Theranostics
13.3
Impact Factor
- Current Issue
- Advance Articles
- Volume 16; 2026
- Volume 15; 2025
- Volume 14; 2024
- Volume 13; 2023
- Volume 12; 2022
- Archive
- Cover Images
- Cover Suggestion
- Special Issues
Top
Introduction
Results
Discussion
Methods
Abbreviations
Supplementary Material
Acknowledgements
References
Introduction
Results
Discussion
Methods
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2018; 8(11):3138-3152. doi:10.7150/thno.21693 This issue Cite
Research Paper
Negative regulation of cationic nanoparticle-induced inflammatory toxicity through the increased production of prostaglandin E2 via mitochondrial DNA-activated Ly6C+ monocytes
Li Liu*, Yantong Liu*, Bocheng Xu, Chuyu Liu, Yanpeng Jia, Ting Liu, Chunju Fang, Wei Wang, Jun Ren, Zhiyao He, Ke Men, Xiao Liang, Min Luo, Bin Shao, Ye Mao, Henyi Xiao, Zhiyong Qian, Jia Geng, Birong Dong, Peng Mi, Yu Jiang, Yuquan Wei, Xiawei Wei ![]()
Lab of Aging Research and Nanotoxicology, State Key Laboratory of Biotherapy, West China Hospital, Sichuan University and Collaborative Innovation Center, No. 17, Block 3, Southern Renmin Road, Chengdu, Sichuan 610041, PR China
*These authors contributed equally to this work.
Received 2017-6-29; Accepted 2018-4-13; Published 2018-5-6
Citation:
Liu L, Liu Y, Xu B, Liu C, Jia Y, Liu T, Fang C, Wang W, Ren J, He Z, Men K, Liang X, Luo M, Shao B, Mao Y, Xiao H, Qian Z, Geng J, Dong B, Mi P, Jiang Y, Wei Y, Wei X. Negative regulation of cationic nanoparticle-induced inflammatory toxicity through the increased production of prostaglandin E2 via mitochondrial DNA-activated Ly6C+ monocytes. Theranostics 2018; 8(11):3138-3152. doi:10.7150/thno.21693. https://www.thno.org/v08p3138.htm
Other stylesAbstract

Rationale: Cationic nanocarriers present with well-known toxicities, including inflammatory toxicity, which limit their clinical application. How the cationic nanocarrier-induced inflammatory response is negatively regulated is unknown. Herein, we found that following a sublethal dose of cationic nanocarriers, the induced inflammatory response is characterized by early neutrophil infiltration and spontaneous resolution within 1 week.
Methods: C57BL/6 mice were intravenously injected with a dosage of 1-100 mg/kg cationic DOTAP liposomes as well as other cationic materials. Cell necrosis was detected by flow cytometry. Release of mitochondrial DNA was quantified by qPCR via Taqman probes. Signal proteins were detected by Western blotting. PGE2 production in the supernatant was quantitated using an enzyme immunoassay (EIA). The infiltrated inflammatory cells were observed in WT mice, Ccr2-/- mice, Sting-/-mice and Tlr9-/-mice.
Results: The early stage (24-48 h) inflammatory neutrophil infiltration was followed by an increasing percentage of monocytes; and, compared with WT mice, Ccr2-/- mice presented with more severe pulmonary inflammation. A previously uncharacterized population of regulatory monocytes expressing both inflammatory and immunosuppressive cytokines was identified in this model. The alteration in monocyte phenotype was directly induced by mtDNA release from cationic nanocarrier-induced necrotic cells via a STING- or TLR9-dependent pathway. Neutrophil activation was specifically inhibited by PGE2 from Ly6C+ inflammatory monocytes, and intravenous injections of dual-phenotype monocytes beneficially modified the immune response; this inhibitory effect was abolished after treatment with indomethacin. Moreover, we provide clear evidence that mitochondrial DNA activated Ly6C+ monocytes and increased PGE2 production through TLR9- or STING-mediated MAPK-NF-κB-COX2 pathways.
Conclusion: Our findings suggest that Ly6C+ monocytes and mtDNA-induced Ly6C+ monocyte PGE2 production may be part of a feedback mechanism that contributes to the resolution of cationic nanocarrier-induced inflammatory toxicity and may have important implications for understanding nanoparticle biocompatibility and designing better, safer drug delivery systems.
Keywords: cationic nanocarriers, inflammatory toxicity, prostaglandin E2, monocytes, mitochondrial DNA (mtDNA)
Introduction
Materials-based nanotherapeutics substantially impact clinical medicines such as cancer therapy, vaccine development, and tissue repair and regeneration [1]. Nanoparticle-based experimental therapeutics are currently being investigated in numerous human clinical trials [2]. The potential adverse effects of nanoparticle delivery systems have always been the major concern in preclinical studies and clinical evaluations. Surface charge is one of the important characteristics of nanoparticles [3], which influences their cellular uptake and cytotoxicity [4-7]. Due to their positively charged surface, cationic nanocarriers load and condense nucleic acids simply via electrical interactions with the anionic nucleic cargo [8]. Thus, cationic nanoparticles, including cationic liposomes, polyethylene imines (PEI), and chitosan, are most commonly used in gene delivery as non-viral vectors for nucleic acids [9-13]. It is generally accepted that cationic nanoparticles are more cytotoxic and inflammatory than neutral or negative nanoparticles [14, 15].
Numerous cationic nanoparticles have emerged as potential delivery alternatives in the basic research and preclinical studies; however, a limited number of nanoparticles have been clinically evaluated [16-20]. Nanoparticle toxicity following systemic administration has always been a barrier to the successful translation of cationic nanoparticles into the clinic [8, 21, 22]. In fact, most clinical studies based on cationic liposomal or cationic polymer delivery systems have reported adverse events, such as fever, fatigue, and chills, related to inflammatory toxicity [23-25]. For example, in a study with nineteen patients, the most common treatment-related adverse events of any grade were fatigue (n=12), chills (n=12), and fever (n=10). Importantly, a total of five (21%) patients withdrew from the study due to an adverse event. In some clinical trials, before the systemic administration of a cationic liposomal or cationic polymer drug delivery system, a pretreatment regimen, including oral diphenhydramine, oral famotidine, and intravenous methylprednisolone, was required to reduce the risk of adverse events related to inflammatory toxicity. Cytokines such as tumour necrosis factor-α (TNF-α), interleukin-6 (IL-6), interleukin-1β (IL-1β) and interferon-γ (IFN-γ) are increased in the serum after treatment with cationic delivery systems [26]. In addition, local administration, such as through the airway, also appears to induce an inflammatory response [27]. Although the apparent inflammatory response in these patients may be caused by the drugs in the cationic nanoparticles, the response appeared transient, often lasting 12-24 h, indicating that the inflammatory response triggered by the cationic nanoparticles might be negatively regulated by the immune system.
Inflammatory toxicity has also been found in animal testing, and the mechanism underlying this toxicity has been explored [28-30]. Local lipoplex administration (by nebulization or intratracheal instillation) triggered apparent inflammation for hours in the lungs, as evidenced by an increase in several kinds of proinflammatory cytokines and by the accumulation of leukocytes [30]. Immediately after systemic administration, cationic nanocarriers, such as liposomes and PEI, have also been reported to accumulate in the lungs to a great extent, which has gained attention [31-33]. Recently, our group has reported that cationic nanoparticles, such as liposomes, PEI and chitosan, cause a severe pulmonary inflammatory response that is characterized by early neutrophil infiltration following systemic administration [30]. This inflammation was also observed in the liver and other tissues to a lesser degree. Furthermore, we explored the possible mechanism by which inflammatory toxicity is triggered by cationic nanoparticles. We found that the cationic nanoparticles induced acute cell necrosis via interacting with Na+/K+-ATPase and subsequently caused the release of mitochondria and mitochondrial contents, such as mtDNA, from necrotic cells as damage associated molecular patterns (DAMPs) to further stimulate the innate immune response [30]. Our unpublished preliminary findings during these observations revealed that within 1 week following a sublethal dose, cationic nanocarriers induced an inflammatory response that was characterized by early neutrophil infiltration and that was gradually, spontaneously resolved.
On the basis of the clinical and experimental findings mentioned above, it is conceivable that cationic nanoparticles not only trigger an inflammatory response but also simultaneously negatively regulate the inflammatory response, which in turn leads to inflammation resolution. However, how the cationic nanocarrier-induced inflammatory response is negatively regulated and the mechanisms underlying the inflammation resolution are still unknown. Therefore, research investigating the regulation and resolution of cationic nanoparticle-induced inflammation is important for identifying the possible mechanisms underlying this toxicity, for providing new strategies that prevent toxicity and for promoting the application of this nanotechnology in biomedical fields.
Results
Inflammation resolution and Ly6C+ monocyte recruitment in cationic nanoparticle-induced inflammation
To investigate the regulation and remission of cationic nanocarrier-induced acute inflammation, we continuously monitored the inflammation for days and found that the inflammation was often resolved within 1 week following the injection of a single sublethal dose of typical cationic nanocarriers, including DOTAP liposomes, PEI and chitosan, as shown by H&E staining (Figure 1A). At 24 h after mice were treated with cationic nanoparticles, notable pathological changes were observed, and the severe inflammatory response was accompanied by an influx of an abundance of inflammatory cells (Figure 1A-B). The complex with cargo DNA also induced inflammation response and inflammatory cells infiltrating (Figure S5 and Figure S16). To characterize the cationic nanocarrier-induced inflammation, flow cytometric analysis was performed to determine the percentage of CD45+CD11b+Ly6G+ inflammatory neutrophils and CD45+CD11b+Ly6C+ inflammatory monocytes in the lung tissues; an increased number of inflammatory neutrophils and monocytes were recruited to the lungs 48 h after the intravenous injection of cationic nanocarriers (Figure 1C-F). Next, we investigated the dynamics between inflammatory neutrophils and monocytes to identify the predominant cells that regulate the cationic nanocarrier-induced inflammation in the lung. The results revealed that many inflammatory neutrophils infiltrated during the early stage (24-48 h) of inflammation, and then, an increasing percentage of inflammatory monocytes infiltrated from 48-72 h (Figure 1G), suggesting that the number of inflammatory neutrophils decreases after the inflammatory monocyte infiltration.
To investigate whether the infiltrating monocytes play a role in the regulation of the cationic liposome-induced pulmonary inflammation, we utilized Ccr2-/-mice, which lack the chemokine receptor CCR2 and cannot recruit Ly6C+ inflammatory monocytes from the bone marrow during inflammation. Interestingly, 48 h after the administration of cationic liposomes, the pulmonary inflammation was more severe in the Ccr2-/- mice than in the WT mice (Figure 1H-J). The number of Ly6C+MHCII+ monocytes and inflammatory neutrophils (Ly6G+) in the lungs was significantly different between the Ccr2-/-mice and WT mice: more inflammatory neutrophils were detected in the Ccr2-/-mice than in the WT mice (Figure S2A-C). Monocytes isolated from WT mice bone marrow were injected into the cationic nanocarrier-treated Ccr2-/- mice, and the pulmonary inflammation was reduced (Figure 1K-L). Thus, these results suggest a possible immunosuppressive effect of infiltrating monocytes in acute pulmonary inflammation, which might contribute to negative regulation of the neutrophil-dominated immune response.
The dual monocyte phenotypes are induced by necrotic cells and mtDNA during the inflammatory response
To further investigate the possible immunosuppressive effect of infiltrating monocytes, the infiltrating monocyte phenotype was characterized during acute pulmonary inflammation. In the present study, during acute pulmonary inflammation, we detected a regulatory monocyte population, and this population and IL-10 expression were significantly increased from 48-72 h following the cationic nanoparticle treatment, as characterized by flow cytometry (Figure 2A-B and Figure 3A). The Ly6C+ monocyte phenotypes were analysed by intracellular cytokine staining for IL-10 in combination with TNF-α, IL-6 and IFN-γ. The results revealed that DOTAP liposomes and other cationic nanocarriers including PEI and chitosan induced the dual monocyte phenotype during the pulmonary inflammatory response 48 h after injection (Figure 3B-C, E), which caused us to investigate the mechanism underlying this monocyte phenotype alteration.
We have previously reported that cationic nanoparticles induce pulmonary inflammation by triggering cell necrosis, and the resulting necrotic cells, specifically the released cytoplasmic damage-associated molecular patterns (DAMPs) such as mtDNA, are responsible for the inflammatory response. The induction of cell necrosis by cationic nanocarriers, the release of mtDNA from cationic particle-induced necrotic cells and the promotion of inflammation via the injection of necrotic cells or mtDNA were also confirmed in this study (Figure S8 and Figure 3D). Inflammatory neutrophils and monocytes were significantly increased after the treatment with necrotic, cationic nanoparticle-induced lung cells (Figure 2C-D). To further investigate how inflammatory monocytes elicit the dual phenotype, we investigated whether the regulatory phenotype is related to the necrotic cells or mtDNA release caused by cationic nanocarriers. Bone marrow- or lung-derived monocytes were incubated with necrotic cells prepared from mice treated with cationic nanoparticles and were detected via flow cytometry. The monocytes were characterized by an increase in the expression of inflammatory cytokines, including TNF-α, IL-6 and IFN-γ, as well as an increased regulatory, IL-10-expressing monocyte population after the co-culture with the necrotic cells (Figure 2E).
Similarly, the number of dual-phenotype monocytes was significantly increased after the monocytes were treated with mitochondrial DNA both in vitro and in vivo (Figure 3F-I). Moreover, the level of PGE2, another effective immunosuppressive factor produced by monocytes, was also increased in the monocyte supernatant after co-culturing with mtDNA (Figure 3J). As a control, monocytes were stimulated with artificially synthesized CpG1668, which also increased the intracellular cytokines IL-10, TNF-α, IL-6 and IFN-γ to a similar level (Figure 3K). Together, these results revealed a population of dual-phenotype monocytes with possible immunesuppressive effects in the cationic nanoparticle-induced pulmonary inflammation model; in addition, mtDNA, an immunostimulatory factor in cationic nanoparticle-induced inflammation, also plays a key role in reprogramming monocytes into an immunoregulatory phenotype during this process.
Figure 1
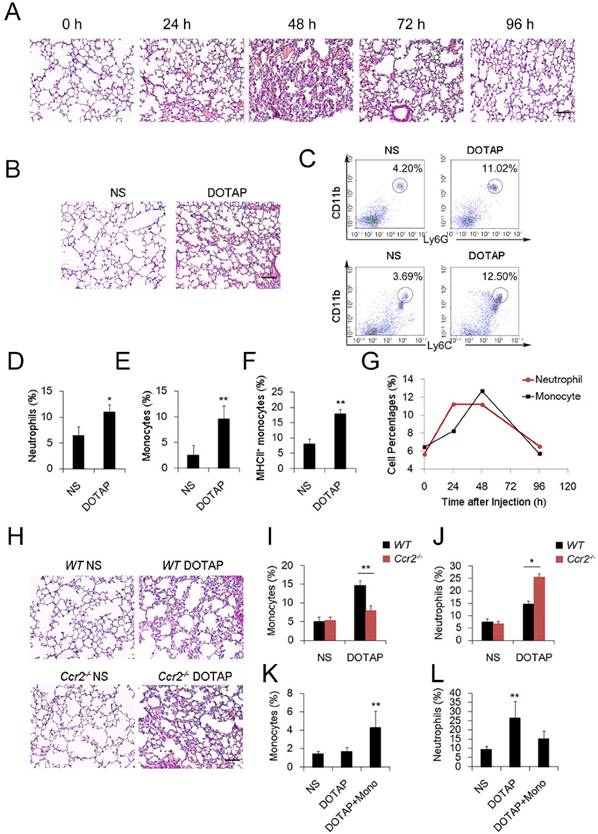
Inflammatory monocyte and neutrophil recruitment and regulation of cationic nanoparticle-induced pulmonary inflammation. C57BL/6 mice were treated with intravenous injections of DOTAP liposomes (25 mg/kg) or with normal saline as a control treatment. (A) Representative haematoxylin-eosin (H&E)-stained sections of lung tissue at 24, 48, and 96 h after mice were treated with DOTAP liposomes (n=5); scale bars=50 μm. (B) Representative H&E-stained sections of lung at 24 h after DOTAP liposome treatment (n=5 per group); scale bars=50 μm. (C) Flow cytometric analysis of recruited myeloid cells in the lung at 48 h after the DOTAP liposome dose. Numbers indicate either CD45+CD11b+Ly6C+ monocytes or CD45+CD11b+Ly6G+ neutrophils in the quadrant and are expressed as the percentage of total cells within the same lung (n=5). (D-E) Bar graph summarizing the average percentage of inflammatory monocyte and neutrophil influx into the lung (n=3). (F) Flow cytometric assessment of recruited MHCII+ inflammatory monocytes. Cells were gated on CD45+CD11b+Ly6C+ monocytes. (G) Infiltrating inflammatory monocytes and neutrophils were detected by flow cytometry at 24, 48 and 96 h after DOTAP liposome administration (n=4). (H) Representative H&E-stained sections of lung from C57BL/6 or Ccr2-/- mice treated with DOTAP liposomes (n=4); scale bar=50 μm. (I-J) Percentage of infiltrating neutrophils and monocytes in the lungs of C57BL/6 and Ccr2-/- mice treated with DOTAP liposomes. (K-L) Monocytes isolated from C57BL/6 mice bone marrow were injected into the cationic liposome-treated Ccr2-/- mice, and the monocytes and neutrophils were detected by flow cytometry. Data are representative of three independent experiments, and the results are expressed as the mean ± S.E.M. Statistical comparisons were performed using Student's t-test or Dunnet's t-test (*P<0.05; **P<0.01).
Figure 2
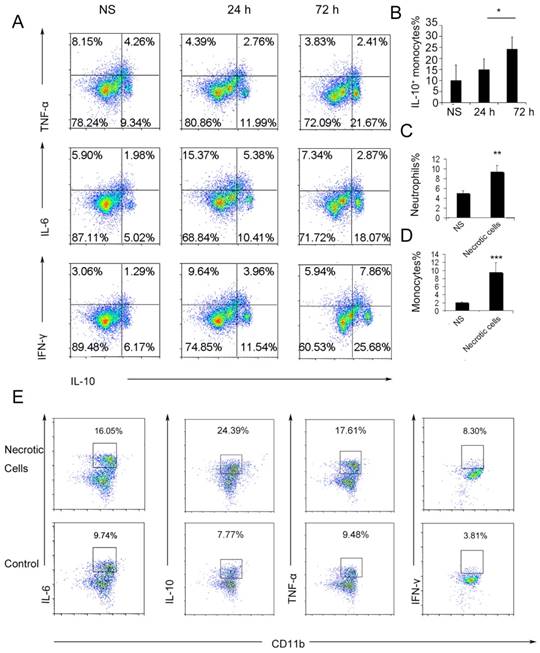
Ly6C+ inflammatory monocytes adopted a dual phenotype after being stimulated with necrotic lung cells. (A) DOTAP liposomes or normal saline as a control were injected into C57BL/6 mice, and the mice were sacrificed 24 or 72 h after the injection. The intracellular cytokines of Ly6C+ inflammatory monocytes were analysed by flow cytometry. First, the cells were gated on CD45+CD11b+Ly6C+ monocytes. Intracellular cytokine staining for IL-10 in conjunction with TNF-α, IL-6 and IFN-γ was conducted. Numbers represent the percentage of cells in each gate (n=9). (B) Average percentage of IL-10+ inflammatory monocytes in the control and at 24 and 72 h after dosing (n=9). (C-D) Analysis of inflammatory neutrophils and monocytes 48 h after an intravenous injection of 1×106 necrotic lung cells (n=3). (E) Monocytes isolated from bone marrow were stimulated with necrotic lung cells for 4 h in the presence of brefeldin A. Intracellular cytokine staining for IL-10, TNF-α, IL-6 and IFN-γ; first, the cells were gated on CD45+CD11b+Ly6C+ monocytes (n=3). Data are representative of three independent experiments, and the results are expressed as the mean ± S.E.M. Statistical comparisons were performed using Student's t-test or Dunnet's t-test (*P<0.05; **P<0.01; ***P<0.005).
Regulatory monocytes inhibit inflammation and suppress neutrophils via PGE2 production
After characterizing the phenotypic changes of monocytes in cationic particle-induced pulmonary inflammation and demonstrating that the acquisition of a regulatory monocyte phenotype is the result of mtDNA-driven activation, we next investigated how these regulatory monocytes affect neutrophil behaviour during inflammation. The suppressive effect of dual-phenotype Ly6C+ monocytes on neutrophil activation was observed by co-culturing neutrophils and monocytes together with either mtDNA or necrotic lung cells; stimulation with necrotic cells or mtDNA significantly reduced the percentage of TNF-α+ neutrophils (Figure 4A-C).
Small intestine lamina propria Ly6C+ monocytes and macrophages can reduce inflammation by producing PGE2 in T. gondii infections and in caecal ligation and puncture (CLP) models [34, 35]. In our study, the increased release of PGE2 from monocytes was observed following mtDNA stimulation (Figure 3J). Likewise, we confirmed the ability of PGE2 to inhibit neutrophil activation and inflammation both in vitro and in vivo (Figure 4D-F). To further study whether monocytes regulate and attenuate the pulmonary inflammatory response via producing PGE2, we pretreated monocytes with indomethacin, a COX-1 and COX-2 inhibitor, to inhibit PGE2 production. We found that indomethacin abolished the capability of monocytes to inhibit neutrophil activation both in vitro and in vivo (Figure 4G-H and Figure S8). This suggests that PGE2 plays an important role in the regulation of cationic particle-induced acute pulmonary inflammation via infiltrating monocytes.
Since IL-10 expression in monocytes might also be essential for regulating the inflammatory response, we utilized IL-10-/-mice to study whether IL-10 affects neutrophil activation. Monocytes isolated from WT and IL-10-/- mouse bone marrow were primed with mtDNA and were injected into cationic liposome-treated mice to observe the effect on pulmonary inflammation. WT monocytes and IL-10-/-monocytes both efficiently reduced pulmonary inflammation, as evidenced by the decreased number of neutrophils, specifically, infiltrating inflammatory TNF-α+ neutrophils (Figure S3C and Figure 4J-K). PGE2 was found in the supernatant of monocytes isolated from IL-10-/- mouse bone marrow and co-cultured with mtDNA (Figure 4I). However, this inhibitory effect of monocytes was abolished after treatment with indomethacin (Figure 4L and Figure S3B), demonstrating that monocyte production of PGE2, but not IL-10, plays a key role in the regulation of cationic nanocarrier-induced pulmonary inflammation. In addition to inhibiting inflammatory neutrophil activation, the IL-10 released by monocytes might participate in the regulation of cationic particle-induced inflammation in other ways.
Figure 3
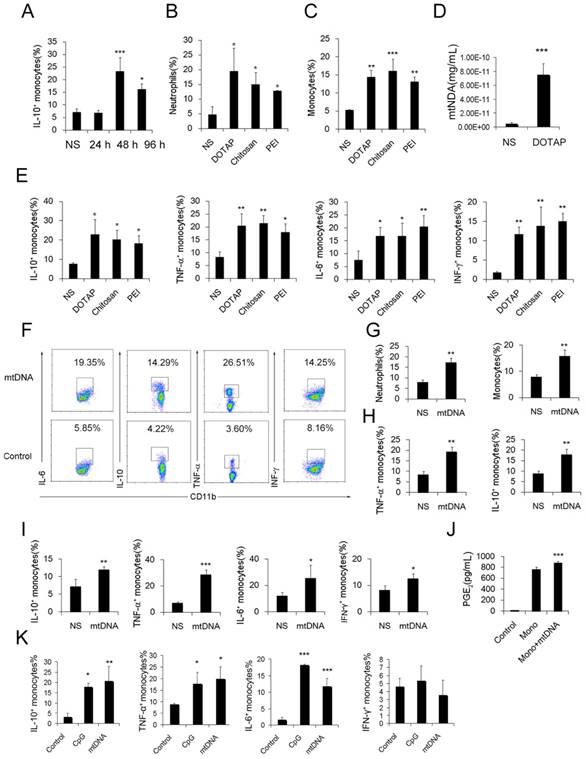
Regulatory Ly6C+ inflammatory monocytes are increased in the lungs after the cationic liposome dose, and mtDNA-stimulated Ly6C+ inflammatory monocytes adopted a dual phenotype. (A) C57BL/6 mice were injected with DOTAP liposomes or normal saline as a control and were sacrificed 24, 48 or 96 h after the injection. Cells were first gated on CD45+CD11b+Ly6C+ monocytes, and intracellular cytokine staining for IL-10 was performed. (B-C, E) C57BL/6 mice were treated with DOTAP liposomes (25 mg/kg), PEI (5 mg/kg) or chitosan (25 mg/kg); 48 h after the injection, the number of neutrophils and monocytes and the intracellular levels of IL-10, TNF-α, IL-6 and IFN-γ in inflammatory monocytes were determined by flow cytometry (n=5). (D) The mtDNA level in the sera of mice treated with DOTAP liposomes was determined by qPCR at 6 h after the injection (n=3). (F) Monocytes isolated from bone marrow were cultured with mtDNA (5 μg/mL) for 4 h in the presence of 4 μg/mL brefeldin A, a Golgi-blocking agent. Intracellular cytokine staining for IL-10, TNF-α, IL-6 and IFN-γ; the cells were first gated on CD45+CD11b+Ly6C+ monocytes. (G-H) C57BL/6 mice were injected with mtDNA (5 μg/mouse) or normal saline as a control and sacrificed 48 h after injection. Flow cytometric analysis was conducted. Neutrophils and inflammatory monocytes in the lungs of the mice treated with mtDNA or normal saline; IL-10 and TNF-α expression in inflammatory monocytes (CD45+CD11b+Ly6C+) (n=4). (I) Monocytes isolated from bone marrow were stimulated with mtDNA (5 μg/mL) for 4 h in the presence of brefeldin A. Intracellular cytokine staining for IL-10, TNF-α, IL-6 and IFN-γ (n=3). (J) EIA for PGE2 in the supernatants of bone marrow monocytes cultured alone or with mtDNA for 20 h (n=3). (K) Monocytes were stimulated with mtDNA or CpG1668 in the presence of brefeldin A; intracellular cytokine staining for IL-10, TNF-α, IL-6 and IFN-γ (n=3). Data are representative of three independent experiments, and the results are expressed as the mean ± S.E.M. Statistical comparisons were performed using Student's t-test or Dunnet's t-test (*P<0.05; **P<0.01; ***P<0.005).
Increased PGE2 production through TLR9- or STING-mediated MAPK-NF-κB-COX2 pathway activation via mtDNA alone
To demonstrate the pathways required for mtDNA to induce the PGE2-expressing inhibitory monocyte phenotype, Sting-/-mice and Tlr9-/-mice were used. The stimulation of interferon genes is essential for the host defence against DNA pathogens [36], and we hypothesized this might also be important for the recognition of mtDNA as a DAMP in the cationic nanocarrier-induced pulmonary inflammation model. The toll-like receptor 9 (TLR9) pathway has already been shown to play a key role in neutrophil activation in mtDNA-induced inflammation and was used as a control in this study. Interestingly, mtDNA-induced pulmonary inflammation was also alleviated in Sting-/-mice, suggesting that STING pathways might contribute to the ability of immune cells to recognize mtDNA (Figure 5A). In addition, Sting-/- mice and Tlr9-/-mice presented with a decreased percentage of infiltrating neutrophils and monocytes after the cationic liposome injection (Figure 5B-C). Sting-/- mice also presented with a decreased percentage of infiltrating TNF-α+neutrophils and IL-10+ monocytes following the cationic liposome injection (Figure 5D-E). The reduced inflammation was also confirmed by injecting necrotic cells and mtDNA into WT and Sting-/-mice (Figure 4F-G).
Figure 4
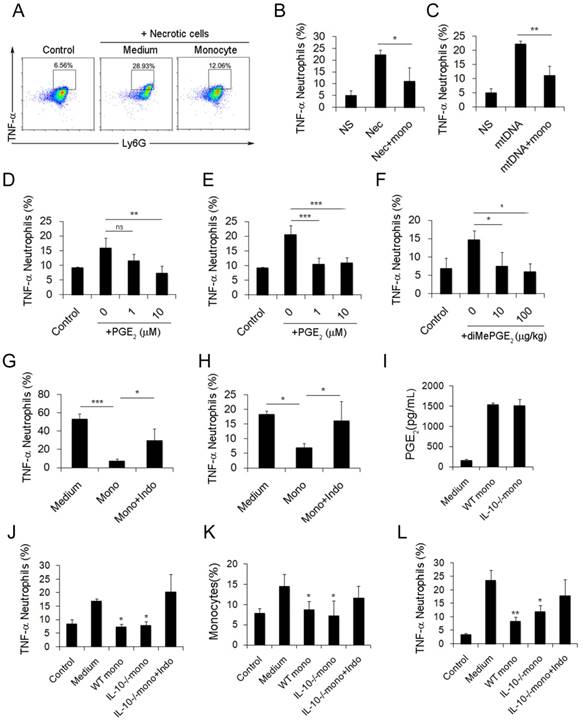
Monocytes reduce the inflammatory response induced by necrotic lung cells and DOTAP liposomes via PGE2 production. (A-C) Intracellular cytokine staining for TNF-α produced by neutrophils (CD45+CD11b+Ly6G+) stimulated with necrotic lung cells or mtDNA and cultured with control medium or monocytes. Numbers represent the percentage of cells in each gate. (D) TNF-α produced by neutrophils stimulated with necrotic lung cells and treated with increasing concentrations of purified PGE2 (n=3). (E) TNF-α produced by neutrophils stimulated with mtDNA and treated with increasing concentrations of purified PGE2 (n=3). (F) TNF-α produced by neutrophils (CD45+CD11b+Ly6G+) in the lungs of mice injected with DOTAP liposomes and treated with increasing concentrations of diMePGE2 (n=3). (G) TNF-α produced by neutrophils stimulated by necrotic lung cells and cultured with monocytes or indomethacin (indo; 10 μM) (n=3). (H) Monocytes from bone marrow were stimulated with mtDNA for 20 h in the presence of indomethacin or vehicle and were injected into DOTAP liposome-treated mice. TNF-α production by neutrophils (CD45+CD11b+Ly6G+) in the lungs was analysed by flow cytometry (n=3). (I) WT and IL-10-/- monocytes were isolated from C57BL/6 and IL-10-/-mouse bone marrow. EIA test for PGE2 in the supernatants from WT monocytes, IL-10-/- monocytes and control medium (n=3). (J) TNF-α expression in inflammatory neutrophils (CD45+CD11b+Ly6G+) stimulated with mtDNA for 4 h and cultured with WT monocytes, IL-10-/- monocytes and IL-10-/- monocytes treated with indomethacin (10 μM), as determined by flow cytometry (n=3). (K-L) After being stimulated with mtDNA for 20 h, WT monocytes, IL-10-/- monocytes and IL-10-/- monocytes treated with indomethacin (10 μM) were injected into C57BL/6 mice after DOTAP liposome administration. Mice were sacrificed 48 h after the injection; the number of monocytes (CD45+CD11b+Ly6C+) in the lung and the TNF-α produced by neutrophils were determined by flow cytometry (n=3). Data represent three independent experiments, and the results are expressed as the mean ± S.E.M. Statistical comparisons were performed using Student's t-test or Dunnet's t-test (*P<0.05; **P<0.01; ***P<0.005)
Next, we investigated whether the mtDNA-induced activation of regulatory monocytes was also associated with the STING and TLR9 pathways. Sting-/- and Tlr9-/- monocytes presented with decreased PGE2 release in response to mtDNA treatment (Figure 5H). To confirm the roles of the STING and TLR9 pathways in producing PGE2 in mtDNA-stimulated monocytes, we determined the expression of cyclooxygenase (COX-2) and COX1 by Western blotting; Sting-/- and Tlr9-/- monocytes presented with a decreased expression of COX2, and mtDNA did not affect COX1 expression (Figure 5I). Next, we investigated the involvement of the p38 MAPK and p65 NF-κB signalling pathways, which are important components of TLR9-induced signalling pathways; mtDNA induced the phosphorylation of p38 MAPK and p65 NF-κB. As shown in Figure 5I, the phosphorylation of p38 MAPK and p65 NF-κB was significantly decreased in Sting-/- and Tlr9-/- monocytes.
Figure 5
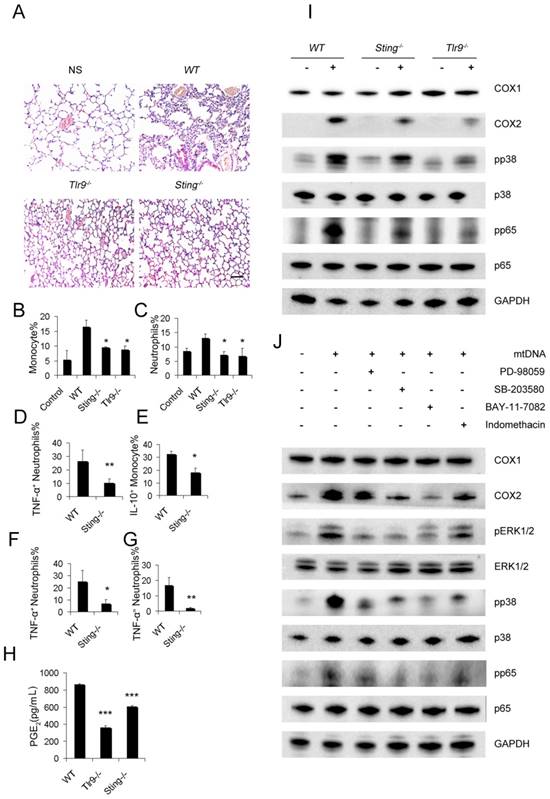
STING and TLR9 pathways are essential for the cationic liposome-induced activation of pulmonary inflammation. WT, Sting-/- and Tlr9-/- mice were treated with DOTAP liposomes for 48 h. (A) Representative H&E staining of lung sections of mice treated with DOTAP liposomes (n=5); scale bar=50 μm. (B) Inflammatory monocyte (CD45+CD11b+Ly6C+) influx into the lungs of mice treated with DOTAP liposomes (n=5). (C) Inflammatory neutrophil (CD45+CD11b+Ly6G+) influx into the lungs of mice treated with DOTAP liposomes (n=5). (D) TNF-α expression in the inflammatory neutrophils (CD45+CD11b+Ly6G+) in the lungs of WT and Sting-/-mice (n=3). (E) IL-10 production in inflammatory monocytes (CD45+CD11b+Ly6C+) in the lungs of WT and Sting-/-mice (n=3). (F) Neutrophils were isolated from the bone marrow of WT and Sting-/-mice and were stimulated with necrotic lung cells or mtDNA (G) for 4 h in the presence of brefeldin A. TNF-α expression in inflammatory neutrophils (CD45+CD11b+Ly6G+) was determined by flow cytometry, (n=3). (H) Monocytes were isolated from the bone marrow of WT, Tlr9-/- and Sting-/-mice and were cultured with mitochondrial DNA (5 μg/mL) at a concentration of 5×106 cells/mL; EIA assays were conducted for PGE2 expression in supernatants (n=3). (I) Western blot analysis was performed. (J) The freshly isolated monocytes were cultured with mtDNA (5 μg/mL) at a concentration of 5×106 cells/mL. The inhibitors PD98059 (30 μM), SB203580 (10 μM), BAY117082 (10 μM) and indomethacin (10 μM) were added to the cells, and the cells were incubated for 2 h at 37 °C, then, Western blot analysis was performed. Data are representative of three independent experiments, and the results are expressed as the mean ± S.E.M. Statistical comparisons were performed using Student's t-test or Dunnet's t-test (*P<0.05; **P<0.01; ***P<0.005).
To determine the role of mtDNA-induced p38 MAPK and p65 NF-κB activation in COX-2 and PGE2 expression, the p44/42 MAPK inhibitor PD98059, p38 MAPK inhibitor SB203580, p65 NF-κB inhibitor BAY117082 and indomethacin were used. Western blotting assays showed that mtDNA-induced COX2 expression was significantly inhibited by PD98059, SB203580, BAY117082 and indomethacin (Figure 5J). We also assessed the phosphorylation of p44/42 MAPK, p38 MAPK and p65 NF-κB and evaluated the effects of these inhibitors on mtDNA-induced p38 MAPK and p65 NF-κB activation. Taken together, these data clearly revealed that mtDNA-induced COX2 expression was mediated by p38 MAPK- and p65 NF-κB-dependent signalling pathways (Figure 5J).
Thus, we conclude that the STING and TLR9 pathways are responsible for inducing both the inflammatory and regulatory phenotypes of monocytes in cationic nanoparticle-induced pulmonary inflammation. Together, these results demonstrated that mtDNA triggers suppressive monocyte phenotypes via the simultaneous activation of the STING and TLR9 pathways, which are also essential for the initiation of cationic-nanoparticle (mtDNA)-induced acute inflammation. An outline of the regulation of inflammation induced by the immunomodulatory complex released from cationic nanocarrier-induced necrotic cells is shown in Figure 6.
Figure 6
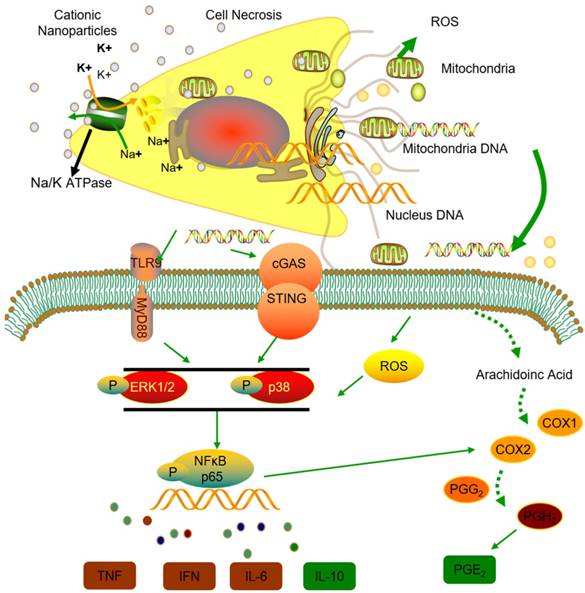
Model of the regulation of inflammation induced by the immunomodulatory complex released from cationic nanocarrier-induced necrotic cells. The cationic nanoparticles induced acute cell necrosis via interacting with Na+/K+-ATPase and caused the subsequent release of mitochondria and mitochondrial contents such as mtDNA from the necrotic cells as damage associated molecular patterns (DAMPs) to further stimulate the innate immune response. Here, we show the mechanism involved in the regulation of Ly6C+ monocyte inflammation. Mitochondrial DNA activated the p38 MAPK and p65 NF-κB signalling responses via the STING and TLR9 pathways and increased PGE2 production in monocytes through STING- or TLR9-mediated MAPK-NF-κB-COX2 pathways. In addition, p38 MAPK and NF-kB activation are involved in COX-2 synthesis. The increased production of PGE2 in mtDNA-induced Ly6C+ inflammatory monocytes contributes to the regulation and resolution of the inflammatory response.
Discussion
The negative regulation of the inflammatory response that protects the host from tissue damage in a cationic nanoparticle-induced inflammatory environment and the mechanisms underlying the inflammation resolution remain largely unexplored. In the current study, several observations were made concerning cationic nanoparticles, inflammation resolution, immunosuppressive monocytes, PGE2 production and mtDNA. We found that the inflammatory response is negatively regulated to protect the host from tissue damage in a cationic nanoparticle-induced inflammatory environment. Moreover, Ly6C+ inflammatory monocytes and their increased mtDNA-induced production of PGE2 may contribute to the regulation and resolution of the inflammatory response caused by cationic nanoparticles. mtDNA-induced activation of TLR9 or STING signalling may upregulate the genes involved not only in the promotion of inflammation but also in the resolution of inflammation. Thus, the increased production of PGE2 induced by mtDNA may be a part of a feedback mechanism that attenuates the inflammatory toxicity caused by TLR stimulation or STING. This possibility was supported by our findings. The current findings revealed that, following a sublethal dose of cationic nanocarriers, the induced inflammatory response is characterized by early neutrophil infiltration that is gradually resolved within 96 h or 1 week; inflammatory neutrophil infiltration in the early stage (24-48 h) is followed by an increasing percentage of monocytes in the acute inflammation of the lung; and, after cationic nanoparticle treatment, pulmonary inflammation is more severe in Ccr2-/- mice than in WT mice, due to the inability of Ccr2-/- mice to recruit bone-marrow monocytes during inflammation. Our work addressed a previously uncharacterized population of regulatory monocytes in a model of cationic nanoparticle-induced acute pulmonary inflammation, which is characterized by the expression of both inflammatory (TNF-α, IL-6 and IFN-γ) and immunosuppressive cytokines (IL-10 and PGE2). The alteration in the monocyte phenotype was directly induced by mtDNA released from cationic carrier-induced necrotic cells, and the STING and TLR9 pathways are responsible for inducing the dual phenotype in monocytes. Neutrophil activation is specifically inhibited by PGE2 from Ly6C+ inflammatory monocytes, and intravenous injections of dual-phenotype monocytes beneficially modified the immune response. The inhibitory effect of monocytes was abolished after the indomethacin-induced inhibition of PGE2 production. Increased PGE2 production was induced through TLR9- or STING-mediated MAPK-NF-κB-COX2 pathway activation via necrotic cells or mtDNA alone. In summary, our findings above suggest that the cationic nanocarrier-induced inflammatory response was negatively regulated and that this inflammatory response may be resolved via the increased production of PGE2 by Ly6C+ inflammatory monocytes that are activated by the mtDNA released from necrotic cells, which is triggered by cationic nanocarriers.
Mitochondria DNA is generally known as an immunostimulatory molecule and is released from necrotic cells after cell injury to activate the innate immune response [37]. The cationic polysaccharide chitosan also stimulates mitochondrial stress, ROS production, and mtDNA release and promotes cellular immunity via the DNA sensor cGAS-STING-dependent induction of type I interferon [38, 39]. Previously, we found that mtDNA released from necrotic cells triggered by cationic nanoparticles could activate neutrophils to induce acute inflammation in the lung [30]. The regulatory function of Ly6C+ inflammatory monocytes is activated by immunostimulatory factors, such as lipopolysaccharides (LPS) [40] or infections [41, 42]. However, the negative regulation of the immune response triggered by mtDNA has not been reported.
To our knowledge, this is the first study to demonstrate that mtDNA from damaged cells stimulates a dual phenotype in Ly6C+ inflammatory monocytes, which acquire both inflammatory and regulatory functions. Our study also revealed that the TLR9 and STING pathways are responsible for both the activation of neutrophils and the induction of the dual monocyte phenotype via mtDNA. Furthermore, our findings indicate that the negative regulation of Ly6C+ inflammatory monocytes may result from their increased production of PGE2 through the STING- or TLR9-mediated MAPK-NF-κB-COX2 pathways. Although P38 or NF-kB can be activated following exposure to STING ligands, such as cytosolic dsDNA or mtDNA, or TLR-9 ligands, such as mtDNA [30, 42, 43], COX2 activation and the subsequent increased production of PGE2 by these ligands have not been reported. In this study, we found that mtDNA induced COX2 expression in the monocytes via STING- and TLR9-dependent pathways, but mtDNA was not involved in COX1 activation. Additionally, mtDNA activated the p38 MAPK and p65 NF-κB signalling responses involved in the STING and TLR9 pathways. Moreover, we found that the p38 MAPK-specific inhibitor SB203580 and p65NF-kB-specific inhibitor BAY117082 significantly reduced p38 and NF-kB activation, respectively, and both inhibitors reduced COX-2 expression, indicating that p38 MAPK and NF-kB activation are involved in COX-2 synthesis. We also pretreated monocytes with indomethacin, a COX-1 and COX-2 inhibitor, to inhibit PGE2 production. We found that indomethacin abolished monocytes' capability to inhibit neutrophil activation both in vitro and in vivo. Thus, we suggest that mitochondrial DNA may activate Ly6C+ monocytes and increase the production of PGE2 through TLR9- or STING-mediated MAPK-NF-κB-COX2 pathways. These molecular events may be part of a feedback mechanism that contributes to the regulation and resolution of the inflammatory response caused by cationic nanocarriers.
Systemic treatments with DNA nanoparticles (DNPs) has been reported to induce potent immune regulatory responses via STING signalling, produce the immune suppressive enzyme indoleamine 2,3 dioxygenase (IDO) and suppress experimental autoimmune encephalitis (EAE) in a mouse model [44]. In addition, prostaglandin E2 (PGE2) production is related to indoleamine 2,3-dioxygenase (IDO) expression [45, 46]; PGE2 released by cancer cells unregulated IDO expression in fibroblasts through an EP4/signal transducer and activator of transcription 3 (STAT3)-dependent pathway [47]. These findings also support our results.
In summary, our work describes the previously uncharacterized negative regulation of the inflammatory response, which protects hosts from tissue damage in a cationic nanoparticle-induced inflammatory environment. Ly6C+ inflammatory monocytes and their increased PGE2 production induced by mtDNA via TLR9- or STING-mediated MAPK-NF-κB-COX2 pathways may contribute to the regulation and resolution of the cationic nanoparticle-induced inflammatory response. We have provided insight into a new mechanism for the resolution of cationic nanoparticle- or mtDNA-induced inflammatory toxicity, and, in addition to other applications, these findings may have important implications for understanding nanoparticle biocompatibility and for designing better, safer drug delivery systems and therapies that control dsDNA-mediated inflammatory responses.
Methods
Mice
Myd88-/-, Ccr2-/-, IL-10-/-and Sting-/- mice with a C57BL/6 background were obtained from Jackson Laboratory. Tlr9-/-mice with a C57BL/6 background were obtained from Bioindustry Division Oriental Yeast Co., Ltd. (Tokyo, Japan). Female C57BL/6 WT mice were purchased from Vital River (Beijing, China). The mice were housed and maintained under SPF conditions in an animal facility. Eight- to sixteen-week-old female mice were used. All animal experiments were performed according to the guidelines of the Institutional Animal Care and Use Committee of Sichuan University (Chengdu, Sichuan, China), and the protocols were approved by the Institutional Animal Care and Use Committee of Sichuan University (Chengdu, Sichuan, China).
Liposome preparation
Cationic liposomes were composed of N-[1-(2,3-dioleoyloxy) propyl]-N, N, N-trimethylammonium chloride (DOTAP-CI). The film was prepared by a conventional evaporation method and was dispersed in sterile normal saline. The concentration of DOTAP liposomes was 1 mg/mL, and the particle size ranged from 100-130 nm with a 40-50 mV zeta potential. The liposome suspension was sterilized through 0.22 μm microporous membranes (Millipore). The size distribution and zeta potential of the prepared liposomes were determined by Malvern Nano-ZS 90 laser particle size analyser. The endotoxin levels were under 0.25 EU/mL for all samples.
Primary lung cell isolation and necrotic lung cell preparation
Mouse primary lung cells were isolated and cultured with a modified method as described previously [48]. The tissues were cut into approximately 1 mm pieces and then digested in 5 mL of 0.25% trypsin at 37 °C for 45 min. Culture medium containing 10% FBS was used to terminate the digestion, and the suspensions were filtered through 70 μm nylon mesh, centrifuged at 218 ×g for 5 min, then, the supernatant was removed. The cells were cultured in RPMI 1640 culture medium containing 10% FBS. The next day, the supernatant was removed, new culture medium was added, and the cells were passaged for 1-4 generations. The cells were incubated with the DOTAP liposomes (50 μg/mL) in RPMI 1640 medium (Gibco) supplemented with penicillin, streptomycin, HEPES and glutamine but without foetal bovine serum (FBS) at 37 °C for 2 h. After removing the liposomes, the cells were suspended in RPMI 1640 basic medium at a density of 5×106 cells/mL.
Mitochondrial DNA preparation
Mitochondrial DNA was isolated using a mitochondrial DNA isolation kit (Abcam) under sterile conditions. Mitochondrial DNA was isolated from the muscle tissue of C57BL/6 mice, and the mtDNA concentration was determined by a spectrophotometer; no protein contamination was found. The mitochondrial DNA was diluted in sterile water and stored at -80 °C. The endotoxin levels were under 0.25 EU/mL for all samples.
Endotoxin detection
The endotoxin content in the liposomes and reagents was determined according to the instructions (Xiamen Horseshoe Reagent Factory, China Co., Ltd.) and a previous report [49]. The Limulus Amoebocyte Lysate(LAL) gel-clot assay with a sensitivity of 0.25 EU/mL was used. After 30 min of incubation, the 0.25 EU/mL endotoxin standard reacted with the gel, but all the test reagents did not produce a reaction, indicating that the reagent endotoxin levels were less than 0.25 EU/mL.
Detection of inflammatory neutrophils and monocytes
Mice were treated with intravenous injections of DOTAP liposomes (25 mg/kg), necrotic lung cells (1×106 cells/mouse) or mitochondrial DNA (5 μg/mouse). Mouse lung tissues were dissected and cut into small pieces and dissociated by 1 mg/mL collagenase Type II in RPMI 1640 basic medium for 2 h in 37 °C. The suspensions were filtered through 70 μm nylon mesh. The cell suspensions were treated with red blood cell lysate buffer and were washed twice with phosphate-buffered solution (PBS). Cells were counted, dispersed in PBS at 1×106 cells/mL and stained with fluorescence-conjugated antibodies for 30 min in PBS at 4 °C, then, the cells were washed with PBS twice. APC-labelled rat anti-mouse CD45 (BD Biosciences, 1:100), PerCP-Cy5.5-labelled rat anti-mouse CD11b (BD Biosciences, 1:100), PE-labelled rat anti-mouse Ly6C (BD Biosciences, 1:100), FITC-labelled rat anti-mouse Ly6G (BD Biosciences, 1:100) and Brilliant Violet 421TM rat anti-mouse MHCII (I-A/I-E) (BD Biosciences, 1:100) antibodies were used. Data acquisition was performed on a FACS AriaIII flow cytometer and was analysed by FlowJo software.
Intracellular neutrophil and monocyte cytokine detection
For neutrophil intracellular cytokine detection, the cells were stained with APC-labelled rat anti-mouse CD45 (BD Biosciences, 1:100), PerCP-Cy5.5-labelled rat anti-mouse CD11b (BD Biosciences, 1:100) and FITC-rat anti-mouse Ly6G antibodies (BD Biosciences, 1:100) for 30 min in PBS at 4 °C and then were washed twice with PBS. The cells were fixed in 2% paraformaldehyde solution for 20 min, permeabilized using 1% Triton X-100 for 30 min at 4 °C and washed with PBS. The cells were stained with PE-labelled rat anti-mouse TNF-α antibody (BD Biosciences, 1:100) for 2 h in 4 °C and then were washed. For monocyte intracellular cytokine detection, the cells were stained with APC-labelled rat anti-mouse CD45 (BD Biosciences, 1:100), PerCP-Cy5.5-labelled rat anti-mouse CD11b (BD Biosciences, 1:100) and PE-labelled rat anti-mouse Ly6C antibodies (BD Biosciences, 1:100) for 30 min at 4 °C and were treated as previously described. The intracellular cytokines were stained with the following antibodies or their appropriate isotype controls (rat IgG1 or IgG2b) for 2 h in PBS at 4 °C: FITC-labelled rat anti-mouse TNF-α (BD Biosciences, 1:100), Brilliant Violet 421TM rat anti-mouse IL-10 (BD Biosciences, 1:100), FITC-labelled rat anti-mouse IL-6 (BD Biosciences, 1:100) and FITC-labelled rat anti-mouse IFN-γ (BD Biosciences, 1:100). Data acquisition was performed on a FACS AriaIII flow cytometer, and the results were analysed by FlowJo software.
Neutrophil and monocyte isolation
All bone marrow cells were obtained, and the suspensions were filtered through 70 μm nylon mesh. The cells were carefully layered onto Histopaque-1077/1119 gradient solutions (Sigma) and were centrifuged at 700 ×g for 30 min. After centrifugation, the monocytes were aspirated from the “mononuclear” layer, and neutrophils were found in the “granulocyte” layer. Cells were collected and washed twice with sterile PBS. The cells were cultured in RPMI 1640 medium supplemented with 10% FBS, penicillin, streptomycin, HEPES and glutamine.
Monocyte stimulation and intracellular cytokine detection
Bone marrow monocytes (1×106 cells/well) were stimulated with necrotic lung cells (1×105 cells/mL), mitochondrial DNA (5 μg/mL) or CpG1668 (1 μg/mL) (Invitrogen) in the presence of brefeldin A (GolgiPlug, BD Biosciences) in a 24-well plate at 37 °C for 4 h. For intracellular cytokine detection, the cells were stained with APC-labelled rat anti-mouse CD45 antibody (BD Biosciences, 1:100), PerCP-Cy5.5-labelled rat anti-mouse CD11b antibody (BD Biosciences, 1:100) and PE-labelled rat anti-mouse Ly6C antibody (BD Biosciences, 1:100) for 30 min at 4 °C and then were fixed and permeabilized as described. The intracellular cytokines were stained with FITC-labelled rat anti-mouse TNF-α antibody (BD Biosciences, 1:100), Brilliant Violet 421TM rat anti-mouse IL-10 antibody (BD Biosciences, 1:100), FITC-labelled rat anti-mouse IL-6 antibody (BD Biosciences, 1:100) and FITC-labelled rat anti-mouse IFN-γ antibody (BD Biosciences, 1:100) or their appropriate isotype controls, including rat IgG1 and IgG2b, for 2 h in PBS at 4 °C. Data acquisition was performed on a FACS AriaIII flow cytometer, and the results were analysed by FlowJo software.
Quantitative determination of PGE2 levels
Monocytes isolated from bone marrow were cultured (2×106 cells/well) and were stimulated with necrotic lung cells (1×105 cells/mL) or mitochondrial DNA (5 μg/mL) for 20 h in a 6-well plate at 37 °C. PGE2 production in the supernatant was quantitated using an enzyme immunoassay (EIA) kit (Enzo Life Science). The concentration was determined according to manufacturer's instructions.
Stimulation and suppression of neutrophil inflammation in vitro
Neutrophils (1×106 cells/well) were co-cultured with monocytes (5×105 cells/well) and stimulated with necrotic lung cells (1×105 cells/mL) or mitochondrial DNA (5 μg/mL) in a 24-well plate at 37 °C for 4 h. Neutrophils were stained with APC-labelled rat anti-mouse CD45 (BD Biosciences, 1:100), PerCP-Cy5.5-labelled rat anti-mouse CD11b (BD Biosciences, 1:100) and FITC-labelled rat anti-mouse Ly6G antibodies (BD Biosciences, 1:100) for 30 min in PBS at 4 °C and then were washed twice with PBS. Cells were fixed in 2% paraformaldehyde solution for 20 min, permeabilized using 1% Triton X-100 for 30 min at 4 °C and washed with PBS. The cells were stained with PE-labelled rat anti-mouse TNF-α antibody (BD Biosciences, 1:100) for 2 h at 4 °C and then were washed. Data acquisition was performed on a FACS AriaIII flow cytometer, and the results were analysed by FlowJo software.
Monocytes suppression assay in vivo
Monocytes were cultured with mitochondrial DNA (5 μg/mL) in a 24-well plate for 20 h at 37 °C. The cells were harvested and intravenously injected into mice (1×106 cells/mouse), then, the mice were treated with DOTAP liposomes (25 mg/kg), necrotic lung cells (1×106 cells/mouse) or mitochondrial DNA (5 μg/mouse). Forty-eight hours after treatment, the cells in the mouse lung tissues were analysed by flow cytometry. APC-labelled rat anti-mouse CD45 (BD Biosciences, 1:100), PerCP-Cy5.5-labelled rat anti-mouse CD11b (BD Biosciences, 1:100), FITC-labelled rat anti-mouse Ly6G (BD Biosciences, 1:100) and PE-labelled rat anti-mouse TNF-α antibodies (BD Biosciences, 1:100) were used for staining as previously described. The percentage of inflammatory neutrophil influx into the lung and the expression of TNF-α in neutrophils were determined. To detect the expression of cytokines in Ly6C+ inflammatory monocytes, APC-labelled rat anti-mouse CD45 (BD Biosciences, 1:100), PerCP-Cy5.5-labelled rat anti-mouse CD11b (BD Biosciences, 1:100), PE-labelled rat anti-mouse Ly6C (BD Biosciences, 1:100), FITC-labelled rat anti-mouse TNF-α (BD Biosciences, 1:100), Brilliant Violet 421TM rat anti-mouse IL-10 (BD Biosciences, 1:100), FITC-labelled rat anti-mouse IL-6 antibody (BD Biosciences, 1:100) and FITC-labelled rat anti-mouse IFN-γ antibodies (BD Biosciences, 1:100) were utilized as described previously. Data acquisition was performed on an AriaIII flow cytometer, and the results were analysed by FlowJo software.
Prostaglandin E2, 16, 16-Dimethyl prostaglandin E2 and indomethacin treatments
Neutrophils were treated with PGE2 (Sigma) at different concentrations and simultaneously stimulated with necrotic lung cells (1×105 cells/mL) or mitochondrial DNA (5 μg/mL). Mice were injected with DOTAP liposomes (25 mg/kg), necrotic lung cells (1×106 cells/mouse) or mitochondrial DNA (5 μg/mouse) and were treated with intravenous injections of diMePGE2 (10 or 100 μg/kg body weight) (Sigma). The COX-1 and COX-2 inhibitor indomethacin (Sigma) was used at a concentration of 10 μM when needed.
Quantitative real-time PCR for mitochondrial DNA
The mtDNA in the plasma was purified using a QIAamp DNA Blood Mini kit (Qiagen) and was quantified by qPCR via Taqman probes. The PCR primers and probes were designed as described previously [50]. Primers were designed and synthesized by Invitrogen. The following sequences were used: the sense primer 5′-ACCTACCCTATCACTCACACTAGCA-3′, antisense primer 5′- GAGGCTCATCCTGATCATAGAATG-3′, and the FAM-labelled TAMRA-quenched probes 5′-ATGAGTTCCCCTACCAATACCACACCC-3′. The standard curve was created by analysing serial dilutions of plasmid DNA with inserts of the target PCR product (J01420, positions 2891- 3173).
Detection of cell necrosis in lungs
DOTAP liposomes (25 mg/kg) were injected into mice through the tail vein. At 6 h after treatment, the mice were killed, and bronchoalveolar lavage fluid (BAL) was prepared with a modified method as descried previously [51]. For BAL, the trachea was cannulated and lavaged three times with 0.8 mL sterile PBS at room temperature. Samples were centrifuged at 491 ×g for 5 min and the cells were collected. The cells were counted, dispersed in PBS at 1×106 cells/mL and stained with FITC-Annexin-V and PE-PI (Roche) in binding buffer. Data acquisition was performed on an AriaIII flow cytometer, and the results were analysed by FlowJo software; FITC+ PE+ cells were gated.
Lung perfusion
After the mice were anaesthetized as described [52], the murine lung was perfused with a modified method as described previously [53, 54]. Lung perfusion with PBS solution was performed to remove residual blood by inserting a needle into small incision in the left ventricle and perfusing 20 mL of PBS through the circulatory system. Perfusion was deemed successful when the lungs expanded and became pale. After perfusion, the lungs were removed carefully.
Western blot assay
Freshly isolated monocytes were cultured and simultaneously stimulated with mitochondrial DNA (5 μg/mL) at a concentration of 5×106 cells/mL. The inhibitors PD98059 (EMD Chemicals), SB203580 (EMD Chemicals), BAY117082 (EMD Chemicals) and indomethacin (Sigma) were added to the cells, and the cells were incubated for 2 h at 37 °C. After the incubation, the monocytes were lysed, and protein samples were prepared. Western blot analysis was performed: equal amounts of protein (50 μg) were electrophoresed on 10% Bis-Tris gels (Invitrogen Nupage Novex), transferred onto a PVDF membrane and then incubated in TBST buffer (150 mM NaCl, 20 mM Tris-HCl, 0.02% Tween 20 (pH 7.4)) containing 5% nonfat milk. Antibodies to phospho-p38 MAPK (Thr 180/Tyr 182; Cell Signalling Technology), p38 MAPK (Cell Signalling Technology) and phospho-p44/42 extracellular signal-regulated kinase (ERK) 1/2 (Thr 202/Tyr 204) (Cell Signalling Technology), p44/42 ERK 1/2 (Cell Signalling Technology), NF-κB p65 (Cell Signalling Technology), phospho-NF-κB p65 (Cell Signalling Technology), GAPDH (Abcam), COX1 (Cell Signalling Technology) and COX2 (Cell Signalling Technology) were used. Proteins were visualized with specific primary antibodies and then were incubated with HRP-conjugated secondary antibodies. Immunoreactivity was detected using a Super Signal West Dura Substrate (Thermo Fisher Scientific).
Statistical analyses
Groups were compared with Prism software (GraphPad) using a two-tailed unpaired Student's t-test or Dunnet's t-test. Data are presented as the mean ± S.E.M.
Abbreviations
COX-2: cyclooxygenase 2; CCR2: CC chemokine receptor 2; DAMPs: damage-associated molecular patterns; DOTAP-Cl: N-[1-(2,3-Dioleoyloxy) propyl] -N, N, N-trimeth- ylammonium chloride; IFN-γ: interferon-γ; IL-1β: interleukin-1β; IL-6: interleukin-6; MAPKs: mitogen-activated protein kinases; mtDNA: mitochondrial DNA; PEI: polyethylene imines; PGE2: prostaglandin E2; STING: stimulator of interferon genes; TLR9: toll-like receptor 9; TNF-α: tumor necrosis factor-α.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
This work is supported by the National Key Research and Development Program of China (No. 2016YFA0201402) and the Natural Science Foundation of China (No. 81602492).
Author contributions
L.L., Y.L., B.C., C.L., and X.W. designed the in vivo and ex vivo studies and analysed the data. Z.H., K.M., Y.J., Z.Q. and P.M. designed and developed the nanoparticles. L.L., T.L., J.R., Y.L., C.F., W.W., X.L., M.L., B.S., Y.M., Y.W., J.G., B.D., Y.J. and H.X. developed the flow cytometry methods and analysed the data. L.L. and X.W. wrote the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Hubbell JA, Langer R. Translating materials design to the clinic. Nat Mater. 2013;12:963-6
2. Wang Y, Zhou K, Huang G, Hensley C, Huang X, Ma X. et al. A nanoparticle-based strategy for the imaging of a broad range of tumours by nonlinear amplification of microenvironment signals. Nature Materials. 2014;13:204-212
3. Torchilin VP. Recent advances with liposomes as pharmaceutical carriers. Nat Rev Drug Discovery. 2005;4:145-60
4. Xiao K, Li Y, Luo J, Lee JS, Xiao W, Gonik AM. et al. The effect of surface charge on in vivo biodistribution of PEG-oligocholic acid based micellar nanoparticles. Biomaterials. 2011;32:3435-46
5. Lv H, Zhang S, Wang B, Cui S, Yan J. Toxicity of cationic lipids and cationic polymers in gene delivery. J Control Release. 2006;114:100-9
6. Wang H-X, Zuo Z-Q, Du J-Z, Wang Y-C, Sun R, Cao Z-T. et al. Surface charge critically affects tumor penetration and therapeutic efficacy of cancer nanomedicines. Nano Today. 2016;11:133-44
7. Frohlich E. The role of surface charge in cellular uptake and cytotoxicity of medical nanoparticles. Int J Nanomedicine. 2012;7:5577-91
8. Davis ME, Chen ZG, Shin DM. Nanoparticle therapeutics: an emerging treatment modality for cancer. Nat Rev Drug Discovery. 2008;7:771-82
9. Vega-Villa KR, Takemoto JK, Yanez JA, Remsberg CM, Forrest ML, Davies NM. Clinical toxicities of nanocarrier systems. Adv Drug Deliv Rev. 2008;60:929-38
10. Kirtane AR, Panyam J. Polymer nanoparticles: Weighing up gene delivery. Nat Nanotechnol. 2013;8:805-6
11. Kanasty R, Dorkin JR, Vegas A, Anderson D. Delivery materials for siRNA therapeutics. Nat Mater. 2013;12:967-77
12. Shim G, Kim M-G, Park JY, Oh Y-K. Application of cationic liposomes for delivery of nucleic acids. Asian J Pharm. 2013;8:72-80
13. Luo D, Carter KA, Razi A, Geng J, Shao S, Giraldo D. et al. Doxorubicin encapsulated in stealth liposomes conferred with light-triggered drug release. Biomaterials. 2016;75:193-202
14. Shao XR, Wei XQ, Song X, Hao LY, Cai XX, Zhang ZR. et al. Independent effect of polymeric nanoparticle zeta potential/surface charge, on their cytotoxicity and affinity to cells. Cell Prolif. 2015;48:465-74
15. Knudsen KB, Northeved H, Kumar PE, Permin A, Gjetting T, Andresen TL. et al. In vivo toxicity of cationic micelles and liposomes. Nanomedicine. 2015;11:467-77
16. Ballarin-Gonzalez B, Howard KA. Polycation-based nanoparticle delivery of RNAi therapeutics: adverse effects and solutions. Adv Drug Deliv Rev. 2012;64:1717-29
17. Blanco E, Shen H, Ferrari M. Principles of nanoparticle design for overcoming biological barriers to drug delivery. Nat Biotechnol. 2015;33:941-51
18. Petersen GH, Alzghari SK, Chee W, Sankari SS, La-Beck NM. Meta-analysis of clinical and preclinical studies comparing the anticancer efficacy of liposomal versus conventional non-liposomal doxorubicin. J Control Release. 2016;232:255-64
19. Chen J, Guo Z, Tian H, Chen X. Production and clinical development of nanoparticles for gene delivery. Mol Ther Methods Clin Dev. 2016;3:16023
20. Davis ME, Zuckerman JE, Choi CH, Seligson D, Tolcher A, Alabi CA. et al. Evidence of RNAi in humans from systemically administered siRNA via targeted nanoparticles. Nature. 2010;464:1067-70
21. Zuckerman JE, Davis ME. Clinical experiences with systemically administered siRNA-based therapeutics in cancer. Nat Rev Drug Discovery. 2015;14:843-56
22. Ginn SL, Alexander IE, Edelstein ML, Abedi MR, Wixon J. Gene therapy clinical trials worldwide to 2012 - an update. J Gene Med. 2013;15:65-77
23. Senzer N, Nemunaitis J, Nemunaitis D, Bedell C, Edelman G, Barve M. et al. Phase I study of a systemically delivered p53 nanoparticle in advanced solid tumors. Mol Ther. 2013;21:1096-103
24. Lu C, Stewart DJ, Lee JJ, Ji L, Ramesh R, Jayachandran G. et al. Phase I clinical trial of systemically administered TUSC2(FUS1)-nanoparticles mediating functional gene transfer in humans. PloS one. 2012;7:e34833
25. Yin H, Kanasty RL, Eltoukhy AA, Vegas AJ, Dorkin JR, Anderson DG. Non-viral vectors for gene-based therapy. Nat Rev Genet. 2014;15:541-55
26. Tabernero J, Shapiro GI, LoRusso PM, Cervantes A, Schwartz GK, Weiss GJ. et al. First-in-humans trial of an RNA interference therapeutic targeting VEGF and KSP in cancer patients with liver involvement. Cancer Discov. 2013;3:406-17
27. Zuckerman JE, Gritli I, Tolcher A, Heidel JD, Lim D, Morgan R. et al. Correlating animal and human phase Ia/Ib clinical data with CALAA-01, a targeted, polymer-based nanoparticle containing siRNA. Natl Acad Sci USA. 2014;111:11449-54
28. Hwang TL, Aljuffali IA, Lin CF, Chang YT, Fang JY. Cationic additives in nanosystems activate cytotoxicity and inflammatory response of human neutrophils: lipid nanoparticles versus polymeric nanoparticles. Int J Nanomedicine. 2015;10:371-85
29. Uchida S, Itaka K, Chen Q, Osada K, Ishii T, Shibata MA. et al. PEGylated polyplex with optimized PEG shielding enhances gene introduction in lungs by minimizing inflammatory responses. Mol Ther. 2012;20:1196-203
30. Wei X, Shao B, He Z, Ye T, Luo M, Sang Y. et al. Cationic nanocarriers induce cell necrosis through impairment of Na(+)/K(+)-ATPase and cause subsequent inflammatory response. Cell Res. 2015;25:237-53
31. Kunzmann A, Andersson B, Thurnherr T, Krug H, Scheynius A, Fadeel B. Toxicology of engineered nanomaterials: focus on biocompatibility, biodistribution and biodegradation. Biochim biophys Acta. 2011;1810:361-73
32. Fadeel B, Garcia-Bennett AE. Better safe than sorry: Understanding the toxicological properties of inorganic nanoparticles manufactured for biomedical applications. Adv Drug Deliv Rev. 2010;62:362-74
33. Minami K, Okamoto K, Doi K, Harano K, Noiri E, Nakamura E. siRNA delivery targeting to the lung via agglutination-induced accumulation and clearance of cationic tetraamino fullerene. Sci Rep. 2014;4:4916
34. Grainger JR, Wohlfert EA, Fuss IJ, Bouladoux N, Askenase MH, Legrand F. et al. Inflammatory monocytes regulate pathologic responses to commensals during acute gastrointestinal infection. Nat Med. 2013;19:713-21
35. Németh K, Leelahavanichkul A, Yuen PST, Mayer B, Parmelee A, Doi K. et al. Bone marrow stromal cells attenuate sepsis via prostaglandin E2-dependent reprogramming of host macrophages to increase their interleukin-10 production. Nat Med. 2008;15:42-9
36. Ishikawa H, Ma Z, Barber GN. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature. 2009;461:788-92
37. Gu X, Wu G, Yao Y, Zeng J, Shi D, Lv T. et al. Intratracheal administration of mitochondrial DNA directly provokes lung inflammation through the TLR9-p38 MAPK pathway. Free Radic Bio Med. 2015;83:149-58
38. Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, Junger W. et al. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature. 2010;464:104-7
39. Carroll EC, Jin L, Mori A, Munoz-Wolf N, Oleszycka E, Moran HB. et al. The vaccine adjuvant chitosan promotes cellular immunity via DNA sensor cGAS-STING-dependent induction of type I interferons. Immunity. 2016;44:597-608
40. Riteau N, Sher A. Chitosan: an adjuvant with an unanticipated STING. Immunity. 2016;44:522-4
41. Hohenhaus DM, Schaale K, Le Cao KA, Seow V, Iyer A, Fairlie DP. et al. An mRNA atlas of G protein-coupled receptor expression during primary human monocyte/macrophage differentiation and lipopolysaccharide-mediated activation identifies targetable candidate regulators of inflammation. Immunobiology. 2013;218:1345-53
42. Lastrucci C, Benard A, Balboa L, Pingris K, Souriant S, Poincloux R. et al. Tuberculosis is associated with expansion of a motile, permissive and immunomodulatory CD16(+) monocyte population via the IL-10/STAT3 axis. Cell Res. 2015;25:1333-51
43. Chatterjee M, von Ungern-Sternberg SN, Seizer P, Schlegel F, Buttcher M, Sindhu NA. et al. Platelet-derived CXCL12 regulates monocyte function, survival, differentiation into macrophages and foam cells through differential involvement of CXCR4-CXCR7. Cell Death Dis. 2015;6:e1989
44. Lemos H, Huang L, Chandler PR, Mohamed E, Souza GR, Li L. et al. Activation of the STING adaptor attenuates experimental autoimmune encephalitis. J Immunol. 2014;192:5571-8
45. Wong JL, Obermajer N, Odunsi K, Edwards RP, Kalinski P. Synergistic COX2 induction by IFNgamma and TNFalpha self-limits type-1 immunity in the human tumor microenvironment. Cancer Immunol Res. 2016;4:303-11
46. Li M, Sun X, Kuang X, Liao Y, Li H, Luo D. Mesenchymal stem cells suppress CD8+ T cell-mediated activation by suppressing natural killer group 2, member D protein receptor expression and secretion of prostaglandin E2, indoleamine 2, 3-dioxygenase and transforming growth factor-beta. Clin Exp Immunol. 2014;178:516-24
47. Chen J-Y, Li C-F, Kuo C-C, Tsai KK, Hou M-F, Hung W-C. Cancer/stroma interplay via cyclooxygenase-2 and indoleamine 2,3-dioxygenase promotes breast cancer progression. Breast Cancer Res. 2014;16:410
48. Tager AM, Kradin RL, LaCamera P, Bercury SD, Campanella GS, Leary CP. et al. Inhibition of pulmonary fibrosis by the chemokine IP-10/CXCL10. Am J Respir Cell Mol Biol. 2004;31:395-404
49. Ong KG, Leland JM, Zeng K, Barrett G, Zourob M, Grimes CA. A rapid highly-sensitive endotoxin detection system. Biosens Bioelectron. 2006;21:2270-4
50. Masuyama M, Iida R, Takatsuka H, Yasuda T, Matsuki T. Quantitative change in mitochondrial DNA content in various mouse tissues during aging. Biochimica Biophysica Acta. 2005;1723:302-8
51. Dong Y, Geng Y, Li L, Li X, Yan X, Fang Y. et al. Blocking follistatin-like 1 attenuates bleomycin-induced pulmonary fibrosis in mice. J Exp Med. 2015;212:235-52
52. Aeffner F, Woods PS, Davis IC. Activation of A1-adenosine receptors promotes leukocyte recruitment to the lung and attenuates acute lung injury in mice infected with influenza A/WSN/33 (H1N1) virus. J Virol. 2014;88:10214-27
53. Peng X, Hassoun PM, Sammani S, McVerry BJ, Burne MJ, Rabb H. et al. Protective effects of sphingosine 1-phosphate in murine endotoxin-induced inflammatory lung injury. Am J Respir Crit Care Med. 2004;169:1245-51
54. Crawford SE, Patel DG, Cheng E, Berkova Z, Hyser JM, Ciarlet M. et al. Rotavirus viremia and extraintestinal viral infection in the neonatal rat model. J Virol. 2006;80:4820-32
Author contact
![]() Corresponding author: Xiawei Wei, Ph.D. Phone: +8618081954096, E-mail addresses: xiaweiweiedu.cn
Corresponding author: Xiawei Wei, Ph.D. Phone: +8618081954096, E-mail addresses: xiaweiweiedu.cn
Citation styles
APA
Liu, L., Liu, Y., Xu, B., Liu, C., Jia, Y., Liu, T., Fang, C., Wang, W., Ren, J., He, Z., Men, K., Liang, X., Luo, M., Shao, B., Mao, Y., Xiao, H., Qian, Z., Geng, J., Dong, B., Mi, P., Jiang, Y., Wei, Y., Wei, X. (2018). Negative regulation of cationic nanoparticle-induced inflammatory toxicity through the increased production of prostaglandin E2 via mitochondrial DNA-activated Ly6C+ monocytes. Theranostics, 8(11), 3138-3152. https://doi.org/10.7150/thno.21693.
ACS
Liu, L.; Liu, Y.; Xu, B.; Liu, C.; Jia, Y.; Liu, T.; Fang, C.; Wang, W.; Ren, J.; He, Z.; Men, K.; Liang, X.; Luo, M.; Shao, B.; Mao, Y.; Xiao, H.; Qian, Z.; Geng, J.; Dong, B.; Mi, P.; Jiang, Y.; Wei, Y.; Wei, X. Negative regulation of cationic nanoparticle-induced inflammatory toxicity through the increased production of prostaglandin E2 via mitochondrial DNA-activated Ly6C+ monocytes. Theranostics 2018, 8 (11), 3138-3152. DOI: 10.7150/thno.21693.
NLM
Liu L, Liu Y, Xu B, Liu C, Jia Y, Liu T, Fang C, Wang W, Ren J, He Z, Men K, Liang X, Luo M, Shao B, Mao Y, Xiao H, Qian Z, Geng J, Dong B, Mi P, Jiang Y, Wei Y, Wei X. Negative regulation of cationic nanoparticle-induced inflammatory toxicity through the increased production of prostaglandin E2 via mitochondrial DNA-activated Ly6C+ monocytes. Theranostics 2018; 8(11):3138-3152. doi:10.7150/thno.21693. https://www.thno.org/v08p3138.htm
CSE
Liu L, Liu Y, Xu B, Liu C, Jia Y, Liu T, Fang C, Wang W, Ren J, He Z, Men K, Liang X, Luo M, Shao B, Mao Y, Xiao H, Qian Z, Geng J, Dong B, Mi P, Jiang Y, Wei Y, Wei X. 2018. Negative regulation of cationic nanoparticle-induced inflammatory toxicity through the increased production of prostaglandin E2 via mitochondrial DNA-activated Ly6C+ monocytes. Theranostics. 8(11):3138-3152.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.