Theranostics
13.3
Impact Factor
- Current Issue
- Advance Articles
- Volume 16; 2026
- Volume 15; 2025
- Volume 14; 2024
- Volume 13; 2023
- Volume 12; 2022
- Archive
- Cover Images
- Cover Suggestion
- Special Issues
Top
Introduction
Materials and Methods
Results
Discussion
Acknowledgements
Supplementary Material
References
Introduction
Materials and Methods
Results
Discussion
Acknowledgements
Supplementary Material
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2018; 8(3):735-745. doi:10.7150/thno.23491 This issue Cite
Research Paper
Evans Blue Attachment Enhances Somatostatin Receptor Subtype-2 Imaging and Radiotherapy
Rui Tian1,2, Orit Jacobson2 ![]() , Gang Niu2, Dale O. Kiesewetter2, Zhantong Wang2, Guizhi Zhu2, Ying Ma2, Gang Liu1, Xiaoyuan Chen2
, Gang Niu2, Dale O. Kiesewetter2, Zhantong Wang2, Guizhi Zhu2, Ying Ma2, Gang Liu1, Xiaoyuan Chen2 ![]()
1. State Key Laboratory of Molecular Vaccinology and Molecular Diagnostics & Center for Molecular Imaging and Translational Medicine, School of Public Health, Xiamen University, Xiamen 361102 China;
2. Laboratory of Molecular Imaging and Nanomedicine, National Institute of Biomedical Imaging and Bioengineering, National Institutes of Health, Bethesda, Maryland, USA.
Received 2017-10-25; Accepted 2017-11-25; Published 2018-1-1
Citation:
Tian R, Jacobson O, Niu G, Kiesewetter DO, Wang Z, Zhu G, Ma Y, Liu G, Chen X. Evans Blue Attachment Enhances Somatostatin Receptor Subtype-2 Imaging and Radiotherapy. Theranostics 2018; 8(3):735-745. doi:10.7150/thno.23491. https://www.thno.org/v08p0735.htm
Other stylesAbstract

Purpose: Radionuclide therapy directed against tumors that express somatostatin receptors (SSTRs) has proven effective for the treatment of advanced, low- to intermediate-grade neuroendocrine tumors in the clinic. In clinical usage, somatostatin peptide-based analogs, labeled with therapeutic radionuclides, provide an overall response rate of about 30%, despite the high cumulative activity injected per patient. We set out to improve the effectiveness of somatostatin radiotherapy by preparing a chemical analog that would clear more slowly through the urinary tract and, concomitantly, have increased blood circulation half-life and higher targeted accumulation in the tumors. Experimental Design: We conjugated a common, clinically-used SST peptide derivative, DOTA-octreotate, to an Evans blue analog (EB), which reversibly binds to circulating serum albumin. The resulting molecule was used to chelate 86Y and 90Y, a diagnostic and a therapeutic radionuclide, respectively. The imaging capabilities and the radiotherapeutic efficacy of the resulting radioligand was evaluated in HCT116/SSTR2, HCT116, and AR42J cell lines that express differing levels of SST2 receptors. Results: The synthesized radiopharmaceutical retained affinity and specificity to SSTR2. The new molecule also retained the high internalization rate of DOTA-octreotate, and therefore, showed significantly higher accumulation in SSTR2-positive tumors. Labeling of our novel EB-octreotate derivative with the therapeutic, pure beta emitter, 90Y, resulted in improved tumor response and survival rates of mice bearing SSTR2 xenografts and had long term efficacy when compared to DOTA-octreotate itself. Conclusions: The coupling of a targeted peptide, a therapeutic radionuclide, and the EB‑based albumin binding provides for effective treatment of SSTR2-containing tumors.
Keywords: SSTR2, PET, peptide radiotherapy, Evans blue, albumin.
Introduction
Gastroenteropancreatic neuroendocrine tumors are low-incidence tumors that typically are well differentiated and slow growing, though some are quite aggressive (1). Most of these tumors are only discovered after metastatic spread where treatment is more difficult (2). They are often noted for the production and release of neuroamines and hormonal peptides depending on tumor origin, including serotonin, kinins, vasoactive peptides, corticotropic hormone, etc. (3). Nearly 80% of these tumors express somatostatin receptors (1). Treatment of patients, especially those with carcinoid syndrome, with octreotide, a somatostatin ligand, is used clinically to ameliorate symptoms related to the excess hormonal production (1, 2, 4).
Somatostatin (SST) peptide hormone, originally isolated from the hypothalamus, is a growth hormone inhibitory substance. The peptide has two naturally occurring biologically active forms, SST14 and SST28; both act as inhibitors of exocrine, endocrine, paracrine and autocrine activity (5-7). SST is produced by endocrine-like cells mainly found in the nervous systems, the endocrine pancreas, and the gastrointestinal system (5, 6). SST function is mediated specifically by five distinct subtypes of G-protein coupled receptors, namely somatostatin receptors 1-5 (SSTR1-5) (1, 7). These SSTRs are widely expressed in various tumors (8, 9).
SSTR2, the most ubiquitous and abundant subtype in tumor tissues, can activate or suppress various signal transduction pathways (2). Specific SSTR subtypes are expressed by a given tumor depending on tumor type, and some cancers were reported to lose or gain expression of SSTRs (1, 10, 11). For example, loss of the SSTR2 expression in some human adenocarcinomas was implicated in the loss of cell regulation and subsequent proliferation. Because the receptors have been lost, no SST targeted therapy is possible (12). Another layer of complexity observed in some tumors is that the SSTRs form both homo- and heterodimers on the membrane, which confer new and/or different functional features (1, 8, 10, 11, 13, 14).
Therapeutics directed primarily at SSTR2 and SSTR5 employ synthetic analogs of the rapidly degraded natural ligand. Hexapeptide and octapeptide SST analogs were developed that incorporate the biologically active core of SST14, possess superior metabolic stability, and have longer circulation half-life (1.5-2 h) compared to the natural SST (1, 2, 5-8). Preclinical studies with SST analogs showed antitumor and anti-secretory effects in vitro and in vivo; however, the clinical efficacy was relatively limited (8, 9). One approach to improve this therapy was to develop SSTR2-targeted peptide-receptor radionuclide therapy (PRRT) that takes advantage of the fact that SSTR2 binding leads to ligand internalization. The rapid internalization and accumulation of the radiopharmaceutical inside tumor cells in proximity to the nucleus resulted in radioactivity-induced cell death (1, 4). The PRRT ligand was prepared by conjugation of a metal-binding complex to the peptide and then chelation of high energy β-emitting isotopes such as 90Y and 177Lu. 90Y is a pure beta-emitter with a half-life of 64.1 hours, maximum energy of 2.28 MeV, and penetration range of 11 mm. SSTR2-targeted PRRT has been used in the clinic for more than two decades as a treatment of neuroendocrine tumors (NETs). Several Phase I and II clinical trials showed favorable results in NETs in general and even better results in patients with gastroenteropancreatic neuroendocrine tumors (GEPNETs) and bronchial NETs, where as much as 30% objective response was observed (4).
The most frequently used somatostatin analogs in PRRT clinical practice are octreotide and octreotate (TATE); the latter one has higher affinity and selectivity towards SSTR2. Both peptides were conjugated to the bifunctional chelator 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid (DOTA) for chelation of either 177Lu or 90Y. Both peptides clear from the blood through the kidneys, which is the critical organ in terms of radiation toxicity, and limits maximum dosage such that exposure is in the range of 23 to 29 Gy (15-17). The toxicity to the bone marrow is usually mild and transient.
To improve the pharmacokinetics of SSTR2 analogs and reduce PRRT toxicity, we conjugated a truncated Evans Blue (EB) molecule onto octreotate (EB-TATE). The EB moiety provided reversible binding of EB-TATE to albumin in vivo to extend the half-life in the blood. When the EB-TATE was slowly released into the tumor microenvironment, tumor uptake and internalization into SSTR positive cells resulted in delivery of radioactive particles and tumor cell killing. EB-TATE displayed significantly more favorable pharmacokinetics than TATE by achieving higher tumor to non-tumor contrast for both diagnostic positron emission tomography (PET) imaging with 86Y and radiotherapy using 90Y.
Materials and Methods
Syntheses, radiosyntheses, purification methods, and characterization of TATE, EB-TATE, and radiolabeled analogs are described in the Supplementary Materials.
Cell culture
HCT116 (human colon cancer) and AR42J (Rat amphicrine pancreatic) cells were purchased from American Type Culture Collection (ATCC). HCT116/SSTR2+ transfected cells were kindly provided to us by Dr. Carolyn Anderson, University of Pittsburgh Cancer Institute. Cells were cultured in McCoy's 5A medium (Hyclone) containing 10% fetal bovine serum (Gibco) in a humidified atmosphere containing 5% CO2 at 37 °C.
Immunofluorescence and flow cytometry
SSTR2 levels in all three cell lines were determined by flow cytometry and immunofluorescence. 105 cells/well were seeded in either 8-well chamber slides (Lab-Tek, immunofluorescence) or 24-well plates (Corning, flow cytometry). The medium was removed and the cells were washed twice with phosphate buffered saline (PBS, 500 µL). Then bovine serum albumin (2% BSA, 500 µL) was added and the cells were incubated for 1 h at 4 °C, followed by addition of anti-SSTR2 monoclonal antibody or isotype control (R&D Systems, 1:100 dilution). The cells, with the antibody, were incubated for an additional 1 h at room temperature followed by three PBS washes. Subsequently, goat anti-mouse antibody (R&D Systems, 500 µL, 1:100 in 2% BSA) was added and the cells were incubated for additional 1 h at room temperature, followed by three PBS washes. Cells were mounted with UltraCruz mounting medium containing DAPI (Santa Cruz Biotechnology, TX) and fluorescence images were determined by an epifluorescence microscope (200×; Olympus, X81) or a confocal microscope (ZEISS). For flow cytometry, the cells were quantitatively analyzed by LSR II (BD Biosciences) using FlowJo (Tristar).
Cell binding assay
105 HCT116/SSTR2 cells per well were seeded in a 96-well membrane plate (Whatman) with 7.4 KBq of 86Y-TATE and increasing concentrations of TATE or EB-TATE peptides ranging from 0 - 5000 nM. After 1 h incubation, the plate was washed three times with PBS. Subsequently, the cells were assayed in a gamma-counter. Binding affinity was calculated using GraphPad Prism by nonlinear regression. Experiments were performed in triplicate.
Kinetic binding assay by biolayer interferometry (BLI)
Binding assay of EB-TATE to biotinylated BSA/streptavidin biosensors was performed by BLI using an Octet Red96 system (fortéBio) in black 96-well plates (Geiger Bio-One). The assay protocol was as follows: 1) baseline (1 x PBS) 60 s; 2) loading (biotin-labeled BSA, 1 µg/mL) 600 s; 3) baseline (1 x PBS) 60 s; 4) quenching (1 µg/mL Biocytin (Thermo Scientific)) 180 s; 5) baseline (1 x PBS) 60 s; 6) association 600 s; 7) dissociation 600 s. Nonspecific binding calculated from measuring binding of BSA-loaded biosensor to buffer alone and blank biosensor to analytes was subtracted from the binding values. Data collected was analyzed using Octet Analysis software 7.0.
Cell uptake/internalization
Twenty-four h before the assay, 105 cells/well were distributed into 24-wells plate. Cells were washed with PBS X3 and then 18.5 KBq/well of 86Y-EB-TATE or 86Y- TATE were added in 0.5 mL of medium containing 1% (w/v) human serum albumin (HSA). At each indicated time point, the cells were washed X2 with PBS and lysed with 0.1M NaOH. Internalization was measured after removal of membrane-bound tracer by 1 min incubation with 0.5 mL of acid buffer (50 mM glycine, 100 mM NaCl, pH 2.8), wash X2 and lysis. Radioactivity of cell lysate was measured by a γ-counter (Perkin Elmer). The cell uptake and internalization values were normalized as a percent of added radioactivity. Each time point was measured in triplicate.
Mouse xenografts model
Animal protocols were approved by the NIH Clinical Center Animal Care and Use Committee (ACUC). Female athymic nude/nude mice (6-7 weeks, 20-23g) (Harlan) were inoculated on their right shoulder with 5×106 cells of either; HCT116/SSTR2+, HCT116 or AR42J cells in Matrigel (Sigma) 1:1.
In vivo PET and biodistribution studies
PET assays were performed at 25-28 days post tumor cells inoculation when the tumor volume reached about 200-350 mm3. Mice were injected intravenously with 0.92 MBq (4 nmol) of either 86Y-EB-TATE (n = 6, HCT116/SSTR2+; n = 5, HCT116) or 86Y-TATE (n = 4) and scanned for 10-20 min at 1, 4, 24 and 48 h post injection (p.i.). Blocking studies were performed by co-injection with 50-fold excess of unlabeled EB-TATE (n = 3). PET studies were acquired on Nanoscan PET/CT (Mediso) and Inveon (Siemens) scanners. Images were reconstructed using a 3D ordered subset expectation maximum algorithm, and ROI were drawn using IRW (Siemens) and VivoQuant (INVICRO). After the last scan at 48h time point, tumor, heart, lung, liver, spleen, stomach, intestine, pancreas, kidney, muscle, bone and blood were collected from euthanized mice, weighed and measured in a gamma counter. Results are normalized as percentages of the injected dose per gram of tissue (%ID/g).
Tumor radiotherapy studies
Tumor treatment studies were performed in three different cell lines to evaluate the therapeutic efficacy of intravenous injection/s of 90Y-EB-TATE vs. saline and 90Y-TATE in both SSTR2+ and SSTR2- xenografts. The study was commenced 20 days post inoculation of HCT116/SSTR2+ and HCT116 (SSTR2-) xenografts, when all the mice had tumor volume of about 150 mm3. HCT116/SSTR2+ xenografts were divided into 5 groups as followed; 1) saline (n = 6), 2) 7.4 MBq 90Y-EB-TATE (n = 9), 3) 7.4 MBq 90Y-EB-TATE (n = 5) (one dose), 4) 3.7 MBq 90Y-EB-TATE (n = 8), and 5) 7.4 MBq 90Y-TATE (n = 5). HCT116 (SSTR2-) xenografts were divided into additional 2 groups; 6) saline (n=5) and 7) 7.4 MBq 90Y-EB-TATE (n=6). All the mice received the first dose at day 20 (start of treatment). Excluding group 3, which received only one dose by design, animals in all the other groups in which the tumor volume had not exceeded the experimental endpoint, received an additional injection at day 48. All the living mice were monitored for 180 days.
Figure 1
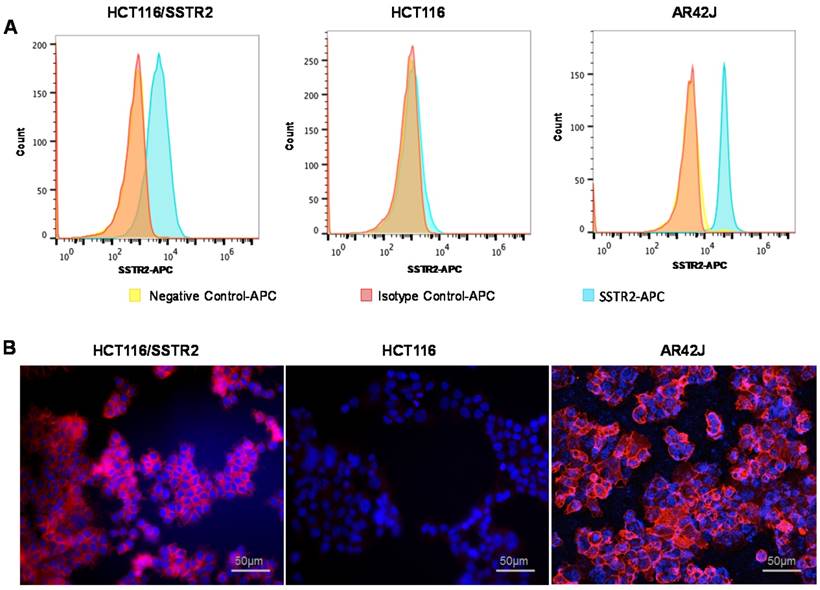
Evaluation of SSTR2 expression levels by cell lines. (A) Representative histograms of SSTR2 expression evaluated by flow cytometry showing unstained cells (negative control), an isotype control antibody and anti-SSTR2 antibody of HCT116 (low levels), HCT116/SSTR2 (medium levels) and AR42J (high levels). (B) Representative immunofluorescence staining (X20) of Anti-SSTR2 (red) and nucleus (DAPI, blue).
Another radiotherapy study was done in AR42J xenografts and started 15 days post-inoculation with tumor volumes at about 100 mm3. In this study, we had 4 groups; 1) saline (n = 3), 2) 7.4 MBq 90Y-EB-TATE (n = 3), 3) 3.7 MBq 90Y-EB-TATE (n = 3) and 4) 7.4 MBq 90Y-TATE (n = 11). All the groups received only one radiotherapy dose at day 15. All the living mice were monitored for up to 90 days. Mice body weight and tumor volume were monitored every 2 days throughout the experiment. The formula used for calculation of tumor volume was V = width2 × length/2.
Endpoint criteria defined by the institute ACUC was weight loss of more than 15%, a tumor volume > 1800 mm3, active ulceration of the tumor or abnormal behavior indicating pain or unease. These definitions were used for Kaplan-Meier analysis as well.
Histopathologic staining
Tumor tissues from each group described above were collected at different time points and frozen or kept at room temperature in Z-FIX (Anatec Ltd). 10 µm-thick sections were mounted on slides using a cryo-microtome. Ki-67, TUNEL and H&E staining were done according to our previous work (18). Number of Ki67-positive nuclei was analyzed by visual counting on 5-6 fields of view per slide, 5 slides for each mouse and 3 mice per group. Quantification was done using Image J software (NIH).
Statistical Analysis
Unless described otherwise, results are mean ± SD. Two-tailed paired and unpaired Student's t tests were used to determine differences within groups and between groups, respectively. P values <0.05 were considered statistically significant.
Results
Cell characterizations and cell binding of EB-TATE
SSTR2 surface expression by HCT116/SSTR2 and AR42J was validated using Flow cytometry and compared to the parental cell line HCT116 that was SSTR2 negative (Fig. 1A). These results were also confirmed by immunofluorescence staining in vitro and in tumor sections ex vivo (Fig. 1B and Supplementary Fig. S1). EB-TATE (Fig. 2A) was synthesized as described in the supplementary materials and was successfully labeled with Yttrium radioisotopes. The conjugation of TATE to truncated EB can affect the binding of each molecule to its target, therefore we tested the binding of EB-TATE to both SSTR2 and albumin in vitro. EB-TATE displayed similar binding characteristics to SSTR2 when compared to TATE alone (Fig 2B and Supplementary Fig. S2A), its binding affinity to serum albumin gave high association constant, Kon of 58233 and low dissociation constant, Koff of 0.2843 (Fig. 2C). EB-TATE affinity to serum albumin was in the range of µM (KD of 4.8 µM) and IC50 of 1.92 ± 0.34 µM (Supplementary Fig. S2C), which is comparable to the published binding affinity of Evans blue to albumin (19). To further ensure that the conjugation of EB to TATE does not reduce the peptide biological activity, we compared cell uptake and internalization studies in vitro in SSTR2 positive and negative cells between 86Y-EB-TATE and 86Y-TATE. Cell uptake of both tracers in HCT116/SSTR2 was similar at all time points and increased over time to reach values of 12.05±0.67% and 10.38±1.82%, respectively, at 24 h. The uptake/internalization of both 86Y-EB-TATE and 86Y-TATE was significantly higher in SSTR2 positive than SSTR2 negative cells (p < 0.001, Fig. 2D and Supplementary Fig. S2B). Using excess amount of unlabeled EB-TATE and/or TATE reduced the uptake in SSTR2-positive cells to the low levels observed in SSTR2-negative cells (p < 0.001, Fig. 2D and Supplementary Fig. S2B). 86Y-TATE and 86Y-EB-TATE both internalized rapidly into SSTR2 positive cells in a comparable manner, confirming that EB conjugation did not reduce the biological activity of the peptide (Fig. 2D and Supplementary Fig. S2B).
86Y Radiolabeled EB-TATE for tumor PET imaging
Next, we evaluated 86Y-EB-TATE uptake in vivo in tumor xenografts and compared it to 86Y-TATE (Fig. 3). 86Y-EB-TATE uptake in SSTR2 positive tumors (HCT116/SSTR2) gradually increased over time. As designed, conjugation of TATE to EB increased its half-life in the blood when compared to 86Y-TATE (p < 0.001, Figs. 3A and 3C). It is important to note that although the half-life in the blood was only increased by 2-fold, the amount of 86Y-EB-TATE retained in the blood is 4-fold compared to 86Y-TATE at 1 h and remained available for SSTR2 binding for longer time (Supplementary Fig. S3A). At 1 h p.i., most of 86Y-EB-TATE was bound to serum albumin and the tracer was slowly released over time, the blood uptake decreased and tumor uptake increased in SSTR2+ tumors but not in SSTR2- tumors (Figs. 3A and 3C). At 24 h p.i., 86Y-EB-TATE accumulated in the tumor to give high tumor-to-muscle and tumor-to-blood ratios (Fig. 3C). A representative co-registration of PET/CT at 24 h p.i. is shown in Fig. 3B. 86Y-TATE on the other hand, showed a significantly less favorable pharmacokinetics, rapidly cleared through the urinary tract (Fig. 3A), with peak uptake in the tumor at 1 h p.i. and much lower accumulation in SSTR2+ tumors of about 5%ID/g (Figs. 3A and 3C). Biodistribution of the two tracers confirmed the PET studies showing a significantly higher tumor uptake of 86Y-EB-TATE in comparison to 86Y-TATE (Supplementary Fig. S3B).
86Y-EB-TATE pharmacokinetics in vivo and specificity to SSTR2 was also evaluated in AR42J xenografts (Figs. 3D and 3E and Supplementary Fig. S4). Similar to the results observed in transfected HCT116/SSTR2 xenografts, 86Y-EB-TATE at 1 h p.i. was mainly bound to serum albumin and circulated in the blood. 86Y-EB-TATE was released from albumin over time and accumulated in the tumor, reaching high tumor uptake of 60.54 ± 8.03 %ID/g (p = 0.0067), and 65.77±7.59 %ID/g (p = 0.0059) at 24 and 48 h p.i., respectively, and displayed high tumor-to-background ratios (Figs. 3D and 3E). 86Y-EB-TATE accumulation in the tumor was efficiently blocked by co-injection of 50-fold excess of unlabeled EB-TATE (Figs. 3D and 3E and Supplementary Fig. S4), indicating that tumor accumulation was SSTR2 specific.
Radionuclide therapy of SSTR2+ tumors
The encouragingly high tumor uptake of 86Y-EB-TATE, coupled with high internalization into the tumor cells resulted in high exposure to the radioactive isotope, as evident from area under the curve (AUC) calculation from non-decay corrected data (Supplementary Fig. S5). This led us to hypothesize that it would be an excellent SSTR2-targeted radiotherapy agent using the beta-emitting radionuclide 90Y. Moreover, 86Y-EB-TATE activity accumulated in the tumor, remained steady from 24 h time point (Fig. 3), suggesting a significantly higher tumor exposure from 90Y than 86Y because of the half-life difference of 64.1 h compared to 14.7h respectively. We tested the therapeutic efficacy in the three models that we had studied with imaging: HCT116/SSTR2 (medium SSTR2 level), HCT116 (SSTR2 negative) and AR42J (high SSTR2 level). Treatment with 90Y-EB-TATE (3.7 and 7.4 MBq) had dramatic inhibitory growth effects on established tumors expressing SSTR2 such as HCT116/SSTR2 and AR42J. In both models, tumor volumes significantly decreased (p < 0.001) and mice survival significantly extended (p < 0.001) when compared to saline and 90Y-TATE groups or to injection of 90Y-EB-TATE into HCT116 xenografts (Figs. 4A and 4B). 90Y-EB-TATE exhibited dose dependency on both radioactivity amount as well as SSTR2 expression. Dosage dependency of 90Y-EB-TATE was evident in the HCT116/SSTR2 tumor model, where the group which received 3.7 MBq had transient tumor regression for about two weeks post treatment, while an injection of 7.4 MBq displayed near-complete tumor regression (Fig. 4A and Supplementary Fig. S6). Unexpectedly, a second injection of 90Y-EB-TATE to the 3.7 MBq group did not have any effect on tumor growth. To evaluate if the recurring tumors still express SSTR2, we imaged the mice with 68Ga-DOTA-TATE and compared it to untreated mice with HCT116/SSTR2 tumors. Surprisingly the PET imaging suggested that the recurrent tumors no longer expressed SSTR2 (Supplementary Fig S7). The loss of SSTR2 in these tumors was also validated by immunofluorescent staining of tumor sections from mice of both 3.7 MBq and 7.4 MBq groups (Supplementary Fig. S8). The loss of SSTR2 expression and/or in vivo selection of cells that do not express SSTR2 explain the lack of response to the second injection of 90Y-EB-TATE (Fig. 4A). Interestingly, the loss of SSTR2 expression by HCT116/SSTR2 tumors occurred over 2 weeks (Supplementary Fig. S9).
In a subsequent therapy study using high SSTR2 expression model, AR42J xenografts, we observed complete regression of the SSTR2+ tumors and full survival for the mice treated with one dose of 3.7 MBq or 7.4 MBq 90Y-EB-TATE. A single dose of 7.4 MBq 90Y-TATE was completely effective (p < 0.001, Figs. 4C and 4D).
Staining for markers of tumor cell proliferation (Ki-67) and DNA damage/ cell apoptosis (TUNEL) showed that mice that received 7.4 MBq had significantly lower Ki-67 positive levels and higher TUNEL signal than the other groups (Fig. 5). H&E staining for the tumors revealed extensive necrotic areas for the 7.4 MBq 90Y-EB-TATE HCT116/SSTR2 treated group (Fig. 5).
Figure 2
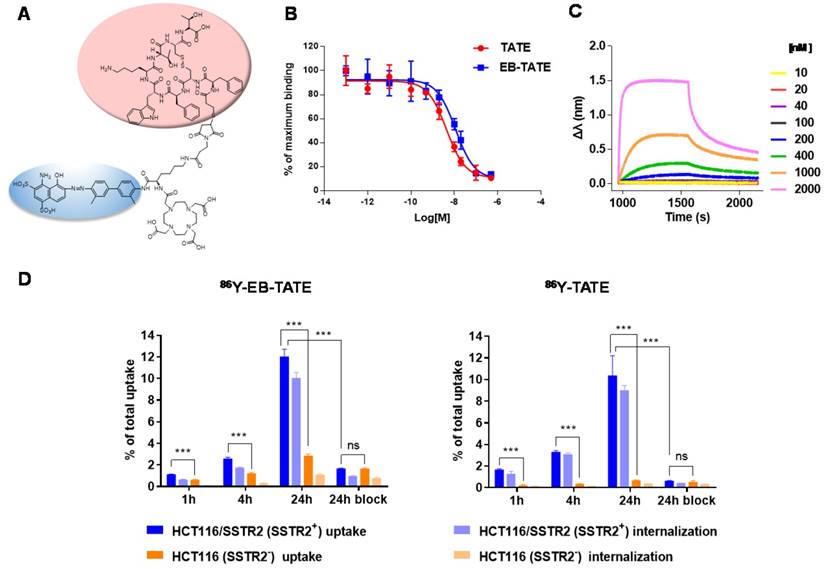
In vitro characterization of EB-TATE. (A) Chemical structure of EB-TATE. Truncated Evans blue, which binds to serum albumin, is marked in blue circle. The SSTR2-specific TATE peptide is shown in a pink circle. (B) Cell binding affinity assays of TATE vs. EB-TATE in HCT116/SSTR2 cells. IC50 of TATE (red curve): 10.21 ± 1.42 nM, and IC50 of EB-TATE (blue curve): 16.50 ± 1.36 nM. Results are shown as average ± SD of triplicates. (C) Kinetic binding assay of EB-TATE to albumin measured by biolayer interferometry. EB-TATE-albumin complex had a Kon of 58233, Koff of 0.2843 and a KD of 4.8 µM. (D) Uptake and internalization in HCT116/SSTR2 (SSTR2+) and HCT116 (SSTR2-) of 86Y-EB-TATE and 86Y-TATE in the presence of 1% (w/v) of human serum albumin. Blocking studies were evaluated at 24 h. Results are shown as average ± SD of triplicates. ***P < 0.001.
Figure 3
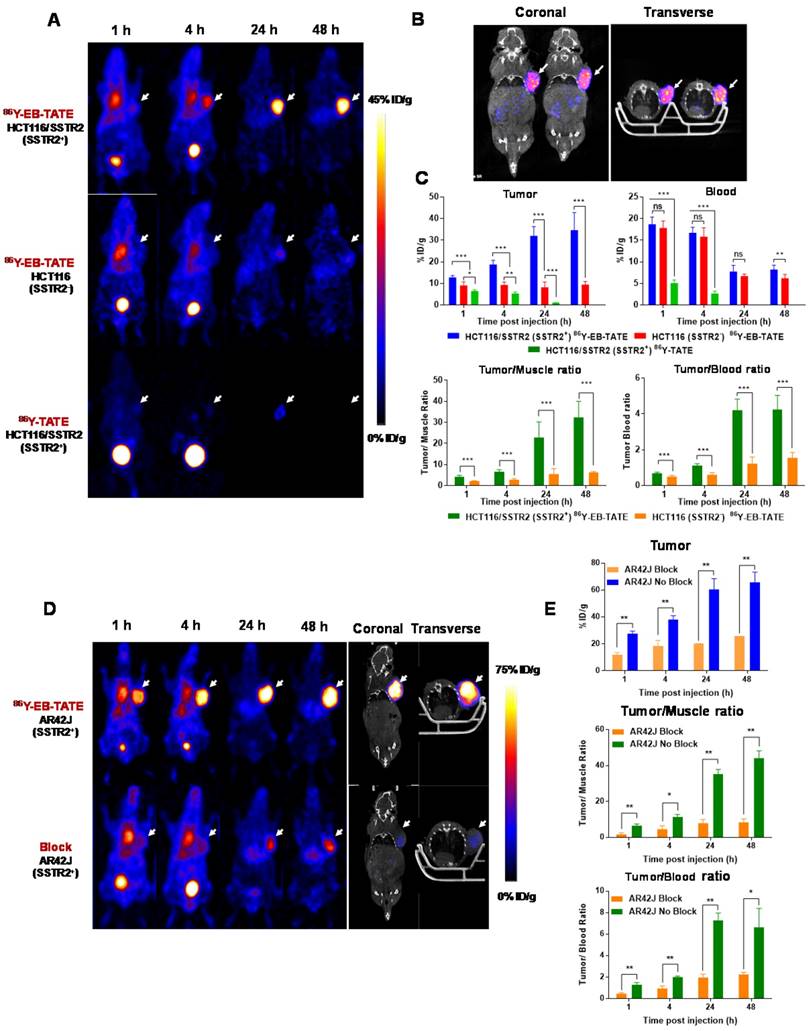
In vivo distribution of EB-TATE. (A) Representative projection PET of HCT116/SSTR2 (SSTR2+) and HCT116 (SSTR2-) tumor models, injected with 86Y-EB-TATE or 86Y-TATE over time. (B) Co-registration of coronal PET/CT of HCT116/SSTR2 injected with 86Y-EB-TATE at 24 h p.i. White arrows indicate the tumor location. (C) Quantification of %ID/g of indicated tracer in tumor and blood (left) and tumor-to-muscle and tumor-to-blood ratios calculated from PET over time in HCT116/SSTR2 tumor model. (D) Representative projection PET (left) and PET/CT (right) of AR42J (SSTR2+) xenografts injected with 86Y-EB-TATE (upper panel) or 86Y-EB-TATE co-injected with unlabeled EB-TATE (>50 fold excess) for blocking. (E) Quantification of %ID/g of indicated tracer in tumor, and tumor-to-muscle and tumor-to-blood ratios calculated from PET over time in AR42J tumor model. White arrows indicate the tumor location. **P < 0.01, ***P < 0.001.
Figure 4
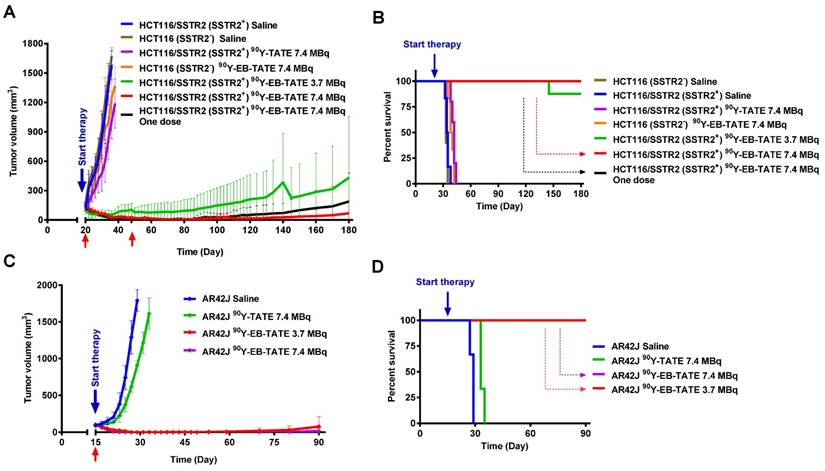
Radionuclide tumor therapy using 90Y. (A) Tumor volume (mm3) of mice bearing HCT116/SSTR2 or HCT116 tumors, injected with either saline, 7.4 MBq 90Y-TATE, 7.4 MBq 90Y-EB-TATE (one or two doses) or 3.7 MBq 90Y-EB-TATE. Red arrows indicate day of injection of the radionuclide to the corresponding group. (B) Survival analysis of mice after radiotherapy study described in A. (C) Tumor volume (mm3) of mice bearing AR42J tumors, injected with either saline, 7.4 MBq 90Y-TATE, 7.4 MBq 90Y-EB-TATE or 3.7 MBq 90Y-EB-TATE. Red arrows indicate day of injection of the radionuclide to the corresponding group. (D) Survival analysis of mice after radiotherapy study described in C. P < 0.001 for 90Y-EB-TATE groups (3.7 and 7.4 MBq) compared to all other groups at all time points.
To test for possible systemic toxicity of the radiotherapy, the body weight of all the mice was monitored throughout the radiotherapy studies (Supplementary Figs. S10 and S11). Although mice treated with 3.7 MBq and 7.4 MBq 90Y-EB-TATE showed slight weight loss, they quickly regained the weight back to normal levels. In accordance, pathologic examination of H&E staining of selected non-cancerous organs and blood analysis revealed no obvious differences between the groups (Supplementary Fig. S12-S13 and Table 1). Biodistribution studies of 90Y-EB-TATE, using a lower dose of 1.85 MBq, was conducted to observe longer term radioactivity uptake (Supplementary Fig. S14). Unexpectedly, at such a low dose, 90Y-EB-TATE still had significant radiotherapeutic efficacy on SSTR2+ tumors, and the tumors were significantly smaller at day 4 post-injection. As a result, the amount of radioactivity in the tumor was significantly reduced (Supplementary Fig. S14). To ensure that the radioactivity accumulation in the tumor was not due to transchelation, the tumors were extracted at days 1, 4, 7 and 12 post-injection and evaluated by radioTLC, which indicated no “free” 90Y form in the tumor (Supplementary Fig. S15).
Discussion
Optimal pharmacokinetics is the Achilles' heel of many drugs, imaging tracers, and radiotherapeutic agents. We recently reported the development of EB-RGD tracer that provided improved pharmacokinetics compared to RGD, and was successfully used as imaging tracer and radiotherapy agent in mouse tumor models (18). Here, we applied the same strategy to improve the pharmacokinetics of TATE peptide, which is a radiotherapeutic agent employed in the clinic. TATE PRRT against somatostatin receptors (SSTRs) has revealed significant potential for the treatment of advanced, low- to intermediate-grade NETs (17). 177Lu-DOTA-TATE is currently the most widely used SST analog in the clinic (17). Nevertheless, its efficacy is limited to about 30% overall response rate despite administration of the high cumulative activity of 29 GBq (17). We hypothesized that improvement of pharmacokinetics of radioactive TATE derivatives would improve tumor uptake, increase treatment efficacy, and, because of reduced renal excretion, reduce the radiation exposure to the kidneys.
Our strategy to improve TATE pharmacokinetics was to synthesize an analog with affinity for human serum albumin, which would be expected to decrease its clearance rate. Human serum albumin, the most abundant protein in blood plasma (~ 50 mg/ml), is the chief carrier protein in the blood and has been exploited as a drug carrier since the mid-1990s (20,21). In the present study, we designed a new molecule, EB-TATE, by conjugation of TATE with an analog of the azo dye, Evans blue (EB) (Fig. 3). Evans blue and its analogs exhibit modest affinity for serum albumin.
Figure 5
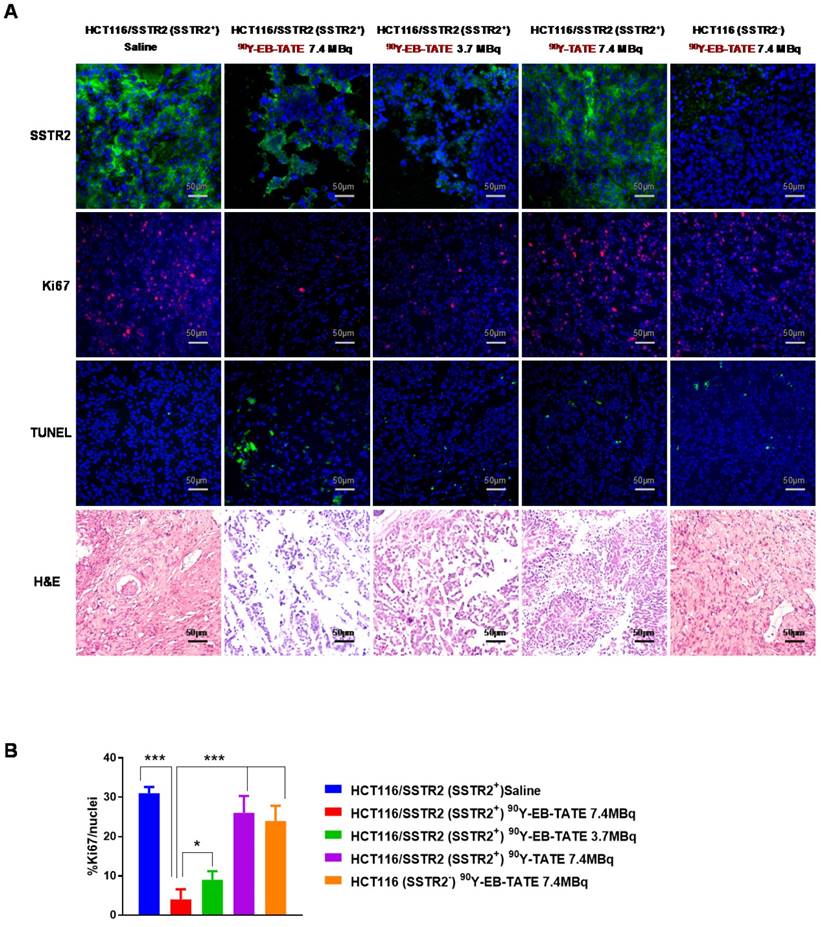
(A) Tumor biology evaluation after 90Y radiotherapy. Immunofluorescence staining (X20) of excised HCT116/SSTR2 tumors for SSTR2 levels (green), Ki-67 (red), TUNEL (green) and H&E after radiotherapy treatment was terminated. In all immunofluorescent staining, blue is nuclear staining. (B) Mean percentage of cells stained for Ki-67 quantified from immunofluorescence staining of excised HCT116/SSTR2 tumors; Saline (blue bar 31 ± 1.6), 7.4 MBq 90Y-EB-TATE (red bar 4 ± 2.6), 3.7 MBq 90Y-EB-TATE (green bar 9 ± 2.2), 7.4 MBq 90Y-TATE (purple bar 26 ± 4.3) and excised HCT116 tumors (orange bar 24 ± 3.8). * P < 0.05, *** P < 0.001.
Table 1
Complete blood counts from mice injected with 7.4 MBq 90Y-EB-TATE (treated) and saline (control) at 90-120 days post-injection.
| Treated | Control | ||||
|---|---|---|---|---|---|
| Average | STD | Average | STD | Units | |
| WBC count | 2.7 | 0.8 | 2.8 | 0.8 | K/µL |
| RBC count | 7.7 | 0.8 | 8.3 | 0.3 | M/µL |
| Hemoglobin | 12.6 | 1.0 | 14.3 | 0.9 | g/dL |
| Hematocrit | 40.5 | 3.7 | 43.8 | 2.1 | % |
| MCV | 53.0 | 1.2 | 53.2 | 1.8 | fL |
| Platelets | 955.5 | 189.1 | 934.5 | 149.7 | K/µL |
| Polys | 24.0 | 7.9 | 21.8 | 2.8 | % |
| Lymphocytes | 57.3 | 11.7 | 64.6 | 4.1 | % |
| Polys Absolute | 0.7 | 0.4 | 0.7 | 0.3 | K/µL |
| Lymphocytes Absolute | 1.37 | 0.40 | 1.81 | 0.50 | K/µL |
During circulation, EB-TATE will be in equilibrium binding with serum albumin. EB-TATE released from albumin in the vicinity of the higher affinity SSTR2, will bind to the receptor expressing cells and subsequently be internalized. The improved pharmacokinetics of EB-TATE, demonstrated by both longer-half life and higher retention in the blood pool, resulted in significantly higher accumulation of 86Y-EB-TATE in SSTR2+ tumor (Fig. 3) and enhanced target to background ratios.
When 90Y was used in place of 86Y, the improved pharmacokinetics provided unparalleled radiotherapy of established SSTR2-positive tumors. AUC calculation comparing 86Y-EB-TATE to 86Y-TATE showed approximately 6-fold higher exposure to the radioisotope (Supplementary Fig. S5.). This difference is a low estimation because 90Y has longer half-life and the decline in activity seen in tumors from mice injected with 86Y-EB-TATE was only due to radioactive decay and not biological clearance. In accordance, in SSTR2 mouse xenografts a single injection of 7.4 MBq was enough to eradicate SSTR2+ tumors (Fig. 4), in contrast to the multiple injection employed in the clinic. A rough dosimetry calculation using the NCI recommended formula (22) showed that treatment of a mouse at an average weight of 27 g with 7.4 MBq is equivalent to treatment of a human subject that weighs 60 Kg with 1.3 GBq, which is an order of magnitude lower than the cumulative 29 GBq currently being used in the clinic. Similarly, other published studies with 90Y-labeled modified octreotide (23) also had to use very high dose (370 MBq 90Y- octreotide for a 300 g rat), and only small but not large tumors had favorable response to one cycle of radiotherapy.
Previous studies have reported that the treatment of some SSTR2-positive tumors resulted in loss of receptor expression and regrowth of tumors (1,24). We have witnessed this same result following the sub-optimal therapy dose of 3.7 MBq (Fig. 4A). The recurring tumors did not respond to additional 90Y-EB-TATE, and showed low uptake of 68Ga-DOTA-TATE on diagnostic imaging. The higher dose of 90Y-EB-TATE, 7.4 MBq, led to eradication of tumors without the emergence of SSTR2-negative tumors thereafter.
In conclusion, we have designed a theranostic agent, based on TATE peptide, that targets SSTR2-expressing tumors and demonstrates improved pharmacokinetics. Utilizing a PET radionuclide, 86Y, and a therapeutic radionuclide, 90Y, we obtain improved contrast for imaging and effective radiotherapy of SSTR2-positive tumors. The results described herein are highly encouraging and suggest that the EB-TATE tracer should be evaluated in human trials.
Acknowledgements
The authors gratefully acknowledge the Intramural Research Program, National Institute of Biomedical Imaging and Bioengineering, National Institutes of Health. The authors are particularly thankful to Dr. Lawrence P. Szajek of the NIH Cyclotron Facility for his prompt assistance in 86Y production. Rui Tian was partially funded by the China Scholarship Council (CSC).
Authors Contributions
- Conception and design: Orit Jacobson, Gang Niu, Xiaoyuan Chen
- Development of methodology: Orit Jacobson, Xiaoyuan Chen
- Acquisition of data: Rui Tian, Orit Jacobson, Zhantong Wang
- Analysis and interpretation of data: Orit Jacobson, Rui Tian, Gang Niu, Dale Kiesewetter, Xiaoyuan Chen
- Writing, review and /or revision of the manuscript: Orit Jacobson, Rui Tian, Dale Kiesewetter, Xiaoyuan Chen
- Administrative, technical, or material support: Gang Liu, Guizhi Zhu, Ying Ma
- Study supervision: Xiaoyuan Chen
Supplementary Material
Supplementary figures.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Appetecchia M, Baldelli R. Somatostatin analogs in the treatment of gastroenteropancreatic neuroendocrine tumours, current aspects and new perspectives. J Exp Clin Cancer Res. 2010;29:19
2. Kharmate G, Rajput PS, Lin YC, Kumar U. Inhibition of tumor promoting signals by activation of SSTR2 and opioid receptors in human breast cancer cells. Cancer Cell Int. 2013;13:93
3. Modlin IM, Oberg K, Chung DC. et al. Gastroenteropancreatic neuroendocrine tumours. Lancet Oncol. 2008;9:61-72
4. Bodei L, Cremonesi M, Kidd M. et al. Peptide receptor radionuclide therapy for advanced neuroendocrine tumors. Thorac Surg Clin. 2014;24:333-49
5. Patel YC, Greenwood MT, Panetta R, Demchyshyn L, Niznik H, Srikant CB. The somatostatin receptor family. Life Sci. 1995;57:1249-65
6. Panetta R, Patel YC. Expression of mRNA for all five human somatostatin receptors (hSSTR1-5) in pituitary tumors. Life Sci. 1995;56:333-42
7. Stengel A, Rivier J, Tache Y. Modulation of the adaptive response to stress by brain activation of selective somatostatin receptor subtypes. Peptides. 2013;42:70-7
8. Keskin O, Yalcin S. A review of the use of somatostatin analogs in oncology. Onco Targets Ther. 2013;6:471-83
9. Tartarone A, Lerose R, Aieta M. Somatostatin Analog Therapy in Small Cell Lung Cancer. Semin Nucl Med. 2016;46:239-42
10. Reubi JC, Laissue J, Krenning E, Lamberts SW. Somatostatin receptors in human cancer: incidence, characteristics, functional correlates and clinical implications. J Steroid Biochem Mol Biol. 1992;43:27-35
11. Reubi JC, Kvols L. Somatostatin receptors in human renal cell carcinomas. Cancer Res. 1992;52:6074-8
12. Buscail L, Saint-Laurent N, Chastre E. et al. Loss of sst2 somatostatin receptor gene expression in human pancreatic and colorectal cancer. Cancer Res. 1996;56:1823-7
13. Rocheville M, Lange DC, Kumar U, Patel SC, Patel RC, Patel YC. Receptors for dopamine and somatostatin: formation of hetero-oligomers with enhanced functional activity. Science. 2000;288:154-7
14. Rocheville M, Lange DC, Kumar U, Sasi R, Patel RC, Patel YC. Subtypes of the somatostatin receptor assemble as functional homo- and heterodimers. J Biol Chem. 2000;275:7862-9
15. Del Prete M, Buteau FA, Beauregard JM. Personalized 177Lu-octreotate peptide receptor radionuclide therapy of neuroendocrine tumours: a simulation study. Eur J Nucl Med Mol Imaging. 2017
16. Kwekkeboom DJ, de Herder WW, Kam BL. et al. Treatment with the radiolabeled somatostatin analog [177 Lu-DOTA 0,Tyr3]octreotate: toxicity, efficacy, and survival. J Clin Oncol. 2008;26:2124-30
17. Cives M, Strosberg J. Radionuclide Therapy for Neuroendocrine Tumors. Curr Oncol Rep. 2017;19:9
18. Chen H, Jacobson O, Niu G. et al. Novel "Add-On" Molecule Based on Evans Blue Confers Superior Pharmacokinetics and Transforms Drugs to Theranostic Agents. J Nucl Med. 2017;58:590-7
19. Peters TJ. All About Albumin. Academic Press. 1995:1 st Edition: 76-132
20. Topchieva IN, Kurganov BI, Teplova MV, Gorskaya IA, Rudakova IP. Use of conjugates of bovine serum albumin with poly(alkylene oxide)s for solubilization of riboflavin ester. Biotechnol Appl Biochem. 1993;17( Pt 3):337-48
21. Gabor F, Wollmann K, Theyer G, Haberl I, Hamilton G. In vitro antiproliferative effects of albumin-doxorubicin conjugates against Ewing's sarcoma and peripheral neuroectodermal tumor cells. Anticancer Res. 1994;14:1943-50
22. Nair AB, Jacob S. A simple practice guide for dose conversion between animals and human. J Basic Clin Pharm. 2016;7:27-31
23. de Jong M, Breeman WA, Bernard BF, Bakker WH, Visser TJ, Kooij PP. et al. Tumor response after [90Y-DOTA0,Tyr3]octreotide radionuclide therapy in a transplantable rat tumor model is dependent on tumor size. J Nucl Med. 2001;42(12):1841-6
24. Scarpignato C, Pelosini I. Somatostatin analogs for cancer treatment and diagnosis: an overview. Chemotherapy. 2001;47(Suppl 2):1-29
Author contact
![]() Corresponding authors: Orit Jacobson, LOMIN/NIBIB/NIH, 10 Center Drive 10/B2C315, Bethesda, MD 20892 301-435-2229 orit.jacobsonweissgov Xiaoyuan Chen, LOMIN/NIBIB/NIH, 35A Convent Drive 35A/GD937, Bethesda, MD 20892 301-451-4246 Shawn.chengov
Corresponding authors: Orit Jacobson, LOMIN/NIBIB/NIH, 10 Center Drive 10/B2C315, Bethesda, MD 20892 301-435-2229 orit.jacobsonweissgov Xiaoyuan Chen, LOMIN/NIBIB/NIH, 35A Convent Drive 35A/GD937, Bethesda, MD 20892 301-451-4246 Shawn.chengov
Citation styles
APA
Tian, R., Jacobson, O., Niu, G., Kiesewetter, D.O., Wang, Z., Zhu, G., Ma, Y., Liu, G., Chen, X. (2018). Evans Blue Attachment Enhances Somatostatin Receptor Subtype-2 Imaging and Radiotherapy. Theranostics, 8(3), 735-745. https://doi.org/10.7150/thno.23491.
ACS
Tian, R.; Jacobson, O.; Niu, G.; Kiesewetter, D.O.; Wang, Z.; Zhu, G.; Ma, Y.; Liu, G.; Chen, X. Evans Blue Attachment Enhances Somatostatin Receptor Subtype-2 Imaging and Radiotherapy. Theranostics 2018, 8 (3), 735-745. DOI: 10.7150/thno.23491.
NLM
Tian R, Jacobson O, Niu G, Kiesewetter DO, Wang Z, Zhu G, Ma Y, Liu G, Chen X. Evans Blue Attachment Enhances Somatostatin Receptor Subtype-2 Imaging and Radiotherapy. Theranostics 2018; 8(3):735-745. doi:10.7150/thno.23491. https://www.thno.org/v08p0735.htm
CSE
Tian R, Jacobson O, Niu G, Kiesewetter DO, Wang Z, Zhu G, Ma Y, Liu G, Chen X. 2018. Evans Blue Attachment Enhances Somatostatin Receptor Subtype-2 Imaging and Radiotherapy. Theranostics. 8(3):735-745.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.