Theranostics
13.3
Impact Factor
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Special Issues
Top
Introduction
Experimental section
Results
Discussion and Conclusion
Supplementary Material
Acknowledgements
References
Introduction
Experimental section
Results
Discussion and Conclusion
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2018; 8(8):2217-2228. doi:10.7150/thno.24041 This issue Cite
Research Paper
Polyamine-Targeting Gefitinib Prodrug and its Near-Infrared Fluorescent Theranostic Derivative for Monitoring Drug Delivery and Lung Cancer Therapy
Xinyu Song1,2, Xiaoyue Han2,4, Fabiao Yu2,3 ![]() , Xiaoyu Zhang1,2, Lingxin Chen2,3
, Xiaoyu Zhang1,2, Lingxin Chen2,3 ![]() , Changjun Lv1,3
, Changjun Lv1,3 ![]()
1. Department of Respiratory Medicine, Binzhou Medical University Hospital, Binzhou 256603, China
2. Key Laboratory of Coastal Environmental Processes and Ecological Remediation, Yantai Institute of Coastal Zone Research, Chinese Academy of Sciences, Yantai 264003, China
3. Medicine Research Center, Institute of Molecular Medicine, Binzhou Medical University, Yantai 264003, China.
4. University of Chinese Academy of Sciences, Beijing 100049, China.
Received 2017-11-27; Accepted 2018-1-26; Published 2018-3-8
Citation:
Song X, Han X, Yu F, Zhang X, Chen L, Lv C. Polyamine-Targeting Gefitinib Prodrug and its Near-Infrared Fluorescent Theranostic Derivative for Monitoring Drug Delivery and Lung Cancer Therapy. Theranostics 2018; 8(8):2217-2228. doi:10.7150/thno.24041. https://www.thno.org/v08p2217.htm
Other stylesAbstract

The therapy of non-small-cell lung cancer (NSCLC) is challenging because of poor prognosis. There are urgent demands for targeting anti-tumor drugs with reliable efficacy and clear pharmacokinetics.
Methods: We designed and synthesized an active tumor-targeting prodrug for the precision therapy of NSCLC. The prodrug polyamine analog Gefitinib (PPG) was derived from the conjugation between a tumor-targeting ligand polyamine analog (PA) and an epidermal growth factor receptor tyrosine kinase inhibitor Gefitinib via a cleavable disulfide linker. Furthermore, the integration of the near-infrared azo-BODIPY fluorophore into the structure of the prodrug PPG yielded an activatable fluorescent theranostics (TPG), which could be used to monitor the in real-time delivery of prodrug PPG and initiate precise medicine in vivo.
Results: PPG efficiently delivered the anti-tumor drug to cancer cells and reduced the serious side effects of the drug to normal cells, thereby increasing the potent of the anti-tumor drug. PPG was not only efficacious for killing Gefitinib-sensitive PC9 cells, but also for inhibiting the growth of Gefitinib-resistant H1650 cells. We provided a new evidence that the tumor-targeting PA ligand could inhibit the Akt pathway in H1650 cells, and had a synergistic effect with Gefitinib for anticancer efficacy. The in vivo results on nude mice bearing tumors of NSCLC cell lines demonstrated that PPG could target tumor lesions and had the expected therapeutic effects. Finally, we used TPG for fluorescent labeling of transbronchial lung biopsy (TBLB) specimens. The results indicated that TPG could provide rapid diagnosis for lung cancer within 4 h.
Conclusion: Our work had identified that PPG could be effectively used for the treatment of Gefitinib-resistance NSCLC in cells and in mice models. The theranostic TPG emerged as a promising fluorescent imaging tool for the application in the therapy and diagnosis of NSCLC.
Keywords: Lung cancer, Drug delivery, Targeting prodrug, Fluorescence theranostics, Precision medicine
Introduction
Non-small-cell lung cancer (NSCLC) is highly malignant and has poor-prognosis worldwide [1]. To counter high metastasis and relapse of NSCLC, patients receive systemic administration of non-selective antitumor drugs causing serious side effects [2]. Recently approved drugs in oncology mainly focus on small molecule inhibitors and receptor antagonists [3] as well as monoclonal antibodies (mAb), which, due to their high molecular weight, are limited by prolonged clearance time [4]. Gefitinnib is a inhibitor of epidermal growth factor receptor tyrosine kinase (EGFR-TKIs) [5], which benefits NSCLC patients with EGFR-activating mutations. Gefitinib blocks tyrosine kinase and interrupts the phosphorylation of the mutant EGFRs on the cell membrane, terminating the downstream cell signal transduction of EGFR [5]. However, the activation of the EGFR pathway does not follow a linear pattern and is a rather complex process. For example, patients with a loss of phosphatase and tensin homolog (PTEN) can incorrectly activate the downstream Akt pathway when the EGFR pathway is blocked [6]. For this reason, not all patients with mutant EGFRs benefit from Gefitinib [7, 8]. Thus, the initial drug-resistance has become a challenging issue that needs to be addressed.
Targeted drug delivery systems decorated with cancer cell-specific small-molecule ligands are expected to improve the prognosis of cancers because these systems can guide antitumor drugs to cancer lesions while leaving healthy tissues mostly unaffected [9]. This approach relies on the selection of receptor and its targeting ligand. Receptors overexpressed on cancer cells are considered to be potential candidates for targeted delivery of drugs. Compared with the macromolecular targeting ligands, such as antibodies, aptamers, small protein scaffolds, and peptides, the small molecular weight ligands offer many advantages including good pharmacokinetics, non-antigenicity, excellent biological stability, ease of chemical synthesis, and strong ability to penetrate solid tumors [10]. The targeting drugs are delivered specifically to cancer cells, and their high accumulation in cancer cells can contribute to improved therapeutic efficacy and reduced toxicity. Drug effects on tumor lesions rely on its pharmacokinetics (PKs) i.e., absorption, distribution, metabolism, and elimination. Furthermore, the targeted therapeutic systems can carry multiple inhibitors impairing signal transduction in cancer cells [11, 12].
Imaging technologies, such as magnetic resonance, nuclear tomography, X-ray computed tomography, and positron emission tomography-computed tomography, have become indispensable in personalized medicine [12]. Advances in imaging technologies have extended to drug development by visualizing the pharmacokinetics [13, 14]. However, these techniques lack sensitivity or specificity for the early assessment of cancer. Furthermore, there are often conflicting clinicopathological and metabolic results [10, 13, 14] that need to be addressed. Compared to other imaging modalities, fluorescence imaging is of particular significance for investigating specific molecular events in live cells or whole organisms [15-18]. Also, it is inexpensive, fast, noninvasive, has high spatial and temporal resolution, and does not involve radionuclides [19]. However, the fluorescence imaging technique is predominantly obstructed by tissue penetration and tissue auto-fluorescence and also causes phototoxicity. The near-infrared (NIR, 650-900 nm) fluorescence can maximize tissue penetration while minimizing the absorbance of heme in hemoglobin and myoglobin, water, and lipids [20]. For in vivo imaging, it is preferable to choose fluorophores with excitation and emission spectra in the NIR region [21, 22]. In this regard, synthetic methods can be used to develop small-molecule fluorescently labeled drugs with similar PKs to their therapeutic non-labeled forms [23-27]. These fluorescent reagents can be utilized not only to directly indicate the distribution of the administered drug, but also to illustrate the effect on tumor lesions. This emerging field of the combination of diagnosis and therapy, theranostics, is gaining credence in personalized medicine [21, 28-32].
We designed and synthesized a new prodrug polyamine analog Gefitinib (PPG) by conjugating a polyamine-targeting ligand for the therapy of NSCLC (Scheme 1). PPG enhanced the efficacy of antitumor activity against murine solid tumor models comparable to the clinical effects of Gefitinib. We fluorescently labeled PPG to be used as a theranostic agent (TPG) and validated the targeting ability and efficacy of the new prodrug in cells. Furthermore, by using in vivo imaging, we analyzed in real-time drug targeting and distribution in a xenograft model. Our results indicated that the new prodrug PPG exhibited high antitumor effects on the drug-resistant H1650 cell line and in the xenografted tumor model. Thus, our findings have validated that the newly designed theranostic is not only a powerful platform for the real-time noninvasive visualization of therapeutic efficacy both in vitro and in vivo but is also valuable for the diagnosis of NSCLC.
Experimental section
Ethics statement
All surgical procedures were conducted in conformity with National Guidelines for the Care and Use of Laboratory Animals. Experimental protocols were approved by the Institutional Animal Care and Use Committee in Binzhou Medical University, Yantai, China. Approval Number: No.BZ2014-102R.
Cell culture
NCI-H1650 and IMR-90 cell lines were purchased from the Committee on Type Culture Collection of Chinese Academy of Sciences (Shanghai, China). PC9 cell line was purchased from European Collection of Authenticated Cell Cultures. Human non-small-cell lung cancer cell lines, PC9 cells and NCI-H1650 cells, were cultured in high glucose Dulbecco's Modified Eagle's Medium (DMEM) (Hyclone, USA) and RPMI (Roswell Park Memorial Institute)-1640 medium (Gibco, USA), respectively, supplemented with 10% (v/v) fetal bovine serum (Gibco, Australia) and 100 units/mL penicillin and streptomycin. The human diploid fibroblast-like cell line, IMR-90, was cultured in complete medium comprised of 87 mL Minimum Essential Medium (MEM) (Gibco, USA), 10 mL FBS, 1 mL Gluta-max (Gibco, USA), 1 mL NEAA (Gibco, USA) and sodium pyruvate (Gibco, USA). All cells were maintained at 37°C in a humidified atmosphere with 5% CO2. All cells tested negative for mycoplasma with a PCR-based detection method.
Animal models
All animal experiments were performed in accordance with the guidelines established by the Committee on Animal Research Policy of Binzhou Medical University. 5-week-old female specific pathogen free (SPF) athymic nude mice and BALB/c mice were obtained from Changzhou Cavens Lab Animal Co. Ltd. The animals were housed in individually ventilated cages and fed a SPF laboratory diet and water ad libitum. 2 × 106 cells were suspended in media and implanted subcutaneously into nude mice.
Patient samples
The transbronchial lung biopsy (TBLB) tissues were obtained from the Department of Respiratory Medicine, Affiliated Hospital of Binzhou Medical University. CT scanning was performed before TBLB to ensure the suspected locations. Informed written consents were provided by all patients. The experiments were approved by the Ethics Committee of Binzhou Medical University.
Cancer cells detection in clinical tissues
TBLB tissues were separated with a bronchoscope inside the lung and placed into 10 mL cold Hank's Balanced Salt Solution (HBSS) (Gibco, USA). The tissues were cut into pieces as small as possible and washed three times by HBSS. 200 U/mL type I collagenase (Gibco, USA) was heated to 37°C in HBBS. The finely cut small pieces of tissues were placed into the collagenase solution at 37°C for 3 h to obtain the single cell suspension. The screen mesh was used to filter the cell masses, and the collagenase was discarded by centrifugation. The cells were washed with PBS three times and treated by FACS Lysing Solution (BD Bioscience, USA). The cells were again washed three times with PBS and counted before suspending in 1 mL RMPI-1640 medium. 5 μM TPG was added to the single cell suspension to stain for 15 min at 37°C. After washing twice with PBS, the cells were treated with ice-cold ethanol at -20°C overnight. Subsequently, CEA Antibody, FITC (CI-P83-1) (Invitrogen, USA) and Cytokeratin 19 Monoclonal Antibody (A53-B/A2), or Alexa Fluor 488 (Invitrogen, USA) were used to stain the cells for 30 min. After three washes with PBS, 104 cells were used for flow cytometry.
Statistical analysis
Statistical Product and Service Solutions (SPSS) software 19.0 was used for the statistical analysis. The error bars shown in the figures represent the mean ± s.d. Differences were determined with a one-way, two-way analysis of variance (ANOVA) followed by the LSD test. Differences between two groups were assessed by using the Student's t-test. The Kaplan-Meier method was used to evaluate survival. Statistical significance was assigned at *P < 0.05, **P < 0.01. The sample size was chosen empirically based on our previous experience and pre-test results. No statistical method was used to predetermine sample size and no data were excluded. The numbers of animals or samples in every group are described in the corresponding figure legends. The distributions of the data were normal. All experiments were done with at least three biological replicates. Experimental groups were balanced regarding animal age, sex, and weight. All animals were caged together and treated in the same way. Appropriate tests were chosen according to the data distribution. The variance was comparable between groups in experiments described throughout the manuscript. The investigator was not blinded to the group allocation during the experiment or to outcome assessments. No randomization was used to allocate animals to experimental groups.
Results
Molecular design and synthesis of the prodrug and its theranostic capabilities. There is an urgent need to rationally design targeted prodrugs for cancers. The general design strategy of this class of prodrugs involves a targeting ligand that is linked to an antitumor drug via a cleavable bond (Scheme 1a). The targeting ligand delivers the drug into cancer cells. The overexpressed receptors on cancer cells are excellent candidates for targeting ligands [33-35]. To date, there has been little effort spent identifying specific targeting ligands for NSCLC. In this respect, polyamines, including putrescine, spermidine, and spermine, can accumulate in lungs [36] and cause the differentiation of type II alveolar epithelial cells (AECIIs) [37]. The polyamine uptake system (PUS) is an ATP-dependent transmembrane transport system that is preferentially expressed on AECII cells [36]. The overexpression of polyamines can, therefore, be considered as a new therapeutic target for NSCLC [38, 39]. Polyamine analogs (PA) can competitively bind the PUS and accumulate in AECIIs, serving as a potential targeting moiety for NSCLC.
We modified polyamine to yield a low-toxicity derivative as the desirable targeting ligand (Scheme 1a). The cleavable bond is used for drug release in the tumor microenvironment. Given the high concentration of glutathione (GSH) in tumor cells, we chose disulfide bond as the cleavable linker. After conjugating the targeting ligand with Gefitinib, we obtained a targeted prodrug PPG (Scheme 1a). The rationale is when the prodrug is targeted to tumor cells, the high level of intracellular GSH will trigger the activation of the disulfide bond (Figure S1), resulting in sustained release and higher local concentration of Gefitinib for killing tumor cells.
The molecular structure of the theranostic was divided into four components (Scheme 1b). The targeting ligand was conjugated with a fluorophore for diagnosis. The antitumor drug was introduced via a cleavable bond for therapy [40]. As a proof of concept, we selected azo-BODIPY as the NIR fluorophore due to its excellent fluorescence properties and biocompatibility [41]. As illustrated in Figure S2, we obtained the fluorescent theranostic TPG in which the fluorescence emission was blocked before the cleavage of the disulfide bond. Once the disulfide bond was cleaved, azo-BODIPY emitted fluorescent signals for tracking the pharmacokinetics of Gefitinib and illustrating the therapy of NSCLC in real-time. The details of the synthetic process and characterization of the compounds are shown in Supplementary Material.
Spectral dynamics for GSH. We employed the theranostic TPG to examine the targeting performance and pharmacokinetics of the prodrug PPG. TPG exhibited a maximum absorption band at 671 nm (Figure S3A). Addition of GSH (10 mM) cleaved the disulfide bond, which was followed by sustained release of Gefitinib. A new absorption peak appeared at 706 nm with accompanying fluorescence emission at 730 nm (Figure S3B). The gradual increase in fluorescence intensity reached a plateau after 17 h (Figure S3C), which could be used to trace the release of Gefitinib (Figure S4). We also monitored the stability and selectivity of TPG in the presence of various bioactive species (Figure S3D-F). The results showed that only high concentrations of biothiols could trigger fluorescence. Other bioactive species, such as amino acids, peptides, reactive oxygen species, reactive nitrogen species, and metal ions barely induced fluorescence changes. This observation was promising in that both the prodrug and theranostic did not release Gefitinib until they were delivered into tumor cells containing a high concentration of biothiols, such as GSH. These results demonstrated that TPG could be used to detect the delivery of Gefitinib to cancer tissues because of fluorescence changes.
Scheme 1
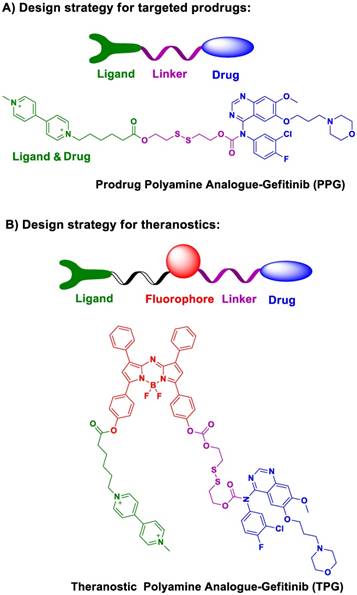
Overview of the proof of concept and chemical structure of the potential prodrug and theranostic TPG.
Efficacy evaluation in cells. To validate the efficacy of our prodrug and theranostic, we selected two human NSCLC cell lines as cell models, PC9 and H1650 cell lines. As is known, the PC9 cell line is sensitive to Gefitinib, while the H1650 cell line is resistant to Gefitinib as a result of activation of the Akt pathway [6]. We incubated both NSCLC cell lines with TPG and collected fluorescent signals starting at 1 h. As shown in Figure S5, after the administration of TPG, the fluorescence intensity reached its highest value at 16 h, indicating a process of sustained drug release. The fluorescence lasted for more than 23 h, indicating a long intracellular retention time that would facilitate long-term monitoring and imaging analysis. The exogenous addition of cysteine could markedly increase the fluorescence intensities in cells, indicating acceleration of the drug release process (Figure S6). If the cells were pretreated with N-ethylmaleimide (NEM) for 30 min to eliminate the biothiols, almost no fluorescence was detected (Figure S7). The results demonstrated that the drug release was controlled by intracellular biothiols (mainly GSH).
Since the transmembrane transport of a targeted prodrug is mediated by the relevant receptors that are overexpressed on cell membranes, we next examined the cellular transport of the prodrug. Complete saturation of receptors resulted in the obstruction of transport of the extracellular prodrug into cells. The energy-dependent transport process could also be suspended at low temperature because of the reduced respiration intensity. The two cell lines were divided into four groups. Cells in two groups were pretreated with excessive PA for the inhibition of its receptors. The other two groups of cells were placed in a milieu at 4°C for 1 h to reduce physiological activities. As shown in Figure S8, no fluorescence was detected under these conditions and TPG could not be transported into cells. There was faint fluorescence around the cells (Figure S9), providing strong evidence that the accumulated TPG could not cross the cell membrane. These results also demonstrated that the entry of our prodrug into cells was dependent on receptor-mediated transmembrane transport.
The cleaved-caspase 3 (c-caspase 3) is considered to be a marker of irreversible cell apoptosis [42]. We exploited the commercial Caspase-3/7 Green Detection Reagent for visual assessment of the cytotoxicity of our prodrug. PC9 and H1650 cells were treated with TPG for 24 h. Then the cells were co-stained with Caspase-3/7 Green Detection Reagent and nuclear dyes Hoechst 33258. As shown in Figure 1A, the red fluorescence of TPG displayed drug release in cells. Cell apoptosis resulted in an increase in green fluorescence originating from Caspase-3/7 Green Detection Reagent. Co-localization assay with Hoechst 33258 proved caspase 3-dependent apoptosis. For the Gefitinib-resistant H1650 cell line, TPG performed well for killing H1650 cells (Figure 1A). Cytotoxicity tests of PPG provided similar results as TPG. The results confirmed that the theranostic TPG could be used to investigate the pharmacokinetics of the prodrug PPG, and the prodrug PPG was effective for the Gefitinib-resistant H1650 cell line.
We further examined the superior efficacy of our prodrug PPG towards H1650 cells using the cell viability assay. For the inhibition of PC9 cells, the prodrug and its corresponding theranostic agent provided a slight advantage over Gefitinib (Figure 1B). The IC50 values of Gefitinib, PPG, and TPG were 0.74, 0.07 and 0.05 μM, respectively. However, when examined in H1650 cells, only PPG and its corresponding theranostic TPG offered the desirable result of inhibiting proliferation of H1650 cells (Figure 1B). The IC50 values for Gefitinib, PPG, and TPG were 16.9, 0.58 and 0.78 μM, respectively. Furthermore, the consistent behaviors of the prodrug and its corresponding theranostic TPG make them competent tools for assessing pharmacokinetics. We also performed an Annexin V/PI assay to assess the apoptotic rate induced by the prodrug and corresponding theranostic for 72 h. The control group was treated with Gefitinib. As displayed in Figure 1C, treatment with TPG and PPG resulted in the corresponding apoptotic rates of 58% and 56.1% in PC9 cells, which were much higher than that caused by Gefitinib (39.2%). The apoptosis rates induced by TPG and PPG in H1650 cells were 69.1% and 72.6%, while the apoptosis rate of H1650 cells following exposure to Gefitinib was 24.5% (Figure 1C). These data were consistent with the CCK-8 assays. The expression of c-caspase 3 was also examined using Western blot analysis. The prodrug and its corresponding theranostic induced a higher expression of c-caspase 3 in PC9 cells than Gefitinib (Figure 1D). In H1650 cells, PPG and TPG exclusively upregulated expression of c-caspase 3 (Figure 1D). These results demonstrated that the efficacies of our new prodrug PPG surpassed Gefitinib in PC9 cells. The distinct behavior of PPG prompted us to examine the underlying mechanism for the stronger cytotoxicity of PPG compared to Gefitinib. We hypothesized that the PA not only served as a ligand for NSCLC but also participated in damaging cancer cells. To confirm the hypothesis, we performed migration assays on PC9 and H1650 cells by the scratch method. The cells were treated with 1 μM Gefitinib, PPG and TPG for 24 h. The migration distances represented the adhesive and invasive abilities of cells (Figure 1E). PPG and TPG decreased the migration distances of PC9 and H1650 cells significantly, indicating their weakened adhesion and invasion to the adjacent cells.
Figure 1
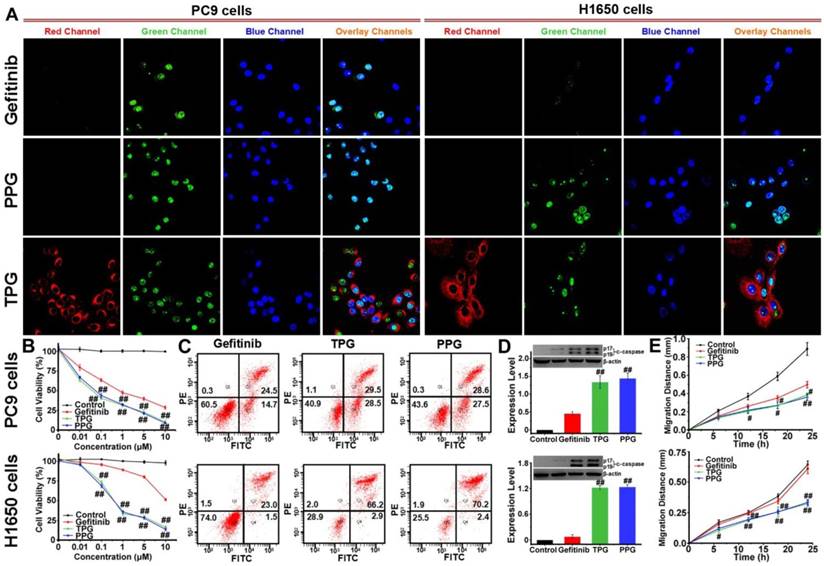
Antitumor imaging and efficacy of TPG and PPG in NSCLC cells. (A) Fluorescence imaging of apoptosis in PC9 and H1650 cells. The cells were treated with 1 μM Gefitinib, TPG, or PPG (red channel: λex/em = 635/670-770 nm) for 24 h, co-stained with Hoechst 33258 (blue channel: λex/em = 405/410-490 nm) for 30 min and Caspase-3/7 Green Detection Reagent (green channel: λex/em = 488/500-570 nm) for 1 h and examined by confocal laser scanning microscopy (n = 3 independent experiments). The overlay images are the combination of red, blue and green channels. Bright fields are shown in Supplementary Material. Scale bar: 20 μm. (B) Cell viabilities of PC9 and H1650 cells. The cells were treated with Gefitinib, TPG, or PPG at 0, 0.01, 0.1, 1, 5 or 10 μM for 72 h, and then measured using CCK-8 kit. The error bars shown in the figures represent the mean ± s.d. (n = 6 independent experiments). (C) Apoptosis of PC9 and H1650 cells. Cells were treated with 1 μM Gefitinib, TPG, or PPG at 37°C for 72 h and detected by Annexin V/PI assay with flow cytometry (n = 3 independent experiments). (Q1: necrotic cells, Q2: late apoptotic cells, Q3: survival cells, Q4: early apoptotic cells). (D) The expression levels of cleaved-caspase 3 in PC9 and H1650 cells (n = 3 independent experiments). Inset: results of Western blot analysis. Untreated cells were used as controls, and β-actin served as the loading control. (E) The migration distance of PC9 and H1650 cells treated with 1 μM Gefitinib, TPG or PPG for 24 h (n = 6); untreated cells were used as controls. The error bars shown in the figures represent the mean ± s.d. Differences were determined with a one-way ANOVA. #P < 0.05, # #P < 0.01 vs. Gefitinib group.
Mechanism of Akt inhibition. As an excellent EGFR-TKI, Gefitinib has benefited NSCLC patients. However, the problem of drug resistance is not yet resolved. As described above, PPG can induce apoptosis in H1650 cells and may also alleviate drug resistance. H1650 cells have a delE746-A750 deletion in exon 19 of PTEN, inactivating the gene [43]. Akt (serine-threonine protein kinase) is the most important downstream pathway of EGFR. The loss of PTEN contributes to unrestrained activation of the Akt signal transduction pathway, which is a pivotal mechanism for the survival of H1650 cells in the presence of Gefitinib [44]. Since phosphorylation of Ser473 is important for activation of the Akt pathway [45], it is possible that inhibition of the overactive Akt pathway might be crucial for drug-resistant NSCLC. As we have shown earlier, the targeting ligand of prodrug PPG transferred Gefitinib across the cell membrane in H1650 cells and was highly proapoptotic. It was reasonable to assume that the PA ligand of PPG might participate in the inhibition of p-Akt.
We next examined the potential pharmacological effects of PA on H1650 cells using CCK-8 and Annexin V/PI assays. The human embryonic lung IMR-90 cell line was used as a control. The cell viability of IMR-90 cells was unaffected by PA (Figure 2A). As displayed in Figure 2B, the survival rate of H1650 cells remained almost 100% after treatment with 10 μM PA for 24 h. However, the 48 h survival rate was altered in a concentration-dependent manner, indicating that PA bound to the cancer cells with high affinity and caused long-term effects on H1650 cells. The results were further verified by an Annexin V/PI assay. Flow cytometry analysis revealed late apoptosis in H1650 cells at 48 h (Figure S11). The polyamine derivative paraquat, a poisonous herbicide [46], was applied as a control and compared with PA treatment. PA could cause destructive injury to cells, as seen under transmission electron microscopy (TEM). Upon exposure to PA for 48 h, subcellular changes in morphology, such as mitochondria swelling and endoplasmic reticulum dilatation, were observed, causing acute cytotoxicity. As displayed in Figure 2C, the toxicological effects of PA on cells were distinct from those caused by paraquat. We examined the expression levels of ornithine decarboxylase (ODC), the key enzyme for the metabolism of polyamines [47], in PC9, H1650, and IMR-90 cells (Figure 2D). The overexpression of ODC in lung cancer cells indicated that the PUS was a feasible receptor for the targeted delivery of the prodrug PPG, illustrating that PA ligand could offer potential tumor targeting ability.
Figure 2
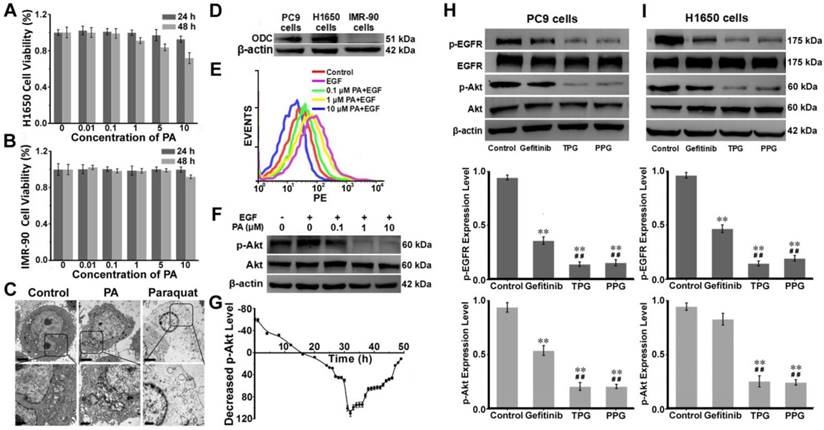
Mechanism of apoptosis induction by TPG and PPG. Cell viabilities of (A) IMR-90 cells and (B) H1650 cells. Cells were treated with 0, 0.01, 0.1, 1, 5 and 10 μM of PA and measured using the CCK-8 assay. The error bars shown in the figures represent the mean ± s.d. (n = 6 independent experiments). (C) H1650 cells treated with 1 μM PA and 1 μM paraquat for 48 h and observed by transmission electron microscopy (n = 3 independent experiments). Scale bars: 2 μm (upper) and 1 μm (lower). (D) The expression levels of ornithine decarboxylase (ODC) in PC9, H1650, and IMR-90 cells (n = 3 independent experiments). (E) Flow cytometry analysis and (F) Western blot analysis of the levels of p-Akt (Ser 473) and total Akt (n = 3 independent experiments). H1650 cells were treated with PA for 48 h. (G) Decreased levels of p-Akt in H1650 cells treated with 1 μM PA from 0 to 48 h (n = 3 independent experiments). Samples were subjected to stimulation with 10 ng/mL EGF for 5 min. (H) The expression levels of p-EGFR and p-Akt in PC9 cells and (I) H1650 cells (n = 3 independent experiments). Total EGFR and Akt were also tested. The cells were treated with 1 μM Gefitinib, TPG, and PPG for 72 h. The error bars shown in the figures represent the mean ± s.d. Differences were determined with a one-way ANOVA. *P < 0.05, **P < 0.01 vs. control group. # P < 0.05, # # P < 0.01 vs. Gefitinib group.
Using flow cytometry, we verified the changes in the phosphorylation (Ser 473) level of Akt. H1650 cells were treated with epidermal growth factor (EGF) and different concentrations of PA. EGF was an activator for p-Akt, and the increasing concentrations of PA enhanced the inhibition of p-Akt (Figure 2E). These results were consistent with those obtained from Western blot analysis (Figure 2F). We next studied the inhibition of p-Akt by PA. H1650 cells were incubated with 1 μM PA during the assay. H1650 cells were first induced by EGF for 5 min to activate p-Akt. As shown in Figure 2G, the level of p-Akt was strongly inhibited by PA after 16 h. The inhibition of p-Akt reached its maximum at 32 h. There was a sudden decrease from 30 h to 32 h, following which the degree of inhibition recovered gradually. This was probably the reason why apoptosis occurred at the time interval between 24 h and 48 h (Figure 2G). These results indicated the PA ligand's synergistic effect with Gefitinib on the inhibition of p-Akt in H1650 cells. It mainly caused cell apoptosis rather than necrosis in resistant H1650 cells.
We further investigated the drug effects of TPG and PPG in PC9 cells and H1650 cells. The two cell lines were treated with Gefitinib, TPG, and PPG at 1 μM. As illustrated in Figure 2H, the inhibition of p-EGFR downregulated the level of p-Akt in PC9 cells. The effects of the two agents were greater than Gefitinib. H1650 cells suffered from decreased p-EGFR following the downregulation of p-Akt when treated with TPG and PPG (Figure 2I). The results demonstrated that the PA ligand could deliver Gefitinib to cancer cells. Furthermore, in H1650 cells, the p-Akt downstream of p-EGFR, which is the resistant mechanism of H1650 cells for Gefitinib, could be inhibited. As expected, TPG and PPG could inhibit the phosphorylation pathway of EGFR and Akt, leading to cell apoptosis.
Figure 3
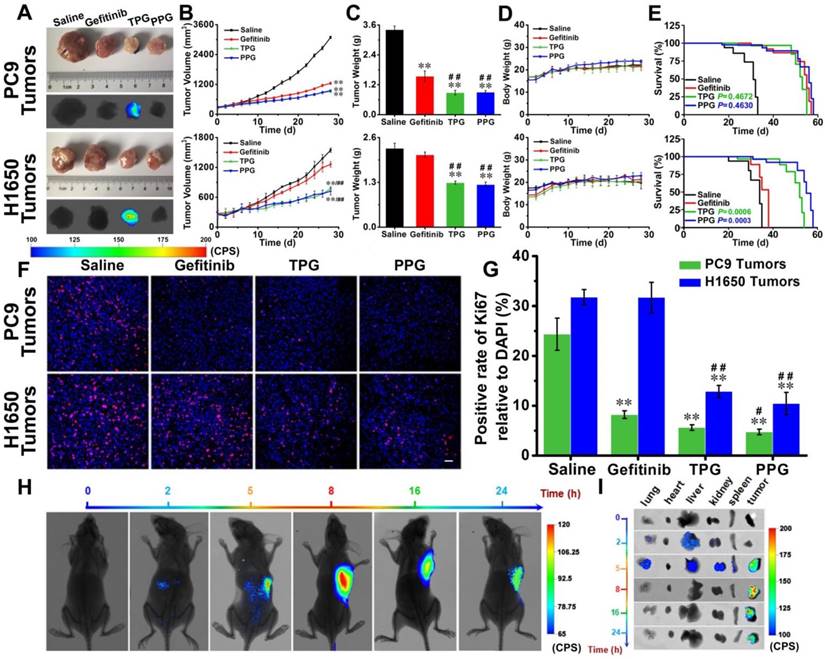
Efficacy evaluation in vivo. Nude mice with PC9 cells and H1650 cells subcutaneous tumor xenografts were established and treated with saline, Gefitinib, TPG, or PPG (0.5 mM in 0.2 mL, DMSO/saline, 1:1/v/v, qod. iv.) (n = 7 per group). (A) Tumor masses and fluorescence images. λex = 610 nm, filter: 710 nm. (B) Tumor sizes. The calculation of the volume was according to the formula: volume = length × width2 × 0.5. (C) Tumors mean weights. (D) Body weights. The error bars shown in the figures represent the mean ± s.d. (E) Survival curves of the tumor-bearing nude mice following treatment. (F) Immunofluorescence staining of proliferation markers Ki67 (red channel: λex/em = 635/670-770 nm) with anti-Ki67 mAb (Alexa Fluor 647 conjugate) and nuclei (blue channel: λex/em = 405/410-490 nm) with DAPI of PC9 and H1650 tumor sections (n = 3 independent experiments). Scale bar: 50 μm. (G) Statistical analysis of the data derived from (F). The error bars shown in the figures represent the mean ± s.d. Differences were determined with a one-way ANOVA except the tumor sizes with a two-way ANOVA. *P < 0.05, **P < 0.01 vs. control group. # P < 0.05, # # P <0.01 vs. Gefitinib group. The Kaplan-Meier method was used to evaluate survival. (H) Imaging of the subcutaneously implanted H1650 tumor xenografts of nude mice at 2, 5, 8, 16 and 24 h after tail vein injection of a single-dose of 0.2 mL of TPG (DMSO/saline 1:1/v/v) (n = 3 independent experiments). (I) Images of the excised organs (lung, heart, liver, kidney, spleen) and tumors of the mice from (G) (n = 3 independent experiments).
Efficacy evaluation in vivo. Since our prodrug and theranostic were shown to be efficacious in cultured cells, we attempted to test their in vivo efficacy. Tumor-bearing (PC9 cells and H1650 cells) nude mice were divided into four groups. The mice were injected via tail vein with saline, Gefitinib, TPG, and PPG every other day. Tumor volumes and nude mouse weights were measured every two days for 28 days. Subsequently, all tumors were dissected and weighed. As illustrated in Figure 3A, except saline, all the agents provided effective treatment for PC9 tumor-bearing mice. The growth of tumors in H1650 mice was significantly inhibited by systemic administration of TPG and PPG, but no noticeable difference was observed in tumor size of the mice administered Gefitinib (Figure 3B). The inhibition rate curves and tumor weights are displayed in Figure 3B and 3C. The control group and the therapeutic groups showed no obvious difference in body weights (Figure 3D), indicating low systemic side effects of the therapeutic agents. The survival curves of PC9 tumor-bearing nude mice indicated that TPG and PPG prolonged the life span compared with the saline group. However, both prodrugs did not show an advantage compared with Gefitinib. We observed that both TPG and PPG benefited the H1650 tumor-bearing nude mice compared with Gefitinib (Figure 3E). We used Ki67, the nuclear protein indicative of cell proliferation and a common immunological biomarker in clinical diagnosis, to monitor tumor cell proliferation. As displayed in Figure 3A, tumor tissues were sectioned and co-stained by Ki-67 (D3B5) Rabbit mAb (Alexa Fluor 647 conjugate) and DAPI. Compared with the control group, significantly decreased numbers of Ki67-positive cells revealed that Gefitinib, TPG, and PPG could effectively inhibit the cell proliferation in PC9 cells-derived tumors (Figure 3F, G). In contrast, the number of Ki67-positive cells was hardly reduced in H1650 cell-derived tumors unless the tumors were treated with TPG and PPG. The results indicated that TPG and PPG were efficacious for the nude mice bearing drug-resistant H1650 tumors.
Since PPG exhibited excellent efficacy in both H1650 and PC9 tumors, we examined its targeting ability in vivo by using its corresponding theranostic TPG. To observe the organ-targeting ability of PA, we obtained Probe-PA by conjugating PA and azo-BODIPY. Probe-PA and TPG were injected in a single dose 100 μM (200 μL DMSO: saline 1:1 v/v) via the tail vein. The mice were imaged in real-time via a small animal in vivo imaging system. As shown in Figure S12, Probe-PA could accumulate in the lungs within 4 h, following which the probe was quickly eliminated from the lungs within 6 h. The result showed the excellent targeting ability of the probes to the lungs. The H1650-derived tumors overexpress the polyamine uptake system. As illustrated in Figure 3H and I, TPG was mainly delivered to the tumor mass and released Gefitinib. The emitted fluorescence indicated the drug release process and the tumor treatment status in real-time. The fluorescence mainly accumulated in the tumor tissue and the drug release occurred in the cytoplasm (Figure S13). There was no fluorescence in the lung, liver, and kidneys 8 h after the intravenous administration of TPG. However, fluorescence signal in the tumor lesion could be maintained for more than 24 h. We next checked the pulmonary toxicity of TPG and PPG. As shown in Figure S14, TPG and PPG did not induce pulmonary fibrosis during 28 days of treatment. These results demonstrated that PA enabled the targeted delivery and efficacy of the prodrug PPG to tumor lesions. The increasing concentrations of Gefitinib and PA in situ could synergistically kill resistant H1650 cells. Using the theranostic TPG, we could not only visually study its pharmacokinetics but could also image tumors for treatment effects in real-time.
Figure 4
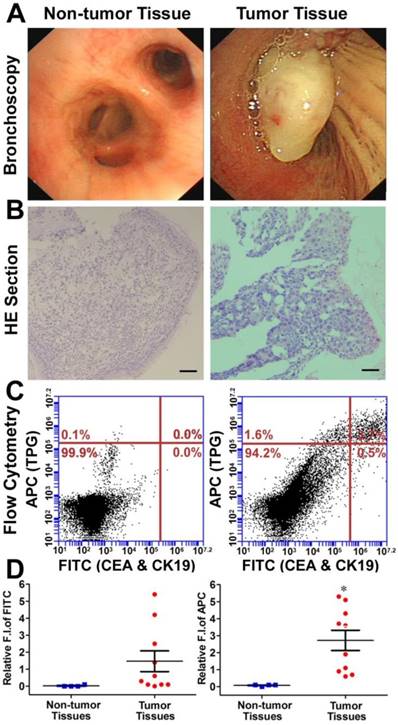
Diagnosis of TBLB specimens by TPG. (A) Representative non-tumor tissue and tumor tissue specimens were acquired by TBLB. (B) H&E staining of (A). Scale bar: 50 μm. (C) Flow cytometry analysis of TPG and immunofluorescence using anti-CEA and anti-CK19. The cells in the suspension were treated with 1 μM TPG and fixed to process immunofluorescence of CEA and CK19. The results are shown as density plots with CEA and CK19 in the FITC channel and TPG in the APC channel. (D) Statistical data of the FITC and APC channels of the non-tumor tissues (n = 4) and tumor tissues (n = 10). All tissues were confirmed by pathological examination. The data are presented as plot diagrams and the comparisons between two groups were determined using Student's t-test. *P < 0.05 was statistically significant.
Diagnosis of transbronchial lung biopsy (TBLB) samples. The theranostic TPG could be used to target tumor cells both in vitro and in vivo. We further evaluated the practical application of TPG in clinical diagnosis via flow cytometry. Test samples of TBLB were obtained from 14 patients. All tissue specimens were diagnosed by pathological biopsy (Table S1). Two commercial serum biomarkers, carcinoembryonic antigen (CEA) for adenocarcinomas and cytokeratin 19 (CK19) for squamous carcinomas, were selected as comparisons for NSCLC [50]. The detection channel was FITC and the NIR fluorescence of TPG was collected in the APC channel. Fresh TBLB specimens were dissociated by type I collagenase and cell suspensions were tested after co-staining with commercial agents and the theranostic TPG. The testing results were obtained using the relative fluorescence intensities in two channels. According to the results of non-tumor tissues shown in Figure S15, the reference value was 0.1 as the criterion for distinguishing whether TBLB tissue was cancerous. We verified the diagnostic ability of TPG by screening all 10 tumor tissues. As illustrated in Figure 4, the commercial biomarkers CEA and CK19 failed in screening 4 samples. Additionally, the changes in serum biomarker were consistent with the results of flow cytometry analysis. (Table S1). The false negative results could be attributed to the complexity of NSCLC. Therefore, the biomarker detection was limited in its specificity and sensitivity. Our results showed that TPG had better specificity and sensitivity compared to the other biomarkers. There was a significant difference between non-tumor and tumor samples. The diagnosis of NSCLC using TPG could avoid ambiguities derived from tumor heterogeneity and individual differences, and the results could be rapidly obtained within 4 h. Thus, the preliminary diagnosis of NSCLC by TPG was reliable and rapid, suggesting that TPG can be potentially utilized for the accurate diagnosis of NSCLC.
Figure 5
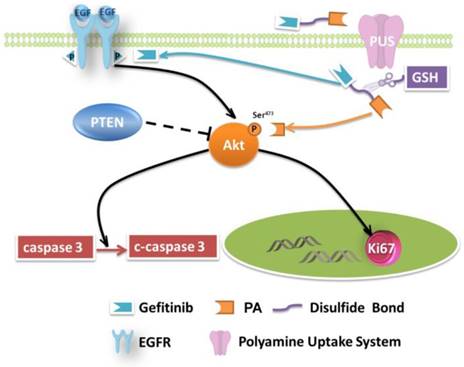
Mechanisms of growth inhibition of NSCLC cell lines by TPG.
Discussion and Conclusion
NSCLC involves several pathological patterns, such as squamous carcinoma, adenocarcinoma, and large cell carcinoma. Its early metastasis is the main reason for the poor prognosis. Once metastases occur in ipsilateral mediastinal or subcarinal lymph nodes, aggressive local therapy provides little survival advantage [51], and the patients suffer from severe side effects caused by medications. In this respect, tumor-specific small-molecule ligand prodrugs can enhance efficacy and avoid unwanted toxicities. The targeted prodrugs deliver therapeutic agents specifically to tumor cells, thereby reducing the exposure of normal cells to therapeutic agents. One example is our newly designed targeted prodrug PPG, which is efficacious for NSCLC. Another significant advantage is the possibility of using the PA ligand as a new chemotherapy drug (Figure 5). The PA ligand is used to deliver antitumor drugs and simultaneously behaves as an inhibitor of Akt. The inhibition, a potential strategy for multiple-pathway inhibitors of the Akt pathway, enhances the therapeutic killing of drug-resistant H1650 cells. The enhanced inhibition is due to the upregulated c-caspase 3 and the downregulated Ki67.
The emerging advances in theranostics may play a dynamic role in precision medicine. This is exemplified by an approach wherein the targeted prodrugs are integrated with diagnostic imaging modalities that include radionuclides for PET, contrast agents for CT, and fluorophores for optical imaging. Fluorescence imaging is useful for tracking specific molecular events both in vitro and in vivo. In particular, fluorescence imaging in the distinct NIR region has the advantage of maximizing tissue penetration, minimizing the absorbance of heme in hemoglobin and myoglobin, water, and lipids while preventing the interference of biological autofluorescence, which will improve the target-to-background ratio. The adoption of fluorescence imaging in theranostics permits direct tracking of drugs in real time instead of time-consuming dissection and histology. This is valuable for determining the necessary dose and timing, biodistribution, pharmacokinetics, drug delivery kinetics, local response, and efficacy.
Herein, we describe the design and synthesis of a fluorescence imaging technology platform TPG, which can preferentially deliver an anticancer chemotherapy drug to the tumor and monitor its antitumor activity in real time. TPG is nonfluorescent in the inactive state but becomes fluorescent when it is turned on following activation by GSH in tumor cells (Figure S2). Theranostic TPG can rapidly target tumor tissues, and the surplus agent can be quickly eliminated from circulation (Figure 3h). The retention time is more than 24 h in tumor lesions, and its chemotherapeutic efficacy, i.e., changes in tumor volume and temporal drug distribution, can be directly measured.
Despite the availability of multidisciplinary diagnostic systems, molecular pathology, and advanced medical imaging, diagnosis of NSCLC is a challenging task for physicians [2]. TBLB is a less invasive procedure for the pathological examination of NSCLC, which is the gold standard for confirmed diagnosis. However, it is time-consuming. The clinical medical imaging of biomarkers can offer clues for suspected diagnosis and pathological examination. We employed flow cytometry analysis to examine TBLB samples using TPG. The technique could accurately provide quantitative data to distinguish between tumor and non-tumor tissues of the TBLB samples within 4 h. The results demonstrated that TPG utilizes the PUS of lung cancer cells, which is not limited by pathological patterns. Our theranostic agent TPG could also track the delivery of drugs and guide the distribution of tumor lesions for diagnosis in real-time. Such a technology can potentially advance precision medicine by identifying NSCLC early and accurately while minimizing testing time.
In conclusion, we have described the design of a targeted prodrug PPG for the precision therapy of NSCLC. The targeting ligand PA exclusively delivers the prodrug to tumor cells, enhancing the efficacy of Gefitinib and reducing adverse effects. In particular, prodrug PPG can effectively kill drug-resistant H1650 tumor cells because of the inhibition of EGFR and Akt signaling pathways targeted by Gefitinib and PA ligand, respectively. Thus, PA can be considered a new potential chemotherapy drug. We also developed the fluorescent theranostic TPG to directly examine the targeting ability and pharmacokinetics of the prodrug. The NIR fluorescence allowed monitoring of tumor targeting and drug release in real-time. The fluorescent signals in tumor lesions could be maintained for more than 24 h. Significantly, the labeling of TBLB samples with TPG can offer reliable and rapid data for the diagnosis of NSCLC within 4 h. The results suggest that TPG is an efficient tool for screening tumor tissues as well as a targeted therapy in precision medicine.
Supplementary Material
Additional detailed figures and tables, mass spectrum, and compound characterization.
Acknowledgements
We thank the National Nature Science Foundation of China (No. 21775162, No. 31470415, No. 81670064, No. 21575159, and No. 41776110), the program of Youth Innovation Promotion Association, CAS (Grant 2015170), Medical Science and Technology Development Project of Shandong Province (Grant 2017WS800), State Key Laboratory of Environmental Chemistry and Ecotoxicology, Research Center for Eco-Environmental Sciences, CAS (Grant KF2016-22), and the Instrument Developing Project of the Chinese Academy of Sciences (YZ201662).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015;65:5-29
2. Wender R, Fontham ET, Barrera E Jr, Colditz GA, Church TR, Ettinger DS, Etzioni R, Flowers CR, Gazelle GS, Kelsey DK, LaMonte SJ, Michaelson JS, Oeffinger KC, Shih YC, Sullivan DC, Travis W, Walter L, Wolf AM, Brawley OW, Smith RA. American Cancer Society lung cancer screening guidelines. CA Cancer J Clin. 2013;63:106-117
3. Mullard AA. 2015 FDA drug approvals. Nat Rev Drug Discov. 2016;15:73-76
4. Kue CS, Kamkaew A, Burgess K, Kiew LV, Chung LY, Lee HB. Small Molecules for Active Targeted in Cancer. Med Res Rev. 2016;36:494-575
5. Herbst RS, Fukuoka M, Baselga J. Gefitinib—a novel targeted approach to treating cancer. Nat Rev Cancer. 2004;4:979-987
6. Sos ML, Koker M, Weir BA, Heynck S, Rabinovsky R, Zander T, Seeger JM, Weiss J, Fischer F, Frommolt P, Michel K, Peifer M, Mermel C, Girard L, Peyton M, Gazdar AF, Minna JD, Garraway LA, Kashkar H, Pao W, Meyerson M, Thomas RK. PTEN loss contributes to erlotinib resistance in EGFR-mutant lung cancer by activation of Akt and EGFR. Cancer Res. 2009;69:3256-3261
7. Kokubo Y, Gemma A, Noro R, Seike M, Kataoka K, Matsuda K, Okano T, Minegishi Y, Yoshimura A, Shibuya M, Kudoh S. Reduction of PTEN protein and loss of epidermal growth factor receptor gene mutation in lung cancer with natural resistance to Gefitinib (IRESSA). Br J Cancer. 2005;92:1711-1719
8. Yamamoto C, Basaki Y, Kawahara A, Nakashima K, Kage M, Izumi H, Kohno K, Uramoto H, Yasumoto K, Kuwano M, Ono M. Loss of PTEN expression by blocking nuclear translocation of EGR1 in Gefitinib-resistant lung cancer cells harboring epidermal growth factor receptor-activating mutations. Cancer Res. 2010;70:8715-8725
9. Srinivasarao M, Galliford CV, Low PS. Principles in the design of ligand-targeted cancer therapeutics and imaging agents. Nat Rev Drug Discov. 2015;14:203-219
10. Rudin M, Weissleder R. Molecular imaging in drug discovery and development. Nat Rev Drug Discov. 2003;2:123-131
11. Blair JA, Rauh D, Kung C, Yun CH, Fan QW, Rode H, Zhang C, Eck MJ, Weiss WA, Shokat KM. Structure-guided development of affinity probes for tyrosine kinases using chemical genetics. Nat Chem Biol. 2007;3:229-238
12. Todorova MI, Larroque AL, Dauphin-Pierre S, Fang YQ, Jean-Claude BJ. Subcellular distribution of a fluorescence-labeled combi-molecule designed to block epidermal growth factor receptor tyrosine kinase and damage DNA with a green fluorescent species. Mol Cancer Ther. 2010;9:869-882
13. Weissleder R, Nahrendorf M. Advancing biomedical imaging. Proc Natl Acad Sci USA. 2015;112:14424-14428
14. Kulkarni A, Rao P, Natarajan S, Goldman A, Sabbisetti VS, Khater Y, Korimerla N, Chandrasekar V, Mashelkar RA, Sengupta S. Reporter nanoparticle that monitors its anticancer efficacy in real time. Proc Natl Acad Sci USA. 2016;113:E2104-E2113
15. Han X, Yu F, Song X, Chen L. Quantification of cysteine hydropersulfide with a ratiometric near-infrared fluorescent probe based on selenium-sulfur exchange reaction. Chem Sci. 2016;7:5098-5107
16. Wang CK, Cheng J, Liang XG, Tan C, Jiang Q, Hu YZ, Lu YM, Fukunaga K, Han F, Li X. A H2O2 responsive theranostic probe for endothelial injury imaging and protection. Theranostics. 2017;7:3803-3813
17. Liang XG, Chen B, Shao LX, Cheng J, Huang MZ, Chen Y, Hu YZ, Han YF, Han F, Li X. A fluorogenic probe for ultrafast and reversible detection of formaldehyde in neurovascular tissues. Theranostics. 2017;7:2305-2313
18. Xia J, Zhang Y, Zhao H, Wang J, Gao X, Chen J, Fu B, Shen Y, Miao F, Zhang J, Teng G. Non-invasive monitoring of CNS MHC-I molecules in schemic stroke mice. 2017; 7:2837-2848.
19. Chan J, Dodani SC, Chang CJ. Reaction-based small-molecule fluorescent probes for chemoselective bioimaging. Nat Chem. 2012;4:973-984
20. Han X, Yu F, Song X, Chen L. A ratiometric near-infrared fluorescent probe for quantification and evaluation of selenocysteine-protective effects in acute inflammation. Adv Funct Mater. 2017;27:201700769
21. Han X, Yu F, Song X, Chen L. A ratiometric fluorescent probe for imaging and quantifying antiapoptotic effects of GSH under temperature stress. Chem Sci. 2017;8:6991-7002
22. Hilderbrand SA, Weissleder R. Near-infrared fluorescence: application to in vivo molecular imaging. Curr Opin Chem Biol. 2010;14:71-79
23. Gao M, Yu F, Lv C, Choo J, Chen L. Fluorescent chemical probes for accurate tumor diagnosis and targeting therapy. Chem Soc Rev. 2017;46:2237-2271
24. Vetter ML, Zhang Z, Liu S, Wang J, Cho H, Zhang J, Zhang W, Gray NS, Yang PL. Fluorescent visualization of Src by using dasatinib-BODIPY. Chembiochem. 2014;15:1317-1324
25. Rachid Z, Brahimi F, Domarkas J, Jean-Claude BJ. Synthesis of half-mustard combi-molecules with fluorescence properties: correlation with EGFR status. Bioorg Med Chem Lett. 2005;15:1135-1138
26. Shreder KR, Wong MS, Nomanbhoy T, Leventhal PS, Fuller SR. Synthesis of AX7593, a quinazoline-derived photoaffinity probe for EGFR. Org Lett. 2004;6:3715-3718
27. Zhang F, Zhu G, Jacobson O, Liu Y, Chen K, Yu G, Ni Q, Fan J, Yang Z, Xu F, Fu X, Wang Z, Ma Y, Niu G, Zhao X, Chen X. Transformative nanomedicine of an amphiphilic camptothecin prodrug for long circulation and high tumor uptake in cancer therapy. ACS Nano. 2017;11:8838-8848
28. Huang Y, Zhou J, Luo S, Wang Y, He J, Luo P, Chen Z, Liu T, Tan X, Ou J, Miao H, Liang H, Shi C. Identification of a fluorescent small-molecule enhancer for therapeutic autophagy in colorectal cancer by targeting mitochondrial protein translocase TIM44. Gut. 2018;67:307-319
29. Wu X, Sun X, Guo Z, Tang J, Shen Y, James TD, Tian H, Zhu W. In vivo and in situ tracking cancer chemotherapy by highly photostable NIR fluorescent theranostic prodrug. J Am Chem Soc. 2014;136:3579-3588
30. Kumar R, Han J, Lim HJ, Ren WX, Lim JY, Kim JH, Kim JS. Mitochondrial induced and self-monitored intrinsic apoptosis by antitumor theranostic prodrug: in vivo imaging and precise cancer treatment. J Am Chem Soc. 2014;136:17836-17843
31. Vineberg JG, Wang T, Zuniga ES, Ojima I. Design, Synthesis, and Biological Evaluation of Theranostic Vitamin-Linker-Taxoid Conjugates. J Med Chem. 2015;58:2406-2416
32. Kumar R, Shin WS, Sunwoo K, Kim WY, Koo S, Bhuniya S, Kim JS. Small conjugate-based theranostic agents: an encouraging approach for cancer therapy. Chem Soc Rev. 2015;44:6670-6683
33. Russell-Jones G, McTavish K, McEwan J, Rice J, Nowotnik D. Vitamin-mediated targeting as a potential mechanism to increase drug uptake by tumours. J Inorg Biochem. 2004;98:1625-1633
34. Liang J, Shang Y. Estrogen and cancer. Annu Rev Physiol. 2013;75:225-240
35. Zhang L, Wan S, Jiang Y, Wang Y, Fu T, Liu Q, Cao Z, Qiu L, Tan W. Molecular Elucidation of Disease Biomarkers at the Interface of Chemistry and Biology. J Am Chem Soc. 2017;139:2532-2540
36. Hoet PH, Nemery B. Polyamines in the lung: polyamine uptake and polyamine-linked pathological or toxicological conditions. Am J Physiol Lung Cell Mol Physiol. 2000;278:417-433
37. Smith LL, Lewis CP, Wyatt I, Cohen GM. The importance of epithelial uptake systems in lung toxicity. Environ Health Perspect. 1990;85:25-30
38. Gerner EW, Meyskens FL Jr. Polyamines and cancer: old molecules, new understanding. Nat Rev Cancer. 2004;4:781-792
39. Min JZ, Matsumoto A, Li G, Jiang YZ, Yu HF, Todoroki K, Inoue K, Toyo'oka T. A quantitative analysis of the polyamine in lung cancer patient fingernails by LC-ESI-MS/MS. Biomed Chromatogr. 2014;28:492-499
40. Lee MH, Sessler JL, Kim JS. Disulfide-Based Multifunctional Conjugates for Targeted Theranostic Drug Delivery. Acc Chem Res. 2015;48:2935-2946
41. Gao M, Yu F, Chen H, Chen L. Near-infrared fluorescent probe for imaging mitochondrial hydrogen polysulfides in living cells and in vivo. Anal Chem. 2015;87:3631-3638
42. Elmore S. Apoptosis: a review of programmed cell death. Toxicol Pathol. 2007;35:495-516
43. Guo A, Villén J, Kornhauser J, Lee KA, Stokes MP, Rikova K, Possemato A, Nardone J, Innocenti G, Wetzel R, Wang Y, MacNeill J, Mitchell J, Gygi SP, Rush J, Polakiewicz RD, Comb MJ. Signaling networks assembled by oncogenic EGFR and c-Met. Proc Natl Acad Sci USA. 2008;105:692-697
44. Havasi A, Li Z, Wang Z, Martin JL, Botla V, Ruchalski K, Schwartz JH, Borkan SC. Hsp27 inhibits Bax activation and apoptosis via a phosphatidylinositol 3-kinase-dependent mechanism. J Biol Chem. 2008;283:12305-12313
45. Liu P, Cheng H, Roberts TM, Zhao JJ. Targeted the phosphoinositide 3-kinase pathway in cancer. Nat Rev Drug Discov. 2009;8:627-644
46. Dinis-Oliveira RJ, Duarte JA, Sánchez-Navarro A, Remião F, Bastos ML, Carvalho F. Paraquat poisonings: mechanisms of lung toxicity, clinical features, and treatment. Crit Rev Toxicol. 2008;38:13-71
47. Mahmoud AA, Farouk A, Goneim A, Hafez MFA, Saleem TH. Ornithine decarboxylase gene expression and activity in lung cancer. Gene Rep. 2016;5:126-129
48. Ponti D, Costa A, Zaffaroni N, Pratesi G, Petrangolini G, Coradini D, Pilotti S, Pierotti MA, Daidone MG. Isolation and in vitro propagation of tumorigenic breast cancer cells with stem/progenitor cell properties. Cancer Res. 2005;65:5506-5511
49. Marsden CG, Wright MJ, Pochampally R, Rowan BG. Identification of a cancer stem cell in human brain tumors. Cancer Res. 2003;63:5821-5828
50. Chantapet P, Riantawan P, Lebnak P, Getngern P. Utility of serum cytokeratin 19 fragment (CYFRA 21-1) and carcinoembryonic antigen (CEA) as tumour markers for non-small cell lung cancer. J Med Assoc Thai. 2000;83:383-391
51. Detterbeck FC, Lewis SZ, Diekemper R, Addrizzo-Harris D, Alberts WM. Executive summary: diagnosis and management of lung cancer: American College of Chest Physicians evidence-based clinical practice guidelines. Chest. 2013;143:7S-37S
Author contact
![]() Corresponding authors: E-mails: fbyuac.cn, lxchenac.cn, lucky_lcjcom. Phone/fax number: +86-0535-2109130
Corresponding authors: E-mails: fbyuac.cn, lxchenac.cn, lucky_lcjcom. Phone/fax number: +86-0535-2109130
Citation styles
APA
Song, X., Han, X., Yu, F., Zhang, X., Chen, L., Lv, C. (2018). Polyamine-Targeting Gefitinib Prodrug and its Near-Infrared Fluorescent Theranostic Derivative for Monitoring Drug Delivery and Lung Cancer Therapy. Theranostics, 8(8), 2217-2228. https://doi.org/10.7150/thno.24041.
ACS
Song, X.; Han, X.; Yu, F.; Zhang, X.; Chen, L.; Lv, C. Polyamine-Targeting Gefitinib Prodrug and its Near-Infrared Fluorescent Theranostic Derivative for Monitoring Drug Delivery and Lung Cancer Therapy. Theranostics 2018, 8 (8), 2217-2228. DOI: 10.7150/thno.24041.
NLM
Song X, Han X, Yu F, Zhang X, Chen L, Lv C. Polyamine-Targeting Gefitinib Prodrug and its Near-Infrared Fluorescent Theranostic Derivative for Monitoring Drug Delivery and Lung Cancer Therapy. Theranostics 2018; 8(8):2217-2228. doi:10.7150/thno.24041. https://www.thno.org/v08p2217.htm
CSE
Song X, Han X, Yu F, Zhang X, Chen L, Lv C. 2018. Polyamine-Targeting Gefitinib Prodrug and its Near-Infrared Fluorescent Theranostic Derivative for Monitoring Drug Delivery and Lung Cancer Therapy. Theranostics. 8(8):2217-2228.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.