Theranostics
13.3
Impact Factor
- Current Issue
- Advance Articles
- Volume 16; 2026
- Volume 15; 2025
- Volume 14; 2024
- Volume 13; 2023
- Volume 12; 2022
- Archive
- Cover Images
- Cover Suggestion
- Special Issues
Top
Introduction
Methods
Results
Discussion
Acknowledgements
Abbreviations
Supplementary Material
References
Introduction
Methods
Results
Discussion
Acknowledgements
Abbreviations
Supplementary Material
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2018; 8(9):2387-2406. doi:10.7150/thno.23620 This issue Cite
Research Paper
Resveratrol counteracts bone loss via mitofilin-mediated osteogenic improvement of mesenchymal stem cells in senescence-accelerated mice
Ya-Jie Lv1,4*, Yi Yang1,3*, Bing-Dong Sui1,2*, Cheng-Hu Hu1,2*, Pan Zhao1,2, Li Liao1,2, Ji Chen1, Li-Qiang Zhang1,2, Tong-Tao Yang5, Shao-Feng Zhang3 ![]() , Yan Jin1,2
, Yan Jin1,2 ![]()
1. State Key Laboratory of Military Stomatology, Center for Tissue Engineering, School of Stomatology, Fourth Military Medical University, Xi'an, Shaanxi 710032, China.
2. Xi'an Institute of Tissue Engineering and Regenerative Medicine, Xi'an, Shaanxi 710032, China.
3. State Key Laboratory of Military Stomatology, Department of Prosthodontics, School of Stomatology, Fourth Military Medical University, Xi'an, Shaanxi 710032, China.
4. Department of Dermatology, Tangdu Hospital, Fourth Military Medical University, Xi'an, Shannxi, 710069, China.
5. Department of Orthopaedics, Tangdu Hospital, Fourth Military Medical University, Xi'an, Shannxi, 710069, China.
*These authors contributed equally to this manuscript.
Received 2017-11-1; Accepted 2018-2-18; Published 2018-3-23
Citation:
Lv YJ, Yang Y, Sui BD, Hu CH, Zhao P, Liao L, Chen J, Zhang LQ, Yang TT, Zhang SF, Jin Y. Resveratrol counteracts bone loss via mitofilin-mediated osteogenic improvement of mesenchymal stem cells in senescence-accelerated mice. Theranostics 2018; 8(9):2387-2406. doi:10.7150/thno.23620. https://www.thno.org/v08p2387.htm
Other stylesAbstract

Rational: Senescence of mesenchymal stem cells (MSCs) and the related functional decline of osteogenesis have emerged as the critical pathogenesis of osteoporosis in aging. Resveratrol (RESV), a small molecular compound that safely mimics the effects of dietary restriction, has been well documented to extend lifespan in lower organisms and improve health in aging rodents. However, whether RESV promotes function of senescent stem cells in alleviating age-related phenotypes remains largely unknown. Here, we intend to investigate whether RESV counteracts senescence-associated bone loss via osteogenic improvement of MSCs and the underlying mechanism.
Methods: MSCs derived from bone marrow (BMMSCs) and the bone-specific, senescence-accelerated, osteoblastogenesis/osteogenesis-defective mice (the SAMP6 strain) were used as experimental models. In vivo application of RESV was performed at 100 mg/kg intraperitoneally once every other day for 2 months, and in vitro application of RESV was performed at 10 μM. Bone mass, bone formation rates and osteogenic differentiation of BMMSCs were primarily evaluated. Metabolic statuses of BMMSCs and the mitochondrial activity, transcription and morphology were also examined. Mitofilin expression was assessed at both mRNA and protein levels, and short hairpin RNA (shRNA)-based gene knockdown was applied for mechanistic experiments.
Results: Chronic intermittent application of RESV enhances bone formation and counteracts accelerated bone loss, with RESV improving osteogenic differentiation of senescent BMMSCs. Furthermore, in rescuing osteogenic decline under BMMSC senescence, RESV restores cellular metabolism through mitochondrial functional recovery via facilitating mitochondrial autonomous gene transcription. Molecularly, in alleviating senescence-associated mitochondrial disorders of BMMSCs, particularly the mitochondrial morphological alterations, RESV upregulates Mitofilin, also known as inner membrane protein of mitochondria (Immt) or Mic60, which is the core component of the mitochondrial contact site and cristae organizing system (MICOS). Moreover, Mitofilin is revealed to be indispensable for mitochondrial homeostasis and osteogenesis of BMMSCs, and that insufficiency of Mitofilin leads to BMMSC senescence and bone loss. More importantly, Mitofilin mediates resveratrol-induced mitochondrial and osteogenic improvements of BMMSCs in senescence.
Conclusion: Our findings uncover osteogenic functional improvements of senescent MSCs as critical impacts in anti-osteoporotic practice of RESV, and unravel Mitofilin as a novel mechanism mediating RESV promotion on mitochondrial function in stem cell senescence.
Keywords: resveratrol, osteoporosis, mesenchymal stem cells, senescence-accelerated mice, osteogenesis, Mitofilin/IMMT/Mic60
Introduction
Stem cell senescence and functional decline have emerged among the core features of aging, underlying deficient regenerative potential and lost tissue homeostasis in various organs/systems [1-3]. Among these, the best characterized include age-related disorders of the skeletal system, where adult mesenchymal stem cells (MSCs) reside and mediate senile osteoporosis, which is attributed to their senescence-associated dysfunction, particularly the impairment of osteogenesis [4, 5]. Currently, the most promising anti-aging strategy is dietary restriction, the effects of which can be safely mimicked by small molecules, such as resveratrol (RESV) [6-8]. Conclusively, RESV has been documented to extend lifespan in lower organisms [9-11] and improve health of aging rodents against multiple alterations [12-14]. However, although a pioneering report in a genetic progeroid model documenting that RESV replenishes the adult stem cell pool in bone [15], whether RESV rejuvenates the function of senescent stem cells in alleviating age-related phenotypes remains largely unknown, potentially contributing to the controversial or conditional findings regarding RESV effects on longevity of mammals [12, 16, 17]. Specifically, despite preliminary reports on RESV protecting bone mass in a variety of pathological conditions [18-20], whether RESV improves osteogenesis of senescent MSCs and counteracts senescence-associated bone loss are not understood. Therefore, functional evidence of RESV impacts on senescent stem cells/MSCs is of great demand to further determine its translational potential in aging.
RESV, also known as 3,5,4'-trihydroxystilbene, is basically a natural polyphenolic compound found in red wine and is well known for its phytoestrogenic and antioxidant properties, not surprisingly being applied originally in estrogen-deficient and oxidative bone deteriorations [21-23]. Molecularly, RESV was further discovered as an activator of the protein deacetylase sirtuin1 (Sirt1), which modulates aging under nutrient control by metabolic regulation via peroxisome proliferator-activated receptor gamma coactivator-1 (Pgc-1) [9, 24]. Accordingly, RESV has been shown effective in increasing Pgc-1 activity by decreasing Pgc-1 acetylation depending on Sirt1 [13]. Together with its promotion of the activity of another critical metabolic regulatory factor, the adenosine 5'-monophosphate-activated protein kinase (Ampk), RESV improves mitochondrial function and preserves metabolic homeostasis in a variety of experimental models [12, 13, 25]. Notably, mitochondrial metabolic failure is also acknowledged as a vital hallmark of aging [26-28], and mitochondrial functional status crucially determines functional fate decisions of stem cells, including osteogenesis and senescence of MSCs [27-30]. Hence, it would be safe to assume protective effects of RESV against senescence-associated osteogenic defects of MSCs through mitochondrial improvements in senile osteoporosis.
In this study, to evaluate effects of RESV on stem cell function in aging, according to the above information and based our previous work basis, we selected bone marrow-derived MSCs (BMMSCs) as a model and analyzed their osteogenesis in close relation to bone formation in osteoporosis development and treatment [31-33]. The bone-specific, senescence/bone loss-accelerated mice (the SAMP6 strain) were accordingly chosen, which develop a low turnover senile osteopenia primarily attributed to a defective osteoblastogenesis/osteogenesis without an elevated bone resorption or systemic alterations, making them the ideal experimental model for this study [34-37]. We aimed to investigate: (1) Whether RESV rescues senescence-associated osteogenic decline of BMMSCs in ameliorating the accelerated bone loss in SAMP6 mice; and (2) whether and how RESV improves mitochondrial metabolism in improving senescent BMMSC osteogenesis. As expected, we proved that chronic intermittent application of RESV enhances bone formation and alleviates osteopenia in SAMP6 mice, with RESV promoting osteogenic differentiation of senescent BMMSCs through restoring mitochondrial function and autonomous gene transcription. Furthermore, in ameliorating senescence-associated mitochondrial disorders of BMMSCs, particularly the mitochondrial morphological alterations, we surprisingly discovered non-canonically that RESV upregulates Mitofilin, also known as inner membrane protein of mitochondria (Immt) or Mic60, the core component of the mitochondrial contact site and cristae organizing system (MICOS, or mitochondrial inner membrane organizing system, MINOS) [38, 39]. We further revealed that insufficiency of Mitofilin is detrimental for mitochondrial stability and osteogenesis of BMMSCs as well as the bone homeostasis, and that Mitofilin mediates resveratrol-induced mitochondrial and osteogenic improvements of BMMSCs in senescence. Collectively, our findings uncover osteogenic functional improvements of senescent MSCs as critical impacts in anti-osteoporotic practice of RESV, and unravel Mitofilin as a novel mechanism mediating RESV promotion on mitochondrial function in stem cell senescence.
Methods
Animals
Animal experiments were approved by Fourth Military Medical University and were performed following the Guidelines of Intramural Animal Use and Care Committee of Fourth Military Medical University. For the bone-specific senescence-accelerated mouse model and its control [34, 37], female senescence-accelerated mice-prone 6 (SAMP6) and senescence-accelerated mice-resistant 1 (SAMR1) strains at 4-6 month of age (weight, 20-22 g) were used, which were AKR/J background and were obtained from our colony provided by the Council for SAM Research of Kyoto University, Japan. According to previous works by us and others, SAMP6 mice at this age develop accelerated bone loss with senescence-associated osteogenic decline of BMMSCs, while SAMR1 mice demonstrate normal phenotypes [32, 40]. In detail, for in vivo experiments of RESV and shImmt treatments, mice received interventions started at 4 months of age and were sacrificed after a 2-month experimental period. Alternatively, for ex vivo experiments on BMMSCs, 4 month-old mice were sacrificed for bone marrow sampling. For BMMSCs from the natural aging model, female C3H mice 22 months old were used for bone marrow sampling with the 4-month-old young control. The mice were maintained with good ventilation and a 12 h light/dark cycle, and were kept with feeding and drinking ad libitum before being sacrificed.
Isolation and culture of BMMSCs
Isolation and culture of murine BMMSCs were according to our previous protocol [41, 42]. Briefly, bone marrow cells were sampled from the hindlimbs of mice and were seeded for incubation overnight at 37 ℃ in a humidified atmosphere of 5% CO2. The non-adherent cells were then removed by rinsing with PBS, while the adherent cells were cultured with alpha-minimum essential medium supplemented with 20% fetal bovine serum, 2 mM L-glutamine, 100 U/mL penicillin, and 100 g/mL streptomycin (all from Invitrogen, USA) at 37 ℃ in a humidified atmosphere of 5% CO2. The media were changed every 2 days, and primary MSC colonies were passaged with 0.25% trypsin. The passaged BMMSCs were then seeded into culture plates for further experimental tests.
RESV preparation and in vivo experimental designs
RESV used in this study was purchased from Sigma-Aldrich (Sigma-Aldrich, USA) [8]. For in vivo treatments, RESV was firstly dissolved in dimethyl sulfoxide (DMSO) [43] at 200 mg/mL, and was then diluted with saline to 10 mg/mL (containing 5% DMSO). For in vitro treatments, RESV was firstly dissolved in DMSO [43] at 1 M, and was then diluted with media to 1 mM (containing 0.1% DMSO). In vitro working solution was set at 10 μM according to previous dose-determining papers on osteogenic promotion in young MSCs [44-46]. Equal amount of DMSO was applied into culture media as the control at a final concentration of 0.001%.
RESV for in vivo application was injected at 100 mg/kg intraperitoneally via the right lower quadrant of the abdominal area with mice in a head-down position, once every other day for 2 months. Control groups were injected intraperitoneally with an equal amount of 5% DMSO in saline solvent. The dosage was chosen based on previous documents and our preliminary tests below the potential highest dose not to induce obvious systemic alterations, and reports applying this dosage to affect stem cell activity in response to chemical injury in vivo [47, 48]. The application route was selected considering the better control of applied dose by intraperitoneal injection over feeding for a proof-of-concept study (instead of a preclinical study), and the reported efficacy of mitochondrial intervention by intraperitoneal injection of RESV [49]. The experimental duration was determined given the time needed for chronic changes of bone mass by bone remodeling according to our observations [50]. The method of chronic intermittent application every other day was based on previous reports on feeding-based RESV delaying age-related deterioration in mice [16].
As the recipients of RESV administration, littermate SAMR1 and SAMP6 mice were randomly allocated into 8 groups, i.e., 4 groups of each strain: (1) the SAMR1 4-month control group (n = 5), (2) the SAMR1 6-month control group (n = 5), (3) the SAMR1+DMSO group (started at 4 months old and sacrificed at 6 months old) (n = 4), and (4) the SAMR1+RESV group (started at 4 months old and sacrificed at 6 months old) (n = 4), (5) the SAMP6 4-month control group (n = 5), (6) the SAMP6 6-month control group (n = 5), (7) the SAMP6+DMSO group (started at 4 months old and sacrificed at 6 months old) (n = 4), and (8) the SAMP6+RESV group (started at 4 months old and sacrificed at 6 months old) (n = 5).
Lentivirus-based gene knockdown experiments
Lentiviral vector construction and transfection were performed according to a previous protocol [31, 51]. Mouse Immt (Mitofilin) was amplified by polymerase chain reaction from genomic DNA with targeting sequences for short hairpin RNA (shRNA) as denoted below. Forward: 5'- CCGGCCGTCCTTACACTGCTATCATCTCGAGATGATAGCAGTGTAAGGACGGTTTTTG -3'; Reverse: 5'- AATTCAAAAACCGTCCTTACACTGCTATCATCTCGAGATGATAGCAGTGTAAGGACGG -3'. The PCR product was digested with restriction enzymes AgeI and EcoRI, inserted into the pLKO.1 vector (Invitrogen, USA), and verified by Sanger sequencing. Negative control (NC) was performed with a lentiviral construct containing a scrambled sequence. The lentivirus constructs were then produced by co-transfecting 293T cells with a transfer vector and two packaging vectors, psPAX2 and pMD2.G. After transfection for 48 h, media containing virus were collected and the virus was purified by centrifugation at 500 rcf for 10 min followed by filtering through 0.45 μm polyethersulfone low protein-binding filters. For in vitro experiments, 1st-passaged BMMSCs plated in 6-well plates were transfected with constructs for either NC or shRNA for Immt (shImmt) (titer > 5×105 IFU/mL) and 10 μg/mL polybrene (Sigma-Aldrich, USA) for 48 h. Transfected BMMSCs were then evaluated for function and the expression of Immt was examined at both mRNA and protein levels as described below.
For in vivo experiments, intra-femoral bone marrow injection of lentiviral constructs was performed following published procedure [52]. Littermate SAMR1 mice (n = 3) were used, and the left femora were designed for NC injection while the right femora for shImmt. Briefly, mice were anesthetized with 1% pentobarbital sodium (5 μL/g body weight) and the hairs around the knee joint were shaved off. The joint was inflexed and a sterile scalpel blade was used to make a small deep incision along one side of the knee joint. The kneecap was then exposed by sterile forceps without damaging any blood vessels. A needle of a 1 mL syringe was used to gently drill a hole through the groove of the kneecap in the femur about 0.5 cm deep. 20 μL lentiviral suspension (2×106 IFU/μL) containing either NC or shImmt was injected through the hole gently with a Hamilton syringe (Hamilton, Bonaduz, Switzerland). After removing the needle, the hole was immediately closed with bone wax using a sterile scalpel blade. Finally, the surgical area was washed with PBS and the wound was closed with an appropriate suture.
Micro-computed tomography (micro-CT) analysis
Trabecular bone mass evaluation by micro-CT analysis was performed based on our previous reports [41, 42, 53]. Briefly, at sacrifice, the left femora were removed, fixed overnight in 4% paraformaldehyde, and prepared into 1 mm blocks with the distal metaphysis included. The specimens were then scanned at a resolution of 8 μm, a voltage of 80 kV, and a current of 80 μA by a desktop micro-CT system (eXplore Locus SP; GE Healthcare, USA). After image reconstruction, quantification was performed at a region-of-interest in the distal metaphysis, from 0.3 mm to 0.8 mm away from the epiphysis. Data were analyzed using parameters of bone volume over tissue volume (BV/TV) and bone mineral density (BMD) with the Micview V2.1.2 software [54].
Calcein labeling assay
For bone histomorphometric analysis on bone formation rates, double-time calcein labeling was performed according to previous studies [53, 55]. At 16 days and 2 days prior to sacrifice, mice received intraperitoneal injections of 20 mg/kg calcein (Sigma-Aldrich, USA) dissolved at 2 mg/mL in PBS supplemented with 1 mg/mL NaHCO3 (Sigma-Aldrich, USA). Calcein was injected at 10 μL/g each time away from light via the right lower quadrant of the abdominal area with mice in a head-down position. At sacrifice, the right femora were isolated, fixed in 80% ethanol, embedded in methyl methacrylate, and sagittally sectioned into 30 μm slices using a hard tissue slicing machine (SP1600; Leica, Germany) away from light. Cortical endosteum surfaces were evaluated by a fluorescence microscope (STP6000; Leica, Germany) with an excitation wavelength of 488 nm. Quantification was performed using the parameters of mineral apposition rate (MAR) and bone formation rate (BFR) by the ImageJ 1.47 software and the related calculation [56].
Functional evaluation of BMMSCs
For osteogenic differentiation analysis [57], BMMSCs at the 1st passage in 6-well plates were induced in normal culture media supplemented with 100 μg/mL ascorbic acid (MP Biomedicals, USA), 2 mM β-glycerophosphate (Sigma-Aldrich, USA) and 10 nM dexamethasone (Sigma-Aldrich, USA). The media were changed every 3 days. After induction for 7 days, alkaline phosphotase (ALP) staining was performed using the BCIP/NBT ALP color development kit (Beyotime, China) and the quantification was determined with the ImageJ 1.47 software based on grey values, while ALP activity was determined using the ALP (AKP/ALP) detection kit (BioVision, USA) [55]. After induction for 14 days, alizarin red staining was performed to determine the mineralization status. Quantitative parameters of number of mineralized nodules and the percentage of mineralized area were determined with the ImageJ 1.47 software [32].
To determine cell senescence, senescence-associated beta-galactosidase (SA-β-gal) activity was evaluated in the 1st-passaged BMMSCs in 12-well plates using the SA-β-gal Staining Kit (Beyotime, China), according to the manufacturer's instructions. The percentage of SA-β-gal positive cells was determined using the ImageJ 1.47 software [32]. For the colony-forming assay, after treatment with ACK lysis buffer (Lonza, Basel, Switzerland) to remove red blood cells, murine primary bone marrow cells were plated in 5 cm culture dishes at a density of 1×105 cells/cm2 and cultured in normal media. The formation of colonies was evaluated after 14 days of culture after fixation with 4% paraformaldehyde for 30 min and stained with crystal violet for 5 min [43]. For proliferation analysis, BMMSCs at the 1st passage were plated at 2×103 cells/well in 96-well plates. Cell viability at indicated time points was determined by incubation with 20 μL 5 mg/mL methyl thiazolyl tetrazolium (MTT) (MP Biomedicals, USA) for 4 h. The precipitates were extracted with 180 μL DMSO and the absorbance was measured at 490 nm [58]. Cell cycle analysis was performed after obtaining single cell suspensions of 1st-passaged BMMSCs, and the cells were fixed in ice-cold 75% ethanol at 4 ℃ overnight, washed twice with PBS, and stained with 100 mg/mL propidium iodide (Sigma-Aldrich, USA) at 4 ℃ for 30 min at dark. The stained BMMSCs were then subjected to cell cycle analysis using a flow cytometer (CytoFLEX; Beckman Coulter, USA) and the percentages of cells in the S-phase were determined with the CXP 2.1 software [59].
Metabolic analysis
The ratio of adenosine triphosphate (ATP) over adenosine diphosphate (ADP) was measured according to recent papers using the ADP/ATP Ratio Assay Kit (Abcam, UK) following the manufacturer's instructions [60]. BMMSCs were evaluated at 48 h post to RESV or DMSO treatments and shImmt or NC transfection. ATP and ADP levels were determined by measuring luminescence in the absence or presence of ADP-converting enzyme using a luminometer (Tecan, Switzerland) and the ATP/ADP ratio was then calculated.
Total intracellular reactive oxygen species (ROS) contents of BMMSCs was measured using the fluorescent probe 2',7'-dichlorofluorescin diacetate (DCFDA) [31]. Briefly, 25 mM DCFDA was added to BMMSCs at 48 h post RESV or DMSO treatments and shImmt or NC transfection, and was incubated for an additional 30 min at 37 ℃. After washing with PBS twice to eliminate the unlabeled DCFDA, BMMSCs were examined under a fluorescence microscope (Olympus, Japan) with excitation at 488 nm. For quantification of ROS, BMMSCs were then digested and measured using the flow cytometer (CytoFLEX; Beckman Coulter, USA) with the CXP 2.1 software.
Mitochondrial membrane potential (Ψm) of BMMSCs was measured using the fluorescent probe Rhodamine 123 [61]. Briefly, 0.4 M Rhodamine 123 was added to BMMSCs at 48 h post RESV or DMSO treatments and shImmt or NC transfection, and was incubated for 15 min at 37 ℃. After washing with PBS twice, BMMSCs were examined under a fluorescence microscope (Olympus, Japan) and the fluorescence signal was measured using the Synergy™ H1 Hybrid Multi-Mode Reader (BioTek, USA) at 490/535 nm. The transmembrane distribution of the Rhodamine 123 is proportional to the strength of Ψm.
Mitochondrial oxidative phosphorylation (OXPHOS) activity was measured using Mito-ID® O2 Extracellular Sensor Kit (Enzo Life Sciences, USA) according to the manufacturer's protocol, in which oxygen consumption is detected based on a fluorescent probe [62]. Briefly, 10 μL probe was added to BMMSCs at 48 h post RESV or DMSO treatments, and was read immediately using the Synergy™ H1 Hybrid Multi-Mode Reader (BioTek, USA) at 380/650 nm kinetically for 90 minutes, at an interval of 1 minute. The mean value of fluorescence intensity of 3 wells/group was determined for the oxygen consumption curves.
Transmission electron microscopy (TEM) analysis
TEM observation of BMMSC mitochondria was performed accordingly at 48 h post RESV or DMSO treatments and shImmt or NC transfection [63]. Briefly, 1×106 BMMSCs were digested, washed with PBS 3 times and fixed by 1% osmic acid at 4 ℃ for 2 h. After dehydration in ethanol, embedding in resin and solidifying, ultrathin sections at 70 nm were obtained using an ultramicrotome (CM3050; Leica, Germany). The slices were then stained with 3% uranyl acetate and citric acid, and were viewed by a transmission electron microscope (JEM-1230; JEOL, Japan). Mitochondrial parameters were quantified by measuring the mitochondrial number and area in random cells using Adobe Photoshop software. The ratio of mitochondrial inner membrane (IM) over outer membrane (OM) (IM:OM ratio) was determined using ImageJ 1.47 software by tracing and quantitating the membrane profiles in transmission electron microscopy images, as stated [64].
Quantitative real-time polymerase chain reaction (qRT-PCR) analysis
Total RNA was collected from the 1st-passaged BMMSCs during osteogenic induction or at 48 h post RESV or DMSO treatments and shImmt or NC transfection. RNA was extracted by adding Trizol Reagent (Takara, Japan) to the culture plates and was purified by phenol-chloroform extraction. cDNA synthesis and PCR procedures were performed as previously described [8, 32]. The primer sequences of the mouse genes detected in this study are listed in the Supplementary material (Table S1). Relative expression level of each gene was obtained by normalizing against β-actin abundance.
Immunoprecipitation (IP)
Pgc-1 acetylation was analyzed after IP of Pgc-1 by an acetyl-lysine (Ac-Lys) antibody (Cell Signaling, USA) according to previous methods [13, 24]. Briefly, IP of Pgc-1 was performed in nuclear lysates from BMMSCs at 48 h post RESV or DMSO treatments. Nuclear extracts were incubated with a rabbit anti-Pgc-1 antibody (Santa Cruz Biotechnology, USA) or the control IgG at concentrations of 1:50 for 2 h on a rocking platform at 4 ℃. Protein A/G (Merke Millipore, USA) was then added and incubated at 4 ℃ overnight. The beads were treated with RIPA buffer and the protein was collected from the supernatant for western blot analysis, as described below.
Western blot analysis
Western blot was performed as previously described [8, 65]. Total protein samples were collected from the 1st-passaged BMMSCs during osteogenic induction or at 48 h post RESV or DMSO treatments and shImmt or NC transfection. Cell lysates were prepared using the RIPA Buffer (Beyotime, China), and the protein was extracted by centrifuging at 13000 rcf for 15 min at 4 ℃. Protein samples were then loaded on sodium dodecyl sulfate-polyacrylamide gels, transferred to polyvinylidene fluoride membranes (Millipore, USA), and blocked with 5% bovine serum antigen (Sigma-Aldrich, USA) in PBS with 0.1% Tween for 2 h at room temperature. The membranes were incubated overnight at 4 ℃ with the following anti-mouse primary antibodies: for Alp, Pgc-1, runt-related transcription factor 2 (Runx2) (all from Santa Cruz Biotechnology, USA) at a concentration of 1:1000; for Immt (Mitofilin), osteocalcin (Ocn), p16INK4α, p21 (all from Abcam, USA) at a concentration of 1:1000; for p-Ampk, Ampk, acetylated p53 (Ac-p53), Ac-Lys (all from Cell Signaling, USA) at a concentration of 1:1000; and for β-actin (Abcam, USA) at a concentration of 1:4000. The membranes were then incubated with peroxidase-conjugated secondary antibodies (Boster, China) at a concentration of 1:40000 for 1 h at room temperature. The blotted bands were visualized using an enhanced chemiluminescence kit (Amersham Biosciences, USA) and a gel imaging system (5500; Tanon, China). The gray values of the bands were analyzed using the ImageJ 1.47 software.
Statistical analysis
Data are represented as the mean ± standard deviation (SD). Statistical significance was evaluated by two-tailed Student's t test for two-group comparison, or by one-way analysis of variation (ANOVA) followed by Newman-Keuls post-hoc tests for multiple comparisons in the GraphPad Prism 5.01 software. Values of P < 0.05 were considered statistically significant.
Results
RESV enhances bone formation and counteracts accelerated bone loss in SAMP6 mice
To investigate whether RESV improves osteogenesis of senescent MSCs in ameliorating osteoporosis, we applied the bone-specific, senescence-accelerated, osteoblastogenesis/osteogenesis-defective SAMP6 mice [34-37], together with the SAMR1 control. We confirmed that while 4-month-old SAMP6 mice demonstrated lower peak bone mass compared to age-matched SAMR1 mice, 6-month-old SAMP6 mice showed more severe bone loss, while SAMR1 mice maintained bone mass in the 2-month period (Figure 1A-C). These results verified the characteristics of accelerated bone loss in SAMP6 mice even at a relatively young age. We further discovered that the accelerated bone loss in SAMP6 mice was indeed attributed to a defective bone formation, which at least occurred at as early as 4 months of age and continued to 6 months of age (Figure 1D-F). Therefore, these experimental models are appropriate to be used in the current study.
As a proof-of-concept study, we applied RESV intraperitoneally once every 2 days within the 2-month experimental period, and RESV effects on both SAMP6 and SAMR1 mice were evaluated to understand whether the potential impacts are senescence-dependent or not. Micro-CT data demonstrated that while RESV administered following this method was not effective in further promoting bone mass in SAMR1 mice, chronic intermittent application of RESV indeed significantly retarded the accelerated trabecular bone loss in SAMP6 mice (Figure 1A), as quantified by the indexes BV/TV (Figure 1B) and BMD (Figure 1C). Importantly, RESV-treated SAMP6 mice at 6 months of age demonstrated paralleled, or slightly higher but not significant, trabecular bone mass compared to the 4-month-old SAMP6 mice, resulting in still lower, but not significant, trabecular bone mass compared to age-matched RESV-treated SAMR1 mice (Figure 1A-C). Further analysis by calcein labeling illustrated that RESV ameliorated the impairment in bone formation of SAMP6 mice (Figure 1D), as shown by the MAR (Figure 1E) and BFR (Figure 1F) quantification. Also, the bone formation of SAMP6 mice after RESV treatments exhibited a still lower, but not significant, rate compared to age-matched RESV-treated SAMR1 mice, while RESV was not effective in further promoting bone formation in SAMR1 mice (Figure 1D-F). Collectively, these data suggested that RESV enhances bone formation and counteracts accelerated bone loss in SAMP6 mice.
Figure 1
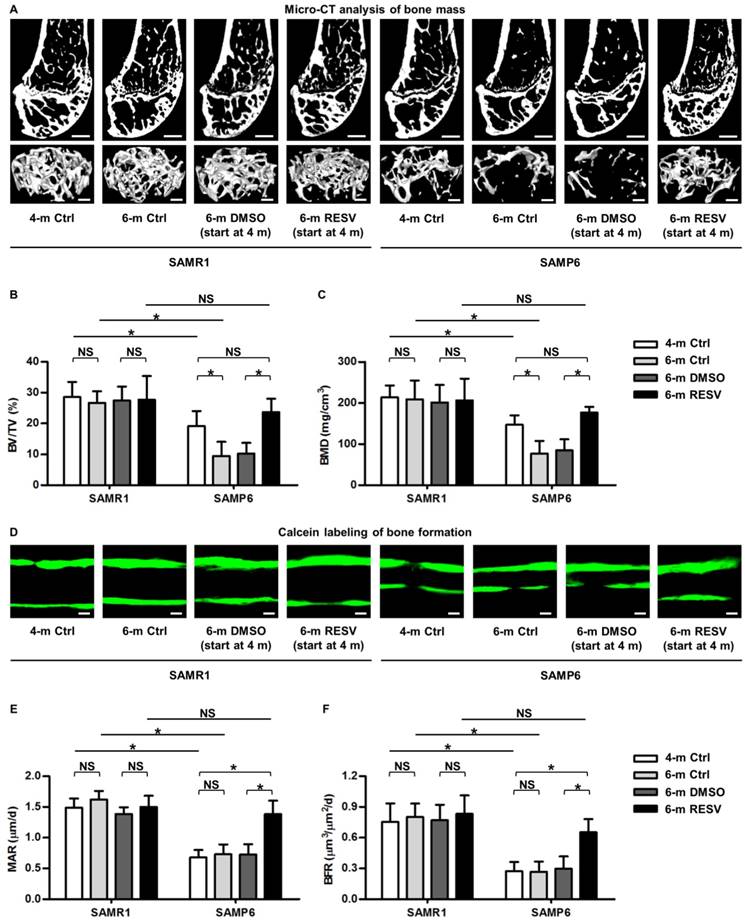
Resveratrol enhances bone formation and counteracts accelerated bone loss in SAMP6 mice. (A-C) Representative 2D section and 3D reconstruction of micro-CT images (A) and quantitative analysis of trabecular bone volume (B) and bone mineral density (C) in distal femora. Bars: 500 μm (2D images) and 50 μm (3D images). (D-F) Representative images of calcein double labeling (D) with quantification of mineral apposition rates (E) and bone formation rates (F) in distal femora. Bars: 25 μm. 4-month-old SAMR1 and SAMP6 mice were treated with either resveratrol (100 mg/kg i.p., every other day for 2 months) or the DMSO (5%) solvent control. n = 4-5 per group. Data represent mean ± SD. *P < 0.05; NS, not significant (P > 0.05). Data were analyzed using ANOVA followed by Newman-Keuls post-hoc tests.
RESV improves osteogenic differentiation of senescent BMMSCs
It has been recognized that BMMSCs are putatively responsible for postnatal bone homeostasis and regeneration [5, 66], and that osteogenic decline of BMMSCs is critical for osteoporosis with advanced age [4, 29]. To directly evaluate osteogenic changes of BMMSCs upon RESV treatment, we isolated BMMSCs from 4-month-old SAMP6 and SAMR1 mice. As exhibited, BMMSCs from SAMP6 mice at this age already demonstrated the typical cell senescent phenotypes ex vivo, with a higher SA-β-gal positive cell percentage (Figure S1A-B), a weaker colony-forming capability, a lower cell percentage in the S-phase of the cell cycle (Figure S1C), and a decreased proliferation rate (Figure S1D). Importantly, these cells were dramatically defective in osteogenic differentiation, as quantified by less than half of the ALP activity (Figure 2A-C) and mineralization capability (Figure 2D-F) during osteogenic induction compared to their SAMR1 counterparts, underlying the impaired osteoblastogenesis in vivo. Notably, RESV treatments substantially rescued the osteogenic decline of BMMSCs from SAMP6 mice (Figure 2A-F), as confirmed also by it promoting the expression of osteogenic marker genes Runx2 and Ocn [67] at both mRNA (Figure 2G) and protein (Figure 2H) levels.
Figure 2
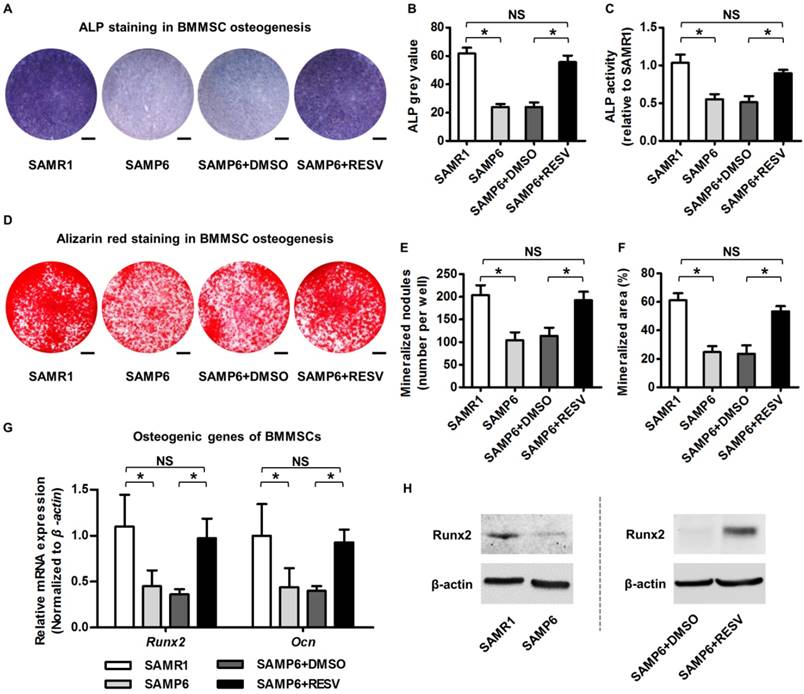
Resveratrol improves osteogenic differentiation of BMMSCs derived from SAMP6 mice. (A-C) Representative images of ALP staining (A) with quantification of ALP grey value of staining (B) and ALP activity (C) in osteogenic differentiation of BMMSCs. Bars: 5 mm. (D-F) Representative images of alizarin red staining (D) with quantification of mineralized nodules (E) and area (F) in osteogenic differentiation of BMMSCs. Bars: 5 mm. (G, H) qRT-PCR analysis of mRNA expression (G) and western blot analysis of protein expression (H) levels of osteogenic marker genes in osteogenic differentiation of BMMSCs. BMMSCs from 4-month-old SAMP6 mice were treated with either resveratrol (10 μM) or the DMSO (0.001%) solvent control. n = 3 per group. Data represent mean ± SD. *P < 0.05; NS, not significant (P > 0.05). Data were analyzed using ANOVA followed by Newman-Keuls post-hoc tests.
RESV improvements on osteogenesis of senescent BMMSCs were further verified in naturally aged BMMSCs. As shown, BMMSCs derived from 22-month-old naturally aged mice also demonstrated an increased SA-β-gal positive percentage (Figure S2A-B) with a decreased proliferation rate (Figure S2C). Furthermore, the naturally aged BMMSCs were impaired in ALP activity (Figure S2D-E) and mineralization capability (Figure S2F-G) during osteogenic differentiation. Importantly, RESV treatments in vitro could remarkably rescue the osteogenic impairment (Figure S2D-G). Taken together, these data suggested that RESV improves osteogenic differentiation of senescent BMMSCs.
RESV ameliorates mitochondrial functional and transcriptional disorders in BMMSC senescence
In identifying the underlying reason for RESV protection against osteogenic decline of senescent BMMSCs, it should be noticed that metabolic compromise is proposed to be an important mechanism underlying senescence of stem cells including BMMSCs [28, 29], and that mitochondrial improvements serve as one of the most prominent RESV influences [13, 68]. Accordingly, we evaluated the effects of RESV on the metabolic profiles of senescent BMMSCs and the related mitochondrial changes. As expected, we discovered that BMMSCs from SAMP6 mice demonstrated >50% reduction in ATP production versus ADP contents compared to those from SAMR1 mice, which could be rescued by RESV treatments (Figure 3A). Furthermore, SAMP6 BMMSCs also suffered from oxidative stress, as shown by DCFDA fluorescence (Figure 3B) and flow cytometric quantification (Figure 3C) confirming an increase in the total cellular ROS level. Notably, the oxidative stress of SAMP6 BMMSCs was also significantly ameliorated by RESV (Figure 3B-C).
These metabolic changes of SAMP6 BMMSCs after RESV treatments could be attributed to mitochondrial contributions. As shown by Rhodamine 123 detection of mitochondrial membrane potential (Figure 3D) and its quantification (Figure 3E), BMMSCs derived from SAMP6 mice demonstrated impaired mitochondrial membrane potential, which was restored by RESV treatments. Importantly, RESV also restored the suppressed OXPHOS activity of senescent BMMSCs, as analyzed by the oxygen consumption in culture (Figure 3F), indicating mitochondrial functional improvements underlie the alleviation of metabolic disorders.
To further examine whether the improved OXPHOS activity was due to recovery of impaired mitochondrial OXPHOS electron transport chain (ETC) complexes, considering that the complex subunits are encoded by either the nuclear genome or the mitochondrial DNA (mtDNA) [69, 70], we separately investigated expression levels of genes of these 2 categories to see whether RESV facilitated mitochondrial non-autonomous and/or autonomous recovery. Gene representatives encoding subunits of each of the 5 ETC complexes were selected for analysis accordingly [26]. Results showed that despite downregulation of nuclear-encoded mitochondrial genes for complex I, II and ATP synthase in BMMSCs from SAMP6 mice, RESV was not effective in rescuing this decline (Figure 3G). Alternatively, RESV restored expression levels of most mtDNA-encoded genes in BMMSC senescence, leading to substantial recovery, particularly in complex I and III, but also in certain subunits of complex IV and ATP synthase (Figure 3H and Figure S3). These data collectively suggested that RESV ameliorates mitochondrial functional and transcriptional disorders in BMMSC senescence.
RESV upregulates Mitofilin in alleviating senescence-associated mitochondrial morphological alterations of BMMSCs
Next, we intended to figure out how RESV improved mitochondria in senescent BMMSCs. Currently, Ampk and Sirt1/Pgc-1 pathways are known to be primary targets mediating RESV direction of mitochondrial metabolism [12, 13]. Nevertheless, unexpectedly, although RESV indeed stimulated Ampk and Pgc-1 activities in BMMSCs from SAMP6 mice, as demonstrated by increased Ampk phosphorylation (Figure S4A) and a decreased Pgc-1 acetylation (Figure S4B), effects of RESV between SAMR1 and SAMP6 BMMSCs were equally efficient, as shown by RESV induction of comparable changes of these signaling proteins in BMMSCs from SAMR1 mice (Figure S4A-B). Thus, while activation of Ampk and Pgc-1 pathways might still participate in RESV effects on BMMSCs, there were probably more specific mechanisms in the senescent situation, considering the profound alleviation of skeletal deficiency specifically in SAMP6 mice in vivo (Figure 1). Correspondingly, Pgc-1 downstream transcripttional factors regulating mitochondrial metabolism [71] showed a different tendency in RESV-treated BMMSCs from SAMR1 and SAMP6 mice; in particular, they were not restored by RESV in BMMSC senescence (Figure S4C). These results indicated that RESV improves mitochondria in senescent BMMSCs potentially through non-canonical pathways.
To elucidate how RESV restored mtDNA transcription and mitochondrial metabolism in SAMP6 BMMSCs, we decided to observe in detail the ultrastructural morphology of mitochondria, hoping to gather some information to help us decipher what changes occurred. Surprisingly, we found that, compared to BMMSCs from SAMR1 mice, BMMSCs from SAMP6 mice did not increase mitochondria quantity, but instead possessed larger and swollen mitochondria, with remarkably increased IM:OM ratio (Figure 4A-D). Interestingly, RESV treatments in SAMP6 BMMSCs rescued this altered mitochondrial morphology, indicating that the development and RESV-directed amelioration of mitochondrial compromise in BMMSC senescence were based on modulation of the morphological stability.
Figure 3
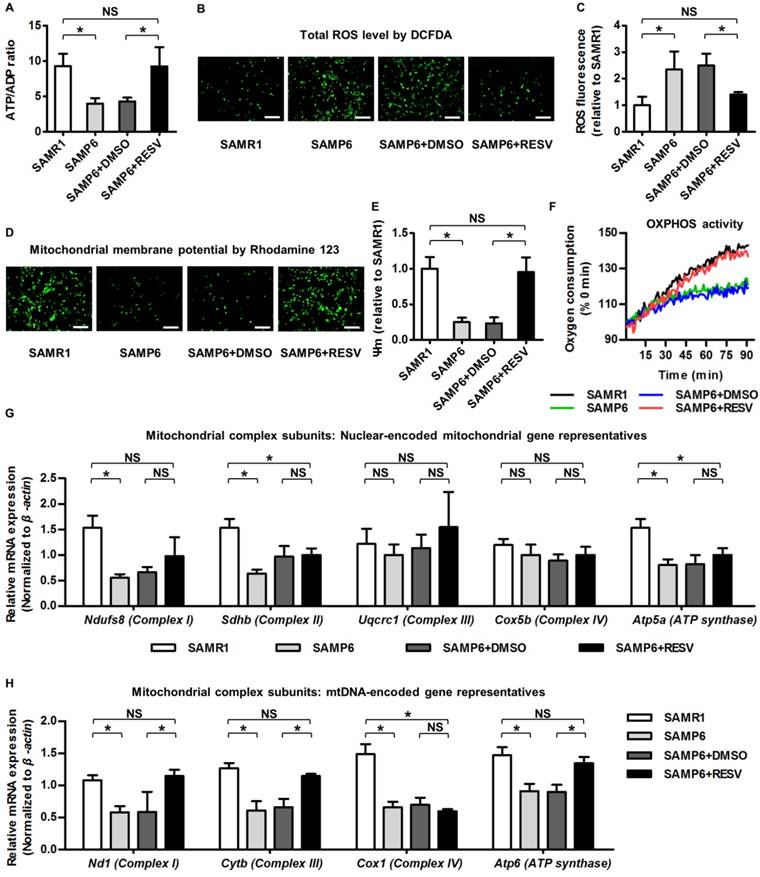
Resveratrol improves mitochondrial functionality and transcription in BMMSCs derived from SAMP6 mice. (A) Quantitative analysis of ATP production versus ADP ratio in BMMSCs. (B, C) DCFDA detection of total ROS level in BMMSCs (B) with flow cytometric quantitative analysis of fluorescence intensity (C). Bars: 10 μm. (D, E) Rhodamine 123 detection of mitochondrial membrane potential of BMMSCs (D) with quantitative analysis of fluorescence intensity (E). Bars: 10 μm. (F) Kinetic analysis of oxygen consumption as an index of mitochondrial OXPHOS activity in cultured BMMSCs. (G, H) qRT-PCR analysis of mRNA expression levels of nuclear-encoded (G) and mtDNA-encoded (H) gene representatives for mitochondrial complex subunits of BMMSCs. BMMSCs from 4-month-old SAMP6 mice were treated with either resveratrol (10 μM) or the DMSO (0.001%) solvent control. n = 3 per group. Data represent mean ± SD. *P < 0.05; NS, not significant (P > 0.05). Data were analyzed using ANOVA followed by Newman-Keuls post-hoc tests.
Figure 4
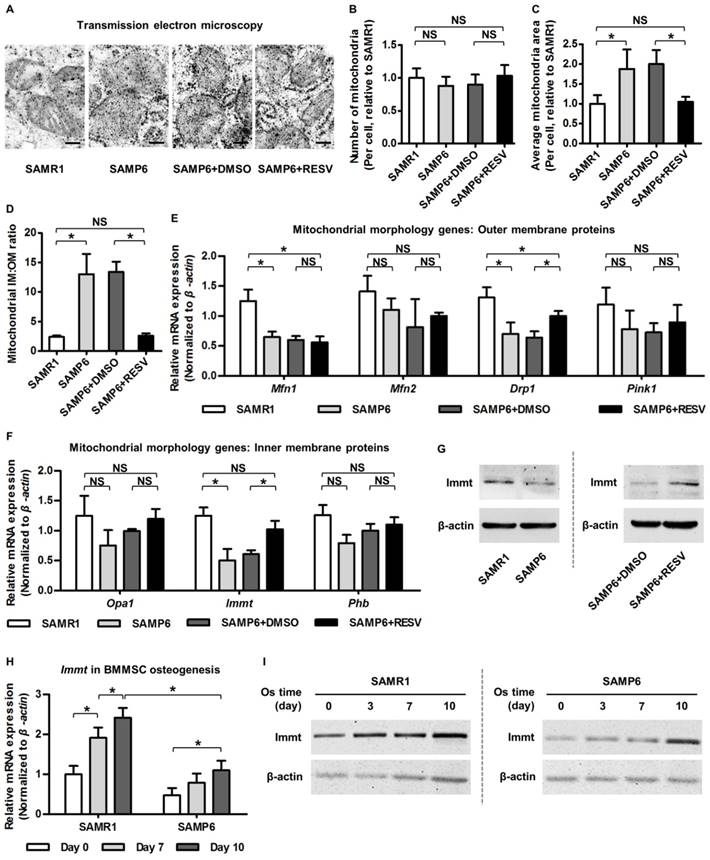
Resveratrol promotes Mitofilin expression and restores mitochondrial morphology in BMMSCs from SAMP6 mice. (A-D) Representative transmission electron microscopy images (A) and quantitative analysis of mitochondrial number (B) and average area (C) per cell as well as the ratio of mitochondrial inner membrane versus outer membrane (D) in BMMSCs. Bars: 125 nm. (E, F) qRT-PCR analysis of mRNA expression levels of mitochondrial morphological genes encoding proteins on outer membrane (E) and inner membrane (F) in BMMSCs. (G) Western blot analysis of protein expression levels of Immt (Mitofilin) in BMMSCs. BMMSCs from 4-month-old SAMP6 mice were treated with either resveratrol (10 μM) or the DMSO (0.001%) solvent control. (H, I) qRT-PCR analysis of mRNA expression (H) and western blot analysis of protein expression (I) levels of Immt (Mitofilin) in osteogenic differentiation of BMMSCs from 4-month-old SAMR1 and SAMP6 mice. n = 3 per group. Data represent mean ± SD. *P < 0.05; NS, not significant (P > 0.05). Data were analyzed using ANOVA followed by Newman-Keuls post-hoc tests.
Given the changes in relative area of mitochondrial IM versus OM, we next investigated outer membrane protein representatives (mainly mitochondrial dynamic regulators [72, 73]) and inner membrane protein representatives (primarily modulators of organization of cristae where the OXPHOS ETC exists [74, 75]) reported to regulate mitochondrial morphology. Data demonstrated that while RESV ameliorated expression decline of a fission-related gene encoding dynamin-related protein 1 (Drp1) in BMMSCs from SAMP6 mice, the mitochondrial outer membrane proteins were largely not influenced by RESV (Figure 4E). On the other hand, we discovered that mRNA expression of the Immt gene, which encodes the inner membrane protein Mitofilin/Mic60, the core component of MICOS [38, 39], was significantly downregulated in senescent BMMSCs and was restored by RESV application (Figure 4F). The promotion of Mitofilin by RESV in BMMSCs from SAMP6 mice was further confirmed at the protein expression level (Figure 4G). These data suggested that RESV upregulates Mitofilin in alleviating senescence-associated mitochondrial morphological alterations of BMMSCs.
Figure 5
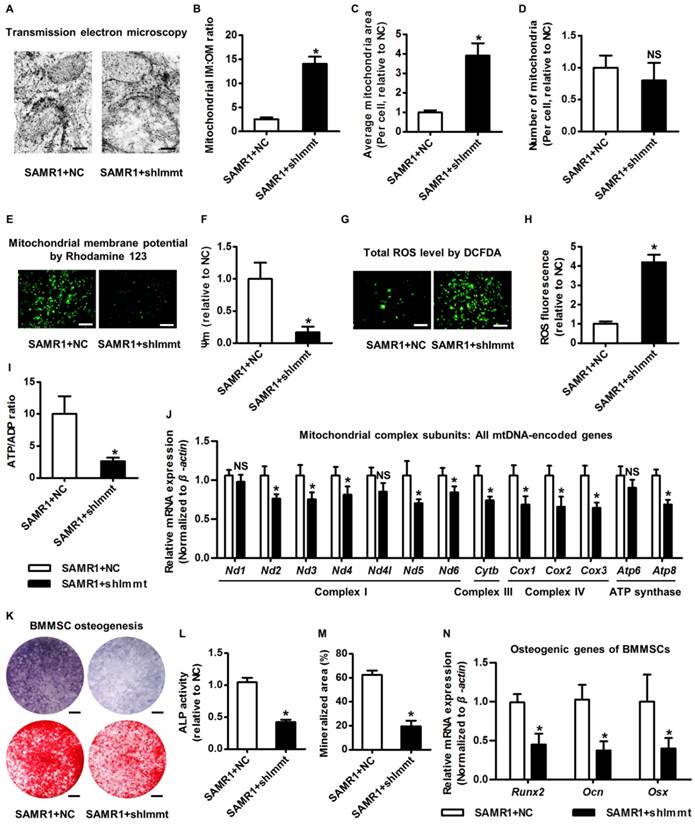
Mitofilin is indispensable for mitochondrial homeostasis and osteogenesis of BMMSCs. (A-D) Representative transmission electron microscopy images (A) and quantitative analysis of the ratio of mitochondrial inner membrane versus outer membrane (B) as well as mitochondrial average area (C) and number (D) per cell in BMMSCs. Bars: 125 nm (high magnification). (E, F) Rhodamine 123 detection of mitochondrial membrane potential of BMMSCs (E) with quantitative analysis of fluorescence intensity (F). Bars: 10 μm. (G, H) DCFDA detection of total ROS level in BMMSCs (G) with flow cytometric quantitative analysis of fluorescence intensity (H). Bars: 10 μm. (I) Quantitative analysis of ATP production versus ADP ratio in BMMSCs. (J) qRT-PCR analysis of mRNA expression levels of all 13 mtDNA-encoded mitochondrial complex subunits in BMMSCs. (K-M) Representative images of ALP and alizarin red staining (K) with quantification of ALP activity (L) and mineralization (M) in osteogenic differentiation of BMMSCs. Bars: 5 mm. (N) qRT-PCR analysis of mRNA expression levels of osteogenic marker genes in osteogenic differentiation of BMMSCs. BMMSCs from 4-month-old SAMR1 mice were transfected with either the shRNA for Immt (Mitofilin) or the negative control (a scrambled sequence, NC) by a lentiviral vector. n = 3 per group. Data represent mean ± SD. *P < 0.05; NS, not significant (P > 0.05). Data were analyzed using the two-tailed Student's t test.
To further establish a rationale to investigate the roles of Mitofilin in mitochondrial and osteogenic function of BMMSCs in senescence, we examined whether Mitofilin participates in osteogenesis of BMMSCs and, more importantly, if it serves as a covariant factor in declined osteogenesis of SAMP6 BMMSCs. As analyzed by osteogenic induction, both mRNA and protein expression levels of the Immt gene (Mitofilin) gradually increased with advancing osteogenic time, while its expression in BMMSCs from SAMP6 mice was lower at each time point compared to BMMSCs from SAMR1 mice (Figure 4H-I), indicating a close relationship between Mitofilin and the osteogenic capability of BMMSCs in differentiation and in senescence-associated decline. Therefore, Mitofilin was selected afterwards for mechanistic experiments.
Mitofilin is required for mitochondrial homeostasis and osteogenesis of BMMSCs
The above clues prompted us to clarify the function of Mitofilin in modulating mitochondrial and osteogenic phenotypes of BMMSCs. Accordingly, we knocked down expression of the Immt gene using shRNA (Figure S5A-B). As expected, knockdown of Immt in BMMSCs from SAMR1 mice induced giant mitochondria (Figure 5A) with increased IM:OM ratio (Figure 5B) and average area (Figure 5C), but without changing the mitochondrial number (Figure 5D). Importantly, BMMSCs deficient for Mitofilin developed impaired mitochondrial membrane potential (Figure 5E-F) and cellular metabolic disorders involving increased total ROS (Figure 5G-H) but less ATP production (Figure 5I). Furthermore, Immt knockdown generally suppressed mtDNA transcripttion (Figure 5J), while the genes for nuclear-encoded mitochondrial complex subunits remained uninfluenced (Figure S5C). These results indicated that Mitofilin indeed regulates mitochondrial stability of BMMSCs via facilitating mitochondrial autonomous gene transcription.
With regard to the functional phenotypes of BMMSCs after Immt knockdown, interestingly, we discovered that Mitofilin deficiency in BMMSCs from SAMR1 mice mimicked senescent phenotypes observed in BMMSCs from SAMP6 mice, including a dramatic increase in SA-β-gal-positive cell percentage (Figure S5D-E), a decrease of S-phase cell percentage (Figure S5F), and a declined proliferation rate (Figure S5G). Importantly, insufficiency of Mitofilin resulted in defective osteogenesis of BMMSCs from SAMR1 mice, as proved by weaker ALP activity, impaired mineralization capability and downregulated expression levels of osteogenic marker genes (Figure 5K-N). Furthermore, knockdown of Immt in vivo in the bone marrow space lead to osteoporosis in SAMR1 mice (Figure S5H-J). These data highlighted that Mitofilin is necessary for mitochondrial homeostasis and osteogenesis of BMMSCs.
Mitofilin mediates resveratrol-induced mitochondrial and osteogenic improvements of BMMSCs in senescence
Finally, we examined whether upregulation of Mitofilin mediated the protective effects of RESV against the mitochondrial and osteogenic compromise of senescent BMMSCs. As shown by the expression of mtDNA-encoded mitochondrial complex subunits, Immt knockdown remarkably inhibited RESV promotion on most complex I subunits (Figure 6A) and blocked positive effects of RESV on subunits of complex III (Figure 6B), IV (Figure 6C) and ATP synthase (Figure 6D). This general suppression of RESV facilitating mitochondrial autonomous gene transcription in Mitofilin deficiency resulted in blockade of RESV improvement of osteogenesis of senescent BMMSCs (Figure 6E), leading to maintenance of defective ALP activity (Figure 6F), mineralization capability (Figure 6G) and osteogenic marker gene expression (Figure 6H-I) in BMMSCs from SAMP6 mice. These findings collectively suggested that Mitofilin mediates resveratrol-induced mitochondrial and osteogenic improvements of BMMSCs in senescence.
Discussion
Bone aging brings about many degenerative skeletal diseases such as primary osteoporosis and osteoarthritis, and may also exert adverse effects on other systems such as the central nervous system, the hematopoietic and the related immune system, and the endocrinium [76-78]. RESV, a small molecule compound that safely mimics the effects of dietary restriction, has been well documented to extend the lifespan of lower organisms [9-11] and improve the health of aging rodents [12-14]. However, although it is a critical pathogenesis of osteoporosis in aging [4, 79], whether senescence-associated osteogenic functional decline of BMMSCs is rescued by RESV remains unknown. In the present study, using the bone-specific, senescence-accelerated, osteoblastogenesis/osteogenesis-defective SAMP6 mouse strain as an experimental model, we show that application of RESV in a chronic intermittent methodology specifically enhances bone formation and counteracts accelerated bone loss and, importantly, improves osteogenic differentiation of senescent BMMSCs. Furthermore, while we have proved that the protective effects of RESV against osteogenic decline in BMMSC senescence are through restoring cellular metabolism through mitochondrial functional recovery and autonomous gene transcription, we unexpectedly discovered a non-canonical molecular target, the MICOS core component Mitofilin/Immt/ Mic60 [38, 39], that RESV uses as the key mediator of its influences on mitochondrial morphological and functional homeostasis and osteogenesis in senescent BMMSCs. We have also revealed that Mitofilin is indispensable for mitochondrial stability and osteogenesis of BMMSCs in bone integrity. Taken together, our findings uncover osteogenic functional improvements of senescent MSCs as critical impacts in the anti-osteoporotic practice of RESV, and unravel Mitofilin as a novel mechanism mediating RESV promotion of mitochondrial function in stem cell senescence.
Figure 6
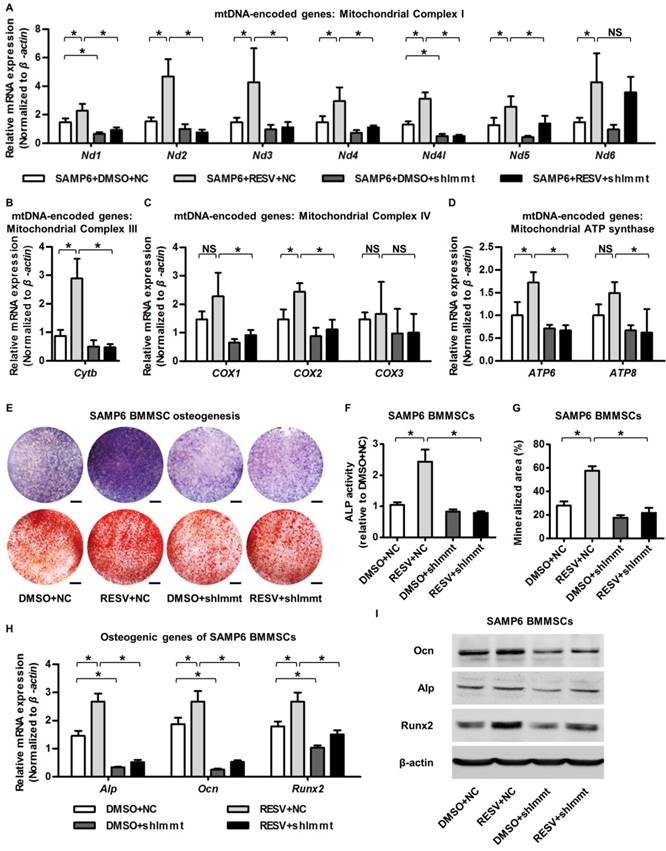
Mitofilin mediates resveratrol-induced mitochondrial and osteogenic improvements of SAMP6 BMMSCs. (A-D) qRT-PCR analysis of mRNA expression levels of all 13 mtDNA-encoded mitochondrial complex subunits in complex I (A), complex III (B), complex IV (C) and ATP synthase (D) in BMMSCs. (E-G) Representative images of ALP and alizarin red staining (E) with quantification of ALP activity (F) and mineralization (G) in osteogenic differentiation of BMMSCs. Bars: 5 mm. (H, I) qRT-PCR analysis of mRNA expression (H) and western blot analysis of protein expression (I) levels of osteogenic marker genes in osteogenic differentiation of BMMSCs. BMMSCs from SAMP6 mice treated with resveratrol (10 μM) and the DMSO (0.001%) solvent control were transfected with either the shRNA for Immt (Mitofilin) or the negative control (a scrambled sequence, NC) by a lentiviral vector. n = 3 per group. Data represent mean ± SD. *P < 0.05; NS, not significant (P > 0.05). Data were analyzed using ANOVA followed by Newman-Keuls post-hoc tests.
Current understanding of RESV impacts on adult stem cells mainly comes from in vivo conditions of tissue injury and regeneration as well as in vitro situations of proliferation and induced differentiation. It has been reported that RESV ameliorates irradiative injury of hematopoietic stem cells (HSCs) [80], oxidative injury of neural stem cells (NSCs) [81] and chemical injury of spermatogonial stem cells (SSCs) [48], while RESV further promotes recovery of HSC populations from genetic defects [82] and facilitates endogenous myocardial regeneration of cardiac stem cells (CSCs) [83, 84]. Furthermore, for the functional potential of stem cells, increasing evidence has highlighted effects of RESV to promote self-renewal [85-87] and multipotency [88-91] in vitro, particularly for osteogenic differentiation of MSCs derived from healthy individuals [44, 46, 92, 93]. However, previous studies did not reach a consensus on RESV impacts on senescence of MSCs: some researchers claimed that RESV could protect adipose-derived MSCs from H2O2- and D-glucose-induced senescence and also attenuated senescence in long-term culture [94], while more reports documented only transient protective effects [95], dose- and duration-dependent positive or detrimental effects [45], and even accelerating effects of RESV on MSC senescence in serial passages [96]. This controversy might be due to differences of the origin of MSCs or application method of RESV, but may further be attributed to the induced in vitro models that cannot replicate real in vivo senescence [97]. To address this issue, we applied the SAMP6 mice and their BMMSCs in this study, the ex vivo findings of which reflect phenotypic changes in situ [32]. Furthermore, we additionally confirmed positive effects of RESV on BMMSCs from natural aging mice. Together with Zhou and colleagues using a laminopathy-based progeroid model (the Zmpste24-/- mice) [15], we have added a dimension to this field showing that RESV indeed improves senescent stem cells. Moreover, while Zhou's group demonstrated RESV recovery of the MSC population in progeria [15], we further reveal that RESV rescues osteogenic functional decline of MSCs specifically in accelerated bone loss. Although we did not show general effects of RESV directly on senescence levels of MSCs and senescence-associated phenotypes of impaired cell viability and self-renew [98], misdirected differentiation toward adipocytes [99], or DNA damage [100] and altered secretion [101], the current findings, particularly on RESV improving osteogenesis and bone formation in osteoporosis, is of translational significance as RESV may be developed as a remedy for various skeletal diseases caused by defective osteoblastogenesis, such as glucocorticoid-induced bone loss [102] and osteogenesis imperfecta [103]. Our findings open an avenue for future research on the application of RESV to rejuvenate stem cells in the natural aging process.
The in vivo application of RESV has been extensively investigated in preclinical experimental models. Although most studies have conducted low-dose dietary RESV delivery to mimic caloric restriction and delay age-related deteriorations in rodents, this methodology does not increase the longevity of ad libitum-fed animals when started midlife, with no observations on stem cell rejuvenation [14, 16]. Also, despite recent in vivo reports on low-dose dietary RESV promoting adipogenesis of adipose progenitors/stromal vascular cells, its implications in the aging process are unknown [104]. As a proof-of-concept study, we here applied RESV in vivo intraperitoneally, considering the better control of applied dose by intraperitoneal injection over feeding, and the reported efficacy of mitochondrial intervention by intraperitoneal injection of RESV [49]. The dosage was then selected at 100 mg/kg, which is according to reports applying this dosage to affect stem cell activity in response to chemical injury in vivo, and is based on previous documents below the potential highest dose not to induce obvious systemic alterations [47, 48]. The experimental duration was determined given the time needed for chronic changes of bone mass by bone remodeling according to our observations [50], and the method of chronic intermittent application every other day was based on previous reports on feeding-based RESV retarding aging parameters in mice [16]. Importantly, the high-dose intraperitoneal administration of RESV chronically intermittently in the 2-month period did not induce any obvious alterations of mice by our observation, which maintained good physiological conditions as the control mice, suggesting the safety of this experimental methodology. Thus, further translational researches may consider using this method for a stem-cell based rejuvenative strategy.
Mitochondria are highly specialized and dynamic double-membrane organelles of bacterial origin that play crucial roles in multiple processes in eukaryotic cells, such as their fundamental function in energy production and metabolism [105, 106]. Particularly, recent discoveries have suggested critical participation of mitochondria in the pluripotent maintenance and differentiation of stem cells [106, 107]. Accordingly, while anaerobic glycolysis is required at the pluripotent state, a metabolic switch to mitochondrial OXPHOS is necessary for differentiation of stem cells [108, 109], including osteogenesis of MSCs [110, 111]. Notably, mitochondrial dysfunction critically contributes to senescence of stem cells, in which OXPHOS metabolism suffers from impairments, leading to less energy production but increased oxidative stress [27, 112]. In MSCs, this mitochondrial compromise results in declined self-renewal and misdirected differentiation, underlying age-related osteoporosis [31, 112, 113]. Correspondingly, it has emergingly been noticed that function of stem cells can be revitalized by metabolic reprogramming under nutrient control [106]. For example, a recent report has confirmed that a reduction of glucose by 10%-30% in culture conditions of BMMSCs significantly prevented replicative senescence with an accelerated osteogenic commitment due to higher activities of ETC, and more synthesis of ATP and antioxidants [114]. Nevertheless, despite the discovery of dietary restriction mimetic reagents like RESV, metformin and rapamycin [7, 8], feasible pharmacological strategies to reprogram the mitochondrial metabolism of senescent stem cells are still lacking. Further complicating matters, certain anti-aging pharmacology may exert detrimental side effects, such as the well-known immunosuppression by rapamycin [115, 116], in spite of its efficacy to improve MSC function in vivo [117, 118]. In this regard, RESV exhibits a particular advantage as it safely mimics wide-ranging positive effects of dietary restriction with easy administration but no adverse impacts on rodents even at very high concentrations, which indicates it is well-tolerated and non-toxic [47]. Here, we further uncover that chronic intermittent application of RESV enhances bone formation and counteracts accelerated bone loss in vivo, providing an optimal solution to revitalize mitochondrial function and improve the function of stem cells in senescence. Additionally, it is noticeable that RESV failed to enhance bone formation in SAMR1 mice, which do not show mitochondrial defect, suggesting that the anabolic effects of RESV might be restricted to the senescent status. Further long-term in vivo experiments in aged mice are needed to clarify whether RESV impacts are restricted to the SAMP6 mouse strain, which might be strain-independent based on the in vitro osteogenic findings of BMMSCs in this study.
In order to meet different metabolic needs in distinct situations, cells modulate mitochondrial function through biogenesis regulation as well as dynamic morphological changes [72, 119]. Importantly, mitochondria are known as semi-autonomous organelles that possess their own genome of mtDNA, encoding 13 essential protein subunits of ETC complexes [120]. Therefore, mitochondrial metabolism is regulated by the coordinated signals from the nuclear and mitochondrial genomes, the nuclear compartment of which include the mitochondrial transcription factor A (Tfam) and general biogenesis regulators like Pgc-1 and Ampk [71, 121, 122]. With advanced age, nuclear-mitochondrial communication is disrupted under genetic or metabolic stresses, thus mediating loss of mitochondrial homeostasis through either the Pgc-1 or Ampk pathways to inhibit mitochondrial biogenesis, or a Pgc-1-independent pathway suppressing Tfam to induce deficiency of mitochondrial autonomous gene transcription, both of which are under common control of Sirt1 [26, 27]. Typically, as a Sirt1 activator, RESV improves mitochondrial function by activating Sirt1 and Ampk [12, 13, 25]. Here, we surprisingly discover that RESV may selectively activate a non-canonical target in senescent stem cells, leading to rescue of mtDNA transcription, with the most substantially recovered OXPHOS genes (Nd1, Cytb, Atp6) being just the reported Tfam downstream-regulated subunits in aging [26]. Although we did not rule out the contributions of the typical Pgc-1 and Ampk pathways, or investigate Tfam participations in mediating RESV impacts, the promoted mitochondrial autonomous gene transcription suggests a potential novel mechanism underlying RESV effects on senescent stem cells. How the decision for pathway selection was made and whether crosstalk exists remain to be elucidated, which might be attributed to distinctive characteristics of stem cells in senescence. As shown, this decision could be greatly influenced by cellular energetic states [26].
Mitofilin/Immt/Mic60, originally identified as heart muscle protein (Hmp) [123] and named as formation of crista junction protein 1 (Fcj1) in yeast [124], is a nuclear-encoded mitochondrial inner membrane protein controlling mitochondrial morphology [64, 125]. In detail, as the core component of MICOS [38], Mitofilin functions as a multifunctional regulator of mitochondrial architecture: on one hand, Mitofilin is required for shaping crista junctions to maintain the integrity of the mitochondrial inner membrane [124, 126]; on the other hand, Mitofilin is also coupled to the outer membrane and promotes protein import into the mitochondrial intermembrane space [125, 127]. Based on its vital role in establishing mitochondrial structure, Mitofilin is known to regulate a wide range of mitochondrial functions, including coordination with ETC to maintain OXPHOS [128], preservation of mitochondrial membrane potential and ROS production at physiological levels [64], keeping mtDNA transcription [129], facilitating fission and fusion dynamics [39], and regulating cytochrome c release during apoptosis [130]. Consistent with our findings, Mitofilin deficiency is reported to result in giant mitochondria induced by a massive increase of large concentric stacks of cristae membranes, leading to impaired mitochondrial metabolism with reduced mtDNA transcription [39, 64, 129]. In this study, we further reveal significant functions of Mitofilin in stem cell senescence and functional improvements, in that suppression of Mitofilin is enough to mimic a senescent phenotype in SAMR1 BMMSCs, and Mitofilin upregulation is indispensable for the positive effects of RESV on senescent BMMSCs from SAMP6 mice by modulating mitochondrial autonomous gene transcription. These findings highlight that the presence or absence of Mitofilin could be acting as a bottleneck on the downstream effects of RESV, at least in the SAM strains. Nevertheless, it is still not clear if the BMMSCs from SAMP6 and aged mice share the same mechanism in developing the senescent phenotypes, and whether RESV enhances osteogenic performance of BMMSCs from aged mice by targeting pathways other than Mitofilin, which require future functional studies. Mechanistically, Mitofilin regulates mtDNA via interacting with both mtDNA itself and the transcription factor Tfam, respectively promoting maintenance of mtDNA architecture and binding of Tfam to mtDNA promoters [39, 129]. Additionally, RESV might still restore nuclear-mitochondrial communication in senescent stem cells, given that Mitofilin facilitates import of cytoplasmic protein involving Tfam into mitochondria [125, 131]. Also, as the critical regulator controlling mitochondrial morphology, Mitofilin might also modulate effects of the canonical Pgc-1 and Ampk pathways through a similar bottleneck function, affecting nuclear-mitochondrial communication. The detailed mechanisms underlying RESV-promoted Mitofilin stimulating mtDNA transcription in senescent stem cells is worth exploring in future studies. In addition, how RESV upregulates Mitofilin expression should also be deciphered in future work, which might be attributed to activation of RESV by various signaling in MSCs, such as the Wnt/β-catenin and extracellular signal-regulated kinase (ERK) pathways [46, 86, 96].
In summary, we reveal that RESV enhances bone formation and counteracts accelerated bone loss via Mitofilin-mediated mitochondrial and osteogenic improvement of senescent BMMSCs. Our findings uncover osteogenic functional improvements of senescent MSCs as critical impacts in anti-osteoporotic practice of RESV, and unravel Mitofilin as a novel mechanism mediating RESV promotion of mitochondrial function in stem cell senescence.
Acknowledgements
This work was supported by grants from the National Key Research and Development Program of China (2016YFC1102900 and 2016YFC1101400), the General Program of National Natural Science Foundation of China (81570937 and 81470710), and the State Scholarship Fund of China (201603170205). We thank Dr. Chi-Der Chen for important intelligent and material support.
Author contributions
Ya-Jie Lv, Yi Yang, Bing-Dong Sui and Cheng-Hu Hu contributed equally to the study design, experimental work, data analysis, data interpretation and manuscript preparation. Pan Zhao and Li Liao contributed to the experimental work and revised the manuscript. Ji Chen and Li-Qiang Zhang contributed to the experimental work. Tong-Tao Yang contributed to the data interpretation. Shao-Feng Zhang and Yan Jin conceived and supervised the study. All authors have reviewed and approved the final version of the manuscript.
Abbreviations
Ac-Lys: acetylated lysine; Ac-p53: acetylated p53; ADP: adenosine diphosphate; ALP: alkaline phosphatase; Ampk: adenosine 5'-monophosphate-activated protein kinase; ANOVA: analysis of variance; ATP: adenosine triphosphate; BFR: bone formation rate; BMD: bone mineral density; BMMSCs: bone marrow mesenchymal stem cells; BV/TV: bone volume over tissue volume; Cox: cytochrome c oxidase subunit; CSCs: cardiac stem cells; Ctrl: control; Cytb: cytochrome b; DCFDA: 2',7'-dichlorofluorescin diacetate; DMSO: dimethyl sulfoxide; Drp1: dynamin-related protein 1; ERK: extracellular signal-regulated kinase; Errα: estrogen-related receptor alpha; ETC: electron transport chain; Fcj1: formation of crista junction protein 1; Fis1: fission 1; HMP: heart muscle protein; HSCs: hematopoietic stem cells; IM: inner membrane; Immt: inner membrane protein of mitochondria; IM:OM ratio: the ratio of mitochondrial inner membrane over outer membrane; i.p.: intraperitoneally; IP: immunoprecipitation; MAR: mineral apposition rate; Mfn: mitofusion; MICOS: mitochondrial contact site and cristae organizing system; Micro-CT: micro-computed tomography; MINOS: mitochondrial inner membrane organizing system; MSCs: mesenchymal stem cells; mtDNA: mitochondrial DNA; MTT: methyl thiazolyl tetrazolium; NADH: nicotinamide adenine dinucleotide; NC: negative control; Nd: NADH dehydrogenase subunit; Ndufs8: NADH:ubiquinone oxidoreductase core subunit S8; Nrf1: nuclear respiratory factor 1; NSCs: neural stem cells; Ocn: osteocalcin; OM: outer membrane; Opa1: optic atrophy 1; Os: osteogenesis; Osx: osterix; OXPHOS: oxidative phosphorylation; Pgc-1: peroxisome proliferator-activated receptor gamma coactivator-1; Phb: prohibitin; Pink1: phosphatase and tensin homolog-induced putative kinase 1; Pparα: peroxisome proliferator-activated receptor alpha; qRT-PCR: quantitative real-time polymerase chain reaction; RESV: resveratrol; ROS: reactive oxygen species; Runx2: runt-related transcription factor 2; SA-β-gal: senescence-associated beta-galactosidase; SAMP6: senescence-accelerated mice-prone 6; SAMR1: senescence-accelerated mice-resistant 1; SD: standard deviation; Sdhb: succinate dehydrogenase b; shImmt: short hairpin RNA for the Immt gene; Sirt1: sirtuin1; SSCs: spermatogonial stem cells; TEM: transmission electron microscopy; Tfam: mitochondrial transcription factor A; Uqcrc1: ubiquinol-cytochrome c reductase core protein I; Ψm: mitochondrial membrane potential.
Supplementary Material
Supplementary figures and tables.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Lopez-Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. 2013;153:1194-217
2. Schultz MB, Sinclair DA. When stem cells grow old: phenotypes and mechanisms of stem cell aging. Development. 2016;143:3-14
3. Goodell MA, Rando TA. Stem cells and healthy aging. Science. 2015;350:1199-204
4. Sui BD, Hu CH, Zheng CX, Jin Y. Microenvironmental Views on Mesenchymal Stem Cell Differentiation in Aging. J Dent Res. 2016;95:1333-40
5. Kfoury Y, Scadden DT. Mesenchymal cell contributions to the stem cell niche. Cell Stem Cell. 2015;16:239-53
6. Fontana L, Partridge L. Promoting health and longevity through diet: from model organisms to humans. Cell. 2015;161:106-18
7. Vaiserman AM, Lushchak OV, Koliada AK. Anti-aging pharmacology: Promises and pitfalls. Ageing Res Rev. 2016;31:9-35
8. Zhao P, Sui BD, Liu N, Lv YJ, Zheng CX, Lu YB. et al. Anti-aging pharmacology in cutaneous wound healing: effects of metformin, resveratrol, and rapamycin by local application. Aging Cell. 2017;16:1083-93
9. Howitz KT, Bitterman KJ, Cohen HY, Lamming DW, Lavu S, Wood JG. et al. Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan. Nature. 2003;425:191-6
10. Wood JG, Rogina B, Lavu S, Howitz K, Helfand SL, Tatar M. et al. Sirtuin activators mimic caloric restriction and delay ageing in metazoans. Nature. 2004;430:686-9
11. Valenzano DR, Terzibasi E, Genade T, Cattaneo A, Domenici L, Cellerino A. Resveratrol prolongs lifespan and retards the onset of age-related markers in a short-lived vertebrate. Curr Biol. 2006;16:296-300
12. Baur JA, Pearson KJ, Price NL, Jamieson HA, Lerin C, Kalra A. et al. Resveratrol improves health and survival of mice on a high-calorie diet. Nature. 2006;444:337-42
13. Lagouge M, Argmann C, Gerhart-Hines Z, Meziane H, Lerin C, Daussin F. et al. Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1alpha. Cell. 2006;127:1109-22
14. Barger JL, Kayo T, Vann JM, Arias EB, Wang J, Hacker TA. et al. A low dose of dietary resveratrol partially mimics caloric restriction and retards aging parameters in mice. PLoS One. 2008;3:e2264
15. Liu B, Ghosh S, Yang X, Zheng H, Liu X, Wang Z. et al. Resveratrol rescues SIRT1-dependent adult stem cell decline and alleviates progeroid features in laminopathy-based progeria. Cell Metab. 2012;16:738-50
16. Pearson KJ, Baur JA, Lewis KN, Peshkin L, Price NL, Labinskyy N. et al. Resveratrol delays age-related deterioration and mimics transcriptional aspects of dietary restriction without extending life span. Cell Metab. 2008;8:157-68
17. da Luz PL, Tanaka L, Brum PC, Dourado PM, Favarato D, Krieger JE. et al. Red wine and equivalent oral pharmacological doses of resveratrol delay vascular aging but do not extend life span in rats. Atherosclerosis. 2012;224:136-42
18. Feng J, Liu S, Ma S, Zhao J, Zhang W, Qi W. et al. Protective effects of resveratrol on postmenopausal osteoporosis: regulation of SIRT1-NF-kappaB signaling pathway. Acta Biochim Biophys Sin. 2014;46:1024-33
19. Ornstrup MJ, Harslof T, Kjaer TN, Langdahl BL, Pedersen SB. Resveratrol increases bone mineral density and bone alkaline phosphatase in obese men: a randomized placebo-controlled trial. J Clin Endocrinol Metab. 2014;99:4720-9
20. Habold C, Momken I, Ouadi A, Bekaert V, Brasse D. Effect of prior treatment with resveratrol on density and structure of rat long bones under tail-suspension. J Bone Miner Metab. 2011;29:15-22
21. Baur JA, Sinclair DA. Therapeutic potential of resveratrol: the in vivo evidence. Nat Rev Drug Discov. 2006;5:493-506
22. Zhao L, Wang Y, Wang Z, Xu Z, Zhang Q, Yin M. Effects of dietary resveratrol on excess-iron-induced bone loss via antioxidative character. J Nutr Biochem. 2015;26:1174-82
23. Zhao H, Li X, Li N, Liu T, Liu J, Li Z. et al. Long-term resveratrol treatment prevents ovariectomy-induced osteopenia in rats without hyperplastic effects on the uterus. Br J Nutr. 2014;111:836-46
24. Rodgers JT, Lerin C, Haas W, Gygi SP, Spiegelman BM, Puigserver P. Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature. 2005;434:113-8
25. Zang M, Xu S, Maitland-Toolan KA, Zuccollo A, Hou X, Jiang B. et al. Polyphenols stimulate AMP-activated protein kinase, lower lipids, and inhibit accelerated atherosclerosis in diabetic LDL receptor-deficient mice. Diabetes. 2006;55:2180-91
26. Gomes AP, Price NL, Ling AJ, Moslehi JJ, Montgomery MK, Rajman L. et al. Declining NAD(+) induces a pseudohypoxic state disrupting nuclear-mitochondrial communication during aging. Cell. 2013;155:1624-38
27. Sahin E, Colla S, Liesa M, Moslehi J, Muller FL, Guo M. et al. Telomere dysfunction induces metabolic and mitochondrial compromise. Nature. 2011;470:359-65
28. Sahin E, Depinho RA. Linking functional decline of telomeres, mitochondria and stem cells during ageing. Nature. 2010;464:520-8
29. Sui B, Hu C, Jin Y. Mitochondrial metabolic failure in telomere attrition-provoked aging of bone marrow mesenchymal stem cells. Biogerontology. 2016;17:267-79
30. Chen H, Liu X, Chen H, Cao J, Zhang L, Hu X. et al. Role of SIRT1 and AMPK in mesenchymal stem cells differentiation. Ageing Res Rev. 2014;13:55-64
31. Liao L, Su X, Yang X, Hu C, Li B, Lv Y. et al. TNF-alpha Inhibits FoxO1 by Upregulating miR-705 to Aggravate Oxidative Damage in Bone Marrow-Derived Mesenchymal Stem Cells during Osteoporosis. Stem Cells. 2016;34:1054-67
32. Sui B, Hu C, Liao L, Chen Y, Zhang X, Fu X. et al. Mesenchymal progenitors in osteopenias of diverse pathologies: differential characteristics in the common shift from osteoblastogenesis to adipogenesis. Sci Rep. 2016;6:30186
33. Liu S, Liu D, Chen C, Hamamura K, Moshaverinia A, Yang R. et al. MSC Transplantation Improves Osteopenia via Epigenetic Regulation of Notch Signaling in Lupus. Cell Metab. 2015;22:606-18
34. Jilka RL, Weinstein RS, Takahashi K, Parfitt AM, Manolagas SC. Linkage of decreased bone mass with impaired osteoblastogenesis in a murine model of accelerated senescence. J Clin Invest. 1996;97:1732-40
35. Silva MJ, Brodt MD, Ko M, Abu-Amer Y. Impaired marrow osteogenesis is associated with reduced endocortical bone formation but does not impair periosteal bone formation in long bones of SAMP6 mice. J Bone Miner Res. 2005;20:419-27
36. Kajkenova O, Lecka-Czernik B, Gubrij I, Hauser SP, Takahashi K, Parfitt AM. et al. Increased adipogenesis and myelopoiesis in the bone marrow of SAMP6, a murine model of defective osteoblastogenesis and low turnover osteopenia. J Bone Miner Res. 1997;12:1772-9
37. Takeda T, Hosokawa M, Takeshita S, Irino M, Higuchi K, Matsushita T. et al. A new murine model of accelerated senescence. Mech Ageing Dev. 1981;17:183-94
38. Tarasenko D, Barbot M, Jans DC, Kroppen B, Sadowski B, Heim G. et al. The MICOS component Mic60 displays a conserved membrane-bending activity that is necessary for normal cristae morphology. J Cell Biol. 2017;216:889-99
39. Li H, Ruan Y, Zhang K, Jian F, Hu C, Miao L. et al. Mic60/Mitofilin determines MICOS assembly essential for mitochondrial dynamics and mtDNA nucleoid organization. Cell Death Differ. 2016;23:380-92
40. Mirsaidi A, Kleinhans KN, Rimann M, Tiaden AN, Stauber M, Rudolph KL. et al. Telomere length, telomerase activity and osteogenic differentiation are maintained in adipose-derived stromal cells from senile osteoporotic SAMP6 mice. J Tissue Eng Regen Med. 2012;6:378-90
41. Sui BD, Hu CH, Zheng CX, Shuai Y, He XN, Gao PP. et al. Recipient Glycemic Micro-environments Govern Therapeutic Effects of Mesenchymal Stem Cell Infusion on Osteopenia. Theranostics. 2017;7:1225-44
42. Sui B, Hu C, Zhang X, Zhao P, He T, Zhou C. et al. Allogeneic Mesenchymal Stem Cell Therapy Promotes Osteoblastogenesis and Prevents Glucocorticoid-Induced Osteoporosis. Stem Cells Transl Med. 2016;5:1238-46
43. Shuai Y, Liao L, Su X, Yu Y, Shao B, Jing H. et al. Melatonin Treatment Improves Mesenchymal Stem Cells Therapy by Preserving Stemness during Long-term In Vitro Expansion. Theranostics. 2016;6:1899-917
44. Tseng PC, Hou SM, Chen RJ, Peng HW, Hsieh CF, Kuo ML. et al. Resveratrol promotes osteogenesis of human mesenchymal stem cells by upregulating RUNX2 gene expression via the SIRT1/FOXO3A axis. J Bone Miner Res. 2011;26:2552-63
45. Peltz L, Gomez J, Marquez M, Alencastro F, Atashpanjeh N, Quang T. et al. Resveratrol exerts dosage and duration dependent effect on human mesenchymal stem cell development. PLoS One. 2012;7:e37162
46. Zhou H, Shang L, Li X, Zhang X, Gao G, Guo C. et al. Resveratrol augments the canonical Wnt signaling pathway in promoting osteoblastic differentiation of multipotent mesenchymal cells. Exp Cell Res. 2009;315:2953-62
47. Williams LD, Burdock GA, Edwards JA, Beck M, Bausch J. Safety studies conducted on high-purity trans-resveratrol in experimental animals. Food Chem Toxicol. 2009;47:2170-82
48. Wu C, Zhang Y, Shen Q, Zhou Z, Liu W, Hua J. Resveratrol changes spermatogonial stem cells (SSCs) activity and ameliorates their loss in busulfan-induced infertile mouse. Oncotarget. 2016;7:82085-96
49. Kim SK, Joe Y, Zheng M, Kim HJ, Yu JK, Cho GJ. et al. Resveratrol induces hepatic mitochondrial biogenesis through the sequential activation of nitric oxide and carbon monoxide production. Antioxid Redox Signal. 2014;20:2589-605
50. Yang N, Wang G, Hu C, Shi Y, Liao L, Shi S. et al. Tumor necrosis factor alpha suppresses the mesenchymal stem cell osteogenesis promoter miR-21 in estrogen deficiency-induced osteoporosis. J Bone Miner Res. 2013;28:559-73
51. Jing H, Liao L, An Y, Su X, Liu S, Shuai Y. et al. Suppression of EZH2 Prevents the Shift of Osteoporotic MSC Fate to Adipocyte and Enhances Bone Formation During Osteoporosis. Mol Ther. 2016;24:217-29
52. Mohanty ST, Bellantuono I. Intra-femoral injection of human mesenchymal stem cells. Methods Mol Biol. 2013;976:131-41
53. Hu CH, Sui BD, Du FY, Shuai Y, Zheng CX, Zhao P. et al. miR-21 deficiency inhibits osteoclast function and prevents bone loss in mice. Sci Rep. 2017;7:43191
54. Bouxsein ML, Boyd SK, Christiansen BA, Guldberg RE, Jepsen KJ, Muller R. Guidelines for assessment of bone microstructure in rodents using micro-computed tomography. J Bone Miner Res. 2010;25:1468-86
55. Chen N, Sui BD, Hu CH, Cao J, Zheng CX, Hou R. et al. microRNA-21 Contributes to Orthodontic Tooth Movement. J Dent Res. 2016;95:1425-33
56. Dempster DW, Compston JE, Drezner MK, Glorieux FH, Kanis JA, Malluche H. et al. Standardized nomenclature, symbols, and units for bone histomorphometry: a 2012 update of the report of the ASBMR Histomorphometry Nomenclature Committee. J Bone Miner Res. 2013;28:2-17
57. Chen X, Hu C, Wang G, Li L, Kong X, Ding Y. et al. Nuclear factor-kappaB modulates osteogenesis of periodontal ligament stem cells through competition with beta-catenin signaling in inflammatory microenvironments. Cell Death Dis. 2013;4:e510
58. Yang H, Gao LN, An Y, Hu CH, Jin F, Zhou J. et al. Comparison of mesenchymal stem cells derived from gingival tissue and periodontal ligament in different incubation conditions. Biomaterials. 2013;34:7033-47
59. Liu W, Konermann A, Guo T, Jager A, Zhang L, Jin Y. Canonical Wnt signaling differently modulates osteogenic differentiation of mesenchymal stem cells derived from bone marrow and from periodontal ligament under inflammatory conditions. Biochim Biophys Acta. 2014;1840:1125-34
60. Julien SG, Kim SY, Brunmeir R, Sinnakannu JR, Ge X, Li H. et al. Narciclasine attenuates diet-induced obesity by promoting oxidative metabolism in skeletal muscle. PLoS Biol. 2017;15:e1002597
61. Beckervordersandforth R, Ebert B, Schaffner I, Moss J, Fiebig C, Shin J. et al. Role of Mitochondrial Metabolism in the Control of Early Lineage Progression and Aging Phenotypes in Adult Hippocampal Neurogenesis. Neuron. 2017;93(e6):560-73
62. Hayakawa K, Esposito E, Wang X, Terasaki Y, Liu Y, Xing C. et al. Transfer of mitochondria from astrocytes to neurons after stroke. Nature. 2016;535:551-5
63. Srivastava A, McGinniss J, Wong Y, Shinn AS, Lam TT, Lee PJ. et al. MKK3 deletion improves mitochondrial quality. Free Radic Biol Med. 2015;87:373-84
64. John GB, Shang Y, Li L, Renken C, Mannella CA, Selker JM. et al. The mitochondrial inner membrane protein mitofilin controls cristae morphology. Mol Biol Cell. 2005;16:1543-54
65. Zhou HS, Li M, Sui BD, Wei L, Hou R, Chen WS. et al. Lipopolysaccharide impairs permeability of pulmonary microvascular endothelial cells via Connexin40. Microvasc Res. 2017;115:58-67
66. Bianco P, Cao X, Frenette PS, Mao JJ, Robey PG, Simmons PJ. et al. The meaning, the sense and the significance: translating the science of mesenchymal stem cells into medicine. Nat Med. 2013;19:35-42
67. He YD, Sui BD, Li M, Huang J, Chen S, Wu LA. Site-specific function and regulation of Osterix in tooth root formation. Int Endod J. 2016;49:1124-31
68. Higashida K, Kim SH, Jung SR, Asaka M, Holloszy JO, Han DH. Effects of resveratrol and SIRT1 on PGC-1alpha activity and mitochondrial biogenesis: a reevaluation. PLoS Biol. 2013;11:e1001603
69. Boengler K, Heusch G, Schulz R. Nuclear-encoded mitochondrial proteins and their role in cardioprotection. Biochim Biophys Acta. 2011;1813:1286-94
70. Neupert W. Mitochondrial Gene Expression: A Playground of Evolutionary Tinkering. Annu Rev Biochem. 2016;85:65-76
71. Houten SM, Auwerx J. PGC-1alpha: turbocharging mitochondria. Cell. 2004;119:5-7
72. Chan DC. Mitochondria: dynamic organelles in disease, aging, and development. Cell. 2006;125:1241-52
73. Sui BD, Xu TQ, Liu JW, Wei W, Zheng CX, Guo BL. et al. Understanding the role of mitochondria in the pathogenesis of chronic pain. Postgrad Med J. 2013;89:709-14
74. Harner M, Korner C, Walther D, Mokranjac D, Kaesmacher J, Welsch U. et al. The mitochondrial contact site complex, a determinant of mitochondrial architecture. EMBO J. 2011;30:4356-70
75. Pfanner N, van der Laan M, Amati P, Capaldi RA, Caudy AA, Chacinska A. et al. Uniform nomenclature for the mitochondrial contact site and cristae organizing system. J Cell Biol. 2014;204:1083-6
76. Gibon E, Lu L, Goodman SB. Aging, inflammation, stem cells, and bone healing. Stem Cell Res Ther. 2016;7:44
77. Pritz T, Weinberger B, Grubeck-Loebenstein B. The aging bone marrow and its impact on immune responses in old age. Immunol Lett. 2014;162:310-5
78. Karsenty G, Olson EN. Bone and Muscle Endocrine Functions: Unexpected Paradigms of Inter-organ Communication. Cell. 2016;164:1248-56
79. Bonyadi M, Waldman SD, Liu D, Aubin JE, Grynpas MD, Stanford WL. Mesenchymal progenitor self-renewal deficiency leads to age-dependent osteoporosis in Sca-1/Ly-6A null mice. Proc Natl Acad Sci U S A. 2003;100:5840-5
80. Zhang H, Zhai Z, Wang Y, Zhang J, Wu H, Wang Y. et al. Resveratrol ameliorates ionizing irradiation-induced long-term hematopoietic stem cell injury in mice. Free Radic Biol Med. 2013;54:40-50
81. Cheng W, Yu P, Wang L, Shen C, Song X, Chen J. et al. Sonic hedgehog signaling mediates resveratrol to increase proliferation of neural stem cells after oxygen-glucose deprivation/reoxygenation injury in vitro. Cell Physiol Biochem. 2015;35:2019-32
82. Zhang QS, Marquez-Loza L, Eaton L, Duncan AW, Goldman DC, Anur P. et al. Fancd2-/- mice have hematopoietic defects that can be partially corrected by resveratrol. Blood. 2010;116:5140-8
83. Ling L, Gu S, Cheng Y. Resveratrol activates endogenous cardiac stem cells and improves myocardial regeneration following acute myocardial infarction. Mol Med Rep. 2017;15:1188-94
84. Gorbunov N, Petrovski G, Gurusamy N, Ray D, Kim DH, Das DK. Regeneration of infarcted myocardium with resveratrol-modified cardiac stem cells. J Cell Mol Med. 2012;16:174-84
85. Pearce VP, Sherrell J, Lou Z, Kopelovich L, Wright WE, Shay JW. Immortalization of epithelial progenitor cells mediated by resveratrol. Oncogene. 2008;27:2365-74
86. Dai Z, Li Y, Quarles LD, Song T, Pan W, Zhou H. et al. Resveratrol enhances proliferation and osteoblastic differentiation in human mesenchymal stem cells via ER-dependent ERK1/2 activation. Phytomedicine. 2007;14:806-14
87. Yoon DS, Choi Y, Jang Y, Lee M, Choi WJ, Kim SH. et al. SIRT1 directly regulates SOX2 to maintain self-renewal and multipotency in bone marrow-derived mesenchymal stem cells. Stem Cells. 2014;32:3219-31
88. Joe IS, Jeong SG, Cho GW. Resveratrol-induced SIRT1 activation promotes neuronal differentiation of human bone marrow mesenchymal stem cells. Neurosci Lett. 2015;584:97-102
89. Backesjo CM, Li Y, Lindgren U, Haldosen LA. Activation of Sirt1 decreases adipocyte formation during osteoblast differentiation of mesenchymal stem cells. J Bone Miner Res. 2006;21:993-1002
90. Caldarelli I, Speranza MC, Bencivenga D, Tramontano A, Borgia A, Pirozzi AV. et al. Resveratrol mimics insulin activity in the adipogenic commitment of human bone marrow mesenchymal stromal cells. Int J Biochem Cell Biol. 2015;60:60-72
91. Buhrmann C, Busch F, Shayan P, Shakibaei M. Sirtuin-1 (SIRT1) is required for promoting chondrogenic differentiation of mesenchymal stem cells. J Biol Chem. 2014;289:22048-62
92. Ornstrup MJ, Harslof T, Sorensen L, Stenkjaer L, Langdahl BL, Pedersen SB. Resveratrol Increases Osteoblast Differentiation In Vitro Independently of Inflammation. Calcif Tissue Int. 2016;99:155-63
93. Shakibaei M, Shayan P, Busch F, Aldinger C, Buhrmann C, Lueders C. et al. Resveratrol mediated modulation of Sirt-1/Runx2 promotes osteogenic differentiation of mesenchymal stem cells: potential role of Runx2 deacetylation. PLoS One. 2012;7:e35712
94. Lei LT, Chen JB, Zhao YL, Yang SP, He L. Resveratrol attenuates senescence of adipose-derived mesenchymal stem cells and restores their paracrine effects on promoting insulin secretion of INS-1 cells through Pim-1. Eur Rev Med Pharmacol Sci. 2016;20:1203-13
95. Yuan HF, Zhai C, Yan XL, Zhao DD, Wang JX, Zeng Q. et al. SIRT1 is required for long-term growth of human mesenchymal stem cells. J Mol Med. 2012;90:389-400
96. Yoon DS, Choi Y, Choi SM, Park KH, Lee JW. Different effects of resveratrol on early and late passage mesenchymal stem cells through beta-catenin regulation. Biochem Biophys Res Commun. 2015;467:1026-32
97. de Magalhaes JP. From cells to ageing: a review of models and mechanisms of cellular senescence and their impact on human ageing. Exp Cell Res. 2004;300:1-10
98. Chen PM, Lin CH, Li NT, Wu YM, Lin MT, Hung SC. et al. c-Maf regulates pluripotency genes, proliferation/self-renewal, and lineage commitment in ROS-mediated senescence of human mesenchymal stem cells. Oncotarget. 2015;6:35404-18
99. Li CJ, Cheng P, Liang MK, Chen YS, Lu Q, Wang JY. et al. MicroRNA-188 regulates age-related switch between osteoblast and adipocyte differentiation. J Clin Invest. 2015;125:1509-22
100. Galderisi U, Helmbold H, Squillaro T, Alessio N, Komm N, Khadang B. et al. In vitro senescence of rat mesenchymal stem cells is accompanied by downregulation of stemness-related and DNA damage repair genes. Stem Cells Dev. 2009;18:1033-42
101. Gao L, Bird AK, Meednu N, Dauenhauer K, Liesveld J, Anolik J. et al. Bone Marrow-Derived Mesenchymal Stem Cells From Patients With Systemic Lupus Erythematosus Have a Senescence-Associated Secretory Phenotype Mediated by a Mitochondrial Antiviral Signaling Protein-Interferon-beta Feedback Loop. Arthritis Rheumatol. 2017;69:1623-35
102. Weinstein RS, Jilka RL, Parfitt AM, Manolagas SC. Inhibition of osteoblastogenesis and promotion of apoptosis of osteoblasts and osteocytes by glucocorticoids. Potential mechanisms of their deleterious effects on bone. J Clin Invest. 1998;102:274-82
103. Gioia R, Panaroni C, Besio R, Palladini G, Merlini G, Giansanti V. et al. Impaired osteoblastogenesis in a murine model of dominant osteogenesis imperfecta: a new target for osteogenesis imperfecta pharmacological therapy. Stem Cells. 2012;30:1465-76
104. Wang S, Liang X, Yang Q, Fu X, Rogers CJ, Zhu M. et al. Resveratrol induces brown-like adipocyte formation in white fat through activation of AMP-activated protein kinase (AMPK) alpha1. Int J Obes (Lond). 2015;39:967-76
105. Dyall SD, Brown MT, Johnson PJ. Ancient invasions: from endosymbionts to organelles. Science. 2004;304:253-7
106. Xu X, Duan S, Yi F, Ocampo A, Liu GH, Izpisua Belmonte JC. Mitochondrial regulation in pluripotent stem cells. Cell Metab. 2013;18:325-32
107. Lonergan T, Bavister B, Brenner C. Mitochondria in stem cells. Mitochondrion. 2007;7:289-96
108. Folmes CD, Nelson TJ, Martinez-Fernandez A, Arrell DK, Lindor JZ, Dzeja PP. et al. Somatic oxidative bioenergetics transitions into pluripotency-dependent glycolysis to facilitate nuclear reprogramming. Cell Metab. 2011;14:264-71
109. Chung S, Dzeja PP, Faustino RS, Perez-Terzic C, Behfar A, Terzic A. Mitochondrial oxidative metabolism is required for the cardiac differentiation of stem cells. Nat Clin Pract Cardiovasc Med. 2007;4(Suppl 1):S60-7
110. Chen CT, Shih YR, Kuo TK, Lee OK, Wei YH. Coordinated changes of mitochondrial biogenesis and antioxidant enzymes during osteogenic differentiation of human mesenchymal stem cells. Stem Cells. 2008;26:960-8
111. Pietila M, Lehtonen S, Narhi M, Hassinen IE, Leskela HV, Aranko K. et al. Mitochondrial function determines the viability and osteogenic potency of human mesenchymal stem cells. Tissue Eng Part C Methods. 2010;16:435-45
112. George SK, Jiao Y, Bishop CE, Lu B. Mitochondrial peptidase IMMP2L mutation causes early onset of age-associated disorders and impairs adult stem cell self-renewal. Aging Cell. 2011;10:584-94
113. Pietila M, Palomaki S, Lehtonen S, Ritamo I, Valmu L, Nystedt J. et al. Mitochondrial function and energy metabolism in umbilical cord blood- and bone marrow-derived mesenchymal stem cells. Stem Cells Dev. 2012;21:575-88
114. Lo T, Ho JH, Yang MH, Lee OK. Glucose reduction prevents replicative senescence and increases mitochondrial respiration in human mesenchymal stem cells. Cell Transplant. 2011;20:813-25
115. Mills RE, Taylor KR, Podshivalova K, McKay DB, Jameson JM. Defects in skin gamma delta T cell function contribute to delayed wound repair in rapamycin-treated mice. J Immunol. 2008;181:3974-83
116. Lamming DW, Ye L, Sabatini DM, Baur JA. Rapalogs and mTOR inhibitors as anti-aging therapeutics. J Clin Invest. 2013;123:980-9
117. Chen C, Akiyama K, Wang D, Xu X, Li B, Moshaverinia A. et al. mTOR inhibition rescues osteopenia in mice with systemic sclerosis. J Exp Med. 2015;212:73-91
118. Liu Y, Kou X, Chen C, Yu W, Su Y, Kim Y. et al. Chronic High Dose Alcohol Induces Osteopenia via Activation of mTOR Signaling in Bone Marrow Mesenchymal Stem Cells. Stem Cells. 2016;34:2157-68
119. Nunnari J, Suomalainen A. Mitochondria: in sickness and in health. Cell. 2012;148:1145-59
120. Anderson S, Bankier AT, Barrell BG, de Bruijn MH, Coulson AR, Drouin J. et al. Sequence and organization of the human mitochondrial genome. Nature. 1981;290:457-65
121. Larsson NG, Wang J, Wilhelmsson H, Oldfors A, Rustin P, Lewandoski M. et al. Mitochondrial transcription factor A is necessary for mtDNA maintenance and embryogenesis in mice. Nat Genet. 1998;18:231-6
122. Canto C, Gerhart-Hines Z, Feige JN, Lagouge M, Noriega L, Milne JC. et al. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature. 2009;458:1056-60
123. Icho T, Ikeda T, Matsumoto Y, Hanaoka F, Kaji K, Tsuchida N. A novel human gene that is preferentially transcribed in heart muscle. Gene. 1994;144:301-6
124. Rabl R, Soubannier V, Scholz R, Vogel F, Mendl N, Vasiljev-Neumeyer A. et al. Formation of cristae and crista junctions in mitochondria depends on antagonism between Fcj1 and Su e/g. J Cell Biol. 2009;185:1047-63
125. von der Malsburg K, Muller JM, Bohnert M, Oeljeklaus S, Kwiatkowska P, Becker T. et al. Dual role of mitofilin in mitochondrial membrane organization and protein biogenesis. Dev Cell. 2011;21:694-707
126. Zick M, Rabl R, Reichert AS. Cristae formation-linking ultrastructure and function of mitochondria. Biochim Biophys Acta. 2009;1793:5-19
127. Hessenberger M, Zerbes RM, Rampelt H, Kunz S, Xavier AH, Purfurst B. et al. Regulated membrane remodeling by Mic60 controls formation of mitochondrial crista junctions. Nat Commun. 2017;8:15258
128. Friedman JR, Mourier A, Yamada J, McCaffery JM, Nunnari J. MICOS coordinates with respiratory complexes and lipids to establish mitochondrial inner membrane architecture. Elife. 2015:4
129. Yang RF, Sun LH, Zhang R, Zhang Y, Luo YX, Zheng W. et al. Suppression of Mic60 compromises mitochondrial transcription and oxidative phosphorylation. Sci Rep. 2015;5:7990
130. Yang RF, Zhao GW, Liang ST, Zhang Y, Sun LH, Chen HZ. et al. Mitofilin regulates cytochrome c release during apoptosis by controlling mitochondrial cristae remodeling. Biochem Biophys Res Commun. 2012;428:93-8
131. Garstka HL, Schmitt WE, Schultz J, Sogl B, Silakowski B, Perez-Martos A. et al. Import of mitochondrial transcription factor A (TFAM) into rat liver mitochondria stimulates transcription of mitochondrial DNA. Nucleic Acids Res. 2003;31:5039-47
Author contact
![]() Corresponding authors: Prof. Yan Jin, State Key Laboratory of Military Stomatology, Center for Tissue Engineering, School of Stomatology, Fourth Military Medical University, 145 West Changle Road, Xi'an, Shaanxi 710032, China. E-mail: yanjinedu.cn; Tel: +86-029-84776472; Fax: +86-029-83218039 and Prof. Shao-Feng Zhang, State Key Laboratory of Military Stomatology, Department of Prosthodontics, School of Stomatology, Fourth Military Medical University, 145 West Changle Road, Xi'an, Shaanxi 710032, China. E-mail: sfzhang1963.fmmucom; Tel: +86-029-82665165/84776468.
Corresponding authors: Prof. Yan Jin, State Key Laboratory of Military Stomatology, Center for Tissue Engineering, School of Stomatology, Fourth Military Medical University, 145 West Changle Road, Xi'an, Shaanxi 710032, China. E-mail: yanjinedu.cn; Tel: +86-029-84776472; Fax: +86-029-83218039 and Prof. Shao-Feng Zhang, State Key Laboratory of Military Stomatology, Department of Prosthodontics, School of Stomatology, Fourth Military Medical University, 145 West Changle Road, Xi'an, Shaanxi 710032, China. E-mail: sfzhang1963.fmmucom; Tel: +86-029-82665165/84776468.
Citation styles
APA
Lv, Y.J., Yang, Y., Sui, B.D., Hu, C.H., Zhao, P., Liao, L., Chen, J., Zhang, L.Q., Yang, T.T., Zhang, S.F., Jin, Y. (2018). Resveratrol counteracts bone loss via mitofilin-mediated osteogenic improvement of mesenchymal stem cells in senescence-accelerated mice. Theranostics, 8(9), 2387-2406. https://doi.org/10.7150/thno.23620.
ACS
Lv, Y.J.; Yang, Y.; Sui, B.D.; Hu, C.H.; Zhao, P.; Liao, L.; Chen, J.; Zhang, L.Q.; Yang, T.T.; Zhang, S.F.; Jin, Y. Resveratrol counteracts bone loss via mitofilin-mediated osteogenic improvement of mesenchymal stem cells in senescence-accelerated mice. Theranostics 2018, 8 (9), 2387-2406. DOI: 10.7150/thno.23620.
NLM
Lv YJ, Yang Y, Sui BD, Hu CH, Zhao P, Liao L, Chen J, Zhang LQ, Yang TT, Zhang SF, Jin Y. Resveratrol counteracts bone loss via mitofilin-mediated osteogenic improvement of mesenchymal stem cells in senescence-accelerated mice. Theranostics 2018; 8(9):2387-2406. doi:10.7150/thno.23620. https://www.thno.org/v08p2387.htm
CSE
Lv YJ, Yang Y, Sui BD, Hu CH, Zhao P, Liao L, Chen J, Zhang LQ, Yang TT, Zhang SF, Jin Y. 2018. Resveratrol counteracts bone loss via mitofilin-mediated osteogenic improvement of mesenchymal stem cells in senescence-accelerated mice. Theranostics. 8(9):2387-2406.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.