Theranostics
13.3
Impact Factor
- Current Issue
- Advance Articles
- Volume 16; 2026
- Volume 15; 2025
- Volume 14; 2024
- Volume 13; 2023
- Volume 12; 2022
- Archive
- Cover Images
- Cover Suggestion
- Special Issues
Top
Introduction
Methods
Results
Discussion
Abbreviations
Acknowledgements
Supplementary Material
References
Introduction
Methods
Results
Discussion
Abbreviations
Acknowledgements
Supplementary Material
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2018; 8(13):3643-3653. doi:10.7150/thno.26021 This issue Cite
Research Paper
Phosphorylation of IRS4 by CK1γ2 promotes its degradation by CHIP through the ubiquitin/lysosome pathway
Xinchun Li1#, Li Zhong1#, Zhuo Wang2#, Huiming Chen1, Dan Liao1, Ruhua Zhang1, Hongyu Zhang3 ![]() , Tiebang Kang1
, Tiebang Kang1 ![]()
1. State Key Laboratory of Oncology in South China, Collaborative Innovation Center for Cancer Medicine, Sun Yat-sen University Cancer Center, Guangzhou, China.
2. Department of Pathology, The First Affiliated Hospital of Sun Yat-Sen University, Guangzhou, China.
3. Department of Medical Oncology, The Fifth Affiliated Hospital of Sun Yat-sen University, Zhuhai, Guangdong, China.
#These authors contributed equally to this work.
Received 2018-3-12; Accepted 2018-5-4; Published 2018-6-7
Citation:
Li X, Zhong L, Wang Z, Chen H, Liao D, Zhang R, Zhang H, Kang T. Phosphorylation of IRS4 by CK1γ2 promotes its degradation by CHIP through the ubiquitin/lysosome pathway. Theranostics 2018; 8(13):3643-3653. doi:10.7150/thno.26021. https://www.thno.org/v08p3643.htm
Other stylesAbstract

IRS4, a member of the insulin receptor substrate protein family, can induce constitutive PI3K/AKT hyperactivation and cell proliferation even in the absence of insulin or growth factors and promote tumorigenesis, but its regulation has only been explored at the transcriptional level.
Methods: Scansite was used to predict the potential protein kinases that may regulate the functions of IRS4, and mass spectrometry was used to identify the E3 ligase for IRS4. The protein interaction was carried out by immunoprecipitation, and protein stability was measured by cycloheximide treatment. In vitro kinase assay was used to determine the phosphorylation of IRS4 by casein kinase 1γ2 (CK1γ2). Colony formation assay and xenograft-bearing mice were employed to assess the cancer cell growth in vitro and in vivo, respectively. Immunohistochemistry was performed to examine protein levels of both IRS4 and CK1γ2 in osteosarcoma specimens and their relationship was evaluated by χ2 test. Two-tailed Student's t-test or the Mann-Whitney U test were used to compare the differences between subgroups.
Results: IRS4 was phosphorylated at Ser859 by CK1γ2 in vitro and in vivo, which promoted the polyubiquitination and degradation of IRS4 through the ubiquitin/lysosome pathway by the carboxyl terminus of Hsc70-interacting protein(CHIP). Using osteosarcoma cell lines, the ectopic nonphosphorylated mutant of IRS4 by CK1γ2 triggered higher level of p-Akt and displayed faster cell proliferation and cancer growth in vitro and in nude mice. In addition, a negative correlation in protein levels between CK1γ2 and IRS4 was observed in osteosarcoma cell lines and tissue samples.
Conclusions: IRS4, as a new substrate of CHIP, is negatively regulated by CK1γ2 at the posttranslational level, and specific CK1γ2 agonists may be a potentially effective strategy for treating patients with osteosarcoma.
Keywords: IRS4, CK1γ2, CHIP, ubiquitin/lysosome, p-Akt.
Introduction
The insulin receptor substrate (IRS) protein family is composed of four closely related members, IRS1-4, and two distant relatives, IRS5/DOK4 and IRS6/DOK5. IRS1 and IRS2, the most extensively studied and most prominent members in this family, are involved in a wide range of cellular functions including cell proliferation, survival, migration, metabolism and differentiation [1-3]. Upon activation, the IRS proteins can be phosphorylated and transmit signals from the IGF1 and insulin receptors to several Src homology 2 (SH2) domain-containing proteins, such as PI3K and GRB2, triggering the activation of the PI3K/AKT and MAPK/ERK signaling pathways, respectively [1, 4, 5].
IRS4, initially identified and characterized in the human embryonic kidney (HEK) 293 cell line, shows rapid tyrosine phosphorylation upon insulin treatment [6, 7]. In contrast to IRS1 and IRS2, IRS4 does not bind tyrosine phosphatase SHP2 [7]. IRS4 was found to specifically interact with Crk-II and Crk-L [8], breast tumor kinase (Brk) [9] and SSH1 [10]. Therefore, compared to IRS1 and IRS2, IRS4 may have distinctive functions. The proliferative effect of Brk, which is overexpressed in most of human breast tumors, is enhanced when IRS4 is present [9]. Overexpression of IRS4 mRNA and activation of Akt pathway are critical for the Adenovirus 5 E1A-mediated cell transformation [11]. In addition, IRS4 levels are elevated in human hepatocellular carcinomas (HCC) compared with hepatocytes [12], and IRS4 plays a pivotal role in proliferation/differentiation of the HepG2 hepatoblastoma cell line [13]. Somatic mutants of IRS4 are frequently detected in T-cell acute lymphoblastic leukemia [14-16] and melanoma [17]. These findings indicated that IRS4 is involved in progression of various human cancers.
Contrary to the relatively ubiquitous expression of IRS1 and IRS2, IRS4 shows a restricted tissue expression, as IRS4 mRNA has only been detected in specific tissues, such as pituitary, thyroid, ovary, prostate, fibroblasts, and some tumor cell lines [18]. This may explain why the IRS4-knockout mice have slight effects on growth, reproduction, and glucose homeostasis [19]. As a constitutively active oncogene, IRS4 has been reported to be primarily regulated at the transcriptional level, and the transcriptional deregulation of IRS4 makes cells tend to be carcinous [15, 16]. However, there are numerous phosphorylation sites in the C-terminal region of IRS4, but the function of these phosphorylation sites has not been explored yet.
The casein kinase 1 (CKI) family of serine/threonine kinases, which consist of seven known isoforms (α, -β, -γ1, -γ2, -γ3, -δ and -ε), are evolutionarily conserved and ubiquitously expressed in eukaryotic organisms [20, 21]. All of CKIs contain a conserved core kinase domain and variable N- and C-terminal non-catalytic domains, which are closely correlated with CKI subcellular localization, substrate specificity and kinase activity [22], and their characterized substrates have been shown to participate in a variety of functions, such as vesicular trafficking, DNA damage repair, ribosome biogenesis, cell cycle progression, cytokinesis, cell differentiation, immune response and inflammation, and circadian rhythms [21, 23, 24]. CK1γ2 selectively binds Smad3 and inhibits the Smad3-mediated TGF-β responses in mammalian cells [25], and it also couples Wnt receptor activation to cytoplasmic signal transduction by phosphorylating LRP6 [26], indicating that CK1γ2 may act as an oncogene. On the other hand, CK1γ2 negatively modulates the function of the short form of metastatic tumor antigen 1 (MTA1) [27], which is oncogenic in breast cancer [28]. Therefore, the roles of CK1γ2 in cancers may be cell type dependent, and new substrates or/and binding proteins of CK1γ2 might help to elucidate the distinctive functions of CK1γ2 in different cancers.
The carboxyl terminus of Hsc70-interacting protein (CHIP), initially identified as a co-chaperone in protein folding, is also named Stub1 and interacts with the molecular chaperones Hsp70 and Hsp90 through the N-terminal tetratricopeptide repeat (TPR) domain [29]. CHIP may also serve as a U-box-type E3 ubiquitin ligase to facilitate the ubiquitylation and degradation of chaperone client proteins and play a pivotal role in the protein quality control system [30-32]. Interestingly, some substrates of CHIP require phosphorylation prior to their degradation. For example, phosphorylation of Slug by GSK3β is required for its ubiquitylation and degradation by CHIP, and the accumulation of nondegradable Slug may promote tumor metastasis [33].
Here, we report that IRS4 can be phosphorylated by CK1γ2 at Ser859 and is subsequently ubiquitylated by CHIP, and then is degraded in lysosomes. Using osteosarcoma cell lines, we find that the dysregulation of CK1γ2/IRS4 axis may affect the growth of cancer cells.
Methods
Cell culture and reagents
Human cancer cell lines (U2OS, 143B, HCT116, MDA-MB-231, Hep3B, HeLa) and HEK293T cells were purchased from American Type Culture Collection(ATCC).U2OS/MTX300 cell line, which is the methotrexate-resistant derivative of U2OS cell line, and ZOS and ZOS-M, syngeneic human osteosarcoma cell lines derived from a patient with metastasis, were described previously [34, 35]. All cell lines were cultured in Dulbecco's modified Eagle medium (Invitrogen) supplemented with 10% fetal bovine serum (Gibco) at 37°C and 5%CO2. Bortezomib, Bafilomycin A and CKI-7 were obtained from APExBIO.
Antibodies
Anti-Flag was obtained from Abigent. Antibodies against C-Myc(sc-289) and Akt(sc-8312) were from Santa Cruz Biotechnology Inc. Antibodies against p-AktSer473(4060), p-AktThr308(13038), p-p70S6KThr389(9234), p-4EBP1Thr37/46(2855) and HA(3724) were obtained from Cell Signaling Technology. Anti-IRS4 (ab52622) and CK1γ2 (GTX33123) were purchased from Abcam and GeneTex, respectively. Antibodies against GAPDH (13937-1-AP) and β-Tublin(10094-1-AP) were from Proteintech. Anti-p-IRS4-S859, an antibody specific for the phosphorylation on Ser859 of IRS4, was generated by immunizing rabbits with the coupled peptide DAASKPS859GEGSFSK.
Plasmids
All transient ectopic expression vectors were constructed using the pCDNA3.1 vector (Invitrogen, Carlsbad, MA, USA). Mutations were introduced using the Mut Express- II Fast Mutagenesis Kit (Vazyme Biotech Co., Ltd), and all mutations were verified by DNA sequencing. IRS4 and CK1γ2 were cloned into the pSIN lentivirus vector to generate stable cells overexpressing IRS4 and CK1γ2, respectively. The pLKO.1-puro vector was inserted with the shRNAs targeting CK1γ2 or CHIP, and the sequences of these shRNAs are as follows:
CHIPshRNA-1:CCGGGAAGAGGAAGAAGCGAGACATCTCGAGATGTCTCGCTTCTTCCTCTTCTTTTT;
CHIPshRNA-2:CCGGCGCGAAGAAGAAGCGCTGGAACTCGAGTTCCAGCGCTTCTTCTTCGCGTTTTT;
CK1γ2shRNA-1:CCGGCCCTCCAAGAGCATTAACTATCTCGAGATAGTTAATGCTCTTGGAGGGTTTTTG;
CK1γ2shRNA-2:CCGGGCACACCAAGAGCCTAATCTACTCGAGTAGATTAGGCTCTTGGTGTGCTTTTTG.
Transfection experiments
Transfection experiments were performed according to the manufacturer's instructions using Lipofectamine 2000 (Invitrogen). Briefly, asynchronously growing cells seeded at 2.5 × 105 cells per well in 6-well plates or at 1 × 106 cells per dish (10 cm) were transfected with 2 μg or 12 μg plasmid DNA for 24 h, respectively, and then the cells were treated as indicated.
RNAi treatment and cycloheximide chase assay
RNAi transfections were performed as previously described using the Lipofectamine RNAiMAX transfection reagent (Invitrogen) and 50 nM siRNA. The siRNA oligonucleotides targeting CK1γ2 were as follows: CK1γ2-siRNA-1(5′-ACACCAAGAGCCUAAUCUAdTdT-3′) and CK1γ2-siRNA-2(5′-GCGCUACAUGAGCAUCAACdTdT-3′). For the cycloheximide (CHX) chase assay, cells were treated with 40 μg/mL CHX for 0, 4, 8, 12 and 16 h, and then cell extracts were analyzed by Western blotting.
Immunoblotting and immunoprecipitation
Cells were collected and lysed in RIPA buffer (50 mM Tris-HCl, 150 mM NaCl, 5 mM EDTA, 0.5% NP-40) and centrifuged at 13,400 rcf and 4°C for 15 min. Proteins were resolved by SDS-PAGE and then transferred onto PVDF membranes (Millipore). The immunoblots were blocked with 5% non-fat milk at room temperature for 2 h and incubated with various primary antibodies and horseradish peroxidase-conjugated secondary antibody. The membranes were then detected using the ECL chemiluminescence system (Pierce).
For coimmunoprecipitation, HEK293T cells were transfected with the indicated plasmids. The supernatants were first incubated with anti-FLAG-agarose or anti-HA-agarose or anti-MYC-agarose (Sigma Chemical Co.) overnight at 4°C, and the precipitates were washed five times with RIPA buffer. For the interaction either between CK1γ2 and CHIP or between CK1γ2 and IRS4 at their endogenous levels, cell supernatants were first incubated with anti-CHIP antibody, anti-CK1γ2 antibody, or anti-IRS4 antibody at 4°C overnight, followed by the addition of protein A/G-agarose beads (Santa Cruz Biotechnology Inc.) for 4 h. The precipitates were washed five times with RIPA and analyzed by Western blotting.
Assessment of cell proliferation and colony formation assay
Cell proliferation was determined by MTT assay. Briefly, cells were seeded at 2,000 cells per well in 96-well plates. 20 μL MTT was added to each well and incubated for 3 h. The absorbance was measured at 490 nm with a microplate reader once per day for 4 days. The results are expressed as the mean±SD of three independent experiments. For colony formation assay, U2OS/MTX300 cells were plated at 2,000 cells per well in 6-well plates in triplicate. After 2 weeks, the plates were stained with 0.2% crystal violet and the colonies containing more than 50 cells were counted.
In vitro kinase assays
His-IRS4(WT) and His-IRS4(S859A) were synthesized in vitro using the S30 T7 High-Yield Protein Expression System (Promega) according to the manufacturer's standard procedures. The lysates of 293T cells overexpressing HA-CK1γ2-WT and HA-CK1γ2-KD were immunoprecipitated using anti-HA agarose. Beads were washed 4 times with RIPA Buffer and then incubated with 3 μg of His-IRS4 (WT) or His- IRS4 (S859A) in 50 μL of kinase buffer (Cell Signaling Technology) containing 10 μM ATP for 30 min at 30°C. Subsequently, the samples were eluted in SDS-PAGE buffer and subjected to immunoblotting analysis.
Immunohistochemical analysis
Immunohistochemical assay was performed using anti-IRS4 antibody or anti-CK1γ2 antibody as previously described. Briefly, formalin-fixed, paraffin-embedded tissue samples were cut onto polylysine-coated slides in 4 μm sections. Slides were deparaffinized by xylene and rehydrated with ethanol of gradient concentrations. Then slides were immersed in 3% hydrogen peroxide in methanol for 20 min to inactivate endogenous peroxidase, followed by the antigen epitope retrieval step in 0.01 M sodium citrate buffer (pH 6.0) using a microwave oven. After pre-incubation with 10% normal goat serum for 30 min at room temperature to block nonspecific binding, the sections were incubated with primary antibodies at 4°C overnight. Subsequently, the slides were treated with the secondary antibody (Dako) for 1 h at 37°C. Detection was achieved with 3,3′-diaminobenzidine. Finally, the sections were counterstained with hematoxylin, dehydrated, cleared and mounted. The score of immunoreactivity was evaluated according to a semi-quantitative scoring criterion. The intensity of positive staining(weak, 1; moderate, 2; strong, 3) and the percentage of positive areas (0%, 0; <10%, 1; 10-50%, 2; 51-80%, 3; >80%, 4) were recorded. The staining index (values 0-12) was calculated as their products. The IRS4 and CK1γ2 levels were classified as high or low expression when their scores were in the range of 8-12 or 0-6, respectively.
Statistical analysis
Data are represented as mean ± SEM. Two-tailed Student's t-test or the Mann-Whitney U test was used to compare the differences between subgroups. The association between IRS4 and CK1γ2 abundance was evaluated using χ2 tests. The SPSS 16.0 software was used for statistical analyses. P values <0.05 were considered as significant and < 0.001 as strongly significant.
Study approval
The animal studies were approved by the Animal Research Committee of Sun Yat-sen University Cancer Center and were conducted in consistence with established guidelines. The use of human osteosarcoma samples was reviewed and approved by the ethical committee of Sun Yat-sen University Cancer Center, and written informed consent was obtained. The samples were retrospectively acquired from the surgical pathology archives of Sun Yat-sen University Cancer Center. The raw data of this paper have been uploaded onto the Research Data Deposit (RDD) with an RDD number of RDDB2018000322.
Results
Down-regulation of IRS4 by CK1γ2 is mediated by lysosomes in cells
The C-terminal tail of IRS4 has numerous tyrosine and serine phosphorylation sites;this promoted us to speculate that phosphorylation of IRS4 might be crucial for the functions of IRS4. Using Scansite, which has been widely used to identify the short protein linear motifs that are likely to be phosphorylated by kinases, we found that Akt, Erk, GSK3β, CK1γ2, CK2B, CDC2, CDK5 and GSK3α are the protein kinases that can potentially phosphorylate IRS4 (Figure S1A). However, CK1γ2 was the only one among these kinases that could alter the IRS4 protein level when each kinase was co-transfected with IRS4 in HEK293T cells (Figure S1B-C), and this down-regulation of IRS4 protein level was dependent upon CK1γ2 kinase activity, as the kinase-depleted mutant (KD) lacked an ability to decrease the IRS4 protein level (Figure 1A). Furthermore, IRS4 was decreased by ectopic expression of WT-CK1γ2, but not by its KD form, in multiple cell lines (Figure 1A and Figure S2A). In contrast, IRS4 was increased under treatment of siRNA against CK1γ2 in different cell lines (Figure 1B and Figure S2B). Moreover, the CK1γ2-mediated down-regulation of IRS4 was blocked by BAF1, a lysosome inhibitor, but not by Bortezomib, a proteasome inhibitor, in HEK293T cells (Figure 1C), and ubiquitylation of Flag-IRS4 was increased under co-transfection of CK1γ2 with the K-63 ubiquitin, but not with the K-48 ubiquitin, in HEK293T cells (Figure 1D), suggesting that the ubiquitin/lysosome degradation pathway is involved in the decrease of IRS4 by CK1γ2. Taken together, our results strongly indicated that IRS4 is regulated at the post-transcriptional level, and that the down-regulation of IRS4 protein by CK1γ2 may be mediated by lysosomes in cells. Interestingly, as shown in Figure S3, besides CK1γ2, other isoforms such as CK1γ3, CK1δ and CK1ε, but neither CK1α, CK1γ1 nor VRK1, could also degrade IRS4, indicating that CK1 family members may have specificity for each substrate to some degree. The differences within CK1 family members to degrade IRS4 are desired to be elucidated in the future.
The phosphorylation of IRS4 at Ser859 by CK1γ2 plays a pivotal role in the protein stability of IRS4
Since the down-regulation of IRS4 by CK1γ2 is dependent upon its kinase activity (Figure 1), we further investigated the relationship between CK1γ2 and IRS4. As shown in Figure 2A, the interaction between CK1γ2 and IRS4 was already detected by IP using anti-HA or anti-FLAG when HEK293T cells were co-transfected with both HA-CK1γ2 and Flag-IRS4. More importantly, this interaction was also reciprocally detected at their endogenous levels in cells (Figure 2B). Moreover, we performed an in vitro binding assay using the purified eukaryotic IRS4 and CK1γ2. As shown in Figure 2C, the interaction between IRS4 and CK1γ2 was also detected in such an assay, indicating that CK1γ2 may directly bind IRS4.
Figure 1
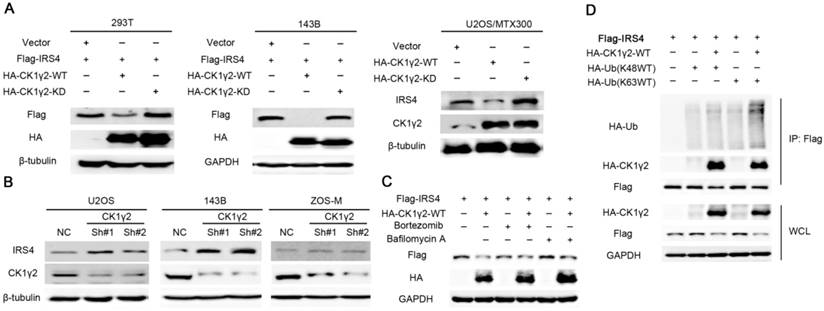
Down-regulation of IRS4 by CK1γ2 is mediated by lysosomes in cells.(A) 293T and osteosarcoma cells were co-transfected with HA-tagged wild type (WT) or kinase-depleted(KD) CK1γ2 plasmids and Flag-IRS4 for 48 h and analyzed by Western blotting. (B) The indicated cells were transfected with the indicated shRNAs and then were analyzed by Western blotting. (C) 293T cells were co-transfected with HA-CK1γ2 and Flag-IRS4 for 48 h and then treated with Bortezomib or Bafilomycin A for 6 h. Cell lysates were then analyzed by Western blotting. (D) 293T cells were co-transfected with Flag-IRS4 and HA-CK1γ2 and HA-Ub(K48WT) or HA-Ub(K63WT). Cell lysates were subjected to immunoprecipitation with anti-Flag-agarose and then analyzed by Western blotting.
Figure 2
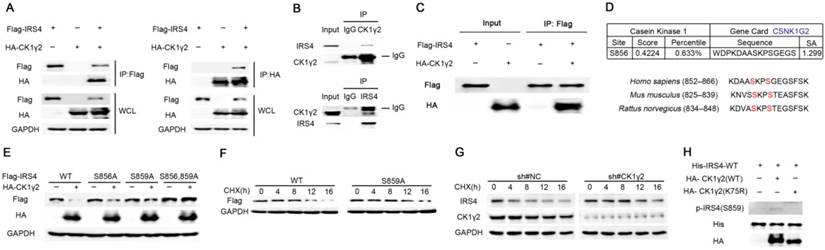
CK1γ2 phosphorylates IRS4 and modulates its protein stability. (A) 293T cells were co-transfected with HA-CK1γ2 and Flag-IRS4 for 48 h, and were lysed and immunoprecipitated using anti-FLAG-agarose or anti-HA-agarose followed by Western blotting. (B) Cell lysates from 293T cells were immunoprecipitated with anti-CK1γ2 or anti-IRS4 and were subsequently detected with anti-IRS4 or anti-CK1γ2 antibody. (C) Flag-IRS4 and HA-CK1γ2 purified from 293T cells were incubated for 4 h at 4°C, and then protein mixtures were immunoprecipitated using anti-Flag agarose overnight. The samples were then analyzed by Western blotting.(D) Cluster alignment of CK1 consensus phosphorylation motif on IRS4 proteins. Arrowheads point to S856 and S859 of human IRS4. (E) 293T cells were co-transfected HA-CK1γ2 with Flag-tagged IRS4-WT, -S856A, -S859A, or both-S856A and -S859A mutants for 48 h, and then were analyzed by Western blotting. (F) 293T cells transfected with Flag-IRS4-WT or -S859A plasmids for 36 h were treated with 40 μg/mL cycloheximide (CHX) for the indicated periods and then analyzed by Western blotting. (G) U2OS cells stably infected with control shRNA or CK1γ2 shRNA were treated with 40 μg/mL CHX for the indicated periods and then analyzed by Western blotting. (H) His-tagged IRS4 WT protein translated in vitro was incubated with HA-CK1γ2 WT or its kinase dead (KD) form, which was IP from 293T cells using anti-HA agarose, in the presence of ATP for 2 h, as indicated. The samples were then analyzed by Western blotting.
Next, we tried to decipher the minimal region of CK1γ2 required for its interaction with IRS4. CK1γ2 contains several pivotal domains involved in its localization, substrate selectivity and kinase activity. Mutants with several N- and C-terminal CK1γ2 deletions were constructed, and were individually co-transfected with Flag-IRS4 and followed by subjection to the Co-IP assay. As shown in Figure S4, the amino acids 192-280 of CK1γ2 are its binding region for IRS4. Consistently, the fragment of 192-415 within CK1γ2 kept its interaction with IRS4 but failed to degrade IRS4 due to the lack of its kinase domain.
Subsequently, we looked into the potential phosphorylation sites of IRS4 by CK1γ2 according to the typical consensus recognition motif of CK1 kinase family that is S/Tp-X-X-S/T, where X refers to any amino acid and the potential CK1 target site is underlined. Phosphorylation by the CK1 kinase family generally requires priming phosphorylation at the -3 position relative to their phosphorylation sites. Therefore, Ser859 may be likely phosphorylated by CK1 in IRS4, whereas Ser856 may be the priming site (Figure 2D). To confirm this speculation, HA-CK1γ2 was co-transfected with IRS4-WT or its mutants, IRS4-S856A, -S859A, or -S856A/S859A, in which the serine residues at positions 856 and/or 859 were replaced by alanine. The protein level of IRS4-WT, but not of its mutants IRS4-S856A, -S859A, or -S856A/S859A, was decreased by CK1γ2 (Figure 2E). Consistently, the half-life of IRS4-WT protein was shorter than that of its IRS4-S859A mutant (Figure 2F), while CK1γ2 knockdown by siRNA resulted in a longer half-life of the IRS4 protein (Figure 2G). Besides the most effective consensus motif S/Tp-X-X-S/T, CK1 family members also prefer acidic substrates in vitro, in particular those with acidic residues in the -3 position, e.g., D-X-X-S/T. Based on this information, we looked into other potential phosphorylation sites in IRS4, and found that S68, S804, T1042, and S1159 may be likely phosphorylated by CK1γ2. As shown in Figure S5, each one of IRS4-S68A, -S804A, -T1042A, -S1159A mutants was also down-regulated by HA-CK1γ2 in cells. Collectively, our results indicate that Ser859 of IRS4 may be the predominant phosphorylation site by CK1γ2 in cells.
Figure 3
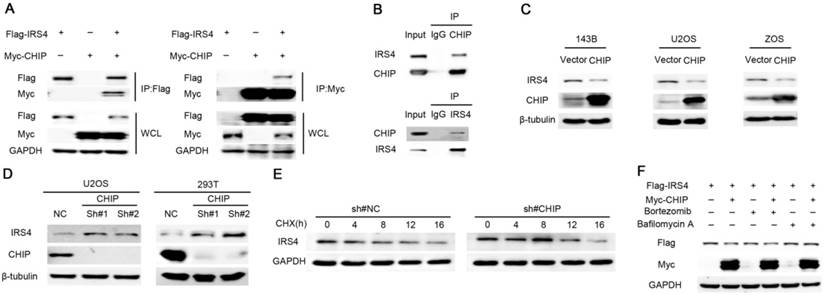
Down-regulation of IRS4 by the E3 ligase CHIP is mediated through lysosomes in cells. (A) 293T cells were co-transfected with Myc-CHIP and Flag-IRS4 for 48 h, and then lysed and immunoprecipitated using anti-Flag-agarose or anti-MYC-agarose followed by Western blotting. (B) Cell lysates from 293T cells were immunoprecipitated with anti-CHIP or anti-IRS4 and were subsequently analyzed with anti-IRS4 or anti-CHIP antibody. (C) The indicated cells were infected with the indicated plasmids and then were analyzed by Western blotting. (D) U2OS and 293T cells were infected with the indicated shRNAs and then were analyzed by Western blotting. (E) U2OS cells stably infected with control shRNA or CHIP shRNA were treated with 40 μg/mL CHX for the indicated periods and then analyzed by Western blotting.(F) 293T cells were co-transfected with Myc-CHIP and Flag-IRS4 for 48 h and then treated with Bortezomib or Bafilomycin A for 6 h. Cell lysates were then analyzed by Western blotting.
Next, we generated anti-p-IRS4-S859, a polyclonal substrate-directed phospho-specific antibody, using the DAASKPS859GEGSFSK peptide. Indeed, as shown in Figure S6A, anti-p-IRS4-S859 antibody was easily detected in the cells exogenously transfected with IRS4-WT, but not with its mutant IRS4-S859A. Similarly, using in vitro CK1γ2 kinase assay, IRS4-WT, but not its mutant IRS4-S859A, was phosphorylated by CK1γ2, but not by its KD from, in vitro (Figure 2H and Figure S6B). Unfortunately, anti-p-IRS4-S859 antibody was not detected at an endogenous level in cells, which may be due to the low affinity of the polyclonal antibody we generated (data not shown). Taken together, our results strongly suggest that the phosphorylation of IRS4 at Ser859 by CK1γ2 may play a pivotal role in the protein stability of IRS4.
E3 ligase CHIP is involved in the CK1γ2-mediated phosphorylation-dependent IRS4 degradation
During the course of seeking the E3 ligase responsible for the degradation of IRS4, we performed mass spectrometry using the IP complexes with Flag-CHIP, a U box-containing E3 ubiquitin ligase that is implicated in cancer progression [36], as CHIP not only has a pivotal role in quality control for unfolded or misfolded proteins, it also regulates protein stability by targeting them for ubiquitination and degradation [30-32]. Interestingly, IRS4 was one of the major proteins interacting with CHIP (data not shown), and the interaction between CHIP and IRS4 was confirmed at both ectopic and endogenous levels in cells (Figure 3A-B). Moreover, significant reduction and elevation of endogenous IRS4 protein levels were observed in cells transfected with ectopic CHIP and CHIP shRNAs, respectively (Figure 3C-D). Likewise, knockdown of CHIP resulted in a longer half-life of the IRS4 protein (Figure 3E), and the CHIP-mediated down-regulation of IRS4 was also blocked by BAF1, but not by Bortezomib (Figure 3F).
Figure 4
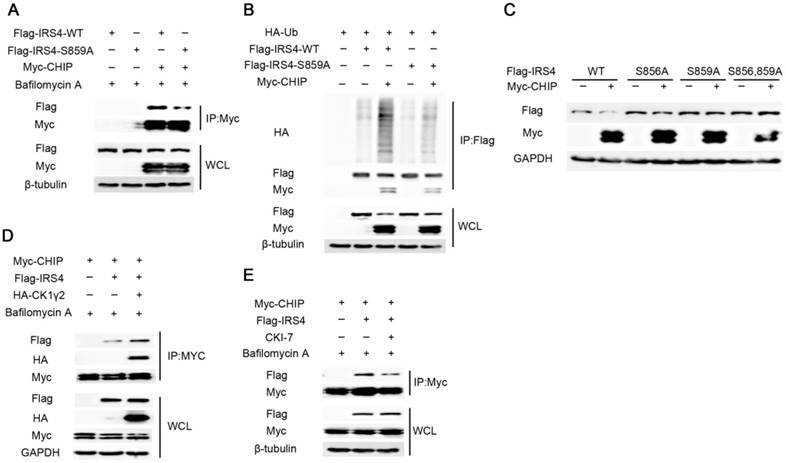
The E3 ligase CHIP is involved in the phosphorylation-dependent degradation of IRS4 by CK1γ2. (A) 293T cells were co-transfected with Flag-tagged IRS4-WT or -S859A with Myc-CHIP for 48 h. Cell lysates were subjected to immunoprecipitation with anti-MYC-agarose and then analyzed by Western blotting. Cells were treated with 200 nM Bafilomycin A for 6 h before harvesting. (B) 293T cells were co-transfected with Flag-IRS4-WT or -S859A with Myc-CHIP and HA-Ub. Cell lysates were subjected to immunoprecipitation with anti-Flag-agarose and then analyzed by Western blotting. (C) 293T cells were co-transfected Myc-CHIP with Flag-tagged IRS4-WT, -S856, or -859A for 48 h and then analyzed by Western blotting. (D) Cell lysates from 293T cells transfected with Myc-CHIP, Flag-IRS4 and HA-CK1γ2 were subjected to immunoprecipitation with anti-MYC-agarose. WCL and immunoprecipitates were then analyzed by Western blotting. Cells were treated with 200 nM Bafilomycin A for 6 h before harvesting. (E) 293T cells were transfected with Myc-CHIP and Flag-IRS4. 24 h after transfection, the cells were treated with 200 nM Bafilomycin A with or without 25 μM CK1 inhibitor CKI-7 for 8 h prior to cell collection. Cell lysates were subjected to immunoprecipitation with anti-MYC-agarose and then analyzed by western blotting.
Next, we explored whether the phosphorylation at Ser859 of IRS4 by CK1γ2 links with the IRS4 degradation by CHIP. As shown in Figure 4A-C, the mutant of IRS4-S859A lacked the ability to bind CHIP, and subsequently was neither ubiquitylated nor degraded by CHIP in cells. Furthermore, the interaction between Flag-IRS4 and Myc-CHIP was enhanced by ectopic HA-CK1γ2, while it was impaired by inhibition of CK1γ2 activity in cells (Figure 4D-E). These results strongly indicate that the phosphorylation at S859 of IRS4 by CK1γ2 enhances its interaction with CHIP and subsequently promotes its degradation.
CHIP consists of three domains: the N-terminal TPR domain that binds Hsp70 and Hsp90 [29], the C-terminal U-box domain, and the intermediate charged region. To identify domains indispensable for the IRS4-CHIP interaction, we performed the Co-IP assay using various CHIP fragments and Flag-IRS4. As shown in Figure S7A, the TPR domain of CHIP is essential and sufficient to interact with IRS4.
Many studies have revealed that CHIP targets substrates for ubiquitination and degradation in a chaperone-dependent manner. To assess the effect of chaperones on CHIP activity, we constructed CHIP (K30A), the mutant failing to interact with chaperones. As shown in Figure S7B, Myc-CHIP (K30A) lost the ability to interact with and to degrade Flag-IRS4. Histidine at position 260 in the U-BOX domain is critical for CHIP to maintain its activity. Similarly, CHIP (H260Q), the mutant failing to bind its cognate E2, and CHIP (P269A), the catalytically-inactive CHIP mutant, still interacted with IRS4 but had an impaired ability to degrade IRS4. Consistently, as shown in Figure S7C, the interaction between IRS4 and CHIP was not observed by in vitro binding assay using the purifiedIRS4 and CHIP, indicating that the interaction between CHIP and IRS4 is not direct and may need other proteins (e.g., chaperones) to form the complex, which is in accordance with the in vivo Co-IP assay. These results suggest that CHIP serves as a direct E3 ubiquitin ligase for IRS4, and that chaperones are indispensable for mediating the activity of CHIP to bind and degrade IRS4.
Figure 5
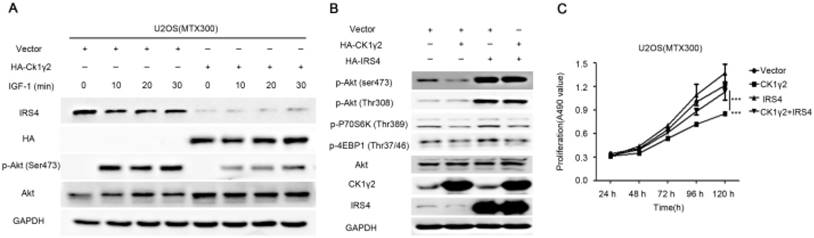
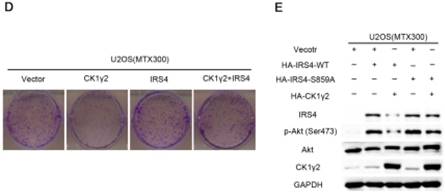
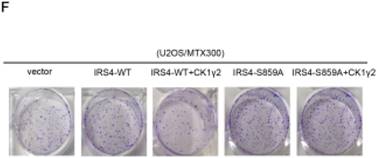
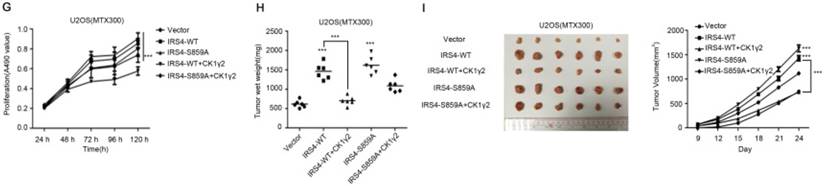
CK1γ2 suppresses cell growth through the Akt signaling pathway by degrading IRS4 in osteosarcoma cell lines in vitro and in vivo. (A) U2OS/MTX300 cells stably infected with vectors containing control and CK1γ2-WT were stimulated to 100 ng/μL IGF-1 after serum starvation for the indicated times and then analyzed by Western blotting. (B) U2OS/MTX300 cells stably expressing control vector or CK1γ2-WT were infected with vectors containing IRS4-WT and then analyzed by Western blotting. (C) U2OS/MTX300 cells were stably infected with the indicated plasmids, as shown in (B), and cell proliferation was measured using MTT assays at the indicated times (n = 6). The dots represent the mean, while the bars indicate the SEM. *, P< 0.05 and ***, P< 0.001. A two-tailed Student's t-test was used for statistical analysis. (D) U2OS/MTX300 stable cell lines generated in (B) were cultured in 6-well plates for 14 days, and representative photographs are shown. (E) U2OS/MTX300 cells that stably expressing IRS4-WT or -S859A were transfected with vectors containing control or CK1γ2-WT and then analyzed by Western blotting. (F) U2OS/MTX300 stable cell lines generated in (E) were cultured in 6-well plates for 14 days, and representative photographs are shown. (G) The proliferative capacities of U2OS/MTX300 stable cell lines generated in (E) were measured using MTT assays at the indicated times (n = 6). The dots represent the mean, while the bars indicate the SEM. *, P< 0.05 and ***, P< 0.001. A two-tailed Student's t-test was used for statistical analysis.(H-I) Nude mice were subcutaneously inoculated with the U2OS/MTX300 stable cell lines generated in (A) (2 × 106 cells per mouse) and were allowed to grow for 24 days(n=8 per group). The tumor volumes were measured every 2 days. At day 24, the xenografts were excised from the mice and weighed. Bars represent SEM.*, P<0.05 and ***, P<0.001. A two-tailed Student's t-test was used for statistical analysis.
CK1γ2 suppresses cell growth through the Akt signaling pathway by degrading IRS4
To further investigate the biological significance of the CK1γ2/IRS4 axis, we generated U2OS/MTX300 cells stably overexpressing vector, CK1γ2, IRS4-WT, IRS4-S859A, CK1γ2 and IRS4-WT, or CK1γ2 and IRS4-S859A. Overexpression of CK1γ2 dramatically inhibited p-Akt induced by IGF-1 in the absence of growth factors (Figure 5A). Under normal condition, overexpression of CK1γ2 also dramatically decreased IRS4 and p-Akt as well as its downstream targets, p-p70S6K and p-4EBP1, and this inhibition was fully reversed by stable overexpression of exogenous IRS4 (Figure 5B-D). More importantly, the CK1γ2-induced inhibition of both proliferation and clone formation were abolished by overexpression of IRS4-S859A, but not of IRS4-WT, in cells (Figure 5F-G). Moreover, U2OS/MTX300 cells stably expressing both CK1γ2 and IRS4-S859A had higher p-Akt levels and tumor weights, and larger tumor sizes than those stably expressing both CK1γ2 and IRS4-WT when they were subcutaneously inoculated into nude mice, although the growth of U2OS/MTX300 cells stably expressing IRS4-WT were similar to those stably expressing IRS4-S859A in nude mice (Figure 5E, 5H-I). Collectively, these results indicate that the inhibitory effect of CK1γ2 on cell growth may probably be through the Akt signaling pathway and is dependent on its phosphorylation of IRS4 at Ser859.
Figure 6
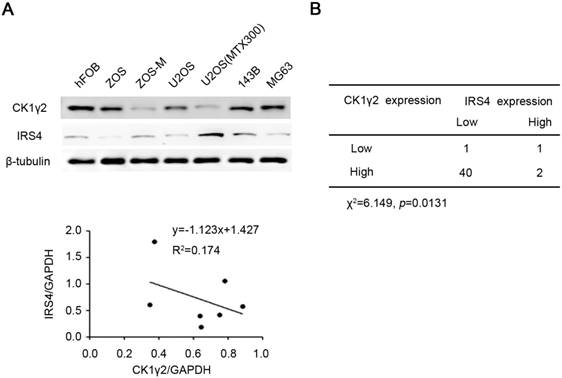
CK1γ2 is inversely correlated with IRS4 in osteosarcomacell lines and tumor samples. (A) The protein levels of both CK1γ2 and IRS4 were determined by Western blotting in seven different osteosarcoma cell lines (upper panel). Protein intensities were normalized to GAPDH levels, and the correlation between CK1γ2 and IRS4 expression levels in these seven cell lines is displayed (lower panel; R: Spearman's correlation coefficient). (B) The inverse correlation between CK1γ2 and IRS4 protein levels was observed by immunohistochemical staining of osteosarcoma specimens (P<0.05, χ2 tests).
Figure 7
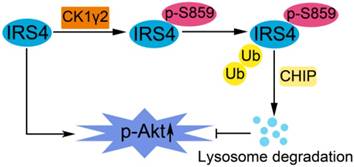
A proposed model for the regulation of IRS4 by CK1γ2. IRS4 generally activates the Akt pathway, and can be phosphorylated at Ser859 by CK1γ2, which in turn promotes the polyubiquitination and subsequent degradation of IRS4 via the CHIP-mediated ubiquitin/lysosome pathway, and consequently impairs the activation of Akt.
CK1γ2 is inversely correlated with IRS4 in osteosarcomacell lines and tumor samples
Finally, we tried to determine the clinical relevance of the CK1γ2/IRS4 axis using osteosarcoma cell lines and tumor samples. As shown in Figure 6A, there was an inverse correlation at the protein level between CK1γ2 and IRS4 in the osteosarcoma cell lines tested (R2=0.417). We further performed immunohistochemistry (IHC) for both CK1γ2 and IRS4 using clinical osteosarcoma samples, and the clinical characteristics of patients with osteosarcoma are presented in Table S1. To our surprise, CK1γ2 showed high expression in most of the tumor samples (42/44; Figure 6B), in disagreement with our speculation that CK1γ2 may be a tumor suppressor in osteosarcoma (Figure 5), whereas IRS4 showed low or no expression in the majority of samples (41/44; Figure 6B). Obviously, CK1γ2 was inversely correlated with IRS4 in osteosarcoma samples, as 40 out of 42 samples with high CK1γ2 levels had low levels of IRS4 (Figure 6B).
Discussion
In this report, as illustrated in Figure 7, we have reported that the phosphorylation of IRS4 at Ser859 by CK1γ2 enhances its polyubiquitination by CHIP and subsequent degradation by lysosomes, and negatively modulates its oncogenic function by down-regulating the Akt pathway. Our results indicate that specific CK1γ2 agonists may be potentially valuable for cancer treatment.
IRSs are crucial intermediators in insulin signaling. The significance of IRS signaling in tumor has been revealed by Chang et al. [37]. Compared with IRS1 and IRS2, IRS4 activation is independent of growth factors due to absence of the tyrosine phosphatase SHP2-binding motif [38]. IRS4 overexpression induces constitutive PI3K/AKT hyperactivation and cell proliferation under the circumstances of insulin or growth factors deficiency, which promotes mammary tumorigenesis and confers resistance to HER2-targeted therapy [39]. As a constitutive active gene, IRS4 expression is generally tightly monitored at the transcriptional level, which is different from that of IRS1 and IRS2. Indeed, IRS4 is frequently targeted in Moloneymurine leukemia virus (MuLV)-induced [40] and mouse mammary tumour virus (MMTV)-induced [41, 42] insertional mutagenesis screens, resulting in the transcriptional deregulation of IRS4, which emphasizes the significance of this gene in tumorigenesis. Chromosomal translocation events can also result in activation of IRS4 transcription in T-cell acute lymphoblastic leukemia [14, 15] and subungual exostosis [16]. IRS4 levels were much higher in HEK293 cells stably expressing the T antigen (293T) and adeno-associated virus (293AAV) than in parental HEK293 cells [38]. In this report, we have revealed that phosphorylation at Ser859 within the C-terminal region by CK1γ2 is a novel regulatory mechanism at the post-translational level to regulate the IRS4 protein stability. More importantly, using osteosarcoma cell lines, accumulation of nondegradable IRS4 by CK1γ2 triggered higher levels of p-Akt and displayed faster cell proliferation in vitro and cancer growth in nude mice, indicating that IRS4 acts as an oncogene by activating Akt in osteosarcoma. However, we found that the majority of osteosarcoma samples showed low or no expression of IRS4 (93%, 41 out of 44) in our cohort examined, indicating that IRS4 expression is primarily regulated at the transcriptional level in osteosarcoma. This is consistent with the report of breast cancer, showing that IRS4-positive samples only accounted for a small proportion in a random set of 27 human primary breast carcinomas (19%, 5 out of 27) and a series of 157 human primary breast cancers (6%, 10 out of 157) [39]. On the other hand, almost all IRS4-negative samples showed an elevated CK1γ2 level (97.5%, 40 out of 41), suggesting that both transcriptional depression and high CK1γ2 level may simultaneously present in osteosarcoma cases, although the former may be predominant. In addition, 2 out of 41 samples positive for IRS4 had high levels of CK1γ2, indicating that, besides CK1γ2, other kinases may also be involved in the turnover of IRS4 in osteosarcoma.
In most human cancers, CK1 isoforms, especially CK1α, δ, ε, may have oncogenic functions by phosphorylating p53 and mdm2, influencing β-catenin stability, inhibiting apoptosis, and by regulating microtubule stability and centrosome-specific functions [43]. The role of CK1γ in tumor development remains to be explored. Increased CK1γ3 expression has been found in renal cell carcinoma [44], and microarray database analyses from tumor cell lines [45] and tissues [46] demonstrated that there are tumor-specific differences on CK1γ2 expression. Here, we have determined that overexpression of CK1γ2 inhibited cell proliferation and growth of osteosarcoma cells by at least partially down-regulating the IRS4 protein level and subsequently impairing Akt activation in vitro and in vivo, indicating that CK1γ2 may act as a tumor suppressor in osteosarcoma by negatively regulating the IRS4/Akt pathway. However, high levels of CK1γ2 were observed by IHC in most of the osteosarcoma cases (40/44), suggesting that CK1γ2 kinase activity should be further investigated in the osteosarcoma samples.
Abbreviations
IRS4: insulin receptor substrate; CK1γ2: casein kinase 1γ2; CHIP: the carboxyl terminus of Hsc70-interacting protein; SH2: src homology 2; TPR: tetratricopeptide repeat; WT: wildtype; KD: kinase-depleted; IHC: immunohistochemistry; IP: immunoprecipitation; siRNA: small interfering RNA; CHX: cycloheximide.
Acknowledgements
This work was supported by the Science and Technology Program of Guangzhou, China (Grant No.201508020102 to T. K.); the National Key Research and Development Program of China [2016YFC0904601 and 2016YFA0500304 to T. K.], and the National Nature Science Foundation in China (NSFC) [81502512 to D. L., 81530081 and 31571395 to T. K.].
Authors' contributions
Kang T conceived the idea. Li X and Zhong L performed the experiments. Li X, Zhong L, Wang Z, Chen H, Liao D, Zhang R and Zhang H analyzed the data. Kang T, Zhang H and Li X wrote the manuscript. All co-authors have seen and approved the manuscript.
Supplementary Material
Supplementary figures.
Supplementary table S1.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Chan BT, Lee AV. Insulin receptor substrates (IRSs) and breast tumorigenesis. J Mammary Gland Biol Neoplasia. 2008;13:415-22
2. Mardilovich K, Pankratz SL, Shaw LM. Expression and function of the insulin receptor substrate proteins in cancer. Cell Commun Signal. 2009;7:14
3. Dearth RK, Cui X, Kim HJ, Hadsell DL, Lee AV. Oncogenic transformation by the signaling adaptor proteins insulin receptor substrate (IRS)-1 and IRS-2. Cell cycle. 2007;6:705-13
4. Taniguchi CM, Emanuelli B, Kahn CR. Critical nodes in signalling pathways: insights into insulin action. Nat Rev Mol Cell Biol. 2006;7:85-96
5. Saltiel AR, Kahn CR. Insulin signalling and the regulation of glucose and lipid metabolism. Nature. 2001;414:799-806
6. Lavan BE, Fantin VR, Chang ET, Lane WS, Keller SR, Lienhard GE. A novel 160-kDa phosphotyrosine protein in insulin-treated embryonic kidney cells is a new member of the insulin receptor substrate family. J Biol Chem. 1997;272:21403-7
7. Fantin VR, Sparling JD, Slot JW, Keller SR, Lienhard GE, Lavan BE. Characterization of insulin receptor substrate 4 in human embryonic kidney 293 cells. J Biol Chem. 1998;273:10726-32
8. Koval AP, Karas M, Zick Y, LeRoith D. Interplay of the proto-oncogene proteins CrkL and CrkII in insulin-like growth factor-I receptor-mediated signal transduction. J Biol Chem. 1998;273:14780-7
9. Qiu H, Zappacosta F, Su W, Annan RS, Miller WT. Interaction between Brk kinase and insulin receptor substrate-4. Oncogene. 2005;24:5656-64
10. Homma Y, Kanno S, Sasaki K, Nishita M, Yasui A, Asano T. et al. Insulin receptor substrate-4 binds to Slingshot-1 phosphatase and promotes cofilin dephosphorylation. J Biol Chem. 2014;289:26302-13
11. Shimwell NJ, Martin A, Bruton RK, Blackford AN, Sedgwick GG, Gallimore PH. et al. Adenovirus 5 E1A is responsible for increased expression of insulin receptor substrate 4 in established adenovirus 5-transformed cell lines and interacts with IRS components activating the PI3 kinase/Akt signalling pathway. Oncogene. 2009;28:686-97
12. Cantarini MC, de la Monte SM, Pang M, Tong M, D'Errico A, Trevisani F. et al. Aspartyl-asparagyl beta hydroxylase over-expression in human hepatoma is linked to activation of insulin-like growth factor and notch signaling mechanisms. Hepatology. 2006;44:446-57
13. Cuevas EP, Escribano O, Chiloeches A, Ramirez Rubio S, Roman ID, Fernandez-Moreno MD. et al. Role of insulin receptor substrate-4 in IGF-I-stimulated HEPG2 proliferation. J Hepatol. 2007;46:1089-98
14. Karrman K, Isaksson M, Paulsson K, Johansson B. The insulin receptor substrate 4 gene (IRS4) is mutated in paediatric T-cell acute lymphoblastic leukaemia. Br J Haematol. 2011;155:516-9
15. Karrman K, Kjeldsen E, Lassen C, Isaksson M, Davidsson J, Andersson A. et al. The t(X;7)(q22;q34) in paediatric T-cell acute lymphoblastic leukaemia results in overexpression of the insulin receptor substrate 4 gene through illegitimate recombination with the T-cell receptor beta locus. Br J Haematol. 2009;144:546-51
16. Mertens F, Moller E, Mandahl N, Picci P, Perez-Atayde AR, Samson I. et al. The t(X;6) in subungual exostosis results in transcriptional deregulation of the gene for insulin receptor substrate 4. Int J Cancer. 2011;128:487-91
17. Shull AY, Latham-Schwark A, Ramasamy P, Leskoske K, Oroian D, Birtwistle MR. et al. Novel somatic mutations to PI3K pathway genes in metastatic melanoma. PLoS One. 2012;7:e43369
18. Sesti G, Federici M, Hribal ML, Lauro D, Sbraccia P, Lauro R. Defects of the insulin receptor substrate (IRS) system in human metabolic disorders. FASEB J. 2001;15:2099-111
19. Fantin VR, Wang Q, Lienhard GE, Keller SR. Mice lacking insulin receptor substrate 4 exhibit mild defects in growth, reproduction, and glucose homeostasis. Am J Physiol Endocrinol Metab. 2000;278:E127-33
20. Ullrich A, Schlessinger J. Signal transduction by receptors with tyrosine kinase activity. Cell. 1990;61:203-12
21. Gross SD, Anderson RA. Casein kinase I: spatial organization and positioning of a multifunctional protein kinase family. Cell Signal. 1998;10:699-711
22. Cheong JK, Virshup DM. Casein kinase 1: Complexity in the family. Int J Biochem Cell Biol. 2011;43:465-9
23. Knippschild U, Gocht A, Wolff S, Huber N, Lohler J, Stoter M. The casein kinase 1 family: participation in multiple cellular processes in eukaryotes. Cell Signal. 2005;17:675-89
24. Vielhaber E, Virshup DM. Casein kinase I: from obscurity to center stage. IUBMB life. 2001;51:73-8
25. Guo X, Waddell DS, Wang W, Wang Z, Liberati NT, Yong S. et al. Ligand-dependent ubiquitination of Smad3 is regulated by casein kinase 1 gamma 2, an inhibitor of TGF-beta signaling. Oncogene. 2008;27:7235-47
26. Davidson G, Wu W, Shen J, Bilic J, Fenger U, Stannek P. et al. Casein kinase 1 gamma couples Wnt receptor activation to cytoplasmic signal transduction. Nature. 2005;438:867-72
27. Mishra SK, Yang Z, Mazumdar A, Talukder AH, Larose L, Kumar R. Metastatic tumor antigen 1 short form (MTA1s) associates with casein kinase I-gamma2, an estrogen-responsive kinase. Oncogene. 2004;23:4422-9
28. Bagheri-Yarmand R, Talukder AH, Wang RA, Vadlamudi RK, Kumar R. Metastasis-associated protein 1 deregulation causes inappropriate mammary gland development and tumorigenesis. Development. 2004;131:3469-79
29. Ballinger CA, Connell P, Wu Y, Hu Z, Thompson LJ, Yin LY. et al. Identification of CHIP, a novel tetratricopeptide repeat-containing protein that interacts with heat shock proteins and negatively regulates chaperone functions. Mol Cell Biol. 1999;19:4535-45
30. McDonough H, Patterson C. CHIP: a link between the chaperone and proteasome systems. Cell Stress Chaperones. 2003;8:303-8
31. Murata S, Chiba T, Tanaka K. CHIP: a quality-control E3 ligase collaborating with molecular chaperones. Int J Biochem Cell Biol. 2003;35:572-8
32. Esser C, Alberti S, Hohfeld J. Cooperation of molecular chaperones with the ubiquitin/proteasome system. Biochim Biophys Acta. 2004;1695:171-88
33. Kao SH, Wang WL, Chen CY, Chang YL, Wu YY, Wang YT. et al. GSK3beta controls epithelial-mesenchymal transition and tumor metastasis by CHIP-mediated degradation of Slug. Oncogene. 2014;33:3172-82
34. Zou CY, Wang J, Shen JN, Huang G, Jin S, Yin JQ. et al. Establishment and characteristics of two syngeneic human osteosarcoma cell lines from primary tumor and skip metastases. Acta Pharmacol Sin. 2008;29:325-32
35. Yin JQ, Shen JN, Su WW, Wang J, Huang G, Jin S. et al. Bufalin induces apoptosis in human osteosarcoma U-2OS and U-2OS methotrexate300-resistant cell lines. Acta Pharmacol Sin. 2007;28:712-20
36. Chen C, Seth AK, Aplin AE. Genetic and expression aberrations of E3 ubiquitin ligases in human breast cancer. Mol Cancer Res. 2006;4:695-707
37. Chang Q, Li Y, White MF, Fletcher JA, Xiao S. Constitutive activation of insulin receptor substrate 1 is a frequent event in human tumors: therapeutic implications. Cancer Res. 2002;62:6035-8
38. Hoxhaj G, Dissanayake K, MacKintosh C. Effect of IRS4 levels on PI 3-kinase signalling. PLoS One. 2013;8:e73327
39. Ikink GJ, Boer M, Bakker ER, Hilkens J. IRS4 induces mammary tumorigenesis and confers resistance to HER2-targeted therapy through constitutive PI3K/AKT-pathway hyperactivation. Nat Commun. 2016;7:13567
40. Uren AG, Kool J, Matentzoglu K, de Ridder J, Mattison J, van Uitert M. et al. Large-scale mutagenesis in p19(ARF)- and p53-deficient mice identifies cancer genes and their collaborative networks. Cell. 2008;133:727-41
41. Klijn C, Koudijs MJ, Kool J, ten Hoeve J, Boer M, de Moes J. et al. Analysis of tumor heterogeneity and cancer gene networks using deep sequencing of MMTV-induced mouse mammary tumors. PLoS One. 2013;8:e62113
42. Theodorou V, Kimm MA, Boer M, Wessels L, Theelen W, Jonkers J. et al. MMTV insertional mutagenesis identifies genes, gene families and pathways involved in mammary cancer. Nat Genet. 2007;39:759-69
43. Schittek B, Sinnberg T. Biological functions of casein kinase 1 isoforms and putative roles in tumorigenesis. Mol Cancer. 2014;13:231
44. Masuda K, Ono M, Okamoto M, Morikawa W, Otsubo M, Migita T. et al. Downregulation of Cap43 gene by von Hippel-Lindau tumor suppressor protein in human renal cancer cells. Int J Cancer. 2003;105:803-10
45. Reinhold WC, Sunshine M, Liu H, Varma S, Kohn KW, Morris J. et al. CellMiner: a web-based suite of genomic and pharmacologic tools to explore transcript and drug patterns in the NCI-60 cell line set. Cancer Res. 2012;72:3499-511
46. Ramaswamy S, Tamayo P, Rifkin R, Mukherjee S, Yeang CH, Angelo M. et al. Multiclass cancer diagnosis using tumor gene expression signatures. Proc Natl Acad Sci U S A. 2001;98:15149-54
Author contact
![]() Corresponding authors: Dr. Tiebang Kang, Tel: 86-20-8734-3183; Fax: 86-20-8734-3170; E-mail: zhhyusysu.edu.cn or kangtborg.cn
Corresponding authors: Dr. Tiebang Kang, Tel: 86-20-8734-3183; Fax: 86-20-8734-3170; E-mail: zhhyusysu.edu.cn or kangtborg.cn
Citation styles
APA
Li, X., Zhong, L., Wang, Z., Chen, H., Liao, D., Zhang, R., Zhang, H., Kang, T. (2018). Phosphorylation of IRS4 by CK1γ2 promotes its degradation by CHIP through the ubiquitin/lysosome pathway. Theranostics, 8(13), 3643-3653. https://doi.org/10.7150/thno.26021.
ACS
Li, X.; Zhong, L.; Wang, Z.; Chen, H.; Liao, D.; Zhang, R.; Zhang, H.; Kang, T. Phosphorylation of IRS4 by CK1γ2 promotes its degradation by CHIP through the ubiquitin/lysosome pathway. Theranostics 2018, 8 (13), 3643-3653. DOI: 10.7150/thno.26021.
NLM
Li X, Zhong L, Wang Z, Chen H, Liao D, Zhang R, Zhang H, Kang T. Phosphorylation of IRS4 by CK1γ2 promotes its degradation by CHIP through the ubiquitin/lysosome pathway. Theranostics 2018; 8(13):3643-3653. doi:10.7150/thno.26021. https://www.thno.org/v08p3643.htm
CSE
Li X, Zhong L, Wang Z, Chen H, Liao D, Zhang R, Zhang H, Kang T. 2018. Phosphorylation of IRS4 by CK1γ2 promotes its degradation by CHIP through the ubiquitin/lysosome pathway. Theranostics. 8(13):3643-3653.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.