Theranostics
13.3
Impact Factor
- Current Issue
- Advance Articles
- Volume 16; 2026
- Volume 15; 2025
- Volume 14; 2024
- Volume 13; 2023
- Volume 12; 2022
- Archive
- Cover Images
- Cover Suggestion
- Special Issues
Top
Introduction
Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References
Introduction
Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2018; 8(9):2565-2582. doi:10.7150/thno.22878 This issue Cite
Research Paper
MicroRNA-378 suppresses myocardial fibrosis through a paracrine mechanism at the early stage of cardiac hypertrophy following mechanical stress.
Jie Yuan1*, Haibo Liu2*, Wei Gao1*, Li Zhang1, Yong Ye2, Lingyan Yuan3, Zhiwen Ding1, Jian Wu1, Le Kang1, Xiaoyi Zhang1, Xiaoyan Wang1, Guoping Zhang1, Hui Gong1, Aijun Sun1, Xiangdong Yang1, Ruizhen Chen1, Zhaoqiang Cui1, ![]() , Junbo Ge1,
, Junbo Ge1, ![]() , Yunzeng Zou1,
, Yunzeng Zou1, ![]()
1. Shanghai Institute of Cardiovascular Diseases, Zhongshan Hospital and Institutes of Biomedical Science, Fudan University, Shanghai 200032, China
2. Department of Cardiology, East Hospital, Tongji University School of Medicine, Shanghai 200120, China
3. Department of kinesiology, Institute of physical education, Shanghai Normal University, Shanghai 200234, China
* equal contribution
Received 2017-9-19; Accepted 2018-2-26; Published 2018-4-3
Citation:
Yuan J, Liu H, Gao W, Zhang L, Ye Y, Yuan L, Ding Z, Wu J, Kang L, Zhang X, Wang X, Zhang G, Gong H, Sun A, Yang X, Chen R, Cui Z, Ge J, Zou Y. MicroRNA-378 suppresses myocardial fibrosis through a paracrine mechanism at the early stage of cardiac hypertrophy following mechanical stress.. Theranostics 2018; 8(9):2565-2582. doi:10.7150/thno.22878. https://www.thno.org/v08p2565.htm
Other stylesAbstract

Rationale: Excessive myocardial fibrosis is the main pathological process in the development of cardiac remodeling and heart failure; therefore, it is important to prevent excessive myocardial fibrosis. We determined that microRNA-378 (miR-378) is cardiac-enriched and highly repressed during cardiac remodeling. We therefore proposed that miR-378 has a critical role in regulation of cardiac fibrosis, and examined the effects of miR-378 on cardiac fibrosis after mechanical stress.
Methods: Mechanical stress was respectively imposed on mice through a transverse aortic constriction (TAC) procedure and on cardiac fibroblasts by stretching silicon dishes. A chemically modified miR-378 mimic (Agomir) or an inhibitor (Antagomir) was administrated to mice by intravenous injection and to cells by direct addition to the culture medium. MiR-378 knockout mouse was constructed. Cardiac fibroblasts were cultured in the conditioned media from the cardiomyocytes with either miR-378 depletion or treatment with sphingomyelinase inhibitor GW4869. Quantitative real-time polymerase chain reaction analysis of gene and miRNA expression, Western blot analysis, immunochemistry and electron microscopy were performed to elucidate the mechanisms.
Results: Mechanical stress induced significant increases in fibrotic responses, including myocardial fibrosis, fibroblast hyperplasia, and protein and gene expression of collagen and matrix metalloproteinases (MMPs) both in vivo and in vitro. All these fibrotic responses were attenuated by treatment with a chemically modified miR-378 mimic (Agomir) but were exaggerated by treatment with an inhibitor (Antagomir). MiR-378 knockout mouse models exhibited aggravated cardiac fibrosis after TAC. Media from the cardiomyocytes with either miR-378 depletion or treatment with sphingomyelinase inhibitor GW4869 enhanced the fibrotic responses of stimulated cardiac fibroblasts, confirming that miR-378 inhibits fibrosis in an extracellular vesicles-dependent secretory manner. Mechanistically, the miR-378-induced anti-fibrotic effects manifested partially through the suppression of p38 MAP kinase phosphorylation by targeting MKK6 in cardiac fibroblasts.
Conclusions: miR-378 is secreted from cardiomyocytes following mechanical stress and acts as an inhibitor of excessive cardiac fibrosis through a paracrine mechanism.
Keywords: mechanical overload, cardiac fibrosis, microRNA-378, extracellular vesicles, p38, MKK6
Introduction
Sustained pressure overload can induce adverse cardiac remodeling accompanying myocardial hypertrophy and fibrosis, which eventually lead to contractile dysfunction and heart failure [1, 2]. Cardiac fibrosis particularly contributes to maladaptive left ventricular remodeling, which is characterized by fibroblast accumulation and the excess deposition of extracellular matrix (ECM) proteins, enhancing collagen levels and the production of matrix metalloproteinases (MMPs) [3, 4]. Excess myocardial fibrosis is the main pathological process underlying the development of cardiac remodeling and heart failure, and, therefore, it is important to prevent its occurrence. Endogenous anti-fibrotic mediators have been found to play beneficial roles in attenuating the adverse cardiac remodeling corresponding with an increased workload [5, 6]. However, the molecular mechanisms underlying the regulation of myocardial fibrosis remain unresolved.
MicroRNAs (miRNAs) are small non-coding RNA molecules of ~18-22 nucleotides in length that are expressed throughout metazoan species and have been recognized as important regulators of post-transcriptional gene control [7]. Growing evidence has indicated that many miRNAs exist that are aberrantly expressed in response to hemodynamic pressure overload in humans and mice and that they control and/or modulate key components of the hypertrophic and fibrotic process during cardiac remodeling [8-12]. Some studies have shown that some miRNAs not only function in their original cells but also can be secreted from donor cells and be taken up and function in recipient cells in patients with cancer [13-15]; similarly in heart disease, some secreted extracellular miRNAs may mediate crosstalk between the heart-hosted cells [16, 17]. Thum et al. identified cardiac fibroblast-derived miR-21* as a potent paracrine-acting RNA mediator of cardiomyocyte hypertrophy [16]. Chaturvedi et al. showed that cardiomyocyte-derived miRNA can be released outside and regulate cardiac remodeling via an MMP9-mediated mechanism in response to exercise [18]. In high pressure-overloaded hearts, only a limited number of secreted miRNAs in association with cardiac remodeling have been explored [19].
Our previous studies have found that heat shock transcription factor 1 (HSF1) is a cardiac protective factor contributing to the preservation of cardiac function during pressure overload. HSF1 knockout (KO) mice display severe cardiac dysfunction and aggravated cardiac fibrotic characteristics after stress overload [20]. We previously found that microRNA-378 (miR-378) was deeply repressed along with the deletion of HSF1 in mouse hearts. Therefore, we speculated that the decreased miR-378 levels in the heart may be related to cardiac dysfunction and that miR-378 may be involved in cardiac remodeling and fibrosis.
The most recent microRNA database (www.mirbase.org) describes the existence of several isoforms of miR-378 in humans. These variants (miR-378a/b/c/d/e/f/g/h/i/j) are encoded by different genomic loci but share identical seed sequences and are thus considered to have common regulating targets. To further explore its potential roles in cardiac remodeling, chemically modified miR-378 mimics or miR-378 antisense oligonucleotides were administered by intravenous injections to pressure-overloaded mice. We found that miR-378 not only had anti-hypertrophic activity, as proposed by others [21], but also caused obvious anti-fibrotic effects in the hearts. In addition, global miR-378 knockout mice generated showed severe cardiac fibrosis compared with the wild-type mice in response to stress. P38 mitogen-activated protein kinase (p38 MAPK), which is involved in a detrimental signaling pathway linked to both hypertrophy and inflammation during cardiac remodeling, has been found to be involved in the regulation of matrix metalloproteinases (MMPs) and collagens in cardiac fibroblasts [22-24]. Mitogen-activated protein kinase kinase 6 (MKK6) phosphorylates and activates p38 MAPK in response to inflammatory cytokines or environmental stress [25]. Here, we demonstrated the critical role of miR-378 in the suppression of pressure overload-induced cardiac fibrosis via the regulation of the p38 MAPK signaling pathways, which was achieved by directly targeting MKK6 in cardiac fibroblasts. Furthermore, we revealed a novel mechanism of paracrine communication between cardiomyocytes and fibroblasts, showing that miR-378 can be released outside of cardiomyocytes under mechanical stimulation and act on cardiac fibroblasts.
Methods
Quantitation of mRNA and miRNA expression, northern blot analyses, Western blot analyses, immunochemistry and electron microscopy were performed according to routine protocols. Detailed information is described in Supplementary Material.
Mouse TAC models and knockout mouse generation
All animal procedures were approved by the Institutional Animal Care and Use committee of Zhongshan Hospital Fudan University in compliance with the guidelines for the Care and Use of Laboratory Animals published by the National Academy Press (NIH Publication No. 85-23, revised 1996). Ten-week-old male mice were subjected to pressure overload by transverse aortic constriction (TAC) or sham surgery under anesthesia, as described [5]. Cardiac morphologic measurements were performed after two weeks. Chemically modified MiR-378 agomirs and antagomirs were purchased from Shanghai GenePharma Co., Ltd. (Figure S3C). Administration of intravenous injections of miR-378 agomirs or antagomirs started from the 1st day after TAC for three days (each 80 mg/kg body weight). Global miR-378 knockout mice were generated and purchased from the Institute of Laboratory Animal Sciences, CAMS. See Supplementary Material for more detailed information. During breeding, the miR-378 homozygous knockout mice showed higher mortality than the heterozygous mice before 8 weeks of age.
Cell culture and mechanical stretching in vitro
Neonatal cardiomyocytes (CMs) and cardiac fibroblasts (CFs) were prepared from anesthetized neonatal 1 to 2-day-old Sprague-Dawley rats (Harlan Sprague-Dawley) and isolated according to routine protocols. CMs or CFs were plated on silicone rubber culture dishes and were stretched 20% with special steel equipment. See Supplementary Material for more detailed information.
Transfection of miRNA mimics and inhibitors
Cells were transfected with mirVana™ miR-378 mimics (35 nM) or mirVana™ miR-378 inhibitors (50 nM) for 48 h as per the manufacturer's instructions. mirVana™ miRNA inhibitor/mimic negative controls were used as mock. See Supplementary Material for more detailed information.
EdU incorporation assay
Cell proliferation was assessed by Cell-Light EdU DNA cell proliferation kit. See Supplementary Material for more detailed information.
Extracellular vesicles extraction
Extracellular vesicles were purified from the conditioned medium of cardiomyocytes cultured in FBS-free Opti-MEM® by the product Total Exosome Isolation (from cell culture media) (Life Technologies). After extracellular vesicles were isolated, total RNA and protein were purified using the Total Exosome RNA and Protein Isolation Kit (Life Technologies). See Supplementary Material for more detailed information.
Statistics
Unless otherwise noted, data are presented as mean ± SE with n≥3. Multiple group comparison was performed by one-way or two-way ANOVA followed by LSD procedure for comparison of means. Comparison between 2 groups under identical conditions was performed by the 2-tailed Student's t-test. A value of p < 0.05 was considered statistically significant.
Results
miR-378 preserves LV function and inhibits hypertrophy and fibrosis during pressure overload-induced cardiac remodeling
To investigate the role of miR-378 during cardiac remodeling in vivo, we constructed high-pressure overload mouse models using TAC. Functional studies of miR-378 were performed by the administration of intravenous injections of chemically modified miR-378 mimics (termed agomir) or antisense RNA oligonucleotides (termed antagomir) for three days after TAC (80 mg/kg) (Figure 1A). Saline-treated mice that underwent TAC were grouped as the vehicle. After 14 days of TAC, we observed a dramatic increase in cardiac miR-378 levels in the mice treated with agomir and a great reduction in miR-378 expression in the antagomir-treated mice compared with the vehicle controls (Figure S2A). An echocardiographic analysis showed that the vehicle group possessed marked increases in key hypertrophic parameters. The overexpression of miR-378 by agomir injection effectively inhibited the TAC-induced cardiac hypertrophy and improved cardiac function, as shown by the decreased ratio of heart weight to body weight and the maintained ejection fraction. However, the silencing of endogenous miR-378 using antagomir led to the aggravation of the cardiac hypertrophy associated with cardiac dysfunction (Figure 1B-C and Table S1). Neither the miR-378 agomir treatment nor the miR-378 antagomir treatment had any obvious effects on heart size in the sham-operated group (Figure S2B). Overexpression of miR-378 greatly repressed fetal genes, while the knockdown of miR-378 was able to reactivate them and still manifested the pathological hypertrophy (Figure 1D).
Masson trichrome staining was performed to show the collagen deposition. The sham agomir-treated and antagomir-treated mice did not differ only with regard to the fibrosis of their cardiac tissues. After 14 days of pressure overload, the amounts of fibrosis in the interstitial and perivascular areas markedly increased in the vehicle mice. The agomir-treated mice showed markedly reduced interstitial and perivascular collagen accumulation in the left ventricular tissue compared with those that underwent TAC. However, the knockdown of endogenous miR-378 by the antagomir treatment led to exaggerated interstitial fibrosis but not perivascular fibrosis compared with the vehicle (Figure 1E).
Figure 1
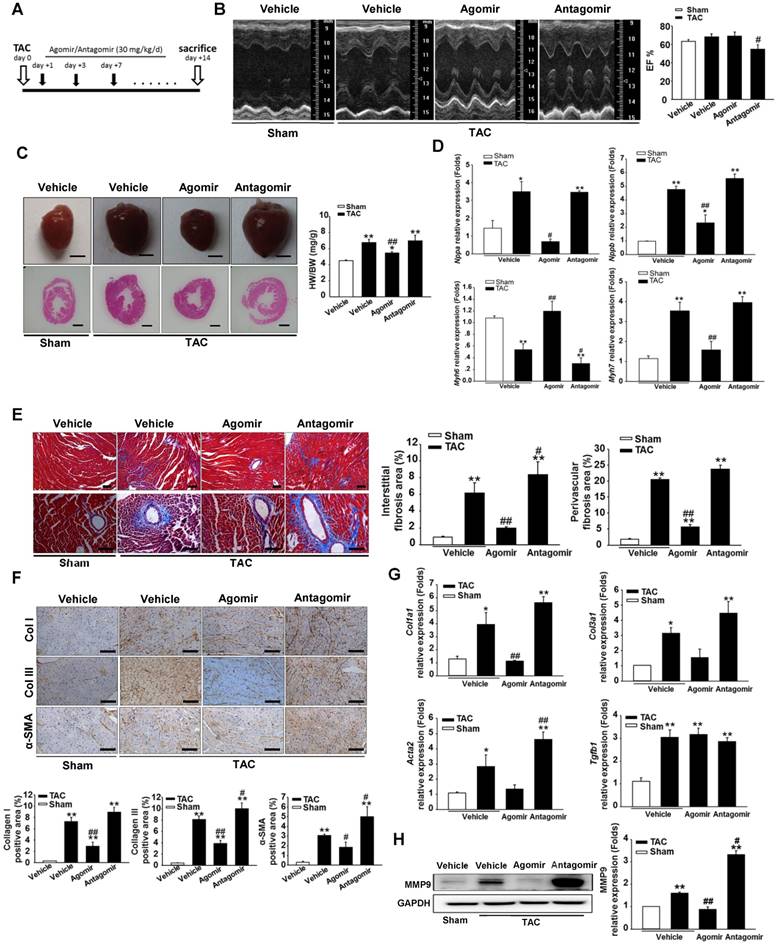
miR-378-regulated pressure overload induced cardiac hypertrophy and fibrosis. Adult male C57B/L6 mice were treated with miR-378 agomir or antagomir for three consecutive days after TAC. Saline-treatment served as the vehicle group. (n≥7/group). (A) Study design: agomir/antagomir targeting miR-378 were administered by three intravenous injections throughout the first 7 days of TAC; 30 mg/kg/injection for each agomir/antagomir. (B) Echocardiographic measurements of mice hearts 2 weeks after TAC or sham operation. Left ventricular ejection fraction values are shown on the right. (C) Representative global heart photographs (scale bar: 2 mm) and hematoxylin and eosin-stained LV transverse sections (magnification ×40). Ratio of heart weight to body weight (HW/BW) is shown 2 weeks after TAC. (D) Quantitative real-time polymerase chain reaction analysis of fetal cardiac genes (Nppa, atrial natriuretic peptide; Nppb, brain natriuretic peptide; Myh6, myosin heavy chain-α; Myh7, myosin heavy chain-β). (E) Representative photomicrographs of the left ventricular perivascular and interstitial fibrosis. For each group, blue staining represents collagen deposition. Original magnification ×100 and ×200 (scale bar: 100 μm). Collagen content was calculated as the percentage of the area in each section that was stained blue. Quantification of perivascular and interstitial fibrosis is shown. (F) Representative immunohistochemical staining of collagen I, collagen III and α -SMA in different groups and their quantification. The expressions of collagen and α-SMA were calculated as the proportion of positive area to total tissue area for all measurements of the section. Original magnification ×200 (scale bar: 100 μm). (G) Total RNA was isolated from cardiac samples. Quantitative real-time polymerase chain reaction analysis of fibrosis marker genes (Col1a1, collagen I α1; Col3a1, collagen IIIα1; Tgfb1, TGFβ1; Acta2, α-SMA). (H) Western blot for MMP9 was performed on heart tissues after different treatments. Relative expression of MMP9 was controlled by GAPDH. Values represent the mean ± SE, n=3-5 per group. **, P< 0.01 vs. sham; *, P< 0.05 vs. sham; ##, P< 0.01 vs. TAC vehicle group; #, P< 0.05 vs. TAC vehicle group.
ECM genes and fibrosis markers were strongly induced following TAC in the saline-treated mice. As shown in Figure 1G, the collagen Iα1 (Col1a1), collagen IIIα1 (Col3a1), TGFβ1 (Tgfb1) and α-SMA (Acta2) mRNA levels were markedly unregulated as measured by quantitative real-time PCR. Similarly, the protein level of MMP9 was also elevated in the hearts (Figure 1H). The overexpression of miR-378 by agomir led to the suppressed expression of these markers following TAC compared with the vehicle, although the downregulation of Col3a1 and Acta2 did not show statistical significance. The downregulation of miR-378 by antagomir enhanced the expression of these markers following TAC compared with the vehicle and only Acta2 displayed significant upregulation. Interestingly, neither the agomir nor the antagomir treatment had any effects on TGFβ1 expression (Figure 1G). Moreover, immunohistochemistry confirmed that the cardiac expressions of collagen I, collagen III and the α-SMA protein were significantly lower in the agomir-378-treated mice compared with the vehicle after TAC. In the hearts of antagomir-378-treated mice, the collagen I, collagen III and the α-SMA protein were all upregulated compared with the vehicle after TAC, and both collagen III and the α-SMA protein particularly showed significant increases. (Figure 1F).
Taken together, we revealed the significant involvement of miR-378 in attenuating the progression of the pressure overload-induced cardiac hypertrophic and fibrotic remodeling.
In vitro delivery of miR-378 mimics substantially inhibits fibrotic response in cardiac fibroblasts
Because miR-378 has been found to be cardiac enriched, we specified the cell type distribution of miR-378 in the heart. By quantitative real-time PCR analysis, we found miR-378 was highly expressed in the cardiomyocytes but not expressed in cardiac fibroblasts isolated from neonatal rat hearts nor from adult mouse hearts (Figure S3), suggesting that miR-378 likely acts as a cardiomyocyte-oriented regulator in the heart and exogenously regulates the fibroblastic response in cardiac fibroblasts.
To determine the anti-fibrotic effects of miR-378 in the cardiac fibroblasts (CFs) during pressure overload, we transfected synthetic miR-378 mimics (30 nM) and scramble nucleotides (mock) into the CFs. We first observed the effects of miR-378 on the proliferation of the CFs and on collagen production in the absence of mechanical stress. Using the MTT assay, we found that delivery of the miR-378 mimic dramatically inhibited proliferation of the CFs compared with transfection of the mock sequences (Figure 2A). Cardiac fibroblasts overexpressing miR-378 or mock scramble nucleotides were then subjected to pressure overload in vitro by mechanical stretching (MS) up to 20% capacity. The results showed that the introduction of miR-378 into the CFs significantly reduced MS-stimulated collagen synthesis, as measured by collagen I and collagen III mRNA expression (Figure 2B). MMP9 expression level in the CFs was also markedly increased by mechanical overload but reduced in the miR-378 mimic-treated cells (Figure 2C). An EdU incorporation assay, which is a more sensitive and specific method, was employed to further define the regulatory effects of miR-378 on the proliferation of the CFs. The number of EdU-positive cells was increased by ~2.5-fold after MS compared with control but was reduced by ~50% when miR-378 was overexpressed (Figure 2D). These results verify that miR-378 also repressed fibrotic activities in vitro.
miR-378 is released from cardiomyocytes under pressure overload and functions in cardiac fibroblasts via paracrine signaling
Because the overexpression of miR-378 in cardiac fibroblasts significantly reduced fibrosis after pressure overload, despite its lack of expression in fibroblasts, we explored the possibility that miR-378 might mediate the expression of collagen in the fibroblasts through paracrine signaling. We first measured the expression levels of miR-378 in the sera of mice subjected to TAC in vivo as well as in the medium of the cultured cardiomyocytes subjected to mechanical stretching in vitro. Quantitative real-time PCR showed that during the adaptive stage, the miR-378 levels continuously increased at 7 days and 14 days after TAC, but during the maladaptive stage, miR-378 was downregulated towards baseline at 28 days after TAC (Figure 3A). Similarly, the in vitro detection of miR-378 in the medium from the cultured cardiomyocytes subjected to mechanical stretching showed that the miR-378 levels gradually increased at 6 h, 12 h and 24 h in a time-dependent manner during the early stage but were decreased at 36 h and 48 h compared to that at 24 h (Figure 3B). These data suggest that pressure overload stimulation may have led to the release of miR-378 outside of the cardiomyocytes.
We next cultured the CFs in the presence of conditioned medium from cardiomyocyte cultures. The CFs were stimulated by MS for 24 h afterwards, as shown in Figure 3C. We observed that the conditioned medium derived from the stretched cardiomyocytes greatly suppressed the upregulation of collagen expression and MMP9 protein levels in the CFs stimulated by mechanical stretching, as compared with the medium from the resting cardiomyocytes. Then, we measured the fibrotic response of CFs cultured in conditioned medium from the miR-378-depleted, stretched cardiomyocytes, which was achieved by introducing miR-378 inhibitors. We found that the anti-fibrotic roles of the medium from the stretched cardiomyocytes were eliminated when miR-378 was knocked down in the cardiomyocytes, as shown by the enhanced MMP9 and collagen expression and the re-invigorated fibrotic response stimulated by mechanical stretching in the CFs. We also found that inhibiting endogenous miR-378 can promote TGFβ1 expression and release from cardiomyocytes (Figure S4), which may be another mechanism for suppression of fibrosis by miR-378. So, we administered pirfenidone, an inhibitor for TGFβ1 production and secretion, along with the miR-378 inhibitor to the cardiomyocytes to exclude the effects of TGFβ1 secretion from cardiomyocytes caused by miR-378 inhibition (shown as in Figure 3C). The conditioned medium from cells that underwent this treatment were used to culture CFs. We found that the collagen gene expression in the CFs still showed reactivation compared with that in the group treated with the conditioned medium from stretched cardiomyocytes alone, but was greatly decreased compared with that in medium from cells with inhibition of endogenous miR-378 alone (Figure 3D-E). These results demonstrated the anti-fibrotic role of miR-378 in cardiac fibroblasts via paracrine signaling.
Figure 2
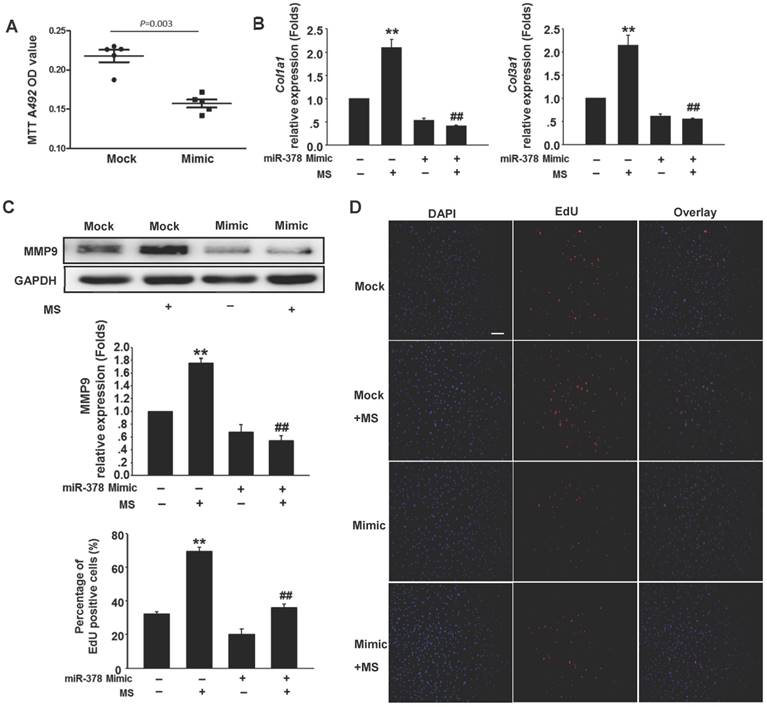
Overexpression of miR-378 in cardiac fibroblasts inhibits collagen deposition and cardiac fibroblasts proliferation. (A) Cultured rat cardiac fibroblasts were transiently transfected with the chemically modified miR-378 mimics (35 nM) or negative scramble control (mock). The effect of miR-378 on fibroblast proliferation was measured by MTT assay. (B) Quantitative real-time polymerase chain reaction analysis of collagen Iα1 and collagen IIIα1 mRNA expression in CFs. (C) Western blot analysis of MMP9 in CFs. Cells were transfected by miR-378 mimics or mock scrambles and subjected to mechanical stretching (MS) for 24 h. (D) Representative photos of CFs analyzed by EdU incorporation assay (scale bar: 50 μm). Red color indicates the EdU-positive cells; blue color indicates DAPI-positive cells. The EdU incorporation rate is shown as the radio of EdU-positive cells to total DAPI-positive cells. Data are from three independent experiments. DAPI indicates 4',6-diamidino-2-phenylindole. Values represent the mean ± SE from three independent experiments. **, P< 0.01 vs. mock; *, P< 0.05 vs. mock; ##, P< 0.01 vs. mock subjected to MS.
Figure 3
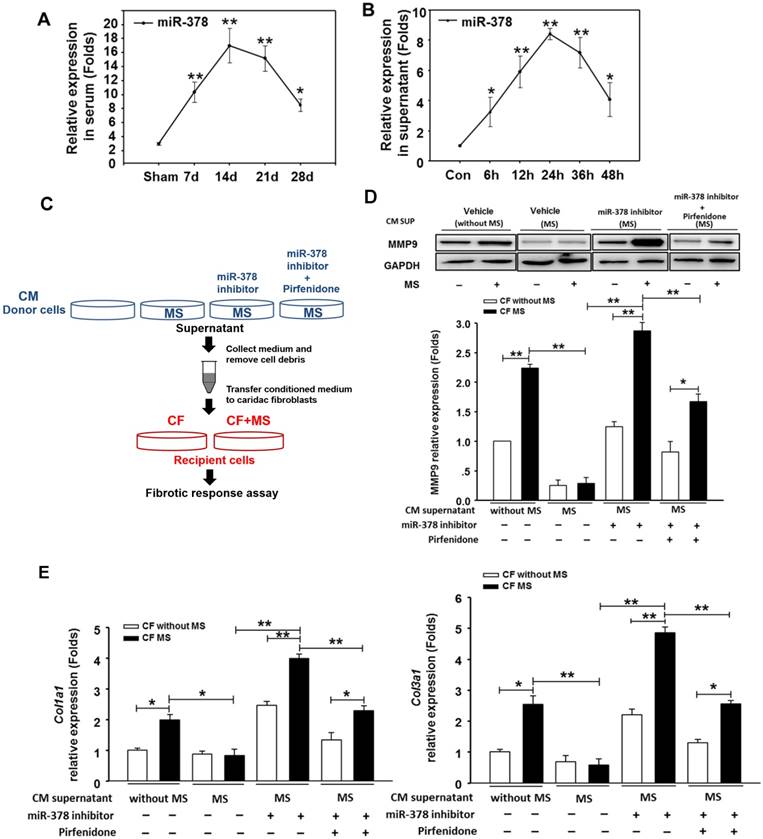
miR-378, secreted by cardiomyocytes under mechanical overload, inhibited fibrotic response of fibroblasts via paracrine signaling. (A) The expression levels of miR-378 in the serum of mice at 7 days, 14 days, 24 days and 48 days after TAC (n=5 per group). Total miRNA was first enriched from the serum, and then, the miR-378 was analyzed by quantitative real-time PCR. Values represent the mean ± SE. **, P< 0.01 vs. sham; *, P< 0.05 vs. sham. (B) The expression level of miR-378 in the supernatant of cardiomyocytes that were stretched at several time points (6, 12, 24, 48 h). Total miRNA was enriched from the supernatant, and then, the miR-378 was analyzed by quantitative real-time PCR. Data were obtained from three different experiments. Values represent the mean ± SE, n=4 per group. **, P< 0.01 vs. control; *, P< 0.05 vs. control. (C) Schematic representation of the workflow to assess the role of miR-378 secreted from cardiomyocytes (CMs) on cardiac fibroblasts (CFs). Briefly, the CMs (as donor cells) were divided into four groups: untreated CMs, CMs subjected to MS for 24 h, miR-378-depleted CMs via transfection of miR-378 inhibitors in the presence of MS for 24 h, miR-378-depleted CMs plus pirfenidone (an inhibitor of TGFβ1 production and secretion) in the presence of MS for 24 h. The CFs (as recipient cells) were cultured in conditioned medium derived from the donor cells for 48 h and were then subjected to MS. After 24 h of MS, the recipient cells were collected for a fibrotic response assay. (D) Western blot analysis of MMP9 in CFs and the quantitative analysis. The relative expression was normalized to GAPDH. (E) Quantitative real-time PCR analysis of collagen type I and III mRNA expression in the CFs. Values represent the mean ± SE from three independent experiments. **, P< 0.01 and *, P< 0.05.
Mechanical stress promotes the secretion of miR-378 from cardiomyocytes in an extracellular vesicles-dependent manner
To investigate the secretory mechanism and biological function of the miR-378 released from the cardiomyocytes, we employed the extracellular vesicles (EVs)-dependent secretory machinery to explore the movement of miR-378 from the cardiomyocytes to the cardiac fibroblasts. Current accumulating evidence suggests that EVs are generally packaged in the intracellular multivesicular bodies (MVBs) and released into the extracellular space after the fusion of the MVBs with the plasma membrane. These secretory vesicles can transfer the miRNAs to neighboring cells, serving as mediators of intercellular communication [15, 16, 26-28]. We first determined whether the cardiomyocytes produced and secreted EVs under stress stimulation. Cultured cardiomyocytes were subjected to mechanical stress for 24 h. The EVs were isolated from the conditioned media containing the cardiomyocytes with and without mechanical stress exposure. Electron microscopy studies were used to identify and characterize the presence of EVs. In the media of the cultured non-stressed cardiomyocytes, we found that the EVs were of the typical sizes of 20 nm to 100 nm with characteristic round morphologies. Interestingly, however, the EVs in the same volume of media containing the stressed cardiomyocytes were present at increased numbers and were of larger sizes (up to 200 nm) (Figure 4A). Western blotting analysis confirmed the presence of the marker protein CD63 in the cardiomyocyte-derived EVs and its greatly increased expression in the mechanically stressed cells (Figure 4B). Then, we blocked EVs production in the cardiomyocytes in vitro using GW4869, which is a well-known inhibitor of neutral sphingomyelinase 2 (nSMase 2) that is responsible for EVs secretion. Accordingly, isolated primary cardiomyocytes were cultured in the serum-free medium for 12 h, then GW4869 was added to the serum-free medium of the cardiomyocytes for 24 h followed by the MS for another 24 h. We found that GW4869 can inhibit the release of EVs by the cardiomyocytes subjected to MS in a dose-dependent manner, which was confirmed with the marker protein CD63 (Figure 4C). After extracting the small RNA from the EVs we found that miR-378 expression was also inhibited by GW4869 in the same dose-dependent manner (Figure 4D).
To confirm that the anti-fibrotic response resulted from the transferred EV miR-378 from the cardiomyocytes, we treated the cardiac fibroblasts with the conditioned cardiomyocyte medium. As schematically depicted in Figure 4E, the supernatants derived from the cultured cardiomyocytes with or without MS or exposure with or without the addition of GW4869 served as the culture media of the fibroblasts. To further observe TGFβ1 effects involved in this process, we added pirfenidone along with GW4869 into the donor cells. Then, the cardiomyocytes were mechanically stretched for 24 h. We found that the conditioned medium from the GW4869-treated donor cells led to a loss of inhibition of the collagen genes and MMP9 protein activation after 24 h of mechanical stretching (Figure 4F-G).When we used the conditioned medium from both GW4869 and pirfenidone-treated donor cells, we found that the expressions of collagens and MMP9 in the CFs were downregulated compared with that of cells treated only by GW4869, which indicated that mechanical stretching also stimulated a certain amount of TGFβ1 secretion from the CMs independent of the EVs. No direct effects of GW4869 on the fibrotic response in the fibroblasts were observed (data not shown). Because miR-378 was not expressed in the cardiac fibroblasts, we measured the internal miR-378 levels in the cardiac fibroblasts by miRNA quantitative real-time PCR. We found higher expression of miR-378 in the CFs in the medium from the mechanically stretched cardiomyocytes compared with that observed in the CFs in the medium from the unstretched cardiomyocytes, but very low levels were observed in the CFs when GW4869 was added to the medium, indicating that miR-378 can be transferred into the cardiac fibroblasts via EVs by a paracrine-associated mechanism (Figure 4H). To ensure that the anti-fibrotic function was mainly caused by miR-378, we first performed a rescue experiment by introducing miR-378 into the recipient cells. When additional miR-378 was transfected into these recipient CFs with GW4869 supplementation, MMP9 was again inhibited (Figure 4I). The other experiment was performed by inhibition of the exogenous miR-378 in the recipient cells when culturing in the medium from the stretched cardiomyocytes. We found that suppression of fibrotic response in the CFs treated by the conditioned medium from the stretched cardiomyocytes was attenuated after introducing the inhibitor of miR-378 (Figure 4J-L). Then, we labeled secreted cardiomyocyte-derived EVs with a green fluorescent marker, PHK67, and incubated cultured cardiac fibroblasts with the labeled EVs. Confocal microscopy revealed the uptake of labeled EVs by the fibroblasts (Figure S5). We also found the number of green-labeled EVs from the stretched cardiomyocytes greatly increased compared with that from the unstretched cells (Figure 4M). These results showed that mechanical stress can promote the increased release of miR-378 from cardiomyocytes in an EVs-dependent manner and the miR-378 in the EVs can be taken up by CFs and act to suppress the fibrotic response.
Figure 4
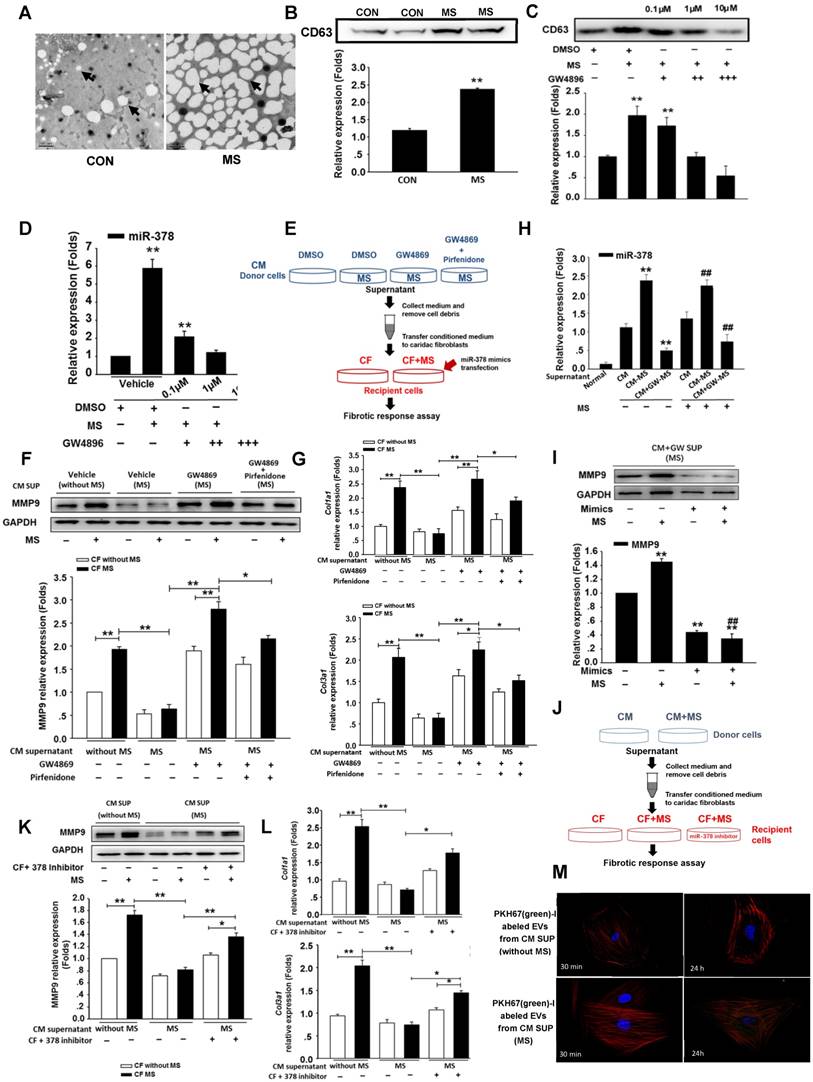
Mechanical stress promotes miR-378 secretion from cardiomyocytes in an EVs-dependent manner. (A) Electron micrographs of rat cardiomyocyte-derived EVs, showing sizes of approximately 20 to 100 nm in diameter for typical cells and larger sizes of approximately 80 to >250 nm for stretched cells. Scale bar: 200 nm (n=5). EVs are indicated by arrows. (B) Western blot analysis of marker protein CD63 from purified EVs secreted from equal numbers of cardiomyocytes with or without mechanical stress. (C) GW4869 treatment inhibited EVs secretion induced by mechanical stress. Cardiomyocytes subjected to mechanical stretching were cultured in dimethyl sulfoxide (DMSO) or with 0.1 μM (+), 1 μM (++) or 10 μM (+++) GW4869. The purified exosomes secreted by equal numbers of control or GW-treated cardiomyocytes were analyzed by Western blotting for the presence of the CD63 protein. (D) Secretion of miR-378 was suppressed by the treatment with GW4869. Values represent the mean ± SE from three independent experiments. **, P< 0.01 and *, P< 0.05 vs. control without MS. (E) Schematic representation of the workflow for investigating the mechanism of miR-378 secretion. (F) Western blot analysis of MMP9 expression in CFs and quantitative analysis. Cardiac fibroblasts were resuspended in conditioned medium and were then stretched or not stretched for 24 h. (G) Quantitative real-time PCR analysis of mRNA expression of collagen types I and III in CFs. Values represent the mean ± SE from three independent experiments. **, P< 0.01 and *, P< 0.05. (H) Quantitative polymerase chain reaction (qRT-PCR) analyses of miR-378 expression in the cardiac fibroblasts after treatment with the supernatant from the stretched cardiomyocytes (CM-MS) or with the supernatant from the stretched cardiomyocytes with GW4869 added (CM+GM-MS); the supernatant from the unstretched cardiomyocytes was used as the negative control (CMs). Values represent the mean ± SE, n=3. **, P< 0.01 and *, P< 0.05 vs. CFs treated with supernatant of unstretched cardiomyocytes without MS; ##, P< 0.01 and #, P< 0.05 vs. CFs treated with supernatant of unstretched cardiomyocytes that were subjected to MS. (I) CFs treated with the supernatant of the stretched cardiomyocytes with GW4869 added were transfected with miR-378 and subsequently subjected to mechanical stretching for 24 h. MMP9 expression was detected by Western blotting and quantitatively analyzed. Values represent the mean ± SE from three independent experiments. **, P< 0.01 vs. control CFs without MS and miR-378 mimics transfection; ##, P< 0.01 vs. CFs without miR-378 mimics transfection that were subjected to MS. (J) Schematic representation of the workflow. (K) Western blot analysis of MMP9 expression in CFs and the quantitative analysis. Cardiac fibroblasts were resuspended in conditioned medium and were then stretched or not stretched for 24 h. (L) Quantitative real-time PCR analysis of mRNA expression of collagen types I and III in CFs. Values represent the mean ± SE from three independent experiments. **, P< 0.01 and *, P< 0.05. (M) Cardiac fibroblasts were incubated with PKH67-labeled (green) EVs from cardiomyocytes for 30 min and 24 h independently (n=5-7) and then fixed for confocal imaging. Scale bar: 10 μm. The exosomes were obtained from the supernatant of CMs without MS or with MS for 24 h.
miR-378 suppresses the p38 MAPK-Smad2/3 pathways in the myocardium under pressure overload
Because miR-378 plays a dual role in suppressing cardiac hypertrophy and fibrosis, we investigated the regulation of the intermediate signaling components involved in these processes. Western blot analysis showed that the phosphorylation (activation) of p38 MAPK (p-p38) was greatly increased in the heart 14 days after TAC. The overexpression of miR-378 by agomir treatment significantly reduced the phosphorylation of p38 MAPK. However, the silencing of endogenous miR-378 with antagomir increased the phosphorylation of p38 MAPK. We also found that the miR-378 agomir treatment repressed the phosphorylation of Smad2/3 (p-Smad2/3) but the antagomir enhanced p-Smad2/3 levels (Figure 5A). To gain insight into the intracellular signaling pathways regulated by miR-378, isolated primary cardiomyocytes and third-passage cardiac fibroblasts were prepared for the introduction of miR-378 mimics or inhibitors. In the cardiomyocytes, miR-378 overexpression significantly repressed the phosphorylation of p38 MAPK, an important kinase activated by mechanical overload in the hypertrophic signaling pathway. Knocking down endogenous miR-378 enhanced the phosphorylation of p38 MAPK by MS, a result consistent with the anti-hypertrophic phenomenon caused by miR-378 (Figure S6). For the CFs, a gain-of-function analysis was performed because of the lack of endogenous miR-378. Overexpression of miR-378 clearly suppressed the phosphorylation of p38 MAPK and Smad2/3 activated by mechanical overload (Figure 5B). After treatment with SB203580, a pharmacological inhibitor of p38MAPK activation, to further inhibit p38 MAPK activation in miR-378-obverexpressing CFs after mechanical stretching, the western blot and quantitative real-time PCR analyses showed that p38 phosphorylation and the expression of MMP9 and collagens were further significantly suppressed compared with that of cells only transfected with miR-378 mimics (Figure 5C-D). These results suggested that the anti-fibrotic activity of miR-378 was closely related to repression of the p38 MAPK /Smad2/3 pathway in cardiac fibroblasts.
MKK6 is a direct target of miR-378
Because overexpression of miR-378 effectively inhibited p38 MAPK phosphorylation but not total p38 MAPK expression, we sought to identify the direct targets of miR-378 involved in phosphorylation activation. We conducted bioinformatics predictions to identify potential targets of miR-378. TargetScan (version 7.0) indicated that the 3'UTR of MKK6 contains the target seed sequence against miR-378, which is conserved among humans, mice and rats. The species-conserved miR-378 seed sequence in the 3' UTR of MKK6 is shown in Figure 6A. MKK6 functions as a MAPK kinase and is an upstream activator of p38 MAPK after stress stimulation. Therefore, we presumed that miR-378 regulates p38 MAPK phosphorylation by targeting MKK6 in the heart. We then detected the MKK6 expression levels in both our in vivo and in vitro models of pressure overload. In cultured cardiac fibroblasts, overexpression of miR-378 significantly suppressed MKK6 in cells that were mechanically stretched (Figure 6B). In the animal models, miR-378 KO (+/-) mice exhibited a significant increase in MKK6 levels in the heart after TAC compared with that in WT mice subjected to TAC (Figure 6C).
We next verified whether miR-378 directly repressed MKK6 expression by using a luciferase reporter system in COS-7 cells. Both a luciferase reporter construct Luc-MKK6 harboring the miR-378-binding site from the rat MKK6 3'UTR and an equivalent luciferase construct Luc-MKK6-MU with the 3'UTR of MKK6 with six mutated bases in the seed sequence (as a negative control) were constructed as illustrated in Figure 6A.
Figure 5
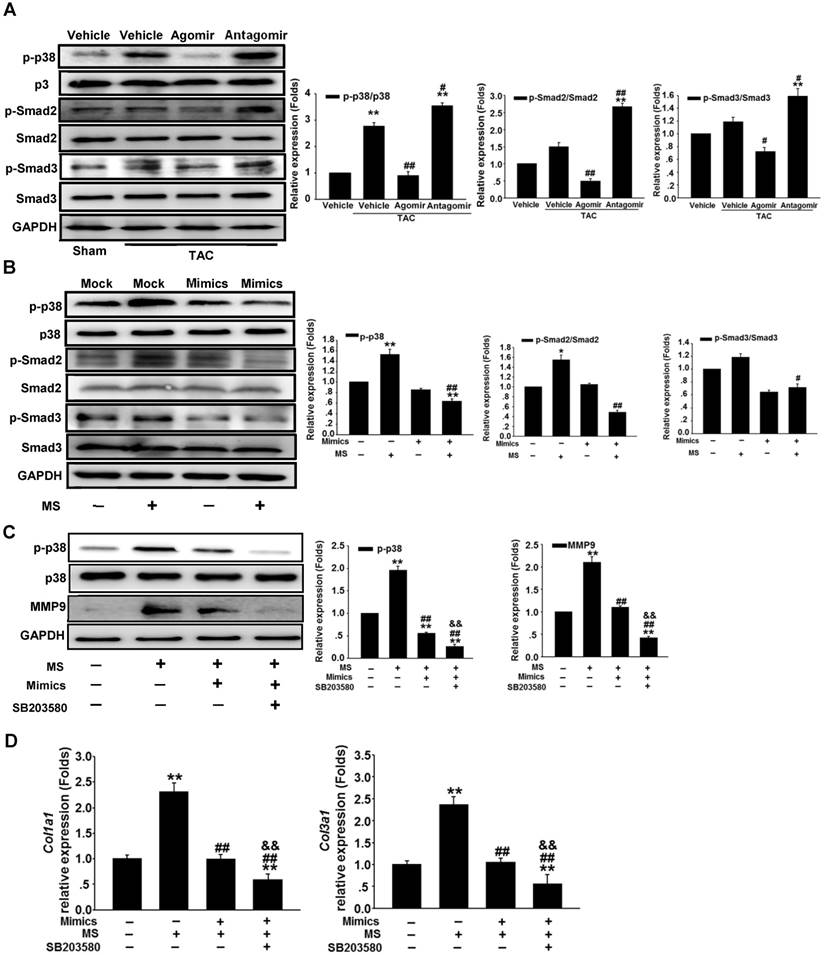
miR-378 regulated the p38 MAPK-Smad2/3 pathway during mechanical overload-induced cardiac remodeling. (A) Western blotting and quantitative analysis of phosphorylated p38 MAPK, total p38, phosphorylated Smad2/3 and total Smad2/3 levels in the protein extracted from mouse hearts subjected to the different treatments as described above. The values represent the mean ± SE, n=3-5 per group. **, P< 0.01 vs. sham; *, P< 0.05 vs. sham; ##, P< 0.01 vs. TAC vehicle group; #, P< 0.05 vs. TAC vehicle group. (B) Western blot and quantitative analysis of phosphorylated p38 MAPK, total p38, the phosphorylated Smad2/3 and total Smad2/3 levels in the CFs. Cells were transfected with either miR-378 mimics or the negative scramble control (mock), and 48 h after transfection, the cells were mechanically stretched (MS) or untreated for 10 min. The values represent the mean ± SE from three independent experiments. **, P< 0.01 and *, P< 0.05 vs. mock; ##, P< 0.01 and #, P< 0.05 vs. mock with MS. (C) Western blot and quantitative analysis of phosphorylated p38 MAPK, total p38 MAPK and MMP9 expression in CFs treated with SB203580 (a p38 MAPK inhibitor). Cells were transfected with miR-378 mimics for 48 h and treated with SB203580 (10 μM) for 1 h prior to MS. Total p38 and p-p38 MAPK in the cells after MS for 10 min as well as MMP9 in the cells after MS for 24 h are shown. The values represent the mean ± SE from three independent experiments. **, P< 0.01 vs. control CFs; ##, P< 0.01 vs. control CFs with MS; &&, P< 0.01 vs. CFs transfected with miR-378 mimics and subjected to MS. (D) Quantitative real-time polymerase chain reaction analysis of collagen type I and III mRNA expression in the CFs. Cells were transfected with miR-378 mimics for 48 h and treated with SB203580 (10 μM) for 1 h before MS. Cells were collected 24 h after MS. The values represent the mean ± SE from three independent experiments. **, P< 0.01 vs. control CFs; ##, P< 0.01 vs. control CFs with MS; &&, P< 0.01 vs. CFs transfected with miR-378 mimics and subjected to MS.
Figure 6
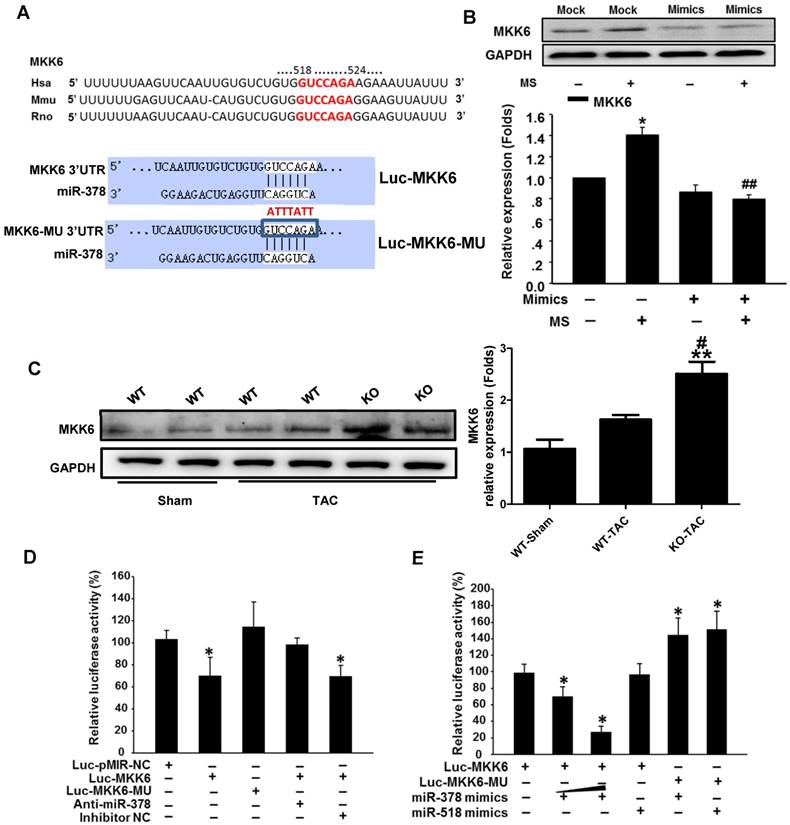
MKK6 is a direct target of miR-378 in cardiac fibroblasts. (A) Sequence conservation of the miR-378 seed region (highlighted in RED) in the 3' UTR of MKK6 mRNA. For the functional assay, the binding region (MKK6 UTR) of miR-378 was cloned downstream of a dual luciferase reporter (Luc-MKK6). The mutant sequence (MKK6-MU UTR) was also constructed as a control(Luc-MKK6-MU). (B) Endogenous MKK6 in the CFs was regulated by miR-378. Western blotting and quantitative analysis of MKK6. Cells were transfected with either miR-378 mimics or the negative scramble control (mock), and 48 h after transfection, the cells were mechanically stretched (MS) or unstretched for 24 h. Values represent the mean ± SE from three independent experiments. **, P< 0.01 vs. mock; *, P< 0.05 vs. mock; ##, P< 0.01 vs. mock with MS. (C) Western blotting and quantitative analysis of MKK6 levels in the WT and KO mice subjected to either a sham operation or TAC. The values represent the mean ± SE, n=3-5 per group. **, P< 0.01 vs. WT sham; #, P< 0.05 vs. WT TAC. (D) Luciferase activity was repressed by endogenous miR-378. COS-7 cells were transiently transfected with a luciferase reporter construct containing the target sequence of miR-378 (Luc-MKK6), a mutant sequence (Luc-MKK6-MU), or a sham pMIR-REPORTTM luciferase reporter vector (Luc-pMIR-NC); these vectors were cotransfected along with the inhibitor of miR-378 (Anti-miR-378) or a mock inhibitor (Anti-NC). Relative luciferase activities were measured 48 h after transfection and standardized with firefly/ beta Gal to eliminate variations due to differences in cell viability and transfection efficiency. Values represent the mean ± SE from three independent experiments. *, P< 0.05 vs. Luc-pMIR-NC. (E) COS-7 cells were transiently transfected with either Luc-MKK6 or Luc-MKK6-MU, as well as with either modified miR-378 mimics (miR-378 mimics) (30 nM and 60 nM) or an unrelated miR, miR-518 (30 nM). Values represent the mean ± SE from three independent experiments. *, P< 0.05 vs. Luc-MKK6 alone
Transfection of the luciferase reporter gene linked to the wild-type 3' UTR of MKK6 Luc-MKK6 resulted in a significant (P <0.01) decrease in luciferase activity compared with that after transfection with either the sham luciferase reporter vector Luc-pMIR-NC or the mutated seeding sequence luciferase reporter vector Luc-MKK6-MU; this difference was due to the endogenous miR-378 expression in COS-7 cells (Figure 6D). Furthermore, cotransfection of a miR-378 inhibitor (anti-miR-378) with Luc-MKK6 attenuated the decrease in luciferase activity observed after transfection with only Luc-MKK6, whereas cotransfection of the inhibitor mock (anti-NC) and Luc-MKK6 had no significant effects. Moreover, COS-7 cells were cotransfected with synthetic miR-378 mimics and the luciferase reporter construct. The luciferase assays showed that miR-378 significantly repressed the luciferase activity of Luc-MKK6, but had no effect on Luc-MKK6-MU. In addition, increasing amounts of the miR-378 resulted in more repression of luciferase activity, whereas this decrease was absent when either the unrelated miR-518 or the mutated luciferase reporter construct was used (Figure 6E). All of these data verified that MKK6 is one of the targets of miR-378.
miR-378 knockout mice exhibit aggravated cardiac fibrosis in response to pressure overload
To assess whether the secreted miR-378 induced by pressure overload is a major mechanism affecting cardiac fibrosis in vivo, we generated miR-378 knockout mice. The miR-378 KO (+/-) mice were used rather than the miR-378 KO (-/-) mice considering the fact that miR-378 KO (-/-) mice showed high mortality before 8 weeks of age. (Figure S7). By using sensitive quantitative real-time PCR assays, we confirmed the decreased expression levels of miR-378 in miR-378 mutant mouse hearts (Figure 7A). The morphology and the ratio of heart weight versus body weight of miR378 mutant mice did not differ significantly from those of the wild-type littermate controls (Figure 7B). Echocardiographic measures of cardiac function did not significantly differ between the miR-378 KO mice and their littermate controls under sham conditions. However, after TAC treatment, miR-378 KO mice showed more severe cardiac dysfunction than did WT mice as observed by the much lower preserved ejection fraction (Figure 7B and Table S2). Masson trichrome staining was performed to identify the collagen deposition in the LV section. The sham wild-type and miR-378 KO mice did not differ significantly regarding the fibrosis of the cardiac interstitium and perivascular tissues. The wild-type mice subjected to 14 days of TAC exhibited increased fibrosis in both the interstitial and perivascular areas. Unsurprisingly, miR-378 KO mice subjected to TAC treatment exhibited increased fibrosis compared with that in wild-type mice treated with vehicle (Figure 7C). Accordingly, the ECM genes and fibrosis markers were also markedly upregulated in miR-378 KO mice compared with the wild-type mice, as shown by the collagen Iα1 (Col1a1), collagen IIIα1 (Col3a1) and α-SMA (Acta2) mRNA levels measured by quantitative real-time PCR (Figure 7D). Western blot analysis of the proteins from the myocardium also showed that MMP9, as well as phosphorylated p38MAPK and Smad2/3 were markedly increased after TAC in KO mice compared with WT mice (Figure 7E).
To verify the anti-fibrotic role of heart-oriented miR-378 in an EVs-dependent manner, we added EVs derived from the serum of WT mice and KO mice under either sham or TAC conditions to the culture medium of the CFs in vitro. After applying mechanical stretching to the CFs for 24 h, we observed that MMP9 and collagen expression in the CFs were greatly upregulated by the treatment of EVs from the sera of wild-type sham-operated mice compared with that in the unstretched cells; however, this upregulation was suppressed by the addition of EVs from the serum of wild-type TAC-operated mice, as shown by western blot and quantitative real-time PCR analyses. When we used conditioned medium containing EVs from the serum of miR-378 KO TAC-operated mice to culture the CFs, we found that the inhibition of collagen gene expression and MMP9 protein activation were lost after 24 h of mechanical stretching (Figure 7G-H). By detecting the expression levels of miR-378 in the EVs from the sera of WT and KO mice, the real-time PCR analysis showed that although the miR-378 levels greatly increased in both the WT and KO mice after TAC, the upregulation in the WT mice was almost three times greater than that in the KO mice (Figure 7F). Therefore, we believe that the aggravated cardiac fibrosis exhibited by miR-378 KO mice was largely due to the reduction of miR-378 levels in the EVs secreted from the cardiomyocytes in response to pressure overload.
Discussion
In this study, miR-378 was assessed to determine whether it plays an important protective role in cardiac remodeling in response to stress based on our former studies of the role of HSF1 in adaptive hypertrophy. The in vivo and in vitro overexpression of miR-378 was shown to not only attenuate cardiac hypertrophy but also suppress myocardial fibrosis in the adaptive stage of pressure-overloaded hearts. Upon inhibiting cardiac fibrosis, miR-378 appeared to suppress p38 MAPK and Smad2/3 signal pathways by directly targeting MKK6 in the cardiac fibroblasts. Furthermore, miR-378 also acted as a mediator of intercellular communication, being released by the cardiomyocytes under pressure overload and entering into the cardiac fibroblasts in an EVs-dependent manner, enabling the cross-talk between the fibroblasts and cardiomyocytes during the hypertrophic response. An illustration of the mechanism of anti-fibrotic effects of miR-378 is shown in Figure 8.
Figure 7
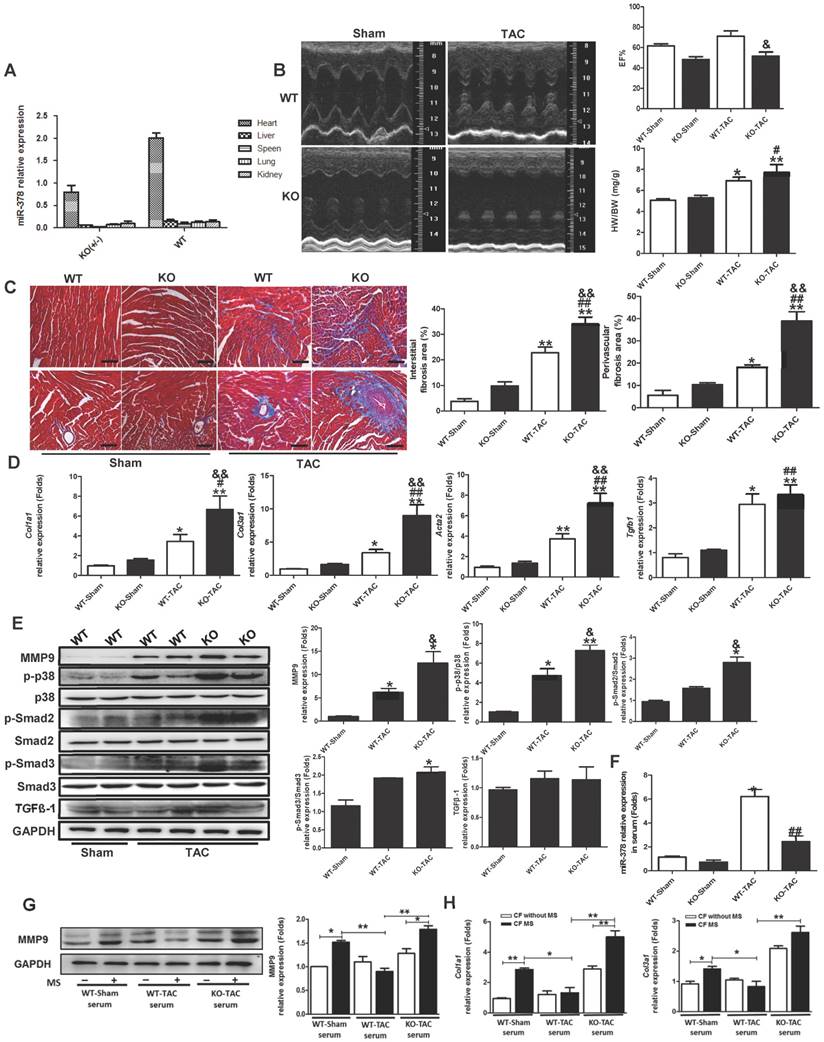
miR-378 knockout mice exhibited aggravated cardiac fibrosis in response to pressure overload. (A) Expression levels of miR-378 in the hearts and other tissues of wild-type (WT) and miR-378 knockout (KO+/-) mice. (B) Echocardiographic measurements of WT and KO mouse hearts 2 weeks after TAC or sham operation. Quantitative analysis of the ratio of heart weight to body weight (HW/BW) and left ventricular ejection fraction value (EF%). (C) Representative photomicrographs of the left ventricular perivascular and interstitial fibrosis. For each group, blue staining indicates collagen deposition. Original magnification ×200 (scale bar: 100 μm). Collagen content was calculated as the percentage of the area in each section that was stained blue. Quantification of interstitial and perivascular fibrosis in the WT and KO mice is shown below. (D) Total RNA was isolated from cardiac samples and quantitative real-time polymerase chain reaction analysis of fibrosis marker genes (Col1a1, collagen I α1; Col3a1, collagen IIIα1; Acta2, α-SMA; Tgfb1, TGFβ1) was performed. (E) Western blotting and quantitative analysis of phosphorylated p38 MAPK, total p38, phosphorylated Smad2/3, total Smad2/3, and TGFβ levels in the protein extracted from the mice. The values represent the mean ± SE (n≥5/group). **, P< 0.01 vs. WT sham; *, P< 0.05 vs. WT sham; ##, P< 0.01 vs. KO sham; #, P< 0.05 vs. KO sham; &, P< 0.05 vs. WT TAC; &&, P< 0.01 vs. WT TAC. (F) Expression of miR-378 in exosomes derived from the serum of WT and KO mice subjected to 2 weeks of TAC or a sham operation, n=5 per group. **, P< 0.01 vs. WT sham; ##, P< 0.01 vs. WT TAC. (G) Western blot and quantitative analysis of MMP9 levels in the CFs. The exosomes derived from the sera of WT and KO mice subjected to either 2 weeks of TAC or a sham operation were added into the culture medium of CFs. The CFs were either subjected to MS for 24 h or untreated. (H) Quantitative real-time polymerase chain reaction analysis of collagen type I and III mRNA expression in the CFs, n=5 per group. **, P< 0.01 and *, P< 0.05.
Figure 8
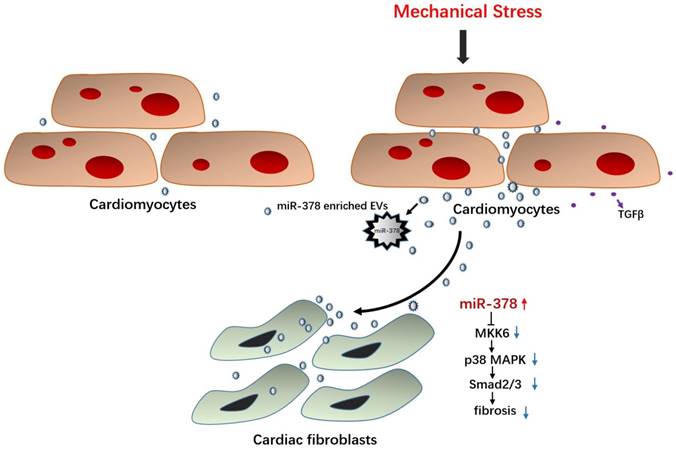
Illustration of the mechanism of anti-fibrotic effects of miR-378 as a paracrine signaling mediator in response to mechanical stress. MiR-378 is preferentially expressed in cardiomyocytes. Cardiac mechanical stress makes cardiomyocytes secrete more exosomes enriched with miR-378, which are transported into the cardiac fibroblasts. In cardiac fibroblasts, miR-378 regulates the p38 MAPK-Smad2/3 signaling pathway by targeting MMK6 and then inhibits fibrosis.
In our previous study, HSF1 KO mice were shown to display severe cardiac dysfunction and aggravated cardiac fibrosis characteristics after stress overload; we found that miR-378 was deeply repressed along with the deletion of HSF1 in the mouse hearts [20]. Therefore, we proposed that the reduction in miR-378 may be related to the impaired cardiac function and that miR-378 may be an endogenously protective modulator in the heart. With regard to pressure overload-induced cardiac remodeling, we demonstrated that cardiac hypertrophy was attenuated by the overexpression of miR-378 agomirs administered through intravenous injection, strongly suggesting that miR-378 is an essential regulator of cardiac hypertrophy. Consistent with our study, both Ganesan et al. and Nagalingam et al. found that miR-378 participates in regulating cardiac hypertrophy [21, 29]. They have found that miR-378 targets key components of the RAS signal pathway, such as GRB2, KSR1 and MAPK1, to regulate hypertrophic activity in cardiomyocytes. Meanwhile, we found that cardiac fibrosis was attenuated after overexpression of an miR-378 agomir administered via intravenous injection but was aggravated after knockdown of endogenous miR-378 expression under pressure overload, suggesting the anti-fibrotic role of miR-378 in the early stage of cardiac remodeling induced by mechanical stress as well.
Castoldi et al. have demonstrated that miR-133a, which is predominantly localized to myocytes and is involved in cardiac hypertrophy, also targets CollA1 and participates in cardiac fibrosis. Their data support the theory that myocardial fibrosis may be modulated not only by miRNAs expressed in CFs but also by muscle-specific miRNAs through synergistic activities [30]. In addition, miR-133a has also been shown to play a role in mediating matrix production in cultured fibroblasts [31]. However, the mechanism underlying the communication between the cardiac myocytes and fibroblasts was not explored. There is growing evidence that secretory miRNAs play roles in a variety of physiological phenomena. Zernecke et al. have shown that the transfer of miR-126 into endothelial apoptotic bodies induces the expression of CXCL12 and the recruitment of progenitor cells, thereby alleviating atherosclerosis [32]. Kosaka et al. have demonstrated that intercellular and extracellular miRNAs packaged in secretory EVs can be delivered into recipient cells and act as physiologically functional molecules to promote gene silencing [13]. In our study, we revealed that miR-378, which is a cardiac-specific miRNA, mediated cardiac fibrosis by regulating collagen and MMP expression in CFs through a new EVs-dependent secretory mechanism. The electron microscopy analysis of the secreted EVs showed that when the cardiomyocytes were stimulated by mechanical stretching, the sizes of the EVs increased, with the biggest diameter being over 200 nm, compared with the sizes of the untreated cells. In addition, mechanical stress promoted the secretion of increased numbers of EVs from the cardiomyocytes containing miR-378, which may account for the increase of miR-378 in the medium in vitro or in the serum in vivo and cause more miR-378 to enter into the recipient cells subsequently. Therefore, when we inhibited the secretion of the EVs from the stretched CMs, we found the conditioned medium from the donor cells strongly enhanced the fibrotic response of the recipient cells. Furthermore, to investigate whether TGFβ1 plays a role in this process, we used the conditioned medium derived from the donor cells with both GW4869 and pirfenidone addition; we found the fibrotic response in the CFs was still enhanced but alleviated compared with only GW4869 addition, which indicated that MS also stimulated a certain amount of TGFβ1 secretion from the CMs independent of the EVs. We speculated that other pro-fibrogenic factors may be secreted from the CMs subjected to MS besides TGFβ1.We then transfected miR-378 inhibitors into the cardiomyocytes and found that endogenous miR-378 inhibition not only suppressed the miR-378 level in the supernatant of stretched cardiomyocytes but also induced TGFβ1 expression and secretion from cardiomyocytes. To exclude the effects of TGFβ1 secretion from the cardiomyocytes, which could aggravate fibrotic response of the CFs, we administered a TGFβ1 inhibitor along with the miR-378 inhibitors to cardiomyocytes in vitro. The conditioned medium from this treatment was used in the culture, and subsequent stretching of the CFs was then performed. We found that the fibrotic response in the CFs was still significantly upregulated compared with that in the control treatment group, but this response was markedly attenuated compared with that of cells cultured with medium from cardiomyocytes treated with only miR-378 inhibitors. In this context, our data suggest that miR-378 may participate in the crosstalk between myocytes and fibroblasts during the cardiac remodeling processes and function as a kind of paracrine factor in the suppression of cardiac fibrosis under mechanical stretching. All of these data verified the existence of miRNAs in cardiomyocytes that serve as mediators of intercellular communication in the heart.
To study the function of miR-378 in vivo, we generated miR-378 KO mice. Generally, generation of cardiomyocyte-specific miR-378 KO would be an ideal model to prove that secreted miR-378 from cardiomyocytes can translocate to and communicate with cardiac fibroblasts to reduce fibrosis after TAC. However, considering the fact that miR-378 may serve as an anti-fibrotic factor via a paracrine mechanism, we constructed global mutant mice rather than cardiomyocyte-specific ones for our study to exclude the possible effect of the secreted miR-378 from other tissues under mechanical stress (miR-378 is also expressed in both muscles and lungs in a small amount). Above all, we found that miR-378 is preferentially expressed in cardiomyocytes and has no expression in cardiac fibroblasts from both neonatal rat hearts and adult wildtype mouse hearts (Figure S3C), which made it feasible to use systemic knockout mice to save time in our study. During breeding, the global miR-378 homozygous knockout mice showed higher mortality than the heterozygous mice before 8 weeks of age, which made us exploit the heterozygous mice in this study (Figure S7A). This phenomenon may account for the important role of miR-378 in the regulation of physiological development, which needs to be investigated further. Because miR-378 is generated from the first intron of the host gene Ppargc1b (PGC-1β) and is coexpressed with PGC-1β [33], we detected PGC-1β expression in the hearts of generated miR-378 KO mouse models with both sham and TAC conditioning. The targeted mutation did not alter the expression of PGC-1β protein in tissues from mutant mice (Figure S7B).
Many studies have shown that the p38 MAPK signaling pathway plays a detrimental role in cardiac remodeling, which is linked to both hypertrophy and inflammation. The activation of the p38 family of stress-activated kinases exacerbates myocardial injury following prolonged ischemia and has been found to be involved in the regulation of MMPs and collagen in cardiac fibroblasts [34, 35]. It has been found that the activation of p38 MAPK is responsible for the proliferation and collagen production of CFs as induced by Ang II [36, 37]. In our study, we found that the phosphorylation of p38 increased in the hearts after TAC. When miR-378 was overexpressed in vivo, the phosphorylation of p38 MAPK was significantly inhibited. However, the silencing of endogenous miR-378 with antagomir aggravated the phosphorylation of p38 MAPK. Moreover, we found miR-378 was able to effectively inhibit the p38 MAPK activation induced by mechanical stretching in cardiomyocytes, which indicates another anti-hypertrophic mechanism by miR-378. In cardiac fibroblasts, miR-378 also negatively regulated the phosphorylation of p38 MAPK, thus inhibiting the mechanical overload-induced fibrotic response by regulating MMPs and collagen production. Our results showed that miR-378 regulates p38 MAPK by directly targeting MKK6, an important MAPK kinase responsible for the phosphorylation of p38 MAPK. To explore the anti-fibrotic signaling pathway in the cells regulated by exogenous miR-378, we overexpressed miR-378 in the CFs and observed that miR-378 suppressed the activation of Smad2/3 as well, but this activity was not associated with the modulation of TGF-β1 levels either at baseline or after exposure to stress, thus suggesting that the anti-fibrotic effects of miR-378 are independent of TGF-β1 signaling but may be associated with p38 MAPK phosphorylation-induced Smad2/3 activation in cardiac fibroblasts.
The relationship between miR-378 and heart disease in humans was also explored preliminarily. We evaluated miR-378 expression in the serum of HF patients and healthy controls and found that circulating miR-378 was significantly elevated in the HF patients. And, the increase of miR-378 level in the patients with moderate HF (LVEF > 45%) was markedly more pronounced than that in the patients with severe HF (LVEF < 45%). In addition, the HF patients with high miR-378 levels showed relatively lower NT-pro BNP values, while those with low miR-378 levels displayed higher NT-pro BNP values (Figure S9). These findings suggested that miR-378 is greatly upregulated at the early stages of HF but is downregulated in subjects with severely abnormal cardiac function, which may be greatly relevant to cardiac compensatory function. This trend is consistent with those observed in our in vitro study and in the mice. Herein, we suggest that the upregulation of miR-378 in circulation is closely associated with cardiac compensatory functions and may play a protective role during bodily stress.
In summary, we revealed that miR-378 plays a dual role in suppressing cardiac hypertrophy and cardiac fibrosis. More importantly, it also serves as a mediator of intercellular communication and can be released by cardiomyocytes under pressure overload, entering into cardiac fibroblasts in a secretory manner. We believed that miR-378 is an endogenous protective factor at the early stage of cardiac hypertrophy following mechanical stress, which is an important compensatory mechanism to heart stress. Therefore, the protective effects of miR-378 imply that the modulation of miR-378 expression by oligonucleotide administration may have future therapeutic applications in the clinical setting.
Abbreviations
WT: wild-type; KO: knockout; TAC: transverse aortic constriction; MS: mechanical stretch; MMP: matrix metalloproteinase; TGF-β: transforming growth factor-beta; α-SMA: alpha-smooth muscle actin; ECM: extracellular matrix; 3' UTR: 3' untranslated region; LVEF: left ventricular ejection fraction; CM: cardiomyocyte; CF: cardiac fibroblast; HF: heart failure; MAPK: Mitogen-ativated protein kinase kinase; MKK6: Mitogen Activated Protein Kinase Kinase 6; HSF1: heat shock transcription factor 1; EVs: extracellular vesicles; DMSO: dimethyl sulfoxside; EDU: 5-ethynyl-20-deoxyuridine; MTT: 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide; RAS: renin-angiotensin system.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
We thank members of the Zou laboratory for advice and technical support. We thank Jianguo Jia for technical assistance with animal models and histological analysis.
Author contributions
J.Y. and Y. Z. originated the central idea, designed the study and wrote the manuscript. Y. Z., J. G. and Z. C. supervised the work and provided guidance. J.Y., H. L., W. G., Z. C., L. Z., X. Z., H. G., A. S., X. Y. and R. C. performed experiments in vitro and analyzed the data. J.Y., Y. Y, L. Y., Z. D., J. W. and L. K. performed mice models experiments. G. Z. contributed the experiment materials supply.
Sources of Funding
This work was supported by National Natural Science Foundation of China (Grant No. 81200185, 30930043, 81521001, 81220108003 and 31371196).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Katz AM. Cardiomyopathy of overload. A major determinant of prognosis in congestive heart failure. N Engl J Med. 1990;322:100-10
2. Borer JS, Truter S, Herrold EM, Falcone DJ, Pena M, Carter JN. et al. Myocardial fibrosis in chronic aortic regurgitation: molecular and cellular responses to volume overload. Circulation. 2002;105:1837-42
3. Creemers EE, Pinto YM. Molecular mechanisms that control interstitial fibrosis in the pressure-overloaded heart. Cardiovasc Res. 2011;89:265-72
4. Bishop JE, Lindahl G. Regulation of cardiovascular collagen synthesis by mechanical load. Cardiovasc Res. 1999;42:27-44
5. Sakamoto M, Minamino T, Toko H, Kayama Y, Zou Y, Sano M. et al. Upregulation of heat shock transcription factor 1 plays a critical role in adaptive cardiac hypertrophy. Circ Res. 2006;99:1411-8
6. Ashrafian H, McKenna WJ, Watkins H. Disease pathways and novel therapeutic targets in hypertrophic cardiomyopathy. Circ Res. 2011;109:86-96
7. Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281-97
8. Divakaran V, Mann DL. The emerging role of microRNAs in cardiac remodeling and heart failure. Circ Res. 2008;103:1072-83
9. Tatsuguchi M, Seok HY, Callis TE, Thomson JM, Chen JF, Newman M. et al. Expression of microRNAs is dynamically regulated during cardiomyocyte hypertrophy. J Mol Cell Cardiol. 2007;42:1137-41
10. Care A, Catalucci D, Felicetti F, Bonci D, Addario A, Gallo P. et al. MicroRNA-133 controls cardiac hypertrophy. Nat Med. 2007;13:613-8
11. Thum T, Gross C, Fiedler J, Fischer T, Kissler S, Bussen M. et al. MicroRNA-21 contributes to myocardial disease by stimulating MAP kinase signalling in fibroblasts. Nature. 2008;456:980-4
12. van Rooij E, Sutherland LB, Thatcher JE, DiMaio JM, Naseem RH, Marshall WS. et al. Dysregulation of microRNAs after myocardial infarction reveals a role of miR-29 in cardiac fibrosis. Proc Natl Acad Sci U S A. 2008;105:13027-32
13. Kosaka N, Iguchi H, Yoshioka Y, Takeshita F, Matsuki Y, Ochiya T. Secretory mechanisms and intercellular transfer of microRNAs in living cells. J Biol Chem. 2010;285:17442-52
14. Fabbri M, Paone A, Calore F, Galli R, Gaudio E, Santhanam R. et al. MicroRNAs bind to Toll-like receptors to induce prometastatic inflammatory response. Proc Natl Acad Sci U S A. 2012;109:E2110-6
15. Li J, Liu K, Liu Y, Xu Y, Zhang F, Yang H. et al. Exosomes mediate the cell-to-cell transmission of IFN-alpha-induced antiviral activity. Nat Immunol. 2013;14:793-803
16. Bang C, Batkai S, Dangwal S, Gupta SK, Foinquinos A, Holzmann A. et al. Cardiac fibroblast-derived microRNA passenger strand-enriched exosomes mediate cardiomyocyte hypertrophy. J Clin Invest. 2014;124:2136-46
17. Fritz JV, Heintz-Buschart A, Ghosal A, Wampach L, Etheridge A, Galas D. et al. Sources and Functions of Extracellular Small RNAs in Human Circulation. Annu Rev Nutr. 2016;36:301-36
18. Chaturvedi P, Kalani A, Medina I, Familtseva A, Tyagi SC. Cardiosome mediated regulation of MMP9 in diabetic heart: role of mir29b and mir455 in exercise. J Cell Mol Med. 2015;19:2153-61
19. Creemers EE, van Rooij E. Function and Therapeutic Potential of Noncoding RNAs in Cardiac Fibrosis. Circ Res. 2016;118:108-18
20. Zou Y, Li J, Ma H, Jiang H, Yuan J, Gong H. et al. Heat shock transcription factor 1 protects heart after pressure overload through promoting myocardial angiogenesis in male mice. J Mol Cell Cardiol. 2011;51:821-9
21. Ganesan J, Ramanujam D, Sassi Y, Ahles A, Jentzsch C, Werfel S. et al. MiR-378 Controls Cardiac Hypertrophy by Combined Repression of Mitogen-Activated Protein Kinase Pathway Factors. Circulation. 2013;127:2097-106
22. Cortez DM, Feldman MD, Mummidi S, Valente AJ, Steffensen B, Vincenti M. et al. IL-17 stimulates MMP-1 expression in primary human cardiac fibroblasts via p38 MAPK- and ERK1/2-dependent C/EBP-β, NF-κB, and AP-1 activation. Am J Physiol Heart Circ Physiol. 2007;293:H3356-H65
23. Tang M, Zhong M, Shang Y, Lin H, Deng J, Jiang H. et al. Differential regulation of collagen types I and III expression in cardiac fibroblasts by AGEs through TRB3/MAPK signaling pathway. Cell Mol Life Sci. 2008;65:2924-32
24. Verma SK, Lal H, Golden HB, Gerilechaogetu F, Smith M, Guleria RS. et al. Rac1 and RhoA differentially regulate angiotensinogen gene expression in stretched cardiac fibroblasts. Cardiovasc Res. 2011;90:88-96
25. Parray AA, Baba RA, Bhat HF, Wani L, Mokhdomi TA, Mushtaq U. et al. MKK6 is Upregulated in Human Esophageal, Stomach, and Colon Cancers. Cancer Invest. 2014;32:416-22
26. Mittelbrunn M, Gutierrez-Vazquez C, Villarroya-Beltri C, Gonzalez S, Sanchez-Cabo F, Gonzalez MA. et al. Unidirectional transfer of microRNA-loaded exosomes from T cells to antigen-presenting cells. Nat Commun. 2011;2:282
27. Pegtel DM, Cosmopoulos K, Thorley-Lawson DA, van Eijndhoven MAJ, Hopmans ES, Lindenberg JL. et al. Functional delivery of viral miRNAs via exosomes. Proc Natl Acad Sci. 2010;107:6328-33
28. Cocucci E, Racchetti G, Meldolesi J. Shedding microvesicles: artefacts no more. Trends Cell Biol. 2009;19:43-51
29. Nagalingam RS, Sundaresan NR, Gupta MP, Geenen DL, Solaro RJ, Gupta M. A cardiac-enriched microRNA, miR-378, blocks cardiac hypertrophy by targeting Ras signaling. J Biol Chem. 2013;288:11216-32
30. Abonnenc M, Nabeebaccus AA, Mayr U, Barallobre-Barreiro J, Dong X, Cuello F. et al. Extracellular Matrix Secretion by Cardiac Fibroblasts: Role of MicroRNA-29b and MicroRNA-30c. Circ Res. 2013;113:1138-47
31. Shan H, Zhang Y, Lu Y, Zhang Y, Pan Z, Cai B. et al. Downregulation of miR-133 and miR-590 contributes to nicotine-induced atrial remodelling in canines. Cardiovasc Res. 2009;83:465-72
32. Zernecke A, Bidzhekov K, Noels H, Shagdarsuren E, Gan L, Denecke B. et al. Delivery of MicroRNA-126 by Apoptotic Bodies Induces CXCL12-Dependent Vascular Protection. Sci Signal. 2009;2:ra81 -
33. Carrer M, Liu N, Grueter CE, Williams AH, Frisard MI, Hulver MW. et al. Control of mitochondrial metabolism and systemic energy homeostasis by microRNAs 378 and 378*. Proc Natl Acad Sci U S A. 2012;109:15330-5
34. Clark JE, Sarafraz N, Marber MS. Potential of p38-MAPK inhibitors in the treatment of ischaemic heart disease. Pharmacol Ther. 2007;116:192-206
35. Sinfield JK, Das A, O'Regan DJ, Ball SG, Porter KE, Turner NA. p38 MAPK alpha mediates cytokine-induced IL-6 and MMP-3 expression in human cardiac fibroblasts. Biochem Biophys Res Commun. 2013;430:419-24
36. Chen J, Mehta JL. Angiotensin II-mediated oxidative stress and procollagen-1 expression in cardiac fibroblasts: blockade by pravastatin and pioglitazone. Am J Physiol Heart Circ Physiol. 2006;291:H1738-H45
37. Kim SI, Kwak JH, Zachariah M, He Y, Wang L, Choi ME. TGF-β-activated kinase 1 and TAK1-binding protein 1 cooperate to mediate TGF-β1-induced MKK3-p38 MAPK activation and stimulation of type I collagen. Am J Physiol Renal Physiol. 2007;292:F1471-F8
Author contact
![]() Corresponding authors: Yunzeng Zou, MD, PhD, Junbo Ge, MD, PhD and Zhaoqiang Cui, MD, PhD, Shanghai Institute of Cardiovascular Diseases, Zhongshan Hospital, Fudan University, 180 Feng Lin Road, Shanghai 200032, China. TEL&FAX: +86-21-54237969; E-mail: zou.yunzengsh.cn
Corresponding authors: Yunzeng Zou, MD, PhD, Junbo Ge, MD, PhD and Zhaoqiang Cui, MD, PhD, Shanghai Institute of Cardiovascular Diseases, Zhongshan Hospital, Fudan University, 180 Feng Lin Road, Shanghai 200032, China. TEL&FAX: +86-21-54237969; E-mail: zou.yunzengsh.cn
Citation styles
APA
Yuan, J., Liu, H., Gao, W., Zhang, L., Ye, Y., Yuan, L., Ding, Z., Wu, J., Kang, L., Zhang, X., Wang, X., Zhang, G., Gong, H., Sun, A., Yang, X., Chen, R., Cui, Z., Ge, J., Zou, Y. (2018). MicroRNA-378 suppresses myocardial fibrosis through a paracrine mechanism at the early stage of cardiac hypertrophy following mechanical stress.. Theranostics, 8(9), 2565-2582. https://doi.org/10.7150/thno.22878.
ACS
Yuan, J.; Liu, H.; Gao, W.; Zhang, L.; Ye, Y.; Yuan, L.; Ding, Z.; Wu, J.; Kang, L.; Zhang, X.; Wang, X.; Zhang, G.; Gong, H.; Sun, A.; Yang, X.; Chen, R.; Cui, Z.; Ge, J.; Zou, Y. MicroRNA-378 suppresses myocardial fibrosis through a paracrine mechanism at the early stage of cardiac hypertrophy following mechanical stress.. Theranostics 2018, 8 (9), 2565-2582. DOI: 10.7150/thno.22878.
NLM
Yuan J, Liu H, Gao W, Zhang L, Ye Y, Yuan L, Ding Z, Wu J, Kang L, Zhang X, Wang X, Zhang G, Gong H, Sun A, Yang X, Chen R, Cui Z, Ge J, Zou Y. MicroRNA-378 suppresses myocardial fibrosis through a paracrine mechanism at the early stage of cardiac hypertrophy following mechanical stress.. Theranostics 2018; 8(9):2565-2582. doi:10.7150/thno.22878. https://www.thno.org/v08p2565.htm
CSE
Yuan J, Liu H, Gao W, Zhang L, Ye Y, Yuan L, Ding Z, Wu J, Kang L, Zhang X, Wang X, Zhang G, Gong H, Sun A, Yang X, Chen R, Cui Z, Ge J, Zou Y. 2018. MicroRNA-378 suppresses myocardial fibrosis through a paracrine mechanism at the early stage of cardiac hypertrophy following mechanical stress.. Theranostics. 8(9):2565-2582.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.