Single-strand cDNA was prepared from the purified RNA using oligo (dT) priming (Thermoscript RT kit; Invitrogen, Carlsbad, CA, USA), followed by SYBR-Green real-time PCR (Qiagen, Hilden, Germany). RT-PCR reactions were performed according to the manufacturer's instructions. Data quantitation was performed by the relative standard curve method with those genes normalized to GAPDH mRNA levels. The sequences of oligonucleotide primers were synthesized by Sangon (Shanghai, China) and are provided in Supplementary Material (Table S2).
Chromatin Immunoprecipitation (ChIP assay)
293FT cells were transfected with indicated plasmids and then cultured for 30 h. After starving for 12 h, the cells were treated with 20 ng/mL human IL-6 for 1 h. The Chromatin Immunoprecipitation (ChIP) assay was conducted according to the protocol of Millipore ChIP kit (#17-371, Millipore Corp, Billerica, MA, USA). Soluble chromatin was sheared to yield fragments of 400-500 base pairs with a Bioruptor Pico sonicator (Diagenode, Denville, NJ, USA). The antibodies utilized for ChIP assay were normal rabbit IgG (Cell Signaling Technology, #2729), HA-Tag (C29F4) rabbit mAb (Cell Signaling Technology, #3724). For standard end-point PCR, KOD-PLUS-NEO kit (KOD-401, TOYOBO, Osaka, JAPAN) was used for amplifications. For real-time quantitative PCR, i Taq Unibersal SYBR Green Supermix (172-5124, BIO-RAD, Hercules, CA, USA) and a CFX96 Touch™ Real-Time PCR Detection System (BIO-RAD, Hercules, CA, USA) were used. The sequences of the ChIP PCR primers are: human MMP2 promoter: forward 5'-TGTTCCCTAAAACATTCCCC-3' and reverse 5'-GTCTCTGAGGAATGTCTTCT-3' (amplicon: -1340 to -1120 bp in the promoter) [37]; and human TWIST promoter: forward 5'-AGTCTCCTCCGACCGCTTCCTG-3' and reverse 5'-CTCCGTGCAGGCGGAAAGTTTGG-3' (amplicon: -364 to -33 bp in the promoter) [36]; and human SOX2 promoter: forward 5'-AGAATGGTTTGTGAGTGGTTAAA-3' and reverse 5'-AGCAGAGTTTGACGACCACC-3' (amplicon: -3913 to -3851 bp in the promoter) [38].
Cell proliferation/viability assay
The protocols and reagents used for the sulforhodamine B assay (detection of cell proliferation/viability) were all according to the document [58].
PET-CT
Every nude mouse (4-5 weeks of age) was injected with 300×104 cells in 200 μL medium via tail vein. After 40 days, nude mice were photographed by micro PET-CT to assess the ability of metastasis. For analysis of liver metastases, livers were removed and fixed in paraformaldehyde for five parts, cut in 3 μm sections, and stained for hematoxylin and eosin. One section every 200 μm throughout the liver was screened histologically, and the number of metastases was counted (diameter > 100 μm).
Statements of Ethical Approval: The protocols for the animal study were approved by the Animal Research Committee at Zhejiang University, with ethical approval number IACUC-15003, and all experimental protocols were conducted in accordance with institutional guidelines.
Immunohistochemical assay
Tissue microarray slides were obtained from US Biomax (LC814a, Alenabio, Xian, China). Paraffin-embedded tissue microarrays were dewaxed, rehydrated, and subjected to microwaving with sodium citrate buffer (pH 6.0) for AKR1C1 (GeneTex, GTX105620), staining and tris-EDTA buffer (pH 9.0) for p-STAT3 (CST, 9145S). Then, an SP Kit (KIT 9710) was used according to the manufacturer's recommendations. The expression scores of AKR1C1 and p-STAT3 in the tissue microarrays were analysed by Image-Pro Plus. (Primary tumour tissue: n = 40; matched lymph node metastatic tumor tissue: n = 40).
In vitro kinase assay
Kinase reaction mixture (pH=7.5) was composed of 20 mM Tris-HCl, 100 μM ATP, 50 mM MgCl2, 1 μg GST-STAT3 and 250 ng corresponding purified protein (His-JAK2, GST-AKR1C1, GST-AKR1C2, GST-AKR1C3) in 30 μL volume. All samples were incubated at 37℃ for 10 min and the same volume of SDS loading buffer was added to stop reactions. The samples were boiled for 15 min, then tested by using Western blotting. GST-STAT3 was bought from Rockland (009-001-T53). His-JAK2 was bought from Abcam (42619). Human recombinant AKR1C1/2/3 was transfected in Escherichia coli BL21, and purified for kinase assay in vitro.
Enzyme activity assay
Human recombinant AKR1C1/2/3 was transfected in Escherichia coli BL21, and purified for enzyme activity assay. The NADP+-linked S-(+)-1,2,3,4-tetrahydro-1-naphthol (S-tetralol) dehydrogenase activity of the enzymes was assayed by measuring the rate of change in NADPH absorbance (at 340 nm) at room temperature. The inhibitor, 3-bromo-5-phenylsalicylic acid (5-BPSA) was dissolved in dimethyl sulphoxide (DMSO). In the reaction mixture, the concentration of DMSO was <1.5%. The reaction mixture consisted of 0.1 M potassium phosphate (pH 6.7), 1 mM S-tetralol, 0.1 mM NADPH and enzyme [6, 59].
Statistical analysis
Experiments were performed at least three times or as indicated. Data are presented as mean ± SD from three independent experiments. Statistical analyses were performed using two-tailed Student's t-test. P values less than 0.05 were considered significant (* P < 0.05, ** P < 0.01 and *** P < 0.001).
Abbreviations
NSCLC: non-small cell lung cancer; AKR1C1: aldo-keto reductase 1, family member, C1; NADPH: reduction of nicotinamide adenine dinucleotide phosphate; STAT3: signal transducer and activator of transcription 3; PET-CT: Positron emission tomography-computed tomography; RT-PCR: real-time polymerase chain reaction; ZEB1: zinc finger E-box binding homeobox 1; CCND1: cyclin D1; MCL1: BCL2 family apoptosis regulator; MMP2: matrix metallopeptidase 2; TWIST: twist family bHLH transcription factor 1; SOX2: SRY-box 2; IL-6: interleukin 6; ChIP: chromatin immunoprecipitation; JAK2: Janus kinase 2; EGFR: epidermal growth factor receptor; 5-BPSA: 3-bromo-5-phenylsalicylic acid; APRE: acute phase response elements; PM2.5: fine particulates; GEO: gene expression omnibus; FBS: fetal bovine serum; SDS-PAGE: sodium dodecyl sulphate-polyacrylamide gel electrophoresis; PVDF: polyvinylidene difluoride; PBS: phosphate buffered saline; HRP: horseradish peroxidase; ECL: enhanced chemiluminescence; DAPI: 4′,6-Diamidine-2′-phenylindole dihydrochloride.
Acknowledgements
We thank Guang-Fa Wang and Jian Chen from First-Affiliated Hospital of Zhejiang University for assisting with PET-CT studies. This work was supported by the National Natural Science Foundation of China (81673458, 81503095), National Natural Science Foundation for Distinguished Young Scholar of China (81625024).
Author contribution statement
HZ, QJH and BY conceived and designed the study. LLC, FJY, YH, CMZ, TYZ and TY performed the experiments. HZ, LLC, MDY and JC performed the data analysis. HZ, LLC, QJH and BY contributed to writing the manuscript. HZ, QJH and BY contributed to the materials. All the authors read and approved the final version of the manuscript.
Supplementary Material
Supplementary methods, figures and tables.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Garon EB, Rizvi NA, Hui R. et al. Pembrolizumab for the treatment of non-small-cell lung cancer. N Engl J Med. 2015;372:2018-2028
2. Hatley ME, Patrick DM, Garcia MR. et al. Modulation of K-Ras-dependent lung tumorigenesis by MicroRNA-21. Cancer Cell. 2010;18:282-293
3. Chen VW, Ruiz BA, Hsieh MC. et al. Analysis of stage and clinical/prognostic factors for lung cancer from SEER registries: AJCC staging and collaborative stage data collection system. Cancer. 2014;120(Suppl 23):3781-3792
4. Yang Z, Guo Q, Wang Y. et al. AZD3759, a BBB-penetrating EGFR inhibitor for the treatment of EGFR mutant NSCLC with CNS metastases. Sci Transl Med. 2016;8:368ra172
5. Hsu CY, Lin CH, Jan YH. et al. Huntingtin-Interacting Protein-1 Is an Early-Stage Prognostic Biomarker of Lung Adenocarcinoma and Suppresses Metastasis via Akt-mediated Epithelial-Mesenchymal Transition. Am J Respir Crit Care Med. 2016;193:869-880
6. Dhagat U, Endo S, Sumii R. et al. Selectivity determinants of inhibitor binding to human 20alpha-hydroxysteroid dehydrogenase: crystal structure of the enzyme in ternary complex with coenzyme and the potent inhibitor 3,5-dichlorosalicylic acid. J Med Chem. 2008;51:4844-4848
7. Fluck CE, Meyer-Boni M, Pandey AV. et al. Why boys will be boys: two pathways of fetal testicular androgen biosynthesis are needed for male sexual differentiation. Am J Hum Genet. 2011;89:201-218
8. Seo DC, Sung JM, Cho HJ. et al. Gene expression profiling of cancer stem cell in human lung adenocarcinoma A549 cells. Mol Cancer. 2007;6:75
9. Woenckhaus M, Klein-Hitpass L, Grepmeier U. et al. Smoking and cancer-related gene expression in bronchial epithelium and non-small-cell lung cancers. J Pathol. 2006;210:192-204
10. Rizner TL. Enzymes of the AKR1B and AKR1C Subfamilies and Uterine Diseases. Front Pharmacol. 2012;3:34
11. Chang HC, Chen YL, Chan CP. et al. Overexpression of dihydrodiol dehydrogenase as a prognostic marker in resected gastric cancer patients. Dig Dis Sci. 2009;54:342-347
12. Hsu NY, Ho HC, Chow KC. et al. Overexpression of dihydrodiol dehydrogenase as a prognostic marker of non-small cell lung cancer. Cancer Res. 2001;61:2727-2731
13. Fukumoto S, Yamauchi N, Moriguchi H. et al. Overexpression of the aldo-keto reductase family protein AKR1B10 is highly correlated with smokers' non-small cell lung carcinomas. Clin Cancer Res. 2005;11:1776-1785
14. Chien CW, Ho IC, Lee TC. Induction of neoplastic transformation by ectopic expression of human aldo-keto reductase 1C isoforms in NIH3T3 cells. Carcinogenesis. 2009;30:1813-1820
15. Matsunaga T, Yamaguchi A, Morikawa Y. et al. Induction of aldo-keto reductases (AKR1C1 and AKR1C3) abolishes the efficacy of daunorubicin chemotherapy for leukemic U937 cells. Anticancer Drugs. 2014;25:868-877
16. Ji Q, Aoyama C, Nien YD. et al. Selective loss of AKR1C1 and AKR1C2 in breast cancer and their potential effect on progesterone signaling. Cancer Res. 2004;64:7610-7617
17. Penning TM, Lerman C. Genomics of smoking exposure and cessation: lessons for cancer prevention and treatment. Cancer Prev Res (Phila). 2008;1:80-83
18. Penning TM. Human aldo-keto reductases and the metabolic activation of polycyclic aromatic hydrocarbons. Chem Res Toxicol. 2014;27:1901-1917
19. Nishizawa M, Nakajima T, Yasuda K. et al. Close kinship of human 20alpha-hydroxysteroid dehydrogenase gene with three aldo-keto reductase genes. Genes Cells. 2000;5:111-125
20. Rizner TL, Smuc T, Rupreht R. et al. AKR1C1 and AKR1C3 may determine progesterone and estrogen ratios in endometrial cancer. Mol Cell Endocrinol. 2006;248:126-135
21. Smuc T, Rizner TL. Aberrant pre-receptor regulation of estrogen and progesterone action in endometrial cancer. Mol Cell Endocrinol. 2009;301:74-82
22. Hanekamp EE, Gielen SC, Smid-Koopman E. et al. Consequences of loss of progesterone receptor expression in development of invasive endometrial cancer. Clin Cancer Res. 2003;9:4190-4199
23. Fan L, Peng G, Hussain A. et al. The Steroidogenic Enzyme AKR1C3 Regulates Stability of the Ubiquitin Ligase Siah2 in Prostate Cancer Cells. J Biol Chem. 2015;290:20865-20879
24. Qi J, Tripathi M, Mishra R. et al. The E3 ubiquitin ligase Siah2 contributes to castration-resistant prostate cancer by regulation of androgen receptor transcriptional activity. Cancer Cell. 2013;23:332-346
25. Gao Y, Liu Y, Meng F. et al. Overexpression of Siah2 Is Associated With Poor Prognosis in Patients With Epithelial Ovarian Carcinoma. Int J Gynecol Cancer. 2016;26:114-119
26. GDS2865. Metastatic prostate tumor model. https://www.ncbi.nlm.nih.gov/sites/GDSbrowser?acc=GDS2865
27. GDS3091. Noncancerous hepatic tissues from hepatocellular carcinoma patients with and without venous metastasis. https://www.ncbi.nlm.nih.gov/sites/GDSbrowser?acc=GDS3091
28. Gene: AKR1C1; Analysis Type: Cancer vs. Normal Analysis; Data Type: mRNA; Sample Type: Clinical Specimen. https://www.oncomine.org/resource/login.html
29. Gene: AKR1C1; Analysis Type: Cancer vs. Normal Analysis; Data Type: mRNA; Sample Type: Clinical Specimen; Yamagata Lung. https://www.oncomine.org/resource/login.html
30. Lung Cancer. http://kmplot.com/analysis/index.php?p=service&cancer=lung
31. Liu CC, Lin SP, Hsu HS. et al. Suspension survival mediated by PP2A-STAT3-Col XVII determines tumour initiation and metastasis in cancer stem cells. Nat Commun. 2016;7:11798
32. Chen J, Lan T, Zhang W. et al. Feed-Forward Reciprocal Activation of PAFR and STAT3 Regulates Epithelial-Mesenchymal Transition in Non-Small Cell Lung Cancer. Cancer Res. 2015;75:4198-4210
33. Yu Y, Zhao Q, Wang Z. et al. Activated STAT3 correlates with prognosis of non-small cell lung cancer and indicates new anticancer strategies. Cancer Chemother Pharmacol. 2015;75:917-922
34. Carpenter RL, Lo HW. STAT3 Target Genes Relevant to Human Cancers. Cancers (Basel). 2014;6:897-925
35. Wang Y, Ning H, Ren F. et al. GdX/UBL4A specifically stabilizes the TC45/STAT3 association and promotes dephosphorylation of STAT3 to repress tumorigenesis. Mol Cell. 2014;53:752-765
36. Lo HW, Hsu SC, Xia W. et al. Epidermal growth factor receptor cooperates with signal transducer and activator of transcription 3 to induce epithelial-mesenchymal transition in cancer cells via up-regulation of TWIST gene expression. Cancer Res. 2007;67:9066-9076
37. Huang W, Yu LF, Zhong J. et al. Stat3 is involved in angiotensin II-induced expression of MMP2 in gastric cancer cells. Dig Dis Sci. 2009;54:2056-2062
38. Foshay KM, Gallicano GI. Regulation of Sox2 by STAT3 initiates commitment to the neural precursor cell fate. Stem Cells Dev. 2008;17:269-278
39. Qi J, Xia G, Huang CR. et al. JSI-124 (Cucurbitacin I) inhibits tumor angiogenesis of human breast cancer through reduction of STAT3 phosphorylation. Am J Chin Med. 2015;43:337-347
40. Oi T, Asanuma K, Matsumine A. et al. STAT3 inhibitor, cucurbitacin I, is a novel therapeutic agent for osteosarcoma. Int J Oncol. 2016;49(6):2275-2284
41. Song J, Liu H, Li Z. et al. Cucurbitacin I inhibits cell migration and invasion and enhances chemosensitivity in colon cancer. Oncol Rep. 2015;33:1867-1871
42. Burger M, Hartmann T, Burger JA. et al. KSHV-GPCR and CXCR2 transforming capacity and angiogenic responses are mediated through a JAK2-STAT3-dependent pathway. Oncogene. 2005;24:2067-2075
43. Lo HW, Hsu SC, Ali-Seyed M. et al. Nuclear interaction of EGFR and STAT3 in the activation of the iNOS/NO pathway. Cancer Cell. 2005;7:575-589
44. Ram PA, Waxman DJ. Interaction of growth hormone-activated STATs with SH2-containing phosphotyrosine phosphatase SHP-1 and nuclear JAK2 tyrosine kinase. J Biol Chem. 1997;272:17694-17702
45. Kamakura S, Oishi K, Yoshimatsu T. et al. Hes binding to STAT3 mediates crosstalk between Notch and JAK-STAT signalling. Nat Cell Biol. 2004;6:547-554
46. Couture JF, Legrand P, Cantin L. et al. Human 20alpha-hydroxysteroid dehydrogenase: crystallographic and site-directed mutagenesis studies lead to the identification of an alternative binding site for C21-steroids. J Mol Biol. 2003;331:593-604
47. Steckelbroeck S, Jin Y, Gopishetty S. et al. Human cytosolic 3alpha-hydroxysteroid dehydrogenases of the aldo-keto reductase superfamily display significant 3beta-hydroxysteroid dehydrogenase activity: implications for steroid hormone metabolism and action. J Biol Chem. 2004;279:10784-10795
48. Rousseaux S, Debernardi A, Jacquiau B. et al. Ectopic activation of germline and placental genes identifies aggressive metastasis-prone lung cancers. Sci Transl Med. 2013;5:186ra166
49. GSE30219. http://bioinformatica.mty.itesm.mx:8080/
50. Wang Y, Wang Y, Zhang Z. et al. Mechanism of progestin resistance in endometrial precancer/cancer through Nrf2-AKR1C1 pathway. Oncotarget. 2016;7:10363-10372
51. Liu C, Guo H, Cheng X. et al. Exposure to airborne PM2.5 suppresses microRNA expression and deregulates target oncogenes that cause neoplastic transformation in NIH3T3 cells. Oncotarget. 2015;6:29428-29439
52. Wang HW, Lin CP, Chiu JH. et al. Reversal of inflammation-associated dihydrodiol dehydrogenases (AKR1C1 and AKR1C2) overexpression and drug resistance in nonsmall cell lung cancer cells by wogonin and chrysin. Int J Cancer. 2007;120:2019-2027
53. Matsumoto R, Tsuda M, Yoshida K. et al. Aldo-keto reductase 1C1 induced by interleukin-1beta mediates the invasive potential and drug resistance of metastatic bladder cancer cells. Sci Rep. 2016;6:34625
54. Tian H, Li X, Jiang W. et al. High expression of AKR1C1 is associated with proliferation and migration of small-cell lung cancer cells. Lung Cancer (Auckl). 2016;7:53-61
55. Yepuru M, Wu Z, Kulkarni A. et al. Steroidogenic enzyme AKR1C3 is a novel androgen receptor-selective coactivator that promotes prostate cancer growth. Clin Cancer Res. 2013;19:5613-5625
56. Li C, Tian ZN, Cai JP. et al. Panax ginseng polysaccharide induces apoptosis by targeting Twist/AKR1C2/NF-1 pathway in human gastric cancer. Carbohydr Polym. 2014;102:103-109
57. Song X, Hao J, Wang J. et al. The cancer/testis antigen MAGEC2 promotes amoeboid invasion of tumor cells by enhancing STAT3 signaling. Oncogene. 2016;36(11):1476-1486
58. Chen Z, Wu Q, Ding Y. et al. YD277 Suppresses Triple-Negative Breast Cancer Partially Through Activating the Endoplasmic Reticulum Stress Pathway. Theranostics. 2017;7(8):2339-2349
59. Carbone V, Chung R, Endo S. et al. Structure of aldehyde reductase in ternary complex with coenzyme and the potent 20alpha-hydroxysteroid dehydrogenase inhibitor 3,5-dichlorosalicylic acid: implications for inhibitor binding and selectivity. Arch Biochem Biophys. 2008;479:82-87
Author contact
![]() Corresponding author: Dr. Qiao-Jun HE and Dr. Bo YANG, Zhejiang Province Key Laboratory of Anti-Cancer Drug Research, Institute of Pharmacology and Toxicology, College of Pharmaceutical Sciences, Zhejiang University, 866#Yuhangtang Rd, Hangzhou, Zhejiang 310058, China, email: yang924edu.cn; qiaojunheedu.cn; fax: 86571-88208400, tel: 86571-88208400
Corresponding author: Dr. Qiao-Jun HE and Dr. Bo YANG, Zhejiang Province Key Laboratory of Anti-Cancer Drug Research, Institute of Pharmacology and Toxicology, College of Pharmaceutical Sciences, Zhejiang University, 866#Yuhangtang Rd, Hangzhou, Zhejiang 310058, China, email: yang924edu.cn; qiaojunheedu.cn; fax: 86571-88208400, tel: 86571-88208400
Citation styles
APA
ZHU, H., CHANG, L.L., YAN, F.J., HU, Y., ZENG, C.M., ZHOU, T.Y., YUAN, T., YING, M.D., CAO, J., HE, Q.J., YANG, B. (2018). AKR1C1 Activates STAT3 to Promote the Metastasis of Non-Small Cell Lung Cancer. Theranostics, 8(3), 676-692. https://doi.org/10.7150/thno.21463.
ACS
ZHU, H.; CHANG, L.L.; YAN, F.J.; HU, Y.; ZENG, C.M.; ZHOU, T.Y.; YUAN, T.; YING, M.D.; CAO, J.; HE, Q.J.; YANG, B. AKR1C1 Activates STAT3 to Promote the Metastasis of Non-Small Cell Lung Cancer. Theranostics 2018, 8 (3), 676-692. DOI: 10.7150/thno.21463.
NLM
ZHU H, CHANG LL, YAN FJ, HU Y, ZENG CM, ZHOU TY, YUAN T, YING MD, CAO J, HE QJ, YANG B. AKR1C1 Activates STAT3 to Promote the Metastasis of Non-Small Cell Lung Cancer. Theranostics 2018; 8(3):676-692. doi:10.7150/thno.21463. https://www.thno.org/v08p0676.htm
CSE
ZHU H, CHANG LL, YAN FJ, HU Y, ZENG CM, ZHOU TY, YUAN T, YING MD, CAO J, HE QJ, YANG B. 2018. AKR1C1 Activates STAT3 to Promote the Metastasis of Non-Small Cell Lung Cancer. Theranostics. 8(3):676-692.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.